

Abstract
Escherichia coli SeqA binds clusters of transiently hemimethylated GATC sequences and sequesters the origin of replication, oriC, from methylation and premature reinitiation. Besides oriC, SeqA binds and organizes newly synthesized DNA at replication forks. Binding to multiple GATC sites is crucial for the formation of stable SeqA–DNA complexes. Here we report the crystal structure of the oligomerization domain of SeqA (SeqA–N). The structural unit of SeqA–N is a dimer, which oligomerizes to form a filament. Mutations that disrupt filament formation lead to asynchronous DNA replication, but the resulting SeqA dimer can still bind two GATC sites separated from 5 to 34 base pairs. Truncation of the linker between the oligomerization and DNA-binding domains restricts SeqA to bind two GATC sites separated by one or two full turns. We propose a model of a SeqA filament interacting with multiple GATC sites that accounts for both origin sequestration and chromosome organization.
Keywords: chromosome segregation, filament, oriC sequestration, SeqA, SeqA–DNA superhelix
Introduction
SeqA plays important roles in both DNA replication and chromosome organization in Gram-negative bacteria. In Escherichia coli, initiation of DNA replication starts from a single origin, oriC. A balance of the positive (DnaA, Dam) and negative (SeqA) effectors leads to the precise timing of replication initiation (Boye et al, 1996; Wold et al, 1998; Torheim and Skarstad, 1999; Ryan et al, 2004). oriC contains binding sites for the initiator protein DnaA, which melts DNA duplex to initiate replication, and multiple GATC sequences, to which SeqA binds and suppresses DNA melting and hence replication initiation (Zyskind and Smith, 1986; Slater et al, 1995; Torheim and Skarstad, 1999). GATC sequences throughout the chromosome are methylated at the N6 position of adenines by DNA adenine methyltranferase (Dam methylase). SeqA binding to fully methylated DNA has been recently shown to introduce positive supercoils, which might preclude open complex formation (Klungsoyr and Skarstad, 2004).
The balance of positive and negative effectors also ensures the replication synchrony and a discrete number of chromosomes in each cell. Upon initiation of DNA replication, the GATC sequences exist in a transient hemimethylated state until the newly synthesized daughter strand is methylated. SeqA binds both fully methylated and hemimethylated DNA with a preference for the latter. Although most of the GATC sequences throughout the chromosome are methylated soon after replication, those within oriC remain hemimethylated for one-third of the cell cycle due to sequestration by SeqA (Campbell and Kleckner, 1990; Lu et al, 1994). Sequestration of hemimethylated oriC prevents replication re-initiation and multiple rounds of replication during each cell cycle (Slater et al, 1995; Skarstad et al, 2000). During rapid cell growth, the replicating chromosome is multiforked and the newborn cells contain more than one copy of oriC. Initiation of DNA replication from these multiple origins is synchronized; in other words all origins fire at the same time. Deletion of the seqA gene abolishes origin sequestration, which leads to premature initiations and asynchronous DNA replication (Slater et al, 1995; Boye et al, 1996). Therefore, SeqA plays a dual role in regulation of DNA replication. It regulates timing and synchrony of DNA replication and prevents premature re-initiation in each cell cycle.
SeqA binding to hemimethylated DNA is not limited to the oriC (Slater et al, 1995). Its affinity for GATC sequences enables it to regulate gene expression (Slominska et al, 2001; Lobner-Olesen et al, 2003; Slominska et al, 2003a, 2003b). In addition, SeqA plays a role in chromosome organization (Weitao et al, 1999; Brendler et al, 2000). The fluorescent foci formed by GFP-fused SeqA appear to follow replication forks. This likely involves binding of SeqA to the hemimethylated sequences that are transiently produced as the replication forks progress around the chromosome and release of SeqA due to methylation by the Dam methylase. The SeqA foci probably represent these moving tracts of bound proteins, rather than the SeqA bound to oriC (Onogi et al, 1999; Brendler et al, 2000). Consistent with this, formation of SeqA foci requires ongoing DNA replication but not the presence of oriC (Hiraga et al, 2000). Also, the cell cycle-dependent migration of SeqA foci is quite different from the migration pattern of oriC (Niki and Hiraga, 1998; Onogi et al, 1999; Hiraga, 2000). seqA null strains exhibit increased negative superhelicity and abnormal localization of nucleoids (Bahloul et al, 1996; Weitao et al, 2000). Overproduction of SeqA delays nucleoid segregation and cell division (von Freiesleben et al, 2000; Bach et al, 2003).
SeqA has two functional domains, an N-terminal oligomerization domain (residues 1–50) and a C-terminal DNA-binding domain (residues 51–181) (Guarné et al, 2002). The structure of the C-terminal domain of SeqA (SeqA–C) in complex with a DNA dodecamer containing a hemimethylated GATC sequence reveals the protein–DNA recognition and the stoichiometry of one SeqA monomer per GATC sequence (Guarné et al, 2002; Fujikawa et al, 2004). However, binding of a SeqA–C monomer to a single GATC sequence has a dissociation constant of 7 μM (Guarné et al, 2002), while wild-type SeqA requires two hemimethylated GATC sequences within three helical turns to form a protein–DNA complex with a kd in the nM range (Brendler and Austin, 1999). Association of SeqA and DNA is cooperative with a Hill coefficient of 4.7 (Slater et al, 1995; Skarstad et al, 2000), and binding of SeqA to a pair of hemimethylated GATC sites attracts additional SeqA molecules to aggregate on hemimethylated DNA (Han et al, 2003; Han et al, 2004). These data strongly suggest that SeqA oligomerization is crucial for its function. However, it is unclear how SeqA forms functional oligomers that are able to trigger and release oriC sequestration and meanwhile organize the newly replicated chromosomes.
To elucidate the nature of the specific interactions that mediate formation of these versatile oligomers, we have determined the crystal structure of the N-terminal domain of SeqA. The minimal structural unit of SeqA is a dimer, which polymerizes by hydrophobic interactions to form helical filaments. DNA binding and flow cytometry assays reveal that oligomerization is required for SeqA function. Based on our structural and functional data, we propose a model in which interaction of the SeqA dimer with two adjacent GATC sequences is sufficient for tight binding, but oligomerization of DNA-bound SeqA dimers into a helical filament is needed to alter DNA topology and regulate replication.
Results
SeqA–N is a dimer
The N-terminal domain of SeqA, SeqA–N (residues 1–50), forms aggregates of 4–12 molecules in solution that remain soluble at high protein concentrations and low ionic strength (Guarné et al, 2002). We have solved the crystal structure of SeqA–N using seleno-methionine (SeMet)-substituted protein and MAD phasing (Table I) (Hendrickson et al, 1990). A SeqA–N polypeptide chain folds into three secondary structures, a β-strand (residues 2–6) and two amphipathic approximately orthogonal α-helices, α1 (residues 8–16) and α2 (residues 24–33) (Figure 1A). The monomeric form of SeqA–N is likely unstable in solution because of the unsatisfied hydrogen-bonding potential of the β-strand and a large exposed hydrophobic surface. SeqA–N is stabilized upon dimerization, where the N-terminal β-strands of the two subunits are swapped and hydrogen bonded to form an antiparallel pair (Figure 1B). The dimer interface is chiefly hydrophobic. Salt bridges between residues Asp7, Glu9 and Arg30 and hydrogen bonds between the side chains of Asp8 and Ser26, and the main chains of Met1 and Asp7 are reciprocated between the two SeqA–N subunits and further stabilize the dimer.
