

Abstract

Despite considerable progress in using lipid nanoparticle (LNP) vehicles for gene delivery, achieving selective transfection of specific cell types remains a significant challenge, hindering the advancement of new gene or gene-editing therapies. Although LNPs have been equipped with ligands aimed at targeting specific cellular receptors, achieving complete selectivity continues to be elusive. The exact reasons for this limited selectivity are not fully understood, as cell targeting involves a complex interplay of various cellular factors. Assessing how much ligand/receptor binding contributes to selectivity is challenging due to these additional influencing factors. Nonetheless, such data are important for developing new nanocarriers and setting realistic expectations for selectivity. Here, we have quantified the selective, targeted transfection using two uniquely engineered cell lines that eliminate unpredictable and interfering cellular influences. We have compared the targeted transfection of Chinese ovary hamster (CHO) cells engineered to express the human transferrin receptor 1 (hTfR1), CHO-TRVb-hTfR1, with CHO cells that completely lack any transferrin receptor, CHO-TRVb-neo cells (negative control). Thus, the two cell lines differ only in the presence/absence of hTfR1. The transfection was performed with pDNA-encapsulating LNPs equipped with the DT7 peptide ligand that specifically binds to hTfR1 and enables targeted transfection. The LNP’s pDNA encoded for the monomeric GreenLantern (mGL) reporter protein, whose fluorescence was used to quantify transfection. We report a novel LNP composition designed to achieve an optimal particle size and ζ-potential, efficient pDNA encapsulation, hTfR1-targeting capability, and sufficient polyethylene glycol sheltering to minimize random cell targeting. The transfection efficiency was quantified in both cell lines separately through flow cytometry based on the expression of the fluorescent gene product. Our results demonstrated an LNP dose-dependent mGL expression, with a 5-fold preference for the CHO-TRVb-hTfR1 when compared to CHO-TRVb-neo. In another experiment, when both cell lines were mixed at a 1:1 ratio, the DT7-decorated LNP achieved a 3-fold higher transfection of the CHO-TRVb-hTfR1 over the CHO-TRVb-neo cells. Based on the low-level transfection of the CHO-TRVb-neo cells in both experiments, our results suggest that 17–25% of the transfection occurred in a nonspecific manner. The observed transfection selectivity for the CHO-TRVb-hTfR1 cells was based entirely on the hTfR1/DT7 interaction. This work showed that the platform of two engineered cell lines which differ only in the hTfR1 can greatly facilitate the development of LNPs with hTfR1-targeting ligands.
1. Introduction
1.1. Gene Delivery
In recent years, gene therapy has become increasingly important in medical research due to its potential to treat a wide array of diseases, ranging from genetic disorders to various types of cancer.1 On December 8, 2023, the U.S. FDA approved the first cell-based gene-editing therapy for sickle cell disease. This therapy involves isolating, modifying, and reinfusing the patient’s own blood stem cells.2 However, many future gene therapies are expected to require targeted delivery of genes to specific tissues or cells in vivo, a task requiring specialized viral or nonviral delivery vectors.3 Clinical trials have tested various viral vectors, such as retroviruses, lentiviruses, and adeno-associated viruses (AAV).3,4 While viral transduction often leads to long-term genome integration,5 it can cause off-target effects, such as undesired immune responses to the vector, hepatotoxicity, and in rare instances, fatalities.6 Moreover, AAVs have limited transgene carrying capacity (∼4.8 kb), restricting their use with larger CRISPR-based editors and other genetic payloads.7 Additional challenges with viral vectors include low production yields, scalability issues, and impurities during manufacturing.6
Recently, nonviral formulations have gained prominence in gene delivery, primarily through engineered lipid nanoparticles (LNPs) to mitigate the immunogenicity and off-target risks associated with viral formulations.8,9 The advantages of nonviral therapies lie in their remarkable capacity for engineering stable nanostructures and the versatility of surface chemistry programmability,10 enabling efficient encapsulation of nucleic acids, cellular delivery, and endosomal release. Engineered nonviral formulations can be tailored for prolonged blood circulation, reduced renal clearance, attenuated immune responses, and for transfecting cells that are typically difficult to transfect.8,11 Furthermore, the covalent attachment of active targeting ligands to LNPs can be achieved through several commercially available functionalized lipids.12
Although LNP formulations with encapsulated mRNA or siRNA have been proven effective in cellular uptake and endosomal escape, LNPs with plasmid DNA (pDNA) encapsulation have yet to be explored more. Notably, pDNA delivery offers the advantage of sustained transgene expression.13,14 Compared to RNA, DNA is more resistant to enzymatic degradation,15 and its production is also more cost-effective.16 One of the most significant limitations of traditional LNPs arises from their physicochemical resemblance to low-density lipoproteins (LDL) and their tendency to adsorb apolipoprotein E (Apo-E) in blood plasma.17 This characteristic leads to their accumulation in the liver and hepatocytic uptake via the low-density lipoprotein receptor (LDL-R). Although traditional LNP technologies effectively target hepatocytes, their specific characteristics significantly limit their application to nonliver tissues. Developing LNPs capable of delivering nucleic acids to tissues beyond the liver remains a significant challenge. Overcoming this barrier is critical to fully realizing the potential of nucleic acid delivery technologies. Successfully addressing this issue will enable the advancement of novel gene therapies in various medical contexts.18
1.2. Selective Targeting
Significant advancements have been made in controlling the size, ζ-potential, nucleic acid encapsulation, and polydispersity index in LNP-mediated gene therapy. However, the primary challenge that remains is the selective targeting of specific tissues or cell types. Selective targeting with LNPs can, in theory, be achieved by passive or active targeting approaches, yet there is a scarcity of reported quantitative data on selectivity.
Passive targeting mechanisms in LNPs are predominantly controlled by their size and charge, which can be modified through changes in the molar compositions and identity of the lipids employed in the formulation.19 For instance, using negatively charged LNPs and increasing quantities of DMG-PEG2000, a synthetic neutral lipid with a polyethylene glycol (PEG) chain of ∼45 ethylene glycol units, resulted in enhanced cellular uptake by CD8+ dendritic cells in lymph nodes.20 In some cases, passive targeting is achieved by introducing an additional lipid, termed selective organ targeting (SORT) lipids.21,22 The resulting LNP effectively targeted organs such as the liver, spleen, and lungs.22,23 The lipid composition and the specific ionizable lipid used in the formulation are key factors influencing the transfection efficiency of pDNA-LNPs.14,24
In the pursuit of active targeting, various ligands have been utilized to modify LNPs. Several research groups have employed antibodies and Fabs by grafting them onto the particles after synthesis using different approaches.25−27 LNPs decorated with antibodies against plasmalemma vesicle-associated protein (PV1) have been utilized to target lung tissue.25 In a separate study, eight LNPs were noncovalently coated with targeting antibodies (against CD44, CD34, Ly6C, CD3, CD4, CD25, CD29, and Itgb7) via a recombinant protein called anchored secondary scFv enabling targeting (ASSET). ASSET, a membrane-anchored lipoprotein, integrates into siRNA-loaded LNPs and interacts with the antibodies’ Fc domain.26 Furthermore, a single intracerebral injection of CRISPR-LNPs targeting the epidermal growth factor receptor (EGFR) has been successfully developed and tested for cancer therapy.27 Carbohydrates have also played a pivotal role in active targeting strategies. For example, DSPE-PEG2000-mannose was incorporated into a formulation to facilitate the selective delivery of LNPs to liver sinusoidal endothelial cells.28 Additionally, small molecules,29 aptamers,30 peptides,31,32 and proteins have been utilized to modify the surface of actively targeting LNPs.12,33
Achieving satisfactory selectivity in active cell targeting remains challenging. We believe that many cellular factors, such as the composition and distribution of different cell types, size, morphology, cell surface makeup, cellular microenvironment, nonspecific binding, and receptor distribution are likely to affect the selectivity. This complexity is in part supported by mechanistic studies of the delivery of nucleic acids complexed with cationic lipids.34,35 To what extent a desired ligand/receptor binding contributes to an observed selectivity is difficult to discern without the influences of other factors. However, such data are important for the development of new nanocarriers and for establishing realistic selectivity expectations. One objective of this work was to gain a better understanding of the selectivity based on receptor/ligand interactions.
The synthesis of LNPs with targeting ligands has been accomplished through two different methods.36 One approach involves the inclusion of lipids modified with targeting ligands as one of the lipid components during LNP assembly.28 In another approach, targeting ligands can be chemically conjugated to the surface of LNPs after LNP assembly.27 Once the LNPs have reached their target, they must be internalized by endocytosis and release their cargo. Fast and efficient internalization occurs via an active transport by an internalizing receptor.
Here we describe the transfection of an engineered Chinese hamster ovary (CHO) cell line that expresses the human transferrin receptor 1 (hTfR1) (CHO-TRVb-hTfR1)37 using LNPs that have a hTfR1-targeting ligand conjugated to their surface and comparison to a CHO cell line that completely lacks transferrin receptors (CHO-TRVb-neo).38 This platform allows for quantitative information on transfection selectivity without the interference of other cellular factors. The LNPs were designed with encapsulated pDNA coding for the fluorescent reporter protein monomeric GreenLantern (mGL) for the determination of transfection. Furthermore, LNPs were assembled with PEG sheltering to increase their stability and reduce cellular uptake unless the cells express hTfR1. The two cell lines studied here are unique true positive and negative controls for hTfR1 expression, which allowed for a unique comparative transfection quantification in vitro.
