

Abstract
UDP-glucose:glycoprotein glucosyltransferase (GT) is a key component of the glycoprotein-specific folding and quality control system in the endoplasmic reticulum. By exclusively reglucosylating incompletely folded and assembled glycoproteins, it serves as a folding sensor that prolongs the association of newly synthesized glycoproteins with the chaperone-like lectins calnexin and calreticulin. Here, we address the mechanism by which GT recognizes and labels its substrates. Using an improved inhibitor assay based on soluble conformers of pancreatic ribonuclease in its glycosylated (RNase B) and unglycosylated (RNase A) forms, we found that the protein moiety of a misfolded conformer alone is sufficient for specific recognition by GT in vitro. To investigate the relationship between recognition and glucosylation, we tested a variety of glycosylation mutants of RNase S-Protein and an RNase mutant with a local folding defect [RNase C65S, C72S], as well as a series of loop insertion mutants. The results indicated that local folding defects in an otherwise correctly folded domain could be recognized by GT. Only glycans attached to the polypeptide within the misfolded sites were glucosylated.
Keywords: calnexin, calreticulin, endoplasmic reticulum, misfolding, protein folding
Introduction
The endoplasmic reticulum (ER) serves as the compartment for the processing, folding and oligomeric assembly of a large number of proteins in the eukaryotic cell. To promote their correct folding and maturation, the ER lumen contains a variety of folding enzymes and molecular chaperones (Gething and Sambrook, 1992). In addition, the ER supports a system, called quality control (QC), that allows it to distinguish proteins that have reached their native conformation from those that have not (Hurtley and Helenius, 1989; Ellgaard and Helenius, 2003). Export is generally limited to correctly folded and assembled conformers, whereas misfolded or unassembled proteins are transported from the ER to the cytosol and degraded in a process called ER-associated degradation (ERAD) (Klausner and Sitia, 1990). In many human diseases that involve expression of defective proteins in the ER, the quality control system is known to play a central role (Amara et al, 1992; Aridor and Balch, 1999).
The best-studied ER quality control system involves two homologous lectin-like chaperones, calnexin and calreticulin (Schrag et al, 2003; Helenius and Aebi, 2004). These interact with glycoproteins that carry monoglucosylated, N-linked glycans (Vassilakos et al, 1998) and promote their proper folding by preventing aggregation, by blocking premature export from the ER and by mediating their interaction with the thiol oxidoreductase ERp57.
The monoglucosylated oligosaccharides are generated either by removal of glucoses from ER-specific glycan structures (Glc1–3Man7–9GlcNAc2) by glucosidases I and II, or by reglucosylation of glucose-free trimming intermediates (Man7–9GlcNAc2) by UDP-glucose:glycoprotein glucosyltransferase (GT) (Parodi, 2000). The alternate action of these enzymes drives a cycle of substrate binding to and release from calnexin and calreticulin (Hammond et al, 1994).
In this study, we have focused on GT, which serves as the folding sensor in the cycle. In vivo and in vitro experiments have shown that it only reglucosylates glycoproteins that have non-native conformations (Trombetta et al, 1989; Ganan et al, 1991; Sousa et al, 1992; Gardner and Kearse, 1999). In doing so, it selectively returns incompletely folded glycoproteins to calnexin and calreticulin.
GT is a soluble, lumenal glycoprotein of 170 kDa ubiquitously expressed in the ER of most species (Trombetta et al, 1989; Fernandez et al, 1994; Parker et al, 1995; Arnold et al, 2000). It consists of an N-terminal part of about 1200 residues presumed to participate in protein recognition, and a C-terminal, catalytic segment of 300 amino acids with homology to members of the glycosyl transferase family 8 (Campbell et al, 1997; Tessier et al, 2000). GT transfers a glucose unit from UDP-glucose to protein-bound Man9GlcNAc2, Man8GlcNAc2 and Man7GlcNAc2 groups with decreasing efficiency (Sousa et al, 1992). It does not need additional factors except for UDP-glucose and Ca2+ ions to selectively reglucosylate non-native glycoproteins (Trombetta et al, 1989).
Experiments using hydrophobic peptides, hydrophobic glycoproteins or neoglycoproteins have shown that GT recognizes exposed hydrophobic patches or peptide elements (Sousa and Parodi, 1995; Parodi, 2000; Caramelo et al, 2003). Longer glycopeptides containing hydrophobic patches can also serve as substrates (Taylor et al, 2003). Isolated N-linked glycans and small glycopeptides are not glucosylated, and misfolded, non-glycosylated proteins have been found not to inhibit glucose transfer when mixed with bona fide substrates (Sousa et al, 1992; Sousa and Parodi, 1995). This has led to the suggestion that GT somehow recognizes the protein moiety and the glycan moiety together. Under the same assay conditions, the presence of only the innermost GlcNAc residue of the N-linked glycan was sufficient to make a denatured protein ‘visible' to the enzyme (Sousa et al, 1992; Sousa and Parodi, 1995).
Glycoproteins must be partially structured to be a substrate for GT (Ritter and Helenius, 2000; Trombetta and Helenius, 2000; Caramelo et al, 2003). In fact, late folding intermediates seem to be the preferred substrate (Caramelo et al, 2004). However, a mostly native-like conformer of ribonuclease B (RNase B) with a minor conformational change induced by a proteolytic ‘nick' in the protein failed to be recognized by GT. In studies with RNase B dimers, we found, furthermore, that GT reglucosylated glycans that were located in misfolded domains while it ignored glycans in nearby folded domains (Ritter and Helenius, 2000).
Here, we have extended our analysis of GT's substrate specificity using a variety of additional conformers and glycosylation mutants of RNase B and of its unglycosylated version, RNase A. The results show that the specificity-generating interactions between GT and non-native RNase molecules do not depend on the presence of carbohydrate moieties. However, reglucosylation is confined to N-linked glycans localized directly within structurally disturbed areas of the protein. As a folding sensor, GT thus labels misfolding in a strictly local context.
Results
Generation of RNase A and B conformers and mutants
Bovine pancreatic RNase A and B are small (124 aa), soluble enzymes with four intrachain disulfide bonds. They are identical except that RNase B has a high-mannose N-linked glycan in position Asn34. Both forms have been well characterized biophysically and functionally (Richards and Wyckow, 1971; Raines, 1998). They fold easily into their native conformation allowing the use of recombinant protein for many of our studies. Conformational variants can be generated in vitro by selective proteolysis with subtilisin using the so-called ribonuclease S system (Richards, 1958; Richards and Vithayathil, 1959; Trombetta and Helenius, 2000). Subtilisin induces a ‘nick' between Ala20 and Ser21, and renders the protein slightly altered but without significant changes in the enzymatic activity or the overall structure (Kim et al, 1992). However, when the 20-amino-acid-long N-terminal S-Peptide is removed to generate the RNase S-Protein, the protein is structurally disturbed and inactive. Rebinding of S-Peptide to form the reconstituted RNase S′ occurs with high affinity, and activity is restored.
A major disadvantage of the standard commercial RNase B samples used so far in GT studies is that they represent mixtures of glycoforms of which only ∼20% have sufficient mannose residues to be a substrate for GT (Zapun et al, 1997). The remaining ∼80% are likely to serve as a competitive inhibitor in the assay (Trombetta and Helenius, 2000). To overcome this problem, we employed an affinity chromatography procedure (Gonzalez et al, 2000) to isolate the Man8GlcNAc2 glycoform of RNase B and used it in a denatured form with scrambled disulfide bonds, which we call RNase B(Man8)sc.
