

Abstract
A substrate for protein kinase B (PKB)α in HeLa cell extracts was identified as methyltransferase-like protein-1 (METTL1), the orthologue of trm8, which catalyses the 7-methylguanosine modification of tRNA in Saccharomyces cerevisiae. PKB and ribosomal S6 kinase (RSK) both phosphorylated METTL1 at Ser27 in vitro. Ser27 became phosphorylated when HEK293 cells were stimulated with insulin-like growth factor-1 (IGF-1) and this was prevented by inhibition of phosphatidyinositol 3-kinase. The IGF-1-induced Ser27 phosphorylation did not occur in 3-phosphoinositide-dependent protein kinase-1 (PDK1)-deficient embryonic stem cells, but occurred normally in PDK1[L155E] cells, indicating that the effect of IGF-1 is mediated by PKB. METTL1 also became phosphorylated at Ser27 in response to phorbol-12-myristate 13-acetate and this was prevented by PD 184352 or pharmacological inhibition of RSK. Phosphorylation of METTL1 by PKB or RSK inactivated METTL1 in vitro, as did mutation of Ser27 to Asp or Glu. Expression of METTL1[S27D] or METTL1[S27E] did not rescue the growth phenotype of yeast lacking trm8. In contrast, expression of METTL1 or METTL1[S27A] partially rescued growth. These results demonstrate that METTL1 is inactivated by PKB and RSK in cells, and the potential implications of this finding are discussed.
Keywords: METTL1, PKB, protein synthesis, RSK, tRNA methylation
Introduction
Protein kinase B (PKB, also called Akt) plays a central role in mediating the intracellular actions of signals that trigger the activation of class 1 phosphatidyinositol (PtdIns) 3-kinases and elevate the intracellular level of PtdIns (3,4,5) trisphosphate. The PtdIns (3,4,5) trisphosphate generated in response to such signals binds to the pleckstrin homology (PH) domains of PKB and its activator 3-phosphoinositide-dependent protein kinase-1 (PDK1), colocalising them at the plasma membrane and allowing PDK1 to activate PKB. PKB then performs multiple functions by phosphorylating many intracellular proteins. In this way, it mediates many of the metabolic actions of insulin, as well as regulating the growth, proliferation and survival of cells, including tumour cells (reviewed by Mora et al, 2004). For these reasons, the identification of its substrates is of great importance for our understanding of both diabetes and cancer.
The discovery that PKB phosphorylates serine and threonine residues that lie in Arg-Xaa-Arg-Xaa-Xaa-Ser/Thr- motifs (where Xaa is any amino acid) (Alessi et al, 1996) has facilitated the identification of potential substrates for PKB. However, such motifs can only be phosphorylated in vivo if they lie on accessible regions of proteins, while other residues in the vicinity of the phosphorylation site may be negative or positive specificity determinants. Moreover, other members of the AGC subfamily of protein kinases, such as SGK (Kobayashi and Cohen, 1999), RSK and S6K isoforms (Alessi et al, 1996), also phosphorylate the same consensus motif as PKB. Indeed, some substrates phosphorylated by PKB, such as glycogen synthase kinase 3 (GSK3), are also phosphorylated by RSK in vivo (Shaw and Cohen, 1999).
We have developed a method for finding new physiological substrates of protein kinases, termed KESTREL (Knebel et al, 2001). This method is especially useful for identifying proteins that are phosphorylated by one particular protein kinase, but not by closely related protein kinases with similar specificity requirements. For example, we have exploited KESTREL to identify elongation factor 2-kinase as a protein that is phosphorylated at Ser359 and inactivated by p38δ MAP kinase, but not by the closely related p38α, p38β and p38γ (Knebel et al, 2001). More recently, we used the same method to identify filamin gamma as a physiological substrate for PKBα and not SGK1 (Murray et al, 2004b) and NDRG isoforms as physiological substrates for SGK1 and not PKBα (Murray et al, 2004a). In this paper, we have detected a further protein that is phosphorylated much more efficiently by PKBα than SGK1 and identify it as methyltransferase-like protein-1 (METTL1) (Bahr et al, 1999), an enzyme that catalyses the 7-methylguanosine modification at position 46 of tRNAPhe in vitro (Alexandrov et al, 2002). We have also found that phosphorylation inactivates METTL1. To our knowledge, the present study is the first to suggest that any tRNA modification is regulated by insulin and growth factors.
Results
PKB phosphorylates a 36 kDa protein in HeLa cell extracts
Desalted HeLa cell extract was fractionated from 0–5% polyethylene glycol 6000 and the 5% supernatant chromatographed on Source Q. Aliquots of each fraction collected were phosphorylated for 4 min with MgATP in the absence or presence of PKBα or SGK1. A protein of apparent molecular mass 36 kDa was detected, eluting at about 0.2 M NaCl, that was phosphorylated by PKBα, but very poorly by SGK1 (Figure 1). This protein was purified further by successive chromatographies on HiTrap-heparin-Sepharose and Mono S. At this stage, the 32P radioactivity comigrated with the major protein-staining band in the preparation, which was excised from the gel and identified by tryptic mass fingerprinting (see Supplementary Table 1) as methyltransferase-like protein-1 (METTL1), the catalytic subunit of an enzyme that catalyses the m7G46 modification of tRNAPhe (Alexandrov et al, 2002). To investigate whether the PKBα substrate and METTL1 were the same protein, the purified material was maximally phosphorylated (Figure 2), digested with trypsin and chromatographed to reveal two phosphopeptides T1 and T2 (Figure 2A). Peptide T2 was examined by mass spectrometry, which revealed it to be a mixture of two peptides comprising residues 25–35 and 23–35 of METTL1, each containing one phosphate group, the latter arising from incomplete tryptic digestion of the Arg–Ala bond between residues 24 and 25. A similar analysis of T1 showed that it also comprised residues 25–35 of METTL1 plus one phosphate, but in which the methionine residue of the peptide had become oxidised. Edman and solid-phase sequencing of T1 and T2 confirmed the identity of each peptide and established that there was a single site of phosphorylation at Ser27 (Figure 2B). This experiment also showed that the 36 kDa substrate for PKBα was indeed METTL1. The identification of Ser27 as the site of phosphorylation was anticipated, as it is the only residue in METTL1 that lies in the canonical consensus sequence for phosphorylation by PKB, Arg-Xaa-Arg-Xaa-Xaa-Ser/Thr- (where Xaa is any amino acid). The amino-acid sequence and domain structure of METTL1 are shown in Figure 3.
Figure 1.
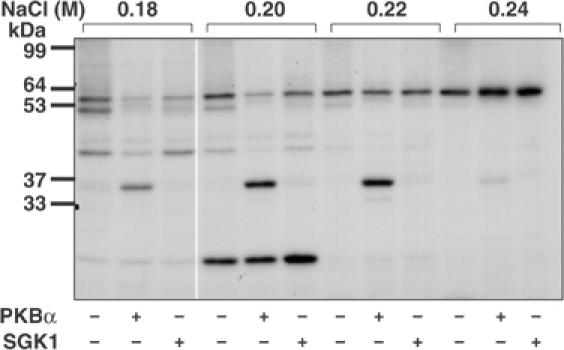
Detection of a 36 kDa substrate for PKBα in HeLa extracts. HeLa cell extracts were fractionated from 0–5% (w/v) PEG 6000, the 5% supernatant desalted and the material (950 mg protein) chromatographed on a 20 ml column of Source Q (see Materials and methods). Aliquots of each fraction were incubated for 4 min at 30°C as described previously (Knebel et al, 2001) at a further 10-fold dilution in the presence of 50 mM Tris–HCl pH 7.5, 1 mM EGTA, 0.1% (v/v) 2-mercaptoethanol, 10 mM MgCl2 and 20 nM [γ-32P]ATP (4 × 106 cpm/pmol) in the presence (+) or absence (−) of PKBα or SGK1, each at 0.4 U/ml (Davies et al, 2000); 1 U is the amount that catalyses the phosphorylation of 1 nmol substrate peptide in 1 min. After SDS–PAGE and autoradiography, a 36 kDa protein eluting at about 0.2 M NaCl was detected that was phosphorylated by PKBα, but very poorly by SGK1. Autophosphorylation of PKB and SGK1 was negligible under the conditions used.
