

Abstract
The endoplasmic reticulum (ER) can elicit proapoptotic signalling that results in transmission of Ca2+ to the mitochondria, which in turn stimulates recruitment of the fission enzyme DRP1 to the surface of the organelle. Here, we show that BH3-only BIK activates this pathway at the ER in intact cells, resulting in mitochondrial fragmentation but little release of cytochrome c to the cytosol. The BIK-induced transformations in mitochondria are dynamic in nature and involve DRP1-dependent remodelling and opening of cristae, where the major stores of cytochrome c reside. This novel function for DRP1 is distinct from its recognized role in regulating mitochondrial fission. Selective permeabilization of the outer membrane with digitonin confirmed that BIK stimulation results in mobilization of intramitochondrial cytochrome c. Of note, BIK can cooperate with a weak BH3-only protein that targets mitochondria, such as NOXA, to activate BAX by a mechanism that is independent of DRP1 enzyme activity. When expressed together, BIK and NOXA cause rapid release of mobilized cytochrome c and activation of caspases.
Keywords: Bax, calcium, caspase, fission, NOXA
Introduction
Elimination of cells that are either damaged or represent a potential threat to an organism, such as pre-cancerous cells, is achieved through a programmed form of cell death called apoptosis. Intrinsic apoptotic stress signals in these cells activate the mitochondrial death pathway through their influence on members of the proapoptotic BH3-only subgroup of the BCL-2 protein family. These proteins induce the release of cytochrome c from the mitochondria, which then binds to Apaf-1, a cytosolic adaptor protein, leading to recruitment and activation of caspase-9 (Kaufmann and Hengartner, 2001). BH3-only BCL-2 members have the potential to gain direct access to the core mitochondrial apoptosis machinery because, in their active conformation, they regulate the ability of the multi-BH domain members, BAX and BAK, to oligomerize in the mitochondrial outer membrane and release intermembrane proteins, including cytochrome c, to the cytosol (Wei et al, 2000, 2001). Antiapoptotic BCL-2 proteins, on the other hand, can form dimers with the activated conformers of proapoptotic members and block this progression of events at the mitochondrial surface (Letai et al, 2002; Ruffolo and Shore, 2003).
Other cellular pathways can cooperate with this core mitochondrial machinery to enhance apoptosis. One example was described by Youle and colleagues who found that recruitment and activation of dynamin-related protein-1 (DRP1) at discrete sites on the mitochondrial tubular network in intact cells contributed to egress of cytochrome c to the cytosol and apoptosis (Frank et al, 2001). DRP1 is a GTPase that causes scission of the mitochondrial outer membrane, resulting in fission of mitochondrial tubules into punctiform fragments (reviewed in Bossy-Wetzel et al, 2003; Karbowski and Youle, 2003). The exact role of DRP1 in mitochondrial apoptosis remains to be determined, however, and the recent findings that multiple members of the mitochondrial fusion–fission machinery are also involved (Olichon et al, 2003; Lee et al, 2004; Sugioka et al, 2004; Szabadkai et al, 2004) suggest a complex interplay between these regulators.
DRP1 recruitment to the mitochondria and subsequent mitochondrial fission can be induced by the release of Ca2+ from endoplasmic reticulum (ER) stores and subsequent uptake by mitochondria, which in one case is initiated by caspase-8 cleavage of BAP31, an integral protein of the ER membrane (Breckenridge et al, 2003b). These calcium- and DRP1-dependent events initiated by the p20 cleavage product of BAP31 cooperate with other caspase-8-initiated pathways, notably conversion of endogenous BID to its active conformer, tBID, to enhance the release of cytochrome c to the cytosol. The mechanism underlying the Ca2+- and DRP1-dependent sensitization of mitochondria to direct proapoptotic insults, however, is poorly understood. Nevertheless, given the probable universality of ER Ca2+ signalling during apoptosis (Ferrari et al, 2002; Breckenridge et al, 2003a; Orrenius et al, 2003), it is likely that such a two-hit mechanism for mitochondrial apoptosis in intact cells extends to other death pathways, and involves regulators at the ER and mitochondria commensurate with the death stimulus in question.
Insights into this question have come from biochemical studies of the cytochrome c release pathway in isolated mitochondria. Treatment of these mitochondria in vitro with recombinant tBID resulted in opening of cristae and mobilization of intracristae stores of cytochrome c, which could then leak out of the organelle by a BAX, BAK-dependent mechanism (Scorrano et al, 2002). Since in the absence of a death stimulus mitochondria typically contain closed cristae, remodelling of these cristae to mobilize intramitochondrial cytochrome c, therefore, is likely to be a hallmark of most pathways that operate through the mitochondrial death mechanism.
Interestingly, BCL-2 homologs can be found at the ER in addition to their well-characterized mitochondrial localization. One such example is the BH3-only protein BIK, which can stimulate a mitochondria-dependent apoptotic pathway from an ER location (Germain et al, 2002). We demonstrate here that BIK initiates early Ca2+-mediated remodelling of inner membrane cristae and fragmentation of mitochondria. Opening of cristae in response to BIK correlates with mobilization of intraorganellar cytochrome c stores. Additionally, BIK can cooperate with a weak mitochondria-targeted BH3-only protein, NOXA, to conformationally activate BAX. Combined, BIK and NOXA stimulate robust release of mobilized cytochrome c from the organelle and activation of effector caspases. The early remodelling of mitochondria cristae that arises in response to BIK expression is inhibited in the presence of the dominant interfering DRP1(K38E), suggesting a novel function for DRP1 in the regulation of cytochrome c release from mitochondria and induction of apoptosis.
Results
BH3-only BIK contains a hydrophobic transmembrane (TM) segment at its extreme C-terminus (Boyd et al, 1995; Han et al, 1996). Upon induction of the endogenous protein or expression from an ectopic vector, BIK is found primarily as an integral protein of the ER membrane and, given sufficient time, it can induce caspase-dependent apoptosis from this location (Germain et al, 2002; Mathai et al, 2002). Substitution of the BIK TM segment with the ER-selective TM of cytochrome b5 (Flag-BIKb5) generated a protein that also induces apoptosis over time, confirming that BIK can function at the ER membrane (Germain et al, 2002). Of note, however, ectopic expression of BIK in H1299 cells utilizing the Adenovirus vector Ad HA-BIK caused induction of effector caspase activity (DEVDase), but robust caspase activity lagged by several hours the appearance of BIK protein (Figure 1A and B). BCL-2 that was selectively targeted to the ER utilizing the cytochrome b5 TM retarded BIK-induced caspase activity and permitted an enhanced accumulation of BIK in these cells (see also Mathai et al, 2002). Given the potential role of early ER signals in sensitizing mitochondria to apoptotic stimuli (Breckenridge et al, 2003a, 2003b), we thus focused on time points shortly after the appearance of BIK protein (10–12 h post-infection with Ad HA-BIK) to explore the influence of BIK on these signals. The levels of BIK that were expressed in response to infection of H1299 cells with Ad BIK were similar to the levels of endogenous BIK that resulted following a cell stress stimulus such as overexpression of p53 (Mathai et al, 2002).
Figure 1.
