

Abstract
We report the crystal structure of a soluble form of human urokinase-type plasminogen activator receptor (uPAR/CD87), which is expressed at the invasive areas of the tumor-stromal microenvironment in many human cancers. The structure was solved at 2.7 Å in association with a competitive peptide inhibitor of the urokinase-type plasminogen activator (uPA)–uPAR interaction. uPAR is composed of three consecutive three-finger domains organized in an almost circular manner, which generates both a deep internal cavity where the peptide binds in a helical conformation, and a large external surface. This knowledge combined with the discovery of a convergent binding motif shared by the antagonist peptide and uPA allowed us to build a model of the human uPA–uPAR complex. This model reveals that the receptor-binding module of uPA engages the uPAR central cavity, thus leaving the external receptor surface accessible for other protein interactions (vitronectin and integrins). By this unique structural assembly, uPAR can orchestrate the fine interplay with the partners that are required to guide uPA-focalized proteolysis on the cell surface and control cell adhesion and migration.
Keywords: cancer, structure, three-finger fold, tissue remodelling, uPAR
Introduction
The involvement of urokinase-type plasminogen activator receptor (uPAR; CD87) in the pathology of human cancers is well documented (Stephens et al, 1999; Ploug et al, 2001; Rosenberg, 2001; Blasi and Carmeliet, 2002; Ploug, 2003; Rømer et al, 2004). In particular, high levels of uPAR in tumour tissues and plasma from patients with various human cancers are associated with poor prognosis and increased risk of tumour recurrence and metastasis (Heiss et al, 1995; Pappot et al, 1997; Stephens et al, 1999; Mustjoki et al, 2000; Rømer et al, 2004). Also, synthetic peptide inhibitors that compete with the urokinase-type plasminogen activator (uPA)–uPAR interaction impair dissemination of aggressive human carcinoma cell lines (Ploug et al, 2001; Sato et al, 2002).
The biochemical properties of uPAR have been extensively characterized (Ploug, 2003). Human uPAR is a glycolipid-anchored (Ploug et al, 1991) modular protein having a single-chain polypeptide (283 amino acids) organized into three extracellular domains (Roldan et al, 1990; Ploug and Ellis, 1994). Each of these comprises approximately 90 residues with four to five disulphide bonds and they are members of the Ly-6/uPAR/α-neurotoxin protein domain family, which are predicted to adopt a three-finger fold (Ploug and Ellis, 1994). The only mammalian members of this family documented so far to contain multiple copies of this domain are uPAR and the metastasis-associated C4.4A (Hansen et al, 2004). Until now, no experimentally determined structures are available for these modular proteins.
The primary function of uPAR is to bind uPA with high affinity, and hence to focalize the cellular conversion of plasminogen to plasmin (Ellis et al, 1989; Ploug, 2003). The potency of this system is elegantly demonstrated by the severe skin pathogenesis that develops in a plasminogen-dependent process in bitransgenic mice having a combined overexpression of both uPA and uPAR by epidermal keratinocytes, but this phenotype is absent in either of the corresponding single-transgenic mice (Zhou et al, 2000; Bolon et al, 2004). Expression of uPAR at the invasive front of various human carcinomas therefore favours a confined uPA-catalysed plasmin degradation of basement membranes and extracellular matrix, which contributes to the intravasation of cancer cells into vascular or lymphatic systems (Stephens et al, 1999; Rømer et al, 2004). This cellular migration is also regulated by a complex molecular interplay between uPAR and other partners, including extracellular matrix-deposited vitronectin (Wei et al, 1994), various integrins (Wei et al, 1996, 2001) and the G protein-coupled chemotactic receptor FPRL1/LXA4R (Fazioli et al, 1997; Blasi and Carmeliet, 2002). Partitioning of the glycolipid-anchored uPAR into membrane lipid rafts represents another regulatory level for controlling the focalized function of this receptor (Cunningham et al, 2003).
To elucidate the structural basis for the complex biochemical properties of human uPAR, we have solved the crystal structure of a soluble form of this receptor at 2.7 Å, in complex with a peptide that has a high affinity for uPAR and inhibits its capacity to bind uPA (Ploug et al, 2001; Ploug, 2003). This peptide was originally selected for its potent antagonist properties using a combination of phage display and combinatorial chemistry (Goodson et al, 1994; Ploug et al, 2001). Based on this crystal structure of the uPAR–peptide complex, we have subsequently constructed a model of the human uPA–uPAR complex revealing, for the first time, how the modular structure of uPAR may guide uPA-focalized proteolysis on the cell surface and control cell adhesion and migration.
Results and discussion
Crystallization of uPAR complexes
The recombinant truncated human uPAR (residues 1–283) was produced as a secreted protein by Drosophila Schneider 2 cells (S2-cells). This host expression system was chosen as it gives rise to a simple and homogenous biantenneary glycosylation, which is advantageous for protein crystallization (Gårdsvoll et al, 2004). We produced uPARwt and several uPAR mutants having their five N-linked glycosylation sites removed individually by site-directed mutagenesis (N52Q, N162Q, N172Q, N200Q or N233Q). All purified uPAR mutants exhibit similar binding kinetics for the interaction with uPA, as assessed by surface plasmon resonance (Gårdsvoll et al, 2004). Complexes between uPAR variants and previously described antagonist peptides (Ploug et al, 2001) were subjected to crystallization trials. To increase solubility, two selected peptides were extended at both termini with polar residues. They were denoted AE147 (K-S-D-Cha-F-s-k-Y-L-W-S-S-K; Cha: L-β-cyclohexyl-alanine) and AE170 (K-S-D-Cha-F-s-k-CHg-L-W-S-S-K). Crystals in monoclinic and cubic forms were obtained for uPARwt and uPARN200Q in complex with AE147 and AE170, respectively. The free thiol group of AE170 was reacted with p-chloromercuribenzoic acid, and the covalently linked mercury atom allowed us to solve the structure of a 1:1 uPAR–AE170 complex by MAD, and to build an initial model at 3.15 Å resolution. The monoclinic crystals contained eight uPAR molecules arranged as an octamer in the asymmetric unit, as revealed after molecular replacement. The eight molecules were refined to 2.7 Å resolution. Three regions (residues 84–90, 131–136 and 276–283) were, however, poorly defined in the electron density map and they were therefore not included in the structure. All eight uPAR molecules present in the unit cell are structurally equivalent with an r.m.s.d. of 0.79 Å for all Cα.
Crystal structure of human uPAR
The determined crystal structure reveals that uPAR comprises three domains (DI, DII and DIII), each of which adopts a typical three-finger fold with three adjacent loops rich in β-pleated sheets and a small C-terminal loop (Figure 1A). In general, this fold possesses four disulphide bonds: three at the base of the loops and a fourth that locks the C-terminal loop (Low et al, 1976). This property is replicated in DII and DIII, but not in DI, which lacks the third consensus cystine (Ploug et al, 1993). The overall folding topology of DI and DII resembles that of bucandin, a snake venom α-neurotoxin from Bungarus candidus (Torres et al, 2001), whereas DIII is reminiscent of CD59, a mammalian glycosylphosphatidylinositol (GPI)-anchored protein that inhibits complement activation (Fletcher et al, 1994), as illustrated in Figure 1B. The secondary structure of uPAR is thus dominated by three consecutive β-sheets—DI and DII provide six β-strands each, whereas DIII donates only five β-strands (Figure 1). Altogether, uPAR possesses 17 antiparallel β-strands, and two short helical stretches at the edge of loop 3 of DIII.