Figure 2.
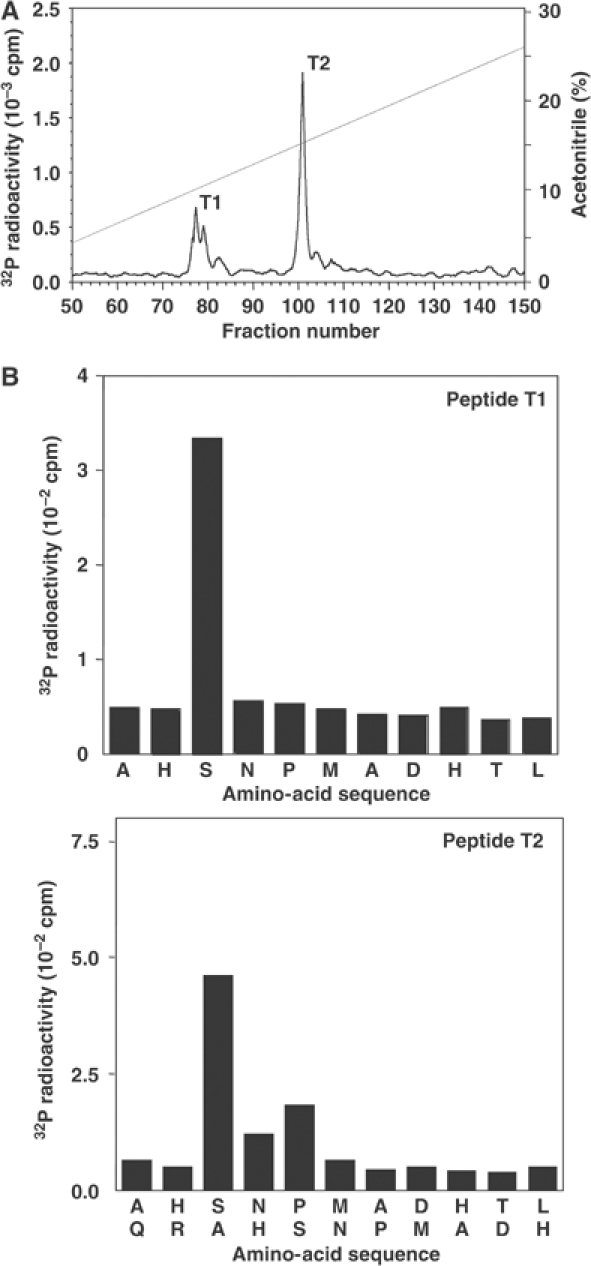
Identification of the residue in METTL1 phosphorylated by PKBα in vitro. (A) Following Mono S purification, METTL1 was phosphorylated with PKBα for 1 h as described in the legend to Figure 1, except that the concentration of [γ-32P]ATP was increased to 0.1 mM and the specific radioactivity was 1000 cpm/pmol. After SDS–PAGE, the gel was stained with Sypro-orange, the phosphorylated band excised and digested with trypsin, and the peptides separated by reverse-phase hydrophobic interaction chromatography on a Vydac C18 column equilibrated in 0.1% (v/v) trifluoroacetic acid. The column was developed with an acetonitrile gradient in 0.1% (v/v) trifluoroacetic acid (diagonal line) at a flow rate of 0.8 ml/min and fractions of 0.4 ml were collected. The two major peaks of 32P radioactivity T1 and T2 were subjected to mass spectrometry (Supplementary Table 1). (B) Peptides T1 and T2 were subjected to Edman degradation to confirm their sequence and to solid-phase sequencing to identify the site(s) of phosphorylation (Stokoe et al, 1992). 32P radioactivity released after each cycle of Edman degradation during solid-phase sequencing was analysed by Cerenkov counting.
Figure 3.
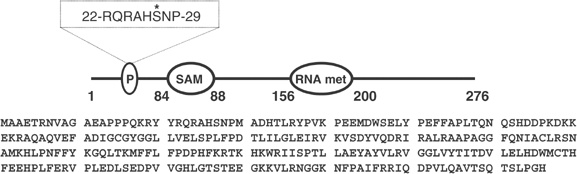
The sequence of human METTL1 and the location of its functional domains. The phosphorylation site is at residue 27, a putative SAM-binding motif (GXGXG) between residues 58 and 65, and a motif similar to that found in an rRNA methylase (rRNA met) of C. trachomatis was found between residues 156 and 200 (Bahr et al, 1999).
Phosphorylation of METTL1 in cells
To examine whether METTL1 became phosphorylated in cells, we generated antibodies capable of immunoprecipitating the human protein and a phospho-specific antibody that recognised METTL1 only when phosphorylated at Ser27 (see Supplementary data). To investigate whether METTL1 was a physiological substrate for PKBα, we stimulated human embryonic kidney (HEK) 293 cells with insulin-like growth factor-1 (IGF-1), a potent activator of this protein kinase, which at 20 ng/ml does not activate extracellular signal-regulated protein kinases 1 and 2 (ERK1/ERK2) of the classical MAP kinase cascade in these cells. METTL1 was not phosphorylated significantly in unstimulated cells, but became phosphorylated at Ser27 after stimulation by IGF-1. Phosphorylation was maximal after about 15 min, a time at which the activation of PKB was also maximal (data not shown). The phosphorylation of METTL1, like the phosphorylation of PKB itself, was prevented by the PtdIns 3-kinase inhibitor wortmannin (Figure 4A) but not by PD 184352, a specific inhibitor of MKK1, the activator of ERK1/ERK2 (Seebolt-Leopold et al, 1999; Davies et al, 2000) (Figure 4C), consistent with METTL1 being phosphorylated by PKB in cells. PD 184352 did not alter the IGF-1-induced phosphorylation of PKB at Thr308 significantly when normalised to the total level of expression of PKB in either this (Figure 4A) or several other similar experiments (data not shown).
Figure 4.
