

Abstract
Proteases and their natural protein inhibitors are among the most intensively studied protein–protein complexes. There are about 30 structurally distinct inhibitor families that are able to block serine, cysteine, metallo- and aspartyl proteases. The mechanisms of inhibition can be related to the catalytic mechanism of protease action or include a mechanism-unrelated steric blockage of the active site or its neighborhood. The structural elements that are responsible for the inhibition most often include the N- or the C-terminus or exposed loop(s) either separately or in combination of several such elements. During complex formation, no major conformational changes are usually observed, but sometimes structural transitions of the inhibitor and enzyme occur. In many cases, convergent evolution, with respect to the inhibitors' parts that are responsible for the inhibition, can be inferred from comparisons of their structures or sequences, strongly suggesting that there are only limited ways to inhibit proteases by proteins.
Keywords: inhibition, protease, protein inhibitor, protein–protein recognition
Introduction
A strong inhibition of an active protease by a protein appears to be a paradox. Nevertheless, proteinaceous inhibitors of proteolytic enzymes comprise the largest and structurally most diverse group of naturally occurring enzyme inhibitors. A comprehensive list of 48 inhibitor families has been recently published (Rawlings et al, 2004) and is available at http://merops.sanger.ac.uk. Inhibitor structures, modes of inhibition, kinetic and thermodynamic parameters, and the nature of the enzyme–inhibitor complexes are surprisingly diversified. On the other hand, in many cases, convergence of structure and/or function can be observed, pointing to the fact that there is a limited number of inhibition modes.
Some protein folds support structural elements that are responsible for unrelated types of inhibition. For example, the β-barrel fold is involved in inhibition of cysteine, serine and metalloproteases (Rzychon et al, 2003), knottins are responsible for inhibition of serine and metalloproteases (Bode et al, 1989), bovine pancreatic trypsin inhibitor (BPTI)-like proteins can inhibit serine proteases through two unrelated mechanisms (Wei et al, 1998) and proteins belonging to Bowman–Birk family can inhibit serine or cysteine proteases (Hatano et al, 2002). The above-mentioned folds are rigid and stable, which might reflect the fact that high resistance to proteolysis of the inhibitor framework is useful in supporting the inhibitory epitopes.
Inhibition of the concave protease active site is usually achieved by docking of an exposed structural element of the inhibitor, like a single loop or a protein terminus, either independently or in combination of two or more such elements. Since inhibitors are proteins, inhibition in many cases is linked to the mechanism of peptide bond cleavage observed in protein substrates. Besides the protein inhibitors discussed in this review, proteases can also be effectively inhibited by prosegments that catalyze folding of mature enzymes (Khan and James, 1998). The inhibitors are usually specific toward one of four mechanistic classes of proteases (serine, cysteine, aspartic or metalloproteases), with protein inhibitors of threonine and glutamyl proteases (Fujinaga et al, 2004) remaining yet to be discovered (Table I). In this review, we discuss well-documented mechanisms of inhibition, supported by the spatial structures of respective complexes. We focus on those features of inhibitors that allow them to escape regular proteolysis.
Table 1.
Structural and mechanistic features of protein inhibitors of proteases
| Protease type | Inhibitor | Representatives | Major features of inhibition | Inhibitor size |
|---|---|---|---|---|
| Serine | Canonical inhibitors | BPTI, OMTKY3, eglin c, CMTI I | Often extremely tight and fast, noncovalent interaction resembling Michaelis complex, direct blockage of the active site, no conformational changes, antiparallel β-sheet between enzyme and inhibitor, similar mode of interaction through canonical protease binding loop in 18 different inhibitor scaffolds, moderate size of interface, utmost role of P1 residue, additive effects on association energy | 3–21 kDa per domain |
| Noncanonical inhibitors | Hirudin, TAP, ornithodorin | Extremely strong, fast and specific interaction so far known for factor Xa and thrombin only, two-step kinetics, inhibition of the active site through inhibitor's N-terminus forming parallel β-sheet with enzyme active site, large interface composed of two interaction areas | 6–8 kDa per domain | |
| Serpins | α-1-Antitrypsin, antithrombin | Irreversible covalent acyl–enzyme complex, mouse-trap mechanism, huge conformational changes in inhibitor, important role of P1 position, suicide inhibition, disruption of protease active site | 45–55 kDa | |
| Cysteine | Cystatins | Chicken cystatin, cystatin C, stefin B, kininogen | Extremely tight but not specific, reversible and noncovalent inhibition, interaction through a wedge formed by two hairpin loops and N-terminus, catalytic Cys25 accessible in complex, important interactions through P2 position | 11–13 kDa, up to 60–120 kDa (kininogen) |
