

Abstract
The inactivation of glycogen synthase kinase (GSK)3 has been proposed to play important roles in insulin and Wnt signalling. To define the role that inactivation of GSK3 plays, we generated homozygous knockin mice in which the protein kinase B phosphorylation sites on GSK3α (Ser21) and GSK3β (Ser9) were changed to Ala. The knockin mice were viable and were not diabetic. Using these mice we show that inactivation of GSK3β rather than GSK3α is the major route by which insulin activates muscle glycogen synthase. In contrast, we demonstrate that the activation of muscle glycogen synthase by contraction, the stimulation of muscle glucose uptake by insulin, or the activation of hepatic glycogen synthase by glucose do not require GSK3 phosphorylation on Ser21/Ser9. GSK3 also becomes inhibited in the Wnt-signalling pathway, by a poorly defined mechanism. In GSK3α/GSK3β homozygous knockin cells, Wnt3a induces normal inactivation of GSK3, as judged by the stabilisation of β-catenin and stimulation of Wnt-dependent transcription. These results establish the function of Ser21/Ser9 phosphorylation in several processes in which GSK3 inactivation has previously been implicated.
Keywords: exercise, glucose transport, GSK3 inhibitor AR-A014418, PI 3-kinase, PKB/Akt
Introduction
Insulin promotes the conversion of glucose to glycogen in skeletal muscle by stimulating glucose uptake and activating glycogen synthase (Roach, 2002; Ferrer et al, 2003). Insulin activates glycogen synthase by inducing its dephosphorylation at a cluster of C-terminal residues (Ser641, Ser645, Ser649 and Ser653), which are phosphorylated by glycogen synthase kinase-3α (GSK3α) and GSK3β (Cohen, 1999). In muscle, insulin is thought to stimulate the dephosphorylation of glycogen synthase at these residues by inducing the inactivation of GSK3α and GSK3β via phosphorylation of an N-terminal Ser residue (Ser21 in GSK3α and Ser9 in GSK3β), which is catalysed by protein kinase B (PKB, also known as Akt) (Cross et al, 1995; Shaw et al, 1997) and reversed by the muscle glycogen-associated protein phosphatase-1. Consistent with this model of regulation, small cell-permeable inhibitors of GSK3 with diverse structures stimulate glycogen synthase activity in cell lines or skeletal muscle (reviewed in Cohen and Goedert, 2004). However, it has also been reported that insulin stimulates the dephosphorylation of glycogen synthase at Ser7, which is not regulated by GSK3, and, furthermore, it has been suggested that GSK3 is not the only kinase which phosphorylates the residues between Ser641 and Ser653 (reviewed in Roach, 2002; Ferrer et al, 2003). Thus, the possibility that insulin may regulate glycogen synthase by a mechanism that is independent of GSK3 has not been excluded.
It has also been proposed that insulin increases the activity of a glycogen-associated form of protein phosphatase-1 that dephosphorylates the residues in glycogen synthase that are phosphorylated by GSK3 (Suzuki et al, 2001; Delibegovic et al, 2003), but the mechanism or importance of this observation is unclear. The relative contributions made by the inactivation of GSK3 versus potential activation of protein phosphatase or regulation via other pathways to the activation of glycogen synthase is unknown.
In the liver, hepatic glycogen synthase is also regulated by phosphorylation of the sites targeted by GSK3. In support of this, GSK3 inhibitors stimulate hepatic glycogen synthase (Coghlan et al, 2000; Cline et al, 2002). However, an important difference between muscle and liver glycogen synthase is that the latter is regulated by the level of blood glucose (Bollen et al, 1998). Glucose has been proposed to stimulate glycogen synthase by binding to phosphorylase-a, thereby promoting its conversion to phosphorylase-b, and so relieve the inhibition that phosphorylase-a exerts on the hepatic glycogen-associated form of protein phosphatase-1, termed PP1GL (Alemany and Cohen, 1986; Bollen et al, 1998). This allows protein phosphatase-1 to dephosphorylate and hence activate hepatic glycogen synthase, thereby stimulating glycogen synthesis. The relative importance of the insulin-induced inactivation of GSK3 versus the glucose-stimulated phosphatase activity in the regulation of hepatic glycogen synthase is unknown.
In order for GSK3 to phosphorylate many of its substrates, including glycogen synthase, the substrate is required to be phosphorylated at a priming site, lying four residues C-terminal to the Ser/Thr residue phosphorylated by GSK3 (Frame and Cohen, 2001; Roach, 2002). In the case of glycogen synthase, CK2 is believed to act as a priming kinase by phosphorylating Ser657 (Roach, 2002; Ferrer et al, 2003). Recent studies have revealed that GSK3 possesses a specific phosphate recognition motif located within its kinase domain, which interacts specifically with the primed phosphorylated residue on the GSK3 substrate (Frame and Cohen, 2001). Following phosphorylation by PKB, the N-terminal phosphorylated sequence of GSK3 interacts with the phosphate recognition motif on the GSK3 kinase domain, thereby inhibiting GSK3 by competing for substrate binding. Mutation to Ala of the sites on GSK3α and GSK3β phosphorylated by PKB prevents the inhibition of GSK3 by insulin.
The inactivation of GSK3 also plays an important role in the Wnt signalling pathway which is critical for embryonic development (Logan and Nusse, 2004). Wnt inhibits GSK3 by a poorly defined mechanism which is thought to involve disruption of a complex containing GSK3, axin, the adenomatous polyposis coli (APC) protein, and β-catenin. This leads to dephosphorylation and stabilisation of β-catenin, stimulating transcriptional processes controlled by β-catenin. In many forms of human colorectal cancer, mutations arise, mainly in APC and β-catenin, that induce the stabilisation of β-catenin, causing uncontrolled activation of the pathway, which leads to loss of cell polarity and increased proliferation (Sancho et al, 2004). Several groups have proposed that Wnt induces phosphorylation of GSK3 isoforms at the same sites phosphorylated by PKB (Yuan et al, 1999; Fukumoto et al, 2001); however, it has also been suggested that Wnt inactivates GSK3 by a distinct mechanism (Ding et al, 2000).
Results
Generation of GSK3 knockin mice
We generated knockin mice in which the codon encoding Ser21 of GSK3α, and Ser9 of GSK3β, was changed to encode a nonphosphorylatable Ala residue, using the methodology described under Materials and methods and in Supplementary Figure 1. Single homozygous GSK3α21A/21A and GSK3β9A/9A, as well as double GSK3α/β21A/21A/9A/9A knockin mice, were bred using the strategy depicted in Table I. Throughout this study, wild-type (WT) littermate animals were employed in control experiments performed with the single GSK3α21A/21A and GSK3β9A/9A knockin mice. In the case of the double GSK3α/β21A/21A/9A/9A knockin mice, this was impractical, as WT littermate and double knockin mice can only be obtained at a ratio of 1 in 16 in the same cross (Table I). In order to breed GSK3α/β21A/21A/9A/9A and appropriate control mice, we interbred GSK3α/β21A/21A/9A/9A and GSK3α/β21A/21A/9A/9A as well as GSK3α/β+/+/+/+ and GSK3α/β+/+/+/+ littermates derived from a 1 in 16 ratio cross.
Table 1.
