

Abstract
Apoptosis-inducing factor (AIF) is a mitochondrial intermembrane flavoprotein that is translocated to the nucleus in response to proapoptotic stimuli, where it induces nuclear apoptosis. Here we show that AIF is synthesized as an ∼67-kDa preprotein with an N-terminal extension and imported into mitochondria, where it is processed to the ∼62-kDa mature form. Topology analysis revealed that mature AIF is a type-I inner membrane protein with the N-terminus exposed to the matrix and the C-terminal portion to the intermembrane space. Upon induction of apoptosis, processing of mature AIF to an ∼57-kDa form occurred caspase-independently in the intermembrane space, releasing the processed form into the cytoplasm. Bcl-2 or Bcl-XL inhibited both these events. These findings indicate that AIF release from mitochondria occurs by a two-step process: detachment from the inner membrane by apoptosis-induced processing in the intermembrane space and translocation into the cytoplasm. The results also suggest the presence of a unique protease that is regulated by proapoptotic stimuli in caspase-independent cell death.
Keywords: AIF, apoptosis, mitochondria, protein export, protein import
Introduction
Mitochondria have a central role in the initiation of apoptosis, releasing apoptosis-related proteins into the cytoplasm that are usually present in the intermembrane space (IMS) in the early stages of apoptotic cell death (Newmeyer and Ferguson-Miller, 2003). Among them are apoptosis-inducing factor (AIF) (Susin et al, 1999), Smac/DIABLO (Du et al, 2000; Verhagen et al, 2000), cytochrome c (Liu et al, 1996), and HtrA2/Omi (Suzuki et al, 2001; Hegde et al, 2002; Verhagen et al, 2002). The exact membrane permeabilization mechanisms, however, are a matter of debate. Irrespective of the exact mechanism of membrane permeabilization, the antiapoptotic members of the Bcl-2 family tend to stabilize the barrier function of mitochondrial membranes, whereas proapoptotic Bcl-2 family proteins such as Bax or Bak tend to antagonize such function and permeabilize the membranes (Cory and Adams, 2002).
AIF is a flavoprotein in the mitochondrial IMS that has a dual nature in controlling cellular life and death; during apoptosis, it is translocated from the mitochondria to the nucleus to function as a proapoptotic factor in a caspase-independent pathway (Joza et al, 2001), while in normal mitochondria, it functions as an antiapoptotic factor via its oxidoreductase activity (Klein et al, 2002). Many aspects of the AIF-associated pro- or antiapoptotic function, however, remain unclear. AIF is synthesized in the cytosol and subsequently imported into the mitochondrial IMS. Under normal circumstances, transcription and translation of the nuclear-coded AIF gene give rise to an ∼67-kDa precursor molecule (613 amino-acid residues) that carries a predicted mitochondrial localization signal (MLS) in its N-terminal presequence. After import into the mitochondrial IMS, it has been suggested that the 101-amino-acid residue presequence segment is cleaved off, leading to the generation of the mature AIF molecule (∼57 kDa), which is confined to the mitochondrial IMS in a soluble form (Susin et al, 1999). The IMS-localized mature form (∼57 kDa) is released to the cytoplasm in response to specific death signals, and translocated to the nucleus, where it induces nuclear apoptosis in a caspase-independent manner (Susin et al, 1999; Loeffler et al, 2001). Release of AIF from the IMS is therefore considered to be a key initial step in the caspase-independent apoptotic process, although the caspase dependency remains controversial (Arnoult et al, 2002, 2003) and the issue of whether it is an IMS-soluble protein is not clear.
To address the underlying mechanisms by which AIF is released from mitochondria dependent on apoptotic stimuli, we first determined the topology of AIF in mitochondrial IMS using the differential cell-permeabilization method and submitochondrial fractionation with HeLa cells transfected with a dual T7- and FLAG-tagged AIF. For correct sorting of AIF to the IMS, a cluster of hydrophobic amino-acid residues (a putative transmembrane segment (TMS)) at residues 66–84 was required. We found, unexpectedly, that the N-terminal 52-amino-acid extra sequence of AIF is cleaved off in the matrix by the mitochondrial processing peptidase (MPP) to form the inner-membrane-anchored mature form. During apoptosis, it is further proteolytically processed at amino-acid position 101 in the IMS. We report that AIF is released from the IMS by a two-step process: proteolytic processing at position 101/102 in the IMS to detach from the inner membrane followed by permeabilization of the outer membrane to be released into the cytoplasm.
Results
Permeabilization and staining
Digitonin is widely used for selective permeabilization of cellular and organellar membranes in animal cells (Knight and Scrutton, 1986). Here, we have established the condition that specifically permeabilizes mitochondrial outer membrane or both outer and inner membranes using cytochrome c and HtrA2 as the IMS markers and Hsp60 and mHsp70 as the matrix markers: 0.4 mg/ml digitonin for permeabilization of the outer membrane, and 2.0 mg/ml for both the outer and inner membranes (Supplementary Figure 1). Using these conditions, we investigated the intracellular localization of AIF and its mutation constructs as described below.
The N-terminal segment of residues 1–40 is necessary for mitochondrial targeting of AIF
The N-terminal portion of full-length AIF bears a region with a predicted MLS at residues 1–30, a hydrophobic TMS at residues 66–84, and the predicted cleavage site at leucine 101–glycine 102 (Susin et al, 1999). Residues 123–262 and 401–480, and 262–400 provide FAD-binding domains and an NADH-binding domain, respectively (Lipton and Bossy-Wetzel, 2002; Ye et al, 2002).
To search for the MLS in AIF experimentally, we constructed AIF-EGFP-FLAG fusions carrying various deletions from the C- or N-terminus, or within the internal segment of AIF (Figure 1A). These constructs were transiently expressed in HeLa cells and FLAG tag and GFP of the expressed proteins were detected by immunofluorescence and fluorescence microscopy after differential permeabilization of the cells (Figure 1B).
Figure 1.
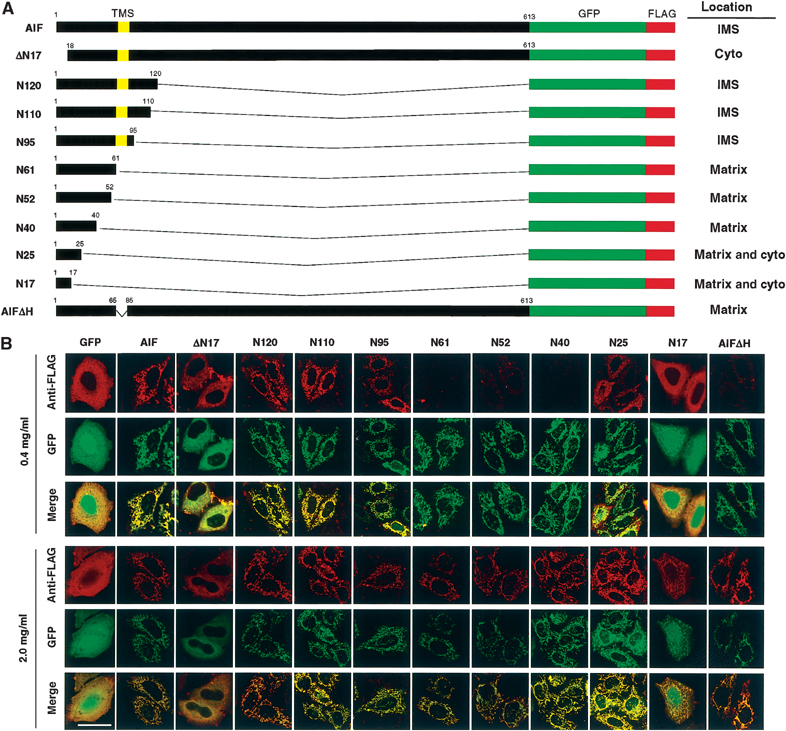
Intracellular and intramitochondrial localization of AIF variants. (A) Schematic representation of AIF variants and their intracellular or intramitochondrial localizations. The N-terminal segments of AIF from the first to the indicated residues were ligated to the N-terminus of GFP-FLAG, or the N-terminal 17 residues or TMS of AIF-GFP-FLAG was deleted (ΔN17 and AIFΔH, respectively). (B) HeLa cells expressing the indicated constructs were permeabilized and the expressed proteins were detected by GFP fluorescence and anti-FLAG antibody under confocal microscopy. Magnification, × 630; bar=20 μm.