The hTfR1, also called CD71, is a crucial transmembrane glycoprotein that mediates iron transport. It is highly expressed on the cell surface of malignant cells due to their rapid proliferation and high iron demand.39,40 In fact, this receptor has been recognized as a universal cancer biomarker,41 and it has become promising for active targeting approaches against cancer and other conditions.39,42 Human transferrin (Tf), the natural ligand for hTfR1, plays a vital role in the iron transport process and exhibits a strong affinity for hTfR1.39,43 It has been widely utilized as a targeting ligand for delivering therapeutic agents to tumors.44 However, its application is hindered by high concentrations of endogenous Tf in human blood.45 The naturally occurring Tf competes with the Tf-modified drug delivery systems, potentially reducing its targeting efficiency in vivo.46 This is a disadvantage of using Tf as a hTfR1-targeting ligand for drug delivery. The T7 peptide (HAIYPRH), discovered by phage display technology,47 has exhibited significant binding affinity to hTfR1.48−50 However, the proteolytic susceptibility of all-L-configured peptides reduces their bioavailability.51 Therefore, the all-D-configured retro-inverso peptide DT7 (hrpyiah) has been developed, displaying enhanced stability and a high affinity to the hTfR1 (KD = 22 ± 1 nM), even surpassing that of the T7 peptide (KD = 120 ± 5 nM).52 Furthermore, DT7 binds to hTfR1 at a different site than Tf; therefore, the two ligands do not compete, further underscoring the potential of the DT7 peptide in targeted delivery applications.52
1.3. LNPs’ Characteristics and Assembly
Different fundamental properties critically influence LNP performance, directly affecting their efficacy and behavior in various applications. Some of the properties impacting the performance of LNPs include (a) Particle size: The size of LNPs is a critical factor in both their in vitro and in vivo performance. LNPs typically exhibit average diameters between 100 and 400 nm. For systemic drug delivery via intravenous (IV) injection, diameters in the 10 to 200 nm range are considered optimal.53,54 Particles smaller than 200 nm are preferred due to their ability to pass through liver sinusoidal endothelial fenestrae,55,56 and to be sterilized by filtration.57 (b) Surface charge: A surface charge on LNPs significantly influences their interactions with cellular membranes. This parameter is experimentally determined by measuring the particles’ zeta (ζ) potential. The ζ-potential values also indicate colloidal dispersion stability by quantifying the degree of repulsion force.58 Typically, an LNP with a ζ-potential exceeding +30 mV or falling below −30 mV exhibits robust electrostatic stability, effectively preventing LNP aggregation.58,59 Moreover, the surface charge on LNPs is crucial in promoting interactions with cell membranes and facilitating endosomal escape. While anionic cell membranes generally repel anionic LNPs, cationic LNPs may induce cytotoxicity by directly disrupting cellular membranes. Preferably, neutral LNPs (−10 mV < ζ-potential <10 mV) are employed to mitigate these issues.54 Incorporating ionizable lipids in LNPs becomes essential as the overall surface charge of LNPs is dependent on the environmental pH.60 This approach helps avoid undesirable electrostatic interactions with the cell membrane.61 (c) Polydispersity index (PDI): A PDI < 0.2 is generally acceptable, indicating a relatively uniform LNP size distribution, contributing to the homogeneity and reproducibility of the formulation.61 For LNPs with nucleic acid encapsulation, a desirable PDI can generally be achieved through the lipid composition, the protocol for mixing the aqueous nucleic acid phase with the organic lipid phase, the total flow rate (TFR), and the flow rate ratio (FRR).62 (d) Surface modification (e.g., PEGylation): Incorporating PEG imparts an external polymeric layer to the outer shell of LNPs, effectively inhibiting the adsorption of serum proteins and components of the phagocytic system. This phenomenon extends the in vivo circulation time of LNPs, prevents particle aggregation, and enhances their stability, even when the LNP is neutral in charge. PEG shields LNPs from serum proteins, such as Apo-E and albumins.63 However, excessive PEG in the LNP formulation can lead to excessive stability, hindering cellular internalization and intracellular release of the nucleic acids, ultimately reducing intracellular delivery.64 It is crucial to understand and optimize these properties to tailor LNPs for specific therapeutic or diagnostic applications, thereby improving their efficacy in various biomedical settings.
The ethanol dilution method is a well-established technique for assembling LNPs. In this process, one volume equivalent of an ethanol solution containing all lipids is promptly combined with three volume equivalents (of an aqueous buffer solution containing the nucleic acid component.54,65 Various methods are employed to swiftly mix these two phases, including pipet mixing, vortex mixing, and microfluidic mixing.22,62 The pipet mixing method, which involves manually pipetting the mixture up and down, is trendy for producing small-scale LNP batches.22 Its simplicity makes it a favored choice for optimization and characterization in in vitro studies, and for low-dose in vivo experiments.22
2. Results and Discussion
2.1. Synthesis of LNPs
LNPs with encapsulated pDNA were prepared using DLin-MC3-DMA as the ionizable cationic lipid. Cationic lipids are a crucial component in the composition of LNPs intended to encapsulate nucleic acids, such as mRNA, siRNA, or pDNA,66 by neutralizing their negative charges.67 In early formulations of LNPs, cationic lipids with a permanent positive charge, such as 1,2-dioleoyl-3-trimethylammoniumpropane (DOTAP), were frequently used,23,68 however, the permanent positive charge of these lipids posed challenges in vivo as these lipids are cytotoxic and result in unspecific targeting.69 In contrast, the charge of ionizable cationic lipids, e.g., DLin-MC3-DMA,70 is pH-dependent. At low pH (∼ pH 4.0), most DLin-MC3-DMA molecules are protonated, enabling electrostatic binding with nucleic acids. However, at physiological pH of 7.4, only a fraction of their amines remain protonated.70 DLin-MC3-DMA stands out as an optimized ionizable lipid employed in Onpattro, the first FDA-approved siRNA therapy.71,72
In addition to using the ionizable lipid, cholesterol was employed to augment LNP stability.73 DSPC served as an overall neutral helper lipid to assist in the formation of LNPs.74 DMG-PEG2000, another neutral lipid, was used to decorate the LNP surface with PEG, stabilizing and counteracting LNP aggregation through steric hindrance.75 Furthermore, DSPE-PEG2000-Mal was introduced at a concentration of 0.5 mol % in the formulation to enable the postassembly conjugation of the DT7 ligand to the LNP surface. Thus, the DT7-modified LNP can selectively target cells that express the hTfR1.
The LNP synthesis started with a molar ratio of DLin-MC3-DMA: cholesterol: DSPC: DMG-PEG2000 and DSPE-PEG2000-Mal of 50:38.5:10:1:0.5, respectively. However, to identify the most favorable lipid composition yielding optimal physicochemical LNP characteristics, various LNPs were synthesized by varying the DMG-PEG2000 mol % (1, 2, 2.5, 3, and 5 mol %) of the total lipid composition. Throughout these experiments, the ionizable cationic lipid to negative charges in the pDNA (N/P) ratio was maintained at 6, and the percentages of DLin-MC3-DMA, DSPC, and DSPE-PEG2000-Mal were held constant. The incremental adjustments in the DMG-PEG2000 mol % were compensated by proportionately reducing the amount of cholesterol. Subsequently, after determining the optimal composition based on the DMG-PEG2000 mol %, different N/P ratios were tested while keeping the lipid formulation constant to enhance the characteristics of LNPs further.
pDNA with the gene encoding for the reporter protein mGL (mGL-pDNA) was dissolved in acetate buffer (pH 4) and mixed with the lipids, previously dissolved in ethanol, using the pipetting mixing method (Figure 1a). mGL was selected as the fluorescent protein due to its superior brightness in cells, exhibiting up to 6-fold greater intensity than its more commonly used counterpart, the enhanced green fluorescent protein (EGFP).76 Upon dilution, buffer exchange, and concentration using an Amicon filter, LNPs functionalized with maleimide (LNP-Mal) were obtained and characterized by determining their average diameter, ζ-potential, PDI, DNA encapsulation efficiency, and DNA recovery yields. With LNP-Mal in hand, they could now be conjugated with freshly prepared DT7 peptide to afford the desired LNP-DT7 particles (Figure 1b), ready to be further characterized and tested for their target capabilities.
Figure 1.

Schematic representation of (a) the preparation of LNPs with maleimide functionalization (LNP-Mal) by the pipetting mixing method (Figure 1a created with BioRender) and (b) postassembly modification with the DT7 peptide to yield the LNP-DT7 particles.