Mice matings reported in this study
| Cross | Genotype | Number (%) | |
|---|---|---|---|
| GSK3α(+/S21A)GSK3β(+/+) | GSK3α(+/+) | GSK3β(+/+) | 50 (25.5%) |
| GSK3α(+/S21A)GSK3β(+/+) | GSK3α(+/S21A) | GSK3β(+/+) | 96 (49%) |
| GSK3α(S21A/S21A) | GSK3β(+/+) | 50 (25.5%) | |
| GSK3α(S21A/S21A)GSK3β(+/+) | GSK3α(S21A/S21A) | GSK3β(+/+) | 11 (100%) |
| GSK3α(S21A/S21A)GSK3β(+/+) | |||
| GSK3α(+/+)GSK3β(+/S9A) | GSK3α(+/+) | GSK3β(+/+) | 48 (23%) |
| GSK3α(+/+)GSK3β(+/S9A) | GSK3α(+/+) | GSK3β(+/S9A) | 104 (50%) |
| GSK3α(+/+) | GSK3β(S9A/S9A) | 56 (27%) | |
| GSK3α(+/+)GSK3β(S9A/S9A) | GSK3α(+/+) | GSK3β(S9A/S9A) | 12 (100%) |
| GSK3α(+/+)GSK3β(S9A/S9A) | |||
| GSK3α(+/S21A)GSK3β(+/S9A) | GSK3α(+/+) | GSK3β(+/+) | 3 (7.5%) |
| GSK3α(+/S21A)GSK3β(+/S9A) | GSK3α(+/+) | GSK3β(+/S9A) | 3 (7.5%) |
| GSK3α(+/+) | GSK3β(S9A/S9A) | 2 (5%) | |
| GSK3α(+/S21A) | GSK3β(+/+) | 5 (12.5%) | |
| GSK3α(+/S21A) | GSK3β(+/S9A) | 19 (47.5%) | |
| GSK3α(+/S21A) | GSK3β(S9A/S9A) | 2 (5%) | |
| GSK3α(S21A/S21A) | GSK3β(+/+) | 1 (2.5%) | |
| GSK3α(S21A/S21A) | GSK3β(+/ S9A) | 3 (7.5%) | |
| GSK3α(S21A/S21A) | GSK3β(S9A/S9A) | 2 (5%) | |
| GSK3α(S21A/S21A)GSK3β(S9A/S9A) | GSK3α(S21A/S21A) | GSK3β(S9A/S9A) | 150 (100%) |
| GSK3α(S21A/S21A)GSK3β(S9A/S9A) | |||
| The indicated matings were set up and the progeny genotyped as described in Materials and methods. The percentage of each genotype observed is indicated in parenthesis. | |||
GSK3 knockin mice develop and grow normally and are not diabetic
The single GSK3α21A/21A, GSK3β9A/9A and double GSK3α/β21A/21A/9A/9A knockin mice were born at the expected Mendelian frequency (Table I) and displayed no overt phenotype. Growth curves from 4 to 16 weeks of age indicated that these animals were of normal size and weight (Figure 1A and Supplementary Figure 2). Glucose tolerance tests indicated that the single (Supplementary Figure 2) as well as double knockin mice (Figure 1B) were able to dispose of injected glucose at the same rate as WT controls, at 20 weeks of age. Double GSK3α/β21A/21A/9A/9A knockin mice also possessed normal fasted and fed glucose and insulin levels (Figure 1C).
Figure 1.
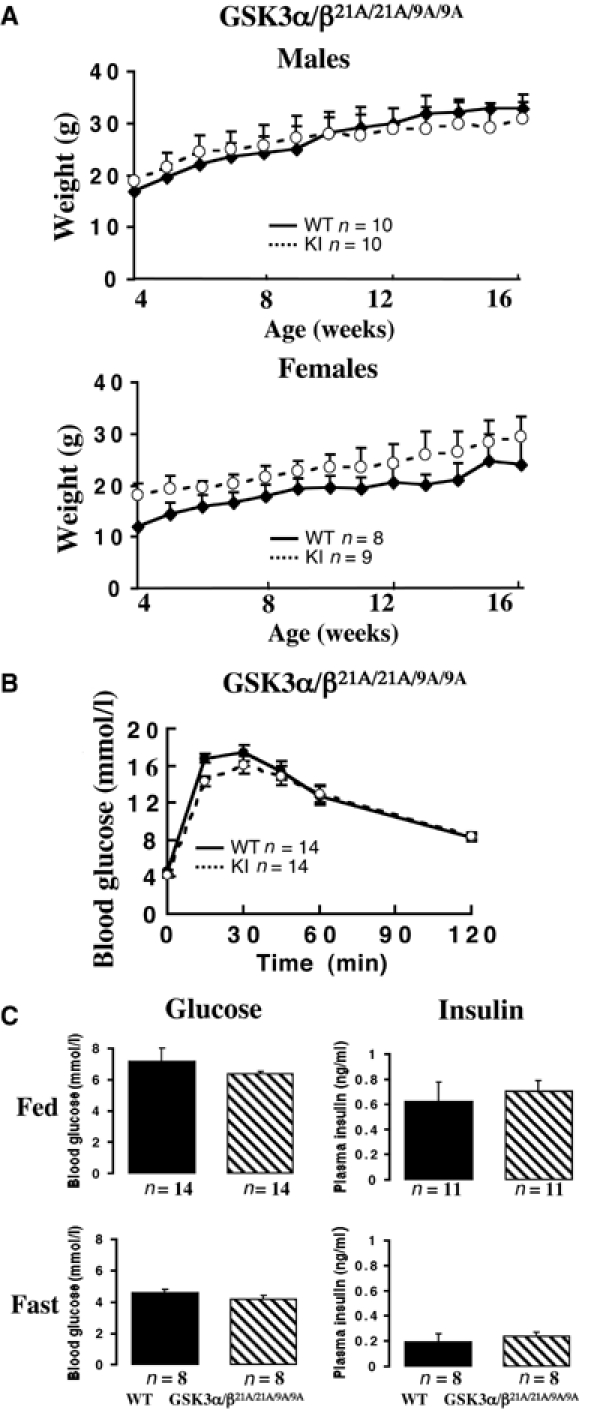
Growth and glucose metabolism in the double knockin mice. (A) The indicated male and female mice were weighed once a week between the ages of 4 and 16 weeks. Each point represents the mean±s.e.m., in which n is the number of mice analysed in each group. No statistical difference in the mean weights was observed for any group using the Student's t-test. (B) Glucose tolerance test of the indicated 20-week-old mice. Mice were injected intraperitoneally with 2 mg/g glucose solution and the blood glucose concentration was determined at the indicated times. n indicates the number of mice in each group. The data are presented as the mean±s.e.m. (C) The indicated mice were fasted overnight for 16 h (Fast) or fed ab libitum (Fed). The blood glucose and plasma insulin levels were measured. The data are presented as the mean±s.e.m., with the number, n, of animals in each group indicated. Approximate equal numbers of male and female mice were used in (B) and (C).
Analysis of PKB and GSK3 in the muscle of knockin mice
Mice were fasted overnight and injected with insulin. In order to verify whether the PI 3-kinase signalling pathway was normally active in the knockin mice, PKBα activity was measured. In the skeletal muscle of GSK3α21A/21A, GSK3β9A/9A and double GSK3α/β21A/21A/9A/9A knockin mice, insulin induced a robust activation of PKB, as well as phosphorylation at one of its activating residues (Ser473), similar to that seen in WT control mice (Figure 2A). The levels of GSK3α and GSK3β protein in the skeletal muscle of GSK3α21A/21A, GSK3β9A/9A and double GSK3α/β21A/21A/9A/9A knockin mice were normal (Figure 2B). We also found that the level of tyrosine phosphorylation in the ‘T-loop' of GSK3 isoforms was similar in WT and knockin mice and not affected by insulin treatment (Figure 2B). Consistent with this, the specific activities of GSK3α derived from muscle extracts of WT and GSK3α21A/21A knockin mice not injected with insulin were similar (Figure 2B). The GSK3β activity in muscle extracts from GSK3β9A/9A knockin mice that had not been injected with insulin was slightly higher than that of WT mice (Figure 2B). A low level of phosphorylation of GSK3β at Ser9, in WT non-insulin-stimulated muscle, could explain this. Insulin was found to induce a marked phosphorylation of both GSK3 isoforms in WT control mice (Figure 2B). In the GSK3α21A/21A knockin mice, no phosphorylation of GSK3α at Ser21 was observed, while phosphorylation of GSK3β at Ser9 was normal. Similarly, in the GSK3β9A/9A knockin mice, no phosphorylation of GSK3β at Ser9 was detected, while phosphorylation of GSK3α at Ser21 occurred normally. In the double knockin GSK3α/β21A/21A/9A/9A mice, phosphorylation of neither isoform of GSK3 was observed. In muscles isolated from WT mice, insulin induced ∼40% inhibition of GSK3α and GSK3β activity, which was maintained for 40 min (Figure 2B). Dephosphorylation of immunoprecipitated GSK3 with protein phosphatase-1γ (PP1γ) largely restored the specific activities of GSK3α and GSK3β to those observed in unstimulated muscle after phosphatase treatment, indicating that inhibition of GSK3 results from Ser/Thr phosphorylation (data not shown). GSK3β in the GSK3α21A/21A muscle and GSK3α in the GSK3β9A/9A muscle were normally inactivated by insulin (Figure 2B). In contrast, insulin did not induce any inactivation of the S21A or S9A mutants of GSK3 in the knockin mice (Figure 2B), nor was the activity of the immunoprecipitated GSK3 increased by treatment with PP1γ (data not shown).
Figure 2.