Table 1.
Data collection and refinement
| Data collection | ||||
|---|---|---|---|---|
| Space group | P3121 | P3121 | ||
| Cell parameters | a=b=112.2, c=62.0, α=β=90, | a=b=112.3, c=61.7, α=β=90, γ=120 | ||
| γ=120 | ||||
| Wavelength (Å) | 1.5418 | 0.9832 | 0.9794 | 0.9686 |
| Resolution (Å)a | 25–2.15 (2.19–2.15) | 30–2.1 (2.14–2.1) | ||
| Completeness (%)a | 98.5 (84.6) | 94.5 (95.1) | 94.7 (95.0) | 95.0 (96.3) |
| Rmergea,b | 7.6 (46.0) | 4.3 (41.2) | 6.2 (47.2) | 7.0 (43.5) |
| I/σ(I)a | 26.1 (2.1) | 26.5 (2.6) | 16.4 (2.7) | 15.8 (2.5) |
| Refinement | ||||
| Unique reflection | 24 365 | |||
| Protein atoms | 1152 | |||
| Solvent atoms | 176 | |||
| R-factor (Rfree) (%)c | 22.2 (23.7) | |||
| Average B (Wilson) (Å2) | 36.54 | |||
| R.m.s. deviations | ||||
| Bonds (Å) | 0.006 | |||
| Angles (deg) |
0.90 |
|
|
|
| aData of the highest resolution shell are shown in parenthesis. | ||||
| bRmerge=∑h∑i∣Ihi−〈Ih〉∣/∑〈Ih〉, where Ihi is the intensity of the ith observation of reflection h, and 〈Ih〉 is the average intensity of redundant measurements of the h reflections. | ||||
| cR-value=∑∣∣Fo∣−∣Fc∣∣/∑∣Fo∣, where Fo and Fc are the observed and calculated structure–factor amplitudes. Rfree is monitored with 1642 reflections excluded from refinement. | ||||
Figure 1.
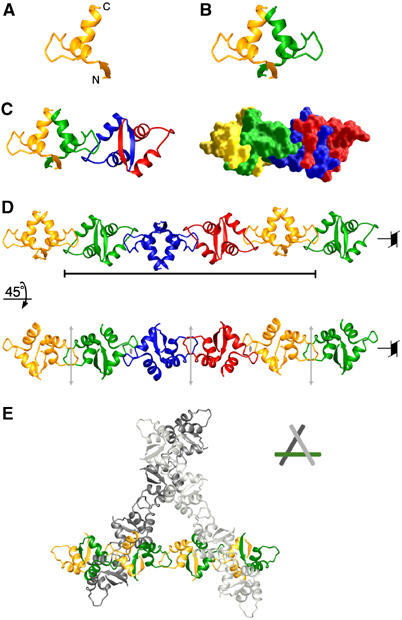
Oligomerization of the SeqA–N dimer. (A) Ribbon diagram of a single SeqA–N subunit. (B) A SeqA–N dimer. The two subunits are shown as yellow and green ribbon diagrams. (C) The asymmetric unit contains two SeqA–N dimers related by a noncrystallographic dyad axis and a 43 screw axis. The two SeqA–N dimers colored yellow–green and blue–red, respectively, are shown in a ribbons diagram (left) and molecular surface representation (right). (D) Two views of the SeqA–N filament. The black bar indicates a complete helical turn consisting of four dimers. The 43 axis and the noncrystallographic (gray arrows) and crystallographic (gray ovals) dyad axes are indicated. (E) Crystal packing of the SeqA filaments shown as a ribbon diagram. Filaments pack according to the crystallographic 31 axis. The central filament is shown with the SeqA monomers colored yellow and green. The top and bottom filaments are shown in light and dark gray, respectively.
SeqA–N dimers form a filament
SeqA–N was crystallized in space group P3121. Each asymmetric unit of the crystal contains two SeqA–N dimers related by a noncrystallographic dyad axis as well as an orthogonal fourfold screw axis (Figure 1C and D). This dimer of dimers is held together by reciprocal hydrophobic interactions between the loops connecting helices α1 and α2 of adjacent subunits, thus creating an elongated tetramer (Figure 1C). Due to the crystallographic dyad axis, the dimer–dimer interaction in an asymmetric unit is repeated and extended throughout the crystal, forming a left-handed SeqA–N helical filament (43 symmetry, Figure 1D). SeqA–N filaments are packed according to the threefold screw axis of the crystal (Figure 1E). As a result of the nonglobular shape of SeqA–N filament, the SeqA–N crystal contains 75% solvent.
The loop connecting helices α1 and α2 (residues 17–23), which consists mainly of hydrophobic residues, is exposed to the solvent in a single SeqA–N dimer (Figure 2A). The α1−α2 loop is locked in an extended conformation (Figure 2A and B) owing to Arg31 and Glu23, which form bidentate salt bridges and buttress the α1−α2 loop via the hydrogen bonds with the main-chain atoms (Figure 2B). In addition, a type I, tight turn at Ile21–Gly22 further rigidifies the extended loop between helices α1 and α2. As a result, the side chain of Ile21 is fully extended from one dimer to a neighboring dimer and nucleates the formation of a SeqA filament (Figure 2B and C). An α1−α2 loop of one SeqA–N dimer interacts with both subunits of a neighboring dimer. For example, Ile21 (B subunit) contacts Ile4 of the D subunit and Ile14, Ala15, Ala25, Ile28 of the C subunit (Figure 2C). The main chain of Ile21 (B subunit) is also hydrogen-bonded to the side chain of Tyr11 from the C subunit (Figure 2A and C).
Figure 2.
