

Abstract
The Nova family of neuron-specific RNA-binding proteins were originally identified as targets in an autoimmune neurologic disease characterized by failure of motor inhibition. Nova-1 regulates alternative splicing of pre-mRNAs encoding the inhibitory neurotransmitter receptor subunits GABAARγ2 and GlyRα2 by directly binding intronic elements, resulting in enhancement of exon inclusion. Here we identify exon E4 in the Nova-1 pre-mRNA itself, encoding a phosphorylated protein domain, as an additional target of Nova-dependent splicing regulation in the mouse spinal cord. Nova binding to E4 is necessary and sufficient for Nova-dependent exon exclusion. E4 harbors five repeats of the known Nova-binding tetranucleotide YCAY and mutation of these elements destroys Nova-dependent regulation. Furthermore, swapping of the sites from Nova-1 and GABAARγ2 indicates that the ability of Nova to enhance or repress alternative exon inclusion is dependent on the position of the Nova-binding element within the pre-mRNA. These studies demonstrate that in addition to its previously described role as a splicing activator, Nova autoregulates its own expression by acting as a splicing repressor.
Keywords: alternative splicing, exonic splicing silencer, KH domain, neuron specific, RNA-binding protein
Introduction
RNA-binding proteins are of fundamental importance for the coordination and regulation of post-transcriptional gene expression in eukaryotes, from pre-mRNA splicing to 5′-end capping, cleavage and polyadenylation, nuclear mRNA export, localization, translation and degradation. The contemporary view that the human genome contains approximately 30 000 genes is remarkably different from that anticipated 10 years ago, when numbers of genes higher than 105 were commonly estimated. One of the key mechanisms for creating protein diversity from a limited number of genes is through alternative splicing of pre-mRNAs. Recent array-based studies suggest that at least 75% of multi-exon genes undergo alternative splicing (Johnson et al, 2003). Both ubiquitous and tissue-restricted RNA-binding proteins are critical to this process (Black, 2003), especially in highly complex systems such as the brain, which has been estimated to be the tissue with the largest total number of tissue-specific alternative splicing events (Xu et al, 2002).
The Nova proteins were the first mammalian tissue-specific splicing factors identified. Nova proteins form a family of RNA-binding proteins that are expressed exclusively in neurons of the central nervous system (CNS; Buckanovich et al, 1993, 1996; Yang et al, 1998). They were identified as the target antigens in paraneoplastic opsoclonus-myoclonus ataxia (POMA), a rare immune-mediated disorder associated with ectopic Nova expression in tumors, and characterized by deficits in inhibitory motor control in the eyes, limbs and trunk (reviewed in Darnell and Posner, 2003).
Nova proteins are sequence-specific RNA-binding proteins that harbor three KH-type RNA-binding domains. Extensive genetic, biochemical and crystallographic studies have demonstrated the ability of Nova to bind RNA containing repeats of the sequence YCAY, both in vivo and in vitro (Buckanovich and Darnell, 1997; Yang et al, 1998; Lewis et al, 2000; Jensen et al, 2000b). Nova regulates alternative splicing of the GABAARγ2 and GlyRα2 inhibitory neurotransmitter receptor subunit pre-mRNAs via binding to intronic YCAY-repeat elements (Jensen et al, 2000a; Dredge and Darnell, 2003). A putative Nova-binding element was also predicted 8 nt downstream of the alternatively spliced penultimate exon, E4, of Nova-1 (previously termed exon H; Buckanovich et al, 1993). Nova fusion protein was shown to bind this sequence with high affinity (Kd∼20 nM) in vitro and native Nova protein in mouse brain lysates co-immunoprecipitates Nova-1 pre-mRNA (Buckanovich and Darnell, 1997). This exon encodes a domain harboring a number of potential sites for phosphorylation by serine/threonine protein kinases.
To further our understanding of Nova function and its role in the regulation of alternative splicing in the brain, we sought to determine whether E4 encodes for a phosphorylated protein domain and whether Nova-1 is capable of autoregulation of alternative splicing of E4. We find that E4 encodes an S/T-rich domain within the Nova-1 protein that is phosphorylated in vivo. Moreover, Nova does indeed autoregulate alternative splicing of E4, both in cell transfection assays and in vivo; splicing of E4 is altered in the CNS of Nova haploinsufficient (Nova-1+/−) mice compared to wild type (Nova-1+/+). Surprisingly, Nova inhibits rather than activates E4 inclusion, and mapping the Nova-binding site that mediates this action identifies a necessary and sufficient element within the E4 exon itself. Moreover, we demonstrate that the ability of Nova to activate or inhibit exon inclusion is dependent on the position of the Nova-binding site within the regulated pre-mRNA. This work demonstrates that Nova is able to function as a splicing repressor in addition to its known role as a splicing activator.
Results
Nova-1 domain 4 is phosphorylated in vivo
The physiological consequence of the inclusion of the peptide sequence encoded by exon 4 (henceforth called ‘domain 4') in Nova-1 protein is unknown. E4 encodes a 24-amino-acid protein domain rich in serine and threonine residues (50%), four of which are adjacent to prolines (Figure 1B), which suggests that these residues may be substrates for phosphorylation by serine/threonine protein kinases (Kemp and Pearson, 1990). We tested whether Nova-1 is phosphorylated in vivo and whether the two alternatively spliced isoforms are phosphorylated differentially. Plasmids encoding Nova-1L (E4 included) or Nova-1S (E4 excluded) were transiently transfected into N2A cells. The cells were then labeled with 32P-orthophosphate followed by immunoprecipitation for T7-tagged Nova-1 protein. We found that Nova-1L was robustly phosphorylated, more than 10-fold greater than the level of Nova-1S phosphorylation (Figure 1A and D).
Figure 1.
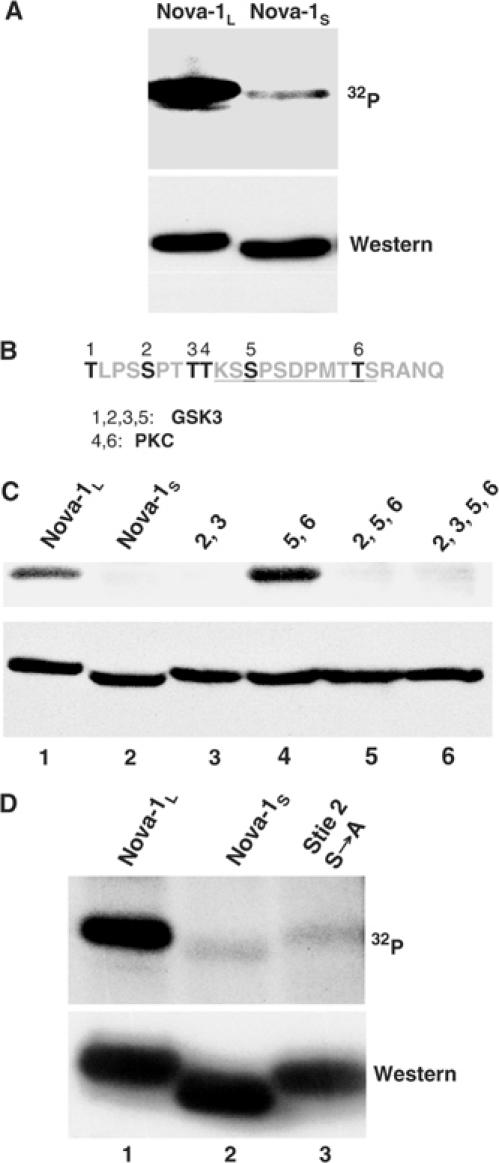
Nova-1 domain 4 is phosphorylated in vivo. (A) N2A cells were transiently transfected with T7-tagged Nova constructs with (Nova-1L) or without (Nova-1S) inclusion of domain 4, and 32P-orthophosphate was added to the medium. After 5 h incubation, cells were harvested, lysed and immunoprecipitated with anti-T7-tag antibody. The pellet was treated with RNase prior to separation by SDS–PAGE, transferred to nitrocellulose and exposed to autoradiography film (upper panel). Subsequently, the membrane was probed by Western blot with anti-T7-tag antibody (lower panel). (B) Amino-acid sequence encoded by E4. The residues in bold and numbered 1–6 are phosphorylation sites predicted by Prosite (http://us.expasy.org/prosite/) and Scansite (http://scansite.mit.edu). The underlined segment is a PEST region predicted by PESTfind (http://emb1.bcc.univie.ac.at/embnet/tools/bio/PESTfind/). (C) Identification of the phosphorylation site by site-directed mutagenesis. Metabolic phosphorylation assays were performed as in (A). Expression constructs encoding Nova-1L (lane 1) or Nova-1S (lane 2), and point mutants in which the indicated residue numbers indicated in (B) were mutated in Nova-1L to alanines (lanes 3–6) were utilized. (D) Expression constructs encoding Nova-1L (lane 1), Nova-1S (lane 2) or Nova-1L with a serine to alanine mutation at site 2 were utilized for the transfection/metabolic labeling assay as in (A).
