

Abstract
The crystal structure of the snake long α-neurotoxin, α-cobratoxin, bound to the pentameric acetylcholine-binding protein (AChBP) from Lymnaea stagnalis, was solved from good quality density maps despite a 4.2 Å overall resolution. The structure unambiguously reveals the positions and orientations of all five three-fingered toxin molecules inserted at the AChBP subunit interfaces and the conformational changes associated with toxin binding. AChBP loops C and F that border the ligand-binding pocket move markedly from their original positions to wrap around the tips of the toxin first and second fingers and part of its C-terminus, while rearrangements also occur in the toxin fingers. At the interface of the complex, major interactions involve aromatic and aliphatic side chains within the AChBP binding pocket and, at the buried tip of the toxin second finger, conserved Phe and Arg residues that partially mimic a bound agonist molecule. Hence this structure, in revealing a distinctive and unpredicted conformation of the toxin-bound AChBP molecule, provides a lead template resembling a resting state conformation of the nicotinic receptor and for understanding selectivity of curaremimetic α-neurotoxins for the various receptor species.
Keywords: acetylcholine-binding protein, crystal structure, high-affinity toxin–receptor complex, nicotinic acetylcholine receptor, snake three-fingered α-neurotoxin
Introduction
The soluble acetylcholine-binding protein (AChBP) from the freshwater snail Lymnaea stagnalis is a structural homolog of the extracellular ligand-binding domain of muscle-type and neuronal nicotinic acetylcholine receptors (nAChRs) (Brejc et al, 2001; Smit et al, 2001) and other pentameric receptors of the Cys-loop members in the ligand gated ionic channel (LGIC) superfamily (Le Novère and Changèux, 1999). AChBP shows ∼24% sequence identity with the neuronal α7 nAChR (Figure 1A) and assembles as a homopentamer that binds the classical alkaloid agonists and antagonists with dissociation constants characteristic of the nAChRs (Taylor et al, 2000; Changeux and Edelstein, 2001; Grutter and Changeux, 2001; Karlin, 2002).
Figure 1.
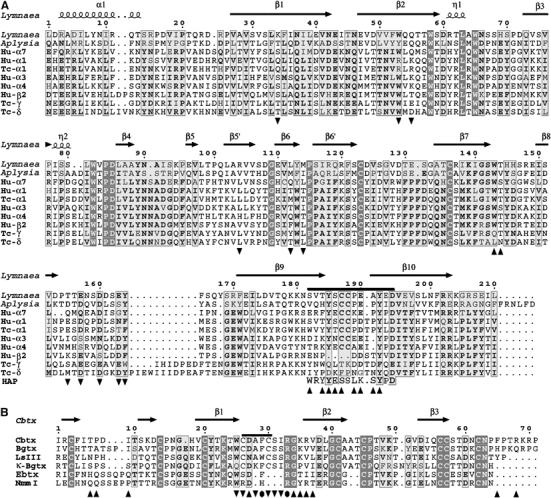
Sequences and numbering of AChBP and Cbtx. (A) Sequence of the L. stagnalis AChBP subunit, aligned with those of the A. californica AChBP subunit (Hansen et al, 2004), of various subunits from human (Hu) and T. californica (Tc) nAChR subtypes (LGIC database), and of the 13-mer high-affinity peptide (Harel et al, 2001). The loop C tip is indicated by a bar above the alignment. Tip up and down triangles respectively denote AChBP residues from the principal and complementary faces of the subunit interface that interact with Cbtx. (B) Structural alignment of the Cbtx sequence with those of the long α-neurotoxins Bgtx and LSIII (bind muscle-type and neuronal nAChRs), of κ-Bgtx (binds neuronal nAChRs), and of the short α-neurotoxins erabutoxin (Ebtx) and NmmI (bind muscle-type nAChRs). The fifth disulfide bridge present in loop II in the long α-neurotoxins and the κ-neurotoxins is indicated by a bar above the alignment. Tip up and down triangles respectively denote Cbtx residues that interact with the AChBP principal and complementary faces of the subunit interface; filled circles denote residues interacting with both faces. A full-color version of this figure is available at The EMBO Journal Online (Supplementary Figure 1).
AChBP also associates with the postsynaptic, curaremimetic α-neurotoxins from snake venom in a manner similar to the skeletal muscle α12βγδ and neuronal α7 nAChRs (Smit et al, 2001; Hansen et al, 2002, 2004). These three-fingered toxins, exemplified by the long α-neurotoxin, α-bungarotoxin (Bgtx), a potent antagonist of the muscle receptor (Chang and Lee, 1963; Changeux et al, 1970), have defined molecular probes and pharmacological tools to investigate the structural and functional biology of the nAChRs. Since then, the long α-cobratoxin (Cbtx) and toxin LSIII and the short erabutoxin, toxin-α, and toxin NmmI (Figure 1B) have also been used as selective ligands for studying the nAChRs (Endo and Tamiya, 1991; Fruchart-Gaillard et al, 2002; Taylor et al, 2002). These peptidic toxins appear unique among the ligands because of their distinctive binding kinetics and remarkably high affinity and selectivity for the various nAChR subtypes; they may also provide lead compounds for the design of clinically useful drugs (Taylor et al, 2002; Tsetlin and Hucho, 2004). Hence, understanding their mode of interaction and defining the interface of the toxin–receptor complexes have been areas of substantial interest in neurobiology for four decades (Nirthanan and Gwee, 2004).
The availability of the primary AChBP structure (Brejc et al, 2001), buttressed by a wealth of pharmacology, mutagenesis, and chemical modification data accumulated over years (Taylor et al, 2000; Karlin, 2002), has prompted an emergence of theoretical models of α-neurotoxin–nAChR complexes, designed from structures of long α-neurotoxins bound to synthetic peptides derived from the α1 subunit sequence (Harel et al, 2001; Moise et al, 2002; Samson et al, 2002; Zeng and Hawrot, 2002) and from mutational analysis of pairwise interactions between Cbtx and an α7 receptor (Fruchart-Gaillard et al, 2002). However, these models, which differ from one another, have been restricted to the AChBP conformation observed in the initial template and only address limited components of the overall toxin–receptor interface.
Early on, the muscle-type nAChR was shown to undergo conformational transitions between resting, desensitized, and open channel states, each with a distinctive affinity for acetylcholine (cf Monod et al, 1965; Changeux and Edelstein, 2001). In turn, α-neurotoxins have been proposed to stabilize the resting state of the nAChR (Moore and McCarthy, 1995). The AChBP conformation observed in the initial structure was thought to reflect primarily the desensitized state of the nAChR, while occurrence of a resting state would require conformational rearrangements in the quaternary structure of the protein (Grutter and Changeux, 2001). Tryptophan fluorescence quenching data have suggested that Bgtx binding induces a unique conformational state of AChBP (Hansen et al, 2002) and that acetylcholine binding induces conformational changes leading to an occluded binding site (Gao et al, 2004; Hibbs et al, 2004). Moreover, channel gating was achieved with a concomitant reduction in agonist affinity from expression of a chimeric cDNA encoding a suitably modified AChBP connected to the transmembrane spans of the 5HT-3 receptor, also a member of the pentameric LGIC family (Bouzat et al, 2004). The ligand-binding and channel gating data are consistent with AChBP retaining the capacity to undergo conformational transitions between states with distinctive agonist affinities and channel conductance properties.
We have solved the crystal structure of the complex formed between the long α-neurotoxin, Cbtx, and Lymnaea AChBP (Table I). This structure, with five toxin molecules bound to the pentameric receptor, reveals conformational changes at the AChBP subunit interfaces that are much larger than those observed for AChBP bound with small nicotinic agonists (Celie et al, 2004). Hence, it establishes how α-neurotoxins bind to the nAChRs and distinguishes between the various models of complexes designed from the primary AChBP structure. Following the lead structure of a related three-fingered toxin, fasciculin Fas2, bound to acetylcholinesterase (AChE) (Bourne et al, 1995), this structure describes a second distinctive, high-affinity complex between a snake three-fingered toxin and a synaptic acetylcholine-recognition protein.
Table 1.