| Thyropins | p41, equistatin | Very tight inhibition, mechanism similar to cystatins but often more specific, unusual inhibition of cysteine and aspartic proteases at different domains of equistatin | 7 kDa per domain | |
| Bromelain inhibitors | BI-VI | Moderately strong inhibition at low pH and no inhibition at neutral pH, structural resemblance to canonical inhibitors of Bowman–Birk family | 6–8 kDa | |
| Staphostatins | Staphostatin B | Moderately strong inhibition, inhibition mechanism resembling that of canonical inhibitors, inhibitor structure different from cystatins, unusual conformation of conserved Gly at P1, substrate-like orientation of inhibitor, large area of interaction, importance of P1′ position | 11 kDa | |
| IAP | XIAP, cIAP1 | Highly specific inhibition, reversible tight binding kinetics, inhibition also through interdomain flexible linker region as nonproductive binding in orientation opposite to that of substrates | 9 kDa per BIR domain | |
| CrmA, PI-9 | Highly specific inhibition, similar to serpin mechanism-based inactivation | 38 kDa | ||
| p35 | Nonspecific inhibition, irreversible acyl-enzyme, distortion of active site, p35 N-terminus shields catalytic Cys360 from water molecules, gross conformational changes in inhibitor | 35 kDa | ||
| Metallo | PCI, LCI | Tight enzyme–product complex, inhibition through C-terminal segment, key role of Val38 (P1), no conformational changes in inhibitor upon complexation | 4 kDa | |
| SMPI | Moderately specific inhibitor, inhibition mechanism resembling standard mechanism of canonical inhibitors of serine proteases, temporary inhibition, rigid protease binding loop | 11 kDa | ||
| P. aeruginosa inhibitor, E. chrysanthemi inhibitor | Both tight and weak inhibition observed, major interactions through five N-terminal residues, N-terminal amino group forms a coordinative bond to catalytic Zn, in analogy to TIMPs | 15 kDa | ||
| TIMP1–4 | Tight but not highly specific noncovalent interaction, N-terminus and five inhibitor loops form wedge contacting the active site, bidental coordination of catalytic Zn through N-terminus, major interactions through P1′ residue, moderate conformational changes in inhibitor upon complexation | 20–22 kDa | ||
| Aspartic | IA3 | Strong, highly specific and fully unique type of inhibition, fully unfolded in free state, forms long helix in the complex comprising only N-terminal half of inhibitor, noncovalent complex | 8 kDa | |
| PI-3 | Strong but not highly specific, antiparallel β-sheet formed between enzyme and inhibitor, no conformational changes | 17 kDa | ||
| BPTI: bovine pancreatic trypsin inhibitor; OMTKY3: turkey ovomucoid third domain; CMTI I: Cucurbita maxima trypsin inhibitor 1; TAP: tick anticoagulant peptide; BI-VI, bromelain inhibitor VI from pineapple; IAP: inhibitor of apoptosis; XIAP: X-linked IAP; cIAP1: cellular inhibitor of apoptosis protein 1; BIR: baculoviral IAP repeat; CrmA: cytokine response modifier A; PI-9: protease inhibitor 9; PCI: potato carboxypeptidase inhibitor; LCI: leech carboxypeptidase inhibitor; SMPI: Streptomyces proteinaceous metalloprotease inhibitor; TIMP: tissue inhibitors of metalloproteases; IA3: inhibitor of aspartic protease from yeast; PI-3, Ascaris suum pepsin inhibitor 3. | ||||
Mechanism-based inhibitors
Inhibition through tight Michaelis complex
A noncovalent protease–inhibitor complex, highly similar to the enzyme–substrate interaction, is a very common way of inhibition. This type of protease inactivation arose many times during the evolution of 18 families of serine protease canonical inhibitors, but there is evidence that it is also utilized to inhibit cysteine and metalloproteases (Table I).
The most intensively studied example of substrate-like interaction is canonical inhibitors of serine proteases (Figure 1A(1)). The majority of the inhibitors are rigid, stable, purely β-sheet or mixed α/β proteins, but they can also be α-helical or irregular proteins rich in disulfide bonds. It is intriguing that in all these families, the loops are of a very similar, canonical conformation, despite completely different amino-acid sequences of the P3–P3′ segments among different families and also between individual members of a family (Bode and Huber, 1992).
Figure 1.
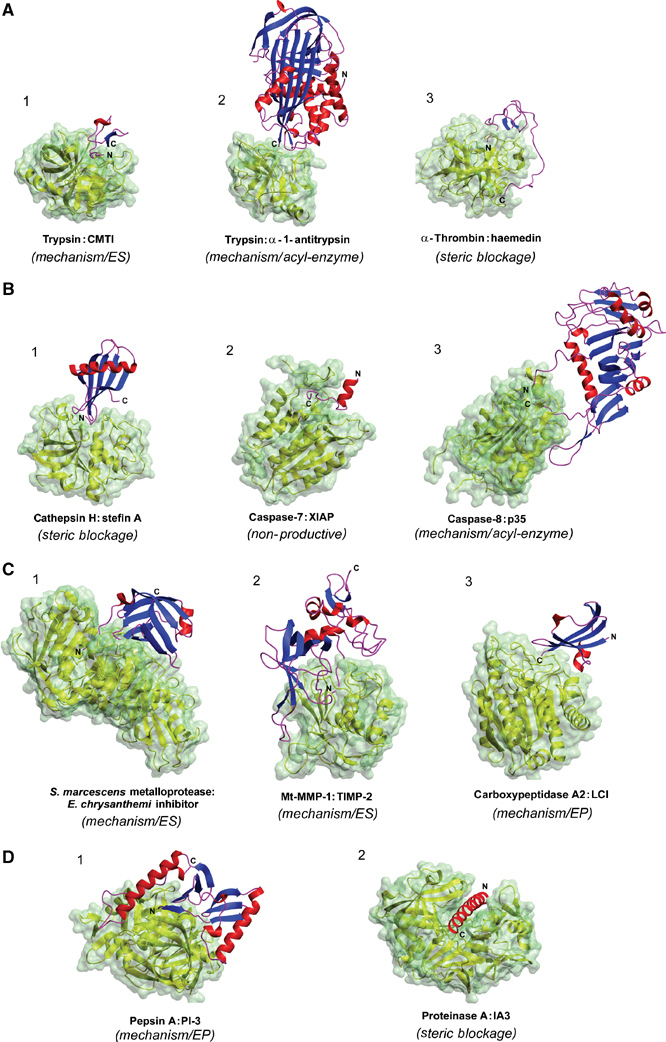
Examples of protease–inhibitor complexes. (A) Serine protease–inhibitor complexes: (1) canonical: trypsin–CMTI (PDB: 1PPE), (2) serpin: trypsin–α-1-antitrypsin (1EZX), (3) noncanonical: α-thrombin–haemedin (1E0F). (B) Cysteine protease–inhibitor complexes: (1) cathepsin H–stefin A (1NB5), (2) caspase-7–XIAP (1I51), (3) caspase-8–p35 (1I4E). (C) Metalloprotease–inhibitor complexes: (1) Serratia marcescens metalloprotease–Erwinia chrysanthemi inhibitor (1SMP), (2) membrane-type MMP-1–TIMP-2 (1BQQ), (3) human carboxypeptidase A2–LCI (1DTD). (D) Aspartic protease–inhibitor complexes: (1) porcine pepsin–PI-3 (1F34), (2) proteinase A–IA3 (1DPJ). Three-dimensional structures of proteases are represented by yellow ribbons with water accessibility surface colored in pale green. Secondary structure elements of inhibitors are marked in blue (β-sheets), red (α-helices) and magenta (coils). The inhibition types of particular enzyme:inhibitor pairs are given in parentheses.