Analysis of the sequence of domain 4 predicted six residues with the potential for phosphorylation by either protein kinase C (PKC) or glycogen synthase kinase 3 (GSK3) (Figure 1B). To test these predictions, we substituted alanine residues for various serine and threonine residues by site-directed mutagenesis. Phosphorylation was unchanged when sites 5 and 6 were mutated but was markedly reduced, to the baseline level seen for Nova-1S, when sites 2 and 3, or sites 2, 5 and 6 were mutated, suggesting that site 2 is necessary for phosphorylation (Figure 1C). Moreover, alanine mutation of site 2 alone was sufficient to reduce the phosphorylation to the level of Nova-1S (Figure 1D). These results indicate that either site 2 is the only site of phosphorylation within domain 4 or that its phosphorylation is required for further phosphorylation to occur, a process termed primed phosphorylation. Primed phosphorylation has been demonstrated for a number of kinases including GSK3 (Fiol et al, 1987; Biondi and Nebreda, 2003). Taken together, these data confirm that Nova-1 domain 4 is phosphorylated in vivo, and identify the second serine as a major site of phosphorylation.
Nova-1 alternative splicing is altered in Nova-1 heterozygous mice
The exon:intron structure of the Nova-1 gene is shown in Figure 2A. Exon 2 encodes the first KH RNA-binding domain, KH1; KH2 and KH3 are both encoded within exon 5. The penultimate exon, E4 (previously referred to as exon H), is alternatively spliced to generate two isoforms of Nova-1 (Buckanovich et al, 1993). The genomic sequence surrounding and including E4 shown in Figure 2A is highly conserved from mouse to human (88% identity, BLAST2) and rich in YCAY repeats: 32 and 28 in mouse and human respectively, compared to 15 expected in a random sequence of this length. Of these, five are present within the 72 nt exon E4; this is significantly higher than the expected frequency of 1.1 for this exon.
Figure 2.
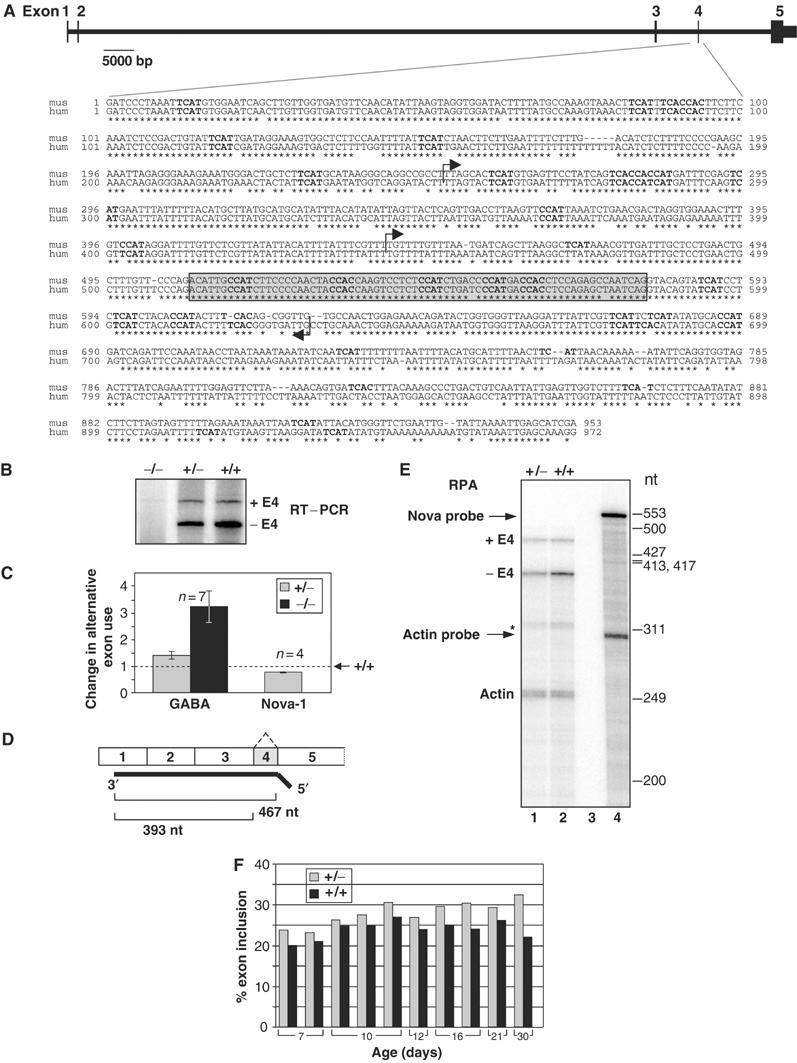
Change in alternative splicing of Nova-1 E4 in Nova-1 mouse spinal cord. (A) Exon structure of the mouse Nova-1 gene, adapted from http://genome.ucsc.edu/. Scale bar represents intronic lengths, and exons are to approximate scale only. Below is a comparison of the mouse (mus) and human (hum) sequence surrounding alternatively spliced E4 (gray box) by ClustalW alignment (MacVector). YCAY motifs are in boldface type, and arrows indicate regions included in minigene constructs pE4307 and pE4116 (see Figure 4). (B) RT–PCR. Spinal cord RNA isolated from P17 Nova-1 null, heterozygous and wild-type mouse littermates was analyzed by semiquantitative RT–PCR using primers to exons 3 and 5. The reverse primer was 32P-labeled. (C) Comparison of the magnitude of splicing defects in heterozygous mice analyzed by RT–PCR. The RT–PCR data from panel B together with additional samples were plotted as the change in alternative exon use (with the ratio of alternative exon included:excluded form from the wild type set to 1) for comparison with previously published RT–PCR data; Jensen et al, 2000a). Error bars indicate the standard deviation, n=number of litters assayed. (D) Schematic representation of the 32P-labeled antisense RNA probe used for RPA and the resulting product sizes used for quantitation. An antisense RNA probe specific for β-actin mRNA was also made. (E) RPA. Spinal cord RNA isolated from P16 Nova-1 heterozygous mouse and wild-type littermate was analyzed by RPA. The specific activities of the probes were adjusted to give protected fragments of approximately equal intensities for each message. Lane 3: yeast RNA control; lane 4: undigested probes. The asterisk marks an unidentified product that was protected by the Nova-1 probe. (F) Quantitation of the data presented in (E) together with additional RNA samples. Products were quantitated by phosphorimager and corrected for the expected number of 32P-labeled uridines per protected RNA fragment.