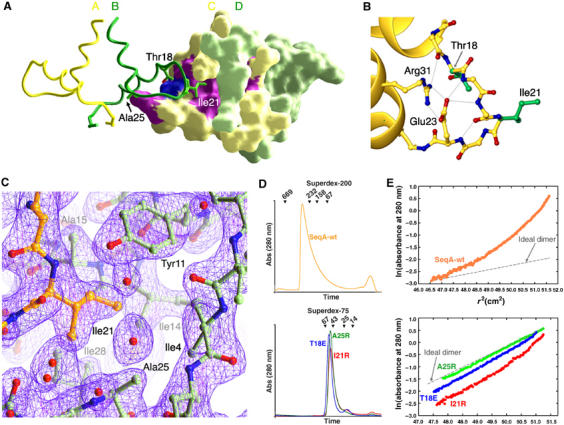
SeqA–N dimer–dimer interactions. (A) Interaction between two SeqA dimers. One dimer (AB) is shown as a ribbon diagram and the other (CD) by molecular surface. The two subunits of each SeqA–N dimer are shown in yellow (A and C) and green (B and D). Residues involved in dimer–dimer interactions are shown as green sticks on one dimer (Ile21, Ala25 and Thr18) and colored purple (Ile21, Ala15 and Ala25) or blue (Thr18) on the surface of the neighboring dimer. (B) The α1–α2 loop is shown in ball-and-stick presentation. Side chains holding the extended conformation of the loop are shown in yellow and side chains involved in dimer–dimer interaction in green. Bidentate salt bridges between Glu23 and Arg31 and the hydrogen bonds that they make are shown as thin gray lines. (C) The 2Fo−Fc electron density map contoured at 1.2σ around Ile21. The refined SeqA–N coordinates are shown as balls-and-sticks in yellow for the B subunit and green for the C and D subunits. (D) Elution profiles of wild-type SeqA from a Superdex-200 column (top panel) and SeqA-I21R (red), SeqA-A25R (green) and SeqA-T18E (blue) from a Superdex-75 column (lower panel). Peak positions of protein standards eluted from the size exclusion columns are indicated with ticks and molecular weights (kDa). (E) Sedimentation equilibrium profiles are plotted as ln(A280) versus square of the radius (r2) in the same color scheme as 2D.
Mutations that disrupt the dimer–dimer interface
To test the functional relevance of the SeqA filament, we chose to mutate Ile21, Thr18 and Ala25 to disrupt the dimer–dimer interface observed in the crystal (Figure 2). Ile21 (B) and Ala25 (C) are in close van der Waals contacts. The minimal side chain of Ala25 at the beginning of helix α2 forms a concave hydrophobic surface to receive Ile21 and the α1–α2 loop from a neighboring dimer (Figure 2). Mutation of Ile21 or Ala25 to Arg introduces bulky charged groups into the hydrophobic interface between neighboring dimers. Thr18 (B) is located at the beginning of the α1–α2 loop and its hydroxyl group is hydrogen bonded to the carbonyl oxygen of Ala15 (C). Mutation of Thr18 to Glu may destabilize the α1–α2 loop and weaken the SeqA–N filament formation.
T18E, I21R and A25R mutant SeqA proteins can be overexpressed and readily purified. Unlike wild-type SeqA, which is only soluble in the presence of 1 M NaCl (Slater et al, 1995), these mutant proteins remain soluble at high protein concentrations regardless of the ionic strength of the buffer. Their elution profiles from a size-exclusion column are consistent with the molecular weight of a dimer (Figure 2D), confirming that the dimer–dimer interface observed in the crystal structure is responsible for aggregation of SeqA in solution.
The aggregation states of wild-type and the mutant SeqA proteins defective in filament formation were quantitatively examined by equilibrium sedimentation (Figure 2E). Wild-type SeqA, whose calculated molecular weight is 21 kDa, was found to be polydisperse with a weight average molecular mass of 350–560 kDa (Figure 2E). SeqA-A25R was found to be monodisperse with a molecular mass of 42 700±1400 Da, confirming that the protein is dimeric (n=2.09±0.07, Figure 2E). Samples of SeqA-I21R and SeqA-T18E were polydisperse with weight average molecular masses, consistent with the presence of both dimer and higher-order species (Figure 2E). Data for SeqA-T18E were best modeled in terms of a reversible dimer–tetramer equilibrium characterized by a dissociation constant of 110 μM. SeqA-I21R can form even larger oligomers, as indicated by the increased slope of observed curvature in Figure 2E. Data were best modeled in terms of a dimer–tetramer–octamer reversible equilibrium with dissociation constants of 100 μM for the dimer–tetramer equilibrium and 35 μM for the dimer–octamer equilibrium.
Filament formation is essential for SeqA function
The abilities of SeqA-I21R (pWY1417), SeqA-T18E (pWY1418) and SeqA-A25R (pWY1421) to complement a seqA defective strain and synchronize DNA replication in vivo were evaluated by flow cytometry. The seqA null strain replicated DNA asynchronously, but synchrony could be restored by introduction of a plasmid encoding wild-type SeqA (pSS1, Figure 3A). The strains harboring SeqA-T18E, SeqA-I21R or SeqA-A25R mutant plasmids exhibited asynchronous DNA replication (Figure 3B). These mutant proteins form stable dimers in solution and bind DNA equally well as wild-type SeqA (Figures 2D and 3C). Besides, the seqA null strains carrying these plasmids have growth curves similar to those of wild-type and seqA null strains (see Supplementary data). Therefore, the defective phenotypes of SeqA-T18E, SeqA-I21R or SeqA-A25R in replication initiation can only be associated with their inability to form filaments. Interestingly, in the presence of 25 μM IPTG, the seqA defective strain carrying SeqA-T18E, SeqA-I21R or SeqA-A25R mutant plasmids showed varying degrees of replication synchrony (Figure 3B). To eliminate any role of IPTG other than induction of SeqA expression, we complemented the seqA null strain with an empty pET11a plasmid. As expected, pET11a did not restore replication synchrony regardless of the presence or absence of IPTG. Additionally, we engineered a DNA-binding defective SeqA by replacing the essential residues for hemimethylated GATC recognition, Asn150 and Asn152, with alanines. The SeqA-N150A/N152A plasmid (pWY1420) also failed to complement the ΔseqA strain and DNA replication remained asynchronous both in the absence and presence of IPTG (Figure 3A). We thus conclude that overexpression of SeqA-T18E, SeqA-I21R or SeqA-A25R overcomes their defects in filament formation.
Figure 3.