Data collection and refinement statistics
| Data collectiona | |
| Beamline (ESRF) | ID14-EH4 |
| Wavelength (Å) | 0.975 |
| Space group | C2221 |
| Cell dimensions (Å) | a=162.6, b=313.4, c=106.5 |
| Resolution range (Å) | 30–4.2 |
| Total observations | 60 284 |
| Unique reflections | 19 232 |
| Multiplicity | 3.1 (2.5) |
| Completeness (%) | 95.1 (86.8) |
| I/σ(I) | 6.7 (2.7) |
| Rsymb | 16.0 (36.6) |
| B from Wilson plot (Å2) | 41.1 |
| Refinementc | |
| R-factor/R-free (%) | 33.1 (35.2)/37.8 (47.1) |
| Average B-factor (Å2) | 39.8 |
| R.m.s.d.d | |
| Bonds (Å)/angles (deg) | 0.012/1.4 |
| Chiral volume (Å3) | 0.081 |
| Validation | |
| Map correlation | |
| Main/side chainse | 0.77 (0.77)/0.68 (0.74) |
| Ramachandran plotf | |
| Outliers (%) | AChBP: 5.1 Cbtx: 9.1 |
| Structure Z-scoresg | |
| Second-generation packing quality | −2.889 |
| Ramachandran plot appearance | −2.112 |
| Chi-1/chi-2 rotamer normality | −2.130 |
| Backbone conformation | 0.004 |
| aValues in parentheses are those for the 4.31–4.2 Å resolution shell. | |
| bRsym=∑hkl∑i∣Ii(hkl)−∣〈Ihkl〉∣∣/∑hkl∑iIi(hkl), where I is an individual reflection measurement and 〈I〉 the mean intensity for symmetry-related reflections. | |
| cR-factor=∑hkl∣∣Fo∣−∣Fc∣∣/∑hkl∣Fo∣, where Fo and Fc are observed and calculated structure factors, respectively. R-free is calculated for 5% of randomly selected reflections excluded from refinement. | |
| dRoot mean square deviation from ideal values, according to Engh and Huber (1991). | |
| eAccording to Branden and Jones (1990). Values in parentheses are those for the interacting residues listed in Table II. | |
| fAccording to Kleywegt and Jones (1996). | |
| gAccording to WHATIF (Hooft et al, 1996). | |
Results and discussion
Preparation and analysis of the Cbtx–AChBP complex in solution
To optimize occupancy of all five binding sites on AChBP, yet minimize excess of unbound toxin that may preclude crystallization, concentration ratios were carefully adjusted in forming the Cbtx–AChBP complex (Figure 2). As expected from the relative masses and net charges of Cbtx and the glycosylated AChBP subunit, and from the nanomolar dissociation constant of the complex (Hansen et al, 2002), the bound Cbtx molecules were found to reduce the mobility of AChBP in native-PAGE. Moreover, titration of the fractional occupancies of the five AChBP subunit interfaces revealed a linear progression in the appearance of all four intermediate complexes before saturation at stoichiometry is achieved. The same reductions in AChBP mobility and intermediate complexes were observed when Bgtx, which also binds AChBP with high affinity (Hansen et al, 2002), was used. By contrast, the cationic short α-neurotoxin of lower affinity, NmmI, yielded a broadened band suggestive of fractional occupation of the sites (not shown).
Figure 2.
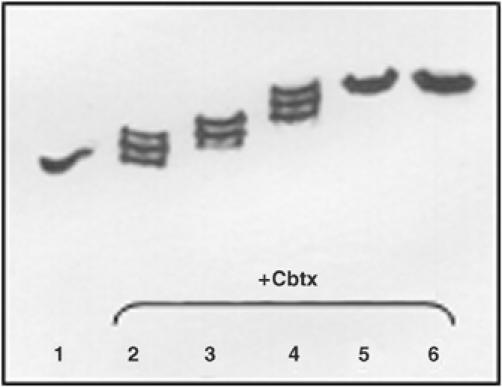
Analysis of Cbtx–AChBP complex formation. Complex formation and stoichiometry were analyzed by native-PAGE with migration toward the anode (bottom). The progressive shift toward the cathode in the positions of the intermediate complexes (molar ratios of 0.3, 0.6, 0.9, 1.2, and 1.5 Cbtx per binding site; lanes 2–6) relative to unliganded AChBP (5 × 30 kDa; pI 5.0; lane 1) denotes increasing fractional occupancies (lanes 2–4) and then full occupancy (lanes 5 and 6) of the AChBP pentamer by Cbtx (8 kDa; pI 8.6). The unbound Cbtx in excess (lanes 5 and 6) migrates toward the cathode and off the gel.
Gel filtration of the immunoaffinity-purified AChBP followed by electrophoretic analyses indicated the presence of monomers and dimers of the pentameric molecule, both yielding the same single band in SDS–PAGE and native-PAGE (not shown). The Cbtx–AChBP complexes prepared from the purified monomers and dimers were both found to elute as dimers of pentameric complexes. This suggests that distinctive dimers of the complex may assemble by end on stacking of the pentameric rings or lateral stacking via the bound toxins.
Crystal packing and quality of the structure
The structure of the Cbtx–AChBP complex was solved by molecular replacement and carefully refined at 4.2 Å resolution (Table I). In the crystal, despite the large solvent channel that biases the overall resolution (cf Materials and methods), two packing interfaces for the pentameric complexes result in very well-ordered bound toxin molecules and binding interfaces (Figures 3 and 4). At the first packing interface, where coaligned dimers of pentamers assemble tail-to-tail as found in the Hepes-bound AChBP crystal (Brejc et al, 2001), the clustered five AChBP C-termini diverge laterally at the protonated Arg206 residues (Figure 1A) but the uncharged 6xHis residues may contribute to the dimer formation (not shown). At the second packing interface, the external, third β-strand in a bound Cbtx molecule closely interacts with the AChBP-exposed antiparallel segment in loop β8–β9 from a symmetry-related pentameric complex (Figure 3C). This interaction results in a Cbtx-mediated lateral association of complexes, where the entrances to the subunit interfaces in two distinct pentamers are separated by 20–24 Å, a distance shorter than that between two ligand-binding sites within a pentamer. Incidentally, this interaction mimics the dimeric assemblies of reversely oriented three-fingered toxin molecules linked by their third β-strands and C-terminal regions observed in structures of α- and κ-neurotoxins (Love and Stroud, 1986; Betzel et al, 1991; Oswald et al, 1991; Dewan et al, 1994). These two arrangements are fully consistent with electron microscopy images of a crystalline Bgtx–nAChR complex (Paas et al, 2003) and with the distinctive dimer assemblies observed by gel filtration (cf above). A third mode of association between complexes in the crystal involves the second β-strand and C-terminal segment of Cbtx.
Figure 3.
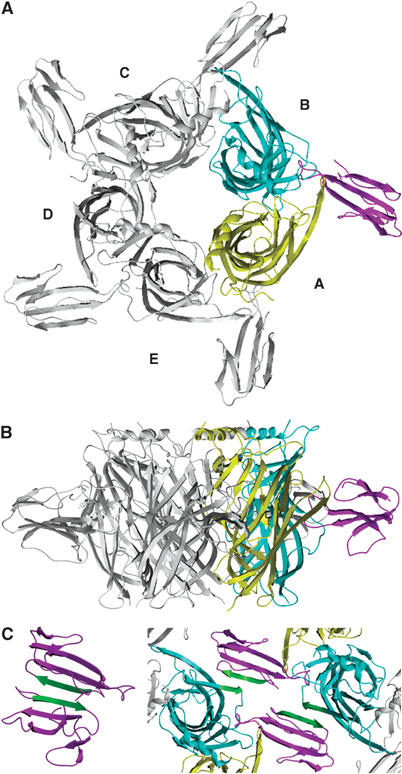
Overall view of the Cbtx–AChBP complex. The pentameric complex is viewed along (A) and perpendicular (B) to the AChBP five-fold axis. AChBP subunits A and B, which respectively contribute to the principal and the complementary faces of the interface, are displayed in yellow and blue; the Cbtx molecule bound at this subunit interface is in purple. (C) Cbtx-mediated assembly of pentameric complexes in the crystal. The AChBP segment Thr155–Ser159 in loop β8–β9 and the Cbtx antiparallel strand β3 (Asp53–Cys57) are shown in green. The crystalline homodimeric assembly of two reversely oriented Cbtx molecules (purple) through their antiparallel strand β3 (green) (structure 2CTX; Betzel et al, 1991) is shown on the right.