The mode of the canonical inhibitor–serine protease interaction is presumed to be adopted also by a productively bound protein substrate. The loop is usually of higher dynamics in the uncomplexed state and becomes significantly rigidified upon complex formation with the protease. Several intermolecular hydrogen bonds of constant pattern are formed between the canonical loop and the enzyme active site, including a short antiparallel β-sheet between P3–P1 and the 214–216 segment (in the chymotrypsin family), two hydrogen bonds between the carbonyl oxygen of P1 and the amides of the oxyanion binding hole and a short contact between the P1 carbonyl carbon and the catalytic serine. In the crystal structures of all enzyme–inhibitor complexes, the latter bond is shorter than the van der Waals distance, however, not short enough to form a tetrahedral adduct.
The conserved mode of recognition between the protease binding loop and the enzyme active site allows many different serine proteases (belonging both to the chymotrypsin and subtilisin families) of different specificities to be inhibited by turkey ovomucoid third domain (Ardelt and Laskowski, 1985). This is true also for other inhibitors. Eglin c inhibits 14 serine proteases with the association constants greater than 108 M−1 (Laskowski and Qasim, 2000). A huge, billion-fold, difference between the association constants exists for the interaction between 13 P1 mutants of BPTI and trypsin, and again the crystal structures of the respective complexes show an identical mode of recognition (Helland et al, 1999, 2003). The complementary character of the convex binding loop of the inhibitor and the concave active site of the enzyme ensures a high level of predicitivity of amino-acid substitutions within the amino-acid sequence of a particular inhibitor for the association energy with the enzyme (Lu et al, 2001). Further, quantitatively similar effects of amino-acid substitutions at the same position of the canonical loop in representatives of different inhibitor families on the association energy with a single protease were found (Qasim et al, 1997; Krowarsch et al, 1999).
Although canonical inhibitors form stable, crystallizable complexes with cognate enzymes, the P1–P1′ peptide bond (the reactive site), located in the center of the canonical loop, can be selectively hydrolyzed by the enzyme. The conformation of the cleaved inhibitor is very similar to the intact form, except for local structural changes near the P1–P1′ peptide bond (Musil et al, 1991). Upon mixing of the cleaved form with the enzyme, resynthesis of the reactive site peptide bond occurs, leading to a complex that is identical to that formed between an intact inhibitor and the enzyme (Ardelt and Laskowski, 1985; Helland et al, 1999). However, the cleavage and resynthesis reactions are surprisingly slow and the hydrolysis equilibrium constant is usually not far away from unity at neutral pH. This means that intact and cleaved inhibitors are of similar thermodynamic stability. Interestingly, while formation of an acyl-enzyme between the enzyme and the inhibitor proceeds fast, a delay occurs at the subsequent deacylation step due to the tightly bound amino (leaving) group, oriented such as to inhibit the deacylation step and favoring the resynthesis of the P1–P1′ peptide bond (Radisky and Koshland, 2002).
A mechanism based on hydrolysis/resynthesis of a single peptide bond, the dominant feature of canonical inhibitors, has been also proposed for Streptomyces metalloproteinase inhibitor (SMPI) (Tate et al, 1998). Its reactive site loop is rigid in the free inhibitor and, in analogy to canonical inhibitors, highly complementary to the active site of the enzyme. The exact nature of the complex, however, must await crystal structure determination of the enzyme–inhibitor complex.