All the constructs, except for N17, N25, and ΔN17, exhibited a tubular pattern of GFP fluorescence, which was superimposable with Mitotracker-Red (Figure 1B), indicating that these constructs were transported to the mitochondria. Both N17 and N25 had both tubular and cytoplasmically diffused staining patterns, whereas N40 exhibited a clear mitochondrial pattern, suggesting that at least the N-terminal 40-amino-acid residue segment functions as an MLS. The N-terminal 17- and 25-amino-acid residue segments of AIF functioned as inefficient MLS.
Residues 66–84 of AIF are required for sorting to the IMS
When the cells expressing the above-described constructs were permeabilized with 0.4 mg/ml of digitonin, full-length AIF, N120, N110, and N95 exhibited tubular staining patterns with anti-FLAG antibody (Figure 1B, upper panel). On the other hand, N61, N52, and N40 were scarcely stained by anti-FLAG antibodies, although there was a tubular staining pattern with GFP fluorescence (upper panel); they were immunostained only when outer and inner membranes were permeabilized by 2.0 mg/ml digitonin (lower panel). These results clearly indicated that full-length AIF, N120, N110, and N95 were correctly localized in the IMS, whereas N61, N52, and N40, but N25 only inefficiently, were transported into the matrix. These results further indicated that the hydrophobic stretch of AIF (residues 66–84) is essential for sorting the protein to the IMS. In this context, AIFΔH, in which the TMS was deleted, was imported into the matrix, since FLAG tag of the protein was accessible to the externally added antibodies only when both outer and inner membranes were permeabilized by 2.0 mg/ml digitonin (AIFΔH in Figure 1B), confirming the importance of the TMS for the IMS sorting.
Taken together, the findings indicate that the N-terminal segment of AIF constitutes a bipartite-type MLS, similar to those that are well defined for several yeast mitochondrial IMS proteins (Jensen et al, 1992; Beasley et al, 1993): residues 1–40 as the MLS, and a hydrophobic stretch of residues 66–84 as the IMS-sorting signal.
Biogenesis of AIF
We then investigated the maturation process of the AIF precursor in vivo in more detail. Kroemer and co-workers (Susin et al, 1999) reported that AIF is synthesized as an ∼67 kDa precursor in the cytoplasm and then imported into the mitochondrial IMS, where the presequence (first 101 amino acids) is cleaved off. To confirm the presequence processing site, we compared the molecular sizes of mature AIF in the mitochondria prepared from rat liver and HeLa cells with that of AIFΔN101, lacking the first 101 residues (Figure 2A). As shown in Figure 2B, mitochondrial AIF had clearly slower mobility on SDS–PAGE than in vitro-synthesized AIFΔN101 (compare lanes 1 and 2 with lane 4). Rather, the size of mature AIF coincided with that of in vitro-synthesized AIFΔN52 (lane 3). Furthermore, the in vitro-synthesized AIF precursor migrating with an apparent size of ∼67 kDa (Figure 2B, lane 5) was processed by purified MPP to the same size as the mitochondrial mature form of AIF (∼62 kDa; lane 6). Import of the 35S-labeled AIF precursor into isolated mitochondria in vitro produced a faster moving band corresponding to the MPP-processed mature AIF (Figure 2C, compare lanes 3 and 4). Protein sequencing of the mature AIF that was immunoisolated from the nonapoptotic cell mitochondria revealed that its N-terminal sequence starts from Ala-54 of the AIF precursor (54ASSRV58). These results clearly indicated that the presequence cleavage occurred during mitochondrial import of the AIF precursor at site M53/A54, but not at site L101/G102.
Figure 2.
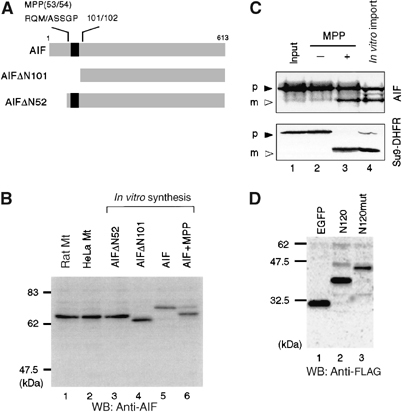
Determination of the processing site of AIF precursor during mitochondrial import. (A) Schematic representation of AIF and its truncation mutants. Numbers represent the positions in the AIF precursor amino-acid sequence. Predicted TMS is shown in black boxes and apoptosis-dependent processing site (101/102) is indicated on the top of the figure. The MPP-processed site deduced by N-terminal sequencing is shown. (B) N-terminal segment of ∼50 amino-acid residues is cleaved by MPP. Mitochondria from rat liver and HeLa cells, cell-free synthesized AIFΔN52, AIFΔN101, and full-length AIF, and cell-free synthesized full-length AIF precursor that had been incubated with purified MPP were subjected to SDS–PAGE and subsequent immunoblot analysis using anti-AIF antibodies. (C) In vitro-synthesized 35S-labeled AIF precursor and pSu9-DHFR (lane 1) were incubated with or without purified MPP (lanes 2 and 3, respectively) or with isolated rat liver mitochondria (lane 4). The reaction mixtures were analyzed by SDS–PAGE and autoradiography. Solid and open arrowheads represent precursor proteins (p) and mature proteins (m), respectively. (D) The MPP-processing site of AIF was probed using the alanine-substituted mutant. N120-GFP-FLAG (N120) and the mutant carrying R51A-M53-A-S55/56A mutations (N120mut) were expressed in HeLa cells and the lysates were analyzed by SDS–PAGE and immunoblotting using anti-FLAG antibody.
Analysis of a large number of sequences around the MPP cleavage site of the mitochondrial precursor proteins revealed no consensus amino-acid sequence for the processing, except that basic amino acids, usually arginine, are often observed at position −2 or −3 from the cleavage site. In addition, a hydrophobic residue at position +1, serine or threonine residues at position +2 and/or +3, and distal basic residues around position −10 are important for MPP recognition (Taylor et al, 2001). It is noteworthy that the sequence M49ARQMASS56 in AIF contains a highly probable MPP-cleavage site. N120-GFP-FLAG (N120) and the construct carrying the R51A-M53A-S55/56A mutations (N120mut) were expressed in HeLa cells and analyzed by immunoblot of the cell lysates (Figure 2D). As expected, this mutant failed to be processed to its mature form compared with wild type (lanes 2 and 3). Accordingly, these data clearly indicated that the presequence of AIF is cleaved off at M49ARQM/ASS56 by MPP on the matrix side during or just after mitochondrial import.
AIF topology
As described above, the presequence of the AIF precursor was cleaved at site M53/A54, thereby leaving a hydrophobic TMS (residues 66–84) in the mature AIF. If this is the case, mature AIF should be associated with either of the mitochondrial membranes. We therefore examined submitochondrial localization and membrane topology of AIF using HeLa cells expressing N120-GFP (N120) and N120ΔH-GFP (N120ΔH). The expressed proteins as well as the endogenous AIF were recovered from mitochondria but not from the postmitochondrial supernatant (Figure 3A, lanes 1 and 2). N120, N120ΔH, and the endogenous AIF recovered in the mitochondria were all resistant to the externally added proteinase K (lane 3). Upon disruption of the outer membrane, N120 and the endogenous AIF were completely degraded by proteinase K, whereas N120ΔH remained undigested (lane 4). As the control, all these proteins were digested by proteinase K when mitochondrial membranes were solubilized by Triton X-100 (lane 5).