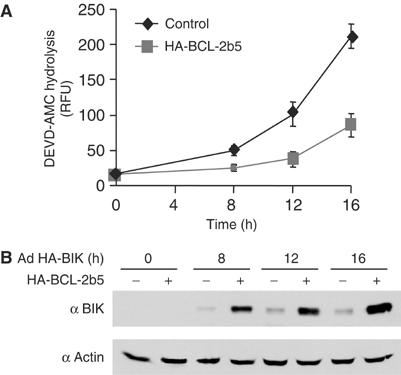
Time course of caspase induction by Ad HA-BIK. (A) H1299 cells stably transfected with vector or vector expressing HA-BCL-2b5 were infected with Ad HA-BIK for the indicated times, after which caspase activity was measured using the fluorescent substrate DEVD-AMC. The results of three independent determinations±s.d. are shown. (B) Expression of BIK in Ad HA-BIK-infected cells. Cells were treated as in (A), proteins were resolved by SDS–PAGE and probed with antibodies against BIK and actin (as a loading control).
Induction of mitochondrial fission by BIK through Ca2+ signals
Since the ER is a major store of intracellular Ca2+ and BCL-2 homologs have been shown to influence ER Ca2+ stores, the early effect of BIK expression on these stores was analyzed. To avoid the potential influences of downstream effector caspases, resulting in possible feedback amplification of responses to BIK, all subsequent experiments were carried out in the presence of the broad-spectrum caspase inhibitor zVAD-fmk, which completely abrogates caspase-3-like (DEVDase) activity induced by BIK (data not shown; see also Germain et al, 2002). H1299 cells were infected with Ad HA-BIK for 12 h in the presence of zVAD-fmk, and then loaded with the Ca2+-sensitive dye Fura-2 to measure cytosolic Ca2+ levels. ER Ca2+ stores were measured as the difference in [Ca2+]cytosolic before and after the addition of thapsigargin (TG), an inhibitor of the SERCA pump that rapidly depletes ER of releasable Ca2+ (Mery et al, 1996; Breckenridge et al, 2003b). Compared to mock-infected cells, expression of HA-BIK resulted in a reduction of ER Ca2+, while the disabled BH3 mutant HA-BIK(L61G), which was expressed at levels higher than wild-type (wt) BIK (Mathai et al, 2002), had a much reduced effect (Figure 2A).
Figure 2.
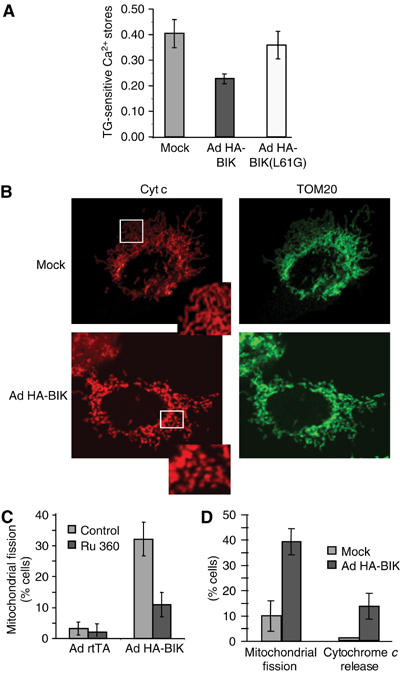
BIK induces changes in the morphology of mitochondria dependent on Ca2+ signals. (A) H1299 cells were infected for 12 h with Ad HA-BIK in the presence of zVAD-fmk, then loaded with the Ca2+-sensitive dye Fura-2. Cytopasmic Ca2+ was measured before and after addition of 2 μM TG, utilizing the 340/380 nm excitation ratio at 510 nm emission wavelength. Results are expressed as the difference in the 510 nm ratio before and after addition of TG. The results of at least five independent determinations±s.d. are shown. (B) Mitochondrial fragmentation and cytochrome c release in Ad HA-BIK-infected cells. Cells were treated with Ad HA-BIK for 12 h in the presence of zVAD-fmk, double-stained with antibodies against mitochondrial TOM20 (Alexa-Fluor 488 (Green)) and cytochrome c (Alexa-Fluor 495 (red)) and examined by immunofluorescence. Representative images are shown. (C) The mitochondrial Ca2+-uptake inhibitor Ru360 prevents BIK-induced mitochondrial fragmentation. H1299 cells were infected for 12 h with either a control adenovector (Ad rtTA) or Ad HA-BIK in the presence of zVAD-fmk and either the absence or presence of 20 μM Ru360. Cells were fixed, stained with the mitochondrial marker TOM20 and the percentage of cells with mitochondrial fission was determined. The results of three independent experiments are shown. (D) Quantification of mitochondrial fission and cytochrome c release from the immunofluorescence shown in (B). The results of three independent determinations±s.d. are shown.
In the Fas death pathway, cleavage of BAP31 at the ER by caspase-8 induces ER Ca2+ release and concomitant uptake of Ca2+ by mitochondria, leading to DRP1-mediated mitochondrial fission (Breckenridge et al, 2003b). Following infection with Ad HA-BIK, mitochondria that stained with an antibody against either outer membrane TOM20 or intermembrane cytochrome c were likewise seen to undergo conversion from a tubular network to a fragmented pattern (Figure 2B). Fission was blocked by Ru360, an inhibitor of mitochondrial Ca2+ uptake (Figure 2C), indicating a dependence of these morphological changes on Ca2+ transmission into the organelle. Moreover, Ru360 inhibited longer term BIK-induced caspase (DEVDase) activation (data not shown), suggesting that the Ca2+ component of the BIK pathway is important for BIK's ability to ultimately trigger mitochondrial apoptosis in intact cells. Of note, expression of HA-BIK in mouse cells deleted of Bap31 (Breckenridge et al, 2002) caused activation of caspases (not shown) and, under the conditions employed here in human H1299 cells (i.e., inclusion of zVAD-fmk), did not result in BAP31 cleavage (Germain et al, 2002), arguing that the mitochondrial fission pathway stimulated by BIK does not involve BAP31. Finally, induction of endogenous cellular BIK in response to overexpression of p53 (Mathai et al, 2002) leads to release of ER Ca2+ and mitochondrial fission, which is inhibited by siRNA knockdown of BIK (Supplementary Figure 1). Thus, the ability of ectopic BIK to initiate this ER pathway is shared by endogenous BIK.
DRP1-mediated dynamic mitochondrial transformation is an early step in response to BIK
Quantification revealed that the mitochondrial fragmentation observed 12 h following Ad HA-BIK infection occurred in about 40% of the cells, while less than 15% of the cells exhibited release of cytochrome c to the cytosol (Figure 2D). While numerous cells with fragmented mitochondria exhibited no cytochrome c release (Figure 2B), all cells that showed cytochrome c release to the cytosol also had fragmented mitochondria (Supplementary Figure 2). This indicates that mitochondrial fragmentation in response to BIK precedes cytochrome c release.
Mitochondrial fission requires the activity of a dynamin-related protein called DRP1 and mutation of its catalytic GTPase active site has been shown to block mitochondrial fission and release of cytochrome c during apoptosis (Frank et al, 2001; Breckenridge et al, 2003b). We thus used dominant-negative DRP1 mutant (CFP-DRP1(K38E); Breckenridge et al, 2003b) in combination with live-cell imaging to assess further the nature of DRP1-regulated changes in mitochondria following infection of H1299 cells with Ad HA-BIK for 12 h. Imaging of mitochondria in live cells was achieved by expressing a pOCT-YFP fusion protein, which is selectively targeted to the mitochondrial matrix. In response to BIK expression, the mitochondria exhibited dramatic fragmentation (Figure 3A) and, of note, these fragmented mitochondria underwent multiple cycles of stretching and contraction. These dynamics are evident in the lower panels of Figure 3A, which capture the image of several fragments over 125 photo frames taken at 30 frames/min and can be seen as a continuous movie in Supplementary video 1. In contrast, cells infected with Ad HA-BIK in the presence of CFP-DRP1(K38E) showed a fused mitochondrial morphology (Figure 3B) (Smirnova et al, 1998; Yoon et al, 2001) and the dynamic mitochondrial stretching and contractions seen with BIK alone were not observed (Figure 3B, bottom panel; see also Supplementary video 2).