To analyze mutants of RNase B carrying the proper N-linked glycan, we synthesized recombinant protein in a cell-free system. By subjecting synthetic mRNA to in vitro translation in the presence of dog pancreas microsomes and [35S]methionine, we could generate small amounts of radioactive, folded RNase B and mutants that contained the correct high-mannose oligosaccharides.
GT is inhibited by misfolded non-glycoproteins
To analyze whether bound carbohydrates are needed for GT to interact with non-native proteins, we modified the in vitro assay of Sousa et al (1992). In this assay, the rate of [3H]glucose transfer from UDP-[3H]glucose to an acceptor glycoprotein is measured. We used the RNase B(Man8)sc described above, and adjusted the concentrations of GT and RNase B(Man8)sc so that the assay could be performed with saturating amounts of substrate glycoprotein. RNase B(Man8)sc was present as monomers (>95%) and as disulfide-linked dimers (<5%). Ultracentrifugation verified that no large aggregates formed throughout the assay, indicating that the proteins remained as monomers or small oligomers typical for the size range of GT's substrates.
The rate of RNase B(Man8)sc glucosylation was measured in the presence of different conformers of RNase A and glycosidase-modified RNase B (see Materials and methods). While these could not serve as glucose acceptors, some of them were found to be efficient inhibitors of GT (Figure 1). The most effective inhibitors were RNase Asc and RNase B(GlcNAc1)sc, whose protein moieties were identical to the substrate RNase B(Man8)sc. A group of inhibitors of intermediate potency contained three RNase S-Proteins: RNase AS-Protein, RNase BS-Protein(GlcNAc1) and RNase BS-Protein(Man1GlcNAc2). Native or native-like conformers (RNase A, RNase B and the nicked RNase AS) did not inhibit GT to any significant extent.
Figure 1.
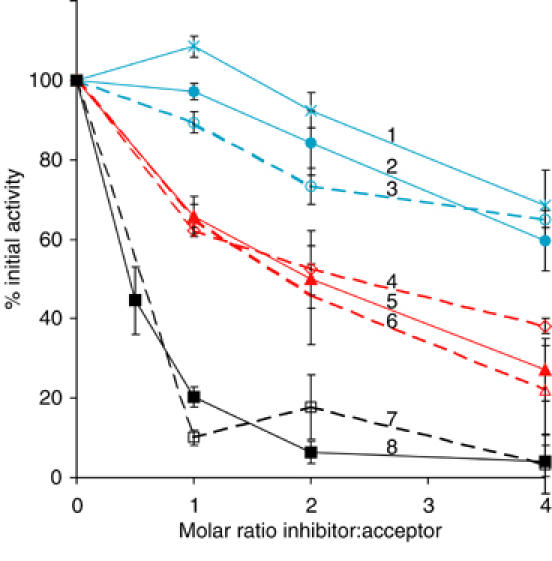
Competitive inhibition of GT by misfolded non-acceptor RNase conformers. Incorporation of [3H]glucose into RNase B(Man8)sc was measured in the presence of a 0- to 4-fold molar excess of inhibitor relative to the uninhibited value for the following inhibitors: 1, RNase AS; 2, RNase A(native); 3, RNase B(native); 4, RNase BS-Protein(Man1GlcNAc2); 5, RNase AS-Protein; 6, RNase BS-Protein(GlcNAc1); 7, RNase B(GlcNAc1)sc; 8, RNase Asc.
Three main conclusions could be drawn from these observations. Firstly, they confirmed published observations that to inhibit GT efficiently, the conformation of a non-substrate protein has to be non-native (Sousa et al, 1992; Sousa and Parodi, 1995). Secondly, it showed that to distinguish between native and non-native proteins, GT does not need glycan moieties to be present. This result differed from reports indicating that various non-glycoproteins fail to inhibit GT (Sousa et al, 1992; Sousa and Parodi, 1995; Trombetta and Helenius, 2000). The reason for the discrepancy is probably the improved assay (see Discussion). Finally, the results suggested that the protein–protein interactions between the incorrectly folded RNases and GT are rather weak. From the curves in Figure 1, the Ki for a scrambled inhibitor could be estimated to be in the range of 50 μM, while the Ki for the RNase S-Proteins was about 250 μM. Weak interactions may be necessary to avoid formation of unproductive, stable complexes between GT and incompletely folded proteins in the ER.
Glycosylation mutants are recognized by GT
In a multidomain glycoprotein, GT reglucosylates only glycans located in a misfolded domain (Ritter and Helenius, 2000). In order to determine whether it makes a difference in which part of such a domain the glycan is located, we analyzed the S-Proteins of RNase B mutants in which the glycan was placed in different parts of the enzyme. We eliminated the wild-type glycosylation site by replacing Asn34 by Gln, and changed a single residue in each mutant to Asn or Thr to generate new glycosylation consensus sequences (Asn-X-Ser/Thr). The new sites were distributed in loops and β-strands over the entire surface of the molecule (Figure 2).
Figure 2.
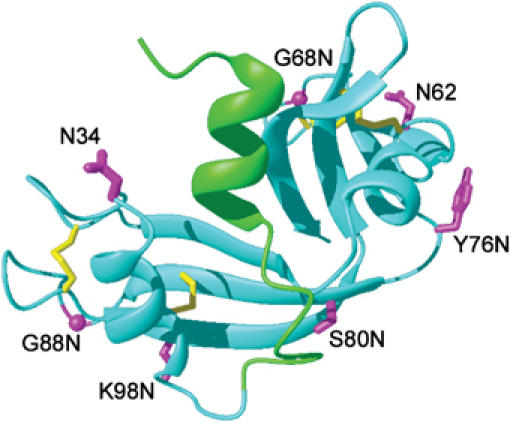
Glycosylation mutants of RNase. The different glycosylation sites introduced into RNase A are depicted in a model of RNase A based on its NMR structure (Santoro et al, 1993). Cyan: protein core (residues 21–124) that forms the S-Protein; green: residues 1–20 corresponding to the S-Peptide; magenta: amino acids turned into an Asn residue to which an N-linked glycan can be attached; yellow: disulfide bonds. The figure was generated with MolMol (Koradi et al, 1996).
Radioactively labeled recombinant RNase B (wild type) and the glycosylation mutants were generated in a cell-free translation–translocation system. SDS–PAGE analysis of the lysed microsomes containing the wild-type RNase (N34) and six mutants showed two bands, of which the upper band corresponded to RNase B (Mr 13.690 kDa) and the lower band to unglycosylated RNase A (Mr 11.542 kDa) (Figure 3A). All mutants were glycosylated to a significant degree.
Figure 3.