Figure 3.
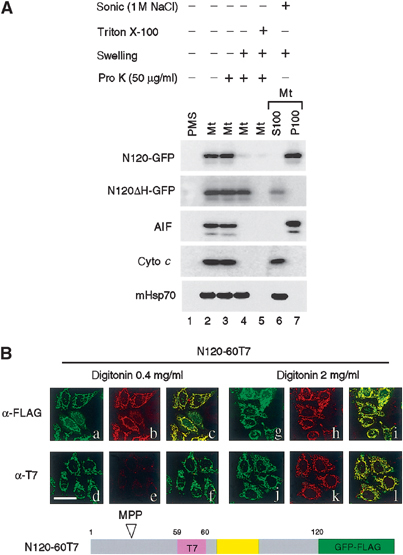
Determination of transmembrane topology of AIF. (A) The isolated mitochondria harboring N120-GFP or N120ΔH-GFP were separated into five aliquots. Four of them were incubated with or without proteinase K under the indicated conditions. The remaining aliquot was sonicated in the presence of 1 M NaCl, followed by ultracentrifugation to separate into the supernatant (S100) and membrane (P100) fractions. All these fractions were subjected to SDS–PAGE and subsequent immunoblot analysis using antibodies against the indicated proteins. (B) HeLa cells transfected with N120-GFP-FLAG carrying an internal T7-tag insertion in the residue position of 59/60 (N120-60T7) were fixed and treated with 0.4 mg/ml digitonin (a–f) or 2.0 mg/ml digitonin (g–l). The expressed proteins were detected by GFP fluorescence (green) and either anti-FLAG (shown in red; b, h) or anti-T7 antibodies (shown in red; e, k) under confocal microscopy. Merged images are also shown (c, f, i, l). Magnification, × 630; bar=20 μm.
When the isolated mitochondria were sonicated in the presence of 1 M NaCl, N120 and the endogenous AIF were recovered exclusively to the membrane fraction, whereas N120ΔH was recovered to the supernatant fraction (lane 6). Furthermore, N120 and the endogenous AIF, but not N120ΔH, were resistant to sodium carbonate (pH 10.5) extraction (Supplementary Figure 2). Taken together, these results indicated that N120 and the endogenous AIF were bound to the inner membrane extruding the bulk portion to the IMS, whereas N120ΔH was imported into the matrix as a soluble form. These results were consistent with those of indirect immunofluorescent staining (see Figure 1B). The behavior of submitochondrial markers, cytochrome c (IMS protein) and mHsp70 (matrix protein), indicated that the submitochondrial fractionation and the protease digestion were well controlled (Figure 3A, lanes 1–7).
We next examined the topology of AIF on the mitochondrial membranes by differential cell permeabilization using HeLa cells transfected with N120-60T7-GFP-FLAG (N120-60T7), the N120-GFP-FLAG fusion construct in which T7 tag was inserted into amino-acid residue position 59/60 upstream of the TMS (Figure 3B). When HeLa cells expressing this construct were treated with 2 mg/ml digitonin, which solubilized both the outer and inner mitochondrial membranes, both FLAG and T7 tags were detected in the mitochondria by immunofluorescence microscopy (h and k). In contrast, in the presence of 0.4 mg/ml digitonin, which selectively permeabilized the outer membrane, N120-60T7 was observed with a mitochondrial staining pattern with anti-FLAG antibody (b), whereas almost no staining signal was detected with anti-T7 antibody (e). Expression of N120-60T7 was verified by GFP fluorescence (a, d, g, and j). Together, these data strongly suggested that the C-terminal part of AIF was exposed to the mitochondrial IMS, while the N-terminal part containing the MPP cleavage site was exposed to the matrix.
Proteolytic processing of AIF during apoptosis
Early during the apoptotic process, the outer mitochondrial membrane becomes permeabilized, and mitochondria release soluble apoptotic proteins that are normally confined to the IMS (Newmeyer and Ferguson-Miller, 2003). Because only the IMS-soluble proteins should be released, membrane proteins such as AIF must be converted to a soluble form prior to export through the outer membrane. To substantiate this assumption, we probed the region of AIF responsible for the apoptosis-dependent mitochondrial release using the AIF-GFP-FLAG constructs with various deletions in the C-terminal segment of AIF (N500, N400, N300, and N200). When expressed in HeLa cells, they were correctly targeted to mitochondria (summarized in Figure 4A and Table I). After induction of apoptosis by the Bax- or Bak-dependent proapoptotic drug actinomycin D in the presence of zVAD-fmk, they were translocated from the mitochondria to the cytoplasm (Table I).
Figure 4.
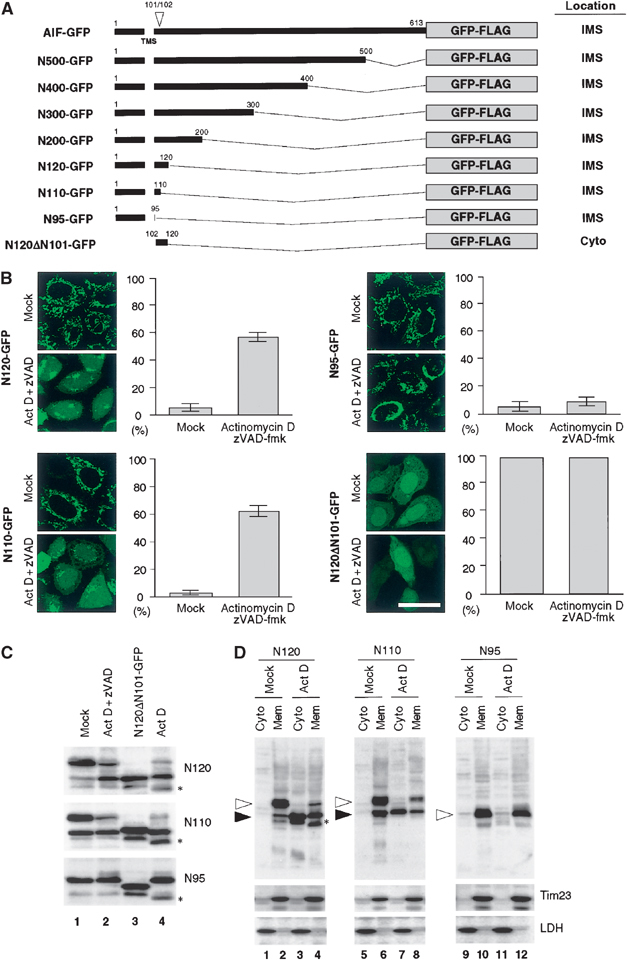
Apoptosis-induced processing and mitochondrial release of AIF. (A) Schematic representation of the AIF constructs and their intracellular location. (B) Subcellular distribution of various AIF constructs in response to actinomycin D treatment. HeLa cells expressing the indicated constructs (48 h after transfection) were treated with or without actinomycin D for 12 h in the presence of zVAD-fmk. The cells were examined by fluorescence confocal microscopy for GFP. The cells with a cytoplasmic GFP pattern were counted. Each histogram indicates mean±s.d. in three fields of at least 100 cells within a representative experiment. Magnification, × 630; bar=20 μm. (C) HeLa cells expressing the indicated constructs were treated with or without actinomycin D in the absence or presence of zVAD-fmk. Total cell lysates were subjected to SDS–PAGE followed by immunoblot analysis using anti-FLAG antibody. The lysate from N120ΔN101-GFP-FLAG-expressing cells was used as a size reference (lane 3). (D) HeLa cells expressing the indicated constructs were treated with or without actinomycin D in the presence of zVAD-fmk. The cells were fractionated into membrane and cytosolic fractions, and each fraction was analyzed by immunoblotting using antibodies against FLAG (for AIF constructs) or against the indicated proteins. Open and closed arrowheads indicate the GFP fusion proteins corresponding to unprocessed and processed forms, respectively. Asterisks represent uncharacterized minor bands detected in the absence of zVAD-fmk. Note that processed forms of N120 and N110 were detectable in mock-treated cells (closed arrowhead in lanes 2 and 6). Apoptosis might be brought about in the cells to a slight extent by mock treatment with Lipofectamine to induce this processing.
