

Abstract
A novel mRNA surveillance for mRNA lacking a termination codon (nonstop mRNA) has been proposed in which Ski7p is thought to recognize stalled ribosomes at the 3′ end of mRNA. Here we report our analysis of translation and decay of nonstop mRNAs in Saccharomyces cerevisiae. Although the reduction of nonstop mRNAs was only 4.5-fold, a level that is sufficient for residual protein synthesis, translation products of nonstop mRNAs were hardly detectable. We show that nonstop mRNAs were associated with polysomes, but not with Pab1p. We also show that ribosomes translating nonstop mRNA formed stable and heavy polysome complexes with mRNA. These data suggest that ribosome stalling at the 3′ end of nonstop mRNA may block further rounds of translation, hence repressing protein synthesis. Furthermore, it was found that the 5′ → 3′ decay pathway was accelerated for nonstop mRNA decay in the absence of Ski7p. We also found that translation of aberrant mRNAs with a shortened 3′-UTR was repressed, suggesting that an improper spatial distance between the termination codon and the 3′ end of mRNA results in translation repression.
Keywords: decapping, nonstop decay, Ski complex, stalled ribosome, translational repression
Introduction
There are many quality control mechanisms to ensure the high fidelity of gene expression. Cells have surveillance systems that recognize and eliminate aberrant mRNAs to avoid the production of potentially harmful protein products. It is well established that mRNA containing a premature termination codon is eliminated by nonsense-mediated mRNA decay (NMD) (Graham, 2003; Singh and Lykke-Andersen, 2003). In mammalian cells, it has been shown that newly transported mRNA associates with exon–exon junction complexes (EJCs) to make an mRNP that recruits Upf complexes essential for NMD (Kim et al, 2001; Lejeune et al, 2002; Gehring et al, 2003). The quality of mRNA is surveyed by a pioneer round of translation, where premature translation termination occurring upstream of the EJC results in the formation of a protein complex on mRNA that initiates the degradation of the aberrant mRNA (Ishigaki et al, 2001; Lejeune et al, 2003). The role of the EJC in NMD has been shown so far only in human cells. In yeast, it has been proposed that ribosomes stalled at an improper termination codon remain associated with mRNA thereby inducing NMD (Muhlrad and Parker, 1999a; Amrani et al, 2004). There are more examples of aberrant mRNA in cells, including mRNA lacking a termination codon (nonstop mRNA). In eubacteria, ribosomes that have stalled at the 3′ end of nonstop mRNA are recycled by tmRNA. The bacterial tmRNA, also called SsrA RNA, is a unique molecule that has properties of both tRNA and mRNA (Keiler et al, 1996; Himeno et al, 1997). In this process, the tmRNA is recruited to the empty A-site of ribosome, in which it acts first as an alanyl-tRNA and then as an mRNA to direct the addition of a short peptide tail to the polypeptide. This cotranslation reaction (trans-translation) terminates at a stop codon contained in the tmRNA reading frame, releasing both the ribosome and the tagged polypeptide (Karzai et al, 2000). The tagged polypeptide is recognized and degraded by several ATP-dependent proteases. An additional important role of the tmRNA system is to facilitate the degradation of truncated mRNAs by removing stalled ribosomes and thus allowing 3′ → 5′ exonucleases to access the free mRNA 3′ end (Yamamoto et al, 2003). Thus, the tmRNA quality control system not only degrades aberrant polypeptides once produced but also prevents production of aberrant polypeptides through a rapid elimination of damaged mRNAs.
How do eukaryotic cells deal with nonstop mRNAs? Recently, a model for nonstop mRNA decay (NSD) in eukaryotes has been proposed (Frischmeyer et al, 2002; van Hoof et al, 2002). In the model, a stalled ribosome is thought to occupy the extreme 3′ end of the mRNA and prevent exonucleases from digesting it. Ski7p, an exosome-associated protein in yeast, might recognize a stalled 80S ribosome at the 3′ end of a nonstop mRNA. It is proposed that this recognition is mediated by the Ski7p carboxyl-terminal domain, which structurally mimics the GTPase domains of translation elongation factors. The amino-terminal domain of Ski7p recruits the exosome complex of 3′ → 5′ exonucleases as well as the Ski complex, resulting in the degradation of nonstop mRNA by a 3′ → 5′ decay pathway (van Hoof et al, 2002). The biological significance of NSD may be to avoid production of potentially deleterious extended products that could have dominant-negative activity against wild-type gene products. Genetic experiments suggested that the degradation of nonstop mRNA mediated by the Ski complex was effective in limiting the production of aberrant protein derived from the nonstop mRNA (van Hoof et al, 2002).
Here we examined translation and decay of nonstop mRNAs in Saccharomyces cerevisiae. Although a sufficient amount of nonstop mRNAs remained associated with polysomes, there was no detectable association with Pab1p and no detectable translation products from these mRNAs. The 5′ → 3′ decay pathway was accelerated for NSD in the absence of Ski7p. We propose that ribosome stalling at the 3′ end of nonstop mRNA may block further rounds of translation, and the removal of Pab1p from the poly(A) tail causes accelerated decapping and degradation of nonstop mRNA, in a process additional to the Ski7p-dependent 3′ → 5′ degradation pathway. In addition, we show that translation was severely repressed when the 3′-UTR was shortened without changing mRNA stability. These results support the view that the proper spatial relationship between termination codon and Pab1p bound to poly(A) is required for proper and efficient translation termination.
Results
Translation product of nonstopHIS3 is not detectable
We constructed a nonstopHIS3 reporter by introducing a frame-shift mutation within the termination codon to produce mRNA lacking a termination codon (Figure 1A). This reporter gene (nonstopHIS3) failed to complement a his3− mutant strain (Figure 1B), in accordance with previous reports (van Hoof et al, 2002). This result suggests that nonstopHIS3 products were either insufficient or lacked activity to complement the his3− mutant strain. Therefore, we first examined the level of HIS3 and nonstopHIS3 reporter mRNAs by Northern blot analysis (Figure 1C). The data indicate that the level of nonstopHIS3 mRNA was about one-fourth of the wild-type HIS3 mRNA, which confirms the instability of nonstop mRNA. However, this reduction in nonstopHIS3 mRNA seems to be insufficient to explain the failure to complement a his3− strain. The most likely possibility is that translation of nonstop mRNA might be repressed.
Figure 1.
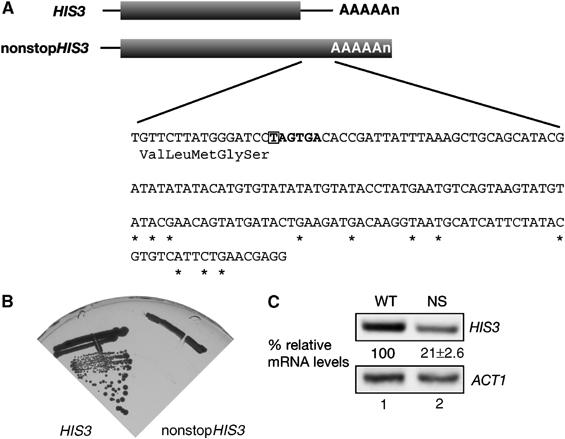
A nonstopHIS3 reporter gene cannot complement his3 mutant. (A) Schematic drawing of HIS3 reporter genes used in this study. The shaded boxes indicate open reading frames (ORFs). DNA sequences of the 3′-UTR region are shown and asterisks represent the poly(A) addition sites determined previously (Mahadevan et al, 1997). Translation termination codon of the HIS3 gene is indicated by bold letters, and the first nucleotide that was deleted to construct nonstopHIS3 reporter gene is boxed. (B) W303 cells were transformed with pIT709 (pHIS3-His6) or pIT711 (phis3-His6-ns), and transformants were streaked on SC-His plate and incubated for 3 days at 30°C. (C) W303 cells harboring pIT709 (pHIS3-His6) or pIT711 (phis3-His6-ns) were grown on SC-Leu medium and total RNAs were prepared. HIS3 or ACT1 mRNAs in strains were detected with Northern blot analysis with DIG-labeled probe. The values under Northern blot show the relative intensity to the amount of wild-type mRNA normalized by control ACT1 mRNA and are shown as the mean values±standard deviations (s.d.), obtained from at least three independent experiments.