Figure 3.
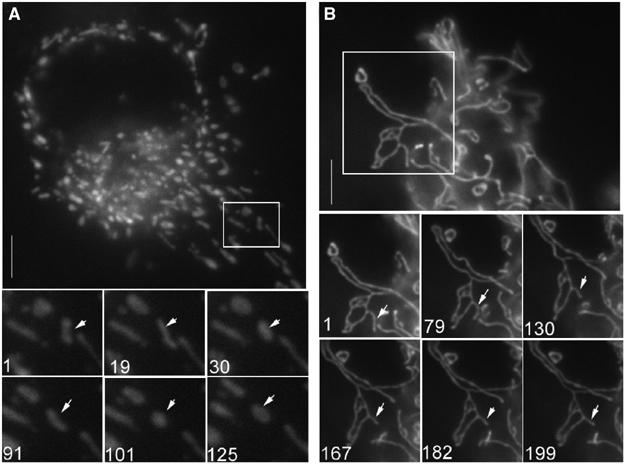
BIK induces DRP1-dependent mitochondrial fission. H1299 cells were infected for 12 h with Ad HA-BIK in presence of zVAD-fmk and in the absence (A) or presence (B) of CFP-DRP1(K38E). Mitochondria were visualized using pOCT-YFP. Images were acquired every 2 s from the boxed region of the cell presented in (A) and (B) to visualize the dynamics of mitochondrial fragments (see video 1 (panel A) and video 2 (panel B)). Bottom images represent stills taken from these movies, the numbers indicating frame number. Representative images are shown.
BIK-induced recruitment of DRP1 to the mitochondria precedes loss of ΔΨ and fragmentation
As shown in Figure 4 and Supplementary video 3, the primarily cytosolic DRP1-YFP was recruited to the mitochondria marked with pOCT-CFP early following infection of COS7 cells with Ad HA-BIK. The first noticeable change in mitochondrial morphology following recruitment was that the nonfragmented organelles began to exhibit dynamic alterations in shape, alternating between tubular and globular structures (Figure 4, upper panel; Supplementary video 3). These morphological changes occurred more gradually relative to the fast stretching and contraction observed in the fragmented organelles imaged at later times after Ad HA-BIK infection (compare Supplementary videos 1 and 3). DRP1-YFP remained quite stably recruited to these structures. Addition of the electrochemical-sensitive dye MitofluorRed589 indicated that DRP1-YFP recruitment clearly preceded the subsequent loss in potential following infection by Ad HA-BIK and that DRP1 remained mitochondria-associated following the loss of ΔΨ (Figure 4, bottom panel), with fragmentation occurring at a later stage.
Figure 4.
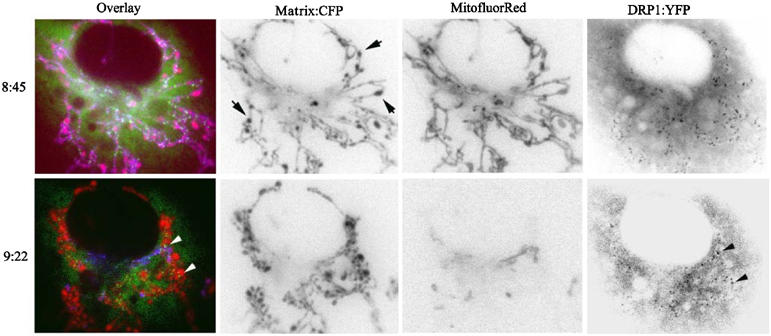
DRP1 recruitment to mitochondria precedes morphological changes and loss of electrochemical potential. COS-7 cells were co-transfected with pOCT-CFP and DRP1-YFP prior to infection with Ad BIK for 8 h. In all, 50 nM of the electrochemical potential sensitive dye, MitofluorRed589, was added and equilibrated at 37°C for 30 min prior to imaging. Three channel videos were taken of the pOCT-CFP (mitochondria, shown red in overlay), MitofluorRed589 (in the presence of ΔΨ, stains the mitochondria purple in overlay), and DRP1-YFP (shown green in overlay). Images were extracted from the series and the time stamps relative to the addition of Ad BIK are shown in hours:minutes. Transition of mitochondria from purple to red indicates a loss of ΔΨ. Arrows indicate areas where the mitochondria appear swollen (upper panel) and point to puncta of mitochondrial-associated DRP1-YFP (lower panel). Black and white images were inverted using Adobe Photoshop.
BIK induces DRP1-regulated remodelling of mitochondria cristae
Although DRP1-mediated transformations of mitochondria are required for cytochrome c release during apoptosis, the reason is poorly understood. It is known, however, that several apoptotic stimuli cause disruption of cristae junctions, resulting in opening of the cristae and an expansion of the intermembrane space (Scorrano et al, 2002; Akao et al, 2003; Kim et al, 2004). As cristae are typically compact tubular structures that trap the majority of cytochrome c molecules within the folds of the intermembrane space, opening of cristae thus increases the accessibility of cytochrome c to channels in the outer membrane (Scorrano et al, 2002).
To examine the state of mitochondrial cristae in intact cells following infection with Ad HA-BIK for 12 h, we employed transmission electron microscopy and measured intracristae cross-sectional distances (ICDs) in the different experimental conditions. In control COS-7 cells, cristae are typically found as compact tubules within mitochondria (Figure 5A; untreated), with the majority (>80%) of the ICDs in mock-infected cells measuring between 20 and 75 nm (Figure 5C). BIK expression caused profound opening of the cristae (seen as an enlarged electron transparent space in Figure 4A; Ad BIK). In these cells, greater than 80% of the ICDs ranged between 75 and 250 nm (Figure 5C). High-resolution immunogold staining of these mitochondria with antibody against YFP-pOCT revealed gold particles associated exclusively with the electron dense matrix compartment (Figure 5A; Matrix YFP-gold), confirming that the enlarged electron transparent spaces did not represent matrix swelling. Importantly, the structural changes in cristae induced by BIK were strongly inhibited by CFP-DRP1(K38E) (Figure 5A; Ad BIK/Ad DRP1(K38E)), where more than 90% of the ICDs were between 20 and 50 nm (Figure 5C). Of note, the inhibitor of Ca2+ uptake by mitochondria, Ru360, also inhibited BIK-induced opening of cristae (Figure 5A; Ad BIK+Ru360). Like controls, the ICDs were found to be predominantly less than 100 nm (Figure 5C), and similar to those recorded following infection of the cells with the disabling BH3 mutant of BIK, Ad BIK(L61G) (Figure 5A and C). Consistent with studies in vitro (Scorrano et al, 2002), an antagonist of the inner membrane permeability transition pore, cyclosporin A, was also inhibitory (Figure 5A and C). BIK-induced changes in cristae morphology, therefore, are dependent on BIK's BH3 domain, Ca2+ transmission to mitochondria, the PTP, and functional DRP1.
Figure 5.