Table 1.
Intracellular localization of various AIF constructs and their response for mitochondrial release upon induction of apoptosis
| Mutant | Location | Mitochondrial release |
|---|---|---|
| AIF | IMS | + |
| ΔN17 | Cytoplasm | ND |
| N500 | IMS | + |
| N400 | IMS | + |
| N300 | IMS | + |
| N200 | IMS | + |
| N120 | IMS | + |
| N110 | IMS | + |
| N95 | IMS | − |
| N61 | Matrix | − |
| N52 | Matrix | − |
| N40 | Matrix | − |
| N25 | Matrix and cytoplasm | ND |
| N17 | Matrix and cytoplasm | ND |
| N120ΔN101 | Cytoplasm | ND |
| AIFΔH | Matrix | − |
| N120mut | IMS | + |
| Various AIF constructs shown in Figure 2A were transiently expressed in HeLa cells and assessed for mitochondrial release using GFP fluorescence microscopy. +, released; −, not released; ND, not determined. | ||
To narrow down the responsible region, we created four more constructs, N120, N110, N95, and N120ΔN101 (Figure 4A), which were transiently expressed in HeLa cells. At 24 h after transfection, N120, N110, and N95 were detected in mitochondrial IMS, as assessed by the differential permeabilization method (see Figure 1B; summarized in Figures 1A and 4A). In contrast, N120ΔN101 was localized in the cytoplasm, consistent with the results that the N-terminal region containing the TMS functions as the mitochondrial IMS-targeting signal. Confirming their intrinsic localization under normal conditions, we then treated cells with actinomycin D in the presence of zVAD-fmk. N120 and N110 were translocated to the cytoplasm and to the nucleus (Figure 4B). In contrast, N95 was not released to the cytoplasm but remained in the mitochondrial IMS. Therefore, the N-terminal region between residues 96 and 110 is most likely to be involved in the apoptosis-dependent mitochondrial release of AIF. It should also be noted that the matrix-targeted mutants N61, N52, and N40 failed to be released into the cytoplasm (Table I).
To investigate why N120 and N110, but not N95, were released from the mitochondrial IMS to the cytoplasm, HeLa cells expressing these constructs were treated with actinomycin D in the absence or presence of zVAD-fmk and the expressed proteins were analyzed by SDS–PAGE and subsequent immunoblotting using anti-FLAG antibodies (Figure 4C). Interestingly, apoptosis-dependent proteolytic processing was observed only for N120 and N110, but not for N95. Furthermore, N120 was processed to the form with the same mobility as N120ΔN101 in SDS–PAGE. The processing site within AIF is suggested to reside between residues 101 and 102, and the amino-acid residue at the latter position represents the start of mature AIF as determined by N-terminal sequencing (Susin et al, 1999). Taken together, it is very likely that the site around residue position 101 of the AIF precursor was cleaved dependent on apoptotic stimuli.
We next examined whether this processing is coupled with mitochondrial release using digitonin-dependent differential cell fractionation (Figure 4D). HeLa cells were treated as above and fractionated into cytoplasmic and membrane fractions, which were analyzed by Western blotting using anti-FLAG antibody. Tim23 was recovered exclusively in the membrane fractions, whereas LDH was recovered in the cytoplasmic fractions, confirming adequate separation of cytoplasmic and membrane fractions. Only the processed products derived from N120 and N110 were detected in the cytoplasmic fractions (Figure 4D, lanes 3 and 7), whereas unprocessed products remained in the membrane fractions (lanes 4 and 8). In contrast, the processing-incompetent construct N95 failed to be released to the cytoplasm and remained exclusively in the membrane fraction (lanes 11 and 12). Thus, it is most likely that the extent of AIF release to the cytoplasm is largely determined by the degree to which AIF is detached from the inner membrane by proteolytic processing in the IMS.
We then examined whether full-length AIF is also exported according to this scenario. When expressed in HeLa cells, full-length AIF/3xFLAG was predominantly localized in the mitochondria, based on its colocalization with the inner membrane marker Tim23 (data not shown). Mock-treated cells revealed a mitochondrial distribution of AIF/3xFLAG (Figure 5Aa and Da). In contrast, after induction of apoptosis by actinomycin D in the presence of zVAD-fmk, both the cytoplasmic and nuclear staining patterns of AIF/3xFLAG were observed (Figure 5Ab). As a control, ΔN101/3xFLAG lacking the IMS-targeting signal exhibited a cytoplasmic staining pattern irrespective of the culture condition (Figure 5Ac and d).
Figure 5.
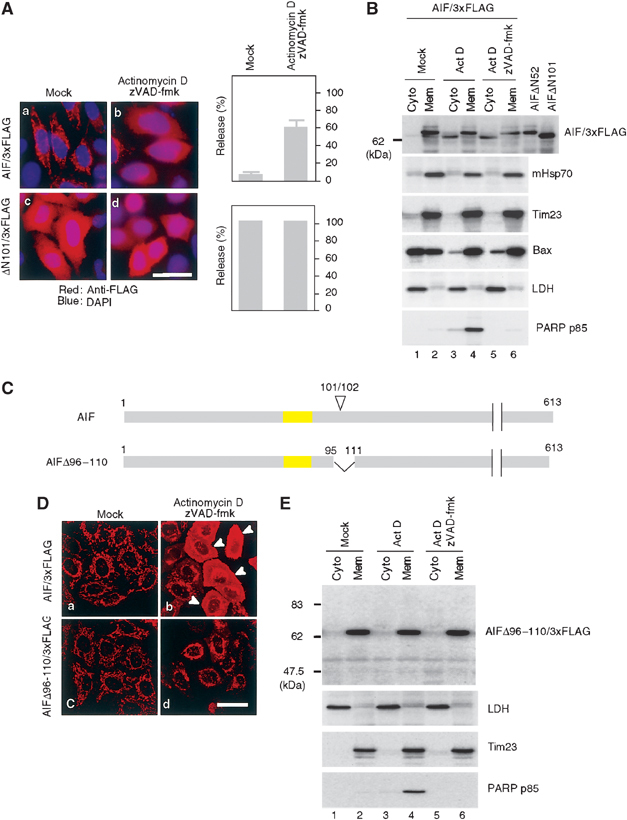
Processing of AIF is essential for the mitochondrial release by proapoptotic stimuli. (A) Subcellular distribution of full-length AIF/3xFLAG and ΔN101AIF/3xFLAG lacking the IMS localization signal. HeLa cells expressing the indicated constructs were treated with or without 20 μM actinomycin D in the presence of zVAD-fmk. The cells were stained with anti-FLAG antibodies (red) and DAPI (blue), and examined by fluorescence confocal microscopy. The number of cells exhibiting cytoplasmic patterns for each construct was counted and is shown in the right panel. Other conditions are same as in (B). (B) HeLa cells expressing AIF/3xFLAG were treated as described in (A), and then fractionated into membrane and cytosolic fractions, and each fraction was analyzed by immunoblotting using antibodies against the indicated proteins. (C) Schematic representation of AIF/3xFLAG and the truncation mutant. TMS is shown in yellow boxes and apoptosis-dependent processing site (101/102) is indicated on the top of the figure. (D) Subcellular distribution of full-length AIF/3xFLAG and AIFΔ96–110/3xFLAG. (E) HeLa cells expressing AIFΔ96–110/3xFLAG were treated with actinomycin D in the presence or absence of zVAD-fmk. They were fractionated into membrane and cytosolic fractions, and each fraction was analyzed by immunoblotting using antibodies against the indicated proteins.