To examine this possibility, we next characterized the protein product of nonstopHIS3 mRNA. In this study, both wild-type and nonstopHIS3 reporter genes were created to contain a hexahistidine-tag sequence at the C-terminus prior to the authentic stop codon of HIS3 and the nonstop sequence of nonstopHIS3. Translation products of the reporter genes were analyzed by Western blotting with anti-His6 antibody. As shown in Figure 2A, hexahistidine-tagged products synthesized from HIS3-His6 mRNA were detected in exponentially growing cells (Figure 2A, lane 2), while no nonstopHIS3-His6 mRNA translation product was detected (Figure 2A, lane 3). The same results were obtained when a FLAG-tag sequence was inserted into the N-termini of wild-type and nonstopHIS3 reporters and detected by anti-FLAG antibody (Figure 2A, lanes 4–6). Therefore, nonstopHIS3 does not complement a his3− strain because of a lack of its protein product. Even when nonstopHIS3 was overproduced, no protein product was detected (Figure 2B, lane 6). Essentially the same results were obtained with N-terminal FLAG-tag constructs (data not shown).
Figure 2.
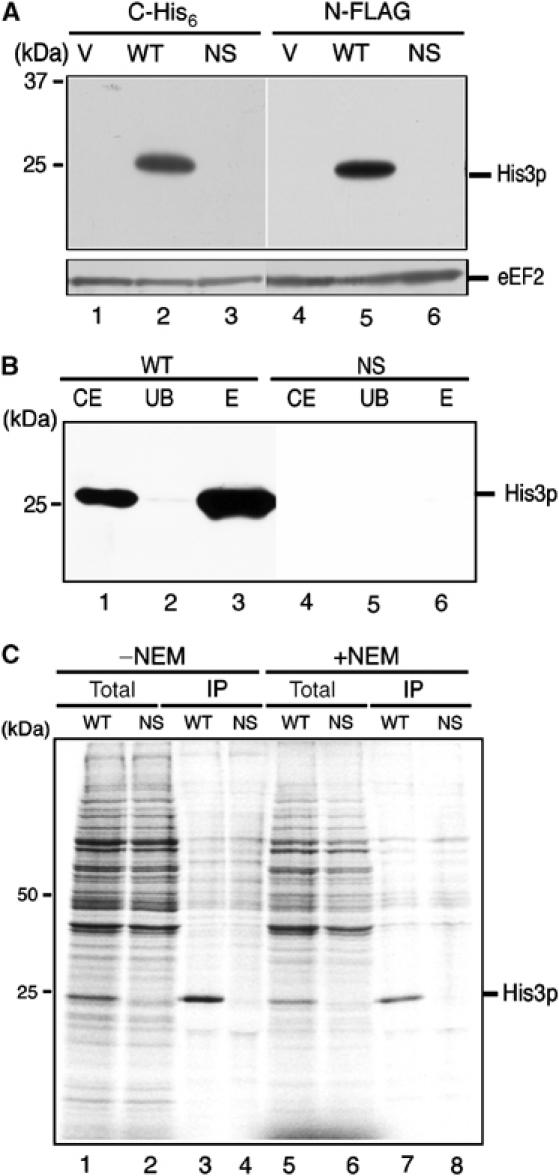
Translation product of nonstopHIS3 mRNA is hardly detectable. (A) W303 cells were transformed with the following plasmids. Lanes 1 and 4: yCplac111 (V); lane 2: pIT709 (pHIS3-His6, WT); lane 3: pIT711 (phis3-His6-ns, NS); lane 5: pIT798 (pFLAG-HIS3, WT); lane 6: pIT799 (pFLAG-his3-ns, NS). Cells were grown on SC-Leu medium, and proteins from cell extracts resolved by SDS–PAGE were blotted to detect His3p or eEF2 protein with anti-His6 antibodies (lanes 1–3), anti-FLAG antibodies (lanes 4–6) or anti-eEF2 antibodies (bottom panel). (B) W303 cells harboring pIT765 (pGAL1p-HIS3-His6, WT) or pIT766 (pGAL1p-his3-His6-ns, NS) were grown on SG-Ura medium. Cell extracts equivalent to 10 OD600 were used for affinity purification with Ni2+-NTA agarose (QIAGEN). Total cell extracts (CE), unbound fractions (UB) and purified samples (E) were resolved by 12% SDS–PAGE and visualized by immunoblot analysis with anti-His6 antibodies. (C) Pulse labeling and immunoprecipitation. W303 cells harboring pIT826 (pGAL1p-FLAG-HIS3, WT) or pIT827 (pGAL1p-FLAG-his3-ns, NS) were grown on SG-UraMet, pulse labeled with [35S]methionine and immunoprecipitated. To inhibit proteasome activity, 0.1 M NEM was added after the pulse label. Total cell extracts (lanes 1, 2, 5 and 6) and immunoprecipitated samples (lanes 3, 4, 7 and 8) were subjected to SDS–PAGE and visualized by autoradiography.
In order to test the possibility that the nonstopHIS3 product might be produced, but in an unstable and quickly degraded form, we performed [35S]methionine pulse-label experiments as shown below. W303 cells harboring pIT826 (pGAL1p-FLAG-HIS3, WT) or pIT827 (pGAL1p-FLAG-his3-ns, NS) were grown on galactose medium and labeled with [35S]methionine before immunoprecipitation and SDS–PAGE analysis. While His3p was clearly detected when translated from FLAG-HIS3 mRNA, no trace of His3p was detected when translated from nonstopFLAG-HIS3 mRNA (Figure 2C, lanes 3 and 4). To exclude the potential involvement of the protein degradation mechanism in eliminating any nonstopHIS3 products, N-ethylmaleimide (NEM), a strong inhibitor of proteasome in vivo (Turner and Varshavsky, 2000), was employed during the expression of nonstopHIS3. A 0.1 M portion of NEM was added after the pulse-label with [35S]methionine and cell extracts were prepared for affinity purification. The addition of NEM failed to recover the expression of nonstopHIS3 (Figure 2C, lanes 7 and 8). These results suggest that the absence of nonstopHIS3 product is not due to degradation but due to translational repression. Although we do not exclude the further possibility that the protein products of nonstop mRNA could be degraded by proteasome-independent protein degradation pathway, we suggest that the first ribosome translating nonstop mRNA may stall at the 3′ end of nonstopHIS3 mRNA, thereby preventing further protein synthesis.
NonstopHIS3 mRNAs are associated with ribosomes
To investigate whether or not ribosomes stall on nonstop mRNAs, we monitored mRNA distribution in polysomes. As shown in Figure 3, both wild-type HIS3 mRNAs (Figure 3A, top-left panel) and nonstopHIS3 mRNAs (Figure 3A, top-right panel) were clearly distributed with the polysome fractions. We also prepared extract in the absence of cycloheximide (CHX) and found that nonstop mRNA was distributed mainly in polysome fractions (Figure 3A, middle panels), indicating that CHX treatment itself did not essentially effect mRNA-polysome distribution. On the other hand, EDTA treatment completely disrupted these polysome complexes (Figure 3A, bottom panels) and mRNAs were distributed in the free fraction, indicating that nonstop mRNAs were associated with ribosome. Together, these data strongly suggest that translation of nonstopHIS3 mRNA is inhibited at a step(s) after initiation.
Figure 3.