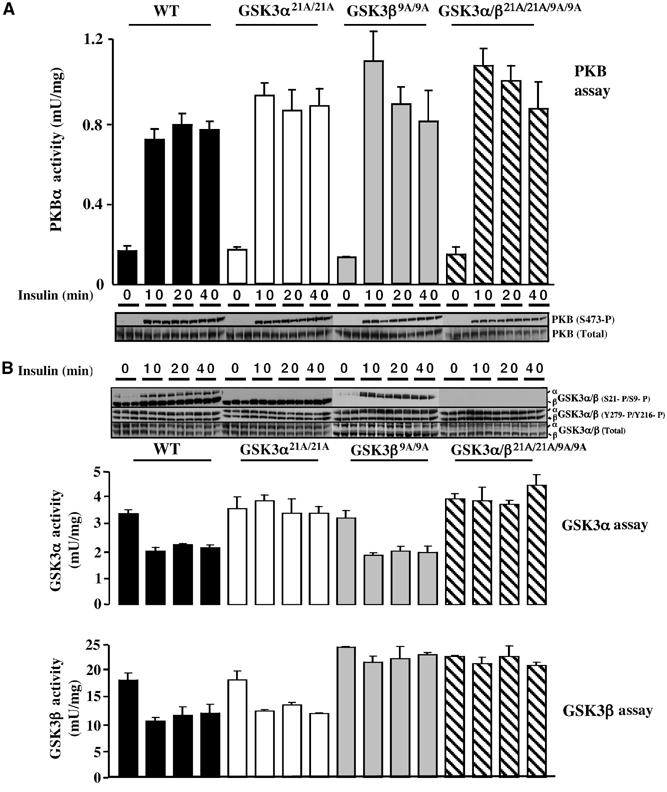
PKB and GSK3 activity in GSK3 knockin mice. (A) The indicated mice were fasted overnight and injected intraperitoneally with insulin (150 mU/g) or saline solution (for the 0 min time point), to anaesthetised mice. At the indicated time (10 min for saline control), skeletal muscle was rapidly extracted and snap frozen in liquid nitrogen. PKBα activity was measured following its immunoprecipitation as described in Materials and methods. The results shown at each time point represent the mean±s.e.m. for three mice each assayed in duplicate for PKBα. The total levels and phosphorylation of PKBα at Ser473 were monitored by immunoblot analysis of tissue extracts. For each time point muscle samples from three separate mice are shown. (B) As above, except that GSK3α and GSK3β were immunoprecipitated from the indicated insulin-stimulated tissues and their activity was measured. The data are presented as the mean±s.e.m. for muscle isolated from three mice each assayed in triplicate. Muscle extracts from the indicated mice were immunoblotted with the indicated antibodies and samples from three separate mice are shown for each time point. The analysis with the three sets of knockin mice was each compared to WT littermate control mice in (A) and (B). Since these data from the sets of WT controls did not differ significantly from each other, only the results obtained for the WT GSK3α21A/21A littermate controls are shown.
Effect of insulin on glycogen synthase in GSK3 knockin muscle
We next assessed the level and activity of glycogen synthase in skeletal muscle extracts. Glycogen synthase protein levels were normal in the muscles of knockin mice (Figure 3A). We measured glycogen synthase activity in muscle extracts by assaying the incorporation of radioactive UDP-glucose into glycogen in the presence or absence of the allosteric activator glucose 6-phosphate (G6P) (Thomas et al, 1968). In WT mice, insulin activated glycogen synthase ∼2-fold, which was sustained for up to 40 min (Figure 3A). This is the normal degree of activation of muscle glycogen synthase that is induced by insulin. Activation was accompanied by dephosphorylation of Ser641 and Ser645, two of the residues phosphorylated by GSK3 that play critical roles in regulating its activity (Roach, 2002; Ferrer et al, 2003). In the single GSK3α21A/21A knockin mice, the basal level of glycogen synthase activity was normal, and insulin still induced an ∼2-fold activation after 10 min that was also accompanied by the dephosphorylation at the sites phosphorylated by GSK3. The activation of glycogen synthase in the GSK3α21A/21A knockin mice was less sustained than in control WT animals (Figure 3A). Although this effect was moderate, it was observed in two independent experiments with three mice being analysed for each time point. In the single GSK3β9A/9A, as well as the double GSK3α/β21A/21A/9A/9A knockin mice, the basal activity and phosphorylation of glycogen synthase at Ser641/Ser645 was comparable to control mice, but, strikingly, insulin failed to induce a significant activation or dephosphorylation of Ser641/Ser645 (Figure 3A).
Figure 3.
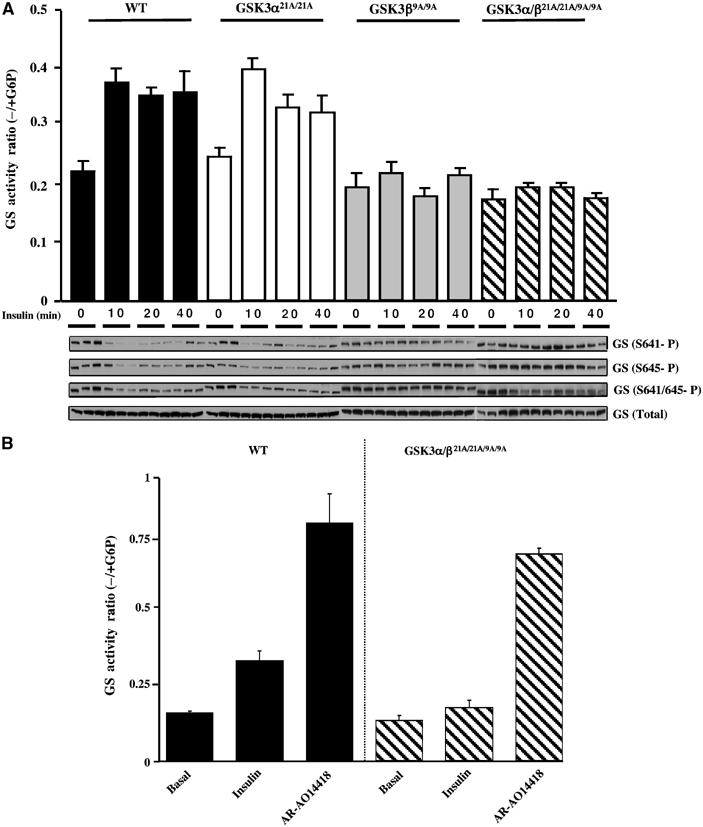
Effect of insulin on glycogen synthase in knockin mice. (A) The indicated mice were fasted overnight and injected intraperitoneally with insulin (150 mU/g) or saline solution (for the 0 min time point), to anaesthetised mice. At the indicated time (10 min for saline control), skeletal muscle was rapidly extracted and snap frozen in liquid nitrogen. Glycogen synthase activity was measured in the absence and presence of G6P. The data are presented as the mean±s.e.m. for muscle isolated from three mice each assayed ±G6P in duplicate. Muscle extracts from the indicated mice were immunoblotted with the indicated antibodies recognising phosphorylated and total glycogen synthase. As for Figure 2, only the WT controls for the GSK3α21A/21A are shown. (B) Soleus muscle was isolated and incubated in the presence or absence of insulin or AR-A014418. After 1 h of incubation, the muscle was rapidly frozen in liquid nitrogen. Glycogen synthase activity was assessed as in (A) above. The results are presented as the mean±s.e.m. for four muscles for each condition.
We next investigated whether activation of glycogen synthase in the knockin mice could be induced by treatment of muscle with AR-A014418, a specific ATP competitive inhibitor of GSK3α and GSK3β (Bhat et al, 2003; Murray et al, 2004). We treated isolated soleus muscle from WT or the double GSK3α/β21A/21A/9A/9A knockin mice with either insulin or AR-A014418. Insulin failed to induce a significant activation of glycogen synthase in the GSK3α/β21A/21A/9A/9A knockin muscle under conditions that resulted in two-fold activation in WT muscle (Figure 3B). In contrast, AR-A014418 induced nearly five-fold activation of glycogen synthase in both the WT and double GSK3α/β21A/21A/9A/9A knockin muscle (Figure 3B).
Relative expression of GSK3α and GSK3β in murine and human muscle
The finding that insulin was unable to stimulate glycogen synthase in the single GSK3β9A/9A knockin mice suggested that GSK3β is the principal isoform phosphorylating glycogen synthase in murine muscle. This observation could be explained if GSK3β was expressed at a higher level than GSK3α in skeletal muscle. To investigate whether this was so, we expressed mouse GSK3α and GSK3β with N-terminal glutathione S-transferase (GST) tags as expression standards for immunoblotting. We carried out a quantitative immunoblot analysis using LI-COR technology, in which equal amounts of recombinant GSK3α and GSK3β were analysed by immunoblotting with an anti-GST antibody and an anti-GSK3 antibody that recognises both GSK3 isoforms. This analysis permitted quantification of the relative abundance of GSK3α and GSK3β isoforms in murine skeletal muscle, and revealed ∼4-fold higher amounts of GSK3β than GSK3α (Figure 4A). Consistent with this, the specific activity of GSK3β is ∼5-fold higher than that of GSK3α in skeletal muscle (Figure 2B). As GSK3 inhibitors are being developed for the treatment of diabetes in humans, we performed a similar analysis employing human skeletal muscle derived from three lean and healthy donors. This revealed that, in human muscle of all three subjects, there was ∼3-fold excess of GSK3β over GSK3α (Figure 4B).