2.2. Characterization of LNPs
Both LNP-Mal and LNP-DT7 groups underwent thorough characterization employing dynamic light scattering (DLS), ζ-potential analysis, and our modified PicoGreen pDNA encapsulation efficiency assay. Maintaining a constant N/P ratio of 6, we investigated five distinct conditions to identify the optimal amount of DMG-PEG2000 for LNP formation and pDNA encapsulation (Figure 2a–c). Starting with a low concentration of 1 mol % of DMG-PEG2000, we obtained LNP-Mal with a size of approximately 115 nm and a PDI of 0.08. However, after the DT7 conjugation, noticeable particle aggregation occurred, showing that only 1 mol % of DMG-PEG2000 compromised the LNP-DT7’s stability (Figure 2a). This issue was resolved by incrementally raising the DMG-PEG2000 concentration in the overall formulation. A progressive reduction in size was observed in all LNPs as the DMG-PEG2000 concentration increased from 2 to 5 mol % (Figure 2a). This correlation between PEG percentage and particle size has been previously documented.77 In all cases, LNP-DT7 particles exhibited a consistent increase of around 59 nm in hydrodynamic size compared to their LNP-Mal precursors, with PDIs remaining below 0.2 in all formulations (Figure 2a).
Figure 2.

Characterization of LNP-Mal and LNP-DT7 at different DMG-PEG2000 concentrations and N/P ratios. (a, b, and c) Size (intensity-weighted), ζ-potential, and EE% at different DMG-PEG2000 concentrations, respectively. (d, e, and f) Size (intensity-weighted), ζ-potential, and EE% at different N/P ratios, respectively. Data indicate mean ± SD of three replicates (see Table S1).
As expected, both LNP-Mal and LNP-DT7 particles were considered neutral in charge before and after DT7 conjugation, displaying ζ-potential values ranging from −10 to 10 mV54 (Figure 2b). ζ-potential measurements were conducted at physiological pH, where the amine moiety in the ionizable lipid DLin-MC3-DMA (pKa = 6.44)70 was mostly deprotonated, placing the LNP ζ-potential in the neutral range. Interestingly, the ζ-potential of all LNP-DT7 particles shifted from positive to negative values compared to their LNP-Mal precursors (Figure 2b), a phenomenon previously observed in DT7-decorated liposomes.52 The dependence of the pDNA EE on the DMG-PEG2000 content was measured by varying the DMG-PEG2000 amount between 2 and 5 mol %. DMG-PEG2000 contents of 2, 2.5, or 3 mol % demonstrated similar EEs of approximately 90%, while LNPs with 5 mol % DMG-PEG2000 exhibited a significantly lower EE (Figure 2c). The higher concentration of PEG appears to impact the LNPs’ assembly mechanism. The overall yield remained consistent across all these preparations, ranging from 82% to 96% (Figure 2c). Our data indicate that LNPs featuring DMG-PEG2000 concentrations of 2, 2.5, and 3 mol % exhibited highly comparable physicochemical properties, with the LNP at 3 mol % showing marginally superior overall characteristics. Preliminary transfection assays revealed that the LNP with 3 mol % of DMG-PEG2000 yielded the highest percentage of transfection (Figure S1, Supporting Information). Additionally, the decision to proceed with the higher DMG-PEG2000 concentration of 3 mol % was influenced by its potential to mitigate protein corona formation and reduce the likelihood of nonspecific uptake in a potential in vivo scenario.61
After establishing the DMG-PEG2000 concentration at 3 mol % in our LNP formulations, we investigated the optimal amount of the ionizable DLin-MC3-DMA for particle formation and pDNA encapsulation. Four different N/P ratios (4, 6, 8, and 10) were tested (Figure 2d,e,f). The transition from an N/P ratio of 4 to 6 resulted in a significant LNP size reduction and a notably improved PDI (Figure 2d). Since the majority of amine groups in DLin-MC3-DMA carry a positive charge in acetate buffer (pH 4),78 the strong electrostatic interaction between positively charged ionizable lipids and negatively charged pDNA at a higher N/P ratio prompted the formation of compacted LNPs with smaller diameters. This observation indicates that at N/P = 4, there are insufficient positively charged lipids to complex with all negatively charged pDNA molecules, allowing a portion of the pDNA to remain unbound in the solution. We measured the pDNA EE% to substantiate this hypothesis, revealing an improvement from approximately 60% at N/P = 4 to around 90% at N/P = 6 (Figure 2f). Further increases in the N/P ratio from 6 to 8 and 10 maintained the size relatively constant while the PDI reached unacceptable levels, particularly after the conjugation of DT7 (Figure 2d). Additionally, higher N/P ratios of 8 and 10 are believed to diminish the pDNA release capacity from endosomes, resulting in reduced transfection efficiency of such LNPs.79 Regarding ζ-potential, all particles can be considered neutral in charge, with ζ-potential values ranging between −10 and 10 mV. Moreover, a consistent shift from positive to negative ζ-potential values was observed when comparing the LNP-Mal group with the LNP-DT7 group (Figure 2e).
LNP-Mal particles generally exhibited more favorable physicochemical properties than the corresponding LNP-DT7 particles. The latter displayed an increase in size and PDI, along with a decrease in EE% and overall yield. This pattern can be attributed to the additional manipulation and purification treatments the LNP-DT7 particles had to undergo during their synthesis. We selected LNP-DT7 with 3 mol % of DMG-PEG2000 and an N/P ratio of 6 for further experiments because LNPs formed under these conditions exhibited particle sizes suitable for in vivo gene delivery (∼150 nm), a low desirable PDI after conjugation (∼0.145), a high encapsulation efficiency (>80%) and a high yield of >90%.
Cryo-electron microscopy (CryoEM) images of selected LNP-Mal and LNP-DT7 particles, both at 3 mol % of DMG-PEG2000 and an N/P ratio = 6, showed their spherical morphology (Figure 3). Remarkably, the lipidic core size of the LNPs, as determined by CryoEM analysis, measured less than 100 nm in both cases. In contrast, dynamic light scattering (DLS) measurements of the same LNPs indicated a size of approximately 94 nm for the LNP-Mal and 150 nm for the LNP-DT7. This discrepancy is attributable to the distinct principles underlying the two measurement techniques.80−82 CryoEM exclusively measures the lipid core of the LNP, thereby rendering the PEG branches attached and the DT7 peptide modification (in the case of LNP-DT7) indiscernible at the employed magnification, resulting in an underestimation of the total LNP diameter. Conversely, DLS assesses the intensity-weighted size calculation based on an equivalent sphere model, representing each particle as a sphere.83 The intensity-weighted average hydrodynamic size of LNPs is notably affected by the temperature, LNP concentration, sedimentation, aggregate presence, pH, buffer viscosity, and surface complexities (i.e., PEGylation and DT7 modification).83 Given that scattering intensity is proportional to the square of the particle molecular weight, any polydispersity or breadth in particle size distributions tends to skew the size average toward larger particle sizes.83,84
Figure 3.
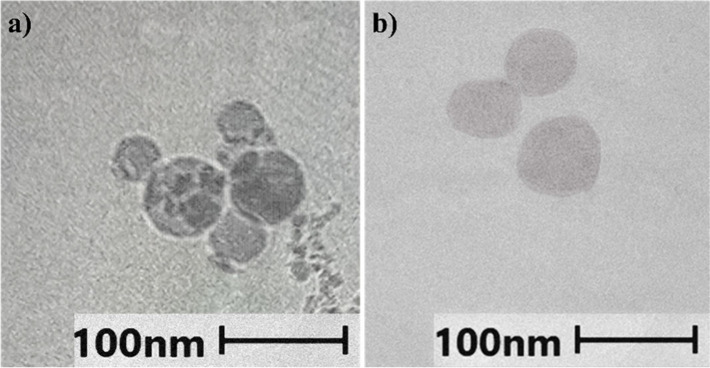
CryoEM images of (a) LNP-Mal and (b) LNP-DT7. Both LNPs were formulated with 3 mol % of DMG-PEG2000 and an N/P ratio = 6.
2.3. In Vitro Transfection Assay
Two genetically modified cell lines were employed to validate the transfection selectivity of LNP-DT7 particles toward hTfR1-expressing cells: CHO-TRVb-neo and CHO-TRVb-hTfR1.37,38 Both cell lines originated from the CHO-TRVb cell line, characterized by the absence of detectable cell surface transferrin receptor.37 The CHO-TRVb-hTfR1 cell line exclusively expresses hTfR1, whereas the CHO-TRVb-neo cells were transfected with an empty vector.38 Both CHO-TRVb (-neo and -hTfR1) cell lines were maintained in F-12 (HAM) medium supplemented with 10% fetal bovine serum and 1 mg/mL G418 as a selection marker.38 These cell lines are an ideal model system for our selectivity experiments, providing a robust positive control (CHO-TRVb-hTfR1) and negative control (CHO-TRVb-neo) regarding transferrin receptor expression. Most commercially available human cell lines exhibit some degree of hTfR1 expression, making an appropriate negative control difficult to find.