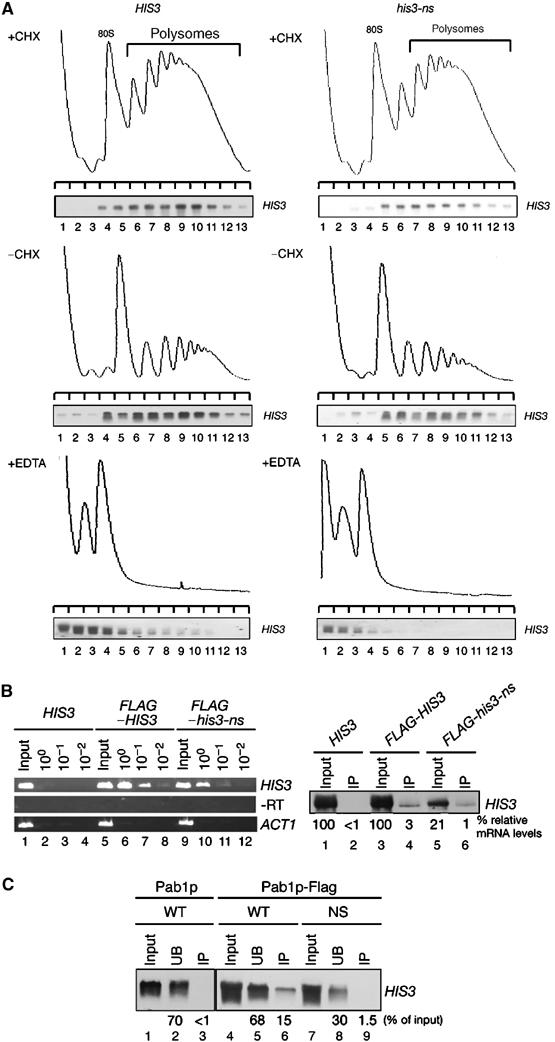
Nonstop mRNAs are associated with ribosomes. (A) W303 cells harboring pIT709 (pHIS3-His6, left panels) or pIT711 (phis3-His6-ns, right panels) were grown on SC-Leu medium. Cell extracts were prepared in the presence (top panels) or absence of (middle panels) CHX. Cell extracts were resolved by velocity sedimentation on 10–50% sucrose gradients. RNA samples prepared from the indicated fractions were analyzed by Northern blotting. Polysome analysis performed in the presence of 30 mM EDTA to separate ribosome subunits is shown in bottom panels. (B) Left: W303 cells harboring the indicated plasmids were grown on SG-Ura medium and cell extracts were prepared in the presence of CHX for affinity purification. RNA samples prepared from purified samples were subjected to RT reaction and cDNA was amplified by PCR with the series of dilutions indicated. Lane 1–4: pIT765 (pGAL1p-HIS3-His6, HIS3); lanes 5–8: pIT826 (pGAL1p-FLAG-HIS3, FLAG-HIS3); lanes 9–12: pIT827 (pGAL1p-FLAG-his3-ns, FLAG-his3-ns). Right: HIS3 mRNAs in samples prepared from total extract or purified samples were detected with Northern blot analysis. (C) W303 cells harboring pIT765 (pGAL1p-HIS3-His6, WT), YIT874 (pab1-FLAGHA TRP1) cells containing pIT765 (pGAL1p-HIS3-His6, WT) or pIT766 (pGAL1p-his3-His6-ns, NS) were grown on SG-Ura medium. An amount of cell extracts equivalent to 40 A260 was used for affinity purification. RNA samples were prepared from cell extracts (lanes 1, 4 and 7), unbound fractions (lanes 2, 5 and 8) and purified fractions (lanes 3, 6 and 9), and analyzed by Northern blotting.
If ribosomes stall on nonstop mRNA, it is crucial to determine whether this stalled ribosome contains a nascent premature product. To address this question, we set out to isolate a FLAG-tagged nascent chain-ribosomal complex and determined whether or not nonstopHIS3 mRNA could be copurified. W303 cells harboring pIT826 (pGAL1p-FLAG-HIS3, WT) or pIT827 (pGAL1p-FLAG-his3-ns, NS) were grown on galactose medium and extracts were prepared for affinity purification of FLAG-tagged protein products. RNA samples were prepared from the purified fractions for reverse transcription–polymerase chain reaction (RT–PCR) detection of mRNA. As shown in Figure 3B, nonstopHIS3 mRNA was found to be associated with FLAG-tagged protein products at 20% of the level found for wild-type mRNA, as determined by semiquantitative RT–PCR (Figure 3B, left panel, lanes 6–8 and 10–12). We also determined the HIS3 mRNA levels in purified fractions by Northern analysis. Both wild-type and nonstopHIS3 mRNA that co-immunoprecipitated with FLAG-tagged products could be detected (Figure 3B, right panel). These results suggest that ribosomes stalled on the nonstopHIS3 mRNA contain nascent polypeptides and that the completion of protein synthesis is repressed.
To see whether Pab1p still binds to the poly(A) tail of nonstop mRNA, we examined whether Pab1p is associated with the nonstop mRNA by co-immunoprecipitation experiments. The PAB1-FLAG strain (YIT874) was transformed with the wild-type or nonstopHIS3 reporters and cell extracts were prepared for co-immunoprecipitation experiments in the absence of CHX. FLAG-tagged Pab1p was affinity purified from extracts with M2-FLAG resin and mRNAs in the fractions were analyzed by Northern blotting. The data clearly showed that in conditions in which wild-type HIS3 mRNA was specifically co-precipitated with Pab1p, essentially no nonstop mRNA was copurified (Figure 3C, lanes 3, 6 and 9). This finding strongly suggests that the first ribosome translating a nonstop mRNA dislodges Pab1p and stalls at the 3′ end of mRNA, leading to translation repression.
The ski mutations have little effect on nonstopHIS3 expression
It has been shown that ski mutations could suppress the His− phenotype of nonstopHIS3 gene (van Hoof et al, 2002). We aimed to confirm this suppression in our system because given it were the case, translational repression of nonstopHIS3 mRNA should be recovered by ski mutation as well. However, we found that none of the ski deletion mutations (i.e. ski2, ski3 and ski7) enabled nonstopHIS3 to complement the his3 mutant (Figure 4A). In these ski mutant backgrounds, there was no apparent change in the level of nonstopHIS3 mRNA (Figure 4B) and the translation product of nonstopHIS3 mRNA was undetectable (Figure 4C). These results suggest that even in the absence of ski gene products, which are thought to be involved in the NSD pathway, the translation of nonstop mRNA is still repressed. Consistent with this finding, we also noticed that the dcp1 deletion strain showed no effect on the translation of nonstopHIS3 mRNA (data not shown).
Figure 4.
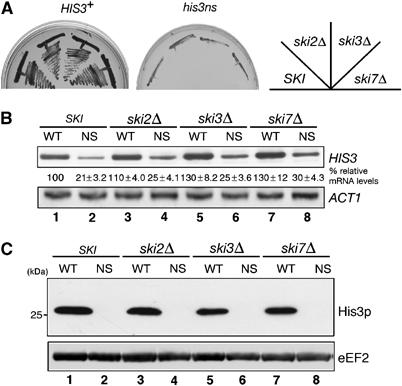
The ski mutations have minimal effect on nonstopHIS3 expression. (A) Yeast strains (WT, W303; ski2Δ, YIT888; ski3Δ, YIT890; ski7Δ, YIT929) were transformed with pIT709 (pHIS3-His6, left panel) or pIT711 (phis3-His6-ns, right panel). Transformants were streaked on SC-His plate and incubated for 3 days at 30°C. (B) Sample preparation and hybridization were performed as described for Figure 1C. Lanes 1 and 2: W303; lanes 3 and 4: YIT888 (ski2Δ); lanes 5 and 6: YIT890 (ski3Δ); lanes 7 and 8: YIT929 (ski7Δ). Cells were transformed with pIT709 (pHIS3-His6, odd lanes) or pIT711 (phis3-His6-ns, even lanes). (C) Yeast cells described in (B) were grown on SC-Leu medium. Protein separation and Western analysis were performed as described for Figure 2A.
Since these results seemed to be inconsistent with those of van Hoof et al (2002), we examined the protein and mRNA levels derived from his3-ns in the strain used in the previous study. We found that the his3-ns plasmid used in this study (pIT799) and the previous study (pAV188) could complement HIS− phenotype of BY4741 strain when ski7 is deleted (Figure 5A). Although the ski7 mutation had little effect on the level of nonstopHIS3 mRNA in BY4741 background (Figure 5B), we detected the protein products when pIT827 (pGAL1p-FLAG-his3-ns) plasmid was introduced into BY4741ski7Δ strain (Figure 5C, lane 12) but not in W303 ski7Δ strain (Figure 5C, lane 8). This increased expression of his3-ns in KO1852 (ski7Δ) strain may account for the suppression shown in Figure 5A. Nevertheless, since we could observe a significant reduction in the amount of detectable protein produced from nonstop mRNAs in two different backgrounds (Figure 5C, lanes 2 and 4), it can be argued here that translational repression of nonstopHIS3 mRNAs occurs independent of the Ski7p-dependent decay machinery.
Figure 5.