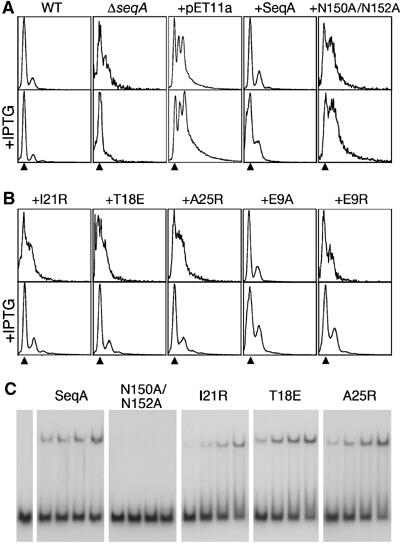
Filament formation is essential for SeqA function in vivo. (A) In a wild-type strain DNA replication is synchronous (WT), whereas in the ΔseqA∷tet strain it is asynchronous (ΔseqA). An empty pET11a plasmid does not affect replication synchrony of the ΔseqA∷tet strain (+pET11a). A pET11a plasmid encoding wild-type SeqA (+pSS1) restores replication synchrony of the ΔseqA∷tet strain (+SeqA), whereas a pET11a plasmid encoding the DNA-binding mutant SeqA-N150A/N152A (pWY1420) does not (+N150A/N152A). Effects of IPTG addition (25 μM) are shown on the bottom panels. The amounts of DNA equivalent to two chromosomes are indicated with arrowheads. (B) pET11a plasmids encoding SeqA mutants with defects in filament formation (SeqA-I21R, SeqA-T18E and SeqA-A25R) do not restore DNA replication synchrony of the ΔseqA∷tet strain (+I21R, +T18E, +A25R). Overexpression of these mutants induced by addition of 25 μM IPTG restores replication synchrony (bottom panels). Mutation of Glu9 to Ala (+E9A) shows a wild-type phenotype in the absence of IPTG but loses synchrony when protein is overexpressed, whereas the SeqA-E9R mutant plasmid (+E9R) behaves similarly to the filament defective mutants. (C) EMSAs of an oligonucleotide containing two hemimethylated GATC sites separated by 12 bp when incubated with increasing quantities (10, 50, 100 and 500 ng) of wild-type SeqA, SeqA-N150A/N152A, SeqA-I21R, SeqA-T18E and SeqA-A25R.
A previous report suggested that mutation of Glu9 to Lys results in dissociation of SeqA aggregates in vitro (Lee et al, 2001). The SeqA–N crystal structure reveals that Glu9 participates in dimer and not the filament (dimer-of-dimers) formation. Glu9 and Arg30 are hydrogen bonded reciprocally across the dimer interface. To clarify the role of Glu9, we engineered SeqA-E9R (pWY1422) and SeqA-E9A (pWY1419) mutants and characterized the behavior of the mutant proteins and their abilities to restore replication synchrony of a seqA defective strain in vivo. Overexpressed SeqA-E9A and SeqA-E9R proteins were largely in the insoluble fraction of cell lysates. Complementation of seqA null strain with the plasmid encoding SeqA-E9A was similar to complementation with wild-type SeqA (Figure 3B). In contrast, complementation of seqA null strain with the plasmid encoding SeqA-E9R showed asynchronous DNA replication that could be reversed by IPTG induction (Figure 3B). These observations suggest that replacing the negatively charged Glu9 by neutral Ala has little functional consequence, but replacing it with a positively charged side chain (Arg or Lys) probably destabilizes SeqA dimer and consequently the filament.
A flexible linker between the N- and C-terminal domains enhances DNA-binding plasticity
Wild-type SeqA binds two GATC sequences separated from 5 to 34 base pairs (bp) and shows binding maxima at separations of 7, 12, 21 and 31 bp (Brendler and Austin, 1999). To assess whether such a binding pattern is achieved by a single SeqA dimer or requires filament formation, we measured the DNA binding by SeqA-I21R, which is impaired in filament formation. With oligonucleotides containing two hemimethylated GATC sequences separated by various spacings, 0.2 μM of SeqA-I21R produced DNA-binding patterns similar to wild-type SeqA (Figure 4A). Therefore, binding of two GATC sequences separated by half to three full turns is an intrinsic property of a SeqA dimer and does not require filament formation.
Figure 4.
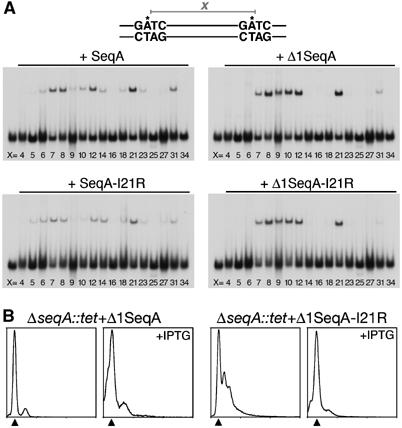
A flexible linker between the N- and C-terminal domains enhances versatile DNA binding. (A) EMSAs of SeqA, SeqA-I21R, Δ1SeqA and Δ1SeqA-I21R with DNAs containing two hemimethylated GATC sequences. For each gel, the left-most lane contains an equimolar mixture of DNAs with 5, 7, 12, 21, 25 and 34 bp between the two GATC sites but no proteins. The following lanes show the interaction of various SeqA proteins with DNAs containing two GATC sequences separated by a variable number of bp (x), where x ranges from 4 to 34 bp. (B) Flow cytometry analysis reveals that the flexible linker between the two domains is partially dispensable for SeqA function in vivo. Basal expression of the plasmid encoding Δ1SeqA (pAG8020) restores replication synchrony of a ΔseqA∷tet strain. The plasmid encoding Δ1SeqA-I21R (pAG8025) with restricted flexibility between domains and unable to form filaments has asynchronous replication. Synchrony of the ΔseqA∷tet strain carrying the pAG8025 plasmid is restored by IPTG-induced protein overexpression. Arrowhead indicates a DNA content equivalent to two chromosomes.
The 28 residues between the N- and C-terminal domains of SeqA are disordered in the SeqA–N and SeqA–C crystal structures, and probably form an unstructured linker that enables SeqA to bind DNA with unusual plasticity. To test this hypothesis, we constructed deletion mutants of SeqA and SeqA-I21R with the linker shortened by 11 residues. These deletions resulted in restricted flexibility between the oligomerization and DNA-binding domains (Δ1SeqA, pAG8020) or both restricted flexibility and impaired filament formation (Δ1SeqA-I21R, pAG8025). Both mutant proteins retain their basic DNA-binding ability but have altered binding preferences. A spacing of 9–10 bp, which resulted in a binding minimum with the full-length protein, produced maximal binding by the linker deletion mutant proteins. Binding to two sites located on opposite faces of DNA separated by 6, 14 or 18 bp was too weak to be observed with the Δ1 mutants of SeqA and SeqA-I21R. In addition, binding to two sites separated by more than two helical turns was very weak for Δ1SeqA and undetectable for Δ1SeqA-I21R (Figure 4A).
The truncated SeqA mutant protein, Δ1SeqA (pAG8020), is functional in vivo, as shown by its ability to complement seqA defective strain and restore synchrony of DNA replication (Figure 4B). As expected, the Δ1SeqA-I21R (pAG8025) double mutant showed an asynchronous DNA replication phenotype, comparable to that of I21R (pWY1417) single mutant (Figures 3B and 4B). Synchrony of Δ1SeqA-I21R was also restored by IPTG-induced protein overexpression.