Figure 4.
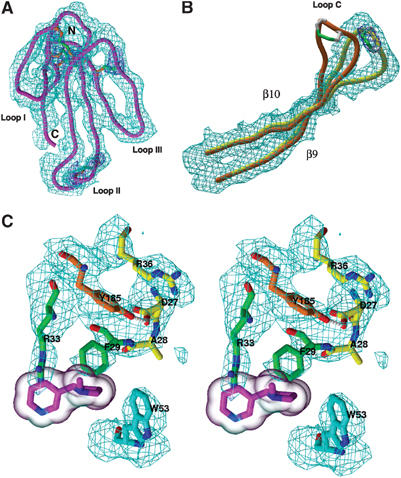
Quality of the Cbtx–AChBP complex structure. (A–C) Views of the 4.2 Å averaged electron density maps for (A) the bound Cbtx molecule, viewed down to the concave face (purple Cα; cyan map contoured at 1σ) and its five disulfide bridges (orange Cα and green bonds; blue map contoured at 2.5σ); (B) the newly open conformation of loop C (yellow Cα; cyan map contoured at 1σ) compared with its conformation in Hepes-bound AChBP (orange Cα) (structure 1I9B; Brejc et al, 2001); (C) the complex interface (stereo view; cyan map contoured at 1σ); Cbtx residues Phe29 and Arg33 partially mimic the nicotine molecule (purple; transparent molecular surface) superimposed as bound to AChBP (structure 1UW6; Celie et al, 2004).
As a result of the full ligand occupation and the tight packing interactions, the quality of the density maps (Figure 4) along with availability of high-resolution structures for each of the two complex partners permits unambiguous positioning of all secondary structure elements in the Cbtx–AChBP complex and most of the side chains at the binding interfaces. Hence, structural comparison of AChBP bound to the antagonist Cbtx with AChBP bound to the agonists, nicotine and carbamoylcholine (Celie et al, 2004), reveals the unique conformational changes induced by the α-neurotoxin (Figures 4, 5 and 6).
Figure 5.
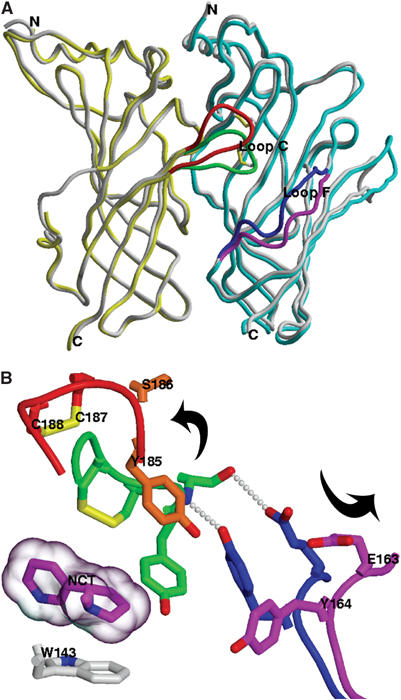
Conformational changes in AChBP associated with Cbtx binding. (A) Superimposition of AChBP subunits A (yellow) and B (blue) in the Cbtx complex with those in the Hepes-bound AChBP structure (gray). AChBP loops C and F, which undergo conformational changes upon Cbtx binding, are respectively displayed in orange and green for the toxin complex and in dark blue and magenta for the Hepes-bound conformation. (B) Close-up view of the conformational changes (indicated by the arrows) within the ligand-binding site, with loop C on left and loop F on right (same colors as in panel A). The likely hydrogen-bonding network is based on interatomic distances. A nicotine molecule (purple; transparent molecular surface) is superimposed as bound to AChBP.
Figure 6.
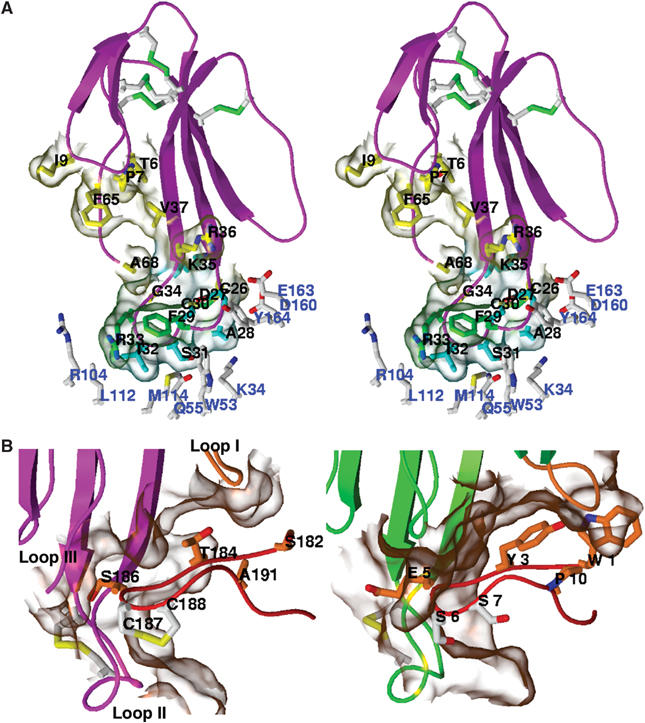
The Cbtx–AChBP complex interface. (A) Close-up view of the bound Cbtx molecule. The Cbtx residues (black labels) that interact with the principal and complementary faces of the AChBP subunit interface are in yellow and cyan/green, respectively. The AChBP residues in the complementary face of the interface are in white (blue labels). (B) Structural comparison of (left) the Cbtx-bound loop Ser182–Glu193 of AChBP and (right) the Bgtx-bound 13-mer peptide (structure 1HC9; Harel et al, 2001). The molecular surfaces of the complex interfaces are shown in transparency.
Overall view of the Cbtx–AChBP complex
The three-fingered Cbtx molecule consists of two antiparallel β-sheets with a central three-stranded β-sheet formed by residues Cys20–Trp25, Arg36–Gly40, and Asp53–Cys57 (Betzel et al, 1991) (Figures 1B and 4A). The Cbtx three loops, loops I, II, and III, that emerge from the dense core containing four disulfide bridges, form a slightly concave disk elongated in the direction of the long loop II that bears the Trp25-Cys-Asp-Ala-Phe-Cys-Ser31 sequence and fifth disulfide bond characteristic of the long α-neurotoxins. In the complex, Cbtx loop II inserts deeply into the ligand-binding pocket, located at the interface between two AChBP subunits (referred to as A for the principal face where loop C resides and B for the complementary face where loop F resides) (Figures 3 and 5). AChBP subunits A and B contribute mean values of 700 and 425 Å2, respectively, to the Cbtx–AChBP interfacial area buried to a 1.6 Å probe radius. An interfacial area in the 1125 Å2 range represents 20–25% of the total accessible surface area of Cbtx and comprises 18 residues of Cbtx, of which four are positively charged (Table II). The buried surface area and number of residues involved are similar to those calculated for the Fas2–AChE complex interface (Bourne et al, 1995). The Cbtx molecular axis, defined by the direction of the central three-stranded antiparallel β-sheet, lies at an ∼45° angle relative to the median axis of the AChBP ring and is near-perpendicular to the cylinder wall (Figure 3). As a result, the center of gravity of Cbtx is located exactly at a midposition in the 62 Å high AChBP cylinder, and the toxin disulfide core resides closer to the ‘membrane' side than the apical side of loop C. Protrusion of the five bound toxins at the outer perimeter of the AChBP pentamer significantly extends, by 50 Å, the radial dimension of the cylinder. The resulting total diameter of 130 Å for the pentameric Cbtx–AChBP complex is consistent with the size of spherical particules observed by electron microscopy from nanocrystals of a Bgtx–nAChR complex (Paas et al, 2003).