We then examined the release of AIF/3xFLAG from the mitochondria (Figure 5B). Again, fidelity of the cell fractionation was verified using marker proteins mHsp70, Tim23, and LDH.
We also confirmed that zVAD-fmk inhibited the cleavage of the caspase substrate PARP (Figure 5B, lanes 4 and 6). In mock-treated cells, AIF/3xFLAG protein was localized exclusively to the membrane fraction (lane 2). In contrast, a significant amount of AIF/3xFLAG was accumulated in the cytosolic fraction after 12 h of actinomycin D treatment, both in the absence or presence of zVAD-fmk (lanes 3 and 5). The release was associated with translocation of Bax from the cytosol to the mitochondrial membrane. As expected, cytosolic AIF/3xFLAG had slightly higher mobility in SDS–PAGE compared with the mitochondrial counterpart (Figure 5B, lanes 3–6), indicating that the cytosolic AIF/3xFLAG was a product processed by an unknown apoptosis-dependent protease. Further, the cytosolic AIF/3xFLAG had the same mobility as AIFΔN101/3xFLAG, suggesting that the sequence required for IMS sorting (residue positions 54–101 in AIF precursor) was cleaved in the IMS at the site around residue 101, and the processed form was selectively released to the cytoplasm. Likewise, the same result was observed with several different apoptosis inducers (staurosporine and etoposide; see Figure 8C).
Figure 8.
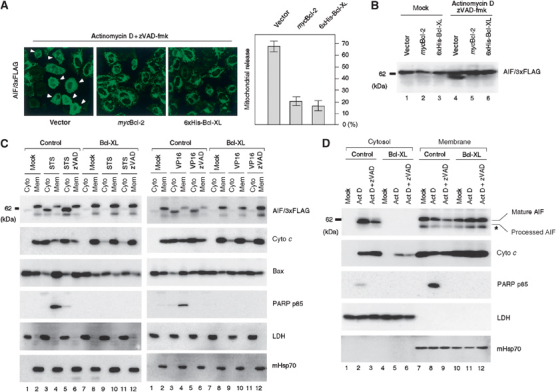
Bcl-2 and Bcl-XL inhibit both proteolytic processing and mitochondrial release of AIF. (A) HeLa cells expressing AIF/3xFLAG were cotransfected with an empty vector, mycBcl-2 vector, or 6xHis-Bcl-XL vector. Cells were treated with actinomycin D in the presence of zVAD-fmk and stained with anti-FLAG antibody. The cells exhibiting the cytoplasmic pattern were counted and are indicated by the histograms. Other conditions are same as in Figure 5. (B) HeLa cells expressing AIF/3xFLAG as described above were analyzed by SDS–PAGE followed by immunoblotting using anti-FLAG antibody. (C) HeLa cells and HeLa/XL cells transiently expressing AIF/3xFLAG were treated with staurosporine (left panel) or etoposide/VP16 (right panel) in the presence or absence of zVAD-fmk. They were then fractionated into membrane and cytosolic fractions, which were analyzed by SDS–PAGE and subsequent immunoblotting using antibodies against the indicated proteins. (D) HeLa and HeLa/XL cells were treated with actinomycin D in the presence or absence of zVAD-fmk. Other conditions are as described in (C). Processing and export of the endogenous AIF were examined using anti-AIF antibodies. The asterisk represents nonspecific bands detected with anti-AIF antibodies.
To corroborate this notion, we used AIFΔ96–110/3xFLAG in which the region covering the apoptosis-dependent processing site was deleted (Figure 5C). When expressed in HeLa cells, it was correctly localized to mitochondria, as is the case for AIF/3xFLAG (Figure 5Dc). In contrast to AIF/3xFLAG, however, this mutant failed to be exported to the cytoplasm after induction of apoptosis by actinomycin D (Figure 5Dd). Cell fractionation revealed that AIFΔ96–110/3xFLAG was not processed and remained exclusively in the mitochondrial fraction (Figure 5E, lanes 3–6), thus confirming the results of immunofluorescent microscopy.
Taken together, these experiments indicated that AIF is released from the mitochondria into the cytoplasm in a two-step process: detachment from the inner membrane by apoptosis-induced processing in the IMS, and translocation to the cytoplasm following permeabilization of the outer mitochondrial membrane. This process was also confirmed with the endogenous AIF (see Figure 8D).
Recently, two groups independently proposed that mitochondrial release of AIF occurs in a caspase activation-dependent (Arnoult et al, 2003) or -independent manner (Cregan et al, 2002). Because the events reported in the present study occurred even in the presence of zVAD-fmk, it is very likely that cleavage of mature AIF and the mitochondrial release involve a caspase-independent process. We cannot rigorously rule out, however, the possibility that zVAD-fmk affects the rate of AIF release, although significant effect in release rate was not obvious in the presence or absence of the inhibitor (data not shown).
Processing site of AIF is oriented in the IMS
We next determined the transmembrane orientation of the apoptosis-dependent processing site of AIF using the differential cell permeabilization with HeLa cells transfected with N120-60T7, N120-90T7, N120-100T7, and N120-110T7, the N120 fusion constructs in which T7 tag was inserted into amino-acid residue positions 59/60, 89/90, 99/100, and 109/110, respectively (Figure 6A). All variants of N120-GFP-FLAG were correctly targeted to the IMS and the membrane topology of all constructs, as judged from the FLAG-tag orientation, was not altered in the variants (data not shown). When HeLa cells expressing these constructs were treated with 2.0 mg/ml digitonin, the T7 tag was detected in the mitochondria by immunofluorescence microscopy (Figure 6B, right panel). In contrast, in the presence of 0.4 mg/ml digitonin (left panel), which selectively permeabilized the outer membrane, N120-100T7 and N120-110T7 had mitochondrial staining patterns with anti-T7 antibody, whereas almost no staining signal was detectable for the N120-60T7 and N120-90T7 constructs. Expression of all constructs was verified by GFP fluorescence. Together, these data strongly suggested that the apoptosis-dependent processing site of AIF is exposed to the IMS.
Figure 6.
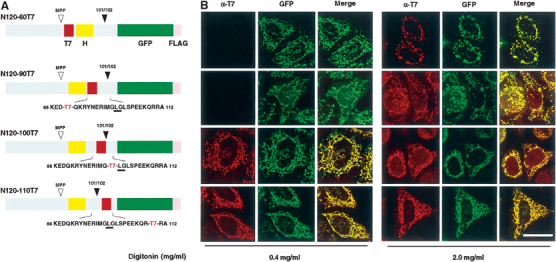
Membrane orientation of apoptosis-dependent processing site of AIF. (A) Schematic representation of the T7-tag-scanned constructs. MPP processing site and apoptosis-dependent processing site (101/102) are indicated. T7-tag sequence was inserted at sites 59/60, 89/90, 99/100, and 109/110 of N120-GFP to create N120-60T7, N120-90T7, N120-100T7, and N120-110T7, respectively. H, predicted TMS. (B) HeLa cells expressing the indicated constructs were fixed and treated with 0.4 mg/ml digitonin (left panel) or 2.0 mg/ml digitonin (right panel). The expressed proteins were detected by GFP fluorescence (shown in green) and anti-T7 antibody (shown in red) under confocal microscopy. Merged images are also shown. Magnification, × 630; bar=20 μm.