Tissue inhibitors of metalloproteases (TIMPs) also interact with their target matrix metalloproteases (MMPs) in a substrate-like manner. They avoid cleavage through an unrelated mechanism based on the displacement of the catalytic water molecule from the enzyme active site. These inhibitors are built of two domains: an N-terminal domain, composed of about 125 amino-acid residues, and a more flexible C-terminal domain of about 65 residues, each domain stabilized by three disulfide bonds and together forming an elongated edge (Gomis-Ruth et al, 1997). This edge, formed by the N-terminus and four loop segments, binds to the active site cleft of the cognate MMP (Fernandez-Catalan et al, 1998) (Figure 1C(2)). About 75% of the contacts are made by the N-terminus and the so-called connector loop. Isolated N-terminal domains are stable and block the activity of various MMPs (Murphy et al, 1991). The N-terminus (Cys1-Pro5) binds to the MMP active site in a substrate-like orientation. The most essential feature of the inhibitory complex is a bidentate coordination of the catalytic Zn2+ by the α-amino and carbonyl groups of Cys1, and interaction of the same amino group and the Thr/Ser2 side chain with the catalytic glutamate. These interactions lead to the displacement of the catalytic water molecule. Thus, although the N-terminal residues 1–4 bind in a productive, substrate-like fashion, the cleavage cannot occur due to the inhibitor-induced distortion of the catalytic apparatus.
The strong interactions between the serralysin group of metalloproteases and their bacterial inhibitors from Pseudomonas aeruginosa (Hege et al, 2001) and Erwinia chrysanthemi (Baumann et al, 1995) resemble those described for the TIMP–MMP complexes (Figure 1C(1)). Although TIMPs and the two bacterial inhibitors are structurally unrelated, both groups of inhibitors form similar substrate-like interactions and coordinative bonds to the catalytic zinc utilizing the N-terminal residue. The length of the N-terminal trunk allows precise and intimate interaction between the N-terminal residue and the zinc ion. A closer approach is prevented by the body of the inhibitors forming an eight-stranded antiparallel β-barrel (Hege et al, 2001).
Enzyme–product complex
Inhibition through the formation of a stable enzyme-product complex is known for potato carboxypeptidase A inhibitor (CPI) (Rees and Lipscomb, 1982) and leech carboxypeptidase inhibitor (LCI) (Reverter et al, 2000) (Figure 1C(3)). Both inhibitors are structurally unrelated (39 versus 66 amino-acid residues, respectively), but their C-terminal tails, which are flexible in free inhibitors (Reverter et al, 2000), are similar (-Pro-Tyr-Val-Gly/Glu), suggesting convergent evolution. Indeed, these inhibitors recognize a carboxypeptidase in a similar way, with the C-terminal residue (Gly and Glu, respectively) cleaved off, but still present in either complex. The strength of both complexes is similar, independently of the presence of the C-terminal residue, suggesting that the inhibitors bind in a similar manner at the pre- (substrate-like) and postcleavage (product-like) stages. In the crystal structures of both complexes, the crucial interaction is made by a Val residue (present at P1′), including the coordination of the catalytic zinc by its carboxylate. There is some analogy to canonical inhibitors of serine proteases: in both types of inhibition, slow cleavage of the P1–P1′ peptide bond occurs, the product of the hydrolytic reaction is active as inhibitor and the amine group dissociation is blocked.
The pepsin inhibitor 3 (PI-3) from the intestinal parasite Ascaris suum is also able to form a stable enzyme–product type of complex. The inhibitor is unspecific as it forms complexes with a number of aspartyl proteases. PI-3 is built of two subdomains, each composed of antiparallel β-sheets flanked by an α-helix (Ng et al, 2000). The N-terminal β-strand forms an antiparallel β-sheet with one strand of the protease active site flap. This leads to the formation of an extensive eight-stranded β-sheet spanning both proteins (Figure 1D(1)). The N-terminal β-strand of the inhibitor is essential for inhibition: Gln1 is positioned near both catalytic aspartates, and it is likely that its α-amino group is close enough to one of the catalytic aspartates to exclude the catalytic water molecule. Gln1 together with Phe2 and Leu3 occupies the S1′–S3′ pockets of the enzyme. Since an additional Thr residue is present at the PI-3 N-terminus prior to incubation with pepsin, it can be inferred that the complex is of the enzyme–product type and the N-terminal threonine is cutoff during complex formation.
Acyl-enzyme intermediate
Compared with the thermodynamically stable Michaelis-like complex, inhibition through formation of an acyl-enzyme is a dynamic and irreversible process leading to a kinetically trapped intermediate. This type of inhibition can only occur for serine and cysteine proteases, which hydrolyze the peptide bond through an acyl- and thioacyl-enzyme intermediate, respectively. Interestingly, cross-class reactivity, covering both classes of proteases, has been demonstrated in a number of cases (Gettins, 2002; Stennicke et al, 2002). Inhibitors that can form a stable acyl-enzyme complex are large, single-domain proteins that undergo a highly cooperative transition destroying the active site of the protease before deacylation can take place. In this group of inhibitors, the reactive center loop (RCL) is flexible, exposed and long to make it a good substrate.