To determine whether Nova-1 may play a role in the regulation of E4 splicing in vivo, we used semiquantitative RT–PCR to analyze alternative splicing of this exon in the spinal cords of wild-type and Nova-1 heterozygous mice, which express reduced levels of Nova-1 protein (Jensen et al, 2000a). RT–PCR revealed a decrease in E4 exclusion in heterozygous mice to 0.75-fold of the wild-type level (Figure 2B and C). This contrasts with previously studied examples of Nova-dependent splicing regulation in which Nova enhances alternative exon inclusion. To confirm these results, we also performed RNase protection assays (RPA), which revealed a 24.6% decrease in steady-state Nova-1 mRNA level relative to actin in heterozygous mice compared to wild-type levels (data not shown). In 14 littermate pairs spanning postnatal day 7 through 30, E4 inclusion was increased 19±10% (P<0.001) in heterozygous mice compared to their wild-type littermates (Figure 2B, C, E and F), demonstrating a relationship between Nova-1 expression and E4 inclusion, and suggesting that Nova-1 could directly mediate E4 exclusion.
The magnitude of the effect on E4 splicing in Nova-1 heterozygous mice, while small, was comparable to that seen in GABAARγ2 splicing between wild type and Nova-1 heterozygotes (Figure 2C). Furthermore, up to a 3.9-fold increase in Nova-1 E4 inclusion has been observed in the brains of Nova-2 null mice compared to their wild-type littermates (M Ruggiu, BK Dredge and RB Darnell, unpublished data), strengthening our conclusion that Nova-1 alternative splicing is altered in Nova-deficient mice, and the prediction that both Nova-1 and Nova-2 proteins likely act through direct inhibition of Nova-1 E4 inclusion.
The presence or absence of domain 4 does not influence Nova-1 regulation of alternative splicing of Nova-1 and GABAARγ2 minigenes
To investigate whether Nova is able to act directly on its own transcript to autoregulate E4 splicing, a minigene construct was generated that contains 957 nt of mouse genomic sequence surrounding and including E4. This was flanked at the extreme 5′ and 3′ ends by donor and acceptor sites derived from the human β-globin gene, which we have previously shown are not themselves subject to Nova-dependent splicing regulation (Dredge and Darnell, 2003). This construct (pE4885) was transiently transfected into N2A and 293T cells in the presence or absence of pNova-1L or pNova-1S plasmid (that is, with or without domain 4) and spliced products were analyzed by primer extension (Figure 3A). Consistent with the analysis of Nova-1 splicing in Nova-deficient mice, cotransfection of either pNova-1L or pNova-1S caused a dose-dependent decrease in E4 inclusion from ⩾95 to ⩽12% in both cell lines. No products were detected in mock-transfected cells (Figure 3A, lanes M). These data confirm that both Nova-1L and Nova-1S can repress E4 inclusion by a similar magnitude and indicate that the sequence surrounding and including E4 is sufficient to direct Nova-mediated splicing regulation. Given the number of YCAY motifs in this fragment, these results are consistent with the hypothesis that sequence-specific interactions between Nova-1 and its own pre-mRNA are necessary for splicing autoregulation.
Figure 3.
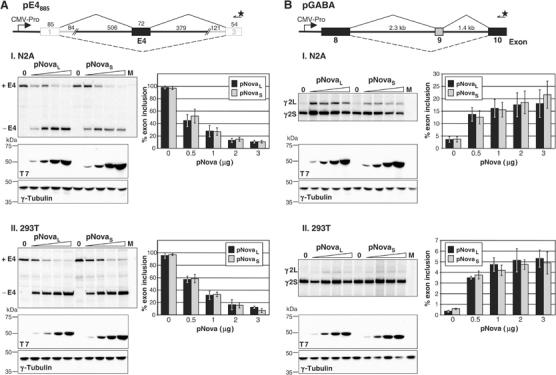
Nova-1 regulates alternative splicing of Nova-1 and GABAARγ2 in cell lines independent of domain E4 inclusion. (A) Nova-1 minigene construct used for transient transfection assays harboring 957 of Nova-1 mouse genomic sequence (black lines, black filled box; sequence is shown in Figure 2) flanked by donor and acceptor sites derived from the human β-globin gene (gray lines, empty boxes). The sizes (in base pairs) are shown above each element. The starred arrow depicts the 32P-labeled primer used for primer extension analysis. Distances are not to scale. RNA was isolated from N2A (I) or 293T (II) cells transiently transfected with pE4885 and increasing amounts of pNova-1L or pNova-1S (0, 0.5, 1, 2 and 3 μg) and analyzed by primer extension (upper panels). Mock transfections were performed using no plasmid DNA (lanes M). Below are Western blots using anti-T7 antibody (middle panels) showing the titration of T7-tagged Nova-1 protein levels. The blots were stripped and reprobed with anti-γ-tubulin antibody (lower panels) as a loading control. The graphs depict quantitation of three independent transfection experiments, displayed as the average percent central exon inclusion [Phosphorimager counts in the exon included product/(counts in the exon included product+counts in the exon excluded product) × 100], ±standard deviation. (B) Transient transfection assays were performed and analyzed as in (A) using a GABAARγ2 (pGABA) minigene containing the full mouse intronic regions surrounding and including alternatively spliced exon 9 plus shortened exons 8 and 10.
We also tested whether domain 4 influences Nova-dependent splicing of GABAARγ2 and GlyRα2 minigenes in cell lines since previous analyses have used only pNova-1L (Jensen et al, 2000a; Dredge and Darnell, 2003). In all cases tested, the absence of domain 4, or mutation of the phosphorylation site identified in Figure 1 had no effect on the direction or magnitude of the Nova-1-dependent splicing effect (Figure 3B and data not shown).
Exonic sequence is necessary and sufficient for Nova-dependent splicing autoregulation of exon 4
We further analyzed the sequence requirements for Nova-dependent splicing autoregulation within the fragment surrounding and including E4 present in pE4885. Initial experiments mutating the four intronic YCAY repeats immediately downstream of E4, predicted earlier to be a Nova-binding site (Buckanovich and Darnell, 1997), showed no effect on Nova-dependent splicing (data not shown). We therefore generated a series of constructs to identify a region necessary and sufficient for Nova-dependent splicing regulation. Figure 4A shows that deletion of the majority of the intronic regions surrounding E4 has no effect on Nova's ability to regulate E4 exclusion (compare constructs pE4885, pE4307, pE4116); pNova-1L transfection resulted in a decrease in E4 inclusion from almost 100 to 10–21% in both N2A and 293T cell lines. Furthermore, the presence of E4 itself in the minigene was sufficient to mediate a robust Nova-dependent effect on splicing; pNova-1L transfection resulted in a decrease in E4 inclusion from almost 100 to 42 or 30% in N2A and 293T cells respectively (construct pE4).
Figure 4.

Exon E4 is necessary and sufficient for regulation of alternative splicing by Nova-1. (A) The minigenes depicted were cotransfected into N2A (I) or 293T (II) cells with increasing amounts of pNova-1L (0, 0.5 and 2.0 μg); spliced products were measured by primer extension and phosphorimage analysis as in Figure 3 and titration of transfected pNova-1L expression was monitored by Western blotting (not shown). The asterisk denotes an uncharacterized product consistently seen with construct pE4307. (B) Mutagenesis of the YCAY repeats in E4 abrogates Nova-dependent regulation of alternative splicing. YAAY mutations were made in the first two (m1, 2), last three (m3, 4, 5) or all five (m1–5) YCAY motifs of E4 in construct pE4. Plasmid pGlo2Δb was used as a Nova-independent control. The minigenes were transfected and analyzed as in (A).