Five different transfection experiments with individual as well as mixed cell types were carried out: (a) CHO-TRVb-neo with LNP-DT7; (b) CHO-TRVb-hTfR1 with LNP-DT7; (c) CHO-TRVb-neo with LNP-2ME; (d) CHO-TRVb-hTfR1 with LNP-2ME; and (e) a mixture of CHO-TRVb-neo and CHO-TRVb-hTfR1 with LNP-DT7. In all transfection experiments, cells were seeded at a concentration of 8,000 cells/well. Subsequently, the cells were treated with five or six different concentrations of LNP-DT7, corresponding to encapsulated pDNA amounts of 100, 200, 300, 400, 500, 600, 800, or 1000 ng/well. This approach enabled the exploration of dose/response effects. The transfections were carried out as described in the Methods Section 4.4. After 48h, in two of the individual transfection experiments (CHO-TRVb-neo with LNP-DT7; and CHO-TRVb-hTfR1 with LNP-DT7) mGL expression was qualitatively assessed by fluorescence microscopy as described in the Methods Section 4.4. The nuclei of live cells were visualized using Hoechst 33342.85 Then, images were acquired by microscopy using the imaging DAPI filter cube for nuclear identification (Hoechst 33342 ex/em: 361/497 nm), the imaging GFP filter cube for mGL expression (ex/em: 503/514 nm), and brightfield. Figure 4 depicts images of both cell lines, CHO-TRVb-neo (top row) and CHO-TRVb-hTfR1 (bottom row), following treatment with 600 ng of mGL-pDNA using LNP-DT7 as the transfection method. A distinct difference is evident in the mGL protein expression between the two cell lines, showing a higher protein expression in CHO-TRVb-hTfR1 than in CHO-TRVb-neo cells. Flow cytometry was employed to assess the percentage of transfected cells quantitatively.
Figure 4.
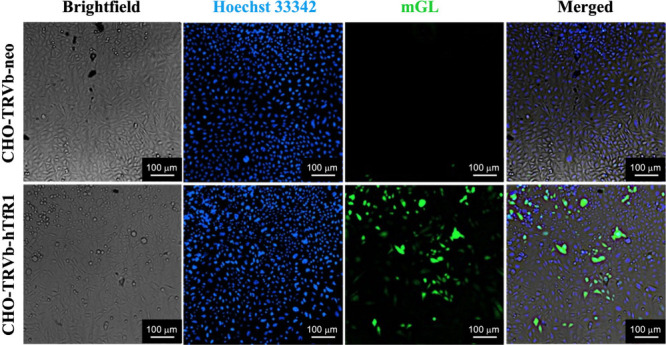
mGL protein expression in CHO-TRVb-neo (top row) and CHO-TRVb-hTfR1 cells (bottom row). At 48 h post-transfection of 600 ng of mGL-pDNA LNP-DT7, cells were stained with Hoechst 33342 (second column) and imaged in the Cytation 7 microscope mode, using the filters for DAPI (Hoechst 33342) and GFP (mGL).
2.4. Transfection Efficiency and Selectivity Determined by Flow Cytometry
After imaging, the cells from each well were detached from the plate using Accutase, which contains collagenolytic and proteolytic enzymes. Compared with trypsin, the Accutase reagent is assumed to be more gentle and less problematic for detecting surface markers in flow cytometry.86 The detached cells were transferred to 1 mL of complete F-12 (HAM) medium. Then, the cells were fixed with FACS fix solution and resuspended in FACS PBS to quantify the transfection efficiency by flow cytometry based on mGL expression. A control group of cells only stained with Hoechst 33342 was used to identify the number of cells by the number of nuclei (Figure S2, Supporting Information). Figure 5 combines the individual transfection results with LNP-DT7 using CHO-TRVb-neo and CHO-TRVb-hTfR1. The percentages of mGL-positive cells of both sets of experiments were compared at various pDNA concentrations. In all transfection experiments using CHO-TRVb-neo cells with LNP-DT7, the results consistently show low mGL expression between approximately 1% and 4%. These findings confirmed the limited affinity of LNP-DT7 particles toward hTfR1-negative CHO-TRVb-neo cells. In contrast, the percentages of mGL-positive CHO-TRVb-hTfR1 cells demonstrated a pronounced and dose-dependent increase, reaching approximately 20% transfection efficiency at a pDNA dose of 1000 ng/well. At higher pDNA doses of 1500 and 2000 ng/well, the percentage of mGL positive cells increased only slightly and appeared to reach a plateau at 22% (Figure S4, Supporting Information). Notably, achieving a 20% transfection efficiency at this dose using pDNA encapsulated in an LNP surpasses the reported outcomes in existing literature, where comparable percentages were only attained with higher pDNA doses (5000 ng pDNA), and only via the ApoE/LDL-R pathway.87,88
Figure 5.

Percentage of mGL-positive cells at different concentrations of pDNA. CHO-TRVb-neo and CHO-TRVb-hTfR1 cells separately transfected with 0, 100, 200, 400, 600, 800, and 1000 ng of mGL-pDNA encapsulated in LNP-DT7. The percentage of mGL transfected cells was evaluated by flow cytometry. Data were represented as mean ± SD of biological replicates (n = 3). Statistical analysis was performed by Bonferroni’s multiple comparisons test: ***p < 0.001; ****p < 0.0001.
Based on flow cytometry analysis, the transfection selectivity of the LNP-DT7 can be quantified at a specific pDNA concentration as the ratio between mGL-positive CHO-TRVb-hTfR1 and mGL-positive CHO-TRVb-neo cells. For example, at a pDNA concentration of 600 ng/well a 5-fold higher transfection of the CHO-TRVb-hTfR1 was achieved, when compared to CHO-TRVb-neo cells under the same conditions, surpassing similar selectivity assessments reported in the literature, most of which are based on cellular uptake.89,90 When considering the sum of all transfected cells as 100% at a pDNA concentration of 600 ng/well, the ratio of transfected CHO-TRVb-neo cells to transfected CHO-TRVb-hTfR1 cells of 1:5 (Figure 5) corresponds to a ratio of 17%: 83%. This suggests that 17% of the transfections occurred nonspecifically.
We also conducted a cell competition experiment, in which equal amounts of CHO-TRVb-hTfR1 and CHO-TRVb-neo cells were seeded together into the same wells prior to being subjected to transfection with different amounts of DT7-LNP. Successfully transfected cells were detected by flow cytometry based on their mGL expression, and CHO-TRVb-hTfR1 and CHO-TRVb-neo cells were distinguished from each other based on the detection of the hTfR1 using the fluorescent mouse anti-hTfR1 (CD71) monoclonal antibody (OKT9 (OKT-9)), PerCP-eFluorTM 710, eBiosciencesTM. Figure 6 shows the quantitative, dose-dependent transfection results using mixtures of CHO-TRVb-hTfR1 and CHO-TRVb-neo cells. As previously observed in the individual transfection experiments (Figure 5), the cells display a dose-dependent transfection efficiency with increasing quantities of pDNA. When compared to the transfection of CHO-TRVb-hTfR1 cells only (Figure 5), an overall lower percentage of mGL positive cells was observed. However, Figure 6 shows that using a 1:1 mixture of CHO-TRVb-hTfR1 and CHO-TRVb-neo cells also resulted in a clear preferential transfection of the CHO-TRVb-hTfR1, albeit with a somewhat lower selectivity when compared to the individual transfection experiments shown in Figure 5. For example, exposing 4000 CHO-TRVb-hTfR1 and 4000 CHO-TRVb-neo cells with 300 ng pDNA, the selectivity for the CHO-TRVb-hTfR1 was only 4-fold. A ratio of transfected CHO-TRVb-neo/transfected CHO-TRVb-hTfR1 of 1:4 corresponds to 20%: 80%, thus, 20% of the transfections occurred nonspecifically. At 600 ng pDNA, the selectivity for the CHO-TRVb-hTfR1 cells decreased to only 3-fold. Despite somewhat lower selectivity in the mixed cell experiment (Figure 6), the observed preferential transfection of the CHO-TRVb-hTfR1 cells has to be attributed to the interaction between hTfR1 and DT7.
Figure 6.

Percentage of mGL-positive cells at different concentrations of pDNA using a 1:1 mixture of CHO-TRVb-neo and CHO-TRVb-hTfR1 cells. A total of 8000 cells, i.e., 4000 cells of CHO-TRVb-neo and 4000 cells of CHO-TRVb-hTfR1 cells, were seeded per well. Cells were transfected with 0, 300, 400, 500, 600, and 1000 ng of mGL-pDNA encapsulated in LNP-DT7. After 48 h, the cells were detached and stained with Hoechst 33342 and the mouse anti-hTfR1 (CD71) monoclonal antibody (OKT9 (OKT-9)), PerCP-eFluorTM 710, eBiosciencesTM (Figure S3, Supporting Information). Data were analyzed in Kaluza for Gallios 1.0 software and represented as mean ± SD of biological replicates (n = 3). Statistical analysis was performed by Bonferroni’s multiple comparisons test: ***p < 0.001; ****p < 0.0001.
To further confirm the transfection dependence on the interaction between the DT7/hTfR1 interaction, CHO-TRVb-neo and CHO-TRVb-hTfR1 cells were separately treated with 2-metcaptoethanol-modified LNP (LNP-2ME) and LNP-DT7 at different pDNA concentrations. Consistent with expectations, CHO-TRVb-neo showed only low average percentage of mGL-expressing cells below 5% across a range of pDNA concentrations with either of the two LNPs (Figure S5, Supporting Information). LNP-2ME was also not able to significantly transfect CHO-TRVb-hTfR1 cells; the percentage of mGL-expressing cells remained below 5% at all pDNA concentrations tested (Figure S6, Supporting Information). At 1000 ng pDNA the LNP-DT7 exhibited almost a 5-fold higher transfection of the CHO-TRVb-hTfR1 cells compared to LNP-2ME (Figure S6, Supporting Information).