Figure 1.
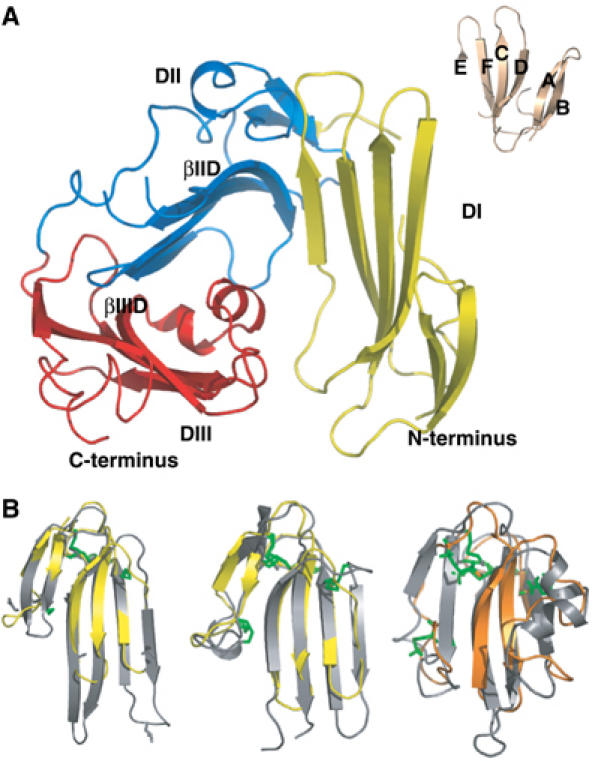
The structure of human uPAR. (A)The overall modular structure of uPAR as a ribbon diagram. The individual uPAR domains are assembled in a right-handed orientation and are coloured yellow (DI), blue (DII) and red (DIII). The inset illustrates the typical β-strand nomenclature (Low et al, 1976) for a three-fingered fold, using bucandin as a model (Torres et al, 2001). Adopting this nomenclature, the 17 β-strands of uPAR encompass the regions 2–6 (βIA), 12–16 (βIB), 23–33 (βIC), 36–46 (βID), 53–57 (βIE) and 64–71 (βIF) in DI; 94–96 (βIIA), 112–114 (βIIB), 121–129 (βIIC), 142–149 (βIID), 155–160 (βIIE) and 164–171 (βIIF) in DII; and 189–199 (βIIIA), 211- 216 (βIIIB), 221–229 (βIIIC), 234–242 (βIIID) and 262–266 (βIIIF) in DIII. The short helical stretches in DIII (αIIIE) encompass residues 244–248 and 250–255. The bended β-strands in DII and DIII are indicated separately. (B) From left to right, superimpositions of bucandin (yellow) on uPAR DI and DII (grey) and of CD59 (orange) on uPAR DIII (grey). Disulphide bonds are coloured green in the superimpositions. For sake of clarity, the positions of the N- and C-termini are indicated in panel A.
The three domains of uPAR are assembled in a right-handed orientation generating an almost globular receptor with a breach between DI and DIII. This topology creates a central cavity (19 Å deep) that accommodates the antagonist peptide AE147. The top of the cavity is widely open (21–25 Å large), surrounded by the tips of eight of the nine loops. The cavity progressively narrows towards the bottom and its floor is delineated by the large turns that connect loops 2 and 3 in the three domains (Figure 2). The same faces of the three β-sheets form the walls of the cavity and establish multiple contacts with the peptide. An inventory of the residues located at this interface is shown in Figure 3, highlighting the 26 residues of uPAR that interact with the peptide AE147. As illustrated in Figure 2A, these residues form a long hydrophobic strip (coloured pale orange) that spans the walls of DI and DIII as well as the floor of the cavity, and is delimited by polar residues (coloured cyan). In agreement with previous biochemical data (Ploug, 1998; Ploug et al, 1998, 2001), DI plays a predominant role in this ligand interaction by providing half of the binding interface. Accordingly, the average temperature factor for DI (39.6 Å2) is more similar to that observed for the peptide (38.0 Å2) than those of DII and DIII (51.0 and 57.3 Å2, respectively), which are therefore more agitated.
Figure 2.
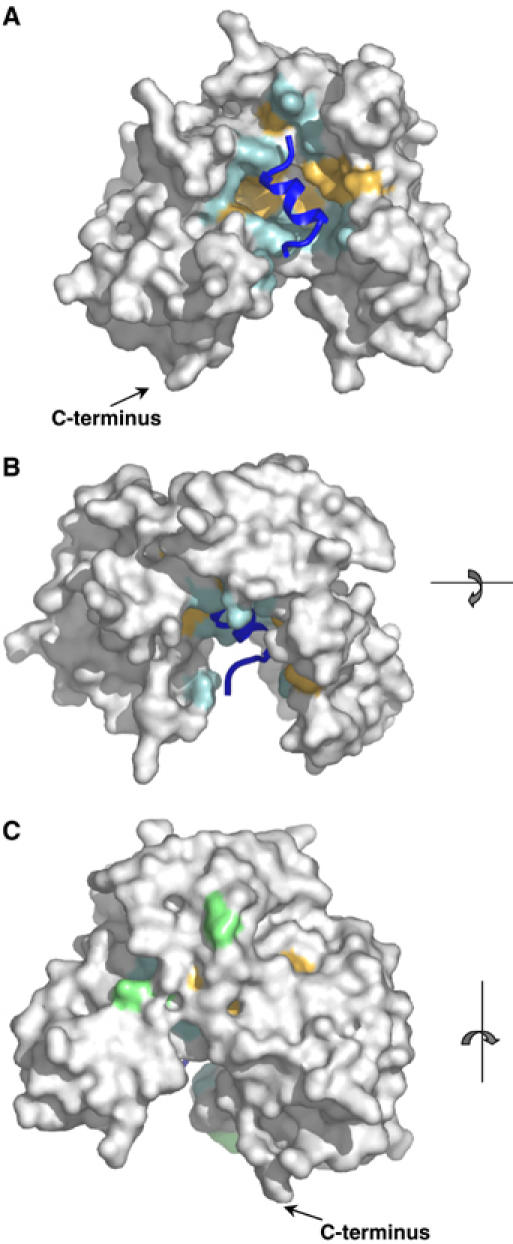
Molecular surface representation of the uPAR–peptide complex. The surface of uPAR is shown in grey, whereas the bound antagonist peptide (AE147) is shown as a ribbon diagram in dark blue. Receptor residues interacting with AE147 are coloured orange (hydrophobic) or cyan (polar). (A) The front side, (B) the upper side and (C) the rear side of the complex, with asparagine-linked glycosylation sites coloured light green. The C-terminal end is shown in panels A and C. In panel B, it is located behind the molecule and hence it cannot be seen by the reader.
Figure 3.