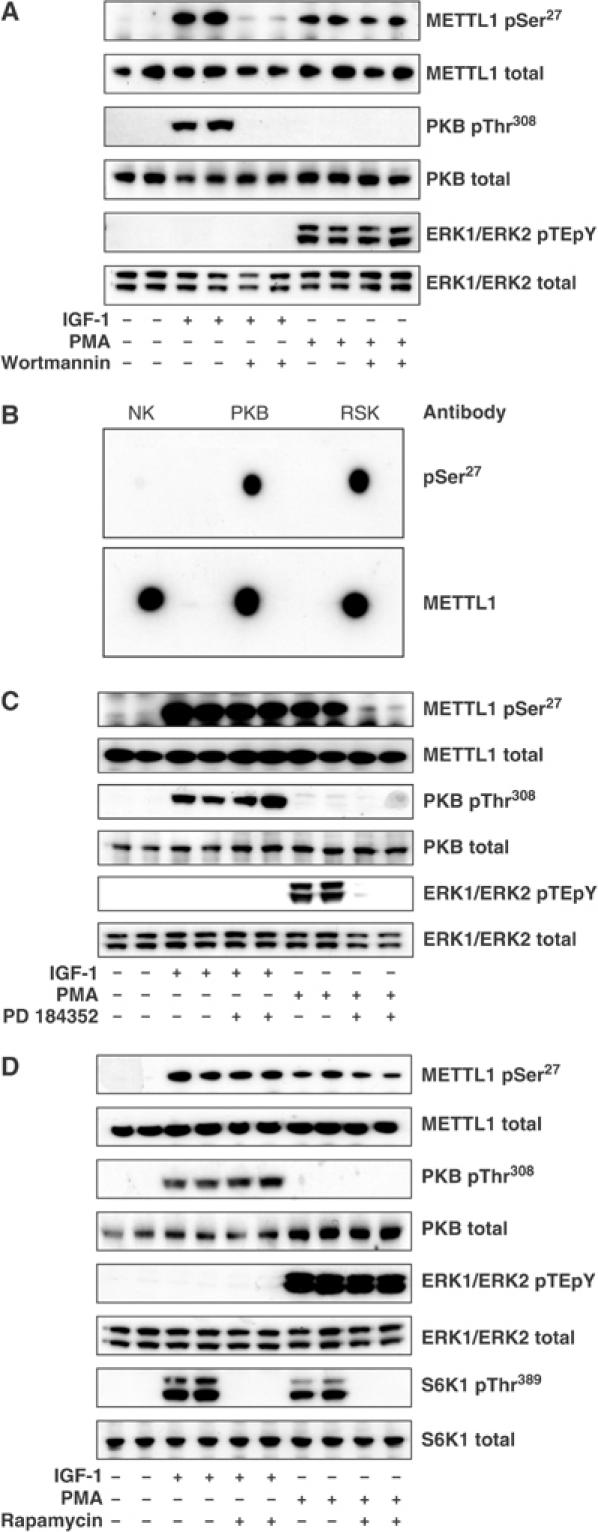
Effects of IGF1 and PMA on the phosphorylation of endogenous METTL1 in HEK293 cells. (A) Cells were serum starved for 8 h and then incubated for 20 min without (−) or with (+) 100 nM wortmannin before stimulation for a further 15 min with 20 ng/ml IGF-1 or for 30 min with 400 ng/ml PMA. METTL1 was immunoprecipitated from 1.5 mg cell lysate protein, denatured in SDS, subjected to SDS–PAGE and, after transfer to Immobilon-P membranes, immunoblotted with the antibody that recognises METTL1 phosphorylated at Ser27 and with the antibody that recognises phosphorylated and unphosphorylated METTL1. A further 20 μg of lysate protein was subjected to SDS–PAGE and immunoblotted with an antibody that recognises PKB phosphorylated at Thr308, an antibody that recognises phosphorylated and unphosphorylated PKBα equally well, an antibody that recognises ERK1 and ERK2 phosphorylated at the Thr-Glu-Tyr motif (pTEpY) and an antibody that recognises all forms of the ERK1 and ERK2 proteins. (B) Bacterially expressed human GST-METTL1 was left unphosphorylated (no kinase, NK) or maximally phosphorylated with PKBα or RSK2 and 100 ng aliquots spotted onto a nitrocellulose membrane and immunoblotted with the antibody that recognises METTL1 phosphorylated at Ser27 and with the antibody that recognises unphosphorylated and phosphorylated METTL1 equally well. (C) Same as panel A, except that after serum starvation for 8 h, the cells were incubated for 1 h in the absence or presence of 2 μM PD 184352 (instead of wortmannin) prior to stimulation with IGF-1 or PMA. (D) Same as panel A, except that following serum starvation the cells were incubated for 1 h in the absence or presence of 100 nM rapamycin (rather than wortmannin) prior to stimulation with IGF-1 or PMA. In addition, 20 μg cell lysate was immunoblotted with an antibody that recognises S6K1 phosphorylated at Thr389 and with an antibody that recognises phosphorylated and unphosphorylated S6K1 equally well.
PKBα and SGK1 are not the only protein kinases that phosphorylate Arg-Xaa-Arg-Xaa-Xaa-Ser- motifs. Other members of the AGC subfamily of protein kinases, such as isoforms of p90 ribosomal protein S6 kinase (RSK) and p70 ribosomal S6 kinase (S6K), also phosphorylate this motif preferentially (Alessi et al, 1996). Indeed, RSK2 can phosphorylate METTL1 at Ser27 in vitro (Figure 4B). To investigate whether RSK phosphorylated METTL1 in cells, we exposed 293 cells to the tumour-promoting phorbol ester phorbol-12-myristate 13-acetate (PMA), which does not activate PKB in these cells but is a potent activator of the classical MAP kinase cascade, and hence the activation of RSK (Figures 4A, C and D). METTL1 became maximally phosphorylated at Ser27 30 min after stimulation with PMA, a time at which the activation of the classical MAP kinase cascade was also maximal, as judged by the phosphorylation of ERK1 and ERK2. The PMA-induced phosphorylation of both ERK1/ERK2 and METTL1 were prevented by PD 184352 (Figure 4C), but not by wortmannin (Figure 4A), consistent with phosphorylation of METTL1 being catalysed by one or more RSK isoforms.
The activation of S6K isoforms requires the protein kinase mTOR (mammalian target of rapamycin), which is potently and specifically inhibited by rapamycin. The activation of mTOR itself requires phosphorylation of the TSC2 component of the tubersclerosis complex, which can be catalysed by either PKB or RSK (Roux et al, 2004). We found that rapamycin prevented the phosphorylation/activation of S6K1 by either IGF-1 or PMA, as expected, but had no significant effect on the phosphorylation of METTL1 or the activation of PKB induced by either agonist in this (Figure 4D) and several other experiments (data not shown). This excluded the involvement of S6K isoforms in the phosphorylation of METTL1 under these conditions.
Further evidence that the IGF-1-induced phosphorylation of METTL1 at Ser27 is catalysed by PKB
To obtain further evidence that the IGF-1-mediated phosphorylation of Ser27 is mediated by PKB, and not by another protein kinase that lies ‘downstream' of PtdIns 3-kinase, we made use of embryonic stem (ES) cells that do not express PDK1 (Williams et al, 2000) or that express the PDK1[L155E] mutant instead of the wild-type enzyme (Collins et al, 2003). This mutation disrupts a hydrophobic pocket in PDK1 required for its interaction with, and activation of, every known PDK1 substrate except PKB (Mora et al, 2004). Thus the PDK1[L155E] mutant can activate PKB normally, but cannot activate substrates such as S6K, SGK and atypical PKCs (Collins et al, 2003). IGF-1 does not activate PKB or induce the phosphorylation of METTL1 at Ser27 in ES cells that do not express PDK1, but activates PKB and induces METTL1 Ser27 phosphorylation similarly in ES cells expressing wild-type PDK1 or PDK1[L155E] (Figure 5). These results indicate the IGF-1-mediated phosphorylation of METTL1 is indeed catalysed by PKB.
Figure 5.
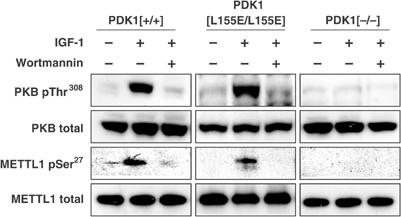
METTL1 is phosphorylated in mouse ES cells expressing wild-type PDK1 or PDK1[L155E], but not in PDK1 null cells. Mouse ES cells expressing PDK1[+/+], PDK1[L155E/L155E] or PDK1[−/−] were serum starved for 4 h, incubated for 20 min without (−) or with (+) 100 nM wortmannin, and then stimulated for a further 15 min with 20 ng/ml IGF-1. METTL1 was immunoprecipitated from 2.5 mg cell lysate protein using the antibody raised against mouse METTL1 residues 258–268, denatured in SDS, subjected to SDS–PAGE and, after transfer to Immobilon-P membranes, immunoblotted as in panel A.
Phosphorylation of Ser27 inactivates METTL1
To examine the effect of METTL1 phosphorylation on its activity, we expressed and purified human METTL1 as a GST fusion protein in 293 cells (see Materials and methods). This preparation was active as a tRNA methylase and activity was not altered by cleavage of the GST tag with PreScission protease (Figure 6A). For this reason, GST-METTL1 was used in subsequent experiments. As a control, GST was expressed and purified, and this preparation was devoid of tRNA methylase activity (Figure 6A). At a concentration of 80 nM GST-METTL1, tRNA methylation was approximately linear for 15 min (Figure 6B), and so the effect of phosphorylation on activity was studied under these conditions.
Figure 6.