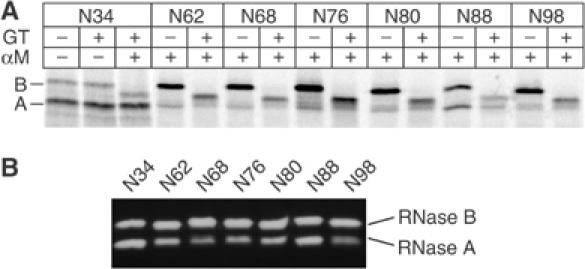
Analysis of glycosylation mutants of RNase. (A) 35S-labeled RNase mutants derived from in vitro translation/translocation were isolated from solubilized microsomes and treated with GT and α-mannosidase where indicated. Differences in molecular weight due to glycan modifications were visualized by SDS–PAGE. The mutants are indicated by the position of their N-linked glycan. A, B: untreated RNase A and B; αM: α-mannosidase. (B) RNase activity of the RNase glycosylation mutants was assayed by zymogram electrophoresis.
That the introduction of novel glycosylation sites had no major effect on the structure of the native proteins was supported by three observations. Firstly, all lysates contained active RNase A and B when analyzed by zymogram electrophoresis (delCardayre et al, 1995) (Figure 3B). Secondly, treatment with subtilisin resulted in a cleavage product with a molecular weight corresponding to that of RNase BS-Protein (for N34 see Figure 4A, lanes 1 and 2, and for N62, lanes 6 and 7). Non-native forms of RNase are fully degraded by this protease. Finally, none of the full-length RNase B glycoforms were reglucosylated by GT prior to the subtilisin digest (Figure 3A). Reglucosylation was assayed by treating the lysates first with GT and subsequently with jack-bean α-mannosidase followed by SDS–PAGE (Hammond et al, 1994; Hebert et al, 1995). α-Mannosidase accentuates mobility differences in SDS–PAGE between glycoproteins containing a terminal glucose residue in their glycan and those that do not, because it trims terminal α-linked mannose residues only (see Figure 4A, lanes 4 and 5, marked GlcNAc2Man4Glc1 and GlcNAc2Man1). The RNase glycoforms in Figure 3A show a large shift in position upon α-mannosidase treatment, indicating that the proteins were not substrates for GT.
Figure 4.
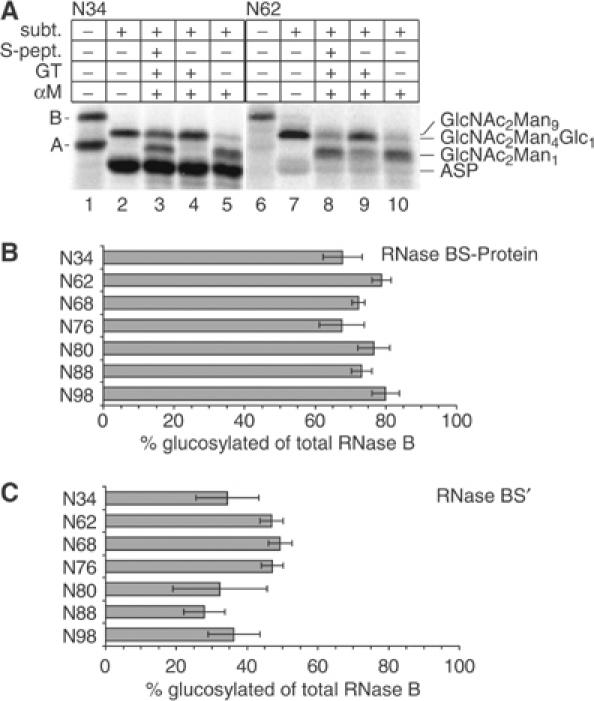
Recognition of glycosylation mutants of RNase S-Protein by GT. (A) 35S-labeled RNase B N34 (lanes 1–5) or RNase B N62 (lanes 6–10) was generated in vitro, and its S-Proteins prepared. Samples where incubated for 40 min with or without 0.1 U GT as indicated and then subjected to α-mannosidase digest. Analysis was performed by 15% SDS–PAGE. A: RNase A; B: RNase B; ASP: RNase AS-Protein; GlcNAc2Man9: untreated RNase BS-Protein with the original mannose content (lanes 2, 7); GlcNAc2Man4Glc1: glucosylated RNase BS-Protein after α-mannosidase treatment; GlcNAc2Man1: unglucosylated RNase BS-Protein after α-mannosidase treatment; subt.: treated with subtilisin. (B) Quantification of GT glucosylation of mutant RNase BS-Proteins. All glycosylation mutants were treated as described in (A). Quantification of each band was performed as described in Materials and methods and the fraction of RNase BS-Protein that became glucosylated relative to the total RNase BS-Protein present in that lane was calculated (BSP(Man4Glc1)/[BSP(Man4Glc1)+BSP(Man1)]). The glycosylation mutants are indicated by the position of the Asn residue to which the glycans were attached. (C) Quantification of GT glucosylation of mutant RNase BS-Proteins reconstituted with S-Peptide. The analysis was performed as in (B).
Next, S-Proteins as well as the S-Proteins reconstituted with S-Peptide were prepared from the various RNase mutants and exposed to GT. SDS–PAGE analysis of the recombinant RNase N34 S-Protein (i.e. the wild-type sequence) is shown in Figure 4A, with the original mixture of RNase A and B in lane 1, and the S-Proteins generated from the same mixture in lane 2. When RNase BS-Protein was treated with α-mannosidase alone (lane 5), the large shift compared to the untreated S-Protein (lane 2) indicated that the majority of the protein lacked glucose residues that would protect the mannoses against α-mannosidase. However, if the RNase BS-Protein was treated first with GT (lane 4), the shift was virtually absent. This confirmed that in vitro-generated RNase BS-Protein with a glycan at position 34 is a substrate for GT.
When the S-Peptide was added back to the RNase BS-Protein (lane 3), about 60% of the RNase BS-Protein was reconstituted, resulting in the appearance of a GT-resistant protein band. This control indicated that a large fraction of our recombinant S-Protein was in a folding-competent state comparable to the well-characterized S-Protein derived from purified RNase.
The same analysis for the mutant N62 is shown in lanes 6–10 (Figure 4A). The pattern is very similar to that of wild-type RNase BS-Protein, indicating that the mutant S-Protein was also a substrate for GT. As shown in the summary of quantified data (Figure 4B and C), all the mutants behaved like the wild-type protein when analyzed in this assay. While none of the original native forms were recognized by GT (Figure 3A), all RNase BS-Proteins were good substrates. Reconstitution with S-Peptide resulted in each case in a significant decrease of recognition. The differences in the residual glucosylation probably reflect the ability of the mutants generated in vitro to refold in the presence of S-peptide rather than recognition of the reconstituted forms. Firstly, RNase BS′ N34 also became partially glucosylated, while this form does not get glucosylated when purified proteins are used. Secondly, it has been shown recently that the degree of glucosylation in a mildly perturbed protein depends on the degree of local hydrophobicity (Taylor et al, 2004). No such correlation between local hydrophobicity and glucosylation could be found for the reconstituted mutants (see Supplementary data). Thus, all mutants behaved similar to RNase B wild type regardless of the location of the glycan.
We concluded that GT not only recognized the conformational difference between native, nicked and S-Peptide-depleted conformations in all these mutants, but it was also able to modify the glycan moiety regardless of its position on the surface of the protein, and of the detailed composition of amino acids in its vicinity. Evidently, to be efficiently glucosylated, a glycan had to be located in a misfolded domain, but not in any defined location within it.