A model of the dynamic SeqA and DNA interactions
In the crystal structure of SeqA-C, two 12 bp DNA molecules with hemimethylated GATC in the middle are stacked end-to-end, forming a pseudo-continuous duplex (Guarné et al, 2002). In this arrangement, the two SeqA–C molecules bound to this pseudo-continuous 24 bp DNA are related by a dyad axis. Based on the crystal structures of SeqA–N and the SeqA–C–DNA complex, we constructed a model of full-length SeqA by placing the SeqA–C and SeqA–N dimers so that they share a common dyad axis (Figure 5A). We then applied the P43 symmetry of SeqA–N filament to the full-length SeqA–DNA dimer, generating a left-handed super helix (Figure 5B, Supplementary data). In this SeqA–DNA super helix, SeqA–N domains form the helical axis, while the SeqA–C domains and DNA modules are radially extended from SeqA–N and wrap around it. The N- and C-terminal domains are placed with a separation of ∼16 Å between their terminal residues to account for the flexible linker of 28 residues and to avoid contacts or clashes between neighboring SeqA–C domains related by the 43 screw axis. DNA molecules are not connected in our model (see Supplementary data), which reflects the fact that GATC sequences are not evenly distributed every 12 bp in genomic DNAs. In reality, the intervening DNAs between GATC sites are likely to loop out, as shown by electron microscopy (Skarstad et al, 2000). Upon binding to DNA, the SeqA filament would constrain one negative supercoil in each filament turn (every eight protomers) over an axial length of 120 Å.
Figure 5.
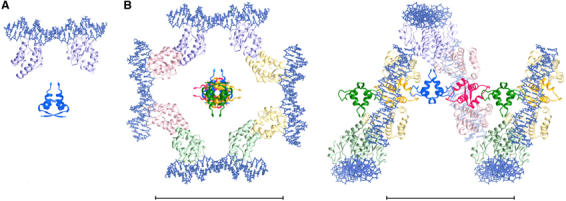
A model of the interactions between a SeqA filament and DNA. (A) A SeqA–N dimer and a pair of crystallographic SeqA–C–DNA complexes are placed to share a common dyad axis. The protein is shown as ribbon diagrams in dark (SeqA–N) and light (SeqA–C) blue and the DNA depicted as stick models. (B) The full-length SeqA dimer–DNA model is allowed to multimerize according to the 43 screw axis of the SeqA–N filament. The four-fold screw axis is perpendicular to the plane. Each SeqA–N dimer and the SeqA–C pair bound to it are shown in dark and light shades of a distinct color. Space between the N- and C-terminal domains accounts for the flexible linker and avoids contacts or clashes between neighboring SeqA–C molecules related by the 43 screw axis. An orthogonal view placing the SeqA–DNA superhelix in plane is shown on the right panel. No artificial coordinates are introduced, and DNAs are left to be discontinuous between adjacent SeqA dimers. Scale bars represent 100 Å.
Discussion
SeqA dimers versus SeqA filaments
SeqA was believed to be a tetramer based on size-exclusion chromatographic experiments (Lee et al, 2001). It was also proposed that a SeqA tetramer interacts with only two hemimethylated GATC sites (Han et al, 2003). However, the SeqA–C/DNA complex structure suggested that should SeqA be a tetramer it would likely interact with four GATC sequences (Guarné et al, 2002). The SeqA–N crystal structure now reveals that the minimal SeqA structural unit is a dimer, consistent with the DNA-binding preference of two GATC sites. The apparent discrepancy between our structural result and the previously reported model can be explained by the nonglobular and polydisperse nature of SeqA. In solution, SeqA exists in equilibrium of dimers and oligomers. Even the filament-formation defective mutant proteins, SeqA-I21R and SeqA-T18E, retain a residual propensity to form tetramers and octamers (Figure 2E). Only the SeqA-A25R mutant protein forms pure dimers under our experimental conditions, probably because the arginine replacement at that position disrupts the dimer–dimer interface most substantially (Figure 2A–C).
The SeqA dimer is not only the structural but also the DNA-binding unit. Indeed, the DNA-binding ability of SeqA mutants with impaired filament formation is comparable to wild-type SeqA. The flexible loop between the oligomerization and DNA-binding domains confers flexibility on the dimeric SeqA, which binds equally well to a pair of GATC sequences separated by 7 or 21 bp (Figure 4A). The linker, however, does not allow complete flexibility. The relative positions of the two DNA-binding domains of the dimer are constrained such that some arrangements, notably those that place the two GATC sites on the same face of the helix when separation is more than one helical turn, are favored (Brendler and Austin, 1999). Our work reveals that shortening of the linker by up to 11 residues does not affect the basic ability of the SeqA dimer to bind DNA nor its ability to regulate replication initiation at oriC in vivo, but this mutant of SeqA binds different subsets of GATC sites, some of which are not favored by the wild-type protein (9–10 bp, Figure 4). The preference for shorter separations between GATC sites (two rather than three full helical turns) and the rejection of the sites located on the opposite faces (5–6, 14 or 18 bp) by the deletion mutant proteins are consistent with the notion of a shorter and less flexible linker. The wild-type-like behavior of the Δ1SeqA mutant in vivo is presumably due to the irregular spacings of GATC sites in oriC and the ability of Δ1SeqA filaments to compensate the shortened and less flexible linker. Supporting this, our recent compilation of SeqA sequences shows that the linker region is the least conserved in length and sequence, and is often four to six residues shorter than that in E. coli. Although DNA binding is essential for SeqA to function in vivo, alteration of the flexible linker between the two functional domains of SeqA is tolerated with little phenotype.
In contrast to the negligible in vivo effect of the linker region, mutations disrupting filament formation cause asynchronous replication even when the mutant protein retains normal DNA binding. Such defect can be overcome by increased SeqA protein expression, suggesting that oligomerization of mutant proteins may still occur if the local concentration of SeqA is sufficiently high. Consistent with this notion, the more the tendency of a mutant SeqA protein to oligomerize in vitro, the better it restores the replication synchrony, and as such I21R and T18E perform better than A25R in vivo. There is likely a synergy between binding of SeqA to multiple GATC sequences and formation of extended filaments. Filament extension from a single bound dimer can induce more SeqA dimers to bind to nearby sites as observed previously (Han et al, 2004). Conversely, association of several individual SeqA dimers to a GATC-rich region increases the local concentration of SeqA and thus promotes filament formation. This would result in the co-operative saturation of SeqA in regions clustered with GATC sites such as oriC (Slater et al, 1995; Skarstad et al, 2000).
The significance of a SeqA filament
Aggregation is normally associated with protein misfolding and loss of function. However, SeqA offers an example of functional aggregation. In vitro, SeqA is isolated as polydisperse aggregates, presumably a state of multiple filaments tangled via the flexible linker and C-terminal DNA-binding domains. The orderly association of SeqA is limited to the interaction of one loop in the SeqA–N domain, which buries 1013 Å2, more than one-fifth of the total dimer surface. Aggregation of full-length SeqA, however, is reversible in vitro (Lee et al, 2001) and must be reversible in vivo upon DNA binding as SeqA foci associated with DNA replication forks constantly assemble and disassemble (Onogi et al, 1999; Brendler et al, 2000).