3. Conclusions
In this study, we synthesized novel LNPs equipped with a cell-targeting ligand and encapsulated pDNA coding for a fluorescent reporter protein (mGL) using the pipet mixing technique. Subsequently, we conjugated the DT7 peptide, a recently developed ligand for the hTfR1, to the LNP surface. Optimization of critical parameters, i.e., LNP diameter, ζ-potential, and pDNA encapsulation, was essential, particularly by adjusting the DMG-PEG2000 mol % and N/P ratios, to ensure efficient cell transfection. This research focused on avoiding random transfection by providing LNPs with PEG sheltering and achieving efficient selective transfection of cells expressing hTfR1 through active targeting. Our optimization experiments indicated that LNPs with 3 mol % DMG-PEG2000 and an N/P ratio of 6 were ideal for pDNA encapsulation and exhibited desirable physicochemical properties.
Transfection experiments were conducted using two unique cell lines, CHO-TRVb-hTfR1 and CHO-TRVb-neo, as positive and negative controls, respectively, for transferrin receptor expression. These transfections were carried out both, separately on individual cell lines, and combined, where cells of both cell lines were mixed together prior to transfection. Fluorescence microscopy showed clear differences in mGL protein expression between the two cell lines, underscoring the superior performance of LNP-DT7 in CHO-TRVb-hTfR1 cells. In the transfections of individual cell lines, flow cytometry results showed a significant, dose-dependent transfection efficiency in CHO-TRVb-hTfR1 cells (up to 20%), compared to a much lower efficiency in CHO-TRVb-neo cells (up to 4%). This demonstrated a 5-fold selectivity for the transferrin receptor-expressing cells, but also revealed that some undesired nonspecific transfection (17%) took place. In the transfection of a 1:1 mixture of CHO-TRVb-hTfR1 and CHO-TRVb-neo cells with LNP-DT7, the best selectivity achieved was 4-fold for the CHO-TRVb-hTfR1 cells over CHO-TRVb-neo cells.
To our knowledge, this is the first report of such an unbiased transfection selectivity study achieved by actively targeting LNPs. The two modified cell lines proved to be a useful platform to obtain true quantitative selectivity data based on flow cytometry. These cells can be used for further optimization of LNPs for the targeted transfection of hTfR1-expressing cells.
4. Methods
4.1. Reagents and Cell Lines
4-(Dimethylamino)butanoic acid, (10Z,13Z)-1-(9Z,12Z)-9,12-octadecadien-1-yl-10,13-nonadecadien-1-yl ester (DLin-MC3-DMA) and α-[(2R)-2,3-bis[(1-oxotetradecyl)oxy]propyl]-ω-methoxy-poly(oxy-1,2-ethanediyl) (DMG-PEG2000) were purchased from Cayman Chemicals, and cholesterol (Cho) and 1,2-distearoyl-sn-glycero-3-phosphatidylcholine (DSPC) from Avanti Polar Lipids (CRODA). 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG2000-Mal) were purchased from Laysan Bio Inc. All lipids were dissolved in absolute ethanol 200 proof (Thermo Fisher Scientific) and stored at −20 °C until used. Bond-Breaker TCEP Solution, Neutral pH (TCEP); Molecular grade Water, Cytiva Hyclone; and Gibco PBS, pH 7.4, were purchased from Thermo Fisher Scientific. The original retro-inverso peptide (hrpyiah) was custom-synthesized with an additional d-cysteine residue at the C-terminus (hrpyiahc) by GenScript. Incorporating cysteine allows for the conjugation of the peptide to a maleimide-derivatized lipid. The plasmid pcDNA3.1-mGreenLantern (mGL-pDNA) was a gift from Gregory Petsko (Addgene plasmid #161912; http://n2t.net/addgene:161912; RRID: Addgene_161912).76Escherichia coli (E. coli) DH5alpha (Top10) (Thermo Fisher Scientific) bacterial cells were transformed and grown in Luria–Bertani (LB) agar supplemented with 50 μg/mL kanamycin. Then, colonies were selected, inoculated in 5 mL LB medium with kanamycin, and incubated under shaking conditions at 37 °C for 12 h. Then, a miniprep (Promega) was performed, followed by EcoRI and ApaI double digestion to confirm a DNA fragment of 739 base pairs corresponding to the mGreenLantern gene. Lastly, the GeneJET plasmid Maxiprep kit (Thermo Fisher Scientific) was used to obtain sufficient quantities of plasmid DNA for the LNP preparation.
Derived from CHO-TRVb cells (TfR-deficient Chinese hamster ovary cells),37 CHO-TRVb-hTfR1 cells, engineered to express hTfR1 (not hTfR2) and CHO-TRVb-neo cells transfected with the empty neomycin vector38 were generously provided by Dr. Phillip Koeffler (Cedars Sinai Medical Center, Los Angeles, CA). Both cell lines were cultured in an F-12 (HAM) medium (Thermo Fisher Scientific), supplemented with 10% fetal bovine serum (Corning) and 1 mg/mL G418 as a selectable marker. Gibco StemPro Accutase Cell Dissociation Reagent (Thermo Fisher Scientific) was used to detach the cells before passages and flow cytometry analysis.
4.2. Synthesis of LNPs
The maleimide-derivatized LNPs with mGL-pDNA encapsulated, coding for the fluorescent protein mGL,76 were synthesized using the pipetting mixing method. To identify conditions that would result in LNPs with desirable properties, initially, lipid mixtures were prepared by combining the ionizable cationic lipid DLin-MC3-DMA, the helper lipid DSPC, cholesterol, the PEG-lipid DMG-PEG2000, and the maleimide-derivatized lipid DSPE-PEG2000-Mal in different molar ratios to achieve a final lipid concentration of 6.2 mg/mL in pure ethanol. The aqueous mixture was prepared by diluting the mGL-pDNA with nuclease-free water to the desired concentration of pDNA. The pH of the aqueous solution was adjusted to pH 4 using 25 mM sodium acetate buffer. The concentration of pDNA in the aqueous solution was determined using NanoDrop (Thermo Fisher Scientific) and adjusted to the desired ionizable cationic lipid to pDNA (N/P) ratio (mol/mol) of the final solution. The aqueous and alcoholic phases in a ratio of 3:1 (v/v) were then rapidly mixed by pipetting up and down for approximately 30 s. Immediately after synthesis, the LNP solution was dissolved in 1X PBS pH 7.4 until the ethanol concentration was approximately 2 vol %. The resulting mixture underwent purification and concentration through Amicon filtration (30 kDa, MilliporeSigma Amicon Ultra-0.5 Centrifugal Filter Units). The volume was adjusted with 1X PBS pH 7.4 to achieve a final lipid concentration of 3 mg/mL, and the samples were stored at 4 °C until use.
For the modification with the DT7 peptide, the maleimide-derivatized LNPs were mixed with 10 equiv of DT7 and the reductant TCEP in 1X PBS pH 7.4 for 2 h, at rt. After the reaction, the excess of DT7 and TCEP was removed by washing with 1X PBS pH 7.4 in an Amicon filter (30 kDa, MilliporeSigma Amicon Ultra-0.5 Centrifugal Filter Units). The volume was adjusted with 1X PBS pH 7.4 to reach a final lipid concentration of 3 mg/mL, and the samples were stored for up to 1 week at 4 °C until use. Concurrently, another batch of maleimide-derivatized LNPs underwent treatment with 2-mercaptoethanol (ME) under identical conditions as those employed for the DT7 modification. These particles (LNP-2ME) served as a negative control in the subsequent transfection experiments (Figure S2, Supporting Information).
4.3. LNP Characterization
4.3.1. Hydrodynamic Size and ζ-Potential
Hydrodynamic size and ζ-potential measurements were conducted using dynamic light scattering (DLS) with a Zetasizer Ultra Red Label instrument (Malvern Instruments). In brief, DLS was employed to determine the average hydrodynamic intensity-weighted size of particles (expressed as the average particle diameter in nm based on light scattering by intensity) using ZEN0040 disposable cuvettes (Malvern Panalytical) in pure water. For ζ-potential measurements, LNPs were diluted 100-fold with nuclease-free water and analyzed using a DTS1070 disposable folded capillary cell (Malvern Panalytical, Malvern, UK).
4.3.2. Encapsulation Efficiency (EE)
The EE of pDNA was determined as a percentage value using the Quant-iT PicoGreen dsDNA assay kit (Thermo Fisher Scientific). Initially, two buffers were prepared: TE-buffer containing Tris (10 mM, pH 7.5), EDTA (1 mM), and Triton/TE-buffer (0.2% v/v Triton X-100 in TE buffer). Both buffers were added in triplicates to a black microplate (Corning 96-Well Solid Polystyrene Microplate). The total pDNA in the LNPs was diluted to approximately 60 ng/mL in TE and added to each TE and TE/Triton well in a 1:1 volume ratio. The EE assay included two standard curves, one containing mGL-pDNA in TE-buffer and the other containing mGL-pDNA in Triton/TE. Each standard curve calculates the pDNA concentration in its respective buffer. The use of two standard curves is essential for accurate EE and pDNA concentration calculation, as a single standard curve in Triton/TE-buffer may overestimate encapsulation by 5–10%, attributed to the higher background fluorescence of PicoGreen in Triton/TE- versus TE-buffers. Microplates were incubated at 40 °C for 15 min with continuous stirring at 200 rpm to extract encapsulated pDNA from LNPs with Triton. Quant-iT PicoGreen reagent in DMSO was diluted 200-fold in TE Buffer and added to each well in a 1:1 volume ratio, resulting in a final volume per well of 200 μL. Microplates were further incubated at room temperature for 5 min under continuous shaking. Subsequently, the end point fluorescence intensity was measured in a BioTek Cytation 7 Cell Imaging Multi-Mode Reader (Agilent) at 485 nm (excitation) and 528 nm (emission) with a bandwidth of 20 nm. To calculate the EE percentage of mGL-pDNA in the LNPs, the following equation was used:
Co represents the concentration of unencapsulated mGL-DNA when only TE-buffer was used, and Ct represents the total mGL-pDNA concentration after LNP lysis with Triton/TE-buffer. Three replicates of each sample were measured to obtain the average EE. The mean EE percentages calculated in this study were obtained from independent experiments to confirm the reproducibility of the LNP formulations.