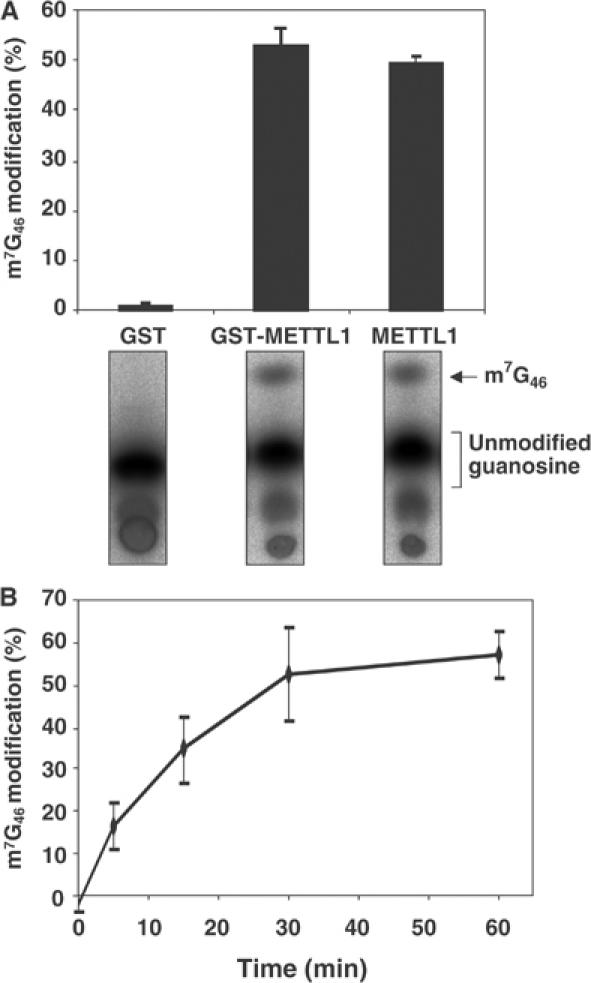
Human METTL1 is a tRNAPhe methylase. Assays were carried out in triplicate and error bars represent the standard error of the mean. Purified GST, GST-METTL1 and METTL1 (each at 80 nM) were incubated for 15 min with [α-32P]GTP-labelled pre-tRNAPhe in the presence of 1 mM SAM. The reactions were terminated by phenol extraction of the tRNA and after complete digestion with P1 nuclease, the m7G46 modification was detected after separation from unmodified guanosine by thin-layer chromatography (TLC), followed by phosphorimager analysis and quantification of the % conversion to m7G46 (see Materials and methods). (B) Same as panel A, except that only GST-METTL1 was used and the assays were performed for the times indicated.
The phosphorylation of METTL1 at Ser27 was found to inactivate its tRNA methylase activity, inhibition correlating well with the extent of phosphorylation (Figure 7A and B). The inactive phosphorylated enzyme could be reactivated by dephosphorylation in vitro catalysed by protein phosphatase 1 (PP1) and reactivation was prevented by microcystin LR, a specific inhibitor of PP1 (Figure 7C).
Figure 7.
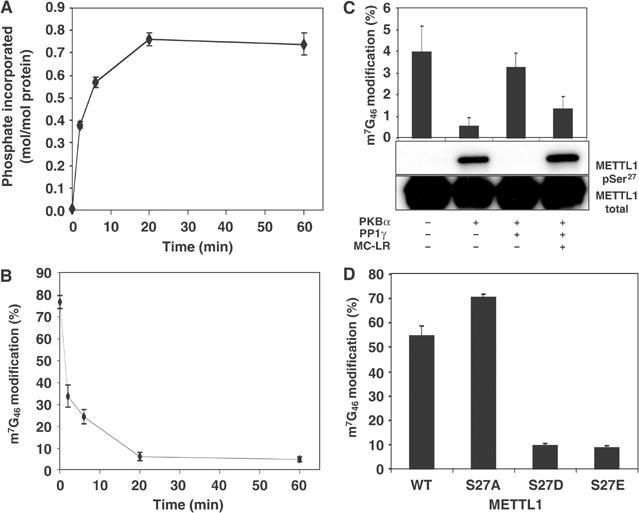
Phosphorylation of GST-METTL1 inhibits its tRNA methylase activity. Assays were carried out in triplicate and error bars represent the standard error of the mean. (A) GST-METTL1 (3 μM) was phosphorylated in the standard assay buffer for the times indicated with 10 mM MgCl2–0.1 mM [γ-32P]ATP (1000 cpm/pmol) and 0.4 U/ml (about 0.01 μM) PKBα. The stoichiometry of phosphorylation was calculated from the 32P radioactivity incorporated (determined after precipitation with trichloroacetic acid), the molecular mass of GST-METT1 and the amount of protein in the assay (estimated by the method of Bradford using BSA as a standard) after correction for the purity of GST-METTL1 determined by densitometric analysis of the Coomassie blue-stained gel. (B) GST-METTL1 was phosphorylated as in panel A, except that unlabelled Mg-ATP replaced Mg-[γ-32P]ATP. At each time point, an aliquot was removed and METTL1 (80 nM) assayed for tRNA methylase activity as in Figure 6B. (C) GST-METTL1 was phosphorylated for 60 min as in panel B, in the presence (+) or absence (−) of PKB. Glutathione-Sepharose (5 μl) was added to each 0.05 ml reaction mix and left for 45 min at 4°C. After brief centrifugation, the supernatant was discarded and the pellet washed twice with 1 ml of 50 mM Tris–HCl, 0.1 mM EGTA, 0.03% (w/v) Brij 35, 0.1% (v/v) 2-mercaptoethanol pH 7.5 and 1 mM MnCl2. The glutathione-Sepharose pellet was then incubated with 0.05 ml of the same buffer containing 20 mM glutathione to elute the GST-METTL1. After brief centrifugation, the supernatant was removed and incubated for 30 min at 30°C in the presence (+) or absence (−) of 50 U/ml PP1γ (where 1 U is the amount that catalyses the dephosphorylation of 1 nmol of phosphorylase a in 1 min). The PP1γ itself had been incubated previously for 10 min in the presence (+) or absence (−) of its inhibitor microcystin LR (MC-LR). Aliquots were then assayed for tRNA methylase activity (uppermost panel) or electrophoresed and immunoblotted with an antibody that recognises METTL1 phosphorylated at Ser27 (middle panel) and with an antibody that recognises all forms of METTL1 (lowest panel). (D) Purified GST-METTL1, GST-METTL1[S27A], GST-METTL1[S27D] and GST-METTL1[S27E], each at 80 nM, were assayed for 15 min as in Figure 6.
The METTL1[S27A] mutant, which had similar activity to the wild-type enzyme, was not phosphorylated at all by PKBα or RSK2 (data not shown), confirming that Ser27 was the only site of phosphorylation. Conversely, the mutation of Ser27 to Asp or Glu to mimic the effect of phosphorylation greatly decreased activity (Figure 7D).
Expression of METTL1 in the presence of WDR4 complements a yeast trm8 growth phenotype in vivo
Saccharomyces cerevisiae express a homologue of METTL1 termed tRNA modifier 8 (trm8) complexed to another protein trm82, which is essential for the stability and function of trm8 (Alexandrov et al, 2005). Human METTL1 can replace trm8 in catalysing the m7G46 modification in yeast and this requires WDR4, the human homologue of trm82 (Alexandrov et al, 2002). We recently showed that yeast trm8 and trm82 mutants have a temperature-sensitive growth defect in minimal media containing glycerol, and that complementation of this phenotype was correlated with m7G methyltransferase activity (Alexandrov et al, 2005). As we also showed that expression of METTL1 and WDR4 in yeast lacking Trm8 and Trm82 yielded extracts with m7G methyltransferase activity (Alexandrov et al, 2002), we speculated that we might be able to demonstrate complementation of yeast mutants with METTL1/WDR4.
As shown in Figure 8A, expression of METTL1/WDR4 under PGAL control complements the temperature-sensitive growth defect of a yeast strain lacking trm8 and containing an additional deletion in trm4 to enhance the phenotype (A Alexandrov and EM Phizicky, unpublished work). In row g of Figure 8A, expression of yeast Trm8p efficiently complements the growth defect at both 33°C (panel II) and 37°C (panel III) in media containing galactose, in which Trm8p is expressed, but not in media containing dextrose (panel V), in which Trm8p is not expressed. In row a, coexpression of wild-type METTL1 and WDR4 also complemented the growth defect at 33 and 37°C, albeit about 10-fold less effectively than Trm8p (row a, panels II and III).
Figure 8.