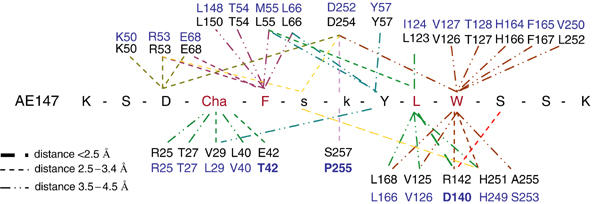
Interacting residues in both the antagonist peptide AE147 and uPAR as observed in the crystal structure of the uPAR–peptide complex. The amino-acid sequence of AE147 is shown in the single-letter code with capitals denoting L-amino acids and lower case D-amino acids. Cha: L-β-cyclohexyl-alanine. Residues in black are from human uPAR (Roldan et al, 1990). The corresponding residues from murine uPAR (see text) are coloured blue (Kristensen et al, 1991). Bold letters in blue highlight nonconservative substitutions among these residues in the cavity of human and murine uPAR.
A characteristic property of the uPAR complex is governed by its large outer surface (Figure 2B and C) that is readily accessible to other uPAR ligands. This external surface is delimited by the opposite face of the three β-sheets and harbours the proximate interdomain linker regions and the five possible N-linked glycosylation sites at N52, N162, N172, N200 and N233. Carbohydrates were observed at four of these sites in the structure, as the mutation N200Q was important for optimal crystal growth. The degree of definition for these carbohydrate structures varied among the eight uPAR molecules in the unit cell, from one β-1,4-N-acetylglucosamine (NAG) to two NAGs and three mannose (Man) moieties in the electron density maps, as illustrated in Figure 4A.
Figure 4.
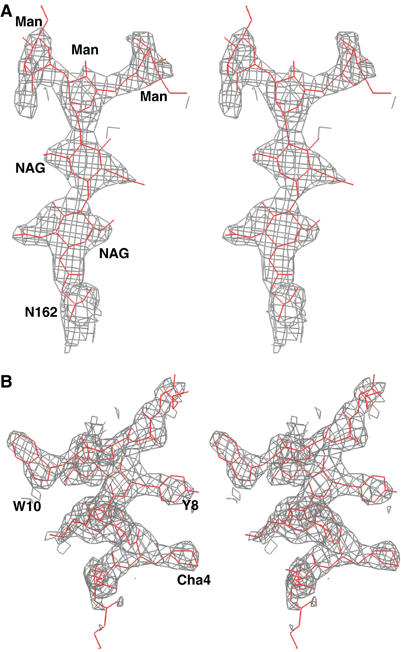
Stereo view of two 2Fo−Fc omit maps. (A) A 1.5σ level contoured map for the carbohydrate linked to N162 in uPAR domain DII. (B) A similar contoured map for the bound AE147 (in grey). The refined atomic model of AE147 is shown in stick representation (red). Cha: L-β-cyclohexyl-alanine; NAG: N-acetylglucosamine; Man: mannose.
The tight assembly of the three domains in uPAR is ensured by two conserved interdomain interfaces that include two analogously bended β-strands in DII (βIID) and DIII (βIIID). The C-terminus of βIID is involved in the formation of an interdomain β-sheet with DI (βIE), whereas the C-terminus of βIIID forms a short interdomain β-sheet with DII (βIIE) (Figure 1A). This unique organization allows the three domains to form a virtually continuous and nearly circular β-sheet. If the eight uPAR molecules of the unit cell are superimposed onto DII, the angle between DI and DII varies from 107.8 to 112.2° and the angle between DII and DIII varies from 115.9 to 121.3°. This flexibility occurs at the conserved RGC sequences in βIID and βIIID, which act as hinges allowing the three successive β-sheets to adopt different relative orientations.
Interaction between human uPAR and the antagonist peptide
Inhibition of the uPA–uPAR interaction by peptides and small molecules has been extensively explored (Rosenberg, 2001; Ploug, 2003; Rømer et al, 2004). Some linear (Appella et al, 1987) or cyclic (Schmiedeberg et al, 2002) peptides were derived from a functionally identified uPA sequence, whereas others resulted from random phage display, followed by affinity maturation (Goodson et al, 1994; Ploug et al, 2001). AE147, which was obtained by the latter approach, binds to uPAR with a Kd of 16.4 nM. When the peptide is bound to uPAR, it adopts a regular right-handed 3.6 α-helical structure from residues 3 to 12 (Figures 4B and 5), showing that the constraints brought by the two D-amino acids affect the side-chain orientations but not the main-chain conformation. The axis of the helix is nearly perpendicular to the major β-sheet of DII, with its N-terminus pointing towards the breach. Superimposition of all peptide antagonists present in the eight complexes of the asymmetric unit cell is shown in Figure 5B. Apart from a slight divergence at the N-terminus, the eight peptides superimpose well, with an r.m.s.d. for their Cα atoms that varied from 0.3 to 0.9 Å in the different peptide pairs. Approximately 2000 Å2 of the overall accessible surface is buried upon complex formation, which is comparable to the average interface of protein–protein interactions (Lo Conte et al, 1999). The peptide buries 66% of its surface and if we consider that an interaction between uPAR and the peptide is effective when observed in at least six of the eight molecules of the asymmetric unit, virtually all the peptide residues interact with uPAR. Residues located at the extremities of the peptide establish less contact with uPAR as compared with F5, L9 and W10 (Figure 3), which are almost entirely buried and cover half of the interface with uPAR. In particular, L9 establishes the shortest hydrogen bond with R142, and W10 occupies the deepest hydrophobic hole of the cavity. Located on the edge of the helix facing the floor of the cavity, these three residues are nearly aligned and constitute a major binding motif for the peptide. Cha4 and Y8 are 70% buried and strengthen the binding contribution of the main motif. These observations provide a unifying structural basis for previous biochemical data obtained by alanine-scanning (Ploug et al, 2001), photoaffinity labelling (Ploug, 1998) and hydrogen–deuterium exchange (Jørgensen et al, 2004) experiments made with highly similar parent peptides.
Figure 5.
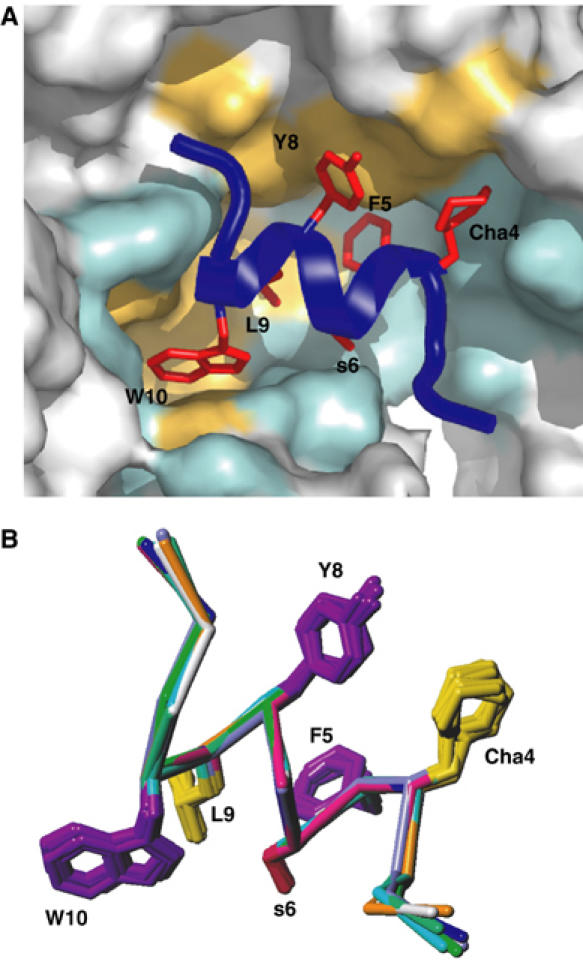
Binding of the antagonist peptide in uPAR cavity. (A) The interface between uPAR (illustrated as surface representation) and AE147 (illustrated by a combined ribbon and stick representation in dark blue) is shown from the front side. The interface provided by uPAR is coloured pale orange (hydrophobic) or cyan (polar), whereas the corresponding side chains in AE147 are coloured red. Note that the indole side chain of W10 efficiently engages the deep and prominent hydrophobic hole of the central cavity of uPAR and the side chain of L9 is located proximate to its entrance. (B) Superposition of all eight peptide molecules observed in the unit cell. The figure shows superposition of their Cα backbones and the side chains of Cha4, F5, s6, Y8, L9 and W10 which are involved in binding to uPAR (see text).