Recognition of local folding defects
Since the structure of RNase S-Protein is not known, it remained a possibility that the glycosylation sites analyzed above were all in regions altered in structural or dynamic properties.
To test whether GT was able to differentiate between glycans attached to native-like and disturbed regions in a single protein domain, we took advantage of the [C65S, C72S] RNase mutant that lacks one of the four native disulfide bonds. Its RNase A form has been extensively compared to wild-type RNase A both in terms of structure and thermodynamic stability in vitro (Shimotakahara et al, 1997). Importantly, a detailed NMR analysis has established that at 20°C, [C65S, C72S] RNase A displays changes only in the spatial vicinity of the disrupted disulfide bond while other parts of the protein are essentially unaffected. As summarized in Figure 5, a short antiparallel β-sheet and the loop between both strands (Gln60–Ser80) are disrupted, and the spatially adjacent β-strands are also influenced. Thermodynamic measurements and 1H/2H exchange data have further revealed a global destabilization of [C65S, C72S] RNase resulting in a significantly reduced Tm (38.5°C compared to 55.5°C for wild-type RNase A). Since the extent of misfolding of this mutant could be adjusted by temperature, it offered itself as a potentially useful tool to analyze the effects of local and global folding defects.
Figure 5.
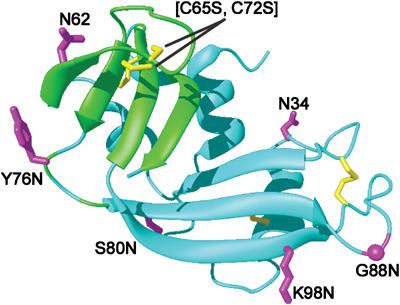
Structural defects in [C65S, C72S] RNase. Differences between the mutant and wild-type RNase A as determined by NMR analysis (Shimotakahara et al, 1997) are depicted in a model of RNase A based on its NMR structure (Santoro et al, 1993). The molecule is rotated 180° around the y-axis relative to Figure 3. Cyan: residues that do not change significantly compared to wild-type RNase A; green: residues displaying significant differences in chemical shifts or 1H/2H exchange rates at 20°C; yellow: native disulfide bonds and Cys residues changed to Ser in the mutant; magenta: amino acids turned into an Asn residue to which an N-linked glycan can be attached. The figure was generated with MolMol (Koradi et al, 1996).
Different glycosylation mutants of [C65S, C72S] RNase with single glycosylation sites at positions described above (Figures 2 and 5) were generated. Since [C65S, C72S] RNase displays only partial enzymatic activity (Shimotakahara et al, 1997), we assayed folding by monitoring trypsin resistance (Lang and Schmid, 1986). All glycosylation mutants of [C65S, C72S] RNase acquired trypsin resistance upon oxidative folding (Figure 6A), while the reduced form of [C65S, C72S] RNase N34 was fully degraded (lane 2). We concluded that the fold of the mutants was not significantly impaired. To further analyze whether the introduction of point mutations and the presence of glycans at different positions in the molecule altered the stability relative to the unglycosylated form characterized by Shimotakahara et al, we subjected [C65S, C72S] RNase N34 and N62 to a series of trypsin pulses with increasing protease concentrations. N34 (the wild-type sequence) is located in the most stable and N62 in the most destabilized region of the molecule. As shown in Figure 6B, trypsin resistance of the two mutants decreased identically with increasing protease concentrations, whereas wild-type RNase B remained resistant. This indicated that the introduction of the point mutation N62 in direct proximity to the disrupted disulfide bond did not destabilize the protein further. Since all mutants were generated as a mix of glycosylated and unglycosylated molecules, we could also show that in each case the glycosylated form was at least as protease resistant as the unglycosylated one (Figure 6B, hatched bars versus solid bars).
Figure 6.
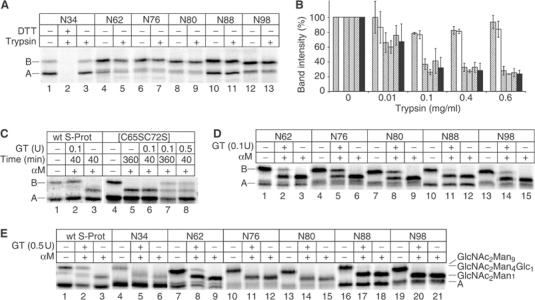
Characterization of [C65S, C72S] RNase and its glycosylation mutants. 35S-labeled wild-type RNase or glycosylation mutants of [C65S, C72S] RNase containing N-linked glycans at the indicated positions were generated in vitro. (A) Glycosylation mutants of [C65S, C72S] RNase were incubated for 5 min at 20°C with or without 0.01 mg/ml trypsin. Where indicated, the samples were first reduced with 50 mM dithiothreitol (DTT) at 95°C. (B) Quantification of trypsin pulses. RNase N34 (white), [C65S, C72S] RNase N34 (gray) and N62 (black) were incubated with the indicated amount of trypsin for 5 min at 20°C and analyzed by SDS–PAGE. The bands corresponding to the undigested proteins with and without glycan were quantified separately. Data from triplicate experiments are shown as percent of the band intensities of the samples treated without trypsin. Hatched: glycosylated protein; solid: unglycosylated protein. (C) Wild-type RNase S-Protein or [C65S, C72S] RNase N34 was incubated at 37°C with different amounts of GT for different times as indicated, and then treated with α-mannosidase. (D) Glycosylation mutants of [C65S, C72S] RNase were incubated with GT at 37°C for 40 min as indicated, and then treated with α-mannosidase. (E) Wild-type RNase S-Protein or glycosylation mutants of [C65S, C72S] RNase were incubated with GT at 20°C for 300 min as indicated, and then treated with α-mannosidase. A: unglycosylated protein; B: glycosylated protein without glycan modifications. Labeling of glycoforms is as in Figure 4.
As shown in Figure 6C and D, the efficiency of glucosylation depended on the distance from the missing disulfide bond. At 37°C, significant glucosylation could be observed only in glycans adjacent to the disrupted disulfide bond, namely glycans in positions N62, N80 and N76. The glycan N98 was only weakly glucosylated, N88 even less and N34 not at all.
According to the biophysical analysis of [C65S, C72S] RNase A, the molecule begins to undergo reversible global denaturation at 37°C. Thus, we expected some glucosylation of all glycans. Indeed, if we extended the incubation to 360 min or increased the amount of GT in the assay five-fold, we could observe significant amount of glucosylation of [C65S, C72S] RNase B N34 (Figure 6C, lanes 7 and 8). This was consistent with the presence of a globally destabilized form of the protein in equilibrium with the mostly folded conformation.
At 20°C, [C65S, C72S] RNase is thermodynamically stable (Shimotakahara et al, 1997). The glucosylation of wild-type RNase BS-Protein N34 demonstrated that GT was still active at this temperature (Figure 6E, lane 2), although incubation times and the amount of enzyme had to be increased. Under these conditions, only [C65S, C72S] RNase N62, in which the glycan is located only three residues from the disrupted disulfide bond, was significantly reglucosylated (Figure 6E, lane 8). Recognition was rather inefficient, suggesting that the structural defect displayed by [C65S, C72S] RNase N62 at 20°C belongs to the smallest detectable by GT.