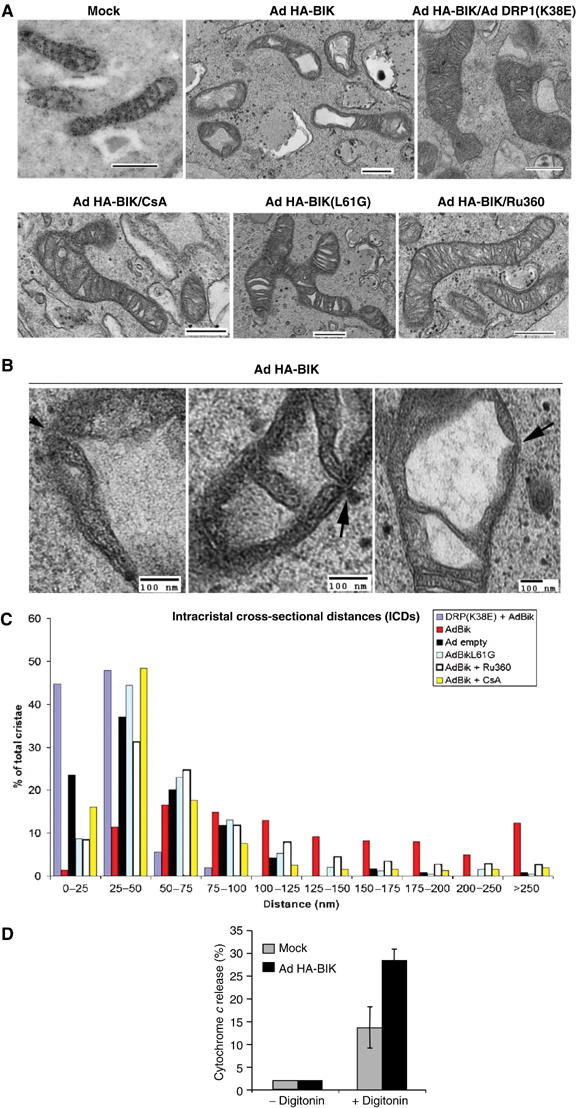
BIK induces DRP1-dependent remodelling of mitochondrial cristae. (A) COS-7 cells were transfected with pOCT-CFP, fixed and prepared for immunolabelling with anti-CFP antibodies decorated with 10 nm gold-labelled secondary antibodies to morphologically distinguish the matrix compartment from the electron translucent intermembrane space (top left). The remaining panels show representative images of COS-7 cells without immunolabelling that were treated for 12 h in the presence of zVAD-fmk with either mock adenovirus, Ad HA-BIK(L61G), or Ad HA-BIK in the presence or absence of Ad DRP1(K38E) or 20 μM Ru360 or 2 μM cyclosporin A (CsA, added 2 h after infection) as indicated. The cells were fixed and mitochondria morphology was examined by electron microscopy. (B) High magnifications of mitochondria in cells infected with Ad HA-BIK for 12 h. Arrows indicate sections where the vacuolized intermembrane space is directly accessible to the outer membrane. (C) Quantification of the intracristal cross-sectional distances expressed as a frequency distribution. Data were acquired using the AMT integrated software. (D) BIK enhances release of cytochrome c by digitonin. H1299 cells were either mock infected (control) or infected for 10 h with Ad HA-BIK in the presence of zVAD-fmk, after which the mitochondria were isolated and incubated with 60 μg/ml digitonin for 5 min at 37°C. The amount of cytochrome c released from the mitochondria was determined by immunoblotting, as described in Materials and methods. The average of three independent determinations±s.d. is shown.
Extensive examination of EM cross sections of mitochondria in Ad HA-BIK-infected COS-7 cells also resulted in numerous images documenting an increased ICD at the otherwise narrow tubular junctions between the cristae and the intermembrane space in Ad BIK-infected cells (Figure 5B). These data indicate that the vacuolated intermembrane space has direct access to the outer membrane. Moreover, if these junctions are indeed enlarged following BIK treatment, it would predict that BIK leads to enhanced mobilization and release of cytochrome c stores following a physical breach of the outer membrane. To assess this possibility, mitochondria were isolated from control or BIK-stimulated cells and examined for their susceptibility to digitonin-mediated release of cytochrome c. Titration of digitonin was conducted with a fixed concentration of mitochondria protein to select a concentration of detergent that permeabilized the outer membrane (OM) without causing significant release of OM TOM20, which would indicate a removal of the OM itself. In multiple independent experiments, mitochondria from unstimulated cells released about 12% of total cytochrome c in response to digitonin (Figure 5D), consistent with estimates that about 10–15% of the cytochrome c content of mitochondria is located in the intermembrane space (Bernardi and Azzone, 1981; Scorrano et al, 2002). On the other hand, mitochondria isolated from BIK-treated cells released 30% of their cytochrome c in the same conditions (Figure 5D), in agreement with a portion of the isolated mitochondria having their cytochrome c potentially mobilized and releasable from the inner membrane (and consistent with a portion of the cells containing fragmented mitochondria but no cytochrome c release, see Figure 2). Moreover, this may be an underestimate of releaseable cytochrome c, since a high-salt buffer was not used in these experiments. These results indicate that an early response to BIK expression is mobilization of cytochrome c stores within the mitochondria, consistent with the effect of BIK on opening of cristae.
BIK and NOXA cooperate to release cytochrome c from the mitochondria
The DRP1-dependent opening of mitochondrial cristae and mobilization of cytochrome c described here could provide an explanation for the requirement of DRP1-mediated events during apoptosis. For example, the DRP1-regulated mobilization of the cytochrome c pool in response to BIK in intact cells might make these mitochondria more sensitive to BAX,BAK-dependent release of cytochrome c induced by a second proapoptotic stimulus operating at the organelle. This could be particularly relevant in situations where the direct mitochondrial stimulus is suboptimal and does not itself lead to both rapid cristae remodelling and activation of BAX,BAK. To this end, we examined NOXA, another BH3-only protein that, like BIK, is induced by overexpression of p53 and has been shown to localize to mitochondria (Oda et al, 2000). Unlike tBID, delivery of NOXA to the mitochondria in vitro does not result in strong release of cytochrome c (Cheng et al, 2001). Adenovectors expressing either the wt human NOXA or a mutant in which the conserved leucine at position 29 in the BH3 domain was changed to an alanine, NOXA(L29A), were generated. Infection of H1299 cells with Ad NOXA for 48 h ultimately resulted in caspase activation (Supplementary Figure 3A), but this was not observed at the early time point of 10–12 h (Figure 6A). In addition, expression of human NOXA for 12 h did not induce mitochondrial fission (Supplementary Figure 3B) and did not cause release of ER Ca2+stores (Supplementary Figure 3C). In contrast, after infection of H1299 cells with both Ad HA-BIK and Ad NOXA for 10 h, there was a significant increase in both cytochrome c release to the cytosol and caspase (DEVDase) activation (Figure 6A). On the other hand, infections with twice as much of either virus alone did not result in a similar increase in caspase activation (Figure 6B). BIK and NOXA, therefore, can cooperate to induce efficient cytochrome c release from mitochondria. The presence of NOXA did not further increase BIK-induced ER Ca2+ release or mitochondrial fission (Supplementary Figure 3). The cooperation between BIK and NOXA was dependent on the presence of an intact BH3 domain in both proteins as either of the BH3 mutants, BIK(L61G) or NOXA(L29A), showed impaired cytochrome c release and caspase activation when co-expressed with its Wt partner (Figure 6A). In contrast, no cooperation was observed when Ad NOXA was co-expressed with Ad tBID for 10 h, but robust activation of caspases was observed (Figure 6C). BID is a potent BH3-only protein that targets the mitochondria and, at least in vitro, can promote both mobilization of internal stores of cytochrome c and its release from the organelle through the activation of BAX,BAK (Wei et al, 2001; Scorrano et al, 2002).