4.3.3. Cryo-Electron Microscopy (CryoEM)
Sample vitrification was conducted by dispensing 6 μL of LNP solution in pure water onto a glow-discharged gold grid with 300 mesh and continuous carbon film. Subsequently, grids were blotted with filter paper for 7 s at 25 °C and 100% relative humidity, followed by immersion into a liquid ethane/propane mixture cooled by liquid nitrogen for rapid freezing. After the vitrification, the frozen grids were stored below −170 °C using liquid nitrogen until analysis. Imaging was performed on a JEOL 3200FS using the Minimum Dose System (MSD) at an accelerating voltage of 300 kV, 80,000× magnification, and 30 e–/Å2 electron dose.
4.4. In Vitro Transfection Assay and Cell Imaging
Both CHO-TRVb-neo and CHO-TRVb-hTfR1 cells, or a mixture thereof, were seeded in 96-well black/clear bottom plates at a density of 8 × 103 cells per well in F-12 (HAM) medium, supplemented with 10% fetal bovine serum (FBS), and 1 mg/mL G418 (neomycin, Corning) as a selecting marker, and incubated at 37 °C and 5% CO2 for 12 h. Then, cells were washed with F-12 (HAM) basal medium before adding the LNPs. mGL-pDNA loaded LNPs modified with DT7 were diluted in a basal medium at varying quantities of encapsulated pDNA in a final volume of 100 μL per well. In parallel, an untreated cell group served as a negative control. Cells were transfected in triplicates for each of the five or six doses, including the untreated control group. After a 4 h transfection, the medium with LNPs was removed, and the wells were washed with F-12 (HAM) basal medium. Lastly, 100 μL complete F-12 (HAM) medium was added and incubated at 37 °C for 48 h. After 48 h, cells were incubated with 100 μL Hoechst 33342 dissolved in PBS at a concentration of 0.5 μg/mL for 20 min at 37 °C. Microplates were imaged in a Cytation 7 (BioTek, Agilent, Santa Clara, CA), using the DAPI and GFP filters.
4.5. Flow Cytometry
Once images were acquired, the cells were washed with PBS, and 100 μL Accutase Enzyme Cell Detachment Medium (Thermo Fisher Scientific) was added to each well. The plate was incubated for 5 min at 37 °C to detach the cells from the wells. The cells were pipetted up and down and transferred to tubes containing 1 mL of complete F-12 (HAM) medium. The cells were centrifuged at 2000 rpm for 2 min, fixed with FACS-Fix buffer (1X PBS, 1% BSA, 2% paraformaldehyde), and incubated for 30 min, followed by washing of the cells with FACS buffer (1X PBS, 1% BSA). The percentage of transfected cells was determined using a Gallios’s Flow Cytometer (Beckman Coulter), counting 2000 events and using the channels FL1 for mGL expression and FL9 for Hoechst 33342. (see Figure S1, Supporting Information) The Flow Cytometry Standard (FCS) files were analyzed using FlowJo software 10.8.1 on a Mac workstation.
4.6. Statistical Analysis
Statistical analyses were performed using GraphPad Prism 9.5.1 (GraphPad Software, Inc., La Jolla, CA). Two-way ANOVA Bonferroni’s multiple comparisons test was used to compare the means of two independent samples against each other. Results were considered significant when p < 0.05.
Acknowledgments
This work was supported by Dr. Keelung Hong and the Yen Chuang Foundation (K.M. and I.C.A.) and by the National Institutes of Health (NIH) grant R21CA283583-01A1 (K.M.). It was partially supported by NIH grant R01CA196266 (M.L.P.). I.V. is grateful for a Dr. Keelung Hong Chemistry Graduate Research Fellowship. I.C.A. was partially supported by NIH grant # 5U54MD007592 (to Dr. Robert A. Kirken) from the National Institute on Minority Health and Health Disparities (NIMHD). The authors thank Dr. Ricardo A. Bernal for his assistance in the CryoEM measurement, Dr. Armando Varela for his assistance in flow cytometry, and the Biomolecule and Omics Core (BAO) Unit and Cellular Characterization & Biorepository (CCB) Unit of the Border Biomedical Research Center (BBRC) at the University of Texas at El Paso, supported by the Research Centers at Minority Institutions (RCMI) grant # 5U54MD007592 (to Dr. Robert A. Kirken) from the NIMHD, a component of the National Institutes of Health (NIH), for full access to flow cytometry instrumentation used in this study. J.C.S.-E. is grateful for a publication cost contribution award from the College of Science (Univ. of Texas at El Paso).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.4c03541.
Diagrams showing transfection percentages at different amounts of DMG-PEG2000 or pDNA in various LNP formulations; characterization data of LNP-Mal and LNP-DT7; flow cytometry data (PDF)
Author Contributions
# I.V. and J.C.S.-E. have contributed equally.
The authors declare the following competing financial interest(s): MLP has a financial interest in Stellar Biosciences, Inc. The Regents of the University of California licensed technologies invented by MLP to this firm. In addition, he has a financial interest in Klyss Biotech, Inc.
Supplementary Material
References
- Sayed N.; Allawadhi P.; Khurana A.; Singh V.; Navik U.; Pasumarthi S. K.; Khurana I.; Banothu A. K.; Weiskirchen R.; Bharani K. K. Gene therapy: Comprehensive overview and therapeutic applications. Life Sci. 2022, 294, 120375. 10.1016/j.lfs.2022.120375. [DOI] [PubMed] [Google Scholar]
- Frangoul H.; Altshuler D.; Cappellini M. D.; Chen Y.-S.; Domm J.; Eustace B. K.; Foell J.; de la Fuente J.; Grupp S.; Handgretinger R.; et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J. Med. 2021, 384 (3), 252–260. 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- Butt M. H.; Zaman M.; Ahmad A.; Khan R.; Mallhi T. H.; Hasan M. M.; Khan Y. H.; Hafeez S.; Massoud E. E. S.; Rahman M. H. Appraisal for the potential of viral and nonviral vectors in gene therapy: A review. Genes (Basel) 2022, 13 (8), 1370. 10.3390/genes13081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulcha J. T.; Wang Y.; Ma H.; Tai P. W.; Gao G. Viral vector platforms within the gene therapy landscape. Signal Transduct Target Ther 2021, 6 (1), 53. 10.1038/s41392-021-00487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotterman M. A.; Schaffer D. V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet 2014, 15 (7), 445–451. 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q.; Polanco A.; Lee Y. S.; Yoon S. Critical challenges and advances in recombinant adeno-associated virus (rAAV) biomanufacturing. Biotechnol. Bioeng. 2023, 120 (9), 2601–2621. 10.1002/bit.28412. [DOI] [PubMed] [Google Scholar]
- Song C.-Q.; Jiang T.; Richter M.; Rhym L. H.; Koblan L. W.; Zafra M. P.; Schatoff E. M.; Doman J. L.; Cao Y.; Dow L. E.; et al. Adenine base editing in an adult mouse model of tyrosinaemia. Nat. Biomed Eng. 2020, 4 (1), 125–130. 10.1038/s41551-019-0357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimondo T. M.; Reed K.; Shi D.; Langer R.; Anderson D. G. Delivering the next generation of cancer immunotherapies with RNA. Cell 2023, 186 (8), 1535–1540. 10.1016/j.cell.2023.02.031. [DOI] [PubMed] [Google Scholar]
- Rajendran A. P.; Ogundana O.; Morales L. C.; Meenakshi Sundaram D. N.; Kucharski C.; Kc R.; Uludağ H. Transfection Efficacy and Cellular Uptake of Lipid-Modified Polyethyleneimine Derivatives for Anionic Nanoparticles as Gene Delivery Vectors. ACS Appl. Bio Mater. 2023, 6 (3), 1105–1121. 10.1021/acsabm.2c00978. [DOI] [PubMed] [Google Scholar]
- Menon I.; Zaroudi M.; Zhang Y.; Aisenbrey E.; Hui L. Fabrication of active targeting lipid nanoparticles: Challenges and perspectives. Mater. Today Adv. 2022, 16, 100299. 10.1016/j.mtadv.2022.100299. [DOI] [Google Scholar]
- DiStasio N.; Arts M.; Lehoux S.; Tabrizian M. IL-10 Gene Transfection in Primary Endothelial Cells via Linear and Branched Poly(β-amino ester) Nanoparticles Attenuates Inflammation in Stimulated Macrophages. ACS Appl. Bio Mater. 2018, 1 (3), 917–927. 10.1021/acsabm.8b00342. [DOI] [PubMed] [Google Scholar]