The only form of biologically functional and polydisperse aggregates known to date is filament. The SeqA filament is reminiscent of the RecA filament, which wraps around DNA and promotes strand exchange in homologous DNA recombination (Story et al, 1992). In the case of RecA, formation of the protein–DNA filament is essential for homologous search and DNA pairing. In the case of SeqA, the filament circularly presents DNA-binding domains around the protein core and thus maximizes its avidity for DNA (Figure 5, Supplementary data). Formation for such protein–DNA helices is dynamic and fluctuates with the local concentrations of both SeqA and hemimethylated GATC sites. The dynamic nature of the SeqA–DNA filamentous complexes plays an essential role in regulating DNA replication and chromosome segregation.
The minimal origin of replication encompasses 11 GATC sites spanning over 245 bp and can be fully bound by as few as six SeqA dimers. The high density of GATC sites in oriC, therefore, critically depends on the aggregation property of SeqA to attain the high-avidity SeqA–oriC complex that is resistant to both DnaA and Dam methyltransferase. For oriC sequestration, formation of a polymer filament enables SeqA to bind hemimethlated GATC sites more tightly than Dam methylase, which functions as a monomer. Therefore, disruptions of the dimer–dimer interface lead to asynchronous replication. It is possible that not all the hemimethylated GATC sites within the origin need to be bound for sequestration from Dam methylase (Slater et al, 1995), but the close spacings between GATC sequences in oriC ensure that sequestration is achieved instantly and only a handful SeqA molecules are required (Figure 6A).
Figure 6.
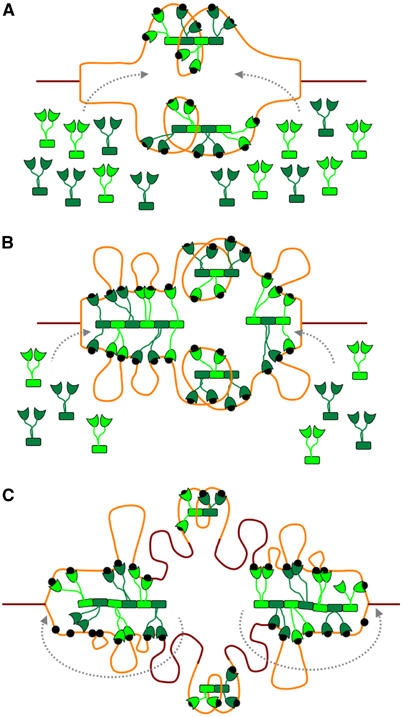
Model of SeqA sequestration and SeqA foci migration following the replication forks. Fully and hemimethylated DNA are shown in brown and orange, respectively. Black dots represent hemimethylated GATC sequences. SeqA dimers are shown in green. (A) As replication initiates, SeqA binds newly generated hemimethylated GATC sequences within oriC, triggering origin sequestration. (B) As the forks progress, more hemimethylated GATC sequences become available. Spacing between the newly occurring GATC sequences favors SeqA filament formation, which in turn favors cooperative binding of distant GATC sequences. (C) As cells have a limited amount of SeqA, dynamic addition and removal of SeqA dimers (indicated by dashed gray arrows) will allow the SeqA filament to bind newly occurring hemimethylated GATC sequences and track forks as they progress. Only few SeqA molecules are required for origin sequestration and, therefore, oriC will remain hemimethylated while the intervening DNA sequences are remethylated.
The origin, however, is not the only chromosomal site of SeqA action. There are almost 4000 GATC sites fairly evenly distributed around the E. coli chromosome, which are able to bind SeqA. As the replication forks progress along the chromosome, hemimethylated GATC sequences are continually produced, but exist only for a short while before being methylated. As SeqA binds hemimethylated better than fully methylated DNA, binding of SeqA to these transiently hemimethylated tracks likely accounts for the SeqA foci observed in cells with ongoing replication (Onogi et al, 1999; Brendler et al, 2000). Therefore, the majority of SeqA molecules in the cell are bound to DNA outside of oriC (Figure 6). We propose that the observed SeqA foci consist of SeqA filaments, whose structures are modeled in Figure 5. Irregular spacing between GATC sites can be accommodated by looping out the intervening DNA and by skipping DNA-binding domains in a SeqA filament (Figure 6B and C). The superhelical structure imposed by the SeqA filament would constrain one negative supercoil with each filament turn if the DNA wrapped continuously on the structure (Figure 5). This appears to contradict the observations that SeqA constrains positive supercoils in relaxed circular DNA whether methylated or unmethylated (Klungsoyr and Skarstad, 2004). However, in reality, natural DNA substrates bound to the SeqA filament contain looped-out DNAs to accommodate irregular spacings between GATC sequences. The form and path of the DNA in these loops can radically influence the overall supercoiling of the DNA region, so that net positive supercoiling could readily be accommodated by the structure that we propose. In fact, it has been recently shown that the SeqA dimer introduces positive supercoils to the DNA, whereas the SeqA filament has an ‘opposite effect' in chromosome topology by introducing negative supercoils (Odsbu et al, submitted). This implies that the flexible linker between the N- and C-terminal domains may have other roles besides providing plasticity to the DNA binding and supports our model of a left-handed superhelical structure.
We envision that SeqA foci formed at replication forks also play an essential role in release oriC sequestration. In addition to condense and organize newly replicated DNAs emerging from a replication forks, an activity which has been proposed to be important for DNA segregation to daughter cells (Brendler et al, 2000; Molina and Skarstad, 2004), foci formation titrates the SeqA concentrations. As each cell has a limited amount of SeqA (Slater et al, 1995), filament formation at the replication forks reduces the free SeqA concentrations. Immediately after replication, oriC with the highest density of GATC sites has the highest affinity for SeqA. With the progression of the replication forks, SeqA dimers are recruited to the heads of replication forks by the presence of new hemimethylated GATC sites (Figure 6). Concurrently, SeqA molecules are chased off from the tails of replication forks by Dam methylase. Therefore, a delayed methylation is unnoticeable outside of oriC (Campbell and Kleckner, 1990). When the free SeqA pool is depleted by the growing SeqA foci, the Dam methylase can finally overcome the oriC sequestration by SeqA and remethylate oriC, thus allowing the next round of replication to initiate.
Materials and methods
Crystallization and structure determination
SeqA–N was obtained as described previously (Guarné et al, 2002). Crystals of SeMet-labeled SeqA–N were grown from a solution containing 30% (v/v) MPD, 0.2 M CaCl2, 0.1 M TRIS (pH=8), and 3% isopropanol as additive. Crystals were cryo-protected by 20% (v/v) glycerol and flash frozen in liquid propane. A three-wavelength MAD data set was collected from a single P3121 crystal that diffracted X-rays to 2.1 Å resolution at X9B of National Synchrotron Light Source (Brookhaven). Data were processed using HKL2000 (Otwinowski and Minor, 1997) (Table I). Seven out of eight selenium sites were readily identified by SOLVE (Terwilliger and Berendzen, 1999). The experimental map after solvent flattening using DM (CCP4, 1994) was easily interpreted. Residues 1–35 of each of the four SeqA–N subunits in an asymmetric unit were traced, whereas residues 36–50 were disordered. Refinement was conducted with a native data set collected in house at 2.15 Å resolution (Table I). The refined model contains residues 1–35 for each subunit (four subunits and a total of 140 residues in each asymmetric unit), 175 water molecules and a well-ordered calcium ion. Over 94% of the residues lie in the most favorable regions of the Ramachandran plot. Figures 1, 2 and 5 were generated with Ribbons (Carson, 1987) and GRASP (Nicholls et al, 1991).