Figure 6.
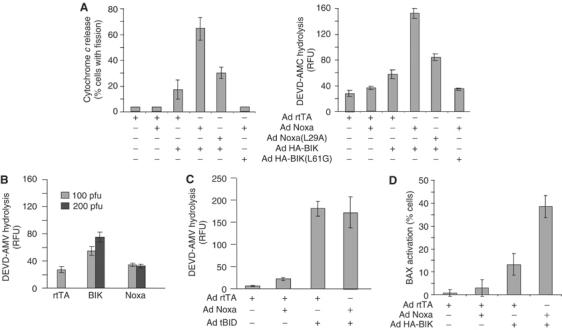
BIK and NOXA can cooperate to induce cytochrome c release from mitochondria. (A) Cytochrome c release and caspase activation by BIK and Noxa. H1299 cells were infected for 10 h with Ad HA-BIK and Ad Noxa or their respective BH3 mutants (100 pfu/cell each virus) in the presence of zVAD-fmk. Cells were then fixed and stained with antibodies against cytochrome c and TOM20. Cells that had their mitochondria fragmented were scored for cytochrome c release. Alternatively, 25 μg lysate from cells treated in the absence of zVAD-fmk was used to measure caspase-3-like activity using the fluorometric substrate DEVD-AMC. The results of at least three independent experiments±s.d. are shown. (B) H1299 cells were infected with either 100 or 200 pfu/cell of the indicated Ad vector and processed as in (A) for measuring caspase-3-like activity. (C) Cells were treated with Ad Noxa and Ad tBID and caspase-3-like activity was evaluated as in (A). (D) H1299 cells were treated and fixed as in (A), after which they were stained with an N-terminal-specific BAX antibody. Cells positive for activated BAX were scored. The result of three independent determinations±s.d. is shown.
BAX stimulation results in the exposure of an epitope at the N-terminus of the protein that can be detected by immunofluorescence using an epitope-specific antibody (Desagher et al, 1999; Wang et al, 2003). Cells infected with Ad HA-BIK and Ad NOXA either alone or in combination for 10 h were fixed, stained with the N-terminal-specific antibody and BAX-positive cells were scored. As with cytochrome c release, BIK and NOXA had an enhanced effect on BAX activation compared to either protein alone (Figure 6D). Thus, BIK on its own initiates rapid DRP1-regulated remodelling of cristae and mobilization of intracristae cytochrome c stores, but in addition BIK appears to work together with NOXA to somehow enhance the activation of BAX. Combined, this provides an explanation for the ability of BIK and NOXA to stimulate rapid egress of mobilized cytochrome c to the cytoplasm.
Involvement of DRP1 in the release of cytochrome c by BIK and NOXA
As shown in Figure 7A, dominant-negative CFP-DRP1(K38E) was able to inhibit cytochrome c release to the cytosol induced by the combined action of BIK and NOXA, whereas CFP and CFP-DRP1 did not, indicating that cytochrome c release in response to the two BH3-only proteins depended on the functional DRP1 enzyme. In contrast, the conformational change in BAX correlating with exposure of its N-terminus was not affected by CFP-DRP1(K38E) (Figure 7B). This is in agreement with previous results (Karbowski et al, 2002) and consistent with the finding that RNAi knockdown of DRP1 does not block BAX translocation to the mitochondria (Lee et al, 2004), indicating that BAX activation is not regulated by catalytically active DRP1. It cannot be excluded, however, that DRP1 plays some role in cytochrome c transport across the outer membrane.
Figure 7.
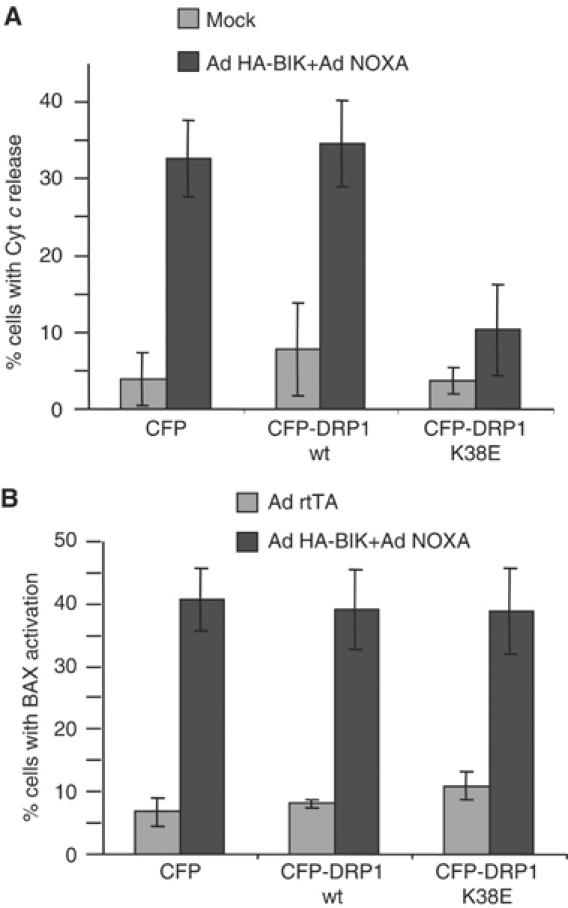
Influence of DRP1 on proapoptotic activities of BIK and Noxa. (A) CFP-DRP1(K38E) prevents the induction of cytochrome c release by BIK and Noxa. H1299 cells were transfected with either CFP, CFP-DRP1 or CFP-DRP1(K38E) and infected with Ad HA-BIK and Ad Noxa. Cells were fixed 10 h post-infection and stained for cytochrome c. Results are expressed as the percent of CFP-positive cells with cytosolic cytochrome c. The results of three independent experiments±s.d. are shown. (B) CFP-DRP1(K38E) does not prevent BAX activation. Cells were treated as in (A) and stained with the N-terminal-specific BAX antibody and scored for the presence of activated BAX. The results of three independent experiments±s.d. are shown. See text for an explanation.
Discussion
DRP-1-mediated transformations of mitochondria during apoptosis are requisite for cytochrome c release to the cytosol in intact cells (Frank et al, 2001). Ca2+ signalling from the ER, and in particular its uptake by mitochondria, provides one signal for this early recruitment of DRP1 to the mitochondrial surface (Breckenridge et al, 2003b; Karbowski and Youle, 2003). Utilizing dominant-negative CFP-DRP1(K38E), we showed here that these DRP1-mediated, dynamic changes in mitochondrial structure that are caused by BIK in fact included cristae remodelling. This DRP1-dependent remodelling requires transmission of Ca2+ to the mitochondria and a functional PTP. Opening of the cristae structure has proven necessary during apoptosis to provide a mobile pool of cytochrome c for BAX/BAK-dependent release of cytochrome c to the cytosol (Scorrano et al, 2002), and BIK expression was indeed associated with mobilization of mitochondrial stores of cytochrome c. Interestingly, the activating conformational change in BAX that occurred in response to the combined actions of BIK and NOXA was not influenced by DRP1(K38E) (see also Karbowski et al, 2002), and this is in agreement with a recent report showing that knockdown of DRP1 by RNAi did not interfere with apoptotic recruitment of BAX to mitochondria (Lee et al, 2004).