Figure 4.
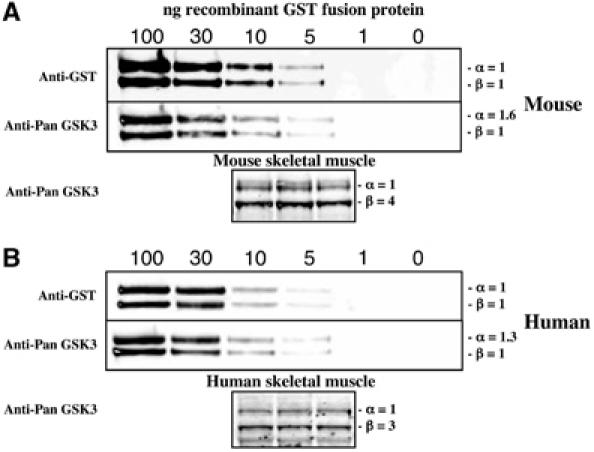
Quantification of the relative levels of GSK3α and GSK3β in muscle. (A) The indicated amounts of the recombinant mouse GST-GSK3α and GST-GSK3β were subjected to immunoblot analysis with anti-GST antibody to verify equal loading of the isoforms or a Pan-GSK3 isoform antibody to determine the relative affinity of this antibody for GSK3α and GSK3β. Quantitation of the immunoblots was performed using LI-COR Odyssey infrared imaging system. Mouse skeletal muscle derived from three WT mice (40 μg total protein) was subjected to quantitative immunoblot analysis with the Pan-GSK3 isoform antibody and from the quantitation data the abundance of GSK3α relative to GSK3β is indicated. (B) As in (A), except that human GST-GSK3α and GST-GSK3β were employed as expression standards and skeletal muscle (30 μg total protein) from three healthy lean human subjects was analysed by immunoblot analysis.
Analysis of glucose uptake and glycogen levels in GSK3 knockin muscle
A major effect of insulin in muscle is to stimulate glucose uptake probably through the activation of PKB. Although the mechanism by which PKB stimulates glucose uptake is unknown, some studies have implicated the PKB-mediated inhibition of GSK3 in this process (Orena et al, 2000; Morfini et al, 2002). We therefore measured glucose uptake in isolated soleus muscle. These studies revealed that, in both the single as well as the double GSK3 knockin mice, insulin stimulated the uptake of glucose four-fold, similar to the effect observed in WT mice (Figure 5A). We next measured the total glycogen levels in the muscle of WT as well as the single and double GSK3 knockin mice. We observed that, in both mice fed ab libitum and fasted for 16 h, that there was no significant reduction in muscle glycogen levels in any of the GSK3 knockin mice compared to WT controls (Figure 5B).
Figure 5.
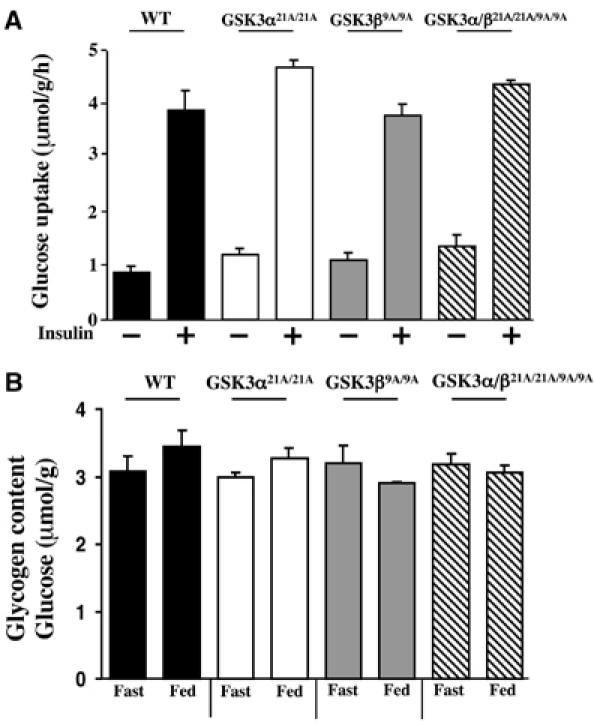
Analysis of glycogen levels and glucose uptake in muscle of the knockin mice. (A) Soleus muscle strips were isolated from the indicated mice and glucose transport activities were measured in the presence or absence of 100 nM insulin. The data are presented as the mean±s.e.m. for muscle isolated from five mice for each genotype. As for Figure 2, only the data for WT control for the GSK3α21A/21A are shown. (B) The indicated mice were fasted overnight for 16 h (Fast) or fed ab libitum (Fed). The mice were killed and the quadricep muscle was rapidly extracted and frozen in liquid nitrogen. The glycogen content in skeletal muscle is expressed as μmol of glycosyl units per gram of muscle. The data are presented as the mean±s.e.m. for muscle isolated from three mice in each group each assayed in triplicate.
Effect of muscle contraction on glycogen synthase and glucose tolerance in GSK3 knockin muscle
Muscle contraction potently activates glycogen synthase by a poorly defined mechanism (reviewed in Nielsen and Richter, 2003). To define whether phosphorylation of GSK3α at Ser21 and GSK3β at Ser9 plays a role in this process, contraction of hind limb muscle was induced in anaesthetised mice in situ by electrical stimulation of the sciatic nerve in one leg, the other leg serving as the noncontracted control. In both the single as well as double GSK3 knockin mice, muscle contraction markedly stimulated the activity of glycogen synthase to the same extent found in the WT control mice (Figure 6A). Consistent with previous findings (Sakamoto et al, 2002, 2003), muscle contraction in WT mice induced a slight phosphorylation of GSK3α at Ser21 and GSK3β at Ser9 and also resulted in a marked dephosphorylation of glycogen synthase, especially at Ser641 and to a lesser extent at Ser645 (Figure 6B). Strikingly however, in both the single and double GSK3 knockin mice, although phosphorylation of the GSK3 knockin isoforms was abolished, exercise still led to dephosphorylation of glycogen synthase at Ser641 and Ser645 (Figure 6B). This does not result from inactivation of GSK3 isoforms by a mechanism distinct from the phosphorylation of Ser21 and Ser9, as the activity of GSK3α or GSK3β isoforms in the double GSK3α/β21A/21A/9A/9A knockin mice was not changed by muscle contraction (Figure 6C). In WT mice, contraction induced a modest inhibition of GSK3α activity consistent with its phosphorylation at Ser21. Contraction induced weak phosphorylation of PKB at Ser473 in WT and knockin mice, that was much weaker than that observed with insulin (Figure 6D). Muscle contraction also induced normal phosphorylation of the ERK1/ERK2 MAP kinases in the single and double GSK3 knockin mice. This could also contribute to the modest phosphorylation of GSK3 through the ERK-stimulated RSK.
Figure 6.