To determine whether the YCAY repeats within E4 are necessary for Nova-dependent repression of this exon, we mutated the first two (m1, 2; Figure 4B), last three (m3, 4, 5) or all five (m1–5) YCAY repeats to YAAY, a sequence that does not bind to Nova (Buckanovich and Darnell, 1997; Jensen et al, 2000a; Lewis et al, 2000). These mutations did not change the baseline level of inclusion of the exon (Figure 4B), indicating that sequences required for the recognition and inclusion of this exon were not disrupted. However, all of the mutations had a dramatic effect on Nova's ability to repress exon inclusion. In both cell lines, mutation of all five YCAY repeats in construct pE4 or pE4116 completely abolished the ability of Nova to regulate E4 splicing (Figure 4B, m1–5, and data not shown). In N2A cells, cotransfection of pNova1L resulted in a decrease in E4 inclusion from almost 100 to 43% (pE4) compared to a decrease from 100% to only 92% (m1, 2) or 87% (m3, 4, 5) upon partial mutation of the exon. Similarly, in 293T cells, addition of pNova1L decreased E4 inclusion from 100 to 32% (pE4) versus 92% (m1, 2) or 82% (m3, 4, 5). We also tested mutation of each YCAY motif individually in 293T cells and found that in a single experiment, Nova reduced E4 inclusion from 100 to 31% (unmutated), 70% (m1), 59% (m2), 47% (m3), 50% (m4) or 72% (m5) upon cotransfection of 2.0 μg of pNova1L (data not shown). Taken together, these data demonstrate that the YCAY repeats in E4 are necessary and sufficient for Nova-dependent splicing regulation of this exon and that all five YCAY motifs contribute to the magnitude of the effect.
The magnitude of Nova's effect on splicing of construct pE4 was smaller than that seen when 47 bp of adjacent downstream Nova-1 intronic sequence was also present in the minigene (Figures 4A and 5), implying that the intronic sequence may add to the Nova-dependent exon repression. Indeed, when the four intronic YCAY repeats downstream of E4 in construct pE4116 were mutated to YAAY, the resulting minigene (pE4116mi) behaved almost identically to one lacking Nova-derived intronic sequence (pE4) (Figure 5). However, inclusion of the intronic regions only, surrounding a heterologous exon (pGlo2Δb116), decreased basal exon inclusion to 74–79%, but was not sufficient to support significant Nova-dependent E4 repression. We conclude that the predicted intronic Nova-binding site immediately downstream of E4 is neither necessary nor sufficient for robust Nova-dependent splicing regulation in this context, but may enhance the ability of Nova-1 to repress E4 inclusion.
Figure 5.
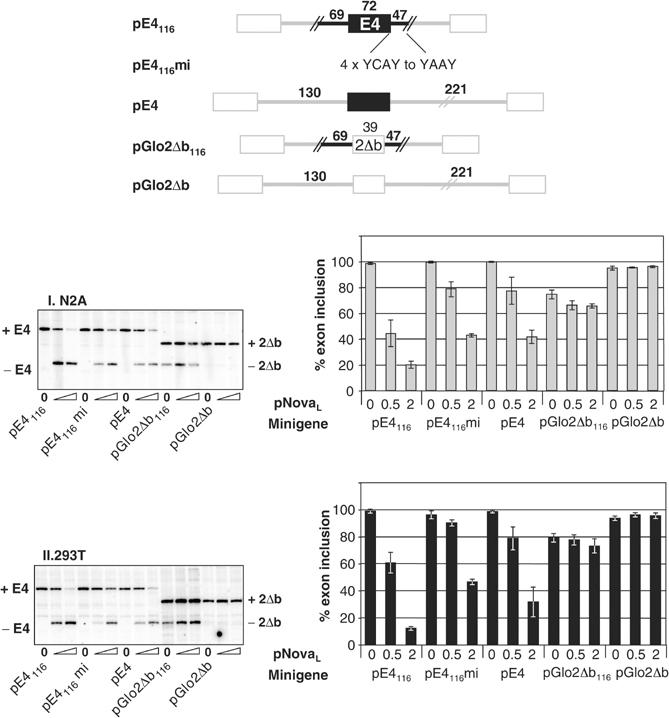
Downstream intronic sequences enhance Nova- dependent splicing of E4, but are neither necessary nor sufficient for the effect. Four YCAY repeats within the Nova-1 intronic sequence downstream of E4 in construct pE4116 were mutated to YAAY to generate construct pE4116mi. Intronic sequences in pGlo2Δb were replaced by the Nova-1 intronic sequences present in pE4116 to generate construct pGlo2Δb116. Constructs pE4 and pGlo2Δb were used as Nova-dependent and -independent controls, respectively. Minigenes were cotransfected into N2A (I) or 293T (II) cells with increasing amounts of pNova-1L (0, 0.5 and 2.0 μg); spliced products were measured by primer extension and phosphorimage analysis as in Figure 3 and titration of transfected pNova-1L expression was monitored by Western blotting (not shown).
To determine which regions of this RNA sequence are necessary for high-affinity Nova-1 binding, the 5′ and 3′ boundaries of the smallest RNA that retains high affinity to Nova-1 were determined. In vitro-transcribed RNA bounding E4 (Figure 6) was labeled with 32P at either the 5′ or 3′ end and subjected to mild alkaline hydrolysis. The RNA was then incubated with Nova-1L fusion protein (NFP), and bound RNAs captured by filtration through nitrocellulose, eluted and analyzed by denaturing PAGE. Two boundaries were detected in each case; when the RNA was 5′ labeled, the 3′ boundaries mapped 1 nt downstream of the third and second YCAY repeats of E4 (Figure 6A (I), 1° and 2°, respectively). When the RNA was 3′ labeled, the 5′ boundaries mapped 1 nt upstream of the seventh (Figure 6A (II), 1°) or 2 nt upstream of the eighth (Figure 6A (I), 2°) YCAY repeat of the test RNA such that three or two YCAY repeats were contained within the smallest RNA bound (Figure 6A (II), 1° and 2°, respectively). In both cases, the secondary boundary was only apparent at a higher protein concentration, suggesting that NFP may bind these shorter RNAs with lower affinity than it does the longer sequences. The results suggest that Nova-1 is able to bind RNA sequences either derived from exon E4 or from the intronic region immediately downstream in vitro. Thus, this experiment did not define a single high-affinity Nova-binding site—rather we can conclude that a minimum of two to three YCAY repeats are necessary for high-affinity Nova binding.
Figure 6.
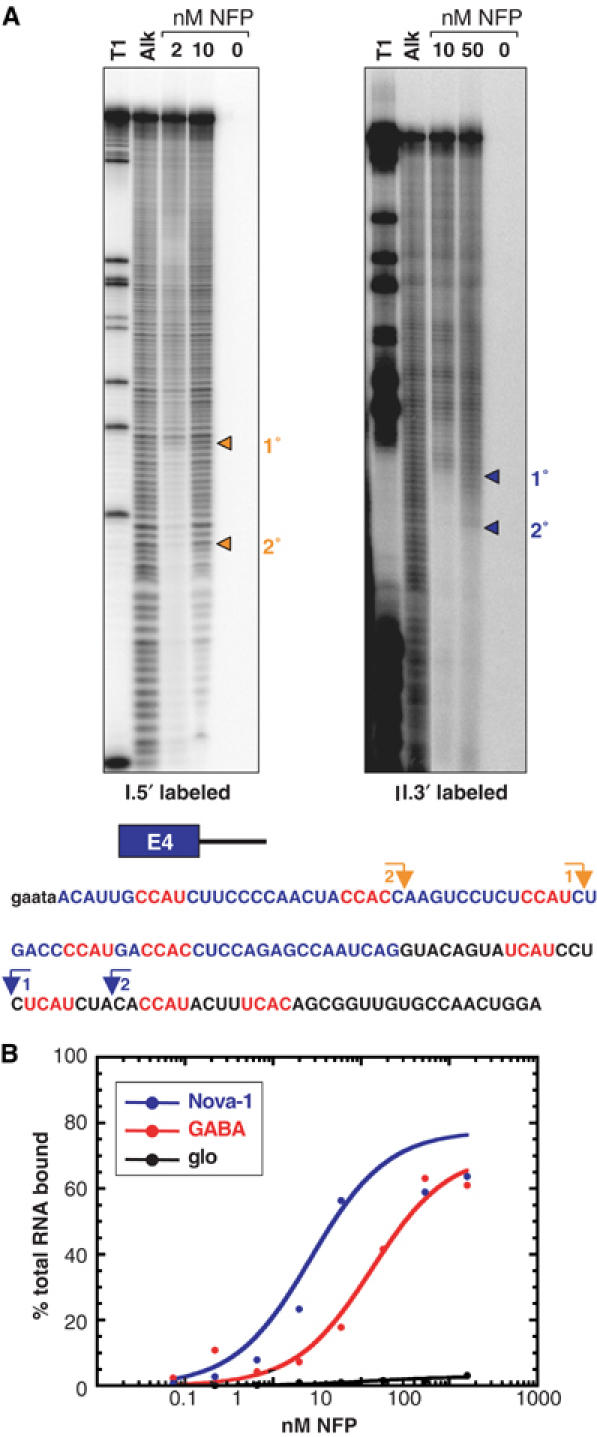
Nova-1 binds with high affinity to its own RNA in vitro. (A) Mapping the boundaries of Nova-1 protein binding to Nova-1 E4 plus downstream intronic RNA. RNA was labeled with 32P at either the 5′ or 3′ end and subjected to mild alkaline hydrolysis (Alk). The RNA was then incubated with Nova-1 fusion protein (NFP) at a final concentration of 2, 10 or 50 nM and protein:RNA complexes were captured by filtration through nitrocellulose. Bound RNAs were eluted and analyzed by denaturing PAGE. RNase T1 digestion (T1) was used to generate size standards. Boundaries are highlighted by arrows and indicated on the transcribed RNA sequence shown at the bottom. A schematic representation of the RNA transcribed is shown above the sequence; exonic sequence is shown in blue, intronic sequence in black and YCAY motifs are highlighted in red. Small letters represent sequences derived from primers. (B) Nitrocellulose filter-binding assays were performed using Nova-1 RNA as in (A) or control RNAs. GABA: 159 nt RNA corresponding to the GABAARγ2 NISE element (Dredge and Darnell, 2003); glo: 175 nt RNA derived from human β-globin, which spans regions of exon 1 and intron 1 and contains no YCAY tetranucleotides.