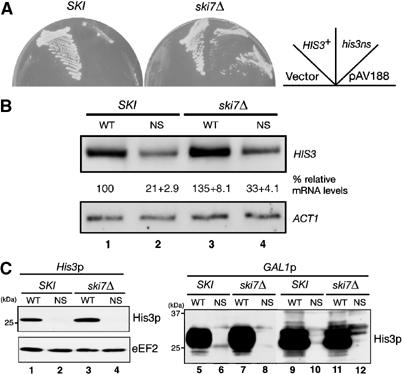
NonstopHIS3 expression in BY4741ski7 mutant. (A) Yeast strains (SKI, BY4741; ski7Δ, KO1852) were transformed with pIT798 (pFLAG-HIS3), pIT799 (pFLAG-his3-ns) or pAV188 (his3-ns). Transformants were streaked on SC-His plates and incubated for 3 days at 30°C. (B) Northern analysis. RNA samples (1 μg) were analyzed as described for Figure 1C. Yeast cells BY4741 (lanes 1 and 2) or KO1852 (ski7Δ; lanes 3 and 4) were transformed with pIT826 (pGAL1p-FLAG-HIS3, odd lanes) or pIT827 (pGAL1p-FLAG-his3-ns, even lanes). (C) Western analysis was performed with anti-FLAG antibodies. Sample preparation was performed as described for Figure 2. Left: Yeast strains (SKI, BY4741; ski7Δ, KO1852) were transformed with pIT798 (pFLAG-HIS3, odd lanes) or pIT799 (pFLAG-his3-ns, even lanes). Right: Yeast cells W303 (SKI, lanes 5–6) or YIT929 (ski7Δ, lanes 7–8), BY4741 (SKI, lanes 9–10) or KO1852 (ski7Δ, lanes 11–12) were transformed with pIT826 (pGAL1p-FLAG-HIS3, odd lanes) or pIT827 (pGAL1p-FLAG-his3-ns, even lanes).
Translation of nonstop mRNA is generally repressed
To test whether translational repression of nonstop mRNA is restricted to HIS3 or is a general property of nonstop mRNAs, we replaced the coding sequence of nonstopHIS3 with that of GFP (nonstopGFP). As shown in Figure 6A, the steady-state level of nonstopGFP mRNA was reduced to one-fourth of the wild-type GFP level and the disruption of any one of SKI genes did not reverse the level. We also found that translation products of nonstopGFP mRNA could not be detected even in the ski mutants (Figure 6B), even though nonstopGFP mRNAs existed in polysome fractions (Figure 6C). These properties of the nonstopGFP are consistent with and corroborate the findings of the nonstopHIS3 mRNA. These results led us to conclude that translational repression is a general phenomenon of nonstop mRNAs and that ribosomal stalling at the 3′ end of nonstop mRNAs is the reason for repressing protein synthesis.
Figure 6.
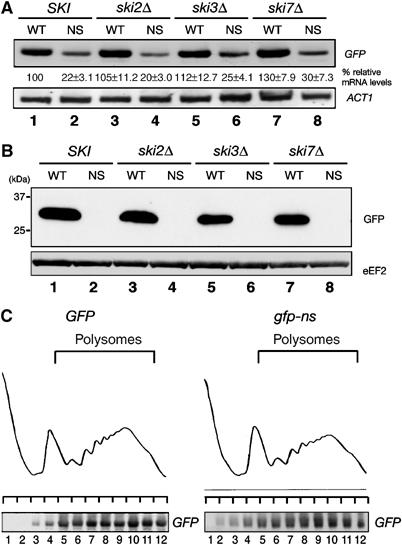
Translation of nonstop mRNA is generally repressed. (A) Yeast cells were grown on SC-Leu medium, and HIS3 mRNAs were detected by Northern blotting with DIG-labeled GFP probe. Lanes 1 and 2: W303; lanes 3 and 4: YIT888 (ski2Δ); lanes 5 and 6: YIT890 (ski3Δ); lanes 7 and 8: YIT929 (ski7Δ). Cells were transformed with pIT810 (pGFP, odd lanes) or pIT811 (pgfp-ns, even lanes). (B) Yeast cells as described in (A) were grown on SC-Leu medium, and proteins from cell extracts equivalent to 0.1 OD600 were subjected to 12% SDS–PAGE and analyzed by immunoblot analysis with anti-GFP antibodies. (C) W303 cells harboring pIT810 (pGFP) or pIT811 (pgfp-ns) were grown on SC-Leu medium and cell extracts were prepared after the addition of 0.1 mg/ml CHX. Cell extracts equivalent to 40 A260 were resolved by velocity sedimentation on 10–50% sucrose gradients. RNA samples prepared from the indicated fractions were analyzed by Northern blotting with DIG-labeled GFP probe.
The 5′ → 3′ decay pathway is accelerated for nonstop mRNA decay in the absence of Ski7p
We found that the steady-state level of nonstop mRNA was reduced four-fold and ski mutations had only slight effect on the level of nonstop mRNA. This suggests that the Ski7p-independent decay pathway is involved in the rapid degradation of nonstop mRNA. To determine the stability of nonstopHIS3 mRNA, we directly measured its decay rate. In this experiment, we monitored the loss of mRNA over time after blocking synthesis of a new transcript by adding glucose in strain harboring GAL1p-HIS3 or GAL1p-nonstopHIS3 plasmid. As shown in Figure 7A, the half-life of wild-type HIS3 mRNA was about 4.5 min, while that of nonstopHIS3 mRNA was less than 1 min. To evaluate the contribution of 3′ → 5′ and 5′ → 3′ degradation pathways to the rapid decay of nonstopHIS3 mRNA, we used mutants carrying dcp1-2 temperature-sensitive (ts) and/or ski7Δ mutation. It is known that decapping activity is severely blocked in the dcp1-2 (ts) mutant at restricted temperature (37°C). We found that the ski7Δ mutation moderately stabilized nonstopHIS3 mRNA (t1/2=1.2 min), while dcp1-2 did not affect the decay rate (t1/2<1 min). These indicate that Ski7p plays an important role in NSD when 5′ → 3′ decay pathway is inactivated. We also found that nonstopHIS3 mRNA was more rapidly degraded than HIS3 mRNA (t1/2=5.2 min) in a ski7Δ mutant. Although the decay rate of nonstopHIS3 mRNA was slightly slowed down by the ski mutation, it was still much faster than wild-type HIS3 mRNA, leading to a little, if any, increase in the bulk transcripts (see Figure 4B). On the other hand, both wild-type and nonstop mRNAs were dramatically stabilized in the dcp1-2 ski7Δ double mutant at restricted temperature (t1/2>16 min). These indicate that nonstopHIS3 mRNA is rapidly degraded by either of the two degradation pathways, Ski7p-dependent 3′ → 5′ decay pathway or decapping-dependent 5′ → 3′ decay pathway.
Figure 7.
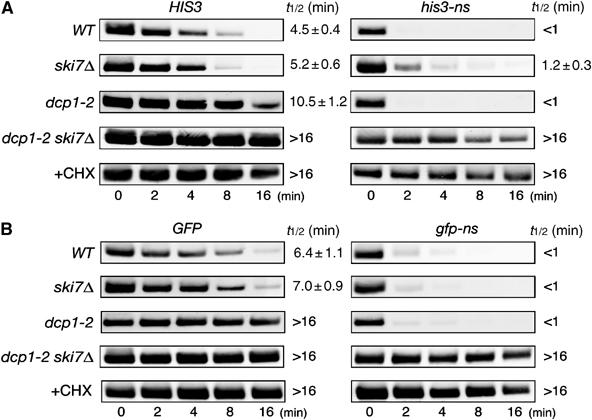
The 5′ → 3′ decay pathway is accelerated for NSD in the absence of Ski7p. (A) Yeast strains (WT, W303; ski7Δ, YS002; dcp1-2, YS088; dcp1-2 ski7Δ, YS091) were transformed with pIT765 (pGAL1p-HIS3-His6, HIS3) or pIT766 (pGAL1p-his3-His6-ns, nonstopHIS3). Cells were grown in SG-Ura. At the beginning of the experiment, glucose was added to inhibit transcription from the GAL1 promoter and samples were harvested at the indicated times. For analysis of mRNA decay in the YS088 (dcp1-2) or YS091 (dcp1-2 ski7Δ) mutant, transcription from the GAL1 promoter was inhibited after incubation at 37°C for 90 min. RNA samples subjected to agarose gel electrophoresis were analyzed by Northern blotting with DIG-labeled HIS3 probe. Where indicated, CHX was added to inhibit translation elongation. The half-lives (t1/2; min) are shown as the mean values±standard deviations (s.d.), which are obtained from at least three independent experiments. (B) Yeast strains (WT, W303; ski7Δ, YS002; dcp1-2, YS088; dcp1-2 ski7Δ, YS091) were transformed with pIT859 (pGAL1p-GFP, GFP) or pIT860 (pGAL1p-gfp-ns, nonstopGFP). Transcriptional repression and hybridization were performed as described for (A), but DIG-labeled GFP probe was used.