Small loop insertions can trigger glucosylation
To address the nature of the structural perturbation needed to make an attached glycan a substrate for GT, we generated mutants of RNase in which we added sequences of amino acids into the loop that contains the site nicked by subtilisin (loop S16–S22) (see Figure 2). These carried a consensus glycosylation site either at the wild-type position (N34) or within the inserted sequences.
One of the elements inserted was a 15-amino-acid polar linker sequence (PL) designed to be mobile and to lack secondary structure (Pelletier et al, 1999). The second (HL) was a 21-amino-acid-long partially hydrophobic sequence from GroES (R14–K34) known to display considerable flexibility when GroES is in solution (Landry et al, 1993). It binds to GroEL via hydrophobic interactions and might therefore mimic a hydrophobic peptide exposed in a misfolded protein. Finally, we introduced the mostly hydrophobic sequence A162–Y181 (2β), which comprises two strands of a β-sheet in the hydrophobic core of soybean agglutinin. The resulting proteins with the glycosylation sites in the loop were named RNasePL-N9L, RNaseHL-N6L and RNase2β-N16L, respectively. The added sequences were short and therefore unlikely to appear as separate domains.
The mutant proteins were generated in radioactive form in the cell-free system. To verify that they were correctly folded, we tested their RNase activity by zymogram electrophoresis (Figure 7A). They all showed RNase activity comparable to that of wild-type RNase. RNase2β N34 turned out to be aggregation-prone and was therefore not analyzed further (data not shown).
Figure 7.
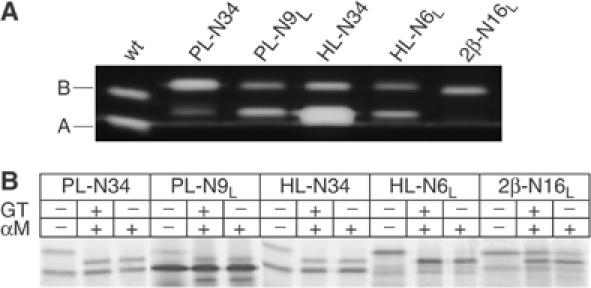
Analysis of small loop insertions into RNase. (A) RNase activity of the RNase loop insertion mutants from a 25 μl translation/translocation mix was assayed by zymogram electrophoresis. For RNaseHL-N6L, more material was loaded in order to visualize the glycosylated form. (B) 35S-labeled RNase loop insertions derived from in vitro translation/translocation were isolated from solubilized microsomes and treated with GT and α-mannosidase where indicated. Differences in molecular weight due to glucosylation were analyzed by 15% SDS–PAGE.
Next, we checked for recognition by GT. When added to the GT assay, the only mutant to become partially glucosylated was RNase2β-N16L, indicating that the 18-amino-acid-long loop could elicit a local signal for GT recognition and reglucosylation albeit not as efficiently as the RNase S-Proteins. The overall hydrophobicity of this loop was significantly higher than that of the other two insertions. Additionally, it might have been partially structured forming a β-hairpin element unlike the other mutants that had flexible loop structures.
Discussion
In many respects, the substrate recognition by GT seems to resemble some of the classical chaperones. However, since GT is an enzyme, it is probably designed for rapid substrate throughput rather than for binding and retention. In vitro characterization indicates that GT is, in fact, a rather slow enzyme with poor catalytic efficiency (Tessier et al, 2000; K Quirin, C Ritter and A Helenius, unpublished results).
The first question addressed in our study was whether recognition of misfolded protein substrates requires the presence of N-linked glycans in the substrate protein using soluble bovine pancreatic RNases as substrates and inhibitors. While the results confirmed that a non-native conformation is essential for interaction, they deviated from previous reports (Sousa et al, 1992; Sousa and Parodi, 1995; Trombetta and Helenius, 2000) in indicating that a glycan moiety was not needed to competitively inhibit the enzyme's interaction with a bona fide glycoprotein substrate. This difference is most likely explained by the improved assay. All protein components used were soluble in contrast to the large, heterogeneous aggregates previously employed by us and others (Sousa and Parodi, 1995). Non-acceptor RNase glycoforms were absent, and the assay was optimized for measurements under steady-state conditions. Since the acceptor and inhibitor proteins had the same amino-acid sequence, differences were limited to conformations and the presence or absence of the N-linked glycan. Pancreatic RNase B had the additional advantage that modifications of the glycan moiety could be performed on the native protein without harsh denaturation conditions.
The observation that GT can sense the folding state of a substrate via the protein moiety alone is not entirely unexpected: Sousa and Parodi, (1995) reported that GT binds to immobilized, hydrophobic peptides devoid of glycans. The finding simplifies the formulation of mechanistic models of substrate recognition, which now can focus on the protein moiety. To allow rapid capture and release, the interaction between GT and misfolded protein domains is probably rather weak. Therefore, the formation of a productive enzyme–substrate complex may require additional contacts with the N-linked glycan. Whether direct protein–protein interactions with non-glycoproteins play any role in the ER remains to be seen.
The second question addressed was how the enzymatic center and the protein-binding sites in GT are functionally and spatially linked. To obtain detailed insight into whether GT can distinguish between glycans in different regions within a single domain, we analyzed three classes of RNase B variants that displayed defined folding defects.
The first comprised glycosylation mutants of RNase BS-Protein in which the location of the glycan was moved to different places on the protein's surface. Regardless of the location, all glycans in the RNase BS-Protein were reglucosylated, while the corresponding native RNase B variants were not. NMR spectroscopy and molecular dynamics simulations indicate that although the main-chain fold is essentially unaltered compared to native RNase A, RNase AS-Protein displays increased backbone dynamics throughout the molecule (V Galius, C Ritter, A Helenius and K Pervushin, unpublished data; S Cotesta and E Di Iorio, personal communication). It is not clear whether RNase AS-Protein can be considered a so-called ‘molten globule' (Graziano et al, 1996), but it seems to have some properties reminiscent of such states. It has been suggested that GT recognizes residues belonging to the hydrophobic core that become exposed by the removal of the N-terminal S-Peptide (Caramelo et al, 2003). This seems unlikely in view of our findings indicating local recognition (see below). Since in our mutants some of the glycans were located in positions distant from this hydrophobic patch, it is more likely that they were reglucosylated because they were all positioned in locally perturbed regions. The main conclusion from these data is that within a globally destabilized protein, GT was able to recognize all glycans whether attached to endogenous or artificially engineered glycosylation sites.
Recently, it has been shown that even subtle structural alterations caused by a single point mutation can result in the recognition of a glycan at the opposite site of the molecule (Taylor et al, 2004). The authors speculated that the point mutation should have caused local conformational changes only, and concluded that GT can modify glycans distant from a structural perturbation. Here we used glycosylation mutants of [C65S, C72S] RNase as model substrates, since [C65S, C72S] RNase A exhibits a localized folding defect that has been precisely characterized by NMR (Shimotakahara et al, 1997). In wild-type RNase, introduction of these glycosylation mutants appeared to have no influence on folding or enzymatic activity. In [C65S, C72S] RNase, we could also not observe differences in trypsin resistance between the glycosylated and non-glycosylated versions of each mutant, nor between the different mutants. These findings corroborate that the glycosylation mutants are likely to exhibit the described local folding defect of [C65S, C72S] RNase, and lead to a consistent picture: the closer a glycan was located to the disrupted region, the better it was glucosylated by GT. Even though RNase is a small protein with a maximum diameter of 4.2 nm, GT selectively glucosylated glycans only when they were attached to the perturbed region of the molecule. We deduced from this observation that (i) GT is able to detect minor folding defects within otherwise properly folded protein domains and (ii) in order to be reglucosylated, a glycan must be located close to or within a perturbed region.