The antagonist peptide and uPA bind to overlapping determinants in the central cavity of uPAR. This is supported by the observation that some of the uPAR residues that interact with AE147 were previously shown to interact with uPA. In particular, four residues in uPAR DI (R53, L55, Y57 and L66) that occupy prominent positions in the binding interface with AE147 were previously emphasized to be important for uPA binding by site-directed mutagenesis (Gårdsvoll et al, 1999). Once the central cavity of uPAR is occupied by an antagonist peptide or a small molecule (Rosenberg, 2001; Ploug, 2003), uPAR cannot simultaneously bind uPA and is therefore unable to focalize proteolysis on the cell surface. Targeting this cavity in uPAR appears therefore as an appealing strategy for designing future anticancer drugs.
The use of various mouse model systems as surrogates for human cancer in preclinical assessment of uPAR-targeted intervention is, however, complicated by an unexpected preference of most antagonists for human uPAR (Rømer et al, 2004). Furthermore, it is well established that a cross-species barrier also exists in the uPAR–uPA interaction between man and mouse (Ploug et al, 2001) but not for the high-affinity interaction between human uPA and cell lines from Chinese hamster, pig and the African green monkey (Estreicher et al, 1989; Fowler et al, 1998; Engelholm and Behrendt, 2001). To explore the basis for the species specificity, we examined whether one (or more) of the residues that differ between murine and human uPAR is (are) contained within the binding interface for AE147. As shown in Figure 3, we found that three of the 26 human uPAR residues that interact with AE147 are indeed nonconservatively changed in the murine receptor (E42T, R142D and S257P). A detailed comparison of the central cavity of human and murine uPAR will be required to unravel the mechanisms underlying the different behaviour of these receptors in terms of recognition of both peptides and their cognate ligands (Ploug et al, 2001).
The antagonist peptide and the binding module of uPA share a common structural motif
The key determinants by which uPA binds uPAR include Y24, F25, I28 and W30 that are located within or close to the tip of a long β-hairpin in the growth factor-like (GFD) receptor-binding module of uPA (Hansen et al, 1994; Magdolen et al, 1996; Ploug, 2003). A constrained uPA-derived cyclic peptide that binds uPAR with high affinity has a Cα backbone that also superimposes well with this β-hairpin of GFD, and the side chains important for uPAR binding occupy almost identical spatial positions as the corresponding residues in uPA (Schmiedeberg et al, 2002). Most strikingly, although the peptide AE147 was originally selected by a random combinatorial approach, its binding residues W10, L9, Y8, s6 and F5 also adopt analogous spatial positions as compared to Y24, F25, W30, S26 and I28 in GFD, respectively (Figure 6A). Therefore, although AE147 and GFD adopt entirely unrelated secondary structures, an α-helix and a β-hairpin, respectively, they seem to have reached a convergent solution for binding to uPAR. This observation has two implications. First, it lends credibility to the view that targeting the central cavity of uPAR with small molecules derived from the hydrophobic binding motif observed in AE147 constitutes an appealing strategy for designing uPAR inhibitors with antitumour effects. Second, assuming that the key residues of the conserved binding motif in the peptide and GFD bind at homologous positions in uPAR, the constraints observed between the peptide and uPAR can be used to elaborate a model of the complex between uPAR and GFD.
Figure 6.
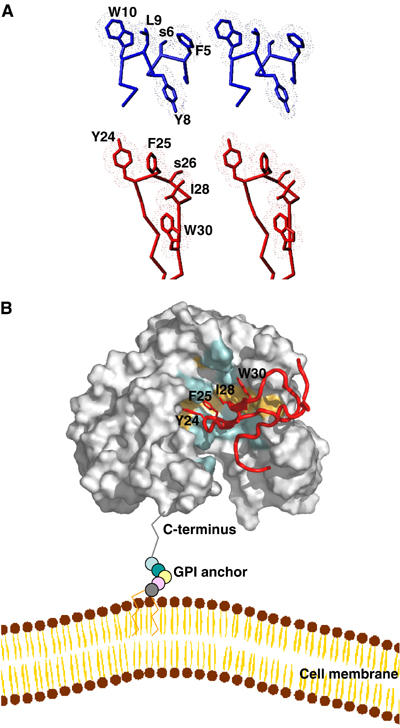
Model of the uPA–uPAR complex. (A) Stereo views of the key residues by which AE147 (blue) and the β-hairpin in GFD of uPA (red) bind to uPAR in our crystal structure and in a GFD–uPAR model, respectively. Note that W10 and L9 of AE147 exhibit a spatial superimposition with Y24 and F25 in uPA. (B) Model for the structure of the human GFD–uPAR complex. The uPAR molecule is shown as a molecular surface representation with the residues interacting with GFD in the same colour code as those used in Figures 2 and 4. The receptor-binding module of uPA is shown in red as a ribbon diagram with the side chains of Y24, F25, I28 and W30, which are highlighted by sticks. A schematic representation of the glycolipid (GPI) anchor is shown to connect the C-terminus of uPAR to a model of the cell membrane. Note that the GPI anchor is not shown to scale and the relative orientation of uPAR to the cell membrane is arbitrarily presented.