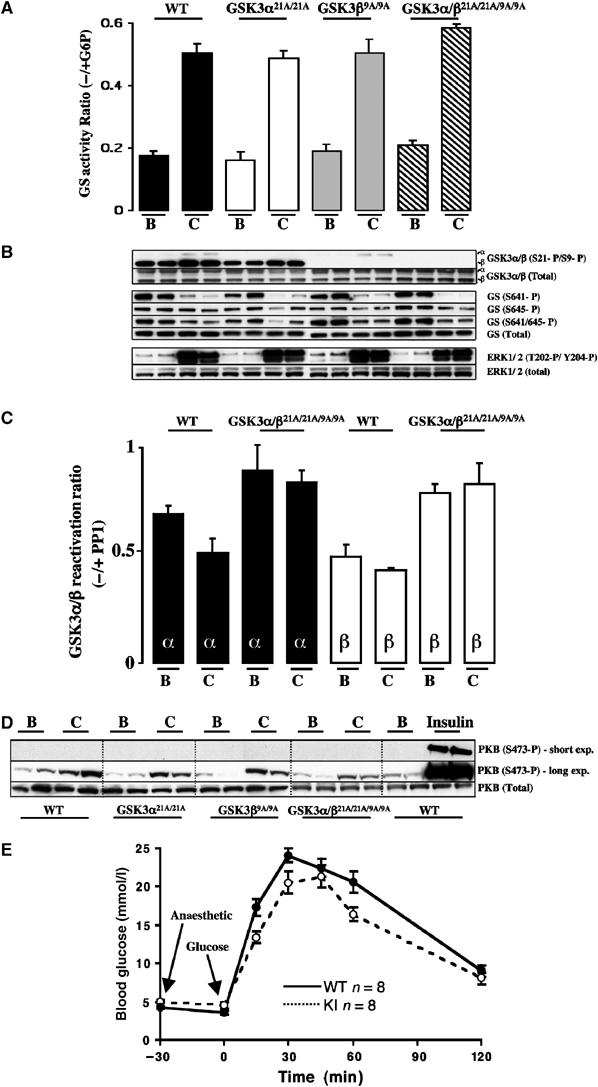
Effect of muscle contraction on glycogen synthase and glucose tolerance in the knockin mice. (A) One leg from anaesthetised WT and knockin mouse was subjected to in situ hindlimb muscle contraction (C, contraction) via sciatic nerve stimulation for 10 min, and the other leg served as noncontracted control (B, basal). Red and white gastrocnemius and extensor digitorum longus (EDL) muscles from both legs were rapidly extracted and snap frozen in liquid nitrogen. Glycogen synthase activity was measured in the absence and presence of G6P. The data are presented as the mean±s.e.m. for muscle isolated from four mice each assayed ±G6P in duplicate. (B) Muscle extracts from the indicated mice were immunoblotted with the indicated antibodies. As for Figure 2, only the WT controls for the GSK3α21A/21A are shown. (C) GSK3α and GSK3β were immunoprecipitated from WT and double knockin GSK3 muscle derived from (A), and activity was measured as a ratio of ±treatment with PP1γ phosphatase as described in Materials and methods. The data are presented as the mean±s.e.m. for muscle isolated from two mice, each assayed in triplicate. (D) Muscle extracts from the WT and knockin mice were immunoblotted with the indicated antibodies. The insulin-treated samples were derived from the muscle of mice that had been injected with insulin as described in Figure 2. In order to compare the levels of phosphorylation of PKB seen in response to insulin and muscle contraction, a short and long exposure of the immunoblot is shown. (E) The indicated mice were fasted overnight and blood glucose levels measured. The mice were then anaesthetised and after 30 min a glucose tolerance test was carried out after mice were injected intraperitoneally with 2 mg/g glucose solution and the blood glucose concentration was determined at the indicated times. n indicates the number of mice in each group. The data are presented as the mean±s.e.m. and similar numbers of female and male mice were present in each group.
To investigate whether physical activity contributed the nondiabetic phenotype observed in the GSK3 knockin mice, as previously observed for mice lacking the muscle insulin receptor (Wojtaszewski et al, 1999), we performed a glucose tolerance test on anaesthetised control and GSK3α/β21A/21A/9A/9A animals. This revealed no significant differences between the WT and knockin mice (Figure 6E).
Effect of glucose on hepatic glycogen synthase in GSK3 knockin mice
We next investigated the role of GSK3 inactivation in regulating hepatic glycogen synthase activity. In contrast to muscle, injection of insulin into fasted mice failed to stimulate hepatic glycogen synthase activity (data not shown). This may be due to the low levels of blood glucose in these insulin-treated mice, as glucose is a key regulator of hepatic glycogen synthase (Bollen et al, 1998). We therefore injected fasted WT and double GSK3α/β21A/21A/9A/9A knockin mice with glucose, which resulted in a two-fold stimulation of hepatic glycogen synthase within 10 min that was sustained for 40 min in both the WT and knockin mice (Figure 7A). Glucose induced a similar marked dephosphorylation of hepatic glycogen synthase at Ser641 and Ser645 in livers of the control and double knockin mice (Figure 7A). Under these conditions, glucose did not significantly increase the basal phosphorylation of PKB at Ser473 or the phosphorylation of GSK3 isoforms at Ser21/Ser9. We also measured hepatic glycogen levels in fed and fasted mice (Figure 7B). In fed WT and knockin mice, the glycogen levels were similar and higher than the levels observed in fasted animals. We consistently noticed that the glycogen levels in the fasted double knockin mice were two- to three-fold higher than those observed in WT control mice (Figure 7B).
Figure 7.
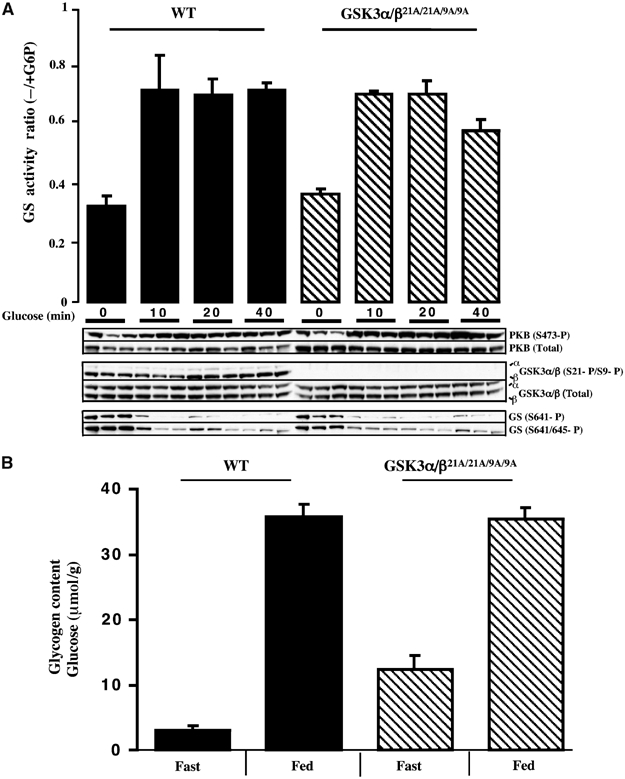
Analysis of liver glycogen synthase activity and glycogen levels in the knockin mice. (A) The indicated mice were fasted overnight and injected intraperitoneally with glucose (2 mg/g) or saline solution (for the 0 min time point), to anaesthetised mice. At the indicated time (10 min for saline control), the liver was rapidly extracted and snap frozen in liquid nitrogen. Glycogen synthase activity was measured in the absence and presence of G6P. The data are presented as the mean±s.e.m. for liver isolated from three mice each assayed ±G6P in duplicate. Liver extracts from the mice were immunoblotted with the indicated antibodies. (B) The WT and knockin mice were fasted overnight for 16 h (Fast) or fed ab libitum (Fed), and liver glycogen content quantified as μmol of glycosyl units per gram of liver. The data are presented as the mean±s.e.m. for liver isolated from six mice in each group each assayed in triplicate.
Wnt3a inactivates GSK3 in knockin cells
To investigate whether Wnt3a-mediated inactivation of GSK3 involves phosphorylation of GSK3α at Ser21 and GSK3β at Ser9, we generated embryonic stem (ES) cells from WT and double GSK3α/β21A/21A/9A/9A knockin mice. We stimulated these with Wnt3a-containing medium and monitored phosphorylation of β-catenin at the sites phosphorylated by GSK3, as well as the total levels of β-catenin (Figure 8A). We found that, in both the WT and GSK3α/β21A/21A/9A/9A knockin ES cells, Wnt3a induced a marked inhibition of phosphorylation of β-catenin on the sites targeted by GSK3 and an ∼2-fold increase in levels of β-catenin (Figure 8A). These Wnt3a-mediated effects were seen within 30 min and sustained for up to 2–4 h. As reported previously (Ding et al, 2000), Wnt3a stimulation failed to induce a detectable increase in the phosphorylation of the total cellular pools of GSK3α at Ser21 and GSK3β at Ser9 in WT ES cells, as measured by immunoblot analysis of whole-cell extracts. We also measured β-catenin-dependent transcription using a luciferase reporter assay. In both the WT and knockin cells, Wnt3a induced an ∼8-fold increase in luciferase gene expression within 8 h (Figure 8B).
Figure 8.