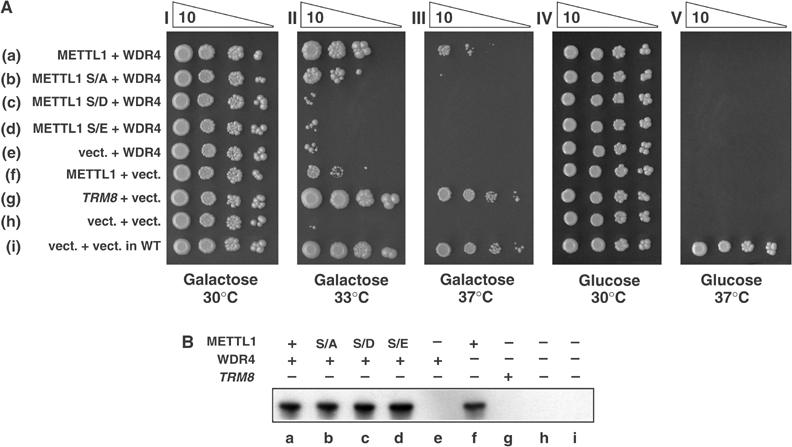
Expression of METTL1[S27A], but not S27D or S27E, complements a yeast trm8-Δ mutant. (A) Complementation of yeast trm8 mutants by METTL1 and Ser27 variants. A trm8-Δ yeast strain AA0636 (Alexandrov et al, 2005), containing a trm4 mutation to enhance its phenotype (see Materials and methods), was transformed with plasmids expressing galactose-inducible (PGAL controlled) WDR4, METTL1, METTL1 variants, TRM8 or vector controls as indicated (Alexandrov et al, 2002), and tested for growth by serial 10-fold dilution of cultures (starting from 4 × 103 cells), followed by incubation at the indicated temperatures on synthetic media containing galactose or glucose, as described in Materials and methods. (B) Analysis of expression of METTL1 and Ser27 variants. Strains described in panel A were grown as described, harvested after 5 h induction in media containing galactose, and crude lysates analysed by immunoblotting using the antibody recognising both phosphorylated and unphosphorylated forms of METTL1. Lanes a–d and f contain the METTL1 variants indicated.
Expression of METTL1[S27A], but not METTL1[S27D] or METTL1[S27E], complements a yeast trm8-Δ mutant in vivo
To begin to address the possibility that phosphorylation of METTL1 at Ser27 inhibits its function in vivo, we mutated this site to Ala, Asp or Glu, and examined complementation of yeast trm8-Δ mutants after expression in yeast. In Figure 8A (row b), expression of METTL1[S27A] complemented the growth phenotype (panel II), similarly to wild-type METTL1 protein (row a). In contrast, the S27D and S27E mutants completely failed to complement (rows c and d). Similar expression of METTL1 variants was confirmed by immunoblot analysis (Figure 8B) demonstrating that expression of each METTL1 variant was similar (lanes a–d).
Lack of effect of METTL1 phosphorylation on its interaction with WDR4
We cloned the human homologue of Trm82 (Alexandrov et al, 2002), termed WD-repeat protein 4 (WDR4) (Michaud et al, 2000), and investigated whether it could interact with METTL1 in cells. These experiments demonstrated that the two proteins do indeed form a complex and that interaction is not disrupted by the IGF-1-stimulated phosphorylation of METTL1 at Ser27 or by the mutation of Ser27 to Asp or Glu (see Supplementary data and Supplementary Figure 2). The PKBα-catalysed phosphorylation of the METTL1–WDR4 complex inhibited the tRNA methylase activity similarly to METTL1. PKBα did not phosphorylate the WDR4 component, consistent with its lack of an Arg-Xaa-Arg-Xaa-Xaa-Ser/Thr- motif (data not shown).
Ser27 of METTL1 lies within a canonical mode I consensus sequence for 14-3-3 binding (Rittinger et al, 1999). However, we have been unable to detect significant binding of 14-3-3 to Ser27-phosphorylated METTL1 in vitro, or in cell extracts (R Cartlidge and C MacKintosh, unpublished results). This is consistent with the inability of IGF-1 to trigger the nuclear exit of METTL1. METTL1 and WDR4 were nuclear in control or IGF-1-stimulated cells (see Supplementary Figure 3).
Discussion
We have identified METTL1 as a protein that is phosphorylated rapidly and stoichiometrically at Ser27 by PKB (Figure 7A), and established that METTL1 becomes phosphorylated at Ser27 in cells in response to agonists that activate PtdIns 3-kinase or the classical MAP kinase cascade. The finding that IGF-1-induced Ser27 phosphorylation is prevented by the PI 3-kinase inhibitors wortmannin (Figure 4A) and LY 294002 (data not shown), but not by PD 184352 or rapamycin, and that it occurs in cells expressing the PDK1[L155E] mutant, but not in PDK1-deficient mouse ES cells (Figure 5), demonstrates that phosphorylation is catalysed by PKB, and not by isoforms of RSK or S6K. We have also shown that PMA, which does not activate PKB, triggers the phosphorylation of METTL1 in 293 cells. In contrast to IGF-1, PMA-stimulated phosphorylation of Ser27 is not prevented by wortmannin or rapamycin, but instead is blocked by PD 184352 (Figure 4C). Since ERK1 and ERK2 do not phosphorylate METTL1 at Ser27, PMA-induced phosphorylation must be catalysed by one or more isoforms of RSK, mitogen and stress-activated protein kinase (MSK) or MAP kinase-integrating kinase (MNK), which are the protein kinases known to be activated by ERK1/ERK2 in cells. Of these, RSK isoforms are most likely to mediate PMA-induced phosphorylation of Ser27 for the following reasons. Firstly, MSKs and MNKs are not only activated by ERK1/ERK2 in response to growth factors but, in contrast to RSKs, are also activated by p38α MAP kinase (Deak et al, 1998); yet METTL1 does not become phosphorylated at Ser27 in response to an agonist (anisomycin) that induces the activation of p38α MAP kinase but not the activation of ERK1/ERK2 (R Cartlidge, unpublished experiments). Secondly, RSK isoforms, like PKB, phosphorylate Arg-Xaa-Arg-Xaa-Xaa-Ser- motifs (Alessi et al, 1996) and we have shown that RSK2 phosphorylates METTL1 at Ser27 in vitro (Figure 4B). Thirdly, a potent and specific cell-permeant inhibitor of RSK isoforms, which we have identified in collaboration with a pharmaceutical company, prevents PMA- but not IGF-1-stimulated METTL1 phosphorylation at Ser27 in 293 cells (R Cartlidge, J Bain and P Cohen, unpublished experiments). Thus, like glycogen synthase kinase-3 (GSK3) (Shaw and Cohen, 1999) and tuberous sclerosis complex protein 2 (TSC2) (Roux et al, 2004), METTL1 can be phosphorylated and inactivated by PKB or RSK in response to agonists that activate either the PI 3-kinase pathway or the classical MAP kinase cascade, respectively.
At the time we identified METTL1 as a substrate of PKB and RSK, it had been noted by others that this protein contained a putative S-adenosylmethionine (SAM)-binding domain and a further domain similar to that found in a protein from Chlamydia trachomatis that had been annotated as an rRNA methylase (Figure 3), suggesting that METTL1 might be an enzyme that methylates a nucleic acid(s) (Bahr et al, 1999). Subsequently, one of our laboratories identified the yeast homologue of METTL1, termed Trm8, as the catalytic subunit of the Trm8–Trm82 complex that catalyses m7G46 modification of tRNAPhe in vitro, and is responsible in vivo for m7G modification of most, if not all, of the 11 tRNA species known to possess this modification (Sprinzl et al, 1998; Alexandrov et al, 2002). Thus, it is likely, but not yet proven, that METTL1 catalyses m7G formation in the nine human tRNAs with this modification.