Nitrocellulose filter-binding assays were performed to determine the affinity of Nova-1 binding to its own pre-mRNA. NFP bound the 133 nt Nova-1 RNA bounding E4 with high affinity (Kd=2.7 nM), approximately five-fold higher affinity than binding to a 159 nt RNA derived from the Nova binding sequence in GABAARγ2 (Kd=13.3 nM; Figure 6B; Dredge and Darnell, 2003). An irrelevant β-globin-derived RNA of similar length (glo, 175 nt) showed no detectable binding (Figure 6B). Thus, Nova-1 binds to its own transcript with physiologically relevant affinity.
Binding site position determines whether Nova enhances or represses alternative exon inclusion
We recently described a Nova-dependent intronic splicing enhancer (NISE) necessary and sufficient for regulation of GABAARγ2 alternative splicing by Nova (Dredge and Darnell, 2003). Aside from harboring repeats of the tetranucleotide YCAY over a similar length of sequence, this site has little in common with the Nova-dependent exonic splicing silencer (NESS) defined here. The NISE element harbors seven YCAY repeats, which span 65 nt of sequence located 80 nt upstream of the downstream splice acceptor site. Of these seven repeats, five are UCAU, one CCAU and one UCAC. This contrasts with the NESS element, which is composed of three CCAU and two CCAC motifs over 50 nt of exonic sequence. We sought to determine whether Nova's ability to enhance or repress exon inclusion was dependent on the sequence of the Nova-binding site or its position within the pre-mRNA. Figure 7 schematizes the constructs made. We replaced exonic sequences in pE4 (now renamed p-eNESS) with the NISE element to generate plasmid p-eNISE (Figure 7A). Alternatively, we replaced intronic sequences in pGlo2Δ with the NISE or NESS elements to generate the plasmids p-iNISE and p-iNESS, respectively (Figure 7B). Corresponding mutant constructs harboring YCAY to YAAY mutations in the introduced segments were also generated (designated with an ‘m').
Figure 7.
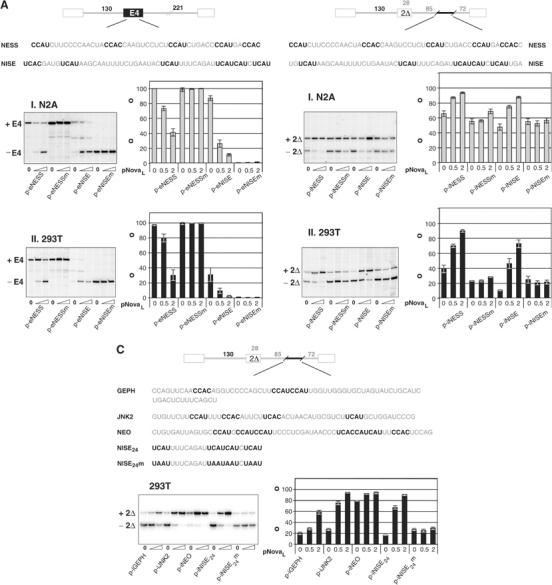
Nova-dependent regulation is dependent on the position of the Nova-binding site within the pre-mRNA. (A) A 50 nt stretch of Nova-1 E4 (NESS) containing all five YCAY repeats was replaced in pE4 (renamed p-eNESS) by a 54 nt stretch of the GABAARγ2 intron 9 NISE region (NISE) encompassing six UCAY repeats to generate plasmid p-eNISE. Corresponding constructs with YCAY to YAAY mutations in the introduced segments (p-eNESSm and p-eNISEm) were also tested. These constructs were cotransfected into N2A (I) or 293T (II) cells with increasing amounts (0, 0.5 and 2.0 μg) of pNova-1L; spliced products were measured by primer extension and phosphorimage analysis as in Figure 3 and titration of transfected pNova-1L expression was monitored by Western blotting (not shown). (B) A total of 51 nt from Nova-1 E4 or GABAARγ2 NISE, each harboring five YCAY repeats, were cloned into pGlo2Δ 72 nt upstream of the downstream splice acceptor site (plasmids p-iNESS and p-iNISE). Corresponding constructs with YCAY to YAAY mutations in the introduced segments (p-iNESSm and p-iNISEm) were also tested. These constructs were transfected and analyzed as in (A). (C) Three additional putative Nova target sequences in gephyrin, JNK2 and neogenin identified by CLIP (Ule et al, 2003) were cloned into pGlo2Δ (plasmids p-iGEPH, p-iJNK2 and p-iNEO). Construct p-iNISE24 harboring a 24 nt stretch of the GABAARγ2 NISE and a corresponding construct with YCAY to YAAY mutations (p-iNISE24m) were used as Nova-dependent and -independent controls, respectively (Dredge and Darnell, 2003). These minigenes were transfected and analyzed as in (A).
When either p-eNISE or p-eNESS was cotransfected with pNova-1L, exon inclusion was repressed (Figure 7A). Conversely, when either Nova-binding site was placed in the intron downstream of the regulated exon, Nova enhanced alternative exon inclusion (Figure 7B). Mutation of the YCAY repeats to YAAY abolished the Nova-dependent effect in all cases. Taken together, these data demonstrate that Nova's ability to enhance or repress exon inclusion is dependent upon the position of the Nova-binding site within the pre-mRNA and upon intact YCAY repeats. Interestingly, introduction of GABAARγ2-derived sequence into Nova-1 E4 (p-eNISE and p-eNISEm) reduced the baseline level of exon inclusion in both cell lines. Furthermore, mutation of the YCAY repeats abolished baseline inclusion of the central exon of p-eNISE. These observations suggest that introduction of the YAAY mutations into the exonic sequences of p-eNISE may have inadvertently generated a splicing silencer, and will be highlighted further in the discussion.