Processing-incompetent AIF construct remains in the IMS even after permeabilization of the outer membrane
Which could be rate limiting in the mitochondrial release reaction of AIF, processing in the IMS or permeabilization of the outer membrane? Taking advantage of inaccessibility of externally added IgGs against IMS-localized protein under 25 μg/ml digitonin that specifically permeabilized the plasma membrane of healthy HeLa cells, we demonstrated that the outer mitochondrial membranes were impermeable to the externally added antibodies at the initial stage of apoptosis. However, the outer membranes became permeable to IgGs with relatively faster kinetics. Expression of BcL-XL clearly blocked the access of anti-FLAG to N95-GFP-FLAG (Supplementary Figure 3). These results indicated that AIF could not be released even after permeabilization of the outer membrane unless its cleavage occurred in the IMS. When kinetics of actinomycin D-induced extramitochondrial release was compared, cytochrome c and Smac/DIABLO, but not AIF, were efficiently released after 4 h incubation; AIF required 11 h for complete release (Figure 7). In this context, no processing of the AIF constructs was observed after 4 h (data not shown) and ∼70% processing was observed after 11–12 h (see Figure 4 for N110 or N120, and Figure 8 for AIF), indicating that permeabilization of the outer membrane precedes the AIF processing and release reactions.
Figure 7.
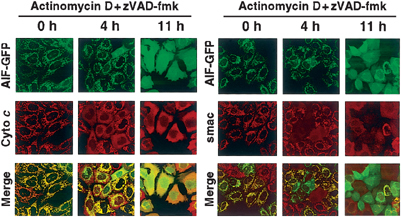
Time course of actinomycin D-induced release of AIF-GFP, cytochrome c, and smac/DIABLO as revealed by fluorescence microscopy. HeLa cells expressing AIF-GFP were treated with actinomycin D in the presence of zVAD-fmk. At the indicated time points, the cells were examined by confocal microscopy for GFP (AIF-GFP) together with immunofluorescence microscopy using antibodies against cytochrome c (left panel) or Smac/DIABLO (right panel). Note that cytochrome c and smac/DIABLO, but not AIF-GFP, were efficiently released to the cytoplasm after 4 h incubation.
Bcl-2 inhibits actinomycin D-induced proteolytic processing and release of AIF
To test whether Bcl-2 inhibits actinomycin D-induced processing and release of AIF, HeLa cells coexpressing mycBcl-2 and AIF/3xFLAG, or 6xHis-Bcl-XL and AIF/3xFLAG were treated with actinomycin D in the presence of zVAD-fmk. Fluorescence microscopy revealed that actinomycin D-induced release of AIF/3xFLAG was inhibited significantly by expression of mycBcl-2 or 6xHis-Bcl-XL (Figure 8A). The inhibitory effect of 6xHis-Bcl-XL on the apoptosis-dependent AIF release was further analyzed by immunoblotting. As shown in Figure 8B, actinomycin D induced processing of AIF/3xFLAG (lane 4), which was blocked by the expression of mycBcl-2 (lane 5) or 6xHis-Bcl-XL (lane 6). We then examined the effect of Bcl-XL on the releasing reaction. HeLa cells expressing AIF/3xFLAG or AIF/3xFLAG plus Bcl-XL were treated with staurosporine (Figure 8C, left panel) or etoposide/VP16 (right panel). They were then fractionated into cytosol and membrane fractions. In mock-treated cells, processing and release of AIF/3xFLAG were only at a background level, and the unprocessed form was recovered to the membrane fraction (Figure 8C, right and left panels, lanes 1 and 2). On the other hand, staurosporine or etoposide/VP16 treatment in the absence or presence of zVAD-fmk induced processing and cytoplasmic release of AIF/3xFLAG, which were both strongly compromised by the expression of Bcl-XL (right and left panels, lanes 3–6 and 9–12). zVAD-fmk prevented staurosporine- or etoposide/VP16-mediated PARP cleavage (left and right panels, lanes 4 and 6) (O'Brien et al, 2001). We then asked whether such efficient processing and release observed with exogenously expressed AIF/3xFLAG reflected in endogenous AIF. HeLa cells were treated with actinomycin D in the presence or absence of zVAD-fmk. They were then fractionated into soluble and mitochondrial fractions, which were subjected to immunoblot analysis for AIF. As shown in Figure 8D, AIF processing occurred in response to actinomycin D treatment and processed AIF was specifically released to the cytosol (lanes 2 and 3). The processing and release reactions were both completely blocked by expressing Bcl-XL (lanes 5 and 6). Altogether, these results indicated that Bcl-2 and Bcl-XL prevent apoptosis-dependent AIF release through the inhibition of proteolytic processing of AIF.
Discussion
AIF was originally identified and characterized as an apoptogenic factor localized in the mitochondrial IMS. The biogenesis of AIF is not yet clear. It has been suggested that AIF is synthesized as an ∼67-kDa protein with an N-terminal presequence of 101 residues that is proteolytically removed upon translocation into the mitochondria to generate the mature ∼57-kDa protein (Susin et al, 1999). With respect to molecular size, our observation that the mitochondrial mature AIF in the healthy condition is ∼62 kDa is not consistent with the previous report by Susin et al (1999), who reported it to be ∼57 kDa. In the size estimation by SDS–PAGE, we used (1) mitochondria prepared from both rat liver and HeLa cells in the healthy condition, (2) cell-free synthesized ΔN52AIF and ΔN101AIF, and (3) cell-free synthesized and MPP-treated AIF as the size references. Furthermore, protein sequencing revealed that alanine 54 in the AIF precursor represented the N-terminus of the mature AIF purified from nonapoptotic mitochondria. Thus, the 53-residue N-terminal segment of the AIF precursor is first proteolytically removed during or just after translocation into the mitochondrial IMS to generate the mitochondrial-type AIF. This processing is executed by MPP on the matrix side. The data obtained led us to conclude that the mitochondrial AIF is the ∼62-kDa mature form in the healthy condition. In contrast, the size of AIF detected in the cytoplasmic fraction in apoptotic conditions is ∼57 kDa. The apoptotic signal-dependent processing of the mitochondrial-type (mature) AIF and mitochondrial release of the processed form could be demonstrated not only for exogenously expressed AIF/3xFLAG but also for the endogenous form of AIF (see Figure 8C and D). According to the report by Susin et al (1999), the molecular sizes of both mitochondrial mature AIF and the cytoplasm-released form of AIF are identical, and the N-terminal sequence of AIF released from the mitochondria after opening the permeability transition pore starts from position 102 of the AIF precursor, which corresponds to the size of the mitochondria-released form in the present study. Why the two-step processing of AIF detected in our study was not observed in the previous report remains unclear.
What is the functional relevance of the two-step release mechanism? Mitochondria contain several apoptogenic factors such as cytochrome c, Smac/DIABLO, AIF, and HtrA2/Omi, and it has been proposed that Bcl-2 family members control apoptosis by regulating the release of these factors (Cory and Adams, 2002). Indeed, the release of cytochrome c from the mitochondria proceeds in a two-step fashion: detachment of cytochrome c from its membrane-anchoring lipid, cardiolipin, followed by release into the cytoplasm during permeabilization of the outer membrane (Ott et al, 2002). Similarly, AIF is anchored to the inner membrane, exposing the bulk of the oxidoreductase domain in the IMS under nonapoptotic conditions. Most likely, a protease facing the IMS is activated by diverse apoptotic signals and cleaves AIF, and the AIF fragment is released outside the mitochondria concomitant with the permeabilization of the outer membrane. In fact, we demonstrated that permeabilization of the outer mitochondrial membranes precedes AIF cleavage during apoptosis induction, that is, AIF release is limited by proteolytic processing of AIF in the IMS. Such a two-step process might function as a self-protective mechanism in the hierarchical progression of apoptosis.