These properties of nonstopHIS3 mRNA in ski7 and dcp1-2 strains were essentially confirmed with a nonstopGFP mRNA as shown in Figure 7B. The nonstopGFP mRNA was more rapidly degraded (t1/2<1 min) than GFP mRNA (t1/2=6.4 min) in the wild-type background. Neither the ski7Δ nor dcp1-2 mutations affected the rapid decay of nonstopGFP mRNA, whereas nonstopGFP mRNA was dramatically stabilized in a dcp1-2 ski7Δ double mutant at restricted temperature. Therefore, we propose that nonstop mRNA can be rapidly degraded by either of the two degradation pathways, Ski7p-dependent 3′ → 5′ decay pathway or decapping-dependent 5′ → 3′ decay pathway. CHX treatment drastically stabilized nonstop mRNAs (Figure 7), indicating that inhibition of translation elongation is required to block rapid degradation of nonstop mRNAs as shown previously (van Hoof et al, 2002).
Stable complexes formation by ribosome and nonstop mRNA in vivo
Since we found that nonstop mRNAs were dramatically stabilized in dcp1-2 ski7Δ double mutant at restricted temperature, we asked whether or not ribosomes translating nonstop mRNA are dissociated from nonstop mRNAs when mRNA degradation was severely inhibited. To see this, we first examined the distribution of nonstop mRNA in polyribosome fractions using extracts of dcp1-2 ski7Δ double mutant expressing HIS3 or nonstopHIS3 mRNA. YS091 (dcp1-2 ski7Δ) cells harboring pIT765 (pGAL1p-HIS3-His6) or pIT766 (pGAL1p-his3-His6-ns) were grown on galactose medium at permissive temperature. After incubation at restrictive temperature (37°C) for 90 min, CHX was added. Cell extracts were resolved by velocity sedimentation on 10–50% sucrose gradients and mRNA distribution of nonstop mRNAs was analyzed by Northern blotting. As shown in Figure 8A, both wild-type HIS3 and nonstopHIS3 mRNAs were distributed in polysome fractions. In addition, we found that nonstopHIS3 mRNAs were distributed in slightly but significantly heavier polysome fractions (Figure 8A, top-right panel) in comparison with HIS3 mRNAs (Figure 8A, top-left panel). We obtained essentially the same results when we prepared extracts in the absence of CHX (Figure 8A, bottom panels). These results suggest that the dissociation of ribosomes from nonstopHIS3 mRNA is defective or much slower than that of HIS3 mRNA, and ribosomes translating nonstop mRNA may form stable and dense complexes with mRNA. We also examined the expression of nonstopHIS3 by pulse labeling with [35S]methionine followed by immunoprecipitation (Figure 8B), and found that the products of nonstop mRNA were still hardly detectable in the condition that products of wild-type HIS3 were detected (Figure 8B, lanes 3 and 4). This suggests that translation of nonstop mRNA is still repressed even when two major decay pathways are inactivated.
Figure 8.
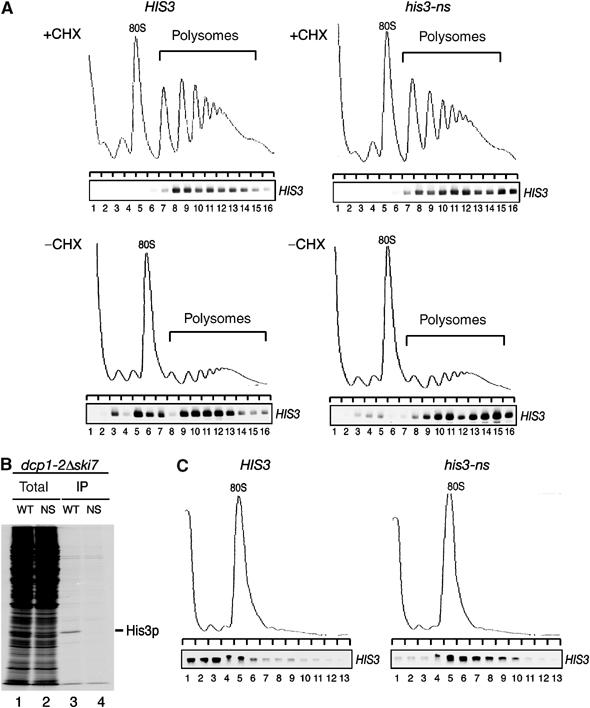
Ribosomes form stable complexes with nonstop mRNA. (A) YS091 (dcp1-2 ski7Δ) cells harboring pIT765 (pGAL1p-HIS3-His6, HIS3, left panels) or pIT766 (pGAL1p-his3-His6-ns, nonstopHIS3, right panels) were grown on SG-UraMet at 25°C. After incubation at 37°C for 90 min, cells were harvested in the presence (top panels) or absence (bottom panels) of CHX. Cell extracts were resolved by velocity sedimentation on 10–50% sucrose gradients. RNA samples prepared from the indicated fractions were analyzed as shown in Figure 3A. (B) YS091 (dcp1-2 ski7Δ) cells harboring pIT826 (pGAL1p-FLAG-HIS3, WT) or pIT827 (pGAL1p-FLAG-his3-ns, NS) were grown on SG-Ura medium at 25°C. After incubation at 37°C for 90 min, cells were labeled with [35S]methionine for 10 min followed by immunoprecipitation. Total cell extracts (lanes 1 and 2) and immunoprecipitated samples (lanes 3 and 4) were subjected to SDS–PAGE and visualized by autoradiography. (C) W303 cells harboring pIT922 (pGPDp-FLAG-HIS3, left panel) or pIT923 (pGPDp-FLAG-his3-ns, right panel) were grown on SC-Ura medium at 30°C. After incubation in medium without glucose for 10 min, cells were harvested. Sample preparation and hybridization were performed as described in Figure 3A.
We next examined the distribution of nonstop mRNA in polyribosome fractions after glucose depletion, such that translation initiation is inhibited, while ribosomes can be released from mRNAs. W303 cells harboring pIT922 (pGPDp-FLAG-HIS3) or pIT923 (pGPDp-FLAG-his3-ns) were grown on SC-Ura medium and then incubated for an additional 10 min in medium without glucose. We harvested cells in the absence of CHX and prepared extracts. Under these conditions, depletion of glucose results in the inhibition of translation initiation but not elongation and termination (Ashe et al, 2000). The distribution of wild-type or nonstop mRNAs was analyzed and as expected, polysomes could not be recovered in these conditions while mRNA for wild-type HIS3 was distributed in the ribosome-free fractions (Figure 8C, left panel). In contrast, nonstopHIS3 mRNAs were distributed mainly in polysome fractions (Figure 8C, right panel) and the distribution was similar to that in normal conditions (see Figure 3A, middle panel). This indicates that ribosomes translating nonstop mRNA stall at the 3′ end of mRNA and form stable complexes with nonstop mRNA even after sufficient time for ribosomes translating wild-type mRNA to be released. Therefore, we conclude that ribosomes translating nonstop mRNA are significantly inhibited in either the elongation and/or the termination steps of protein synthesis.
Translation of aberrant mRNAs with a shortened 3′-UTR is also repressed
Based on the results shown above, we conclude that translation of nonstop mRNA is repressed after initiation. One possibility is that an intact 3′-UTR is required for efficient translation such that aberrant mRNAs with a shortened 3′-UTR are not translated. To address this possibility, we constructed his3 mutant plasmids in which a translation termination codon is placed at several different positions within the 3′-UTR as shown in Figure 9A. We found that the mutations did not affect the HIS3 mRNA levels (Figure 9B, left panel, lanes 1–5). Consistently, the mutations did not affect the stability of those mRNAs in wild-type cells (Figure 9B, right panel) or ski7Δ mutant cells (data not shown). This indicates that translation through the 3′-UTR region of HIS3 gene did not destabilize the mRNA. Western blot analysis revealed that when a translation termination codon is located less than 37 nt downstream from an authentic termination codon (his3-S1, his3-S2), protein levels were almost the same as that of wild-type HIS3. However, when a termination codon is just upstream of the poly(A) addition sites (his3-S3, his3-S4), protein products were hardly detectable (Figure 9C, lanes 4 and 5). This result is consistent with the complementation test of his3 mutant with those mutant plasmids (Figure 9A, right panel). Polysome analysis revealed that both his3-S2 mRNA and his3-S3 mRNA were distributed in polysome fractions (Figure 9D). These results strongly suggest that translation of his3-S3 and his3-S4 is repressed after initiation. We propose that shortened or no spatial distance between the termination codon and the 3′ end of mRNA results in translation repression.