The analysis of the [C65S, C72S] RNase mutant provided additional insights into the fine-tuning of substrate recognition by GT. At 20°C, of the six glycosylation sites in RNase [C65S, C72], only the one attached to the center of the perturbed region was glucosylated. Raising the temperature to 37°C resulted in labeling of glycans at two additional sites in adjacent regions. Glycans in other regions remained virtually unglucosylated. The structural defect displayed by [C65S, C72S] RNase N62 at 20°C seemed to belong to the smallest recognizable by GT, because even this glycan was not as efficiently glucosylated as the one in RNase BS-Protein N34 at this temperature.
A similar behavior toward localized, small folding defects could be observed for our third model system, the RNase loop insertion mutants. In this case, only RNase2β N16L with the glycan attached to a hydrophobic, possibly structured, loop became partially glucosylated. A stretch of hydrophobic residues displayed in RNasePL was not sufficient to trigger glucosylation. This indicates that other features, in addition to the presence of hydrophobic residues, might be important, such as slow dynamic motions of the protein backbone, excessive mobility of amino-acid side chains at the protein surface, or disrupted tertiary structure. Secondary structure elements may, for example, be insufficiently anchored in the protein fold and therefore be more accessible to GT.
Presumably, protein and oligosaccharide binding takes place at distinct sites because at least initial, conformation-dependent binding can occur in the absence of a glycan moiety. To account for the strict selectivity for glycosylation sites in or close to the perturbed regions, protein- and glycan-binding sites of GT are likely to be spatially close to each other. The architecture of the glycan-binding site may require a certain deformability of the immediately surrounding polypeptide chain to accommodate the glycan for a productive interaction. Binding of the protein moiety may be necessary to provide sufficient affinity and specificity. This would avoid tagging of glycans present on naturally occurring flexible regions of a glycoprotein.
Clearly, analysis of other substrate proteins is needed to better understand the molecular characteristics in non-native proteins assessed by GT. The picture that emerges from our results and previous studies indicates that already small, local deviations from the native fold are detected by GT, and that this results in the local glucosylation of glycans. The presence of some type of ordered secondary structure seems to be a prerequisite for recognition.
The specific labeling of glycans attached to protein areas with deviations from the native fold is likely to enable glycoproteins to draw the attention of the ER quality control and folding system to sites that might be critical for the folding and function in each specific protein. At the same time, by eliminating N-linked glycans from a region, it can divert attention to other sites. The distribution of N-linked glycans on the surface of glycoproteins may thus be in part governed by which parts of the molecule need special attention with respect to folding and quality control. Since quality control of glycoproteins plays a central role in the underlying principles of selective recognition of misfolded conformers in the secretory pathway, further analysis of GT is likely to expand our understanding of human diseases with an ER storage phenotype.
Materials and methods
Construction of plasmids
For cloning of the loop mutants, the Nde1/BamHI fragment of the plasmid pF(BGL)RNase_wt was transferred into the pRSET A (Invitrogen) vector. Oligonucleotide cassettes coding for R14–K34 of GroES (RKEVENKSAGGIVLTGSAAAK), a polar linker (RSEASGTSNGTSSTG) (Pelletier et al, 1999) or soybean agglutinin A162–Y181 (RAKVLITYDASTSLLNASLVY) containing a 5′ NotI cleavage site were constructed. The vector pRSet A/RNase was linearized by PCR using a 5′ phosphorylated forward primer starting at the codon for S21 of RNase A and a reverse primer that started at the codon for S15 of RNase A, and contained a 5′ NotI cleavage site. The ligated NotI-derived constructs were transferred back into the original vector. Single amino-acid changes were introduced using the quick change site-directed mutagenesis kit (Stratagene).
Protein preparation
GT and glucosidase II were isolated from bovine and rat liver (Trombetta and Parodi, 1992; Trombetta et al, 1996). RNase B carrying a Man8 oligosaccharide was affinity purified from bovine RNase B (Sigma) as described (Gonzalez et al, 2000). Subtilisin treatment of RNase B was performed as described (Trombetta and Helenius, 2000), and the S-Protein was isolated by precipitation with 4% TCA (Richards and Vithayathil, 1959). To generate RNase B(Man8)sc, the native protein was reduced in 50 mM NaCl, 20 mM HEPES pH 7.0 (HBS) and 20 mM dithiothreitol at 95°C for 20 min, diluted 20-fold into 6 M guanidinium hydrochloride and oxidized by the addition of 7.5 mM diamide (Sigma). After 1 h at room temperature (RT), residual free cysteines were blocked with 50 mM iodoacetamide (Sigma) and RNase B(Man8)sc monomers isolated by gel filtration with Sephadex75 in HBS. Endoglycosidase H treatment of RNase B to generate the GlcNAc1 glycoform was performed without prior denaturation. A 10 mg portion of RNase B was incubated with 5 μl endo Hf (NEB) in NEB-buffer G5 for 16 h at 37°C. endo Hf was removed by gel filtration via Sephadex75. Preparative treatment with α-mannosidase to generate the Man1GlcNAc2 glycoform was performed in 50 mM citrate buffer pH 4.4 for 16 h at 37°C using 0.5 U α-mannosidase (Sigma) for 10 mg RNase B. The proteins were separated via Sephadex75.
In vitro transcription, translation/translocation and oxidation of RNase isoforms and generation of their S-Proteins
To generate mRNA coding for the respective RNase isoforms plus signal sequence, the pF(BGL)-derived plasmids were linearized with EcoRV beyond the 3′ end of the coding region. The DNA fragments were then transcribed in vitro (Weihofen et al, 2000). Translation of mRNA coding for the RNase isoforms was performed in 75 μl reticulocyte lysate (Promega) containing 3 μl 35S-labeled Promix methionine and cysteine (Amersham-Pharmacia) and 3 equiv. of nuclease-treated rough microsomes prepared from dog pancreas (Walter and Blobel, 1983). Samples were incubated for 20 min at 30°C. Translation was stopped with 0.5 mM cycloheximide and 1 mM methionine. Post-translational folding was initiated by adding 4.5 mM oxidized glutathione and carried out for 1 h at 27°C (Rodan et al, 1996). Microsomes were isolated as described (Weihofen et al, 2000) and solubilized with 50 μl of 1% Triton X-100 in the buffer required for the next step. Detergent and membrane fragments were removed with 15 mg bio-beads (Bio-Rad) for 75 min at RT. The volume of the supernatant was adjusted to 100 μl. S-Proteins were generated by treatment with 1.5 μg subtilisin for 4 h on ice in 50 mM TRIS pH 8.0 in the presence of 50 μg RNase A as carrier protein. The digest was stopped with 2 mM PMSF and the S-Protein isolated by precipitation with 4% TCA. Residual TCA was removed by dilution with HBS and concentration to a final volume of 50 μl.