As shown in this study, AIF is a type-I inner membrane protein with its N-terminus in the matrix and the bulk of the C-terminal portion in the IMS. The PSORT II program (Nakai and Horton, 1999) predicts that the 17-amino-acid region between positions 67 and 83 of the AIF precursor functions as the TMS. Our data on the membrane topology of AIF and its behavior during apoptosis induction are best explained by a model in which the IMS portion of AIF, consisting of the oxidoreductase domain, is proteolytically cleaved from the inner membrane and translocated into the cytoplasm (Figure 9). Apoptotic information from the cytoplasm would be transmitted by proapoptotic Bcl-2 family proteins to alter the mitochondrial environment by a yet unidentified mechanism, thus triggering a switch that activates the responsible protease or changes the AIF conformation to make it susceptible to the constitutively active protease. The AIF fragment is then translocated via the cytoplasm to the nucleus (Susin et al, 1999; Joza et al, 2001). Our data indicate that in order to substantiate the proapoptotic function of AIF, the mitochondria-resident unknown protease must first be activated by a diverse set of apoptotic stimuli. Intriguingly, mitochondrial release of AIF is blocked by Bcl-2 at the cleavage step in the IMS. Bcl-2 is a multifunctional inhibitor of apoptosis that has been suggested to act either on mitochondria or on the endoplasmic reticulum to confer cytoprotection (Zhu et al, 1996; Hacki et al, 2000; Annis et al, 2001). How the proteolytic processing of AIF is regulated by Bcl-2 in apoptosis remains to be determined.
Figure 9.
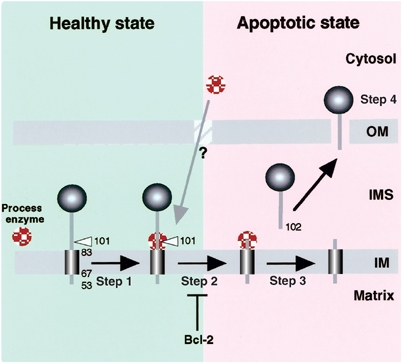
A model for proteolytic processing-dependent release of AIF from mitochondrial IMS upon induction of apoptosis. Apoptotic information from the cytoplasm is transmitted by proapoptotic Bcl-2 family proteins to alter mitochondrial environments (Step 1), thus triggering a switch that activates the responsible protease(s) or renders AIF accessible to constitutively active protease(s) (Step 2). The IMS portion of AIF is proteolytically cleaved from the inner membrane (Step 3). The mature AIF fragment is released from the mitochondrial IMS to the cytoplasm, possibly through a specific channel (Step 4). Bcl-2 blocks not only the initiation of the apoptotic process on the mitochondrial surface by proapoptotic Bcl-2 family proteins, but inhibits AIF processing in the IMS.
The mitochondrial protease responsible for apoptosis-dependent AIF processing has not yet been identified. The IMS-targeted preproteins that contain cleavable bipartite signal sequences undergo maturation in the IMS by the inner membrane protease complex (Imp). In yeast, Imp1p and Imp2p proteases are responsible for the maturation of cytochrome b2 and cytochrome c1, respectively (Schneider et al, 1991; Nunnari et al, 1993). Considering that AIF contains a bipartite-like IMS-sorting signal, the orthologs of yeast Imp1p or Imp2p might function in AIF proteolytic processing in mammals. In this case, the processing site of AIF might be cryptic under normal cell conditions, and, in apoptotic conditions, AIF might change its folding state to be proteolytically processed.
Recent studies demonstrated that yeast mitochondria contain a rhomboid protease (Rbd1/Pcp1) required for the cleavage of two mitochondrial IMS proteins, cytochrome c peroxidase and a dynamin-like GTPase, Mgm1p (Herlan et al, 2003, 2004; McQuibban et al, 2003; Sesaki et al, 2003; Freeman, 2004). It might be involved in the processing of AIF. It is also possible that a yet unidentified protease localized in the inner membrane or IMS is involved in this process. However, we cannot rigorously exclude another possibility that the potential AIF protease localizes in the cytoplasm and enters into the IMS to function upon induction of apoptosis. Polster et al (2004) recently demonstrated in vitro that calpain I induces cleavage and release of AIF from isolated mitochondria. Since they used recombinant AIF lacking the first 100 N-terminal residues (i.e., the mitochondria-released form; Susin et al, 1999; our present paper) as the substrate, calpain I seemed to cleave AIF precursor at downstream of leucine 101–glycine 102. The reason for this discrepancy is not known, and in vivo involvement of calpain I in the AIF processing remains to be analyzed.
Whether the deficiency of proteolytic processing of AIF is physiologically relevant or related to clinical phenotypes of apoptosis-related disorders also remains to be determined.
Materials and methods
Cell fractionation
Cell fractionation was performed using digitonin. Briefly, after washing twice with phosphate-buffered saline, the cultured cells were collected and treated with 0.2 mg/ml digitonin for 3 min at 37°C. The cytoplasmic and organellar fractions were separated by centrifugation and lysed with SDS sample buffer. More than 93% of cytosolic protein was recovered in the supernatant, and more than 98% of the mitochondrial protein was localized to the pellet. Rat liver and HeLa mitochondria were prepared as previously described (Sakaguchi et al, 1992).
Cell culture, cDNA transfection, and morphologic analysis
HeLa cells were cultured in DMEM supplemented with 10% fetal calf serum under 5% CO2, 95% air. DNA transfection to cells was carried out by Lipofectamine or Lipofectamine 2000 (Gibco BRL) as recommended by the manufacturer. A stable transformant of HeLa cells expressing 6xHis-Bcl-XL (termed HeLa/XL) in pIRESpuro3 vector (Clontech) was isolated by selection in the presence of 5 μg/ml puromycin. For immunocytochemistry analysis, HeLa cells were seeded onto glass coverslips. After a 2-day culture, coverslips were placed on a glass slide in the mounting medium and observed by confocal fluorescence microscopy. Antigen–antibody complexes were detected using fluorescein isothiocyanate-labeled goat anti-rabbit IgG antibody and Texas red-labeled goat anti-mouse IgG antibody. EGFP was directly observed by confocal fluorescence microscopy. Cells were fixed with 4% paraformaldehyde and then permeabilized with either 0.4 mg/ml digitonin, 2.0 mg/ml digitonin, or 1% Triton X-100 in PBS at room temperature for 5 min. The nuclei were detected by 1 μg/ml of DAPI. Semipermeabilization of HeLa cell plasma membranes was performed as follows. Cells were permeabilized with 25 μg/ml digitonin in homogenization buffer (20 mM HEPES–KOH (pH 7.5), 0.25 mM sucrose, 2.5 mM magnesium acetate, 25 mM KCl, 2.5 mM EGTA, and 1 μM taxol) at room temperature for 10 min and then immunostained with the desired antibodies.
Induction of apoptosis
HeLa cells were exposed to 20 μM actinomycin D (Sigma), 200 μM etoposide (Sigma), or 1 μM staurosporine (Sigma) with or without 100 μM zVAD-fmk (Peptide Institute, Osaka, Japan).
Supplementary Material
Supplemental Information
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Acknowledgments
We thank M Shirane and K Nakayama (Kyushu University), and A Ito (Kyushu University) for providing Bcl-2 cDNA and purified MPP, respectively. We also thank S Shimizu and Y Tsujimoto (Osaka University), and T Oka for helpful discussions. This work was supported by grants from the Ministry of Education, Science, and Culture of Japan, from the Human Frontier Science Program, Core Research from Evolutional Science and Technology, and Takdea Science Foundation.