Figure 9.
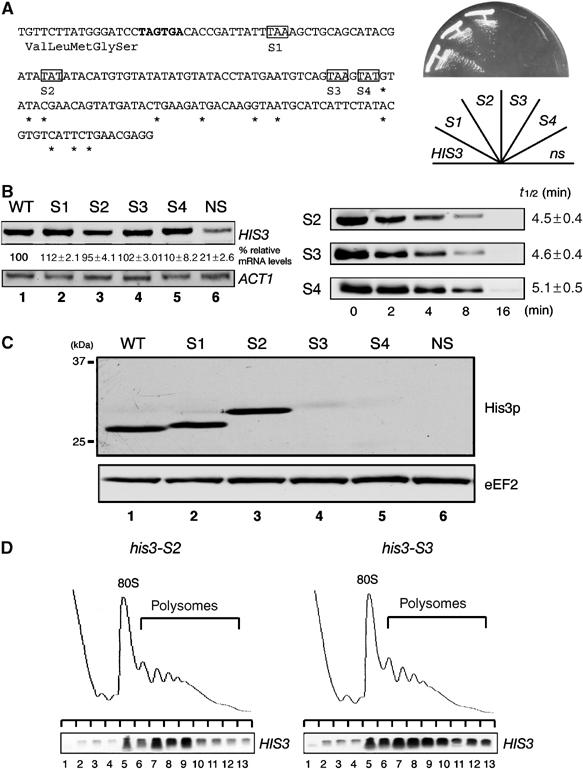
Translation of aberrant mRNA with a shortened 3′-UTR is also repressed. (A) Left: Schematic drawing of HIS3 reporter genes with a termination codon in the 3′-UTR. DNA sequences of 3′-UTR region are shown and asterisks represent the poly(A) addition sites (Mahadevan et al, 1997). An authentic translation termination codon of HIS3 gene is indicated in bold letters, and the shaded boxes indicate termination codons for each HIS3 mutant. Right: The complementation test. W303 cells were transformed with mutant plasmids or pIT798 (WT) or pIT799 (NS), and transformants were streaked on SC-His plate and incubated for 2 days at 30°C. (B) Left: W303 cells harboring pIT938 (pFLAG-his3-S1), pIT939 (pFLAG-his3-S2), pIT940 (pFLAG-his3-S3) or pIT941 (pFLAG-his3-S4) were grown on SC-Leu medium. Sample preparation and hybridization were performed as described for Figure 1C. Right: W303 cells were transformed with pIT927 (pGAL1p-FLAG-his3-S2, S2), pIT928 (pGAL1p-FLAG-his3-S3, S3) or pIT929 (pGAL1p-FLAG-his3-S4, S4). Cells were grown in SG-Ura. Sample preparation and hybridization were performed as described for Figure 7A. (C) Yeast cells as indicated in (B) were grown on SC-Leu medium. Sample preparation and Western blot analysis were performed as described for Figure 5C. (D) W303 cells harboring pIT927 (pGAL1p-FLAG-his3-S2, left panel) or pIT928 (pGAL1p-FLAG-his3-S3, right panel) were grown on SG-Ura medium. Cell extracts were prepared in the absence of CHX. Sample preparation and hybridization were performed as described for Figure 3A.
Discussion
In this study, we have clarified the fate of ribosomes translating nonstop mRNAs. First, translation product of nonstopHIS3 mRNA was not detectable in cell extracts while the level of mRNA was reduced only to one-fourth of the wild-type level (Figures 1 and 2). Translational repression was also confirmed for nonstopGFP mRNA (Figure 6). Second, although nonstopHIS3 products were not detectable in extracts, a nascent peptide synthesized from nonstopHIS3 mRNA was found in a complex pulled down with the nonstopHIS3 mRNA (Figure 3B). Third, essentially no nonstop mRNAs could be copurified with Pab1p in conditions in which wild-type HIS3 mRNA was specifically copurified (Figure 3C). Fourth, nonstop mRNAs existed in polysome fractions (Figures 3A, 6C and 8A) while ribosomes translating nonstop mRNA form stable and heavy polysome complexes when mRNA degradation is inhibited in a dcp1-2 ski7Δ double mutant (Figure 8A). Finally, ribosomes translating nonstop mRNAs were still associated with the mRNA in the same conditions in which ribosomes translating wild-type mRNA completed translation and dissociated from mRNA (Figure 8C). Together, these results indicate that a ribosome stalled at the 3′ end of a nonstop mRNA forms a stable complex with the nascent peptidyl-tRNA, leading to the repression of translation by subsequent ribosomes. Since the disruption of any one of ski complex genes had little effect on repressing translation of nonstop mRNAs in two different genetic backgrounds (Figures 4 and 5), we concluded that translation of nonstop mRNA is repressed even in the absence of Ski7 components.
We observed translation repression of aberrant mRNA with a shortened 3′-UTR, and the condition of ribosomes translating a shortened 3′-UTR mRNA is almost the same as that of ribosomes translating nonstop mRNA (Figure 9D). These results suggest that no translation termination codon or the short distance between the termination codon and the 3′ end of mRNA results in inefficient ribosome release from mRNA. This is consistent with the proposed model suggesting that proper translation termination requires proper spatial relationship between the termination codon and the 3′-UTR (Amrani et al, 2004). We speculate that translation termination of mRNA with a shortened 3′-UTR might be repressed due to an improper spatial distance between Pab1p bound to poly(A) and the eRF1–eRF3 complex. When improper translation termination occurs at a premature translation termination codon, Upf1 is required for ribosomal reverse scanning and reinitiation of translation (Amrani et al, 2004). It would be interesting to investigate the involvement of mRNA surveillance factors in the repression of translation of aberrant mRNAs with no or a shortened 3′-UTR.
It has been suggested by the genetic complementation experiments that the degradation of nonstop mRNA is effective in limiting the production of aberrant protein (van Hoof et al, 2002). Unlike the W303 background, we did observe partial complementation of a his3− mutant by nonstopHIS3 mRNA in the BY4741 background. The reasons for this discrepancy are unclear, but differences in strain background may have permitted limited growth by very limited translation of nonstop mRNA products. In the BY4741 background, we expectedly detected nonstopHIS3 mRNA when ski7 was deleted but importantly observed a strong reduction of its corresponding protein level such that only a minimal amount of protein product was detected upon overexpression of the nonstopHIS3 mRNA.
Our results indicate that nonstop mRNAs lacking a termination codon were efficiently degraded by Ski7p-dependent 3′ → 5′ decay pathway as well as decapping-dependent 5′ → 3′ decay pathway (Figure 7). PGK1 mRNA lacking a termination codon has been shown to be degraded mainly by a 3′ → 5′ decay pathway (van Hoof et al, 2002). It has been shown that PGK1 mRNA is stable because the sequence context of the start codon for PGK1 translation and the coding region function together to stabilize the transcript (LaGrandeur and Parker, 1999). We found that two different nonstop mRNAs, HIS3 and GFP, were degraded in a similar manner. Therefore, we suggest that nonstop mRNAs are generally degraded by Ski7p-dependent 3′ → 5′ decay pathway and the 5′ → 3′ decay pathway. It has been suggested that deadenylation promotes mRNA decapping by the loss of Pab1p (Caponigro and Parker, 1995; Coller and Parker, 2004). Consistently, we found that nonstop mRNA was not associated with Pab1p (Figure 3C). We also found that translation through 3′-UTR did not destabilize mRNA (Figure 9B and C). Therefore, it is likely that the accelerated 5′ → 3′ degradation in NSD is mainly due to the dissociation of Pab1p from nonstop mRNA.
Based on the results shown in this study, we propose a model for the translation status and decay of nonstop mRNA in yeast (Figure 10). The first ribosome translating nonstop mRNA dislodges Pab1p from poly(A) tail. This ribosome stalls at the 3′ end of mRNA and prevents multiround translation by blocking the completion of translation by subsequent ribosomes (Figure 10A). The stable complex formed by the ribosome and nonstop mRNA might be recognized by Ski7p and degraded by the Ski complex–exosome-dependent 3′ → 5′ degradation pathway (Figure 10B). In addition, the removal of Pab1p from the 3′ end of mRNA results in an accelerated 5′ → 3′ decay pathway (Figure 10C). The avoidance of potentially deleterious products is biologically important, and abnormal protein synthesis is eliminated by three quality assurance mechanisms involving translational repression and mRNA degradation by two decay pathways.