Modelling the structure of human uPAR in complex with the uPA-binding module
Based on the above assumption, we built a model of the GFD–uPAR complex using a conventional docking procedure (Brooks et al, 1988). We selected the five complexes showing the most favourable van der Waals energies. These low values (not shown) suggest that the two proteins may interact without requiring major conformational rearrangements. To further validate these models, we subjected residues 21–30 in GFD to a theoretical alanine-scanning and compared the calculated ΔΔG values with those deduced from experimental mutagenesis studies (Magdolen et al, 1996). For all five models, the ΔΔG values were higher than 1 kcal/mol and compared well for the uPA mutants K23A, Y24A and F25A. For example, in the two models showing the best agreement between theoretical and experimental values, the calculated ΔΔG values for K23A, Y24A, F25A and W30A were 1.4±0.2, 1.8±0.2, 1.2±0.4 and 1.2±0.2 kcal/mol, respectively, and the experimental ΔΔG values were 1.0 and 2.2 kcal/mol for K23A and Y24A (M Ploug, unpublished) and 2.2 kcal/mol for both F25A and W30A (calculated from Magdolen et al (1996) after standardization with Kd values from M Ploug). A value of 0.8±0.1 kcal/mol was calculated for I28A while it was experimentally observed to be 1.1 kcal/mol (calculated from Magdolen et al (1996) after standardization with Kd values from M Ploug) or 1.2 kcal/mol (M Ploug, unpublished). For all other single point mutations, the ΔΔG values were lower than 0.5 kcal/mol in both experimental and calculated cases. We found a remarkable spatial superimposition between the bound side chains of L9 and W10 in the peptide and F25 and Y24 in uPA, respectively (Figure 6A). These receptor-binding dyads (L9/W10 and F25/Y24) may similarly anchor the antagonist peptide and uPA tightly to the bottom of the central cavity of uPAR. The situation is slightly different for the remaining two positions in the conserved receptor-binding motif (Y8/F5 and W30/I28). The side chains of Y8 in the peptide and W30 in uPA remain in proximity although they do not really superimpose, whereas those of F5 and I28 are always relatively far apart. Figure 6B shows the model of the GFD–uPAR complex having the best agreement between the calculated and experimental ΔΔG values.
As suggested from this model, the β-hairpin of GFD engages the cavity without generating major clashes with uPAR. The plane formed by its two β-strands is nearly perpendicular to the plane of the major β-sheet in DI. The tip of the β-hairpin of GFD and the first helical turn of AE147 occupy highly similar positions in the cavity proximal to one of the helical stretches in loop 3 of uPAR DIII. The N-terminal part of GFD occupies the upper part of the space between DI and DIII, whereas its C-terminal region resides in proximity to a crenel-like structure observed in the upper part of DI. A large body of biochemical data (Ploug, 2003) provides strong evidence in support of the proposed model for the GFD–uPAR complex. All side chains of R53, L55, Y57 and L66 in loop 3 of DI reside within the binding cavity of uPAR and accordingly these residues were previously shown to be important for the uPA interaction, by site-directed mutagenesis (Gårdsvoll et al, 1999). The same holds true for one or more of the residues R137, K139, R142, H143 and R145 located in loop 2 of DII (Bdeir et al, 2000). In contrast, the fucosylated T18 and N32 in GFD, whose alanine substitutions cause little effect on the complex stability (Hansen et al, 1994; Magdolen et al, 1996), are both located outside the GFD–uPAR interface. Finally, Y57 in uPAR and Y24 in GFD both become inaccessible to chemical modification with tetranitromethane upon complex formation (Ploug et al, 1995), which agrees with our current model where the relative accessible surface areas of both phenolic groups are shielded upon the GFD–uPAR interaction.
The structure of uPAR can orchestrate complex biological events on the cell surface
Expression of uPAR at the invasive front of many human carcinomas is thought to contribute to degradation of the extracellular matrix by focalizing uPA-catalysed plasminogen activation to these areas (Rømer et al, 2004). However, the cellular migration accompanying intravasation into the vascular or lymphatic systems is also regulated by a complex molecular interplay between uPAR and other binding partners, including matrix-deposited vitronectin (Wei et al, 1994; Cunningham et al, 2003), the G protein-coupled chemotactic receptor FPRL1/LXA4R (Fazioli et al, 1997) and various integrins (Wei et al, 1996, 2001; Pluskota et al, 2003). At present, no clear picture of the uPAR determinants that govern vitronectin binding has yet emerged. However, in a model of human vitronectin (Lynn et al, 2005), the N-terminal somatomedin B domain harbours the principal interaction sites for both uPA–uPAR complexes (Deng et al, 1996) and the plasminogen activator inhibitor PAI-1 (Zhou et al, 2003). Our model suggests that uPAR does not undergo dramatic conformational changes when GFD of uPA is embedded in the central cavity and the large outer receptor surface remains accessible to additional binding partners, like vitronectin and integrins. This latter view is supported by a recent study by homologue-scanning mutagenesis, which reports that two regions located on the outer surface of uPAR bind to vitronectin (Li et al, 2003). In other words, formation of a ternary uPA–uPAR–vitronectin complex appears reasonable from a structural point of view and we anticipate that in such a complex, uPAR will adopt a structure that will remain comparable to the one that is reported in this work. Therefore, notwithstanding its relatively small size, the unique modular assembly of the three consecutive three-fingered domains in the intact uPAR enables it to orchestrate the complex events that are required to focalize proteolysis on the cell surface and facilitate a controlled extracellular matrix degradation followed by intravasation and metastasis in various carcinomas.
Traditionally, the three-finger fold has been associated with its ability to exert toxic functions (Ménez et al, 2002), and more recently it has also been recognized for being the single extracellular ligand-binding domain of either the GPI-anchored CD59 (Fletcher et al, 1994) or various cytokine receptors (Greenwald et al, 1999; Kirsch et al, 2000; Hart et al, 2002; Thompson et al, 2003). We have now shown that this domain can be structurally assembled into a multimeric receptor endowed with distinct binding specificities for functionally diverse partners such as a serine protease (uPA), a matrix protein (vitronectin) and cell adhesion receptors (integrins).
Materials and methods
Expression and purification of recombinant soluble uPAR
Drosophila S2-cells were stably transfected by the expression vector pMTC-suPAR, which encodes residues 1–283 of human uPAR along with the native signal sequence of uPAR under the control of an inducible metallothionein promoter (Gårdsvoll et al, 2004). Expression vectors encoding uPARwt as well as the following single-site glycosylation mutants were used (N52Q, N162Q, N172Q, N200Q and N233Q). After 1 week of induction by 0.5 mM CuSO4, the conditioned medium typically contained between 5 and 10 mg/l uPAR and the secreted receptors were subsequently purified using an immunoaffinity column with the immobilized monoclonal anti-uPAR antibody R2 followed by reversed-phase HPLC using a Brownlee Aquapore C4 column as described previously (Gårdsvoll et al, 2004). As judged by SDS–PAGE and Coomassie G250 staining, the purity of these preparations was estimated to be >95%. The glycosylation profiles of the purified uPAR variants were evaluated by MALDI-MS and are dominated by simple Man3GlcNAc2Fuc moieties (Gårdsvoll et al, 2004).