The classic examples of inhibition through this mechanism are serpins, 45–55 kDa proteins that share about 35% sequence homology and a remarkably common fold composed of three β-sheets and eight or nine α-helices forming a single domain (Gettins, 2002). Unlike typical proteins, serpins are metastable in their active state and undergo a huge structural transition to a stable conformation upon complex formation with a target protease. The initial recognition of the exposed RCL is similar as in the case of canonical inhibitors, and the protease attacks the P1–P1′ bond as a potential substrate. At this stage, there are no conformational changes either in the protease or in the serpin, and the conformation of the RCL is canonical (Ye et al, 2001). The subsequent attack by the catalytic Ser residue on the serpin ‘bait' P1–P1′ peptide bond leads to an acyl-enzyme intermediate. In a sharp contrast to canonical inhibitors, the newly formed amino group now easily dissociates from the active site and the fully unconstrained RCL is inserted into β-sheet A, flipping the covalently attached protease to the opposite side of the serpin, over 70 Å from the initial recognition site (Huntington et al, 2000) (Figure 1A(2)). In this covalent and irreversible complex, the acyl linkage between the two macromolecules does not affect serpin, but over one-third of the enzyme molecule is severely deformed, including plucking of its catalytic serine and breakage of the interactions maintaining the active site conformation. Thus, the serpin inhibitory mechanism fully depends on rapid main β-sheet A expansion and subsequent incorporation of the RCL before the hydrolysis of the acyl-enzyme can occur. Biochemical (Huntington et al, 1997) and structural (Aertgeerts et al, 1995) studies have shown that the rate of loop insertion is critical for inhibition. There are many examples of serpins that use overlapping reactive centers to inhibit two or more serine proteases (Potempa et al, 1988; Irving et al, 2002). Accordingly, the length of the inserted RCL can vary depending on the inhibited protease. Even more surprisingly, a single serpin can show dual mechanistic class reactivity encompassing serine and cysteine proteases, using different reactive centers (Al-Khunaizi et al, 2002). This is in a sharp contrast to the constant location of the reactive site of canonical inhibitors, which is precisely defined by the shape and constant length of the canonical loop and always serves as a single recognition site (Ardelt and Laskowski, 1985).
In many aspects, canonical inhibitors and serpins show opposite features. In the former group, the cleavage of the reactive site does not produce conformational changes due to constraints from the neighboring structural elements, like disulfide bond(s), proline(s), hydrogen bonds and/or large hydrophobic side chains that all stiffen the P1–P1′ reactive site. The binding loop is relatively short and enzymatic religation of the newly released amino and carboxyl groups is kinetically favorable. The free energies of intact and cleaved form are comparable. In contrast, the RCL loop of serpins is poorly constrained and long, between 14 and 19 amino acids (Gettins, 2002). The RCL length strongly affects the protease–serpin complex stability: if too long, less plucking stress is applied on the protease active site, but if too short, steric conflicts between the enzyme and β-sheet A effectively block the loop insertion. The sequence restriction of the loop is low in the P7–P1 region, but it is pronounced in the hinge region responsible for the effectiveness of the insertion. Further, proteolysis of a single peptide bond within the RCL loop of serpin leads to a dramatic structural rearrangement and an enormous increase of over 30 kcal mol−1 in the stability of the cleaved form (Im et al, 2000).
The mechanism of inhibition of caspases by the baculovirus p35 protein is similar to that of serpins, as it represents mechanism-based inactivation through the formation of a covalent thiol ester (Xu et al, 2001). In detail, however, the molecular rearrangement of the inhibitor upon cleavage of the peptide bond is much less profound (Figure 1B(3)). The cleavage of the P1–P1′ peptide bond located in the exposed loop allows the amino segment of the cleaved loop to move and bury. As a result, the N-terminal strand of p35 that contains the Cys2 residue is released. In the complex, Cys2 is inserted into the active site, sterically blocking the access of the hydrolytic water molecule. However, compared to serpins, the conformational transition of the inhibitor is less dramatic and the protease structure is not affected.
Inhibition through a nonproductive binding
Nonproductive binding appears to be a relatively simple mechanism of escaping proteolysis. Interestingly, it was developed to control precisely apoptosis (Stennicke et al, 2002). Inhibitors of apoptosis (IAPs) are widely distributed proteins built of tandemly arranged BIR (baculoviral IAP repeat) domains that are able to inhibit specifically caspases. Surprisingly, a direct role in the inhibition is played by a flexible linker connecting the BIR domains, which serves to block the active site of caspase-3 and -7 in the case of the archetypal XIAP (X-linked IAP) inhibitor (Chai et al, 2001) (Figure 1B(2)). Compared with a regular substrate or covalent peptide inhibitor, the linker binds to the active site in a reverse orientation. Again, a case of convergence of inhibition mechanisms can be found, as in the structures of all zymogens of papain-like cysteine proteases, the prosegment covers the active site in a nonproductive orientation (Khan and James, 1998). In the caspase–XIAP complex, the S1 pocket, which determines the selectivity for caspase substrates, remains empty and the crucial interactions are mediated through the P4 residue. The binding is nonproductive, any XIAP carbonyl carbon approaches the catalytic cysteine and the interactions within the oxyanion binding hole are absent. Although the role of BIR domains appears to be passive, the isolated linker is not able to inhibit caspases (Chai et al, 2001).
Steric blockage of the protease active site
In several evolutionarily unrelated cases, the polypeptide chain of the inhibitor is able to block the active site of the protease so that neither of its peptide bonds is in direct contact with the catalytic groups. A classic example is inhibition of papain-like cysteine proteases by inhibitors belonging to the superfamily of cystatins (comprising cystatins, stefins and kininogens) (Turk et al, 1997). However, thyroglobulin type 1 (Guncar et al, 1999) and probably also other inhibitors (Rigden et al, 2002) share clear similarities with cystatins in their mode of interaction with the cysteine enzymes. The protease binding site of cystatins (Bode et al, 1988) and stefins (Stubbs et al, 1990) is composed of two hairpin loops (the first containing the conserved and functionally essential QVVAG sequence) and an N-terminal segment. Together, they form a hydrophobic wedge that is highly complementary to the active site of the archetypal cysteine protease papain, leading to extremely tight, rigid and fast inhibition (Figure 1B). The mechanism of inhibition does not involve the catalytic Cys25, which is too far away from the N-terminal segment to attack it. Interestingly, cystatins are also able to form somewhat weaker complexes with cysteine exopeptidases, as revealed by the crystal structure of the stefin A–cathepsin H complex (Jenko et al, 2003) (Figure 1B(1)). In this case, a number of conformational changes involving in particular the N-terminal segment are required for tight complex formation.