Figure 10.
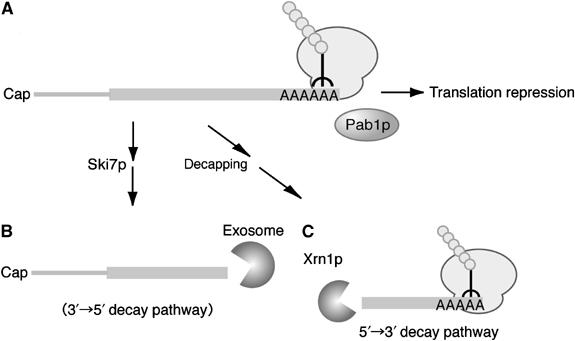
A model for translation repression and degradation pathways of nonstop mRNA in yeast. The first ribosome translating nonstop mRNA dislodges Pab1p from the poly(A) tail and the stalled ribosome at the 3′ end of mRNA represses multiround translation by blocking the completion of translation by subsequent ribosomes (A). The stable complex formed by ribosome and nonstop mRNA might be recognized by Ski7p and degraded by the Ski complex–exosome-dependent 3′ → 5′ degradation pathway (B). The removal of Pab1p from the 3′ end of mRNA results in accelerated 5′ → 3′ decay (C).
In prokaryotes, the tmRNA rescue system is responsible for the quality control by eliminating both aberrant mRNA and its translated peptides (Keiler et al, 1996; Yamamoto et al, 2003). In eukaryotes, no RNA having an equivalent function to tmRNA has been identified. Based on the results of this study, we propose that a stalled ribosome at the 3′ end of mRNA inhibits multiround translation of nonstop mRNA by blocking the movement of subsequent ribosomes. These indicate that the production of abnormal proteins derived from nonstop mRNA might be repressed in prokaryotes and eukaryotes by different mechanisms. It has been shown that a premature nonsense codon leads to a decrease in translation efficiency and blocking mRNA decay can eliminate this repression (Muhlrad and Parker, 1999b). Therefore, we propose that translational repression of aberrant mRNAs and mRNA quality control are mutually important mechanisms to avoid the production of potentially deleterious products in eukaryotes.
Materials and methods
Strains and general methods
Escherichia coli DH5α was used for DNA manipulations. The yeast strains used in this study are described in Supplementary Table 1. Standard procedures were followed for yeast manipulations (Kaiser et al, 1994). The media used in this study included rich medium and synthetic complete medium (SC) containing 2% glucose. SG was identical to SC except that it contained 2% galactose instead of 2% glucose. Recombinant DNA procedures were carried out as described previously (Sambrook et al, 1989). RNA preparation and Western blotting were performed as described.
Plasmids
Plasmids used in this study are described in Supplementary Table 1. A HIS3 XbaI–BamHI fragment reading from its promoter through its entire ORF except the termination codon was amplified by PCR using the primers 5′-GCTCTAGAGTCACTGCCAGGTATCGTT-3′ and 5′-CGGGATCCCATAAGAACACCTTTGGTGGAGG- 3′ and inserted into YCplac111 vector to generate pIT701. BamHI–EcoRI fragments containing the 3′-UTR region of wild-type HIS3 were amplified by PCR using primers 5′-CGGGATCCTAGTGACACCGATTATTTAAAGCT GC-3′ and 5′-GGAATTCCTCGTTCAGAATGACACGTA-3′ while that of nonstopHIS3 was amplified by PCR using the 5′ primer 5′-CGGGATCCAGTGACACCGATTATTTAAAGCTG C-3′ and the 3′ primer 5′-GGAATTCCTCGTTCAGAATGACACGTA-3′. These BamHI–EcoRI fragments were inserted into the corresponding sites of pIT701 to generate pIT702 (HIS3) and pIT703 (nonstopHIS3). To insert hexahistidine-tag sequence, two oligonucleotides (5′-GATCCCATCACCATCACCATCACG-3′ and 5′-GATCCGTGATGGTGATGGTGATGG-3′) were annealed and ligated to BamHI-digested plasmids to generate pIT709 and pIT711. For FLAG-tagged version of HIS3 reporter genes, the SphI–XbaI HIS3 promoter fragments were amplified by PCR using 5′ primer 5′-ACATGCATGCATGTCACTGCCAGGTATCGTTT GAACAC-3′ and 3′ primer 5′-GCTCTAGAGCTTTGCCTTCGTTTATCTTGCCT G-3′ and inserted into YCplac111. Then, XbaI–BamHI fragments containing FLAG-tagged HIS3 ORFs were amplified by PCR using 5′ primer 5′-GCTCTAGATGGACTACAAGGACGACGATGACA AGACAGAGCAGAAAGCCCTAG-3′ and 3′ primer 5′-CGGGATCCCATAAGAACACCTTTGGTGGAGG- 3′ and inserted into the corresponding sites. Finally, the BamHI–EcoRI fragment of pIT702 (HIS3) or/and pIT703 (nonstopHIS3) was inserted to generate pIT798 (FLAG-HIS3) or pIT799 (FLAG-nonstopHIS3). To control HIS3 expression under the GAL1 promoter, pIT765 or pIT766 was generated as follows. XbaI–EcoRI fragments containing the HIS3 ORF of pIT709 or pIT711 were amplified by PCR using 5′ primer 5′-GCTCTAGATGACAGAGCAGAAAGCCCTAG-3′ and 3′ primer 5′-GGAATTCCTCGTTCAGAATGACACGTA-3′. The fragments were inserted into corresponding site of p415 (Mumberg et al, 1994), then the SacI–EcoRI fragments were inserted into YCplac33 to generate pIT765 or pIT766. We replaced the XbaI–EcoRI fragment of pIT765 or pIT766 with that of pIT798 or pIT799 to construct pIT826 (GAL1p-FLAG-HIS3) and pIT827 (GAL1p-FLAG-his3-ns). We replaced the HIS3 ORF of pIT798 and pIT799 with GFP fragment to generate pIT810 and pIT811. XbaI–BamHI fragment containing GFPuv4 ORF was amplified by PCR using 5′ primer 5′-GCTCTAGAGGCCTATGCGGCCGCAGTAAAGGA G-3′, 3′ primer 5′-CGGGATCCTTTGTATAGTTCATCCATGCC-3′ and pGFPgcn4 template. We also replaced HIS3 ORF of pIT765 and pIT766 with GFP fragment to generate pIT859 and pIT860. DNA sequences of all PCR-amplified fragments were confirmed. To construct pIT922 and pIT923, SacI–XbaI fragments of pIT826 and pIT827 were replaced with the fragment containing GPD promoter (Mumberg et al, 1995). For pIT939 and pIT941 plasmids construction, termination codons were introduced into pIT799 plasmid by specific oligonucleotides. For pIT938 plasmid, two nucleotides of the authentic termination codon were deleted by PCR using 5′ primer 5′-CGGGATCCGTGACACCGATTATTTAAAGCTGC -3′ and 3′ primer 5′-GGAATTCCTCGTTCAGAATGACACGTA-3′. For pIT940, two tandem termination codons were deleted by PCR using 5′ primer 5′-CGGGATCCCACCGATTATTTAAAGCTGCAGCA TACG-3′ and 3′ primer 5′-GGAATTCCTCGTTCAGAATGACACGTA-3′. To construct pIT927, pIT928 and pIT929, SacI–XbaI fragments of pIT939, pIT940 and pIT941 were replaced with the fragment containing GAL1p promoter.
Strain construction
The deletions of SKI2, SKI3 and SKI7 were constructed by the PCR-based gene deletion method (Baudin et al, 1993). A FLAGHA-TRP1 3′-UTR transformation module was amplified by PCR and the resultant transformation module was used to transform a haploid strain by selection on TRP plates. The resultant transformants were verified by colony-PCR to confirm that replacement had occurred at the expected locus.