Crystallographic data
Peptide–uPAR crystals were grown at 17–20°C by streak seeding and macroseeding in seating drop vapour diffusion experiments (Stura, 1999, 2001). The concentrations of peptide and uPAR used in crystallization assays were, respectively, 1 and 10 mg/ml. The most suitable crystals were obtained with peptide AE147 in a solution containing 1.29–1.41 M ammonium sulphate in 50 mM imidazole at pH 7.0–7.4 at 20°C. These crystals belong to the monoclinic space group P21 with cell parameters a=106.9 Å, b=136.8 Å, c=140.5 Å and β=97.3 Å. To introduce a heavy metal atom in the crystal, Y8 in AE170 was substituted by a cysteine, the thiol group of which was subsequently derivatized by p-chloromercurybenzoic acid. The resulting peptide conjugate had a mass of 1885.88 Da, as measured by electrospray-ionization mass spectrometry, which is consistent with the covalent incorporation of the mercury atom. Optimized crystals were obtained with this peptide and an N200Q glycosylation site mutant of uPAR in a solution containing 1.2 M ammonium sulphate, 25 mM borate, 5 mM EDTA and 150 mM NaCl (pH 8.0). These crystals grow in the cubic space group I432 with a=b=c=187.0 Å. Crystals for data collection were flash-frozen in a cryo-stream or in liquid ethane using a cryo-solution with 25% of glycerol added to the crystallization solution. Data of the cubic crystal form with the mercury-derivatized peptide were collected at two wavelengths on beam line ID-29 at the European Synchrotron Radiation Facility (ESRF) to 3.15 Å and reduced with HKL (Otwinowski and Minor, 1997); full details are given in Table I. The structure was solved with SHARP (De La Fortelle and Bricogne, 1997). The initial model was built in the cubic crystal form, and used to solve the structure in the monoclinic form. The protein building was then accomplished alternatively between the cubic and the monoclinic form in order to remove the error associated with each crystal form. The refinement was performed in the monoclinic form to 2.7 Å with REFMAC (Collaborative Computational Project number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763). The eight molecules were initially restrained with noncrystallographic symmetry and loosened later during the refinement process. The translation-libration-screw (TLS) method was applied using three rigid groups per uPAR molecule. The three TLS groups were defined according to the two hinges observed in the structure. The first TLS group includes residues 1–77 corresponding to domain I, residues 146–149 from domain II and the peptide residues arbitrarily numbered from 501 to 513. The second group includes residues 92–145 and 150–177 from domain II plus residues 240–255 from domain III. The third group includes residues 192–239 and 256–274 from domain III. After refinement, the R-factor was 0.246 and the Rfree 0.315 for eight uPAR-AE147 molecules, 32 glycosylation sites and 746 water molecules per unit cell, which corresponds to 93 water molecules per uPAR molecule. Phases were further validated by systematic omit maps (Figure 4). In particular, three mannose groups and two NAG were observed at N162 in four of the eight uPAR molecules (Figure 4A). Statistics of the data collection, refinement and model validation are shown in Table I. The coordinates of the structure have been deposited in the Protein Data Bank (PDB) with the code number 1YWH.
Table 1.
Data collection, phasing and refinement statistics
| MAD |
Native cubic | Native monoclinic | |||
|---|---|---|---|---|---|
| Remote | Inflection | ||||
| Wavelength (Å) | 0.9762 | 1.0087 | |||
| Resolution (Å) (highest shell) | 99–3.5 (3.66–3.5) | 99–3.55 (3.68–3.55) | 50–3.15 (3.22–3.15) | 40–2.7 (2.76–2.70) | |
| Space group | I432 | P21 | |||
| Cell constants | a=b=c=187.73 Å | a=106.9. b=136.8. c=140.5 Å | |||
| α=β=γ=90° | α=90°, β=97.3°, γ=90° | ||||
| Reflections | 8068 | 7136 | 7994 | 110 121 | |
| Redundancy | >13 | >13 | 3 | 3 | |
| I/σ | 32.2 (6.2) | 34.8 (5.3) | 11 (1.39) | 12.4 (2.61) | |
| Completeness | 99.7% | 99.7% | 96.9% | 99.5% | |
| Rmerge | 0.097 (0.58) | 0.093 (0.701) | 0.071 (0.543) | 0.09 (0.575) | |
| Phasing | |||||
| FoM (a/c) | 0.3724/0.18807 | ||||
| PhP ano | 1.254 | 0.983 | |||
| PhP iso (a/c) | — | 0.252/0.27 | |||
| Refinement | |||||
| R | 0.245 | ||||
| Rfree | 0.315 | ||||
| Total number of atoms | 18 548 | ||||
| Number of | |||||
| Protein atoms | 15 976 | ||||
| Carbohydrate atoms | 1718 | ||||
| Sulphate ions | 105 | ||||
| Water molecules | 749 | ||||
| Average B-factor | 58.8 Å2 | ||||
| R.m.s.d. from ideality | |||||
| On bonds | 0.011 Å | ||||
| On angles | 1.634° | ||||
| On chirality | 0.112° | ||||
| Percentage of outliers in Ramachandran plot | 0.4% | ||||
Molecular modelling of uPAR–GFD complexes
The NMR-derived structure of the amino-terminal fragment of human uPA (residues 1–135; PDB accession number 1URK) was used for the present modelling of the uPAR–GFD (residues 1–48) complexes. A rigid-body molecular dynamics was first performed to superimpose atoms of residues 30, 24, 25 and 28 in GFD as well as possible with atoms of the residues 8, 10, 9 and 5 of the peptide AE147. A high-temperature molecular dynamics procedure was subsequently applied in which ambiguous constraints were introduced between the side chains of (i) I28 in GFD and R53, L55, L66 and L150 in uPAR; (ii) F25 in GFD and L55, L123, V125, L168, H251 and L150 in uPAR; (iii) Y24 in GFD and V125, L168, H251, L252, A255 and H166 in uPAR; and (iv) W30 in GFD and L55, L66, V29, L31 and Y57 in uPAR. Four-dimensional molecular dynamics at 500 K allowed us to optimize protein–protein interactions during 24 000 steps followed by 8000 steps of regular molecular dynamics at 400 K and 8000 steps at 300 K. Finally, electrostatic terms were introduced during 8000 steps of minimization under harmonic constraints. During the whole docking procedure, the X-ray structure of uPAR was maintained fixed. For the structures with the lowest van der Waals interaction energy, a theoretical alanine-scanning was performed. Binding free energies were calculated using the Poisson–Boltzmann equation (Eriksson and Roux, 2002). All the calculations were performed using the CHARMM program (Brooks et al, 1988) version c28a3 with the force field par_all22_prot_lipid.inp and top_all22_prot_lipid.inp (MacKerell et al, 1998).
Acknowledgments
We thank the staff members of the European Synchrotron Radiation Facility (ESRF) in Grenoble on beam lines BM30A, ID14, ID23 and ID29 for help in collecting data. The technical assistance of Gitte Juhl Funch and Yvonne DeLotto is also acknowledged. This work was supported by CONACyT subventions, John and Birthe Meyer Foundation, The Lundbeck Foundation, The Danish Cancer Society and Eu contract LSHC-CT2003-503297. Figures were made with softwares from DeLano Scientific warren@delanoscientific.com.