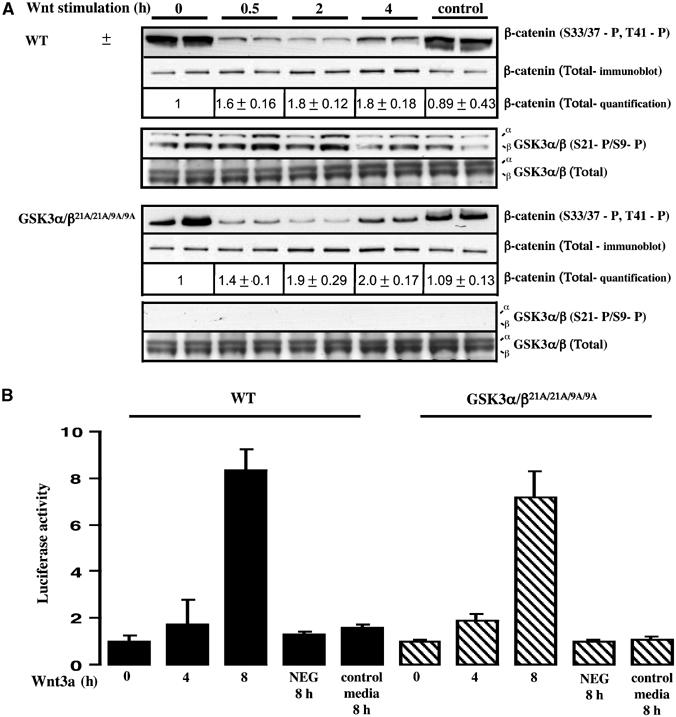
Ability of Wnt to inactivate GSK3 in knockin ES cells. (A) The WT and double knockin GSK3α/β21A/21A/9A/9A ES cells were incubated in medium containing Wnt3a or a control medium obtained from non-Wnt3a-expressing cells. At the indicated times, the phosphorylation state of β-catenin was assessed by immunoblot analysis employing an antibody that recognises β-catenin when phosphorylated on the three residues targeted by GSK3 (S33/37-P, T41-P). Quantitation of the total levels of β-catenin was performed by immunoblot analysis using LI-COR Odyssey infrared imaging system. The extracts were also immunoblotted with antibodies recognising the phosphorylated and total forms of GSK3. For each condition duplicate dishes were analysed and similar results were obtained in two separate experiments. (B) ES cells were co-transfected with a firefly luciferase reporter construct containing LEF/TCF-binding sites as well as a constitutive SV-40 Renilla luciferase construct, to control for transfection efficiency. At 24 h post-transfection, cells were incubated in medium containing Wnt3a or a control medium obtained from non-Wnt3a-expressing cells and luciferase activity was measured. As a control, cells were transfected with a firefly luciferase reportor construct in which the LEF/TCF-binding sites had been mutated (NEG) and were incubated with Wnt3a for 8 h. Luciferase activity was expressed in arbitrary units relative to the activity observed in unstimulated WT cells normalised for Renilla luciferase activity. Four dishes of cells were analysed for each condition and similar results were obtained in two separate experiments. Results shown are mean±s.d. for a single experiment.
Discussion
A key observation in this study was that in the single GSK3β9A/9A knockin mice, insulin failed to stimulate activation of skeletal muscle glycogen synthase and dephosphorylation of Ser641 and Ser645, two residues phosphorylated by GSK3 that are critical for inactivation (Roach, 2002; Ferrer et al, 2003). This was despite GSK3α being inactivated normally by insulin in the skeletal muscle of these mice (Figure 2B). In contrast, pharmacological inhibition of GSK3 with AR-A014418 stimulated glycogen synthase in the soleus muscle of double GSK3α/β21A/21/A9/9A knockin mice (Figure 3B). These results provide conclusive evidence that inactivation of GSK3 is the major regulatory step in the stimulation of muscle glycogen synthase by insulin. They also establish that, in murine muscle, GSK3β rather than GSK3α is the major enzyme phosphorylating glycogen synthase. Thus, inactivation of GSK3β is the crucial mechanism by which insulin stimulates glycogen synthase in mouse skeletal muscle. If activation of a glycogen synthase phosphatase by insulin contributes to activation of glycogen synthase (Suzuki et al, 2001; Delibegovic et al, 2003), then the activity of this enzyme would also have to be controlled by GSK3β. As mentioned in the Introduction, it has also been reported that insulin stimulates the dephosphorylation of glycogen synthase at Ser7, a site that is not regulated by GSK3, and that other kinases may phosphorylate the residues targetted by GSK3 (reviewed in Roach, 2002; Ferrer et al, 2003). Our findings with the GSK3 knockin mice provide firm genetic evidence that GSK3β is the major enzyme that phosphorylates Ser641 and Ser645 and that regulates glycogen synthase activity in response to insulin in muscle. As Ser641 and Ser645 are not dephosphorylated in response to insulin in the double GSK3α/β21A/21A/9A/9A knockin mice, the dephosphorylation of glycogen synthase at Ser7, if it occurs, cannot be of sufficient magnitude to increase glycogen synthase activity significantly in the absence of dephosphorylation of Ser641 and Ser645 residues. Alternatively, the phosphorylation of Ser7 may be dependent on the prior dephosphorylation of Ser641/Ser645.
To our knowledge, a major difference in the substrate specificity of GSK3α and GSK3β has not been reported and both isoforms would be expected to phosphorylate glycogen synthase at a similar rate. The finding that GSK3β is the major regulator of glycogen synthase in skeletal muscle may be accounted for by the four-fold higher expression of GSK3β compared to GSK3α in murine skeletal muscle (Figure 4). The finding that GSK3β is also more abundant than GSK3α in human skeletal muscle suggests that GSK3β may also control human glycogen synthase activity in response to insulin.
The stimulation of glycogen synthase by exercise/muscle contraction may play a role in preventing glycogen particles from becoming too depleted as well as enabling rapid repletion of glycogen in the post exercise period (Nielsen and Richter, 2003). The mechanism by which exercise stimulates glycogen synthase is not fully understood. Previous results indicate that exercise leads to dephosphorylation of the GSK3 phosphorylation site Ser645 on glycogen synthase (Sakamoto et al, 2003). Moreover, in mice lacking the glycogen targeting GM subunit of the PP1 phosphatase, exercise is unable to activate glycogen synthase or induce its dephosphorylation (Aschenbach et al, 2001). We observed that muscle contraction led to normal dephosphorylation of the Ser641 and Ser645 residues of glycogen synthase in the single and double knockin mice (Figure 6B), without inhibiting GSK3α and GSK3β activity (Figure 6C). Taken together, these findings indicate that exercise is exerting its effects by stimulating the dephosphorylation of glycogen synthase through the GM–PP1 complex. At the moment, it is not known whether this effect is mediated through direct activation of the phosphatase complex and/or by glycogen synthase becoming more accessible to the phosphatase, perhaps as a result of depletion of glycogen. It is also possible that exercise promotes the dephosphorylation of glycogen synthase at other residues that might contribute to the activation of this enzyme. The finding that exercise leads to preferential dephosphorylation of Ser641, rather than Ser645 (Figure 6B), may be explained by the presence of a Pro residue C-terminal to Ser645 that renders it more resistant to dephosphorylation by PP1.
Previous studies have indicated that GSK3 phosphorylates the light chains of the kinesin motor protein, resulting in its inhibition (Morfini et al, 2002). As kinesin has been suggested to mediate the translocation of the GLUT4 glucose transporters to the plasma membrane (Emoto et al, 2001), inhibition of GSK3 by insulin could promote glucose uptake. However, our finding that insulin-stimulated glucose uptake occurred normally in soleus muscle of the single and double knockin mice would argue against this notion. Despite insulin not activating glycogen synthase in the muscle of GSK3β9A/9A and GSK3α/β21A/21A/9A/9A knockin animals, muscle glycogen levels were normal (Figure 5B). The normal basal and insulin-stimulated glucose uptake in the muscle of GSK3 knockin mice coupled with the basal activity of glycogen synthase may therefore be sufficient to maintain normal muscle glycogen levels. Allosteric activation of glycogen synthase by G6P might also contribute to maintenance of glycogen levels.
Glucose stimulated a normal activation of hepatic glycogen synthase that was accompanied by dephosphorylation of the Ser641/Ser645 residues in the GSK3 double knockin animals (Figure 7A). These results indicate that the major mechanism by which hepatic glycogen synthase is activated by glucose involves stimulation of a protein phosphatase, rather than Ser21/Ser9-mediated inactivation of GSK3 isoforms. As outlined in the Introduction, glucose binding to phosphorylase-a thereby relieves the inhibition that phosphorylase-a has on the hepatic GL-protein phosphatase-1 complex (Alemany and Cohen, 1986; Bollen et al, 1998). Our findings suggest that this is the major regulatory pathway by which hepatic glycogen synthase is stimulated. In this model, GSK3 isoforms nevertheless still play an important role in maintaining hepatic glycogen synthase in a low-activity state, as shown by the finding that inhibitors of GSK3 are capable of stimulating hepatic glycogen synthase (Coghlan et al, 2000; Cline et al, 2002). Indeed, it is the inhibition of hepatic GSK3, rather than muscle GSK3, that underlies the blood glucose-lowering effects of GSK3 inhibition in animal models of Type II diabetes (Cline et al, 2002).