The discovery that METTL1 is a tRNA methylase allowed us to demonstrate that phosphorylation at Ser27 inactivates this enzyme (Figure 7A–C), and to show that alteration of Ser27 markedly decreases METTL1 function in vivo in yeast (Figure 8). Expression of METTL1/WDR4 complements the temperature-sensitive growth defect of a trm8-deficient yeast strain (Figure 8), consistent with the hypothesis that expression of the human proteins provides m7G methyltransferase activity, as suggested by analysis of complementation of a similar trm8 growth defect (Alexandrov et al, 2005). The data shown in Figure 8 also emphasise a crucial role of Ser27 in this complementation, since METTL1[S27A] complements the growth defect similarly to wild-type METTL1, whereas METTL1[S27D] or METTL1[S27E], which may mimic the effect of phosphorylation on activity in vitro by introducing a negative charge at this position (Figure 7D), do not complement the growth defect. While the data do not themselves prove that phosphorylation regulates the activity of METTL1 in yeast, they do suggest that the presence of a negative charge at position 27 has catastrophic effects on cell growth under the conditions described. Since other work from one of our laboratories indicates that the m7G methyltransferase activity of Trm8 is highly correlated with growth under these conditions (Alexandrov et al, 2005), the simplest explanation is that METTL1 activity in vivo has also been compromised by these mutations. The observation that METTL1[S27A] is marginally less effective than wild-type METTL1 may be a reflection of the critical importance of this residue for the function of the protein in vivo.
It is remarkable that Ser27 and the sequence surrounding it (Tyr-Arg-Gln-Arg-Ala-His-Ser-Asn-Pro) are perfectly conserved in METTL1 in a number of eukaryotes including humans, Drosophila, Caenorhabditis elegans and S. cerevisiae, and the only difference in Schizosaccharomyces pombe is the replacement of the Gln by Ala, supporting the view that the inactivation of this enzyme by phosphorylation is likely to play an important and conserved role in vivo. However, since the precise function of the m7G46 modification is not yet understood, a discussion of how METTL1 phosphorylation might contribute to the regulation of protein synthesis by extracellular signals can only be speculative at the present time.
Insulin and growth factors are known to increase the overall rate of protein synthesis in mammalian cells, as well as stimulating the translation of specific mRNAs. It is tempting to speculate that the PKB-catalysed inactivation of METTL1 is responsible for some of this increased translation, and this would resemble the inactivating effects exerted by the PKB-catalysed phosphorylation of other several substrates. Thus GSK3, TSC2 and FOXO1 all become inhibited as a result of phosphorylation by PKB, resulting in the activation of glycogen synthase, the activation of mTOR and the inhibition of gene transcription, respectively. One hypothesis is that the inactivation of METTL1 decreases the level of m7G46-modified tRNA, which might contribute to the overall increase in protein synthesis induced by insulin or growth factors. If so, it is obvious that rapid response would require a high rate at which m7G46-modified tRNA is replaced by unmodified tRNA, either by an active m7G46-demethylase or by rapid turnover of the existing modified tRNA. The recent finding of the mRNA and tRNA demethylase AlkB (Ougland et al, 2004) lends credence to the hypothesis that a demethylase could exist. Although it is unknown if tRNA is turned over rapidly after insulin stimulation, the recent finding of tRNA turnover from lack of m1A58 in yeast tRNA suggests that tRNA populations are not static (Kadaba et al, 2004).
It is conceivable that under the adverse conditions of nutrient deprivation, m7G46 modification of tRNA is important to maintain a minimum rate of protein synthesis, but when the level of insulin is elevated and the availability of amino acids is high, m7G modification is benign. Phosphorylation of METTL1 may simply reflect turning off of an enzyme that is no longer critical. Our recent elaboration of a distinct temperature-sensitive growth defect of yeast trm8-Δ mutants in glycerol-containing minimal media but not, for example, on rich media (Alexandrov et al, 2005) may bear on this point, since trm8-Δ mutants could be regarded as equivalent to inactivating METTL1 by phosphorylation. It will therefore be important to investigate whether the extent of m7G46 modification of tRNA differs in different metabolic states and to delineate the precise role of m7G modification of tRNA.
The m7G46 modification is one of 25 distinct modifications that have been identified in yeast tRNA and one of 26 so far identified in humans. Although many modifications are highly conserved, and several of them are essential (Gerber and Keller, 1999; Anderson et al, 2000; Gu et al, 2003), the definition of the precise roles of a number of them has been lacking. There is strong evidence that modifications in and around the anticodon loop can enhance the fidelity and efficiency of tRNA in translation (reviewed by Curran, 1998; Bjork et al, 1999), but for many other modifications there is no clear delineation of their role.
Finally, it is possible that METTL1 has further important roles in addition to its presumed role in m7G modification of tRNA. It is known that at least two tRNA-modifying enzymes modify other RNAs (Massenet et al, 1999; Behm-Ansmant et al, 2003), that two other tRNA-modifying enzymes have growth phenotypes that are not linked to their catalytic activities (Gutgsell et al, 2000; Johansson and Bystrom, 2002), that another tRNA processing enzyme has a similar biochemical role in another mRNA processing pathway (Sidrauski et al, 1996) and that alteration of activity of tRNA-modifying enzymes can adversely affect other unrelated pathways (Benko et al, 2000). Thus, it is possible that METTL1 acts alone, or in complex with WDR4 or another partner, to methylate other nucleic acids or other macromolecules. If so, then the phosphorylation of METTL1 would have alternative/additional roles from those that are currently envisaged.
Materials and methods
Materials
See Supplementary data.
Antibodies
To generate a phospho-specific antibody that recognises METTL1 phosphorylated at Ser27, the peptide CYRQRAHpSNPMADH (where pS is phosphoserine), corresponding to residues 20–33 of METTL1, was coupled separately to bovine serum albumin and keyhole limpet haemocyanin. The two conjugates were mixed and injected into a sheep at Diagnostics Scotland (Pennycuik, UK). The antisera were affinity purified on a phospho-peptide antigen-Sepharose column. The antibody was used at 1.0 μg/ml for immunoblotting in the presence of 10 μg/ml unphosphorylated peptide. An antibody that immunoprecipitates METTL1, and recognises the phosphorylated and unphosphorylated forms of the protein equally well, was generated by injecting sheep with glutathione S-transferase (GST)-tagged METTL1. The antisera were affinity purified on GST-Sepharose and then on GST-METTL1-Sepharose. This antibody was also used for immunoblotting at 1.0 μg/ml. To generate an antibody capable of immunoprecipitating mouse METTL1, the peptide KQAVTPNPTLP, corresponding to residues 258–260 of mouse METTL1, was utilised in the same way as for the production of the METTL1 phospho-specific antibody. Antibodies that recognise the phosphorylated and unphosphorylated forms of PKB, S6K1 and ERK2 equally well were generated and affinity purified in the Divison of Signal Transduction Therapy, School of Life Sciences, University of Dundee, UK. For sources of commercial antibodies, see Supplementary data.
DNA constructs
The open reading frame of METTL1 (NCBI Y18643) was amplified from IMAGE clone 1582140 using the GC Rich PCR System (Roche) and oligonucleotides with or without a 5′ FLAG sequence. The PCR products were ligated into pCR2.1 (Invitrogen), sequenced and cloned into the BamHI site of either pGEX6P-1 (Amersham Biosciences), pCMV5 (Andersson et al, 1989) or pEBG6P-1 (parental vector pEBG2t (Sanchez et al, 1994) in which a site that can be cleaved by Precission protease has been added before the BamH1 site). The constructs generated were then mutated with the Quick Change Site-Directed Mutagenesis Kit (Stratagene, Amsterdam, Holland). Constructs for expression in mammalian cells were transformed into Escherichia coli strain DH5α and DNA prepared using the Plasmid Mega Kit (QIAGEN, West Sussex, UK) according to the manufacturer's guidelines.
Intron-containing pre-tRNAPhe was generated by transcription in vitro of a BstNI-digested pUC13phe plasmid (Reyes and Abelson, 1987) using the Ambion MAXIscript T7 in vitro transcription kit (Huntington, UK).
Protein preparations
For bacterial expression, pGEX6P-1 METTL1 was transformed into E. coli strain BL21 pLys S and expression was induced with IPTG for 16 h at 26°C. For expression in mammalian cells, vectors expressing GST-METTL1 were transfected into cells, incubated in serum for 24–36 h, starved for 8 h and lysed as described below. GST fusion proteins were purified from the lysates by affinity chromatography on glutathione-Sepharose, dialysed into 50 mM Tris–HCl pH 7.5, 0.1 mM EGTA, 50% (v/v) glycerol and 0.1% (v/v) 2-mercaptoethanol and stored at −20°C.