References
- Appella E, Robinson EA, Ullrich SJ, Stoppelli MP, Corti A, Cassani G, Blasi F (1987) The receptor-binding sequence of urokinase. A biological function for the growth-factor module of proteases. J Biol Chem 262: 4437–4440 [PubMed] [Google Scholar]
- Bdeir K, Kuo A, Mazar A, Sachais BS, Xiao W, Gawlak S, Harris S, Higazi AA, Cines DB (2000) A region in domain II of the urokinase receptor required for urokinase binding. J Biol Chem 275: 28532–28538 [DOI] [PubMed] [Google Scholar]
- Blasi F, Carmeliet P (2002) uPAR: a versatile signalling orchestrator. Nat Rev Mol Cell Biol 3: 932–943 [DOI] [PubMed] [Google Scholar]
- Bolon I, Zhou HM, Charron Y, Wohlwend A, Vassalli JD (2004) Plasminogen mediates the pathological effects of urokinase-type plasminogen activator overexpression. Am J Pathol 164: 2299–2304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Karplus M, Petit BM (1988) Proteins: A Theoretical Perspective of Dynamics, Structure and Thermodynamics. New York: Wiley [Google Scholar]
- Cunningham O, Andolfo A, Santovito ML, Iuzzolino L, Blasi F, Sidenius N (2003) Dimerization controls the lipid raft partitioning of uPAR/CD87 and regulates its biological functions. EMBO J 22: 5994–6003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Fortelle E, Bricogne G (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction methods. Methods Enzymol 276: 472–493 [DOI] [PubMed] [Google Scholar]
- Deng G, Curriden SA, Wang S, Rosenberg S, Loskutoff DJ (1996) Is plasminogen activator inhibitor-1 the molecular switch that governs urokinase receptor-mediated cell adhesion and release? J Cell Biol 134: 1563–1571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis V, Scully MF, Kakkar VV (1989) Plasminogen activation initiated by single-chain urokinase-type plasminogen activator. Potentiation by U937 monocytes. J Biol Chem 264: 2185–2188 [PubMed] [Google Scholar]
- Engelholm LH, Behrendt N (2001) Differential binding of urokinase and peptide antagonists to the urokinase receptor: evidence from characterization of the receptor in four primate species. Biol Chem 382: 435–442 [DOI] [PubMed] [Google Scholar]
- Eriksson MA, Roux B (2002) Modeling the structure of agitoxin in complex with the Shaker K+ channel: a computational approach based on experimental distance restraints extracted from thermodynamic mutant cycles. Biophys J 83: 2595–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estreicher A, Wohlwend A, Belin D, Schleuning WD, Vassalli JD (1989) Characterization of the cellular binding site for the urokinase-type plasminogen activator. J Biol Chem 264: 1180–1189 [PubMed] [Google Scholar]
- Fazioli F, Resnati M, Sidenius N, Higashimoto Y, Appella E, Blasi F (1997) A urokinase-sensitive region of the human urokinase receptor is responsible for its chemotactic activity. EMBO J 16: 7279–7286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher CM, Harrison RA, Lachmann PJ, Neuhaus D (1994) Structure of a soluble, glycosylated form of the human complement regulatory protein CD59. Structure 2: 185–199 [DOI] [PubMed] [Google Scholar]
- Fowler B, Mackman N, Parmer RJ, Miles LA (1998) Binding of human single chain urokinase to Chinese hamster ovary cells and cloning of hamster u-PAR. Thromb Haemostasis 80: 148–154 [PubMed] [Google Scholar]
- Gårdsvoll H, Danø K, Ploug M (1999) Mapping part of the functional epitope for ligand binding on the receptor for urokinase-type plasminogen activator by site-directed mutagenesis. J Biol Chem 274: 37995–38003 [DOI] [PubMed] [Google Scholar]
- Gårdsvoll H, Werner F, Søndergaard L, Danø K, Ploug M (2004) Characterization of low-glycosylated forms of soluble human urokinase receptor expressed in Drosophila Schneider 2 cells after deletion of glycosylation-sites. Protein Expr Purif 34: 284–295 [DOI] [PubMed] [Google Scholar]
- Goodson RJ, Doyle MV, Kaufman SE, Rosenberg S (1994) High-affinity urokinase receptor antagonists identified with bacteriophage peptide display. Proc Natl Acad Sci USA 91: 7129–7133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald J, Fischer WH, Vale WW, Choe S (1999) Three-finger toxin fold for the extracellular ligand-binding domain of the type II activin receptor serine kinase. Nat Struct Biol 6: 18–22 [DOI] [PubMed] [Google Scholar]
- Hansen AP, Petros AM, Meadows RP, Nettesheim DG, Mazar AP, Olejniczak ET, Xu RX, Pederson TM, Henkin J, Fesik SW (1994) Solution structure of the amino-terminal fragment of urokinase-type plasminogen activator. Biochemistry 33: 4847–4864 [DOI] [PubMed] [Google Scholar]
- Hansen LV, Gårdsvoll H, Nielsen BS, Lund LR, Danø K, Jensen ON, Ploug M (2004) Structural analysis and tissue localization of human C4.4A: a protein homologue of the urokinase receptor. Biochem J 380: 845–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart PJ, Deep S, Taylor AB, Shu Z, Hinck CS, Hinck AP (2002) Crystal structure of the human TbetaR2 ectodomain–TGF-beta3 complex. Nat Struct Biol 9: 203–208 [DOI] [PubMed] [Google Scholar]
- Heiss MM, Allgayer H, Gruetzner KU, Funke I, Babic R, Jauch KW, Schildberg FW (1995) Individual development and uPA-receptor expression of disseminated tumour cells in bone marrow: a reference to early systemic disease in solid cancer. Nat Med 1: 1035–1039 [DOI] [PubMed] [Google Scholar]
- Jørgensen TJ, Gårdsvoll H, Danø K, Roepstorff P, Ploug M (2004) Dynamics of urokinase receptor interaction with peptide antagonists studied by amide hydrogen exchange and mass spectrometry. Biochemistry 43: 15044–15057 [DOI] [PubMed] [Google Scholar]
- Kirsch T, Sebald W, Dreyer MK (2000) Crystal structure of the BMP-2–BRIA ectodomain complex. Nat Struct Biol 7: 492–496 [DOI] [PubMed] [Google Scholar]
- Kristensen P, Eriksen J, Blasi F, Danø K (1991) Two alternatively spliced mouse urokinase receptor mRNAs with different histological localization in the gastrointestinal tract. J Cell Biol 115: 1763–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Lawrence DA, Zhang L (2003) Sequences within domain II of the urokinase receptor critical for differential ligand recognition. J Biol Chem 278: 29925–29932 [DOI] [PubMed] [Google Scholar]
- Lo Conte L, Chothia C, Janin J (1999) The atomic structure of protein–protein recognition sites. J Mol Biol 285: 2177–2198 [DOI] [PubMed] [Google Scholar]
- Low BW, Preston HS, Sato A, Rosen LS, Searl JE, Rudko AD, Richardson JS (1976) Three dimensional structure of erabutoxin b neurotoxic protein: inhibitor of acetylcholine receptor. Proc Natl Acad Sci USA 73: 2991–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn GW, Heller WT, Mayasundari A, Minor KH, Peterson CB (2005) A model for the three-dimensional structure of human plasma vitronectin from small-angle scattering measurements. Biochemistry 44: 565–574 [DOI] [PubMed] [Google Scholar]
- MacKerell AD, Bashford D, Bellott RL, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 102: 3586–3616 [DOI] [PubMed] [Google Scholar]