The functionally important example of direct active site blockage also includes noncanonical inhibitors of serine proteases. These inhibitors insert their N-terminal tail into the enzyme active site forming a short parallel β-sheet with enzyme residues 214–216, in contrast to the canonical inhibitors, which interact through the exposed binding loop and form an antiparallel β-sheet. The noncanonical inhibitors have developed in hematophagous animals as anticoagulants to inhibit either thrombin or factor Xa. Both proteases possess functionally important surface patches that are recognized by additional segments of noncanonical inhibitors: an acidic C-terminal tail or a homologous domain. These extensive secondary interactions significantly increase the contact area and contribute to the surprisingly high strength, speed and specificity of the recognition.
The classic example is recognition of thrombin by the leech inhibitor hirudin. The N-terminus of the globular domain of hirudin contacts the active site through the above-mentioned parallel β-sheet, while the acidic C-terminal tail is recognized by the anionic fibrinogen recognition exosite (Grutter et al, 1990). In haemedin from a land-living leech, the interaction through the N-terminus is similar, but the acidic C-terminal segment interacts with the heparin binding surface (Richardson et al, 2000) (Figure 1A(3)).
In the case of the two-domain ornithodorin, the N-terminus binds the active site in a noncanonical mode while the C-terminal domain is recognized by the fibrinogen recognition exosite (van de Locht et al, 1996). Both domains of ornithodorin resemble a canonical inhibitor, BPTI, but their canonical binding loops are distorted and do not contact the enzyme. The only known noncanonical inhibitor of factor Xa is tick anticoagulant peptide (TAP), again structurally similar to the canonical inhibitor BPTI (Wei et al, 1998).
A small yeast protein IA3 composed of 68 amino acids is able to block the active site of aspartic protease A from the same organism in a highly unusual way. It shows no detectable secondary structure in solution and can be cleaved by many structurally similar aspartic proteases, including pepsin. However, upon complexation with protease A, residues 2–32 of IA3 adopt an almost perfectly α-helical conformation, revealing that the protease body serves as a folding template (Li et al, 2000) (Figure 1D(2)). The nucleophilic water molecule occupies the catalytic position, but no peptide bond of the inhibitor is close enough to be attacked.
This short review attempts to cover the surprisingly diverse group of protein protease inhibitors. With more structural data, we are now starting to better understand the basic molecular mechanisms ensuring inhibitory complex formation. These mechanisms have been classified into just a few types. Can we expect new types of protease complexes in the forthcoming years? The answer to this question is undoubtedly positive, since new proteases are constantly being discovered and protein inhibitors have often coevolved with proteolytic enzymes.
Acknowledgments
Jacek Otlewski thanks the Howard Hughes Medical Institute for generous support.
References
- Aertgeerts K, De Bondt HL, De Ranter CJ, Declerck PJ (1995) Mechanisms contributing to the conformational and functional flexibility of plasminogen activator inhibitor-1. Nat Struct Biol 2: 891–897 [DOI] [PubMed] [Google Scholar]
- Al-Khunaizi M, Luke CJ, Askew YS, Pak SC, Askew DJ, Cataltepe S, Miller D, Mills DR, Tsu C, Bromme D, Irving JA, Whisstock JC, Silverman GA (2002) The serpin SQN-5 is a dual mechanistic-class inhibitor of serine and cysteine proteinases. Biochemistry 41: 3189–3199 [DOI] [PubMed] [Google Scholar]
- Ardelt W, Laskowski M Jr (1985) Turkey ovomucoid third domain inhibits eight different serine proteinases of varied specificity on the same …Leu18-Glu19 … reactive site. Biochemistry 24: 5313–5320 [DOI] [PubMed] [Google Scholar]
- Baumann U, Bauer M, Letoffe S, Delepelaire P, Wandersman C (1995) Crystal structure of a complex between Serratia marcescens metallo-protease and an inhibitor from Erwinia chrysanthemi. J Mol Biol 248: 653–661 [DOI] [PubMed] [Google Scholar]
- Bode W, Engh R, Musil D, Thiele U, Huber R, Karshikov A, Brzin J, Kos J, Turk V (1988) The 2.0 Å X-ray crystal structure of chicken egg white cystatin and its possible mode of interaction with cysteine proteinases. EMBO J 7: 2593–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode W, Greyling HJ, Huber R, Otlewski J, Wilusz Y (1989) The refined 2.0 Å X-ray crystal structure of the complex formed between bovine beta-trypsin and CMTI-I, a trypsin inhibitor from squash seeds (Cucurbita maxima). Topological similarity of the squash seed inhibitors with the carboxypeptidase A inhibitor from potatoes. FEBS Lett 242: 285–292 [DOI] [PubMed] [Google Scholar]