Table 2.
Intermolecular interactionsa
| Cbtxb | AChBP subunit interface |
|
|---|---|---|
| Principal face (subunit A) | Complementary face (subunit B) | |
| Loop I | ||
| Thr6 | Thr184 | |
| Pro7 (Ala7) | Ser182, Thr184, Glu190, Ala191 | |
| Ile9 | Glu190 | |
| Loop II | ||
| Trp25 | Glu163 | |
| Cys26–Cys30 | Glu157, Asp160 | |
| Asp27 | Tyr185 | |
| Ala28 | Lys34, Tyr164 | |
| Phe29 | Tyr185, Tyr192 | Trp53 |
| Ser31 | Gln55, Thr155 | |
| Ile32 (Ser35) | Gln55,Leu112, Met114 | |
| Arg33 | Thr144, Cys187, Tyr192 | Arg104 |
| Gly34 | Ser186 | |
| Lys35 | Ser186 | |
| Arg36 (Val39) | Tyr185 | |
| Val37 | Thr184, Ser186 | |
| C-terminus | ||
| Phe65 (His68) | Thr184, Cys187, Pro189 | |
| Arg68 (Gln71) | Ser186 | |
| aWithin a 4.5 Å distance between atoms from each partner in the complex. | ||
| bResidue substitutions in Bgtx are indicated in parentheses. | ||
| cIndicated in bold are those residues in Cbtx and in the α7 receptor (AChBP numbering) whose mutations cause an affinity decrease of more than five- and seven-fold, respectively (Fruchart-Gaillard et al, 2002). | ||
Superimposition of the Cbtx-bound AChBP pentamer with the Hepes- and agonist-bound pentamers (Brejc et al, 2001; Celie et al, 2004) unambiguously reveals that toxin binding is associated with major positional changes of AChBP loops C and F that border the ligand-binding pocket (Figures 4 and 5). These two loops, which respectively belong to the principal and complementary sides of the subunit interface, are markedly displaced, by up to 10 Å, to uncap the pocket. However, Cbtx binding does not significantly alter the relative orientations of the subunits within the pentamer. This suggests that the antagonist-bound AChBP pentamer does not undergo the 15–16° rigid-body rotation of the inner region of subunits proposed as a mechanism for agonist activation of the nAChR (Unwin et al, 2002).
Detailed view of the Cbtx–AChBP complex interface
Three distinct, separated anchor points of Cbtx on AChBP result in a very well-ordered bound toxin molecule (Figures 4 and 6), consistent with its nanomolar dissociation constant for AChBP (Hansen et al, 2002). The three regions of Cbtx responsible for complex formation, loops I and II and part of the C-terminus, are located in the concave face of the molecule with contributions from one residue and the fifth disulfide bridge located in the convex face of loop II (Table II). Cbtx loop II, which is made of residues Tyr21–Gly40 and forms a narrow hairpin with a bulbous tip containing the fifth disulfide bridge, is central to the complex interface. The Phe29 and Arg33 side chains extending at its tip are well positioned for establishing hydrophobic and aromatic interactions with AChBP Trp53, Tyr185, Tyr192, and perhaps Trp143, in the ligand-binding pocket, 10 Å into the interfacial cleft. In addition, Cbtx Trp25, Asp27, Ala28, and Ile32 that surround the tip of loop II are within contact distance of Tyr185 in AChBP subunit A and Glu163, Glu55, Leu112, Met114, and Tyr164 in subunit B. Significant side-chain interactions may also occur between Cbtx Ser31, Cys26, and Cys30, in the convex face of loop II, and the AChBP segment Ser159–Tyr164 in subunit B; yet part of this region remains disordered as found in previous AChBP structures (Celie et al, 2004).
Remarkably, the position of Cbtx Phe29 in the complex overlays with that of Tyr185 in the closed loop C of Hepes-bound AChBP, as to conform to a filled binding pocket (Figures 4C and 5B). In turn, the newly positioned Tyr185 in the open loop C is sandwiched between the Cbtx Phe29 and Arg36 side chains, where it is most likely hydrogen-bonded to Cbtx Asp27 and in cation-π interaction with Arg36, instead of Arg33 as predicted from docking (Zeng and Hawrot, 2002). Finally, the Cbtx Phe29 and Arg33 side chains, respectively positioned on the principal and complementary faces of the interface, are oriented toward the AChBP binding pocket where they would partially overlap with a bound nicotine molecule; this overlap is consistent with competitive antagonism by α-neurotoxins. Yet in the Cbtx complex, the Arg33 guanidinium nearly overlays with the carbamoyl/acetyl moiety of carbamoylcholine/acetylcholine, while in the agonist-bound structures, the quaternary ammonium of the ligand is in cation-π and electrostatic interactions with the Trp143 indole and backbone carbonyl group, respectively (Celie et al, 2004).
The precise orientation of Cbtx loop II at the binding interface governs the other two interaction points between Cbtx and AChBP (Figures 3 and 6; Table II). At the tip of Cbtx loop I, residues Ile5–Asp8 abut apical to the AChBP newly positioned loop C as to establish interactions with Thr184, Ala191, and Glu190 located within the β9–β10 loop. In the C-terminal region of Cbtx, the Phe65 side chain establishes interactions with AChBP loop C residues. Yet the toxin C-terminus, which contains three vicinal positively charged side chains after the bent hinge of Pro66 (Figure 1B), is disordered and appears nonessential for binding. Similarly, residues at the tip of Cbtx loop III do not contribute interactions, with the closest distance, 8 Å, between the Cα positions of Cbtx Gly51 and AChBP Asp160. This is consistent with structure–activity studies showing that the C-terminal 67–71 stretch and residues within loop III weakly contribute to Cbtx binding (Fruchart-Gaillard et al, 2002).
Overall, this three-point mode of binding closely resembles that previously observed for Fas2 bound to AChE (Bourne et al, 1995). However, while Fas2 sterically occludes substrate entry to the AChE active site gorge, Cbtx prevents binding of small ligands by binding to an overlapping region in the AChBP binding pocket and promoting significant conformational changes.
Conformational changes in AChBP upon toxin binding
AChBP loops C and F are major components of the principal and complementary faces of the subunit interface. In the complex, the loop C tip, comprised of residues Tyr185–Tyr192 and the Cys187–Cys188 disulfide bridge (Figure 1A), is markedly dislodged from its close apposition to the complementary face of the subunit interface seen in Hepes-bound AChBP (Brejc et al, 2001) (Figure 5). The 10 Å distance between the two positions results from a 35° rotation of the loop around an axis that roughly aligns with a vector connecting Ser182 to Glu193. The newly positioned loop C is located midway between Cbtx loops I and II where it is sequestered and almost entirely wrapped by the Cbtx concave face and C-terminal region (Figure 6A). Compared to the closed conformation of loop C, the backbone trace significantly deviates from Ser182 to Tyr192 where two major hinge positions for structural rearrangement are found within the Ser182–Val183 and Ala191–Tyr192 dipeptides.
An additional change in conformation for the Cbtx-bound relative to the Hepes-bound AChBP occurs in loop F where the solvent-exposed Thr155–Tyr164 segment, which faces loop C at the cleft entrance, moves toward strand β9 with the largest deviation, up to 3.5 Å, found for Glu163. Although the Thr155–Ser159 backbone trace is not resolved in detail, the side chains of Glu163 and Tyr164 that help configure the binding pocket architecture are clearly expelled toward the solvent, with deviations up to 8 Å for the carboxyl and hydroxyl groups.