- Kularatne R. N.; Crist R. M.; Stern S. T. The future of tissue-targeted lipid nanoparticle-mediated nucleic acid delivery. Pharmaceuticals (Basel) 2022, 15 (7), 897. 10.3390/ph15070897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni J. A.; Myhre J. L.; Chen S.; Tam Y. Y. C.; Danescu A.; Richman J. M.; Cullis P. R. Design of lipid nanoparticles for in vitro and in vivo delivery of plasmid DNA. Nanomedicine 2017, 13 (4), 1377–1387. 10.1016/j.nano.2016.12.014. [DOI] [PubMed] [Google Scholar]
- Algarni A.; Pilkington E. H.; Suys E. J.; Al-Wassiti H.; Pouton C. W.; Truong N. P. In vivo delivery of plasmid DNA by lipid nanoparticles: the influence of ionizable cationic lipids on organ-selective gene expression. Biomater Sci. 2022, 10 (11), 2940–2952. 10.1039/D2BM00168C. [DOI] [PubMed] [Google Scholar]
- Li Y.; Breaker R. R. Kinetics of RNA Degradation by Specific Base Catalysis of Transesterification Involving the 2‘-Hydroxyl Group. J. Am. Chem. Soc. 1999, 121 (23), 5364–5372. 10.1021/ja990592p. [DOI] [Google Scholar]
- Schlosser K.; Li Y. Biologically inspired synthetic enzymes made from DNA. Chem. Biol. 2009, 16 (3), 311–322. 10.1016/j.chembiol.2009.01.008. [DOI] [PubMed] [Google Scholar]
- Wang S.; Zhang J.; Zhou H.; Lu Y. C.; Jin X.; Luo L.; You J. The role of protein corona on nanodrugs for organ-targeting and its prospects of application. J. Controlled Release 2023, 360, 15–43. 10.1016/j.jconrel.2023.06.014. [DOI] [PubMed] [Google Scholar]
- Huang K.; Zapata D.; Tang Y.; Teng Y.; Li Y. In vivo delivery of CRISPR-Cas9 genome editing components for therapeutic applications. Biomaterials 2022, 291, 121876. 10.1016/j.biomaterials.2022.121876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilliard S. A.; Siegwart D. J. Passive, active and endogenous organ-targeted lipid and polymer nanoparticles for delivery of genetic drugs.. Nat. Rev. Mater. 2023, 8 (4), 282–300. 10.1038/s41578-022-00529-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T.; Kawai M.; Sato Y.; Maeki M.; Tokeshi M.; Harashima H. The effect of size and charge of lipid nanoparticles prepared by microfluidic mixing on their lymph node transitivity and distribution. Mol. Pharmaceutics 2020, 17 (3), 944–953. 10.1021/acs.molpharmaceut.9b01182. [DOI] [PubMed] [Google Scholar]
- Dilliard S. A.; Cheng Q.; Siegwart D. J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (52), e2109256118 10.1073/pnas.2109256118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Liu S.; Sun Y.; Yu X.; Lee S. M.; Cheng Q.; Wei T.; Gong J.; Robinson J.; Zhang D.; et al. Preparation of selective organ-targeting (SORT) lipid nanoparticles (LNPs) using multiple technical methods for tissue-specific mRNA delivery. Nat. Protoc 2023, 18 (1), 265–291. 10.1038/s41596-022-00755-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q.; Wei T.; Farbiak L.; Johnson L. T.; Dilliard S. A.; Siegwart D. J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol 2020, 15 (4), 313–320. 10.1038/s41565-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S.; Khalil I. A.; Elewa Y. H.; Harashima H. Novel lipid combination for delivery of plasmid DNA to immune cells in the spleen. J. Controlled Release 2021, 330, 753–764. 10.1016/j.jconrel.2021.01.005. [DOI] [PubMed] [Google Scholar]
- Li Q.; Chan C.; Peterson N.; Hanna R. N.; Alfaro A.; Allen K. L.; Wu H.; Dall’Acqua W. F.; Borrok M. J.; Santos J. L. Engineering caveolae-targeted lipid nanoparticles to deliver mRNA to the lungs. ACS Chem. Biol. 2020, 15 (4), 830–836. 10.1021/acschembio.0c00003. [DOI] [PubMed] [Google Scholar]
- Kedmi R.; Veiga N.; Ramishetti S.; Goldsmith M.; Rosenblum D.; Dammes N.; Hazan-Halevy I.; Nahary L.; Leviatan-Ben-Arye S.; Harlev M. A modular platform for targeted RNAi therapeutics. Nat. Nanotechnol 2018, 13 (3), 214–219. 10.1038/s41565-017-0043-5. [DOI] [PubMed] [Google Scholar]
- Rosenblum D.; Gutkin A.; Kedmi R.; Ramishetti S.; Veiga N.; Jacobi A. M.; Schubert M. S.; Friedmann-Morvinski D.; Cohen Z. R.; Behlke M. A.; et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci. Adv. 2020, 6 (47), eabc9450 10.1126/sciadv.abc9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.; Jeong M.; Hur S.; Cho Y.; Park J.; Jung H.; Seo Y.; Woo H.; Nam K.; Lee K. Engineered ionizable lipid nanoparticles for targeted delivery of RNA therapeutics into different types of cells in the liver. Sci. Adv. 2021, 7 (9), eabf4398 10.1126/sciadv.abf4398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzysztoń R.; Salem B.; Lee D. J.; Schwake G.; Wagner E.; Rädler J. O. Microfluidic self-assembly of folate-targeted monomolecular siRNA-lipid nanoparticles. Nanoscale 2017, 9 (22), 7442–7453. 10.1039/C7NR01593C. [DOI] [PubMed] [Google Scholar]
- Wilner S. E.; Wengerter B.; Maier K.; de Lourdes Borba Magalhães M.; Del Amo D. S.; Pai S.; Opazo F.; Rizzoli S. O.; Yan A.; Levy M. An RNA alternative to human transferrin: a new tool for targeting human cells. Mol. Ther Nucleic Acids 2012, 1 (5), e21 10.1038/mtna.2012.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagino Y.; Khalil I. A.; Kimura S.; Kusumoto K.; Harashima H. GALA-modified lipid nanoparticles for the targeted delivery of plasmid DNA to the lungs. Mol. Pharmaceutics 2021, 18 (3), 878–888. 10.1021/acs.molpharmaceut.0c00854. [DOI] [PubMed] [Google Scholar]
- Herrera-Barrera M.; Ryals R. C.; Gautam M.; Jozic A.; Landry M.; Korzun T.; Gupta M.; Acosta C.; Stoddard J.; Reynaga R.; et al. Peptide-guided lipid nanoparticles deliver mRNA to the neural retina of rodents and nonhuman primates. Sci. Adv. 2023, 9 (2), eadd4623 10.1126/sciadv.add4623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafech B.; Mohabatpour F.; Hedtrich S. Surface modification of lipid nanoparticles for gene therapy. J. Gene Med. 2024, 26 (1), e3642 10.1002/jgm.3642. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S.; Mandal S. S. Evidence of interlipidic ion-pairing in anion-induced DNA release from cationic amphiphile-DNA complexes.. Mechanistic implications in transfection. Biochemistry 1998, 37 (21), 7764–7777. 10.1021/bi971772j. [DOI] [PubMed] [Google Scholar]
- Muñoz-Úbeda M.; Misra S. K.; Barrán-Berdón A. L.; Aicart-Ramos C.; Sierra M. B.; Biswas J.; Kondaiah P.; Junquera E.; Bhattacharya S.; Aicart E. Why is less cationic lipid required to prepare lipoplexes from plasmid DNA than linear DNA in gene therapy?. J. Am. Chem. Soc. 2011, 133 (45), 18014–18017. 10.1021/ja204693f. [DOI] [PubMed] [Google Scholar]
- Nakamura T.; Sato Y.; Yamada Y.; Abd Elwakil M. M.; Kimura S.; Younis M. A.; Harashima H.. Extrahepatic targeting of lipid nanoparticles in vivo with intracellular targeting for future nanomedicines. Adv. Drug Deliv Rev. 2022, 114417, 114417. 10.1016/j.addr.2022.114417. [DOI] [PubMed] [Google Scholar]
- McGraw T. E.; Greenfield L.; Maxfield F. R. Functional expression of the human transferrin receptor cDNA in Chinese hamster ovary cells deficient in endogenous transferrin receptor. J. Cell Biol. 1987, 105 (1), 207–214. 10.1083/jcb.105.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez J. A.; Helguera G.; Daniels T. R.; Neacato I. I.; López-Valdés H. E.; Charles A. C.; Penichet M. L. Binding specificity and internalization properties of an antibody–avidin fusion protein targeting the human transferrin receptor. J. Controlled Release 2007, 124 (1–2), 35–42. 10.1016/j.jconrel.2007.08.020. [DOI] [PubMed] [Google Scholar]