SeqA mutant preparation
All SeqA mutants were derived from a pET11a plasmid encoding wild-type SeqA (pSS1) using QuikChange site-directed mutagenesis kit (Stratagene). Sequences of the mutants were verified using DNA sequencer PRISM-310 (ABI). Mutant SeqA proteins were overexpressed like wild-type protein (Guarné et al, 2002). A new purification protocol was developed for the mutant proteins that do not precipitate out of solution at low ionic strength. Cell lysates were cleared by centrifugation and loaded onto a heparin column (Hitrap, Amersham Biosciences). Fractions from the heparin column containing SeqA (eluted at ∼0.45 M NaCl) were pooled together and further purified by ionic exchange over a MonoS column (Amersham Biosciences). SeqA mutants were concentrated to 2 mg/ml in a buffer containing 20 mM TRIS, pH=8, 150 mM NaCl, 0.1 mM EDTA, 4.5 mM β-mercaptoethanol and 5% glycerol.
Sedimentation equilibrium analysis
Sedimentation equilibrium experiments with SeqA-T18E, SeqA-I21R and SeqA-A25R proteins were conducted at 20°C on a Beckman Optima XL-A analytical ultracentrifuge. Samples corresponding to loading A280 of 0.8–1.0 were prepared by extensive dialysis against 20 mM TRIS, pH=8, 150 mM NaCl, 0.1 mM EDTA, 4.5 mM β-mercaptoethanol and 5% (v/v) glycerol. Data were acquired as an average of four absorbance measurements at a nominal wavelength of 280 nm and a radial spacing of 0.001 cm at rotor speeds ranging from 8000–16 000 r.p.m. Equilibrium was achieved within 42 h. Data for SeqA-A25R were analyzed in terms of a single ideal solute to obtain the buoyant molecular mass, M(1−vρ). Data for the other two were analyzed in terms of a reversible dimer–tetramer–octamer self-association essentially as described (Jenkins et al, 1996). M(1−vρ) values for SeqA-I21R and SeqA-T18E were calculated using densities, ρ, at 20°C obtained from standard tables and a value of v based on the amino-acid composition (Perkins, 1986).
Flow cytometry
Host strain BL21DE3/pLysS was made ΔseqA∷tet by P1 transduction with lysate from MM294ΔseqA∷tet (a kind gift from Dr Kleckner). This strain was supplemented with pET11a derivatives encoding: SeqA-wt (pSS1), SeqA-E9A (pWY1419), SeqA-E9R (pWY1422), SeqA-T18E (pWY1418), SeqA-I21R (pWY1417), SeqA-A25R (pWY1421), SeqA-N150A/N152A (pWY1420), Δ1SeqA (pAG8020) and Δ1SeqA-I21R (pAG8025). In each case, the average number of origins per cell was determined by the flow cytometry ‘run-off' method with few modifications (Skarstad et al, 1995). Overnight cultures were grown in the absence or presence of 25 μM IPTG at 37°C in M63 minimal media with the appropriate antibiotics. The overnight cultures were diluted to an OD600 of 0.02 and grown to an OD600 ∼0.1 prior to incubation for 3 h with rifampicin (200 μg/ml) and cephalexin (36 μg/ml). After fixing with 77% ethanol, cells were analyzed in a Bryte SH flow cytometer (Biorad) using WinBryte software.
DNA-binding assays
The randomly chosen 72 bp sequence and the design of hemimethylated DNA duplexes with two GATC sites at various spacings used for SeqA protein binding were generated as described earlier (Brendler and Austin, 1999; Brendler et al, 2000). The preparation and radioactive labeling of hemimethylated duplex DNA and the conditions for the SeqA binding electrophoretic mobility shift assay (EMSA) were carried out as described previously (Brendler et al, 1995; Brendler and Austin, 1999). Unless otherwise stated, SeqA protein-binding assays were performed using 0.2 μM of either wild-type or mutant SeqA protein.
Coordinates
Atomic coordinates and structure factors of SeqA–N have been deposited in the Protein Data Bank (accession code 1XRX).
Supplementary Material
Supplementary Figure
Supplementary Movie
Legend to Supplementary Movie
Acknowledgments
We thank Dr Z Dauter for assistance during data collection and Dr J Ortega for critical reading of the manuscript. AG has been supported by the HFSPO (LT00092) and the CIHR (MOP67189).
References
- Bach T, Krekling MA, Skarstad K (2003) Excess SeqA prolongs sequestration of oriC and delays nucleoid segregation and cell division. EMBO J 22: 315–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahloul A, Meury J, Kern R, Garwood J, Guha S, Kohiyama M (1996) Co-ordination between membrane oriC sequestration factors and a chromosome partitioning protein, TolC (MukA). Mol Microbiol 22: 275–282 [DOI] [PubMed] [Google Scholar]
- Boye E, Stokke T, Kleckner N, Skarstad K (1996) Coordinating DNA replication initiation with cell growth: differential roles for DnaA and SeqA proteins. Proc Natl Acad Sci USA 93: 12206–12211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendler T, Abeles A, Austin S (1995) A protein that binds to the P1 origin core and the oriC 13mer region in a methylation-specific fashion is the product of the host seqA gene. EMBO J 14: 4083–4089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendler T, Austin S (1999) Binding of SeqA protein to DNA requires interaction between two or more complexes bound to separate hemimethylated GATC sequences. EMBO J 18: 2304–2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendler T, Sawitzke J, Sergueev K, Austin S (2000) A case for sliding SeqA tracts at anchored replication forks during Escherichia coli chromosome replication and segregation. EMBO J 19: 6249–6258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JL, Kleckner N (1990) E. coli oriC and the dnaA gene promoter are sequestered from dam methyltransferase following the passage of the chromosomal replication fork. Cell 62: 967–979 [DOI] [PubMed] [Google Scholar]
- Carson M (1987) Ribbon models of macromolecules. J Mol Graphics 5: 103–106 [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Fujikawa N, Kurumizaka H, Nureki O, Tanaka Y, Yamazoe M, Hiraga S, Yokoyama S (2004) Structural and biochemical analyses of hemimethylated DNA binding by the SeqA protein. Nucleic Acids Res 32: 82–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarné A, Zhao Q, Ghirlando R, Yang W (2002) Insights into negative modulation of E. coli replication initiation from the structure of SeqA-hemimethylated DNA complex. Nat Struct Biol 9: 839–843 [DOI] [PubMed] [Google Scholar]
- Han JS, Kang S, Kim SH, Ko MJ, Hwang DS (2004) Binding of SeqA protein to hemi-methylated GATC sequences enhances their interaction and aggregation properties. J Biol Chem 279: 30236–30249 [DOI] [PubMed] [Google Scholar]