Overall, these marked conformational changes in the AChBP binding pocket upon toxin binding, notably the expulsion of Tyr185 and the Cys187–Cys188 bridge that both contribute to agonist binding (Figure 5), may reflect a unique conformational state resembling one of the basal resting (activatable) states of the nAChR. This conformation greatly differs from the putative desensitized state seen in Hepes- and agonist-bound AChBP structures, where ligand binding induces only a 2.5–3 Å displacement of the loop C tip (Celie et al, 2004). In fact, in the carbamoylcholine-bound AChBP structure, seven of the 10 subunit interfaces present in the crystalline dimer of pentamers are unliganded and of these, one shows a loop C conformation that resembles that in the Cbtx–AChBP complex. This suggests that, in solution, the functionally important loop C region behaves as a flexible flap and that unliganded AChBP fluctuates between multiple conformational states, of which some were captured in the carbamoylcholine-bound AChBP structure. Binding of the small agonist molecule results in partial closure of the flap. Hence, fluctuations giving rise to transient enlargements of the ligand-binding site appear critical for binding of Cbtx, which, in turn, locks AChBP into a distinctive conformational state. This is in agreement with the slow rates of association for α-neurotoxins compared to the agonist nicotine observed in early and recent studies on Bgtx binding to the nAChR and to AChBP (Weber and Changeux, 1974; Hansen et al, 2002). Moreover, the newly open conformation of loop C is consistent with the increased incorporation of a diazirine photoprobe to the muscle nAChR in the presence of Bgtx, which suggested that toxin binding may shift the receptor conformation toward the resting state (Moore and McCarthy, 1995), and with the distinctive changes in fluorescence of cysteine-substituted fluorophores seen upon Bgtx binding (Hibbs et al, 2004).
Reliability and implications of the Cbtx–AChBP complex structure
The orientation and position of the bound Cbtx relative to the AChBP subunit interface is fully consistent with the biochemical and mutational data available on the nAChRs and/or α-neurotoxins (Ackermann et al, 1998; Malany et al, 2000; Taylor et al, 2000; Fruchart-Gaillard et al, 2002; Sine, 2002). Of these, extensive mutagenesis analysis of Cbtx identified 10 residues as important for binding to the neuronal α7 receptor (Fruchart-Gaillard et al, 2002). In the crystalline Cbtx–AChBP complex, all 10 residues, Trp25, Asp27, Ala28, Phe29, Cys26/Cys30, Arg33, Lys35, Arg36, and Phe65, are positioned within contact distance of residues in the AChBP subunit interface (Figure 6A; Table II). Unexpectedly however, the structure also points to the key roles for Cbtx Ala28, Phe29, and Arg33 near the tip of loop II in conferring toxin specificity.
Residues within loop C of AChBP are largely conserved between members of the pentameric receptor family, except for three residues within this loop, Ser182, Thr184, and Ser186, which interact with Cbtx (Figure 1A) and may play an important role in conferring toxin selectivity for a particular receptor subtype. Indeed, while Ser182 weakly interacts with Cbtx loop I, Thr184 points toward the junction of Cbtx loops I and II and Ser186 anchors midway between the Cbtx β2-strand and C-terminal region. In a related AChBP from Aplysia californica, which shows distinctive agonist/antagonist binding selectivity and a 100-fold lower Bgtx affinity than Lymnaea AChBP, several residues at key positions in the ligand-binding pocket differ (Hansen et al, 2004) (Figure 1A). Of these, the surface-exposed Ser182Val and Thr184His mutations within loop C and the Trp53Tyr, Arg104Val, and Glu163Ser mutations at the binding surface of the complementary subunit represent major determinants contributing to the distinctive ligand selectivity of the two AChBP species. Hence, the substantial shift in fluorescence emission observed upon Bgtx binding to an AChBP Trp53Cys-acrylodan conjugate (Hibbs et al, 2004) might be correlated with its proximity to, and possible steric hindrance with the conserved Phe29 residue that associates at the complementary side of the subunit interface. To validate our structure further, we have explored the Trp53 and Thr184 contributions by structure-guided mutagenesis of the Tyr and His residues found at these positions in Aplysia AChBP. Mutation Tyr53Trp enhanced Bgtx affinity while mutation His184Phe, as found in the neuronal α7 and muscle α1 subunits, enhanced it to an even greater extent (SB Hansen, unpublished data).
Long and short α-neurotoxins and κ-neurotoxins
The binding specificities of the long and short α-neurotoxins for the muscle-type (α1)2βγδ and neuronal α7 nAChRs have been studied in great detail by mutagenesis (Ackermann et al, 1998; Malany et al, 2000; Fruchart-Gaillard et al, 2002). Cbtx Trp25, Asp27, and Arg33 were found to affect comparably the binding to both receptors, whereas Phe29, Arg36, and Phe65 may confer specificity toward the neuronal α7 receptor (Fruchart-Gaillard et al, 2002). These results, along with data from mutagenesis of NmmI Lys23 and Lys49, which affected binding to one site only of the mouse muscle nAChR (Ackermann et al, 1998), led to the conclusion that Cbtx Ala28, Cys26, Cys30, Lys35, and Phe65, which are highly conserved in the long α-neurotoxins but lacking in the short α-neurotoxins (Figure 1B), are α7-specific residues. Instead, the Cbtx–AChBP complex structure shows that Cbtx Phe29 and Arg33 interact with residues in the AChBP binding pocket that are conserved among the nicotinic receptor subtypes (Figures 1A and 4, 5 and 6). Most importantly, the combination of a longer loop II and a shorter loop I in the long α-neurotoxins relative to the short ones is a crucial feature for conferring specificity; this is consistent with the very low affinity of NmmI for AChBP (cf above).
In fact, pairwise interaction analyses between NmmI and the muscle-type nAChR suggested that NmmI Arg33 and Lys27, at the tip of loop II, should be close to Val188/Tyr190 and Asp200 or Tyr190, respectively (Ackermann et al, 1998; Malany et al, 2000). Superimposition of an NmmI model, designed from the structure of its erabutoxin homolog, with the Cbtx molecule bound to AChBP suggests that NmmI His32 and Arg33 are positioned to mimic the orientation and contacts of Cbtx Phe29 and Arg33 within the AChBP binding pocket (not shown). However, NmmI Lys27 is too distant to be in contact with an AChBP residue, and NmmI loop II would have to move laterally while the tip of loop I would have to retract, or swing out as does loop I in Fas2 compared with fasciculin 1 (le Du et al, 1996), to avoid steric hindrance with the open loop C. This suggests a somewhat different orientation of NmmI at the subunit interface of the muscle-type nAChR with Arg33 acting as a pivot. This is consistent with pairwise interaction energies that predicted proximity of NmmI Arg33 to Tyr190 in the α-subunit and Leu119 in the γ-subunit of the α12βγδ receptor (Malany et al, 2000).
The κ-neurotoxins, which differ from the long α-neurotoxins essentially by an Ala/Gln (in κ-Bgtx and variants κ-Bgtx 2 and 3) or Phe (in κ-flavitoxin) substitution for Trp25 (Cbtx numbering) and a short tail of two residues only (Figure 1B), selectively bind neuronal nAChRs of the α3β2 or α4β2 subtypes (Chiappinelli et al, 1996). The main differences between κ-Bgtx and Cbtx within loop II include the Gln26 and Pro36 substitutions for Cbtx Trp25 and Lys35, respectively, and the Lys29 substitution for the conserved Cbtx Ala28 at the loop tip, but the determinants for the distinctive selectivities of α- and κ-neurotoxins remain unclear when based solely on structural alignments, mutagenesis, or chemical modification data (Chiappinelli et al, 1996). Yet, structural superimposition of κ-Bgtx (Dewan et al, 1994) with Cbtx bound to AChBP leads us to identify κ-Bgtx Lys29 at the tip of loop II as a potential major determinant of receptor selectivity, in forming close contacts with the complementary face brought about by the β2 subunit to the subunit interface. Moreover, the shorter C-terminus in κ-Bgtx can accommodate the extended cationic Lys side chain that replaces AChBP Thr184 in loop C of the α3 and α4 subunits and that may, in turn, impair Cbtx binding.