A second putative NESS has been identified in exon 27 (E27) of neogenin by CLIP, a technique developed recently in our laboratory to purify protein–RNA complexes (Ule et al, 2003). Increased levels of E27 inclusion were found in the brains of Nova-2 null mice and E27 is sufficient to confer Nova-dependent exon repression in minigene assays (Ule et al, 2003, and data not shown). In order to test whether this element can act in a position-dependent manner, as an intronic splicing enhancer, we cloned the neogenin CLIP tag into the intron of pGlo2Δ (p-iNEO), and found that the element now conferred Nova-dependent exon inclusion (Figure 7C). Thus, two entirely distinct Nova-responsive exonic elements can be functionally switched to intronic enhancer elements, strongly suggesting that Nova's activity in alternative splicing is directly related to the position of the Nova target elements within the pre-mRNA.
Nova CLIP tags have also been identified in introns 7 and 14 of gephyrin pre-mRNA, and splicing of exon 9 was shown to be enhanced in Nova-2 null mice (Ule et al, 2003). In addition, a Nova-bound CLIP tag was identified, which corresponds to an intronic region of c-jun N-terminal kinase 2 (JNK2), downstream of an alternative exon, 6B, which is spliced in a mutually exclusive manner with exon 6A. Inclusion of exon 6B was shown to be enhanced relative to 6A in Nova-2 null mice (Ule et al, 2003). In both of these cases, Nova appears to bind to intronic sequences to repress exon inclusion, in contrast to the paradigm presented here. However, when these tags were placed into the intron of pGlo2Δ, both mediated Nova-dependent exon inclusion, demonstrating a clear positional effect on alternative splicing in this assay (plasmids p-iGEPH, p-iJNK2; Figure 7C). We conclude that the position of these elements within the native transcripts and possibly additional unidentified sequences in gephyrin and JNK2 pre-mRNAs are likely to be important in mediating Nova's repressive effect on splicing in vivo, and, more generally, that Nova binding sequences confer effects on exon splicing in a position-dependent manner.
Discussion
Nova autoregulates its own splicing
Nova was the first mammalian neuron-specific splicing factor to be identified and modeled in a genetic system (Jensen et al, 2000a). Nova has been shown to regulate the alternative splicing of a number of transcripts in vivo, including the GABAARγ2 and GlyRα2 receptor subunit pre-mRNAs and gephyrin pre-mRNA (Jensen et al, 2000a; Dredge and Darnell, 2003; Ule et al, 2003). Here we have used genetic, biochemical and transient transfection approaches to identify an additional target of Nova-dependent splicing regulation, the Nova-1 pre-mRNA itself. Given that Nova expression is restricted to neurons (Buckanovich et al, 1993), and our demonstration here of altered E4 inclusion in Nova-1 heterozygous mouse spinal cord, we conclude that Nova regulates its own transcript in neurons.
A number of splicing factors have been shown to be capable of autoregulation. For example, Drosophila Sex lethal (Sxl) binds on both sides of an alternatively spliced exon within its own pre-mRNA to induce exon skipping (reviewed in Black, 2003). Similarly, hnRNP A1 binds either side of alternatively spliced exon 7B in its own transcript, resulting in exon skipping (Blanchette and Chabot, 1999). Our results represent the first instance of splicing autoregulation in mammals to be validated in a genetic system.
For Nova-1, splicing of E4 results in the addition of a phosphorylation domain into the protein product. The physiological consequences of inclusion and/or phosphorylation of domain 4 are unclear. Both hyper- and hypophosphorylation have been shown to inhibit the ability of SR proteins to regulate splicing (Prasad et al, 1999). However, in transfection assays, the presence of domain 4 had no effect on Nova's ability to regulate alternative splicing of GABAARγ2, GlyRα2 or Nova-1 minigenes. This implies that domain 4 has no discernable effect on the recognition and binding of these RNAs, nor on protein–protein interactions required for their regulation in tissue culture cells.
Nova-1 domain 4 also harbors a predicted PEST domain. PEST motifs serve as proteolytic signals to target protein for degradation by the 26S proteasome (Rechsteiner and Rogers, 1996). Although we have not directly measured the half-life of the two isoforms in a pulse–chase experiment, we have no indication that Nova-1L is less stable than Nova-1S since the steady-state level of protein after transfection was the same as detected by Western blot against the T7 tag (Figure 3, lower panels). Thus, we do not yet understand the significance of E4 phosphorylation.
Nova as a dual function splicing factor: the NESS element
We have shown that Nova-1 E4 is necessary and sufficient for Nova-dependent repression of alternative exon inclusion, and therefore can be defined as a NESS. This contrasts with previously described examples of Nova regulation of alternative splicing in which Nova enhances exon inclusion via sequence-specific interactions with intronic regions (Jensen et al, 2000a; Dredge and Darnell, 2003). Thus, Nova is a dual function splicing factor, able to both enhance and repress exon inclusion. SR proteins have been shown to function in a similar manner, acting as either positive or negative regulators of alternative splicing depending on the position of the relevant binding site within the pre-mRNA. For example, an SR-protein-binding site positioned just upstream of a branch point causes splicing inhibition in an adenovirus pre-mRNA by blocking access of U2snRNP. The same element can act as an exonic splicing enhancer if it is placed downstream of the 3′ splice site (Kanopka et al, 1996). Interestingly, this inhibition is dependent on phosphorylation of the SR proteins; inhibition is relieved upon dephosphorylation of the bound SR proteins (Kanopka et al, 1998).
We demonstrated that Nova's action on alternative splicing is position dependent by swapping the positions of the Nova-binding sites derived from the Nova-1 and GABAARγ2 pre-mRNAs (Figures 7A, B and 8A), and by transforming an exonic element in neogenin E27 to a splicing enhancer by moving it to an intronic location (Figure 7C). Furthermore, the position of the Nova-binding site within an intron also appears to confer important information, as the gephyrin and JNK2 Nova CLIP tags, which we hypothesize to act as intronic splicing silencers, act as enhancer elements when placed in a location within an intron equivalent to that of the GABAARγ2 NISE. Thus, we can predict that Nova-binding sites within alternatively spliced exons are likely to confer Nova-dependent exon exclusion; however, more examples are needed to define the relationship between the position of Nova-regulatory regions within introns and the outcome of Nova:RNA binding. We note that the single CLIP tags isolated from JNK2 and gephyrin do not preclude the possibility of additional Nova-binding sites in these transcripts.
Figure 8.
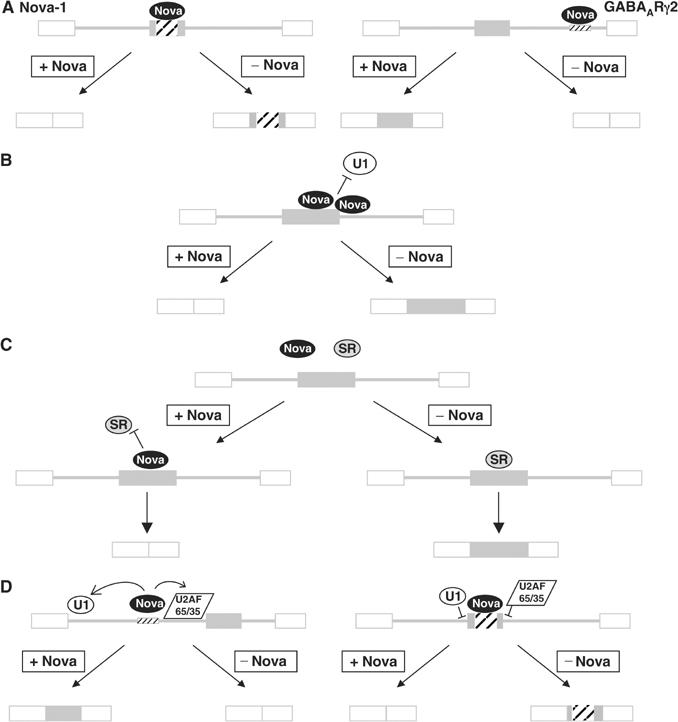
Models for Nova's action on alternative splicing. (A) Position-dependent action of Nova on alternative splicing. Nova binding to a high-affinity site within an alternative exon represses exon inclusion. In contrast, Nova binding within the intron downstream of an alternative exon enhances exon inclusion. (B) Nova-1 binding to exon E4 may inhibit binding and/or assembly of constitutive splicing factors such as U1 as shown, or sterically inhibit bridging interactions across the exon (not shown). (C) Nova-dependent inhibition may occur through competitive inhibition of nonessential splicing factors such as SR protein(s). (D) Nova may inhibit exon inclusion via incorrect recruitment of splicing factors. Empty boxes represent constitutively included exons, and filled boxes represent alternative exons. Hatching represents high-affinity Nova-binding sites. Note that while Nova protein is represented as a single oval, a number of Nova molecules are likely to be bound at each binding site.