- Han JS, Kang S, Lee H, Kim HK, Hwang DS (2003) Sequential binding of SeqA to paired hemi-methylated GATC sequences mediates formation of higher-order complexes. J Biol Chem 278: 34983–34989 [DOI] [PubMed] [Google Scholar]
- Hendrickson WA, Horton JR, LeMaster DM (1990) Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three-dimensional structure. EMBO J 9: 1665–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraga S (2000) Dynamic localization of bacterial and plasmid chromosomes. Annu Rev Genet 34: 21–59 [DOI] [PubMed] [Google Scholar]
- Hiraga S, Ichinose C, Onogi T, Niki H, Yamazoe M (2000) Bidirectional migration of SeqA-bound hemimethylated DNA clusters and pairing of oriC copies in Escherichia coli. Genes Cells 5: 327–341 [DOI] [PubMed] [Google Scholar]
- Jenkins TM, Engelman A, Ghirlando R, Craigie R (1996) A soluble active mutant of HIV-1 integrase: involvement of both the core and carboxyl-terminal domains in multimerization. J Biol Chem 271: 7712–7718 [DOI] [PubMed] [Google Scholar]
- Klungsoyr HK, Skarstad K (2004) Positive supercoiling is generated in the presence of Escherichia coli SeqA protein. Mol Microbiol 54: 123–131 [DOI] [PubMed] [Google Scholar]
- Lee H, Kang S, Bae SH, Choi BS, Hwang DS (2001) SeqA protein aggregation is necessary for SeqA function. J Biol Chem 276: 34600–34606 [DOI] [PubMed] [Google Scholar]
- Lobner-Olesen A, Marinus MG, Hansen FG (2003) Role of SeqA and Dam in Escherichia coli gene expression: a global/microarray analysis. Proc Natl Acad Sci USA 100: 4672–4677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Campbell JL, Boye E, Kleckner N (1994) SeqA: a negative modulator of replication initiation in E. coli. Cell 77: 413–426 [DOI] [PubMed] [Google Scholar]
- Molina F, Skarstad K (2004) Replication fork and SeqA focus distributions in Escherichia coli suggest a replication hyperstructure dependent on nucleotide metabolism. Mol Microbiol 52: 1597–1612 [DOI] [PubMed] [Google Scholar]
- Nicholls A, Sharp KA, Honig B (1991) Protein folding and association: insights from the interfacial and themodynamic properties of hydrocarbons. Proteins Struct Funct Genet 11: 281–296 [DOI] [PubMed] [Google Scholar]
- Niki H, Hiraga S (1998) Polar localization of the replication origin and terminus in Escherichia coli nucleoids during chromosome partitioning. Genes Dev 12: 1036–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odsbu I, Klungsoyr HK, Fossum S, Skarstad K (2005) Specific N-terminal interactions of the E. coli SeqA protein are required to form multimers that restrain negative supercoils and form foci. J Biol Chem submitted [DOI] [PubMed] [Google Scholar]
- Onogi T, Niki H, Yamazoe M, Hiraga S (1999) The assembly and migration of SeqA-Gfp fusion in living cells of Escherichia coli. Mol Microbiol 31: 1775–1782 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Perkins SJ (1986) Protein volumes and hydration effects. The calculations of partial specific volumes, neutron scattering matchpoints and 280-nm absorption coefficients for proteins and glycoproteins from amino acid sequences. Eur J Biochem 157: 169–180 [DOI] [PubMed] [Google Scholar]
- Ryan VT, Grimwade JE, Camara JE, Crooke E, Leonard AC (2004) Escherichia coli prereplication complex assembly is regulated by dynamic interplay among Fis, IHF and DnaA. Mol Microbiol 51: 1347–1359 [DOI] [PubMed] [Google Scholar]
- Skarstad K, Bernander R, Boye E (1995) Analysis of DNA replication in vivo by flow cytometry. Methods Enzymol 262: 604–613 [DOI] [PubMed] [Google Scholar]
- Skarstad K, Lueder G, Lurz R, Speck C, Messer W (2000) The Escherichia coli SeqA protein binds specifically and co-operatively to two sites in hemimethylated and fully methylated oriC. Mol Microbiol 36: 1319–1326 [DOI] [PubMed] [Google Scholar]
- Slater S, Wold S, Lu M, Boye E, Skarstad K, Kleckner N (1995) E. coli SeqA protein binds oriC in two different methyl-modulated reactions appropriate to its roles in DNA replication initiation and origin sequestration. Cell 82: 927–936 [DOI] [PubMed] [Google Scholar]
- Slominska M, Konopa G, Baranska S, Wegrzyn G, Wegrzyn A (2003a) Interplay between DnaA and SeqA proteins during regulation of bacteriophage lambda pR promoter activity. J Mol Biol 329: 59–68 [DOI] [PubMed] [Google Scholar]
- Slominska M, Konopa G, Ostrowska J, Kedzierska B, Wegrzyn G, Wegrzyn A (2003b) SeqA-mediated stimulation of a promoter activity by facilitating functions of a transcription activator. Mol Microbiol 47: 1669–1679 [DOI] [PubMed] [Google Scholar]
- Slominska M, Wegrzyn A, Konopa G, Skarstad K, Wegrzyn G (2001) SeqA, the Escherichia coli origin sequestration protein, is also a specific transcription factor. Mol Microbiol 40: 1371–1379 [DOI] [PubMed] [Google Scholar]
- Story RM, Weber IT, Steitz TA (1992) The structure of the E. coli recA protein monomer and polymer. Nature 355: 318–325 [DOI] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999) Automated structure solution for MIR and MAD. Acta Crystallogr D 55: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torheim NK, Skarstad K (1999) Escherichia coli SeqA protein affects DNA topology and inhibits open complex formation at oriC. EMBO J 18: 4882–4888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Freiesleben U, Krekling MA, Hansen FG, Lobner-Olesen A (2000) The eclipse period of Escherichia coli. EMBO J 19: 6240–6248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitao T, Nordstrom K, Dasgupta S (1999) Mutual suppression of mukB and seqA phenotypes might arise from their opposing influences on the Escherichia coli nucleoid structure. Mol Microbiol 34: 157–168 [DOI] [PubMed] [Google Scholar]
- Weitao T, Nordstrom K, Dasgupta S (2000) Escherichia coli cell cycle control genes affect chromosome superhelicity. EMBO Rep 1: 494–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wold S, Boye E, Slater S, Kleckner N, Skarstad K (1998) Effects of purified SeqA protein on oriC-dependent DNA replication in vitro. EMBO J 17: 4158–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zyskind JW, Smith DW (1986) The bacterial origin of replication, oriC. Cell 46: 489–490 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure
Supplementary Movie
Legend to Supplementary Movie