Experimental structure versus theoretical models
The availability of the first AChBP structure and structures of nAChR-related peptides bound to Cbtx or Bgtx, along with a wealth of mutational data, has prompted molecular modeling of several α-neurotoxin–receptor complexes. Of these, three predicted correct placement of the toxin at the subunit interface and the key role of the sequence-conserved Arg33 and Phe29, at the loop II tip, in toxin binding (Harel et al, 2001; Fruchart-Gaillard et al, 2002; Samson et al, 2002). Yet, the precise toxin orientation and position significantly differ from those observed in the Cbtx–AChBP complex structure. In particular, Cbtx Phe29 and Arg33 in the models are positioned ∼7 Å away from their positions in the crystalline complex and hence are misplaced relative to the complementary and principal faces of the ligand-binding site. Obviously, the concerted large movements of loops C and F could not have easily been predicted. Even the limited changes in the loop C positioning observed in the nicotine- and carbamoylcholine-bound AChBP structures were difficult to predict from the Hepes-bound AChBP structure (Celie et al, 2004).
Modeling of a Bgtx–AChBP complex from the structure of a Bgtx-bound 13-mer high-affinity peptide derived from the α1 subunit sequence (Harel et al, 2001) (Figure 1A) resulted in a perpendicular orientation of the toxin relative to the five-fold axis of AChBP that considerably limits the binding interface surface area. Yet, superimposition of the 13-mer peptide in this complex with the homologous segment 182–193 in the open loop C in our structure results in a close overlay of bound Cbtx and Bgtx molecules (cf deviation values in Materials and methods). Hence, structural comparison of the Cbtx-bound AChBP and Bgtx-bound peptide enables one to visualize directly those mutations within loop C of the nAChRs that can be accommodated at the toxin complex interface (Figure 6B). Of these, substitution of bulkier residues for AChBP Thr184 and Ser186, which tightly interact with Cbtx but are not conserved among the nAChRs, and insertion of two residues in Cbtx loop I as to conform to the Bgtx sequence do not disturb the binding interface. This observation is consistent with the sub-nanomolar affinity of Bgtx for the muscle-type AChR (Changeux et al, 1970) and suggests that the overall conformation of Bgtx as bound to the 13-mer peptide may be retained in the receptor complex. Thus, the Cbtx–AChBP complex structure should serve as a representative template for all the long α-neurotoxin–nAChR complexes. Most importantly, the open conformation of AChBP loop C leads to an alternative strategy for drug design where the distinctive conformational states of the target induced by ligand binding can be used in the design of the drug molecule (Bourne et al, 2004).
In summary, the crystal structure of the pentameric Cbtx–AChBP complex shows a multipoint attachment of the α-neurotoxin to its target and major determinants and interactions at the complex interface that are reminiscent of those involved in the high-affinity Fas2–AChE complex, where the binding site is buried within the subunit rather than at a subunit interface. Comparison with structures of AChBP bound with nonpeptidic ligands reveals inherent flexibility of the AChBP molecule and a distinctive conformation that could not have been predicted simply by docking, and may better resemble a resting state conformation of the nAChR than a desensitized state. Hence, this structure provides a comprehensive template for understanding the structural basis for marked differences observed in ligand binding to Aplysia and Lymnaea AChBPs and for examining further the binding characteristics of the uniquely selective three-fingered toxins at the subunit interfaces of homo- or heteropentameric receptors of the nicotinic receptor family. Given the importance of these receptors in cholinergic signaling and their polymorphisms in neurological diseases, the use of naturally occurring, well-structured peptides has further defined a major target site of drug action and the conformational changes attendant to ligand binding.
Materials and methods
Preparation and analysis of the Cbtx–AChBP complex
AChBP, flanked with an N-terminal, 24-residue 3xFLAG epitope and a C-terminal 6xHis tag, was expressed as a soluble exported protein from stable HEK-293 cells and purified from the culture medium on immobilized anti-FLAG antibody (Hansen et al, 2002). Further purification was by gel filtration FPLC on prepacked Superdex-200 (Amersham Biosciences) equilibrated in 50 mM Hepes, pH 7.0, 50 mM NaCl, and 0.01% (w/v) NaN3. SDS–PAGE and native-PAGE (8–25% gradient or 12.5% homogenous gels) used a PhastSystem apparatus and silver nitrate staining (Amersham Biosciences). MALDI-TOF mass spectrometry was performed on a Voyager-DE™RP BioSpectrometer Workstation (Perseptive Biosystems) in the positive linear mode using sinapinic acid and the dried-droplet method; samples were desorbed with a 337 nm nitrogen laser. Both SDS–PAGE and mass spectrometry yielded an AChBP apparent mass of approximately 30 kDa, that is, 9% higher than the theoretical mass, denoting occupancy of the Asn66 glycosylation site (Figure 1A).
The short α-neurotoxin NmmI was purified from the venom of Naja mossambica mossambica (Marchot et al, 1988). The long α-neurotoxins Cbtx (N. naja siamensis toxin 3) (Latoxan, France) and Bgtx (Sigma-Aldrich) were analyzed by native-PAGE with migration toward the cathode and SDS–PAGE (20% homogenous gels) (Marchot and Bougis, 2000), and by mass spectrometry. They were further purified using reverse-phase HPLC on ultrasphere-octyl and an acetonitrile gradient in 0.1% TFA (Marchot and Bougis, 2000). Fractional occupancies of the five AChBP subunit interfaces and final stoichiometry of toxin binding were analyzed by using increasing toxin-to-pentamer molar ratios (2 h incubation; room temperature) and native-PAGE (12.5% homogenous gels) (Marchot et al, 1996) (Figure 2). The Cbtx–AChBP complex subjected to crystallization was formed at high concentration of purified AChBP (ca 5 mg/ml; 170 μM in binding sites) with a 1.2-fold molar excess of Cbtx (concentration 105 greater than the Kd; Hansen et al, 2002); it was incubated for 2 h at room temperature, then overnight at 4°C; repurified by gel filtration for removal of unbound Cbtx in excess and minor aggregates generated during complexation; concentrated to approximately 10 mg/ml by ultrafiltration; and stored on ice.
Crystallization and data collection
Since protonation of the AChBP N-terminal 3xFLAG and C-terminal 6xHis sequences was likely to preclude crystal growth, crystallization assays were conducted using pH values above 8 leading to uncharged His residues. Crystallization was achieved by vapor diffusion at 4°C using 1 μl hanging drops and a protein-to-well ratio of 1:1 with 0.9 M Na-citrate, pH 8.5, as the well solution. The crystals were flash-cooled in the nitrogen gas stream (100 K) after a short soak in the well solution supplemented with 20% glycerol. The crystals diffracted anisotropically, up to 3.2 Å resolution along the a-axis but 4.5 Å only along the c-axis. They belonged to the orthorhombic space group C2221 with unit cell dimensions a=162.6 Å, b=313.4 Å, and c=106.5 Å, giving a Vm value of 4.1 Å3/Da (70% solvent) for one pentameric Cbtx–AChBP complex in the asymmetric unit (Matthews, 1968). Oscillation images were integrated with DENZO (Otwinowski and Minor, 1997); data were scaled and merged with SCALA (CCP4, 1994).
Structure determination and refinement
The AChBP structure was solved by the molecular replacement method with AMoRe (Navaza, 1994) using, as search model, the coordinates of the AChBP pentamer (accession code 1I9B; Brejc et al, 2001). This procedure yielded a correlation coefficient of 40% and an R-factor value of 45.4% in the 10–4.5 Å resolution range. Rigid-body refinement was then performed with REFMAC (Murshudov et al, 1997) using data between 30 and 4.2 Å. Attempts to determine the position and orientation of the bound Cbtx molecules by molecular replacement using, as alternative search models, either coordinates available for unbound or peptide-bound Cbtx and Bgtx were unsuccessful. Therefore, a single Cbtx molecule (2CTX; Betzel et al, 1991) was manually fitted into the unaveraged SigmaA-weighted 2Fo−Fc and difference Fourier maps with the graphics program TURBO (Roussel and Cambillau, 1989) using, as a starting position, either of those predicted in the models of the Cbtx–α7 (Fruchart-Gaillard et al, 2002) and Bgtx–α12βγδ receptor complexes (Samson et al, 2002). Observation of the electron density maps calculated after rigid-body refinement confirmed the correct positioning of the toxin molecule relative to one AChBP subunit interface. The other four toxin molecules were then positioned similarly at the surface of the other four AChBP subunit interfaces in the pentamer and the whole complex was refined by rigid-body followed by multidomain NCS averaging as implemented in DM (CCP4, 1994). The resulting electron density maps were used, when clearly visible, to correct the backbone traces along the Cbtx and AChBP molecules in the complex as well as to position side chains at the complex interface with TURBO-FRODO. A single restrained refinement step, including tight five-fold NCS restraints for the AChBP (residues Phe1–Pro154 and Glu163–Arg202) and Cbtx molecules, was performed but due to the limited resolution, the remainder of the model was not refined further. Further information on the refinement procedure and structure quality is available at The EMBO Journal Online (Supplementary data: Materials and methods).