How DRP1, which acts at the surface of mitochondria, influences remodelling of cristae, however, is not known. Other dynamins, including dynamins 1 and 2, have also been reported to exhibit activities that are independent of their roles in membrane scission. These include the recruitment of proteins that affect lipid curvature (Schmidt et al, 1999; Farsad et al, 2001; Peter et al, 2004), modulate actin dynamics at the membrane (Lee and De Camilli, 2002; Orth et al, 2002; Orth and McNiven, 2003), and initiate intracellular signalling cascades (Fish et al, 2000; Schlunck et al, 2004). In terms of membrane remodelling activities, Endophilin B, a homolog of the dynamin 1-interacting lipid-modifying protein endophilin, was recently found to affect mitochondrial morphology, where reduction of the protein levels by RNAi led to extended tubules of mitochondrial outer membrane, apparently without any matrix material within the tubes (Karbowski et al, 2004). Like DRP1, translocation of the largely cytosolic Endophilin B to the mitochondria was stimulated following treatment of cells with staurosporine or actinomycin D (Karbowski et al, 2004). Whether or not endophilin B, or another regulator, may function with DRP1 and cause dynamic changes at the outer membrane, which influence remodelling of the cristae, remains to be determined. In addition, there could be an indirect link between DRP1 on the cytoplasmic face of the mitochondria with the intermembrane space GTPase, Opa1, another dynamin-related protein that has been implicated in the regulation of mitochondrial morphology (Guan et al, 1993; Delettre et al, 2000; Misaka et al, 2002; Olichon et al, 2002; Satoh et al, 2003). Interference with OPA1 expression induces changes in the mitochondrial structure such as swelling and stretching (Griparic et al, 2004) as well as cristae reorganization (Olichon et al, 2003; Griparic et al, 2004), that are similar to what is observed during apoptosis. Similarly, interference with OPA1 can indeed enhance apoptotic cytochrome c release from the organelle (Olichon et al, 2003; Lee et al, 2004).
BIK was discovered because of its interaction with cellular and viral homologues of antiapoptoptic BCL-2 proteins (Boyd et al, 1995; Han et al, 1996) and, as with other proapoptotic binding partners, its activity is strictly dependent on the ratio of BIK and BCL-2 (Mathai et al, 2002). Although much attention has been placed on the regulation of mitochondrial integrity by BCL-2 family proteins, it is now clear that this regulation also extends to the ER (Ferrari et al, 2002; Demaurex and Distelhorst, 2003; Germain and Shore, 2003; Orrenius et al, 2003), consistent with a role for the ER in supporting the mitochondrial death program. This is achieved at least in part by the influence of BCL-2 family proteins on ER Ca2+ stores. In addition to their location in the mitochondria, BAX and BAK are also located at the ER. In the mitochondria, activation of proapoptotic BAX and BAK correlates with the formation of oligomeric structures with channel-like properties (Wei et al, 2000) and is required for cytochrome c release to the cytosol (Cheng et al, 2001; Wei et al, 2001). In contrast, recent evidence has revealed that BAX and BAK at the ER membrane contribute to regulation of ER calcium (Scorrano et al, 2003). Moreover, when excess ER-restricted BAK (BAKb5) was delivered into BAX/BAK doubly-deficient cells in the absence of excess BCL-2, it formed oligomers and caused release of Ca2+ from the organelle (Zong et al, 2003). Activation of BAX/BAK at the ER membrane by BIK is thus one possibility to account for ER Ca2+ release induced by BIK.
In summary, our results suggest a model in which BIK stimulates Ca2+ release from ER and DRP1 recruitment to the mitochondria, which in turn induces cristae remodelling in a manner dependent on the transmission of Ca2+ to the mitochondria. The resulting DRP1-dependent opening of cristae mobilizes cytochrome c stores and, upon fragmentation of the organelle, the cytochrome c is potentially available for release, which conceivably could be aided by the dynamic stretching–contraction cycles that these fragments undergo. A second event, independent of DRP1 catalytic activity, is caused by a combination of BIK and another BH3-only protein that targets the mitochondria, such as NOXA, resulting in conformational changes in BAX and a pathway for cytochrome c egress. The combination of cristae remodelling, organelle fragmentation, and activation of BAX results in robust release of cytochrome c and activation of caspases. The mechanism underlying the contribution of ER BIK (and presumably an ER pathway) to BAX activation is presently not known, but it is intriguing that BAX recruitment to mitochondria is influenced by another regulator of mitochondrial fission located at the outer membrane, hFis1 (Lee et al, 2004). Moreover, very recent findings in yeast suggest that Fis1p has an unanticipated prosurvival function that can be substituted by BCL-2 proteins (Fannjiang et al, 2004). Thus, the fission–fusion machinery may be involved in multiple steps of mitochondrial apoptosis. In the present study, we have uncovered a novel role for DRP1 in the regulation of cristae remodelling, and described how a Ca2+-mediated ER signal operating through this pathway enhances cytochrome c release from mitochondria.
Materials and methods
Cell culture and infection with adenoviral vectors
H1299 lung carcinoma cells were cultured in α-minimal essential medium supplemented with 10% heat-inactivated fetal bovine serum and 100 μg/ml streptomycin and penicillin. To generate the HA-BCL-2b5 cell lines, H1299 cells were transfected with pcDNA 3 vector encoding HA-tagged BCL-2 mutant in which the TM domain (amino acids 215–239) was replaced by the TM domain of rabbit cytochrome b5 (amino acids 107–134). G418-resistant colonies were screened for the presence of HA-BCL-2b5. Cells were infected with adenoviral vectors as described (Germain et al, 2002) using 100 plaque-forming units (pfu)/cell of virus. All infections were carried out in the presence of 50 μM zVAD-fmk, except for caspase activity measurements. Adenoviral vectors expressing human Noxa (Oda et al, 2000) and a mutant human Noxa in which the conserved leucine at position 29 in the BH3 domain was mutated to alanine were generated as described for wt BIK (Mathai et al, 2002). Transfections were carried out using Lipofectamine plus reagent™ (Gibco-BRL) using the previously described CFP, DRP1, and DRP1(K38E) constructs (Breckenridge et al, 2003b). Caspase-3 activity essays were conducted as previously described (Breckenridge et al, 2003b).
Antibodies, immunoblots, and confocal immunofluorescence
The following antibodies were used: goat anti-BIK (Santa-Cruz, CA), Hamster anti-BCL-2 (Biomol), rabbit anti-BAX N-terminal epitope (Upstate), mouse anticytochrome c (Pharmingen), mouse antiactin (ICN), and rabbit anti-TOM20 (Germain et al, 2002). For immunoblot analysis, indicated amount of proteins were subjected to SDS–PAGE, transferred to nitrocellulose membranes, and blotted with specific antibodies. Blots were incubated with horseradish peroxidase-conjugated secondary antibodies and visualized by enhanced chemiluminescence (Perkin-Elmer Life Sciences). For immunofluorescence, cells were grown on coverslips and treated as indicated in the figure legends. Cells were then fixed and analyzed by double-label immunofluorescence using AlexaFluor 488 and 594 secondary antibodies and visualized using a Zeiss 510 confocal microscope. Images were captured and overlaid with the accompanying software.
Digitonin treatments
H1299 cells were treated with Ad HA-BIK in the presence of 50 μM zVAD-fmk for 10 h. Mitochondria were isolated as described (Germain et al, 2002) and resuspended in HIM buffer (200 mM mannitol, 70 mM sucrose, 10 mM HEPES, pH 7.4, 1 mM EGTA). Mitochondria (12.5 μg protein) in 25 μl HIM were incubated with 60 μg/ml digitonin for 5 min at 37°C, after which mitochondria were re-isolated and analyzed for the presence of cytochrome c and TOM20 by Western blot.