Inhibitor assay
To ensure assay reproducibility, we ascertained that the incorporation of [3H]glucose was directly proportional to GT activity within the relevant range (4–70 mU). One unit (1 U) corresponds to the transfer of 1 pmol [3H]glucose/min. A 0.12 mM portion of RNase B(Man8)sc was mixed with a 0- to 4-fold molar excess of inhibitor protein and incubated at 37°C for 5 min with 0.5 μM UDP-[3H]glucose (Amersham), 4 mM CaCl2 and 17.5 mU GT in HBS. As baseline, the respective amounts of inhibitor protein were treated identically with GT in the absence of acceptor protein. The retained radioactivity did not exceed 10%. The assay was stopped with 1 ml 10% TCA and 0.08% sodium deoxycholate. The precipitate was washed three times by resuspension in 0.9 ml H2O and reprecipitation with 10% TCA. Incorporated [3H]glucose was quantified by liquid scintillation counting. All assays were repeated at least three times. Protein solubility was assayed by ultracentrifugation after the GT assay. At a four-fold excess of RNase Asc, 90% of the total protein remained in the supernatant as determined by BCA assay (Pierce) and by UV spectroscopy, and was thus considered as soluble.
Assay for the recognition of radiolabeled RNase isoforms by GT
Where indicated, samples were incubated with 0.5 mM S-Peptide for 20 min at RT to form RNase BS′. Aliquots (15 μl) were treated with 0.1 U GT or with HBS for 40 min at 37°C in the presence of 10 mM CaCl2 and 1 mM UDP-glucose in HBS. The assay was stopped and mannose trimming initiated by the addition of 30 μl 50 mM sodium citrate pH 4.4 containing 0.1 U jack-bean α-mannosidase (Sigma). After 1 h at 37°C, protein was precipitated with 10% TCA and 0.04% sodium deoxycholate and analyzed by reducing 15% SDS–PAGE. Labeled proteins were visualized by a STORM PhosphorImager (Molecular Dynamics). To quantify individual bands, a profile of each lane was created using the program ImageQuant and fitted as a sum of Gaussians using Microsoft Excel. One Gaussian function was used for each protein species possibly present in one lane. Control lanes were quantified and used to verify the completeness of the various enzymatic modifications. Optimal definition of each band position was achieved by comparing with control lanes and by comparing corresponding lanes of all glycosylation mutants from one experiment.
Trypsin resistance
A 10 μl portion of translation product was incubated in 50 mM TRIS pH 8.0 for 5 min at 20°C with the indicated amount of trypsin. After precipitation with 10% TCA, samples were analyzed by SDS–PAGE and visualized by a STORM PhosphorImager (Molecular Dynamics). Bands were quantified using the software ‘Quantity One' (Bio-Rad).
Zymogram electrophoresis
RNase activity was confirmed by zymogram electrophoresis (delCardayre et al, 1995). The proteins derived from a 25 μl translation/translocation mix were separated on a 15% SDS–polyacrylamide gel copolymerized with 0.5 mg/ml poly(C), refolded by removing SDS with 20% 2-propanol in 50 mM TRIS pH 7.0 and incubated in 50 mM TRIS pH 7.0 at RT for 30 min. Undigested poly(C) was stained with toluidine blue O, leaving clear bands at the positions corresponding to a Coomassie-stained gel where active RNase was present.
Supplementary Material
Supplementary Data
Acknowledgments
We thank Dr B Martoglio for providing microsomes and A Lingel and L Croci-Torti for help with the cloning of mutants. Funding was obtained from the Swiss National Science Foundation and from the ETHZ.
References
- Amara JF, Cheng SH, Smith AE (1992) Intracellular protein trafficking defects in human disease. Trends Cell Biol 2: 145–149 [DOI] [PubMed] [Google Scholar]
- Aridor M, Balch WE (1999) Integration of endoplasmic reticulum signaling in health and disease. Nat Med 5: 745–751 [DOI] [PubMed] [Google Scholar]
- Arnold SM, Fessler LI, Fessler JH, Kaufman RJ (2000) Two homologues encoding human UDP-glucose:glycoprotein glucosyltransferase differ in mRNA expression and enzymatic activity. Biochemistry 39: 2149–2163 [DOI] [PubMed] [Google Scholar]
- Campbell JA, Davies GJ, Bulone V, Henrissat B (1997) A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem J 326 (Part 3): 929–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramelo JJ, Castro OA, Alonso LG, De Prat-Gay G, Parodi AJ (2003) UDP-Glc:glycoprotein glucosyltransferase recognizes structured and solvent accessible hydrophobic patches in molten globule-like folding intermediates. Proc Natl Acad Sci USA 100: 86–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramelo JJ, Castro OA, De Prat-Gay G, Parodi AJ (2004) The endoplasmic reticulum glucosyltransferase recognizes nearly native glycoprotein folding intermediates. J Biol Chem 279: 46280–46285 [DOI] [PubMed] [Google Scholar]
- delCardayre SB, Ribo M, Yokel EM, Quirk DJ, Rutter WJ, Raines RT (1995) Engineering ribonuclease A: production, purification and characterization of wild-type enzyme and mutants at Gln11. Protein Eng 8: 261–273 [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A (2003) Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4: 181–191 [DOI] [PubMed] [Google Scholar]
- Fernandez FS, Trombetta SE, Hellman U, Parodi AJ (1994) Purification to homogeneity of UDP-glucose:glycoprotein glucosyltransferase from Schizosaccharomyces pombe and apparent absence of the enzyme from Saccharomyces cerevisae. J Biol Chem 269: 30701–30706 [PubMed] [Google Scholar]
- Ganan S, Cazzulo JJ, Parodi AJ (1991) A major proportion of the N-linked glycoproteins are transiently glucosylated in the endoplasmic reticulum. Biochemistry 30: 3098–3104 [DOI] [PubMed] [Google Scholar]
- Gardner TG, Kearse KP (1999) Modification of the T cell antigen receptor (TCR) cpomplex by UDP-glucose:glycoprotein glucosyltransferase. J Biol Chem 274: 14094–14099 [DOI] [PubMed] [Google Scholar]
- Gething M-J, Sambrook J (1992) Protein folding in the cell. Nature 355: 33–45 [DOI] [PubMed] [Google Scholar]
- Gonzalez L, Bruix M, Diaz-Maurino T, Feizi T, Rico M, Solis D, Jimenez-Barbero J (2000) Conformational studies of the Man8 oligosaccharide on native ribonuclease B and on the reduced and denatured protein. Arch Biochem Biophys 383: 17–27 [DOI] [PubMed] [Google Scholar]
- Graziano G, Catanzano F, Giancola C, Barone G (1996) DSC study of the thermal stability of S-protein and S-peptide/S-protein. Biochemistry 35: 13386–13392 [DOI] [PubMed] [Google Scholar]
- Hammond C, Braakman I, Helenius A (1994) Role of N-linked oligosaccharides, glucose trimming and calnexin during glycoprotein folding in the endoplasmic reticulum. Proc Natl Acad Sci USA 91: 913–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert DN, Foellmer B, Helenius A (1995) Glucose trimming and reglucosylation determines glycoprotein association with calnexin. Cell 81: 425–433 [DOI] [PubMed] [Google Scholar]