References
- Annis MG, Zamzami H, Zhu W, Penn LZ, Kroemer G, Leber B, Andrews DW (2001) Endoplasmic reticulum localized Bcl-2 prevents apoptosis when redistribution of cytochrome c is a late event. Oncogene 20: 1939–1952 [DOI] [PubMed] [Google Scholar]
- Arnoult D, Gaume B, Karbowski M, Sharpe JC, Cecconi F, Youle RJ (2003) Mitochondroial release of AIF and Endo G requires caspase activation downstream of Bax/Bak-mediated permeabilization. EMBO J 22: 4385–4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnoult D, Parone P, Martinou J-C, Antonsson B, Esaquier J, Ameisen JC (2002) Mitochondrial release of apoptosis-inducing factor occurs downstream of cytochrome c release in response to several proapoptotic stimuli. J Cell Biol 159: 923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beasley E, Müller MS, Schatz G (1993) The signal that sorts cytochrome b2 to the mitochondrial intermembrane space contains three functional regions. EMBO J 12: 2303–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cory S, Adams JM (2002) The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 9: 647–656 [DOI] [PubMed] [Google Scholar]
- Cregan S, Fortin PA, MacLaurin JG, Callaghen SM, Cecconi F, Yu S-W, Dawson TM, Dawson VL, Park DS, Kroemer G, Slack RS (2002) Apoptosis-inducing factor is involved in the regulation of caspase-independent neuronal cell death. J Cell Biol 158: 507–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, Wang X (2000) Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102: 33–42 [DOI] [PubMed] [Google Scholar]
- Freeman M (2004) Proteolysis within the membrane: rhomboids revealed. Nat Rev Mol Cell Biol 5: 188–197 [DOI] [PubMed] [Google Scholar]
- Hacki J, Egger L, Monney L, Conus S, Rosse T, Fellay I, Borner C (2000) Apoptotic crosstalk between the endoplasmic reticulum and mitochondria controlled by Bcl-2. Oncogene 19: 2286–2295 [DOI] [PubMed] [Google Scholar]
- Hegde R, Srinivasa M, Zhang Z, Wassell R, Mukattsh R, Cilenti L, DuBois G, Lazebnik Y, Zervos A, Fernandes-Alnemri T, Alnemri ES (2002) Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein–caspase interaction. J Biol Chem 277: 432–438 [DOI] [PubMed] [Google Scholar]
- Herlan M, Bornhovd C, Hell K, Neupert W, Reichert AS (2004) Alternative topogenesis of Mgm1 and mitochondrial morphology depend on ATP and a functional import motor. J Cell Biol 165: 167–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlan M, Vogel F, Bornhövd C, Neupert W, Reichert AS (2003) Processing of Mgm1 by the rhomboid-type protease Pcp1 is required for maintenance of mitochondrial morphology and mitochondrial DNA. J Biol Chem 278: 22781–27788 [DOI] [PubMed] [Google Scholar]
- Jensen RE, Schmidt S, Mark RJ (1992) Mutations in a 19-amino acid hydrophobic region of the cytochrome c1 presequence prevent sorting to the mitochondrial intermembrane space. Mol Cell Biol 12: 4677–4686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joza N, Susin SA, Daugas E, Stanfore WL, Cho SK, Li CYJ, Sasaki T, Ella AJ, Cheng HYM, Ravagnan L, Ferri KF, Zamzami N, Wajeham A, Hakem R, Yoshida H, Kong Y-Y, Mak TW, Ziga-Pflicker JC, Kroemer G, Penninger JM (2001) Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature 410: 549–554 [DOI] [PubMed] [Google Scholar]
- Klein JA, Long-Guess CM, Rossmann MP, Seburn KL, Hurd RE, Franke WN, Bronwon RT, Ackerman SL (2002) The harlequin mouse metation down-regulates apaoptosis-inducing factor. Nature 419: 367–374 [DOI] [PubMed] [Google Scholar]
- Knight DE, Scrutton MC (1986) Gaining access to the cytosol: the technique and some applications of electropermeabilization. Biochem J 234: 497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Bossy-Wetzel W (2002) Dueling activities of AIF in cell death versus survival: DNA binding and redox activity. Cell 111: 147–150 [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X (1996) Induction of apoptotic program in cell-free extracts: requiremrnt for dATP and cytochrome c. Cell 86: 147–157 [DOI] [PubMed] [Google Scholar]
- Loeffler M, Daugas E, Susin SA, Zamzami N, Metivier D, Nieminen AL, Brothers G, Penninger JM, Kroemer G (2001) Dominant cell death induction by extramitochondrially targeted apoptosis-inducing factor. FASEB J 15: 758–767 [DOI] [PubMed] [Google Scholar]
- McQuibban GA, Saurya S, Freeman M (2003) Mitochondrial membrane remodelling regulated by a conserved rhomboid protease. Nature 423: 537–541 [DOI] [PubMed] [Google Scholar]
- Nakai K, Horton P (1999) Psort: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci 24: 34–36 [DOI] [PubMed] [Google Scholar]
- Newmeyer DD, Ferguson-Miller S (2003) Mitochondria: releasing power for life and unleashing the machineries of death. Cell 112: 481–490 [DOI] [PubMed] [Google Scholar]
- Nunnari J, Fox TD, Walter P (1993) A mitochondrial protease with two catalytic subunits of nonoverlapping specificities. Science 262: 1997–2003 [DOI] [PubMed] [Google Scholar]
- O'Brien MA, Moravec RA, Riss TL (2001) Poly (ADP-ribose) polymerase cleavage monitored in situ in apoptotic cells. Biotechniques 30: 886–891 [DOI] [PubMed] [Google Scholar]
- Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S (2002) Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA 99: 1259–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polster BM, Etxebarria A, Basanez G, Hardwick JM, Nicolls DG (2004) Calpain I induces cleavage and release of apoptosis inducing factor from isolated mitochondria. J Biol Chem 280: 6447–6454 [DOI] [PubMed] [Google Scholar]
- Sakaguchi M, Hachiya N, Mihara K, Omura T (1992) Mitochondrial porin can be translocated across both endoplasmic reticulum and mitochondrial membranes. J Biochem 112: 243–248 [DOI] [PubMed] [Google Scholar]
- Schneider A, Behrens M, Scherer PE, Pratje E, Michelis G, Schatz G (1991) Inner memebrane protease I, an enzyme mediating intramitochondrial sorting in yeast. EMBO J 10: 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sesaki H, Southard SM, Hobbs AE, Jensen RE (2003) Cells lacking Pcp1p/Ugo2p, a rhomboid-like protease required for Mgm1 processing, lose mtDNA and mitochondrial structure in a Dnm1-dependent manner, but remain competent for mitochondrial fusion. Biochem Biophys Res Commun 308: 276–283 [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G (1999) Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 397: 441–446 [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R (2001) A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell 8: 613–621 [DOI] [PubMed] [Google Scholar]
- Taylor AB, Smith BS, Kitada S, Kojima K, Miyaura H, Otwinowski Z, Ito A, Deisenhofer J (2001) Ctystal structure of mitochondrial procesing peptidase reveal the mode for specific cleavage of import signal sequences. Structure 9: 615–625 [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL (2000) Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to an antagonizing IAP proteins. Cell 102: 43–53 [DOI] [PubMed] [Google Scholar]
- Verhagen AM, Silke J, Ekert PG, Pakusch M, Kaufmann H, Connolly LM, Day CL, Tikoo A, Burke R, Wrobel C, Moritz RL, Simpson RJ, Vaux DL (2002) HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J Biol Chem 277: 445–454 [DOI] [PubMed] [Google Scholar]
- Ye H, Cande C, Stephanou NC, Jiang S, Gurbuxani S, Larochenette N, Daugas D, Garrido C, Kroemer G, Wu H (2002) DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat Struct Biol 9: 680–684 [DOI] [PubMed] [Google Scholar]
- Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DW (1996) Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J 15: 4130–4141 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Information
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3