- Bode W, Huber R (1992) Natural protein proteinase inhibitors and their interaction with proteinases. Eur J Biochem 204: 433–451 [DOI] [PubMed] [Google Scholar]
- Chai J, Shiozaki E, Srinivasula SM, Wu Q, Datta P, Alnemri ES, Shi Y, Dataa P (2001) Structural basis of caspase-7 inhibition by XIAP. Cell 104: 769–780 [DOI] [PubMed] [Google Scholar]
- Fernandez-Catalan C, Bode W, Huber R, Turk D, Calvete JJ, Lichte A, Tschesche H, Maskos K (1998) Crystal structure of the complex formed by the membrane type 1-matrix metalloproteinase with the tissue inhibitor of metalloproteinases-2, the soluble progelatinase A receptor. EMBO J 17: 5238–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga M, Cherney MM, Oyama H, Oda K, James MN (2004) The molecular structure and catalytic mechanism of a novel carboxyl peptidase from Scytalidium lignicolum. Proc Natl Acad Sci USA 101: 3364–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gettins PG (2002) Serpin structure, mechanism, and function. Chem Rev 102: 4751–4804 [DOI] [PubMed] [Google Scholar]
- Gomis-Ruth FX, Maskos K, Betz M, Bergner A, Huber R, Suzuki K, Yoshida N, Nagase H, Brew K, Bourenkov GP, Bartunik H, Bode W (1997) Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature 389: 77–81 [DOI] [PubMed] [Google Scholar]
- Grutter MG, Priestle JP, Rahuel J, Grossenbacher H, Bode W, Hofsteenge J, Stone SR (1990) Crystal structure of the thrombin–hirudin complex: a novel mode of serine protease inhibition. EMBO J 9: 2361–2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guncar G, Pungercic G, Klemencic I, Turk V, Turk D (1999) Crystal structure of MHC class II-associated p41 Ii fragment bound to cathepsin L reveals the structural basis for differentiation between cathepsins L and S. EMBO J 18: 793–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano K, Sawano Y, Tanokura M (2002) Structure–function relationship of bromelain isoinhibitors from pineapple stem. Biol Chem 383: 1151–1156 [DOI] [PubMed] [Google Scholar]
- Hege T, Feltzer RE, Gray RD, Baumann U (2001) Crystal structure of a complex between Pseudomonas aeruginosa alkaline protease and its cognate inhibitor: inhibition by a zinc-NH2 coordinative bond. J Biol Chem 276: 35087–35092 [DOI] [PubMed] [Google Scholar]
- Helland R, Czapinska H, Leiros I, Olufsen M, Otlewski J, Smalas AO (2003) Structural consequences of accommodation of four non-cognate amino acid residues in the S1 pocket of bovine trypsin and chymotrypsin. J Mol Biol 333: 845–861 [DOI] [PubMed] [Google Scholar]
- Helland R, Otlewski J, Sundheim O, Dadlez M, Smalas AO (1999) The crystal structures of the complexes between bovine beta-trypsin and ten P1 variants of BPTI. J Mol Biol 287: 923–942 [DOI] [PubMed] [Google Scholar]
- Huntington JA, Fan B, Karlsson KE, Deinum J, Lawrence DA, Gettins PG (1997) Serpin conformational change in ovalbumin. Enhanced reactive center loop insertion through hinge region mutations. Biochemistry 36: 5432–5440 [DOI] [PubMed] [Google Scholar]
- Huntington JA, Read RJ, Carrell RW (2000) Structure of a serpin–protease complex shows inhibition by deformation. Nature 407: 923–926 [DOI] [PubMed] [Google Scholar]
- Im H, Ahn HY, Yu MH (2000) Bypassing the kinetic trap of serpin protein folding by loop extension. Protein Sci 9: 1497–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irving JA, Pike RN, Dai W, Bromme D, Worrall DM, Silverman GA, Coetzer TH, Dennison C, Bottomley SP, Whisstock JC (2002) Evidence that serpin architecture intrinsically supports papain-like cysteine protease inhibition: engineering alpha(1)-antitrypsin to inhibit cathepsin proteases. Biochemistry 41: 4998–5004 [DOI] [PubMed] [Google Scholar]
- Jenko S, Dolenc I, Guncar G, Dobersek A, Podobnik M, Turk D (2003) Crystal structure of stefin A in complex with cathepsin H: N-terminal residues of inhibitors can adapt to the active sites of endo- and exopeptidases. J Mol Biol 326: 875–885 [DOI] [PubMed] [Google Scholar]
- Khan AR, James MN (1998) Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci 7: 815–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krowarsch D, Dadlez M, Buczek O, Krokoszynska I, Smalas AO, Otlewski J. (1999) Interscaffolding additivity: binding of P1 variants of bovine pancreatic trypsin inhibitor to four serine proteases. J Mol Biol 289: 175–186 [DOI] [PubMed] [Google Scholar]
- Laskowski M, Qasim MA (2000) What can the structures of enzyme–inhibitor complexes tell us about the structures of enzyme substrate complexes? Biochim Biophys Acta 1477: 324–337 [DOI] [PubMed] [Google Scholar]
- Li M, Phylip LH, Lees WE, Winther JR, Dunn BM, Wlodawer A, Kay J, Gustchina A (2000) The aspartic proteinase from Saccharomyces cerevisiae folds its own inhibitor into a helix. Nat Struct Biol 7: 113–117 [DOI] [PubMed] [Google Scholar]