Electron microscopy
Adenoviruses expressing DRP1(K38E)CFP were used to infect one 10 cm2 dish of Cos7 cells at 100 pfu/cell. In order to examine the infection efficiency, a coverslip was included in the dish and, following 24 h, the expression of DRP1(K38E)CFP was confirmed by excitation of CFP at 434 nm and the extent of the interconnected mitochondria was observed following incubation with 50 nM MitofluorRed589 (Molecular Probes) (data not shown). It was determined that ∼95% of the cells were successfully infected with the Ad DRP1(K38E)CFP virus. We then re-infected these cells with Ad HA-BIK (as above) in the presence of zVAD-fmk, to compare the cells infected directly with Ad HA-BIK, Ad HA-Bik+Ru360, Ad:BikL61G, or Ad rtTA (control), all in the presence of zVAD-fmk. Following 12 h, cells were trypsinized, washed in PBS, fixed in 1.6% glutaraldehyde and centrifuged at 3000 g for 15 min. Cell pellets were embedded in SPURR resin (Marivac, Québec), and thin sections were cut with a Leica Ultracut E ultramicrotome and counterstained with lead citrate and uranyl acetate. Digital images were taken using a JEOL 1230 TEM at 60 kV, adapted with a 2 × 2 K bottom-mount CCD digital camera (Hamamatsu, Japan) and AMT software. Random measurements of ICDs were collected with the AMT integrated software, and were recorded into excel spreadsheets. The total intercristal cross-section measurements taken were: mock virus, n=119; Ad BIK, n=423; Ad BIK+Ru360, n=381; Ad BIK+Ad DRP(K38E), n=161; Ad BIK(L61G), n=496. The data were sorted into 25-nm interval ranges and plotted as a distribution profile.
Ca2+ measurements
Measurements of ER Ca2+ stores were carried out as described previously (Mery et al, 1996; Breckenridge et al, 2003b). Briefly, 2 × 106 cells were resuspended in 200 μl Ca2+-free buffer (20 mM HEPES, pH 7.4, 143 mM NaCl, 6 mM KCl, 1 mM MgSO4, 0.1% glucose, 0.1% BSA, 250 μM sulfipyrazone) and loaded with 3 μM Fura-2 (Molecular Probes) in the presence of 0.04% pluronic acid for 30 min at 37°C. Cells were then washed once and resuspended in Ca2+-free buffer. [Ca2+] was determined with the 340/380 nm excitation ratio at 510 nm emission wavelength using an LS 50B Perkin-Elmer luminescence spectrophotometer. ER Ca2+ was determined as the difference in [Ca2+] in the absence and presence of 2 μM thapsigargin.
Online supplementary material
For live imaging presented in supplementary videos, coverslips were mounted in an aluminum chamber in 20 mM HEPES-buffered medium (pH 7.4) and live images were taken using an Olympus IX70 inverted microscope with a Polychrome IV monochrometer and an IMAGO CCD camera from TillPhotonics (GmBH). The cells were maintained at 37°C and images were taken every 2 s (videos 1 and 2) or 4 s (video 3) for 100 or 200 frames. Exposure times were between 200 and 400 ms for the CFP and YFP constructs.
Supplementary Material
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3
Video 1
Video 2
Video 3
Supplemental Figure and Video Captions
Acknowledgments
We thank Peter Rippstein and Rodolfo Zunino for excellent technical help and Ruth Slack for providing Ad DRP1(K38E). MG and JPM are the recipients of Studentships from the Canadian Institutes of Health Research. This work was supported by operating grants from the Canadian Institutes of Health Research (GCS and HMM) and the National Cancer Institute of Canada (GCS).
References
- Akao M, O'Rourke B, Teshima Y, Seharaseyon J, Marban E (2003) Mechanistically distinct steps in the mitochondrial death pathway triggered by oxidative stress in cardiac myocytes. Circ Res 92: 186–194 [DOI] [PubMed] [Google Scholar]
- Bernardi P, Azzone GF (1981) Cytochrome c as an electron shuttle between the outer and inner mitochondrial membranes. J Biol Chem 256: 7187–7192 [PubMed] [Google Scholar]
- Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA (2003) Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol 15: 706–716 [DOI] [PubMed] [Google Scholar]
- Boyd JM, Gallo GJ, Elangovan B, Houghton AB, Malstrom S, Avery BJ, Ebb RG, Subramanian T, Chittenden T, Lutz RJ (1995) Bik, a novel death-inducing protein shares a distinct sequence motif with Bcl-2 family proteins and interacts with viral and cellular survival-promoting proteins. Oncogene 11: 1921–1928 [PubMed] [Google Scholar]
- Breckenridge DG, Nguyen M, Kuppig S, Reth M, Shore GC (2002) The procaspase-8 isoform, procaspase-8L, recruited to the BAP31 complex at the endoplasmic reticulum. Proc Natl Acad Sci USA 99: 4331–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC (2003a) Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 22: 8608–8618 [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Stojanovic M, Marcellus RC, Shore GC (2003b) Caspase cleavage product of BAP31 induces mitochondrial fission through endoplasmic reticulum calcium signals, enhancing cytochrome c release to the cytosol. J Cell Biol 160: 1115–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, Korsmeyer SJ (2001) BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 8: 705–711 [DOI] [PubMed] [Google Scholar]
- Delettre C, Lenaers G, Griffoin JM, Gigarel N, Lorenzo C, Belenguer P, Pelloquin L, Grosgeorge J, Turc-Carel C, Perret E, Astarie-Dequeker C, Lasquellec L, Arnaud B, Ducommun B, Kaplan J, Hamel CP (2000) Nuclear gene OPA1, encoding a mitochondrial dynamin-related protein, is mutated in dominant optic atrophy. Nat Genet 26: 207–210 [DOI] [PubMed] [Google Scholar]
- Demaurex N, Distelhorst C (2003) Apoptosis—the calcium connection. Science 300: 65–67 [DOI] [PubMed] [Google Scholar]
- Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC (1999) Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J Cell Biol 144: 891–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fannjiang Y, Cheng W-C, Lee SJ, Qi B, Pevsner J, McCaffery JM, Hill RB, Basanez G, Hardwick JM (2004) Mitochondrial fission proteins regulate programmed cell death in yeast. Genes Dev 18, (Epub 2004 10 November) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farsad K, Ringstad N, Takei K, Floyd SR, Rose K, De Camilli P (2001) Generation of high curvature membranes mediated by direct endophilin bilayer interactions. J Cell Biol 155: 193–200, (Epub 2001 October 2015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari D, Pinton P, Szabadkai G, Chami M, Campanella M, Pozzan T, Rizzuto R (2002) Endoplasmic reticulum, Bcl-2 and Ca2+ handling in apoptosis. Cell Calcium 32: 413–420 [DOI] [PubMed] [Google Scholar]
- Fish KN, Schmid SL, Damke H (2000) Evidence that dynamin-2 functions as a signal-transducing GTPase. J Cell Biol 150: 145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank S, Gaume B, Bergmann-Leitner ES, Leitner WW, Robert EG, Catez F, Smith CL, Youle RJ (2001) The role of dynamin-related protein 1, a mediator of mitochondrial fission, in apoptosis. Dev Cell 1: 515–525 [DOI] [PubMed] [Google Scholar]
- Germain M, Mathai JP, Shore GC (2002) BH-3-only BIK functions at the endoplasmic reticulum to stimulate cytochrome c release from mitochondria. J Biol Chem 277: 18053–18060 [DOI] [PubMed] [Google Scholar]