A central finding of the present study was that the single and double GSK3 knockin mice displayed normal blood glucose and insulin levels and were not glucose intolerant despite insulin being unable to stimulate glycogen synthase in muscle. This is likely to result from the regulation of hepatic glycogen synthase not being perturbed in the knockin mice, enabling the liver to compensate for the failure of insulin to stimulate glycogen synthase in muscle. Indeed, the moderately higher glycogen levels in the fasted double GSK3 knockin mice (Figure 7B) may be indicative of the existence of such a mechanism. Interestingly, it was recently reported that mice overexpressing GSK3β in skeletal muscle possessed elevated hepatic glycogen levels (Pearce et al, 2004). Exercise is also a major regulator of whole-body glucose homeostasis as muscle contraction stimulates glucose uptake and glycogen synthase (Hayashi et al, 1997). It is therefore also possible that the normal physical activity of the single and double GSK3 knockin mice contributes to the maintenance of glucose homeostasis. Interestingly, it has been shown that mice lacking the insulin receptor in muscle only become glucose intolerant when anaesthetised (Wojtaszewski et al, 1999), suggesting that acute physical activity can compensate for the lack of insulin signalling in these animals. However, our observation that the double GSK3 knockin mice do not display glucose intolerance when anaesthetised (Figure 6E) suggests that acute physical activity does not account for normal glucose tolerance in these animals. We are unable to rule out that the constitutive knockin of GSK3 isoforms leads to compensatory mechanisms during development, resulting in an absence of a diabetic phenotype.
GSK3 is a key component of the Wnt signalling pathway controlling β-catenin levels and transcriptional responses required for development and cell growth (Moon et al, 2002; Nelson and Nusse, 2004). Our finding that Wnt3a induced normal inactivation of GSK3, stabilisation of β-catenin and induction of β-catenin-controlled transcription in the GSK3 double knockin ES cells (Figure 8) shows that the Wnt-mediated inactivation of GSK3 is not dependent upon phosphorylation of GSK3α at Ser21 and GSK3β at Ser9. This conclusion is consistent with previous work indicating that Wnt inhibits GSK3 by a distinct mechanism (Ding et al, 2000). As the GSK3 knockin mice develop normally, this also indicates that the Wnt-mediated inhibition of GSK3 operates by a mechanism that is independent of the phosphorylation of GSK3α at Ser21 and GSK3β at Ser9.
One of the effects of inhibiting components of the insulin signalling pathway in Drosophila and mammals is a marked effect on development and a reduction in the size of the organism caused by a decrease in cell volume and/or cell number (Oldham and Hafen, 2003). Elegant genetic analysis has recently indicated that phosphorylation of the Drosophila homologue of GSK3 plays no role in the regulation of development as well as cell size and number (Papadopoulou et al, 2004). Taken together, all these findings indicate that inactivation of GSK3 isoforms by Ser21/Ser9 phosphorylation does not play a major role in regulating development, cell growth and proliferation. The finding that both the single and double GSK3 knockin mice develop normally and are of normal size (Figure 1A and Supplementary Figure 2) confirms these observations in a mammalian system.
The knockin approach used in this study overcomes the inherent problems associated with studying kinase function by overexpressing dominant-negative or constitutively active mutants. For example, it has been recently been reported that transgenic mice overexpressing GSK3β at four- to seven-fold higher than physiological levels in skeletal muscle were glucose intolerant, possessed elevated plasma insulin levels and had a 50% reduction in muscle glycogen (Pearce et al, 2004). In contrast, the double GSK3 knockin mice in which the GSK3 isoforms are present at normal levels display none of these insulin-resistant phenotypes. Overexpression of the GSK3β[S9A] mutant in a rat skeletal muscle cell line reduced glycogen synthase protein levels (Macaulay et al, 2005), an effect that we do not observe in the muscle of the GSK3 knockin mice (Figure 3A). These examples illustrate the potential danger in employing the more facile overexpression approaches, instead of a more rigorous knockin strategy. We have previously employed a knockin approach to study the roles of the regulatory domains of 3-phosphoinositide-dependent protein kinase-1 (PDK1), in the activation of PKB, as well as other substrates (Collins et al, 2003; McManus et al, 2004). From these analyses and the results obtained from this study, it is our opinion that knockin methodology, coupled with gene knockouts and specific pharmacological inhibitors, are likely to become the methods of choice for dissecting the physiological roles of signal transduction pathways.
Materials and methods
Injection of insulin and preparation of tissue extracts
Following an overnight fast, a bolus of insulin (150 mU/g, Actrapid, Novo Nordisk) or saline solution was injected intraperitoneally to mice that had been anaesthetised with 13.5 mg/kg of Sagatal (pentobarbitone sodium). At the indicated times, the skeletal muscle was rapidly removed and frozen in liquid nitrogen, stored at −80°C and pulverised to a powder in liquid nitrogen. A 10-fold mass excess of ice-cold lysis buffer was added to the powdered tissue, which was briefly vortexed and centrifuged at 4°C for 10 min at 13 000 g to remove insoluble material. The supernatant was collected and protein concentration was measured by the Bradford Method, then snap frozen in aliquots in liquid nitrogen and stored at −80°C.
Glycogen synthase assay
Following an overnight fast, a bolus of insulin (150 mU/g) or saline solution was injected intraperitoneally to terminally anaesthetised mice. At the indicated times, the skeletal muscle was rapidly extracted and freeze clamped in liquid nitrogen, stored at −80°C and homogenised to a powder in liquid nitrogen. A 10-fold mass excess of ice-cold GS assay lysis buffer (50 mM Tris–HCl pH 7.5, 2 mM EGTA, 10 mM EDTA, 1% (by mass) Triton X-100, 1 mM sodium orthovanadate, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 0.27 M sucrose, 0.1 μM microcystin-LR, 0.1% (by vol) 2-mercaptoethanol and ‘Complete' proteinase inhibitor cocktail (one tablet per 50 ml)), was added to the powdered tissue, which was briefly vortexed and centrifuged at 3600 g for 5 min at 4°C to remove insoluble material. The supernatant was snap frozen in aliquots in liquid nitrogen and stored at −80°C. The glycogen synthase activity was measured as described previously (Thomas et al, 1968). Briefly, 300 μg of protein was incubated at 30°C for 20 min in a total volume of 165 μl containing 8.9 mM UDP-glucose (0.07 μCi), 6.7 mg/ml of glycogen, 6.7 mM potassium phosphate, 33.3 mM NaF, 0.67 mM EDTA, 0.1 M KCl, pH 6.8, in the presence or absence of 10 mM G6P. From each assay, aliquots of 100 μl of the reaction were spotted onto a piece (3 × 2 cm) of 31ETCHR paper, which was then washed two times for 20 min in 66% ethanol and once in acetone. After drying, the papers were put in a microfuge tube with 1 ml of scintillation liquid and the amount of UDP-[14C]glucose incorporated to the glycogen quantified. The glycogen synthase activity ratio is defined as activity measured in the absence of G6P divided by activity measured in the presence of G6P.
A detailed description of all other methods employed in this study can be found in the Supplementary data of this paper.
Supplementary Material
Supplementary Section
Acknowledgments
We thank Philip Cohen for helpful discussion, Alfonso Mora for advise, Gail Fraser for genotyping, Maria Deak for cloning of mouse and human GSK3 isoforms, Agnieszka Kieloch for tissue culture assistance, Simon Arthur for advice in generating the targeting construct, Victoria Murray-Tait for help in gene targeting, the protein production and antibody purification teams (Division of Signal Transduction Therapy (DSTT), University of Dundee) co-ordinated by Hilary McLauchlan and James Hastie for expression and purification of PP1γ and affinity purification of antibodies, the Sequencing Service (School of Life Sciences, University of Dundee, Scotland, www.dnaseq.co.uk) for DNA sequencing as well as Freddy Radtke (Lausanne) and Roel Nusse (Stanford University) for providing the Wnt3a-expressing cells and luciferase reporter constructs. Human muscle lysates were collected as part of a study funded by the Chief Scientist Office, Scotland (CZB/4/125) and were kindly supplied by Antonio Ruiz-Alcaraz and Simeen Akhtar, School of Medicine, University of Dundee. EJM is the recipient of a MRC Predoctoral Fellowship. DRA is supported by the Association for International Cancer Research, Diabetes UK, the Medical Research Council UK, the Moffat Charitable Trust, as well as the pharmaceutical companies that support the Division of Signal Transduction Therapy (AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Merck & Co. Inc., Merck KGaA and Pfizer).