- Lu SM, Lu W, Qasim MA, Anderson S, Apostol I, Ardelt W, Bigler T, Chiang YW, Cook J, James MN, Kato I, Kelly C, Kohr W, Komiyama T, Lin TY, Ogawa M, Otlewski J, Park SJ, Qasim S, Ranjbar M, Tashiro M, Warne N, Whatley H, Wieczorek A, Wieczorek M, Wilusz T, Wynn R, Zhang W, Laskowski M Jr (2001) Predicting the reactivity of proteins from their sequence alone: Kazal family of protein inhibitors of serine proteinases. Proc Natl Acad Sci USA 98: 1410–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy G, Houbrechts A, Cockett MI, Williamson RA, O'Shea M, Docherty AJ (1991) The N-terminal domain of tissue inhibitor of metalloproteinases retains metalloproteinase inhibitory activity. Biochemistry 30: 8097–8102 [DOI] [PubMed] [Google Scholar]
- Musil D, Bode W, Huber R, Laskowski M Jr, Lin TY, Ardelt W (1991) Refined X-ray crystal structures of the reactive site modified ovomucoid inhibitor third domains from silver pheasant (OMSVP3*) and from Japanese quail (OMJPQ3*). J Mol Biol 220: 739–755 [DOI] [PubMed] [Google Scholar]
- Ng KK, Petersen JF, Cherney MM, Garen C, Zalatoris JJ, Rao-Naik C, Dunn BM, Martzen MR, Peanasky RJ, James MN (2000) Structural basis for the inhibition of porcine pepsin by Ascaris pepsin inhibitor-3. Nat Struct Biol 7: 653–657 [DOI] [PubMed] [Google Scholar]
- Potempa J, Shieh BH, Travis J (1988) Alpha-2-antiplasmin: a serpin with two separate but overlapping reactive sites. Science 241: 699–700 [DOI] [PubMed] [Google Scholar]
- Qasim MA, Ganz PJ, Saunders CW, Bateman KS, James MN, Laskowski M Jr (1997) Interscaffolding additivity. Association of P1 variants of eglin c and of turkey ovomucoid third domain with serine proteinases. Biochemistry 36: 1598–1607 [DOI] [PubMed] [Google Scholar]
- Radisky ES, Koshland DE Jr (2002) A clogged gutter mechanism for protease inhibitors. Proc Natl Acad Sci USA 99: 10316–10321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Tolle DP, Barrett AJ (2004) Evolutionary families of peptidase inhibitors. Biochem J 378: 705–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DC, Lipscomb WN (1982) Refined crystal structure of the potato inhibitor complex of carboxypeptidase A at 2.5 Å resolution. J Mol Biol 160: 475–498 [DOI] [PubMed] [Google Scholar]
- Reverter D, Fernandez-Catalan C, Baumgartner R, Pfander R, Huber R, Bode W, Vendrell J, Holak TA, Aviles FX (2000) Structure of a novel leech carboxypeptidase inhibitor determined free in solution and in complex with human carboxypeptidase A2. Nat Struct Biol 7: 322–328 [DOI] [PubMed] [Google Scholar]
- Richardson JL, Kroger B, Hoeffken W, Sadler JE, Pereira P, Huber R, Bode W, Fuentes-Prior P (2000) Crystal structure of the human alpha-thrombin–haemadin complex: an exosite II-binding inhibitor. EMBO J 19: 5650–5660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigden DJ, Mosolov VV, Galperin MY (2002) Sequence conservation in the chagasin family suggests a common trend in cysteine proteinase binding by unrelated protein inhibitors. Protein Sci 11: 1971–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzychon M, Filipek R, Sabat A, Kosowska K, Dubin A, Potempa J, Bochtler M (2003) Staphostatins resemble lipocalins, not cystatins in fold. Protein Sci 12: 2252–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stennicke HR, Ryan CA, Salvesen GS (2002) Reprival from execution: molecular basis of caspase inhibition. Trends Biochem Sci 27: 94–101 [DOI] [PubMed] [Google Scholar]
- Stubbs MT, Laber B, Bode W, Huber R, Jerala R, Lenarcic B, Turk V (1990) The refined 2.4 Å X-ray crystal structure of recombinant human stefin B in complex with the cysteine proteinase papain: a novel type of proteinase inhibitor interaction. EMBO J 9: 1939–1947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate S, Ohno A, Seeram SS, Hiraga K, Oda K, Kainosho M (1998) Elucidation of the mode of interaction of thermolysin with a proteinaceous metalloproteinase inhibitor, SMPI, based on a model complex structure and a structural dynamics analysis. J Mol Biol 282: 435–446 [DOI] [PubMed] [Google Scholar]
- Turk B, Turk V, Turk D (1997) Structural and functional aspects of papain-like cysteine proteinases and their protein inhibitors. Biol Chem 378: 141–150 [PubMed] [Google Scholar]
- van de Locht A, Stubbs MT, Bode W, Friedrich T, Bollschweiler C, Hoffken W, Huber R (1996) The ornithodorin–thrombin crystal structure, a key to the TAP enigma? EMBO J 15: 6011–6017 [PMC free article] [PubMed] [Google Scholar]
- Wei A, Alexander RS, Duke J, Ross H, Rosenfeld SA, Chang CH (1998) Unexpected binding mode of tick anticoagulant peptide complexed to bovine factor Xa. J Mol Biol 283: 147–154 [DOI] [PubMed] [Google Scholar]
- Xu G, Cirilli M, Huang Y, Rich RL, Myszka DG, Wu H (2001) Covalent inhibition revealed by the crystal structure of the caspase-8/p35 complex. Nature 410: 494–497 [DOI] [PubMed] [Google Scholar]
- Ye S, Cech AL, Belmares R, Bergstrom RC, Tong Y, Corey DR, Kanost MR, Goldsmith EJ (2001) The structure of a Michaelis serpin–protease complex. Nat Struct Biol 8: 979–983 [DOI] [PubMed] [Google Scholar]