N-terminally His6-tagged PKBα[S473D] residues 118–480 (lacking the PH domain), His-tagged SGK1[S422D] residues 60–431 and His6-tagged S6K1[T412E] residues 1–421 were expressed in the insect cell line Sf21 and activated by incubation with the catalytic domain of His6-PDK1 (residues 52–556). PDK1 was then removed by chromatography on heparin-Sepharose (Alessi et al, 1997). N-terminally His-tagged full-length RSK2 was maximally activated with PDK1 and GST-ERK2. The PDK1 was removed as described above and the GST-ERK2 on glutathione-Sepharose. The catalytic subunit of protein phosphatase 1γ (PP1γ) was expressed in E. coli and purified as described (Alessi et al, 1993).
Cell culture, transfection and lysis
HEK293 cells were cultured and cell extracts prepared as described elsewhere (McNeill et al, 2004). Mouse ES cells were cultured as described previously (Collins et al, 2003). Cells were transfected using the calcium phosphate technique (Cuenda et al, 1997).
Immunoprecipitation of proteins from cell extracts
Untransfected cell extracts (1.5 mg protein) were immunoprecipitated with 5 μg of anti-METTL1 coupled to protein G-Sepharose. Transfected FLAG-METTL1 and HA-WDR4 were immunoprecipitated from 0.5 mg of cell extract protein using 5 μg of anti-FLAG or anti-HA coupled to protein G-Sepharose. After incubation for 1 h at 4°C with shaking, the captured proteins were centrifuged for 1 min at 13 000 g, the supernatants discarded and the beads washed twice in lysis buffer containing 0.5 M NaCl and once in lysis buffer alone. Samples were denatured in SDS, subjected to SDS–PAGE, transferred to Immobilon P membranes and immunoblotted using the ECL detection system (Amersham Pharmacia Biotech).
Purification of a 36 kDa substrate for PKBα from HeLa cell extract
A total of 5 × 1010 HeLa cells (Computer Cell Culture, Seneffe, Belgium) were unfrozen and diluted with 5 volumes of 30 mM Tris–HCl pH 8.0, 0.1 mM EGTA, 0.1 mM EDTA, 1% (v/v) Triton X100, 1% (v/v) 2-mercaptoethanol and complete protease inhibitor cocktail. After mixing for 10 min at 4°C, the cell lysate was centrifuged at 25 000 g for 1 h at 4°C, and the supernatant removed, centrifuged at 50 000 g for 90 min at 4°C and passed through a HiPrep Desalting Sepharose G25 column. Polyethylene glycol (PEG)-6000 (5% (w/v)) was added to the supernatant and the solution mixed for 2 h at 4°C. The suspension was centrifuged at 10 000 g for 30 min at 4°C and the pellet discarded. The supernatant was diluted with 9 volumes of 30 mM Tris–HCl pH 8.0, 10 mM 2-mercaptoethanol, 0.03% (w/v) Brij 35 and 5% (v/v) glycerol and chromatographed on an 8 ml column of Source Q equilibrated in the same buffer. The column was developed with a 160 ml exponential salt gradient to 1 M NaCl in the same buffer. Fractions of 0.4 ml were collected and examined for the presence of substrates of PKBα and SGK1 as described under Results. Fractions containing a 36 kDa protein phosphorylated by PKBα and much more weakly by SGK1 (eluting from 0.18–0.24 M NaCl) were pooled, diluted in 9 volumes of 30 mM Mops–NaOH pH 7.0, 0.1% (v/v) 2-mercaptoethanol, 0.03% (w/v) Brij 35 and 5% (v/v) glycerol and chromatographed on a 1 ml HiTrap Heparin column equilibrated in the same buffer. The column was developed with a 20 ml exponential salt gradient to 1.0 M NaCl in the same buffer. The fractions containing the 36 kDa PKBα substrate (eluting from 0.68–0.72 M NaCl) were pooled, diluted 10-fold into 30 mM MES–NaOH pH 6.0, 0.1% (v/v) 2-mercaptoethanol, 5% (v/v) glycerol and 0.03% (w/v) Brij 35 and chromatographed on a 1 ml column of Mono S equilibrated in the same buffer. The column was developed with a 20 ml exponential gradient salt gradient to 1.0 M NaCl, and fractions containing the 36 kDa PKB substrate (eluting from 0.12–0.16 M NaCl) were pooled and analysed as described in the Results section.
m7G46 methylation activity assay
This was performed as described previously (Alexandrov et al, 2002). 32P-labelled pre-tRNA was methylated and m7G separated subsequently by thin-layer chromatography. The 32P-labelled species were visualised by phosphorimaging and the ratio of m7G46/G quantitated by densitometry. As 26 guanosine residues are present in tRNAPhe, a ratio of 1:26 corresponds to 100% conversion of the guanosine 46 to m7G46.
Complementation of a yeast trm8 growth defect by expression of METTL1/WDR4
METTL1, variants of METTL1, WDR4 and TRM8 were expressed in a homozygous diploid yeast strain AVA0636 (trm8-Δ∷kanMX, trm4-Δ∷nat) (Alexandrov et al, 2005; A Alexandrov and EM Phizicky, unpublished work) under PGAL control using single-copy CEN plasmids AVA0040 and AVA0042 (Alexandrov et al, 2002), containing URA3 or LEU2 selective markers, respectively. The parent yeast strain BY4743 was used as a wild-type control. Cells were grown overnight in synthetic media containing 2% (w/v) raffinose and lacking leucine and uracil, and viability was analysed by serial 10-fold dilutions of cells followed by incubation on synthetic media lacking leucine and uracil and containing either 2% (w/v) galactose, 2% (w/v) raffinose or 2% (w/v) dextrose.
Analysis of expression of METTL1 and variants in yeast
Strains described in Figure 8A were grown on synthetic media containing 2% (w/v) raffinose and lacking leucine and uracil to OD260 nm=0.6 and then induced for 5 h by addition of galactose to a final concentration of 2% (w/v). Crude SDS lysates made with glass beads were analysed by immunoblot using antibody recognising both phosphorylated and nonphosphorylated forms of METTL1.
Supplementary Material
Supplementary Data
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Table 1
Acknowledgments
We acknowledge the award of a Wellcome Trust Studentship (to RAC) and thank colleagues in the MRC Protein Phosphorylation Unit for identifying Ser27 as the site of phosphorylation (Drs David Campbell and Nick Morrice) and mouse ES cells expressing wild-type PDK1, PDK1[L155E] mutants and PDK1-deficient cells (Drs Barry Collins and Dario Alessi). We also thank the protein production and antibody purification teams in the Division of Signal Transduction Therapy (School of Life Sciences, University of Dundee) coordinated by Hilary McLauchlan and James Hastie, for expression and purification of protein kinases and affinity purification of antibodies and the DNA Sequencing Service (School of Life Sciences, University of Dundee; www.dnaseq.co.uk). This study was supported by the UK Medical Research Council, Diabetes UK, The Royal Society, AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Merck and Co, Merck KGaA and Pfizer. AVA and EMP were supported by grant GM52347 from the National Institutes of Health, USA.