References
- Alemany S, Cohen P (1986) Phosphorylase a is an allosteric inhibitor of the glycogen and microsomal forms of rat hepatic protein phosphatase-1. FEBS Lett 198: 194–202 [DOI] [PubMed] [Google Scholar]
- Aschenbach WG, Suzuki Y, Breeden K, Prats C, Hirshman MF, Dufresne SD, Sakamoto K, Vilardo PG, Steele M, Kim JH, Jing SL, Goodyear LJ, DePaoli-Roach AA (2001) The muscle-specific protein phosphatase PP1G/R(GL)(G(M))is essential for activation of glycogen synthase by exercise. J Biol Chem 276: 39959–39967 [DOI] [PubMed] [Google Scholar]
- Bhat R, Xue Y, Berg S, Hellberg S, Ormo M, Nilsson Y, Radesater AC, Jerning E, Markgren PO, Borgegard T, Nylof M, Gimenez-Cassina A, Hernandez F, Lucas JJ, Diaz-Nido J, Avila J (2003) Structural insights and biological effects of glycogen synthase kinase 3-specific inhibitor AR-A014418. J Biol Chem 278: 45937–45945 [DOI] [PubMed] [Google Scholar]
- Bollen M, Keppens S, Stalmans W (1998) Specific features of glycogen metabolism in the liver. Biochem J 336 (Part 1): 19–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline GW, Johnson K, Regittnig W, Perret P, Tozzo E, Xiao L, Damico C, Shulman GI (2002) Effects of a novel glycogen synthase kinase-3 inhibitor on insulin-stimulated glucose metabolism in Zucker diabetic fatty (fa/fa) rats. Diabetes 51: 2903–2910 [DOI] [PubMed] [Google Scholar]
- Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Roxbee Cox L, Mills D, Brown MJ, Haigh D, Ward RW, Smith DG, Murray KJ, Reith AD, Holder JC (2000) Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol 7: 793–803 [DOI] [PubMed] [Google Scholar]
- Cohen P (1999) The Croonian Lecture 1998. Identification of a protein kinase cascade of major importance in insulin signal transduction. Philos Trans R Soc Lond B Biol Sci 354: 485–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Goedert M (2004) GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov 3: 479–487 [DOI] [PubMed] [Google Scholar]
- Collins BJ, Deak M, Arthur JS, Armit LJ, Alessi DR (2003) In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J 22: 4202–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA (1995) Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 378: 785–789 [DOI] [PubMed] [Google Scholar]
- Delibegovic M, Armstrong CG, Dobbie L, Watt PW, Smith AJ, Cohen PT (2003) Disruption of the striated muscle glycogen targeting subunit PPP1R3A of protein phosphatase 1 leads to increased weight gain, fat deposition, and development of insulin resistance. Diabetes 52: 596–604 [DOI] [PubMed] [Google Scholar]
- Ding VW, Chen RH, McCormick F (2000) Differential regulation of glycogen synthase kinase 3beta by insulin and wnt signaling. J Biol Chem 275: 32475–32481 [DOI] [PubMed] [Google Scholar]
- Emoto M, Langille SE, Czech MP (2001) A role for kinesin in insulin-stimulated GLUT4 glucose transporter translocation in 3T3-L1 adipocytes. J Biol Chem 276: 10677–10682 [DOI] [PubMed] [Google Scholar]
- Ferrer JC, Favre C, Gomis RR, Fernandez-Novell JM, Garcia-Rocha M, de la Iglesia N, Cid E, Guinovart JJ (2003) Control of glycogen deposition. FEBS Lett 546: 127–132 [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P (2001) GSK3 takes centre stage more than 20 years after its discovery. Biochem J 359: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto S, Hsieh CM, Maemura K, Layne MD, Yet SF, Lee KH, Matsui T, Rosenzweig A, Taylor WG, Rubin JS, Perrella MA, Lee ME (2001) Akt participation in the Wnt signaling pathway through Dishevelled. J Biol Chem 276: 17479–17483 [DOI] [PubMed] [Google Scholar]
- Hayashi T, Wojtaszewski JF, Goodyear LJ (1997) Exercise regulation of glucose transport in skeletal muscle. Am J Physiol 273: E1039–E1051 [DOI] [PubMed] [Google Scholar]
- Logan CY, Nusse R (2004) The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20: 781–810 [DOI] [PubMed] [Google Scholar]
- Macaulay K, Blair AS, Hajduch E, Terashima T, Baba O, Sutherland C, Hundal HS (2005) Constitutive activation of GSK3 down regulates glycogen synthase abundance and glycogen deposition in rat skeletal muscle cells. J Biol Chem 280: 9509–9518 [DOI] [PubMed] [Google Scholar]
- McManus EJ, Collins BJ, Ashby PR, Prescott AR, Murray-Tait V, Armit LJ, Arthur JS, Alessi DR (2004) The in vivo role of PtdIns(3,4,5)P(3) binding to PDK1 PH domain defined by knockin mutation. EMBO J 23: 2071–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon RT, Bowerman B, Boutros M, Perrimon N (2002) The promise and perils of Wnt signaling through beta-catenin. Science 296: 1644–1646 [DOI] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST (2002) Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J 21: 281–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JT, Campbell DG, Morrice N, Auld GC, Shpiro N, Marquez R, Peggie M, Bain J, Bloomberg GB, Grahammer F, Lang F, Wulff P, Kuhl D, Cohen P (2004) Exploitation of KESTREL to identify N-myc downstream-regulated gene family members as physiological substrates for SGK1 and GSK3. Biochem J 384: 477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson WJ, Nusse R (2004) Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303: 1483–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen JN, Richter EA (2003) Regulation of glycogen synthase in skeletal muscle during exercise. Acta Physiol Scand 178: 309–319 [DOI] [PubMed] [Google Scholar]
- Oldham S, Hafen E (2003) Insulin/IGF and target of rapamycin signaling: a TOR de force in growth control. Trends Cell Biol 13: 79–85 [DOI] [PubMed] [Google Scholar]
- Orena SJ, Torchia AJ, Garofalo RS (2000) Inhibition of glycogen-synthase kinase 3 stimulates glycogen synthase and glucose transport by distinct mechanisms in 3T3-L1 adipocytes. J Biol Chem 275: 15765–15772 [DOI] [PubMed] [Google Scholar]
- Papadopoulou D, Bianchi MW, Bourouis M (2004) Functional studies of shaggy/glycogen synthase kinase 3 phosphorylation sites in Drosophila melanogaster. Mol Cell Biol 24: 4909–4919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce NJ, Arch JR, Clapham JC, Coghlan MP, Corcoran SL, Lister CA, Llano A, Moore GB, Murphy GJ, Smith SA, Taylor CM, Yates JW, Morrison AD, Harper AJ, Roxbee-Cox L, Abuin A, Wargent E, Holder JC (2004) Development of glucose intolerance in male transgenic mice overexpressing human glycogen synthase kinase-3beta on a muscle-specific promoter. Metabolism 53: 1322–1330 [DOI] [PubMed] [Google Scholar]
- Roach PJ (2002) Glycogen and its metabolism. Curr Mol Med 2: 101–120 [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Aschenbach WG, Hirshman MF, Goodyear LJ (2003) Akt signaling in skeletal muscle: regulation by exercise and passive stretch. Am J Physiol Endocrinol Metab 285: E1081–E1088 [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Hirshman MF, Aschenbach WG, Goodyear LJ (2002) Contraction regulation of Akt in rat skeletal muscle. J Biol Chem 277: 11910–11917 [DOI] [PubMed] [Google Scholar]
- Sancho E, Batlle E, Clevers H (2004) Signaling pathways in intestinal development and cancer. Annu Rev Cell Dev Biol 20: 695–723 [DOI] [PubMed] [Google Scholar]
- Shaw M, Cohen P, Alessi DR (1997) Further evidence that the inhibition of glycogen synthase kinase-3beta by IGF-1 is mediated by PDK1/PKB-induced phosphorylation of Ser-9 and not by dephosphorylation of Tyr-216. FEBS Lett 416: 307–311 [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Lanner C, Kim JH, Vilardo PG, Zhang H, Yang J, Cooper LD, Steele M, Kennedy A, Bock CB, Scrimgeour A, Lawrence JC Jr, DePaoli-Roach AA (2001) Insulin control of glycogen metabolism in knockout mice lacking the muscle-specific protein phosphatase PP1G/RGL. Mol Cell Biol 21: 2683–2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JA, Schlender KK, Larner J (1968) A rapid filter paper assay for UDPglucose-glycogen glucosyltransferase, including an improved biosynthesis of UDP-14C-glucose. Anal Biochem 25: 486–499 [DOI] [PubMed] [Google Scholar]
- Wojtaszewski JF, Higaki Y, Hirshman MF, Michael MD, Dufresne SD, Kahn CR, Goodyear LJ (1999) Exercise modulates postreceptor insulin signaling and glucose transport in muscle-specific insulin receptor knockout mice. J Clin Invest 104: 1257–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Mao J, Li L, Wu D (1999) Suppression of glycogen synthase kinase activity is not sufficient for leukemia enhancer factor-1 activation. J Biol Chem 274: 30419–30423 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Section