The final Cbtx–AChBP complex structure comprises AChBP residues Leu1–Arg206 and Leu1–Thr155 and Ser159–Gly205 (in one and four subunits, respectively) and Cbtx residues Ile1–Thr67/Arg68 (in one/four Cbtx molecules, respectively) (Figures 1 and 3) corresponding to 1014 residues for the AChBP pentamer and 339 residues for the five bound toxins. Weak electron densities are associated with the Cbtx molecule bound at the D–E subunit interface of AChBP due to the absence of crystal packing contacts: the Arg68 side chain (truncated to Cα) and the C-terminal residues Lys69–Pro71 in four and all five Cbtx molecules, respectively; the Thr156–Asn158 region in four out of the five AChBP subunits; and the C-terminal residues Arg206–Ala219, which include the 6xHis tag, in all five AChBP subunits. The glycan moiety linked at Asn66 is not visible, suggesting high conformational disorder. The marked anisotropy observed in the diffraction patterns and the limited final resolution result from the presence of a large solvent channel, which extends up to 180 Å along the crystal c-axis and separates, by 60 Å, two AChBP pentamers coaligned head-to-head along their five-fold axis (crystal a-axis). In this channel, clustering of the highly disordered and cationic N-terminal 3xFLAG sequences precludes formation of the head-to-head packing of pentamers found in the Hepes-bound AChBP crystal (Brejc et al, 2001).
The average root mean square deviation between the Cbtx-bound and Hepes-bound AChBP pentamers (1I9B; Brejc et al, 2001) is 0.79 Å for 981 Cα atoms, and that between AChBP subunits from each structure is 0.6 Å for 199 Cα atoms with deviation of up to 10 Å for residue Cys187 located at the tip of loop C. The deviation between the bound and free (2CTX) Cbtx molecules is 1.0 Å for 68 Cα atoms, between Cbtx bound to AChBP and Bgtx bound to the 13-mer peptide (1HC9; Harel et al, 2001) is 1.2 Å for 67 Cα atoms, while between the bound Cbtx and free Bgtx (2ABX; Love and Stroud, 1986) the deviation is 2.1 Å for 52 Cα atoms. The stereochemistry of the structure was analyzed with PROCHECK (Laskowski et al, 1993); no residues were found in the disallowed regions of the Ramachandran plot. Atomic coordinates and structure factors have been deposited with the Protein Data Bank, accession code 1YI5. Corrections to this structure will be made if and when better diffraction data become available. The nAChR subunit sequences and the coordinates of the modeled Cbtx–α7 receptor complex were retrieved from the LGIC database (URL www.pasteur.fr/recherche/banques/LGIC/LGIC.html; Le Novère and Changèux, 1999). Figures were generated with SPOCK (Christopher, 1998) and Raster3D (Merritt and Bacon, 1997).
Supplementary Material
Supplementary Information 1
Supplementary Information 2
Supplementary Figure
Acknowledgments
We are grateful to M Juin and L Renault (CNRS FRE-2738, Marseille) for assistance in protein analysis and crystallogenesis; Z Radić, RE Hibbs (UCSD, La Jolla), G Sulzenbacher and A Roussel (CNRS UMR-6098, Marseille), ME Dodson (YSBL, York) and P Evans (LMB, Cambridge) for fruitful discussions; and the ID14-EH4 staff (ESRF, Grenoble) for assistance in data collection. Supported by grants from the USPHS (R37-GM18360) and the Tobacco-Related Disease Research Program (to PT).
References
- Ackermann EJ, Ang ET, Kanter JR, Tsigelny I, Taylor P (1998) Identification of pairwise interactions in the α-neurotoxin–nicotinic acetylcholine receptor complex through double mutant cycles. J Biol Chem 273: 10958–10964 [DOI] [PubMed] [Google Scholar]
- Betzel C, Lange G, Pal GP, Wilson KS, Maelicke A, Saenger W (1991) The refined crystal structure of α-cobratoxin from Naja naja siamensis at 2.4-Å resolution. J Biol Chem 266: 21530–21536 [DOI] [PubMed] [Google Scholar]
- Bourne Y, Kolb HC, Radić Z, Sharpless KB, Taylor P, Marchot P (2004) Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation. Proc Natl Acad Sci USA 101: 1449–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne Y, Taylor P, Marchot P (1995) Acetylcholinesterase inhibition by fasciculin: crystal structure of the complex. Cell 83: 503–512 [DOI] [PubMed] [Google Scholar]
- Bouzat C, Gumilar F, Spitzmaul G, Wang HL, Rayes D, Hansen SB, Taylor P, Sine SM (2004) Structural network coupling agonist binding to channel gating revealed by ACh-binding protein linked to ion channel. Nature 430: 896–900 [DOI] [PubMed] [Google Scholar]
- Branden CI, Jones TA (1990) Between objectivity and subjectivity. Nature 343: 687–689 [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK (2001) Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 411: 269–276 [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Celie PH, Van Rossum-Fikkert SE, Van Dijk WJ, Brejc K, Smit AB, Sixma TK (2004) Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron 41: 907–914 [DOI] [PubMed] [Google Scholar]
- Chang CC, Lee CY (1963) Isolation of neurotoxins from the venom of Bungarus multicinctus and their modes of neuromuscular blocking action. Arch Int Pharmacodyn Ther 144: 241–257 [PubMed] [Google Scholar]
- Changeux J, Edelstein SJ (2001) Allosteric mechanisms in normal and pathological nicotinic acetylcholine receptors. Curr Opin Neurobiol 11: 369–377, (Review) [DOI] [PubMed] [Google Scholar]
- Changeux J-P, Kasai M, Lee CY (1970) Use of a snake venom toxin to characterize the cholinergic receptor protein. Proc Natl Acad Sci USA 67: 1241–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappinelli VA, Weaver WR, McLane KE, Conti-Fine BM, Fiordalisi JJ, Grant GA (1996) Binding of native κ-neurotoxins and site-directed mutants to nicotinic acetylcholine receptors. Toxicon 34: 1243–1256, (Review) [DOI] [PubMed] [Google Scholar]
- Christopher JA (1998) SPOCK: The Structural Properties Observation and Calculation Kit Program Manual. College Station, TX; The Center for Macromolecular Design, Texas A & M University [Google Scholar]
- Dewan JC, Grant GA, Sacchettini JC (1994) Crystal structure of κ-bungarotoxin at 2.3-Å resolution. Biochemistry 33: 13147–13154 [DOI] [PubMed] [Google Scholar]
- Endo T, Tamiya N (1991) Structure–function relationships of postsynaptic neurotoxins from snake venoms. In Snake Toxins, Harvey AL (ed) pp 165–222. New York: Pergamon Press Inc. [Google Scholar]
- Engh RA, Huber R (1991) Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr A 47: 392–400 [Google Scholar]
- Fruchart-Gaillard C, Gilquin B, Antil-Delbeke S, Le Novere N, Tamiya T, Corringer PJ, Changeux J-P, Ménez A, Servent D (2002) Experimentally based model of a complex between a snake toxin and the α7 nicotinic receptor. Proc Natl Acad Sci USA 99: 3216–3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Bren N, Burghardt TP, Hansen S, Henchman RH, Taylor P, McCammon JA, Sine SM (2004) Agonist-mediated conformational changes in ACh-binding protein revealed by simulation and intrinsic tryptophan fluorescence. J Biol Chem 280: 8443–8451 [DOI] [PubMed] [Google Scholar]
- Grutter T, Changeux J-P (2001) Nicotinic receptors in wonderland. Trends Biochem Sci 26: 459–463 [DOI] [PubMed] [Google Scholar]