References
- Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P (1996) Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett 399: 333–338 [DOI] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PRJ, Reese CB, Cohen P (1997) Characterisation of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bá. Curr Biol 7: 261–269 [DOI] [PubMed] [Google Scholar]
- Alessi DR, Street AJ, Cohen P, Cohen PW (1993) Inhibitor-2 functions like a chaperone to fold three expressed isoforms of mammalian protein phosphatase-1 into a conformation with the specificity and regulatory properties of the native enzyme. Eur J Biochem 213: 1055–1066 [DOI] [PubMed] [Google Scholar]
- Alexandrov A, Grayhack EJ, Phizicky EM (2005) Trm8p/Trm82p m7G methyltransferase: evidence linking activity to a growth phenotype and implicating Trm82p in maintaining levels of active Trm8p. RNA in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov A, Martzen MR, Phizicky EM (2002) Two proteins that form a complex are required for 7-methylguanosine modification of yeast tRNA. RNA 8: 1253–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J, Phan L, Hinnebusch AG (2000) The Gcd10p/Gcd14p complex is the essential two-subunit tRNA(1-methyladenosine) methyltransferase of Saccharomyces cerevisiae. Proc Natl Acad Sci USA 97: 5173–5178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson S, Davis DL, Dahlback H, Jornvall H, Russell DW (1989) Cloning, structure, and expression of the mitochondrial cytochrome P-450 sterol 26-hydroxylase, a bile acid biosynthetic enzyme. J Biol Chem 264: 8222–8229 [PubMed] [Google Scholar]
- Bahr A, Hankeln T, Fiedler T, Hegemann J, Schmidt ER (1999) Molecular analysis of METTL1, a novel human methyltransferase-like gene with a high degree of phylogenetic conservation. Genomics 57: 424–428 [DOI] [PubMed] [Google Scholar]
- Behm-Ansmant I, Urban A, Ma X, Yu YT, Motorin Y, Branlant C (2003) The Saccharomyces cerevisiae U2 snRNA:pseudouridine-synthase Pus7p is a novel multisite–multisubstrate RNA:Psi-synthase also acting on tRNAs. RNA 9: 1371–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benko AL, Vaduva G, Martin NC, Hopper AK (2000) Competition between a sterol biosynthetic enzyme and tRNA modification in addition to changes in the protein synthesis machinery causes altered nonsense suppression. Proc Natl Acad Sci USA 97: 61–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork GR, Durand JMB, Hagervall TG, Leipuviene HKL, Nilsson K, Chen P, Qian Q, Urbonavicius J (1999) Transfer RNA modification: influence on translational frameshifting and metabolism. FEBS Lett 452: 47–51 [DOI] [PubMed] [Google Scholar]
- Collins BJ, Deak M, Arthur JS, Armit LJ, Alessi DR (2003) In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J 22: 4202–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenda A, Cohen P, Buee-Scherrer V, Goedert M (1997) Activation of stress activated protein kinase-3 (SAPK3) by cytokines and cellular stresses is mediated via SAPKK3 (MKK6); comparison of the specificities of SAPK3 and SAPK2 (RK/p38). EMBO J 16: 295–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran J (1998) In Modification and Editing of RNA, Gros-jean H, Benne B (eds) pp 493–516. Washington, DC: American Society for Microbiology [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351: 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deak M, Clifton AD, Lucocq JM, Alessi DR (1998) Mitogen- and stress-activated protein kinase-1 is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J 17: 4426–4441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber AP, Keller W (1999) An adenosine deaminase that generates inosine at the wobble position of tRNAs. Science 286: 1146–1149 [DOI] [PubMed] [Google Scholar]
- Gu W, Jackman JE, Lohan AJ, Gray MW, Phizicky EM (2003) tRNAHis maturation: an essential yeast protein catalyses addition of a guanine nucleotide to the 5′ end of tRNAHis. Genes Dev 17: 2889–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutgsell N, Englund N, Niu L, Kaya Y, Lane BG, Ofengand J (2000) Deletion of the Escherichia coli pseudouridine synthase gene truB blocks formation of pseudouridine 55 in tRNA in vivo, does not affect exponential growth, but confers a strong selective disadvantage in competition with wild-type cells. RNA 6: 1870–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson MJ, Bystrom AS (2002) Dual function of the tRNA(m(5)U54)methyltransferase in tRNA maturation. RNA 8: 324–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadaba S, Krueger A, Trice T, Krecic AM, Hinnebusch AG, Anderson J (2004) Nuclear surveillance and degradation of hypomodified initiator tRNAMet in S. cerevisiae. Genes Dev 18: 1140–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knebel A, Morrice N, Cohen P (2001) A novel method to identify protein kinase substrates: eEF2 kinase is phosphorylated and inhibited by SAPK4/p38δ. EMBO J 20: 4360–4369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Cohen P (1999) Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem J 339: 319–328 [PMC free article] [PubMed] [Google Scholar]
- Massenet S, Motorin Y, Lafontaine DL, Hurt EC, Grosjean H, Branlan C (1999) Pseudouridine mapping in the Saccharomyces cerevisiae spliceosomal U small nuclear RNAs (snRNAs) reveals that pseudouridine synthase pus1p exhibits a dual substrate specificity for U2 snRNA and tRNA. Mol Cell Biol 19: 2142–2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill H, Knebel A, Arthur JS, Cuenda A, Cohen P (2004) A novel UBA and UBX domain protein that binds polyubiquitin and VCP and is a substrate for SAPKs. Biochem J 384: 391–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud J, Kudoh J, Berry A, Bonne-Tamir B, Lalioti MD, Rossier C, Shibuya K, Kawasaki K, Asakawa S, Minoshima S, Shimizu N, Antoarakis SE, Scott HS (2000) Isolation and characterisation of a human chromosome 21q22.3 gene (WDR4) and its mouse homologue that code for a WD-repeat protein. Genomics 68: 71–79 [DOI] [PubMed] [Google Scholar]
- Mora A, Homander D, van Aalten DMF, Alessi DR (2004) PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol 15: 161–170 [DOI] [PubMed] [Google Scholar]
- Murray JT, Campbell DG, Morrice N, Auld GC, Shpiro N, Marquez R, Peggie M, Bain J, Bloomberg GB, Grahammer F, Wulff P, Kuhl D, Cohen P (2004a) Exploitation of KESTREL to identify N-myc downstream regulated gene family members as physiological substrates for SGK1 and GSK3. Biochem J 384: 477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JT, Campbell DG, Peggie M, Mora A, Cohen P (2004b) The identification of Filamin C as a new physiological substrate of PKBα using KESTREL. Biochem J 384: 489–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ougland R, Zhang CM, Liiv A, Johansen RF, Seeberg E, Hou YM, Remme J, Falnes PO (2004) AlkB restores the biological function of mRNA and tRNA inactivated by chemical methylation. Mol Cell 16: 107–116 [DOI] [PubMed] [Google Scholar]
- Reyes VM, Abelson J (1987) A synthetic substrate for tRNA splicing. Anal Biochem 166: 90–106 [DOI] [PubMed] [Google Scholar]
- Rittinger K, Budman J, Zu J, Volinia S, Cantley LC, Smerdon SJ, Gamblin SJ, Yaffe MB (1999) Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol Cell 4: 153–166 [DOI] [PubMed] [Google Scholar]
- Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J (2004) Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci USA 101: 13489–13494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez I, Hughes RT, Mayer BJ, Yee K, Woodgett JR, Avruch J, Kyriakis JM, Zon LI (1994) Role of SAPK/ERK kinase-1 in the stress-activated pathway regulating transcription factor c-Jun. Nature 372: 794–798 [DOI] [PubMed] [Google Scholar]
- Seebolt-Leopold JS, Dudley DT, Herrera R, Becelaere KV, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges A, Przbranowski A, Leopold WR, Saltiel AR (1999) Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med (NY) 5: 810–816 [DOI] [PubMed] [Google Scholar]
- Shaw M, Cohen P (1999) Role of protein kinase B and the MAP kinase cascade in mediating the EGF-dependent inhibition of glycogen synthase kinase 3 in Swiss 3T3 cells. FEBS Lett 461: 120–124 [DOI] [PubMed] [Google Scholar]
- Sidrauski C, Cox JS, Walter P (1996) tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell 87: 405–413 [DOI] [PubMed] [Google Scholar]
- Sprinzl M, Horn C, Brown M, Loudovitch A, Steinberg S (1998) Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res 26: 148–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokoe D, Campbell DG, Nakielny S, Hidaka H, Leevers SJ, Marshall C, Cohen P (1992) MAPKAP kinase-2; a novel protein kinase activated by mitogen-activated protein kinase. EMBO J 11: 3394–3985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MR, Arthur JS, Balendran A, van der Kaay J, Poli V, Cohen P, Alessi DR (2000) The role of 3-phosphoinositide-dependent protein kinase 1 in activating AGC kinases defined in embryonic stem cells. Curr Biol 10: 439–448 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Table 1