Interestingly, the sequences of the Nova-1 E4 and GABAARγ2 Nova regulatory regions are quite different, including a high incidence of Cs (seven of a possible 10) in the first and last positions of the YCAY tetranucleotide in the NESS and a high incidence of Us (12 of a possible 14) in these positions in the NISE. This may simply reflect the tendency for exons to have a higher GC content than introns (reviewed in Bernardi, 1995). However, when the E4 NESS sequence was replaced by GABAARγ2 NISE sequence, the inclusion of the resulting exon was reduced (construct p-eNISE; Figure 7A) or abolished (construct p-eNISEm) even in the absence of Nova protein and despite an increase in length of 4 nt. We hypothesize that the NESS element contains positive signals such as ESEs, which are not present in the central exon of p-eNISE and are blocked by the binding of Nova. Alternatively, the NISE sequence may introduce additional Nova-independent negative sequences. ESE prediction algorithms based on bioinformatic predictors (Fairbrother et al, 2002) and SR protein SELEX results (Cartegni et al, 2003) have been used successfully to predict regulatory elements within exons. However, our attempts to correlate our results with the patterns of ESEs predicted using these programs were unsuccessful. Both the work presented here and that on GABAARγ2 (Dredge and Darnell, 2003) highlight the difficulty of predicting Nova (and other RBPs) regulatory sites bioinformatically, even when many details regarding the sequence specificity and structural details of Nova interactions with RNA have been well established.
Mechanism of Nova action to regulate alternative splicing
Several mechanisms have been proposed to explain exon skipping via exonic splicing silencers (ESSs). In a few cases, exonic sequences are thought to be involved in RNA secondary structures, which prevent access of necessary components of the splicing machinery to splice sites. Chicken β-tropomyosin exon 6B forms RNA–RNA base pairs with the upstream intron, preventing accessibility of this exon in myoblasts and resulting in inclusion of a mutually exclusive exon, 6A (Libri et al, 1991). However, the majority of ESS elements that have been described function through trans-acting protein factors, which bind them.
High-affinity Nova-binding sites exist throughout the exon and extend into the intron downstream of E4. While the intronic elements themselves are not sufficient to mediate Nova-dependent E4 inhibition, when they are present adjacent to E4 we see an enhancement of Nova's action on splicing inhibition. This suggests that Nova may multimerize from an obligate exonic element into the intronic sequences, much like hnRNP A1, initially binding within HIV-1 Tat exon 3, is thought to multimerize to adjacent intronic sequence and block U2AF65 intronic binding (Zhu et al, 2001), and as seen in Rev-RRE multimerization (Malim and Cullen, 1991).
Nova binding to these sites could inhibit splicing in a number of ways. For example, binding of Nova could sterically inhibit the binding and/or assembly of critical spliceosome components such as U1-associated factors at the splice junction(s) of E4, or interfere with bridging interactions across the exon (Figure 8B). This mode of inhibition has been proposed for negative factors such as hnRNP A1 and PTB (reviewed in Cartegni et al, 2002; Black, 2003). Alternatively, Nova binding could competitively inhibit the binding of a positive factor, such as an SR protein, to E4 (Figure 8C). Antagonism between SR proteins such as SF2/ASF, and hnRNP proteins such as hnRNP A1 has been shown to influence splicing of a number of transcripts including HIV-1 tat (Mayeda and Krainer, 1992; Zhu et al, 2001). Evidence for this model comes from our observation that when the E4 NESS sequence was replaced by GABAARγ2 NISE sequence (construct p-eNISE; Figure 7A), the inclusion of the resulting exon was reduced.
An alternative possibility is that Nova binding to E4 could lead to recruitment of constitutive factors, as has been suggested for regulation of GlyRα2 and GABAARγ2 alternative splicing (Dredge and Darnell, 2003), but to the wrong position relative to the RNA and thus in a manner inhibitory for exon inclusion. Nova's orientation to a bound RNA is highly directional, as evidenced by the cocrystal structure of Nova KH3 with a target RNA (Lewis et al, 2000). If Nova interactions with other protein factors are also directional, Nova could dictate the position of these factors relative to the RNA (Figure 8D).
Conclusions
Nova-1 is an important regulator of alternative splicing in the mammalian brain. In addition to previously defined targets, Nova-1 regulates alternative splicing of its own transcript in mouse spinal cord. Nova acts through an exonic splicing enhancer to inhibit inclusion of a 72 nt exon, which encodes a phosphorylation domain. This is the first demonstration that Nova can act directly through an exonic element to mediate exon skipping. Moreover, we have established that the action of Nova to repress or enhance alternative exon inclusion is dependent on the position of the Nova-binding site within the pre-mRNA. Elucidation of the mechanism of Nova-dependent splicing regulation through identification of splicing targets is yielding insight into the general mechanisms of alternative splicing regulation and the exquisite control and complexity of gene expression in the brain.
Materials and methods
Metabolic phosphorylation assay
N2A cells were grown in 10 cm dishes in DMEM supplemented with 10% fetal bovine serum (FBS) and transfected with 5 μg of vector DNA encoding T7-tagged Nova-1 using 15 μl of Fugene (Roche) according to the manufacturer's instructions. When cells reached 70% confluence, usually after 40 h incubation, the medium was substituted with phosphate-free DMEM supplemented with dialyzed FBS. After 1 h incubation in phosphate-free medium, 32P-orthophosphate to a final 0.5 mCi/ml was added. After 5 h incubation at 37°C, the cells were harvested and lysed in 1 ml lysis buffer (1% NP-40, 0.5% DOC, 0.1% SDS, 0.15 M NaCl, 0.01 M Na phosphate pH 7.2, 2 mM EDTA, 50 mM NaF, 0.2 mM Na2OV4 and 1 × complete protease inhibitor cocktail (Roche)). Nova-1 was immunoprecipitated with 5 μg anti-T7-tag monoclonal antibody (Novagen) and treated with RNaseA and T1. Following SDS–PAGE, the products were transferred to nitrocellulose and exposed to autoradiography film. The identity of the bands observed by autoradiography was confirmed by Western blot.
RNase protection assays and RT–PCR
RNA preparation and RNase protection assays were performed as described previously (Dredge and Darnell, 2003). Antisense probes were in vitro transcribed using T7 RNA polymerase from pGEMT-Easy plasmid backbones (Promega) containing PCR products generated with primers Ri28 (5′-CGCCGGACTCGCGGAAAAG) and exH-R (5′-CTGATTGGCTCTGGAGG), and linearized with NdeI; or from plasmid pTRI-β-Actin-Mouse antisense control template (Ambion). Probes were labeled by incorporation of [α-32P]UTP, and the specific activities were adjusted by incorporation of unlabeled UTP to give protected fragments of approximately equal intensities for each message (ratios of unlabeled:labeled UTP were 3:1 and 400:1 for Nova-1 and actin probes, respectively). RT–PCR was performed as previously described (Jensen et al, 2000a) using primers G6 (5′-GGGAGGACAGACAATTGTT) and mRi4 (5′-GCCCCTGACTGCTCCATTA).
Plasmid constructs
Plasmids pNova-1L, pGABA and pGlo2Δ have been described previously (Dredge and Darnell, 2003). Plasmid pNova-1S is identical to pNova-1L, except that exon 4 has been deleted by PCR. Plasmid pE4885 was derived from pGlo2Δ with the insertion of an ∼1 kb BamHI–PstI fragment of mouse Nova-1 genomic sequence. Further chimeric minigenes and mutations were generated by PCR. All DNA segments generated by PCR were sequenced in their entirety. Plasmid DNA for transfection into cell lines was prepared by a modified cesium chloride method (Sambrook et al, 1989), and dialyzed against 1 × TE.