- Hansen SB, Radić Z, Talley TT, Molles BE, Deerinck T, Tsigelny I, Taylor P (2002) Tryptophan fluorescence reveals conformational changes in the acetylcholine binding protein. J Biol Chem 277: 41299–41302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SB, Talley TT, Radić Z, Taylor P (2004) Structural and ligand recognition characteristics of an acetylcholine binding protein from Aplysia californica. J Biol Chem 279: 24197–24202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harel M, Kasher R, Nicolas A, Guss JM, Balass M, Fridkin M, Smit AB, Brejc K, Sixma TK, Katchalski-Katzir E, Sussman JL, Fuchs S (2001) The binding site of acetylcholine receptor as visualized in the x-ray structure of a complex between α-bungarotoxin and a mimotope peptide. Neuron 32: 265–275 [DOI] [PubMed] [Google Scholar]
- Hibbs RE, Talley TT, Taylor P (2004) Acrylodan conjugated cysteine side chains reveal conformational state and ligand site locations of the acetylcholine binding protein. J Biol Chem 279: 28483–28491 [DOI] [PubMed] [Google Scholar]
- Hooft RW, Vriend G, Sander C, Abola EE (1996) Errors in protein structures. Nature 381: 272. [DOI] [PubMed] [Google Scholar]
- Karlin A (2002) Emerging structure of the nicotinic acetylcholine receptors. Nat Rev Neurosci 3: 102–114, (Review) [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Jones TA (1996) Phi/psi-chology: Ramachandran revisited. Structure 4: 1395–1400 [DOI] [PubMed] [Google Scholar]
- Laskowski R, MacArthur M, Moss D, Thornton J (1993) PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr 26: 283–291 [Google Scholar]
- le Du M-H, Marchot P, Bougis PE, Fontecilla-Camps JC (1996) Crystal structure of fasciculin 2 from green mamba snake venom: evidence for unusual loop flexibility. Acta Crystallogr D 52: 87–92 [DOI] [PubMed] [Google Scholar]
- Le Novère N, Changeux J-P (1999) The ligand gated ion channel database. Nucleic Acids Res 27: 340–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love RA, Stroud RM (1986) The crystal structure of α-bungarotoxin at 2.5 Å resolution: relation to solution structure and binding to acetylcholine receptor. Protein Eng 1: 37–46 [DOI] [PubMed] [Google Scholar]
- Malany S, Osaka H, Sine SM, Taylor P (2000) Orientation of α-neurotoxin at the subunit interfaces of the nicotinic acetylcholine receptor. Biochemistry 39: 15388–15398 [DOI] [PubMed] [Google Scholar]
- Marchot P, Bougis PE (2000) The fasciculins, and their interaction with acetylcholinesterase. In Animal Toxins. Facts and Protocols, Rochat H, Martin-Eauclaire M-F (eds) pp 246–275. Basel: Birkhäuser Verlag [Google Scholar]
- Marchot P, Frachon P, Bougis PE (1988) Selective distinction at equilibrium between the two α-neurotoxin binding sites of Torpedo acetylcholine receptor by microtitration. Eur J Biochem 174: 537–542 [DOI] [PubMed] [Google Scholar]
- Marchot P, Ravelli RB, Raves ML, Bourne Y, Vellom DC, Kanter J, Camp S, Sussman JL, Taylor P (1996) Soluble monomeric acetylcholinesterase from mouse: expression, purification, and crystallization in complex with fasciculin. Protein Sci 5: 672–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews BW (1968) Solvent content of protein crystals. J Mol Biol 33: 491–497 [DOI] [PubMed] [Google Scholar]
- Merritt E, Bacon D (1997) Raster3D: photorealistic molecular graphics. Methods Enzymol 277: 505–524 [DOI] [PubMed] [Google Scholar]
- Moise L, Piserchio A, Basus VJ, Hawrot E (2002) NMR structural analysis of α-bungarotoxin and its complex with the principal α-neurotoxin-binding sequence on the α7 subunit of a neuronal nicotinic acetylcholine receptor. J Biol Chem 277: 12406–12417 [DOI] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux J-P (1965) On the nature of allosteric transitions: a plausible model. J Mol Biol 12: 88–118 [DOI] [PubMed] [Google Scholar]
- Moore MA, McCarthy MP (1995) Snake venom toxins, unlike smaller antagonists, appear to stabilize a resting state conformation of the nicotinic acetylcholine receptor. Biochim Biophys Acta 1235: 336–342 [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Navaza J (1994) AMoRe: an automated package for molecular replacement. Acta Crystallogr A 50: 157–163 [Google Scholar]
- Nirthanan S, Gwee MC (2004) Three-finger α-neurotoxins and the nicotinic acetylcholine receptor, forty years on. J Pharmacol Sci 94: 1–17 [DOI] [PubMed] [Google Scholar]
- Oswald RE, Sutcliffe MJ, Bamberger M, Loring RH, Braswell E, Dobson CM (1991) Solution structure of neuronal bungarotoxin determined by two-dimensional NMR spectroscopy: sequence-specific assignments, secondary structure, and dimer formation. Biochemistry 30: 4901–4909 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Paas Y, Cartaud J, Recouvreur M, Grailhe R, Dufresne V, Pebay-Peyroula E, Landau EM, Changeux J-P (2003) Electron microscopic evidence for nucleation and growth of 3D acetylcholine receptor microcrystals in structured lipid-detergent matrices. Proc Natl Acad Sci USA 100: 11309–11314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel A, Cambillau C (1989) TURBO-FRODO. Mountain View, CA: Silicon Graphics [Google Scholar]
- Samson A, Scherf T, Eisenstein M, Chill J, Anglister J (2002) The mechanism for acetylcholine receptor inhibition by α-neurotoxins and species-specific resistance to α-bungarotoxin revealed by NMR. Neuron 35: 319–332 [DOI] [PubMed] [Google Scholar]
- Sine SM (2002) The nicotinic receptor ligand binding domain. J Neurobiol 53: 431–446, (Review) [DOI] [PubMed] [Google Scholar]
- Smit AB, Syed NI, Schaap D, van Minnen J, Klumperman J, Kits KS, Lodder H, van der Schors RC, van Elk R, Sorgedrager B, Brejc K, Sixma TK, Geraerts WP (2001) A glia-derived acetylcholine-binding protein that modulates synaptic transmission. Nature 411: 261–268 [DOI] [PubMed] [Google Scholar]
- Taylor P, Molles B, Malany S, Osaka H (2002) Toxins as probes for structure and specificity of synaptic target proteins. In Perspectives in Molecular Toxinology, Ménez A (ed) pp 271–280. Chichester, UK: John Wiley & Sons [Google Scholar]
- Taylor P, Osaka H, Molles B, Keller SH, Malany S (2000) Contributions of studies of the nicotinic receptor from muscle to defining structural and functional properties of ligand-gated ion channels. In Neuronal Nicotinic Receptors, Vol. 144, pp 79–100. Berlin: Handb. Exp. Pharm., Springer Verlag [Google Scholar]
- Tsetlin V, Hucho F (2004) Snake and snail toxins acting on nicotinic acetylcholine receptors: fundamental aspects and medical applications. FEBS Lett 557: 9–13, (Review) [DOI] [PubMed] [Google Scholar]
- Unwin N, Miyazawa A, Li J, Fujiyoshi Y (2002) Activation of the nicotinic acetylcholine receptor involves a switch in conformation of the α subunits. J Mol Biol 319: 1165–1176 [DOI] [PubMed] [Google Scholar]
- Weber M, Changeux J-P (1974) Binding of Naja nigricollis (3H) α-toxin to membrane fragments from Electrophorus and Torpedo electric organs. Mol Pharmacol 10: 1–40 [PubMed] [Google Scholar]
- Zeng H, Hawrot E (2002) NMR-based binding screen and structural analysis of the complex formed between α-cobratoxin and an 18-mer cognate peptide derived from the α1 subunit of the nicotinic acetylcholine receptor from Torpedo californica. J Biol Chem 277: 37439–37445 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information 1
Supplementary Information 2
Supplementary Figure