- Helenius A, Aebi M (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem 73: 1019–1049 [DOI] [PubMed] [Google Scholar]
- Hurtley SM, Helenius A (1989) Protein oligomerization in the endoplasmic reticulum. Annu Rev Cell Biol 5: 277–307 [DOI] [PubMed] [Google Scholar]
- Kim EE, Varadarajan R, Wyckoff HW, Richards FM (1992) Refinement of the crystal structure of ribonuclease S. Comarison with and between the various ribonuclease A structures. Biochemistry 31: 12304–12314 [DOI] [PubMed] [Google Scholar]
- Klausner RD, Sitia R (1990) Protein degradation in the endoplasmic reticulum. Cell 62: 611–614 [DOI] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wuthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14: 51–55, 29–32 [DOI] [PubMed] [Google Scholar]
- Landry SJ, Zeilstra-Ryalls J, Fayet O, Georgopoulos C, Gierasch LM (1993) Characterization of a functionally important mobile domain of GroES. Nature 364: 255–258 [DOI] [PubMed] [Google Scholar]
- Lang K, Schmid FX (1986) Use of a trypsin-pulse method to study the refolding pathway of ribonuclease. Eur J Biochem 159: 275–281 [DOI] [PubMed] [Google Scholar]
- Parker CG, Fessler LI, Nelson RE, Fessler JH (1995) Drosophila UDP-glucose:glycoprotein glucosyltransferase: sequence and characterization of an enzyme that distinguishes between denatured and native proteins. EMBO J 14: 1294–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parodi A (2000) Protein glucosylation and its role in protein folding. Annu Rev Biochem 69: 69–93 [DOI] [PubMed] [Google Scholar]
- Pelletier JN, Arndt KM, Pluckthun A, Michnick SW (1999) An in vivo library-versus-library selection of optimized protein–protein interactions. Nat Biotechnol 17: 683–690 [DOI] [PubMed] [Google Scholar]
- Raines RT (1998) Ribonuclease A. Chem Rev 98: 1045–1065 [DOI] [PubMed] [Google Scholar]
- Richards FM (1958) On the enzymatic activity of subtilisin modified ribonuclease. Proc Natl Acad Sci USA 44: 162–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards FM, Vithayathil PJ (1959) The preparation of subtilisin-modified ribonuclease and the separation of the peptide and protein components. J Biol Chem 234: 1459–1465 [PubMed] [Google Scholar]
- Richards FM, Wyckow HW (1971) Bovine pancreatic ribonucleases. In The Enzymes, Boyer PD (ed) Vol. IV, pp 647–806. New York: Academic Press [Google Scholar]
- Ritter C, Helenius A (2000) Recognition of local glycoprotein misfolding by the ER folding sensor UDP-glucose:glycoprotein glucosyltransferase. Nat Struct Biol 7: 278–280 [DOI] [PubMed] [Google Scholar]
- Rodan AR, Simons JF, Trombetta ES, Helenius A (1996) N-linked oligosaccharides are necessary and sufficient for association of RNase B with calnexin and calreticulin. EMBO J 15: 6921–6930 [PMC free article] [PubMed] [Google Scholar]
- Santoro J, Gonzalez C, Bruix M, Neira JL, Nieto JL, Herranz J, Rico M (1993) High-resolution three-dimensional structure of ribonuclease A in solution by nuclear magnetic resonance spectroscopy. J Mol Biol 229: 722–734 [DOI] [PubMed] [Google Scholar]
- Schrag JD, Procopio DO, Cygler M, Thomas DY, Bergeron JJ (2003) Lectin control of protein folding and sorting in the secretory pathway. Trends Biochem Sci 28: 49–57 [DOI] [PubMed] [Google Scholar]
- Shimotakahara S, Rios CB, Laity JH, Zimmerman DE, Scheraga HA, Montelione GT (1997) NMR structural analysis of an analog of an intermediate formed in the rate-determining step of one pathway in the oxidative folding of bovine pancreatic ribonuclease A: automated analysis of 1H, 13C, and 15N resonance assignments for wild-type and [C65S, C72S] mutant forms. Biochemistry 36: 6915–6929 [DOI] [PubMed] [Google Scholar]
- Sousa M, Parodi AJ (1995) The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. EMBO J 14: 4196–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa MC, Ferrero-Garcia MA, Parodi AJ (1992) Recognition of the oligosaccharide and protein moieties of glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. Biochemitry 31: 97–105 [DOI] [PubMed] [Google Scholar]
- Taylor SC, Ferguson AD, Bergeron JJ, Thomas DY (2004) The ER protein folding sensor UDP-glucose glycoprotein-glucosyltransferase modifies substrates distant to local changes in glycoprotein conformation. Nat Struct Mol Biol 11: 128–134 [DOI] [PubMed] [Google Scholar]
- Taylor SC, Thibault P, Tessier DC, Bergeron JJ, Thomas DY (2003) Glycopeptide specificity of the secretory protein folding sensor UDP-glucose glycoprotein:glucosyltransferase. EMBO Rep 4: 405–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier DC, Dignard D, Zapun A, Radominska-Pandya A, Parodi AJ, Bergeron JJ, Thomas DY (2000) Cloning and characterization of mammalian UDP-glucose glycoprotein:glucosyltransferase and the development of a specific substrate for this enzyme. Glycobiology 10: 403–412 [DOI] [PubMed] [Google Scholar]
- Trombetta ES, Helenius A (2000) Conformational requirements for glycoprotein reglucosylation in the endoplasmic reticulum. J Cell Biol 148: 1123–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trombetta ES, Simons JF, Helenius A (1996) Endoplasmic reticulum glucosidase II is composed of a catalytic subunit, conserved from yeast to mammals, and a tightly bound non-catalytic HDEL-containing subunit. J Biol Chem 271: 27509–27516 [DOI] [PubMed] [Google Scholar]
- Trombetta S, Bosch M, Parodi AJ (1989) Glucosylation of glycoproteins by mammlian, plant, fungal and trypanosomatid protozoa microsomal membranes. Biochemistry 28: 8108–8116 [DOI] [PubMed] [Google Scholar]
- Trombetta SE, Parodi AJ (1992) Purification to apparent homogeneity and partial characterization of rat liver UDP-glucose:glycoprotein glucosyltransferase. J Biol Chem 267: 9236–9240 [PubMed] [Google Scholar]
- Vassilakos A, Michalak M, Lehrman MA, Williams DB (1998) Oligosaccharide binding characteristics of the molecular chaperones calnexin and calreticulin. Biochemistry 37: 3480–3490 [DOI] [PubMed] [Google Scholar]
- Walter P, Blobel G (1983) Preparation of microsomal membranes for cotranslational protein translocation. Methods Enzymol 96: 84–93 [DOI] [PubMed] [Google Scholar]
- Weihofen A, Lemberg MK, Ploegh HL, Bogyo M, Martoglio B (2000) Release of signal peptide fragments into the cytosol requires cleavage in the transmembrane region by a protease activity that is specifically blocked by a novel cysteine protease inhibitor. J Biol Chem 275: 30951–30956 [DOI] [PubMed] [Google Scholar]
- Zapun A, Petrescu SM, Rudd PM, Dwek RA, Thomas DY, Bergeron JJM (1997) Conformation-independent binding of monoglucosylated ribonuclease B to calnexin. Cell 88: 29–38 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data