Cell transfection and primer extension
N2A and 293T cells (ATCC) were transfected with 3.25 or 2.25 μg of total DNA, comprised of 0.25 μg of the appropriate minigene and variable amounts of pNova-1 and empty pCI-neo vector (Promega), using Fugene6 (Roche) as described by the manufacturer. After 40 h, the cells were washed with 1 × PBS, collected by scraping and halved for RNA extraction and protein extraction. Primer extension was performed using 5 μg of total RNA purified using the RNeasy Mini kit system (Qiagen), 0.5 pmol γ-32P-ATP-labeled primer E3R (5′-CAGCACACAGACCAGCACGTTG, β-globin) or mrp2 (5′-TTGAATGGTTGCTGATCTGGGACG, GABAARγ2) and SuperScript III reverse transcriptase (Invitrogen), at 55°C. Western blots were performed using anti-γ-tubulin monoclonal antibody GTU-88 (Sigma) or HRP-conjugated anti-T7-tag monoclonal antibody (Novagen) and detected by chemiluminescence (Perkin-Elmer).
Boundary mapping and nitrocellulose filter-binding assays
Fusion protein synthesis, boundary mapping and nitrocellulose filter-binding assays were performed as described previously (Dredge and Darnell, 2003). Nova-1 RNA was in vitro transcribed using SP6 RNA polymerase from a gel-purified PCR product generated using primers SP6-exH-F (5′-TCCCGATTTAGGTGACACTATAGAATACATTG CCATCTTCCCC) and intH-R (5′-TCCAGTTGGCACAACCGC). GABA and glo RNAs were generated as described previously (Dredge and Darnell, 2003).
Acknowledgments
We thank Angus Nairn for helpful advice with phosphorylation experiments, Kirk B Jensen and members of the laboratory for useful discussions and critical reading of the manuscript. This work was supported in part by the National Institutes of Health (R01 NS34389 and NS40955). RBD is an investigator of the Howard Hughes Medical Institute.
References
- Bernardi G (1995) The human genome: organization and evolutionary history. Annu Rev Genet 29: 445–476 [DOI] [PubMed] [Google Scholar]
- Biondi RM, Nebreda AR (2003) Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem J 372: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DL (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72: 291–336 [DOI] [PubMed] [Google Scholar]
- Blanchette M, Chabot B (1999) Modulation of exon skipping by high-affinity hnRNP A1-binding sites and by intron elements that repress splice site utilization. EMBO J 18: 1939–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckanovich RJ, Darnell RB (1997) The neuronal RNA binding protein Nova-1 recognizes specific RNA targets in vitro and in vivo. Mol Cell Biol 17: 3194–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckanovich RJ, Posner JB, Darnell RB (1993) Nova, the paraneoplastic Ri antigen, is homologous to an RNA-binding protein and is specifically expressed in the developing motor system. Neuron 11: 657–672 [DOI] [PubMed] [Google Scholar]
- Buckanovich RJ, Yang YY, Darnell RB (1996) The onconeural antigen Nova-1 is a neuron-specific RNA-binding protein, the activity of which is inhibited by paraneoplastic antibodies. J Neurosci 16: 1114–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartegni L, Chew SL, Krainer AR (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 3: 285–298 [DOI] [PubMed] [Google Scholar]
- Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR (2003) ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res 31: 3568–3571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darnell RB, Posner JB (2003) Paraneoplastic syndromes involving the nervous system. N Engl J Med 349: 1543–1554 [DOI] [PubMed] [Google Scholar]
- Dredge BK, Darnell RB (2003) Nova regulates GABA(A) receptor gamma2 alternative splicing via a distal downstream UCAU-rich intronic splicing enhancer. Mol Cell Biol 23: 4687–4700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother WG, Yeh RF, Sharp PA, Burge CB (2002) Predictive identification of exonic splicing enhancers in human genes. Science 297: 1007–1013 [DOI] [PubMed] [Google Scholar]
- Fiol CJ, Mahrenholz AM, Wang Y, Roeske RW, Roach PJ (1987) Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J Biol Chem 262: 14042–14048 [PubMed] [Google Scholar]
- Jensen KB, Dredge BK, Stefani G, Zhong R, Buckanovich RJ, Okano HJ, Yang YY, Darnell RB (2000a) Nova-1 regulates neuron-specific alternative splicing and is essential for neuronal viability. Neuron 25: 359–371 [DOI] [PubMed] [Google Scholar]
- Jensen KB, Musunuru K, Lewis HA, Burley SK, Darnell RB (2000b) The tetranucleotide UCAY directs the specific recognition of RNA by the Nova K-homology 3 domain. Proc Natl Acad Sci USA 97: 5740–5745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD (2003) Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 302: 2141–2144 [DOI] [PubMed] [Google Scholar]
- Kanopka A, Muhlemann O, Akusjarvi G (1996) Inhibition by SR proteins of splicing of a regulated adenovirus pre-mRNA. Nature 381: 535–538 [DOI] [PubMed] [Google Scholar]
- Kanopka A, Muhlemann O, Petersen-Mahrt S, Estmer C, Ohrmalm C, Akusjarvi G (1998) Regulation of adenovirus alternative RNA splicing by dephosphorylation of SR proteins. Nature 393: 185–187 [DOI] [PubMed] [Google Scholar]
- Kemp BE, Pearson RB (1990) Protein kinase recognition sequence motifs. Trends Biochem Sci 15: 342–346 [DOI] [PubMed] [Google Scholar]
- Lewis HA, Musunuru K, Jensen KB, Edo C, Chen H, Darnell RB, Burley SK (2000) Sequence-specific RNA binding by a Nova KH domain: implications for paraneoplastic disease and the fragile X syndrome. Cell 100: 323–332 [DOI] [PubMed] [Google Scholar]
- Libri D, Piseri A, Fiszman MY (1991) Tissue-specific splicing in vivo of the beta-tropomyosin gene: dependence on an RNA secondary structure. Science 252: 1842–1845 [DOI] [PubMed] [Google Scholar]
- Malim MH, Cullen BR (1991) HIV-1 structural gene expression requires the binding of multiple Rev monomers to the viral RRE: implications for HIV-1 latency. Cell 65: 241–248 [DOI] [PubMed] [Google Scholar]
- Mayeda A, Krainer AR (1992) Regulation of alternative pre-mRNA splicing by hnRNP A1 and splicing factor SF2. Cell 68: 365–375 [DOI] [PubMed] [Google Scholar]
- Prasad J, Colwill K, Pawson T, Manley JL (1999) The protein kinase Clk/Sty directly modulates SR protein activity: both hyper- and hypophosphorylation inhibit splicing. Mol Cell Biol 19: 6991–7000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechsteiner M, Rogers SW (1996) PEST sequences and regulation by proteolysis. Trends Biochem Sci 21: 267–271 [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Ule J, Jensen KB, Ruggiu M, Mele A, Ule A, Darnell RB (2003) CLIP identifies Nova-regulated RNA networks in the brain. Science 302: 1212–1215 [DOI] [PubMed] [Google Scholar]
- Xu Q, Modrek B, Lee C (2002) Genome-wide detection of tissue-specific alternative splicing in the human transcriptome. Nucleic Acids Res 30: 3754–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YY, Yin GL, Darnell RB (1998) The neuronal RNA-binding protein Nova-2 is implicated as the autoantigen targeted in POMA patients with dementia. Proc Natl Acad Sci USA 95: 13254–13259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Mayeda A, Krainer AR (2001) Exon identity established through differential antagonism between exonic splicing silencer-bound hnRNP A1 and enhancer-bound SR proteins. Mol Cell 8: 1351–1361 [DOI] [PubMed] [Google Scholar]