- Germain M, Shore GC (2003) Cellular distribution of Bcl-2 family proteins. Sci STKE 2003: pe10. [DOI] [PubMed] [Google Scholar]
- Griparic L, Van Der Wel NN, Orozco IJ, Peters PJ, Van Der Bliek AM (2004) Loss of the intermembrane space protein Mgm1/Opa1 induces swelling and localized constrictions along the lengths of mitochondria. J Biol Chem 279: 18792–18798 [DOI] [PubMed] [Google Scholar]
- Guan K, Farh L, Marshall TK, Deschenes RJ (1993) Normal mitochondrial structure and genome maintenance in yeast requires the dynamin-like product of the MGM1 gene. Curr Genet 24: 141–148 [DOI] [PubMed] [Google Scholar]
- Han J, Sabbatini P, White E (1996) Induction of apoptosis by human Nbk/Bik, a BH3-containing protein that interacts with E1B 19K. Mol Cell Biol 16: 5857–5864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Youle RJ (2003) Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ 10: 870–880 [DOI] [PubMed] [Google Scholar]
- Karbowski M, Jeong SY, Youle RJ (2004) Endophilin B1 is required for the maintenance of mitochondrial morphology. J Cell Biol 166: 1027–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Lee YJ, Gaume B, Jeong SY, Frank S, Nechushtan A, Santel A, Fuller M, Smith CL, Youle RJ (2002) Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J Cell Biol 159: 931–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann SH, Hengartner MO (2001) Programmed cell death: alive and well in the new millennium. Trends Cell Biol 11: 526–534 [DOI] [PubMed] [Google Scholar]
- Kim TH, Zhao Y, Ding WX, Shin JN, He X, Seo YW, Chen J, Rabinowich H, Amoscato AA, Yin XM (2004) Bid-cardiolipin interaction at mitochondrial contact site contributes to mitochondrial cristae reorganization and cytochrome C release. Mol Biol Cell 15: 3061–3072, (Epub 2004 April 3023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, De Camilli P (2002) Dynamin at actin tails. Proc Natl Acad Sci USA 99: 161–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ (2004) Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell 15: 5001–5011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192 [DOI] [PubMed] [Google Scholar]
- Mathai JP, Germain M, Marcellus RC, Shore GC (2002) Induction and endoplasmic reticulum location of BIK/NBK in response to apoptotic signaling by E1A and p53. Oncogene 21: 2534–2544 [DOI] [PubMed] [Google Scholar]
- Mery L, Mesaeli N, Michalak M, Opas M, Lew DP, Krause KH (1996) Overexpression of calreticulin increases intracellular Ca2+ storage and decreases store-operated Ca2+ influx. J Biol Chem 271: 9332–9339 [DOI] [PubMed] [Google Scholar]
- Misaka T, Miyashita T, Kubo Y (2002) Primary structure of a dynamin-related mouse mitochondrial GTPase and its distribution in brain, subcellular localization, and effect on mitochondrial morphology. J Biol Chem 277: 15834–15842 [DOI] [PubMed] [Google Scholar]
- Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, Tokino T, Taniguchi T, Tanaka N (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288: 1053–1058 [DOI] [PubMed] [Google Scholar]
- Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G (2003) Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278: 7743–7746 [DOI] [PubMed] [Google Scholar]
- Olichon A, Emorine LJ, Descoins E, Pelloquin L, Brichese L, Gas N, Guillou E, Delettre C, Valette A, Hamel CP, Ducommun B, Lenaers G, Belenguer P (2002) The human dynamin-related protein OPA1 is anchored to the mitochondrial inner membrane facing the inter-membrane space. FEBS Lett 523: 171–176 [DOI] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium–apoptosis link. Nat Rev Mol Cell Biol 4: 552–565 [DOI] [PubMed] [Google Scholar]
- Orth JD, Krueger EW, Cao H, McNiven MA (2002) The large GTPase dynamin regulates actin comet formation and movement in living cells. Proc Natl Acad Sci USA 99: 167–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth JD, McNiven MA (2003) Dynamin at the actin–membrane interface. Curr Opin Cell Biol 15: 31–39 [DOI] [PubMed] [Google Scholar]
- Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, Evans PR, McMahon HT (2004) BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science 303: 495–499, (Epub 2003 November 2026) [DOI] [PubMed] [Google Scholar]
- Ruffolo SC, Shore GC (2003) BCL-2 selectively interacts with the BID-induced open conformer of BAK, inhibiting BAK auto-oligomerization. J Biol Chem 278: 25039–25045 [DOI] [PubMed] [Google Scholar]
- Satoh M, Hamamoto T, Seo N, Kagawa Y, Endo H (2003) Differential sublocalization of the dynamin-related protein OPA1 isoforms in mitochondria. Biochem Biophys Res Commun 300: 482–493 [DOI] [PubMed] [Google Scholar]
- Schlunck G, Damke H, Kiosses WB, Rusk N, Symons MH, Waterman-Storer CM, Schmid SL, Schwartz MA (2004) Modulation of Rac localization and function by dynamin. Mol Biol Cell 15: 256–267, (Epub 2003 November 2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Wolde M, Thiele C, Fest W, Kratzin H, Podtelejnikov AV, Witke W, Huttner WB, Soling HD (1999) Endophilin I mediates synaptic vesicle formation by transfer of arachidonate to lysophosphatidic acid. Nature 401: 133–141 [DOI] [PubMed] [Google Scholar]
- Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA, Korsmeyer SJ (2002) A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell 2: 55–67 [DOI] [PubMed] [Google Scholar]
- Scorrano L, Oakes SA, Opferman JT, Cheng EH, Sorcinelli MD, Pozzan T, Korsmeyer SJ (2003) BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300: 135–139 [DOI] [PubMed] [Google Scholar]
- Smirnova E, Shurland D-L, Ryazantsev SN, van der Bliek AM (1998) A human dynamin-related protein controls the distribution of mitochondria. J Cell Biol 143: 351–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugioka R, Shimizu S, Tsujimoto Y (2004) Fzo1, a protein involved in mitochondrial fusion, inhibits apoptosis. J Biol Chem 279: 52726–52734 [DOI] [PubMed] [Google Scholar]
- Szabadkai G, Simoni AM, Chami M, Wieckowski MR, Youle RJ, Rizzuto R (2004) Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol Cell 16: 59–68 [DOI] [PubMed] [Google Scholar]
- Wang B, Nguyen M, Breckenridge DG, Stojanovic M, Clemons PA, Kuppig S, Shore GC (2003) Uncleaved BAP31 in association with A4 protein at the endoplasmic reticulum is an inhibitor of Fas-initiated release of cytochrome c from mitochondria. J Biol Chem 278: 14461–14468 [DOI] [PubMed] [Google Scholar]
- Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, Thompson CB, Korsmeyer SJ (2000) tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev 14: 2060–2071 [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon Y, Pitts KR, McNiven MA (2001) Mammalian dynamin-like protein dlp1 tubulates membranes. Mol Biol Cell 12: 2894–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong WX, Li C, Hatzivassiliou G, Lindsten T, Yu QC, Yuan J, Thompson CB (2003) Bax and Bak can localize to the endoplasmic reticulum to initiate apoptosis. J Cell Biol 162: 59–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3
Video 1
Video 2
Video 3
Supplemental Figure and Video Captions