Northern blotting
Total RNAs were resolved by 1.2% agarose gel electrophoresis in the presence of formaldehyde and blotted onto Hybond-N+ membrane (Amersham Pharmacia Biotech Inc., Piscataway) as described. The mRNAs were visualized using digoxigenin (DIG) reagents and kits for nonradioactive nucleic acid labeling by PCR and detection system (Roche) according to the procedure specified by the manufacturer. DIG probes were prepared with the following oligonucleotides: HIS3 (5′-GCTCTAGATGACAGAGCAGAAAGCCCTAG-3′ and 5′-CGGGATCCCATAAGAACACCTTTGGTGGAGG- 3′); ACT1 (5′-TCCCAAGATCGAAAATTTACTG-3′ and 5′-AACATACGCGCACAAAAGCA-3′); GFP (5′-GCTCTAGAGGCCTATGCGGCCGCAGTAAAGGA G-3′ and 5′-CGGGATCCTTTGTATAGTTCATCCATGCC-3′). The intensity of bands was quantified by LAS1000 (Fuji Film, Japan).
RT–PCR analysis
The respective amounts of HIS3 mRNAs in yeast cells were measured by RT–PCR. After affinity purification of FLAG-tagged protein products, RNA samples prepared from purified fractions were treated with DNase, and used for RT. The products obtained were diluted and subjected to 25 cycles of PCR with HIS3- or ACT1-specific oligonucleotides used for the preparation of DIG probe. In all, 10% of the amplified products were run in agarose gels.
Pulse labeling and immunoprecipitation
Yeast cells were grown exponentially at 30°C in minimal media lacking methionine. Cultures (10 ml) were incubated with 100 μCi [35S]methionine (Amersham Pharmacia Biotech Inc., Piscataway), and cell extracts were prepared with lysis buffer (20 mM Tris–HCl pH 7.4, 100 mM KCl, 2 mM MgCl2, 2 mM DTT, 0.5 mM PMSF) by vortexing with beads. Cell extracts were incubated with anti-FLAG M2 resin in IXA-100 buffer (50 mM Tris–HCl pH 7.5, 100 mM KCl, 12 mM Mg(OAc)2, 1 mM DTT, 1 mM PMSF), and then washed three times and eluted with 0.4 mg/ml FLAG peptide.
Yeast extract and sucrose gradient separation
Yeast cells were grown exponentially at 30°C and harvested by centrifugation. Cells were washed once with lysis buffer and extracts were prepared as described previously (Inada et al, 2002). The equivalent of 50 A260 units were then layered onto linear 10–50% sucrose density gradients. Sucrose gradients (10–50% sucrose in 10 mM Tris-acetate pH 7.4, 70 mM ammonium acetate, 4 mM magnesium acetate) were prepared in 25 × 89 mm polyallomer tubes (Beckman Coulter) by gradient master. Crude extracts were layered on top of the sucrose gradients and centrifuged at 27 000 r.p.m. in a P28S rotor (Hitachi Koki) for 3 h at 4°C. Gradients were then fractionated (TOWA lab, Tsukuba). Polysome profiles were generated by continuous absorbance measurement at 254 nm using a single path UV-1 optical unit (ATTO Biomini UV-monitor) connected to a chart recorder (ATTO, digital mini-recorder). Where indicated, equal volume fractions were collected and processed for total RNA purification as described above.
Supplementary Material
Supplementary Table 1
Acknowledgments
We thank Dr Yoshikazu Nakamura and Dr Roy Parker for helpful discussion and critical reading of the manuscript. We also thank Dr Ambro van Hoof, Dr Koichi Ito, Dr Shin-ichi Hoshino and Dr Yasuhiro Araki for strains and plasmids. We also thank Mr Colin Crist for critical reading of the manuscript and valuable comments. This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
References
- Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A (2004) A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 432: 112–118 [DOI] [PubMed] [Google Scholar]
- Ashe MP, De Long SK, Sachs AB (2000) Glucose depletion rapidly inhibits translation initiation in yeast. Mol Biol Cell 11: 833–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C (1993) A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res 21: 3329–3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caponigro G, Parker R (1995) Multiple functions for the poly(A)-binding protein in mRNA decapping and deadenylation in yeast. Genes Dev 9: 2421–2432 [DOI] [PubMed] [Google Scholar]
- Coller J, Parker R (2004) Eukaryotic mRNA decapping. Annu Rev Biochem 73: 861–890 [DOI] [PubMed] [Google Scholar]
- Frischmeyer PA, van Hoof A, O'Donnell K, Guerrerio AL, Parker R, Dietz HC (2002) An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science 295: 2258–2261 [DOI] [PubMed] [Google Scholar]
- Gehring NH, Neu-Yilik G, Schell T, Hentze MW, Kulozik AE (2003) Y14 and hUpf3b form an NMD-activating complex. Mol Cell 11: 939–949 [DOI] [PubMed] [Google Scholar]
- Graham SV (2003) Nonsense-mediated decay breaks the circle? Biochem J 373: e5–e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himeno H, Sato M, Tadaki T, Fukushima M, Ushida C, Muto A (1997) In vitro trans translation mediated by alanine-charged 10Sa RNA. J Mol Biol 268: 803–808 [DOI] [PubMed] [Google Scholar]
- Inada T, Winstall E, Tarun SZ Jr, Yates JR III, Schieltz D, Sachs AB (2002) One-step affinity purification of the yeast ribosome and its associated proteins and mRNAs. RNA 8: 948–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigaki Y, Li X, Serin G, Maquat LE (2001) Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell 106: 607–617 [DOI] [PubMed] [Google Scholar]
- Kaiser CA, Adams A, Gottschling DE (1994) Methods in Yeast Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Karzai AW, Roche ED, Sauer RT (2000) The SsrA–SmpB system for protein tagging, directed degradation and ribosome rescue. Nat Struct Biol 7: 449–455 [DOI] [PubMed] [Google Scholar]
- Keiler KC, Waller PR, Sauer RT (1996) Role of a peptide tagging system in degradation of proteins synthesized from damaged messenger RNA. Science 271: 990–993 [DOI] [PubMed] [Google Scholar]
- Kim VN, Kataoka N, Dreyfuss G (2001) Role of the nonsense-mediated decay factor hUpf3 in the splicing-dependent exon–exon junction complex. Science 293: 1832–1836 [DOI] [PubMed] [Google Scholar]
- LaGrandeur T, Parker R (1999) The cis acting sequences responsible for the differential decay of the unstable MFA2 and stable PGK1 transcripts in yeast include the context of the translational start codon. RNA 5: 420–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune F, Ishigaki Y, Li X, Maquat LE (2002) The exon junction complex is detected on CBP80-bound but not eIF4E-bound mRNA in mammalian cells: dynamics of mRNP remodeling. EMBO J 21: 3536–3545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune F, Li X, Maquat LE (2003) Nonsense-mediated mRNA decay in mammalian cells involves decapping, deadenylating, and exonucleolytic activities. Mol Cell 12: 675–687 [DOI] [PubMed] [Google Scholar]
- Mahadevan S, Raghunand TR, Panicker S, Struhl K (1997) Characterisation of 3′ end formation of the yeast HIS3 mRNA. Gene 190: 69–76 [DOI] [PubMed] [Google Scholar]
- Muhlrad D, Parker R (1999a) Aberrant mRNAs with extended 3′ UTRs are substrates for rapid degradation by mRNA surveillance. RNA 5: 1299–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlrad D, Parker R (1999b) Recognition of yeast mRNAs as ‘nonsense containing' leads to both inhibition of mRNA translation and mRNA degradation: implications for the control of mRNA decapping. Mol Biol Cell 10: 3971–3978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M (1994) Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res 22: 5767–5768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156: 119–122 [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning. A Laboratory Manual, 2nd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press [Google Scholar]
- Singh G, Lykke-Andersen J (2003) New insights into the formation of active nonsense-mediated decay complexes. Trends Biochem Sci 28: 464–466 [DOI] [PubMed] [Google Scholar]
- Takahashi S, Araki Y, Sakuno T, Katada T (2003) Interaction between Ski7p and Upf1p is required for nonsense-mediated 3′-to-5′ mRNA decay in yeast. EMBO J 22: 3951–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner GC, Varshavsky A (2000) Detecting and measuring cotranslational protein degradation in vivo. Science 289: 2117–2120 [DOI] [PubMed] [Google Scholar]
- van Hoof A, Frischmeyer PA, Dietz HC, Parker R (2002) Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science 295: 2262–2264 [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Sunohara T, Jojima K, Inada T, Aiba H (2003) SsrA-mediated trans-translation plays a role in mRNA quality control by facilitating degradation of truncated mRNAs. RNA 9: 408–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1