- Magdolen V, Rettenberger P, Koppitz M, Goretzki L, Kessler H, Weidle UH, König B, Graeff H, Schmitt M, Wilhelm O (1996) Systematic mutational analysis of the receptor-binding region of the human urokinase-type plasminogen activator. Eur J Biochem 237: 743–751 [DOI] [PubMed] [Google Scholar]
- Ménez A, Servent D, Gasparini S (2002) Perspective in Molecular Toxinology. Chichester: Wiley [Google Scholar]
- Mustjoki S, Sidenius N, Sier CF, Blasi F, Elonen E, Alitalo R, Vaheri A (2000) Soluble urokinase receptor levels correlate with number of circulating tumor cells in acute myeloid leukemia and decrease rapidly during chemotherapy. Cancer Res 60: 7126–7132 [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Pappot H, Høyer-Hansen G, Rønne E, Hansen HH, Brünner N, Danø K, Grøndahl-Hansen J (1997) Elevated plasma levels of urokinase plasminogen activator receptor in non-small cell lung cancer patients. Eur J Cancer 33: 867–872 [DOI] [PubMed] [Google Scholar]
- Ploug M (1998) Identification of specific sites involved in ligand binding by photoaffinity labeling of the receptor for the urokinase-type plasminogen activator. Residues located at equivalent positions in uPAR domains I and III participate in the assembly of a composite ligand-binding site. Biochemistry 37: 16494–16505 [DOI] [PubMed] [Google Scholar]
- Ploug M (2003) Structure–function relationships in the interaction between the urokinase-type plasminogen activator and its receptor. Curr Pharm Des 9: 1499–1528 [DOI] [PubMed] [Google Scholar]
- Ploug M, Ellis V (1994) Structure–function relationships in the receptor for urokinase-type plasminogen activator. Comparison to other members of the Ly-6 family and snake venom alpha-neurotoxins. FEBS Lett 349: 163–168 [DOI] [PubMed] [Google Scholar]
- Ploug M, Kjalke M, Rønne E, Weidle U, Høyer-Hansen G, Danø K (1993) Localization of the disulfide bonds in the NH2-terminal domain of the cellular receptor for human urokinase-type plasminogen activator. A domain structure belonging to a novel superfamily of glycolipid-anchored membrane proteins. J Biol Chem 268: 17539–17546 [PubMed] [Google Scholar]
- Ploug M, Østergaard S, Gårdsvoll H, Kovalski K, Holst-Hansen C, Holm A, Ossowski L, Danø K (2001) Peptide-derived antagonists of the urokinase receptor. Affinity maturation by combinatorial chemistry, identification of functional epitopes, and inhibitory effect on cancer cell intravasation. Biochemistry 40: 12157–12168 [DOI] [PubMed] [Google Scholar]
- Ploug M, Østergaard S, Hansen LB, Holm A, Danø K (1998) Photoaffinity labeling of the human receptor for urokinase-type plasminogen activator using a decapeptide antagonist. Evidence for a composite ligand-binding site and a short interdomain separation. Biochemistry 37: 3612–3622 [DOI] [PubMed] [Google Scholar]
- Ploug M, Rahbek-Nielsen H, Ellis V, Roepstorff P, Danø K (1995) Chemical modification of the urokinase-type plasminogen activator and its receptor using tetranitromethane. Evidence for the involvement of specific tyrosine residues in both molecules during receptor–ligand interaction. Biochemistry 34: 12524–12534 [DOI] [PubMed] [Google Scholar]
- Ploug M, Rønne E, Behrendt N, Jensen AL, Blasi F, Danø K (1991) Cellular receptor for urokinase plasminogen activator. Carboxyl-terminal processing and membrane anchoring by glycosyl-phosphatidylinositol. J Biol Chem 266: 1926–1933 [PubMed] [Google Scholar]
- Pluskota E, Soloviev DA, Plow EF (2003) Convergence of the adhesive and fibrinolytic systems: recognition of urokinase by integrin alpha Mbeta 2 as well as by the urokinase receptor regulates cell adhesion and migration. Blood 101: 1582–1590 [DOI] [PubMed] [Google Scholar]
- Roldan AL, Cubellis MV, Masucci MT, Behrendt N, Lund LR, Dano K, Appella E, Blasi F (1990) Cloning and expression of the receptor for human urokinase plasminogen activator, a central molecule in cell surface, plasmin dependent proteolysis. EMBO J 9: 467–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rømer J, Nielsen BS, Ploug M (2004) The urokinase receptor as a potential target in cancer therapy. Curr Pharm Des 10: 2359–2376 [DOI] [PubMed] [Google Scholar]
- Rosenberg S (2001) New developments in the urokinase-type plasminogen activator system. Expert Opin Ther Targets 5: 711–722 [DOI] [PubMed] [Google Scholar]
- Sato S, Kopitz C, Schmalix WA, Muehlenweg B, Kessler H, Schmitt M, Kruger A, Magdolen V (2002) High-affinity urokinase-derived cyclic peptides inhibiting urokinase/urokinase receptor-interaction: effects on tumor growth and spread. FEBS Lett 528: 212–216 [DOI] [PubMed] [Google Scholar]
- Schmiedeberg N, Schmitt M, Rolz C, Truffault V, Sukopp M, Burgle M, Wilhelm OG, Schmalix W, Magdolen V, Kessler H (2002) Synthesis, solution structure, and biological evaluation of urokinase type plasminogen activator (uPA)-derived receptor binding domain mimetics. J Med Chem 45: 4984–4994 [DOI] [PubMed] [Google Scholar]
- Stephens RW, Nielsen HJ, Christensen IJ, Thorlacius-Ussing O, Sørensen S, Danø K, Brünner N (1999) Plasma urokinase receptor levels in patients with colorectal cancer: relationship to prognosis. J Natl Cancer Inst 91: 869–874 [DOI] [PubMed] [Google Scholar]
- Stura EA (1999) Seeding techniques. In Crystllization of Nucleic Acids and Proteins: A Practical Approach, Dueruix A and Giegé R (eds) 2nd edn. Oxford: Oxford University Press pp 177–208 [Google Scholar]
- Stura EA (2001) A simple modification of the Q-plate for parallel screening and combinatorial crystallization. J Cryst Growth 232: 545–552 [Google Scholar]
- Thompson TB, Woodruff TK, Jardetzky TS (2003) Structures of an ActRIIB:activin A complex reveal a novel binding mode for TGF-beta ligand:receptor interactions. EMBO J 22: 1555–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres AM, Kini RM, Selvanayagam N, Kuchel PW (2001) NMR structure of bucandin, a neurotoxin from the venom of the Malayan krait (Bungarus candidus). Biochem J 360: 539–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Eble JA, Wang Z, Kreidberg JA, Chapman HA (2001) Urokinase receptors promote beta1 integrin function through interactions with integrin alpha3beta1. Mol Biol Cell 12: 2975–2986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, Lukashev M, Simon DI, Bodary SC, Rosenberg S, Doyle MV, Chapman HA (1996) Regulation of integrin function by the urokinase receptor. Science 273: 1551–1555 [DOI] [PubMed] [Google Scholar]
- Wei Y, Waltz DA, Rao N, Drummond RJ, Rosenberg S, Chapman HA (1994) Identification of the urokinase receptor as an adhesion receptor for vitronectin. J Biol Chem 269: 32380–32388 [PubMed] [Google Scholar]
- Zhou A, Huntington JA, Pannu NS, Carrell RW, Read RJ (2003) How vitronectin binds PAI-1 to modulate fibrinolysis and cell migration. Nat Struct Biol 10: 541–544 [DOI] [PubMed] [Google Scholar]
- Zhou HM, Nichols A, Meda P, Vassalli JD (2000) Urokinase-type plasminogen activator and its receptor synergize to promote pathogenic proteolysis. EMBO J 19: 4817–4826 [DOI] [PMC free article] [PubMed] [Google Scholar]