- Daniels T. R.; Bernabeu E.; Rodríguez J. A.; Patel S.; Kozman M.; Chiappetta D. A.; Holler E.; Ljubimova J. Y.; Helguera G.; Penichet M. L. The transferrin receptor and the targeted delivery of therapeutic agents against cancer. Biochim. Biophys. Acta 2012, 1820 (3), 291–317. 10.1016/j.bbagen.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels T. R.; Delgado T.; Rodriguez J. A.; Helguera G.; Penichet M. L. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol 2006, 121 (2), 144–158. 10.1016/j.clim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Essaghir A.; Demoulin J.-B. A minimal connected network of transcription factors regulated in human tumors and its application to the quest for universal cancer biomarkers. PLoS One 2012, 7 (6), e39666 10.1371/journal.pone.0039666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X. Current strategies for brain drug delivery. Theranostics 2018, 8 (6), 1481. 10.7150/thno.21254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomme P. T.; McCann K. B.; Bertolini J. Transferrin: structure, function and potential therapeutic actions. Drug Discov Today 2005, 10 (4), 267–273. 10.1016/S1359-6446(04)03333-1. [DOI] [PubMed] [Google Scholar]
- Mojarad-Jabali S.; Mahdinloo S.; Farshbaf M.; Sarfraz M.; Fatahi Y.; Atyabi F.; Valizadeh H. Transferrin receptor-mediated liposomal drug delivery: Recent trends in targeted therapy of cancer. Expert Opin Drug Deliv 2022, 19 (6), 685–705. 10.1080/17425247.2022.2083106. [DOI] [PubMed] [Google Scholar]
- Anderson G. J.; Vulpe C. D. Mammalian iron transport. Cell. Mol. Life Sci. 2009, 66, 3241–3261. 10.1007/s00018-009-0051-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbrich K.; Hekmatara T.; Herbert E.; Kreuter J. Transferrin-and transferrin-receptor-antibody-modified nanoparticles enable drug delivery across the blood–brain barrier (BBB). Eur. J. Pharm. Biopharm 2009, 71 (2), 251–256. 10.1016/j.ejpb.2008.08.021. [DOI] [PubMed] [Google Scholar]
- Smith G. P.; Petrenko V. A. Phage Display. Chem. Rev. 1997, 97 (2), 391–410. 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]
- Lee J. H.; Engler J. A.; Collawn J. F.; Moore B. A. Receptor mediated uptake of peptides that bind the human transferrin receptor. Eur. J. Biochem. 2001, 268 (7), 2004–2012. 10.1046/j.1432-1327.2001.02073.x. [DOI] [PubMed] [Google Scholar]
- Liu D.-z.; Cheng Y.; Cai R.-q.; Wang W.-w.; Cui H.; Liu M.; Mei Q.-b.; Zhou S.-y. The enhancement of siPLK1 penetration across BBB and its anti glioblastoma activity in vivo by magnet and transferrin co-modified nanoparticle. Nanomedicine 2018, 14 (3), 991–1003. 10.1016/j.nano.2018.01.004. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Sun Q.; Peng X.; Gan N.; Zhao L.; Suo Z.; Zhao G.; Li H. A pH-sensitive T7 peptide-decorated liposome system for HER2 inhibitor extracellular delivery: an application for the efficient suppression of HER2+ breast cancer. J. Mater. Chem. B 2021, 9 (42), 8768–8778. 10.1039/D1TB01619A. [DOI] [PubMed] [Google Scholar]
- Guichard G.; Benkirane N.; Zeder-Lutz G.; Van Regenmortel M.; Briand J.-P.; Muller S. Antigenic mimicry of natural L-peptides with retro-inverso-peptidomimetics. Proc. Natl. Acad. Sci. U. S. A. 1994, 91 (21), 9765–9769. 10.1073/pnas.91.21.9765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang J.; Wang Q.; Yu Q.; Qiu Y.; Mei L.; Wan D.; Wang X.; Li M.; He Q. A stabilized retro-inverso peptide ligand of transferrin receptor for enhanced liposome-based hepatocellular carcinoma-targeted drug delivery. Acta Biomater 2019, 83, 379–389. 10.1016/j.actbio.2018.11.002. [DOI] [PubMed] [Google Scholar]
- Patra J. K.; Das G.; Fraceto L. F.; Campos E. V. R.; Rodriguez-Torres M.. et al. Nano based drug delivery systems: recent developments and future prospects. J. Nanobiotechnology 2018, 16 ( (1), ), 1–33. 10.1186/s12951-018-0392-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendonça M. C.; Kont A.; Kowalski P. S.; O’Driscoll C. M.. Design of lipid-based nanoparticles for delivery of therapeutic nucleic acids. Drug Discov Today 2023, 103505. 103505. 10.1016/j.drudis.2023.103505. [DOI] [PubMed] [Google Scholar]
- Witzigmann D.; Kulkarni J. A.; Leung J.; Chen S.; Cullis P. R.; van der Meel R. Lipid nanoparticle technology for therapeutic gene regulation in the liver. Adv. Drug Deliv Rev. 2020, 159, 344–363. 10.1016/j.addr.2020.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon W.; Zhang Y.-N.; Ouyang B.; Kingston B. R.; Wu J. L.; Wilhelm S.; Chan W. C. Elimination pathways of nanoparticles. ACS Nano 2019, 13 (5), 5785–5798. 10.1021/acsnano.9b01383. [DOI] [PubMed] [Google Scholar]
- Tilstra G.; Couture-Senécal J.; Lau Y. M. A.; Manning A. M.; Wong D. S.; Janaeska W. W.; Wuraola T. A.; Pang J.; Khan O. F. Iterative Design of Ionizable Lipids for Intramuscular mRNA Delivery. J. Am. Chem. Soc. 2023, 145 (4), 2294–2304. 10.1021/jacs.2c10670. [DOI] [PubMed] [Google Scholar]
- Tenchov R.; Bird R.; Curtze A. E.; Zhou Q. Lipid nanoparticles— from liposomes to mRNA vaccine delivery, a landscape of research diversity and advancement. ACS Nano 2021, 15 (11), 16982–17015. 10.1021/acsnano.1c04996. [DOI] [PubMed] [Google Scholar]
- Dhiman N.; Awasthi R.; Sharma B.; Kharkwal H.; Kulkarni G. T. Lipid nanoparticles as carriers for bioactive delivery. Front Chem. 2021, 9, 580118. 10.3389/fchem.2021.580118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco M. J.; Alishetty S.; Alameh M.-G.; Said H.; Wright L.; Paige M.; Soliman O.; Weissman D.; Cleveland T. E. IV; Grishaev A. Ionization and structural properties of mRNA lipid nanoparticles influence expression in intramuscular and intravascular administration. Commun. Biol. 2021, 4 (1), 956. 10.1038/s42003-021-02441-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalita T.; Dezfouli S. A.; Pandey L. M.; Uludag H. siRNA functionalized lipid nanoparticles (LNPs) in management of diseases. Pharmaceutics 2022, 14 (11), 2520. 10.3390/pharmaceutics14112520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roces C. B.; Lou G.; Jain N.; Abraham S.; Thomas A.; Halbert G. W.; Perrie Y. Manufacturing considerations for the development of lipid nanoparticles using microfluidics. Pharmaceutics 2020, 12 (11), 1095. 10.3390/pharmaceutics12111095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judge A. D.; Robbins M.; Tavakoli I.; Levi J.; Hu L.; Fronda A.; Ambegia E.; McClintock K.; MacLachlan I. Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice. J. Clin Invest 2009, 119 (3), 661–673. 10.1172/JCI37515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes-da-Silva L. C.; Fonseca N. A.; Moura V.; Pedroso de Lima M. C.; Simões S.; Moreira J. N. Lipid-based nanoparticles for siRNA delivery in cancer therapy: paradigms and challenges. Acc. Chem. Res. 2012, 45 (7), 1163–1171. 10.1021/ar300048p. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Shen R.; Vuong I.; Reynolds R. A.; Shears M. J.; Yao Z.-C.; Hu Y.; Cho W. J.; Kong J.; Reddy S. K. Multi-step screening of DNA/lipid nanoparticles and co-delivery with siRNA to enhance and prolong gene expression. Nat. Commun. 2022, 13 (1), 4282. 10.1038/s41467-022-31993-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; You X.; Wang X.; Cui L.; Wang Z.; Xu F.; Li M.; Yang Z.; Liu J.; Huang P. Delivery of mRNA vaccine with a lipid-like material potentiates antitumor efficacy through Toll-like receptor 4 signaling. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (6), e2005191118 10.1073/pnas.2005191118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilkington E. H.; Suys E. J.; Trevaskis N. L.; Wheatley A. K.; Zukancic D.; Algarni A.; Al-Wassiti H.; Davis T. P.; Pouton C. W.; Kent S. J. From influenza to COVID-19: Lipid nanoparticle mRNA vaccines at the frontiers of infectious diseases. Acta Biomater 2021, 131, 16–40. 10.1016/j.actbio.2021.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Templeton N. S.; Lasic D. D.; Frederik P. M.; Strey H. H.; Roberts D. D.; Pavlakis G. N. Improved DNA: liposome complexes for increased systemic delivery and gene expression. Nat. Biotechnol. 1997, 15 (7), 647–652. 10.1038/nbt0797-647. [DOI] [PubMed] [Google Scholar]
- Gordillo-Galeano A.; Mora-Huertas C. E. Solid lipid nanoparticles and nanostructured lipid carriers: A review emphasizing on particle structure and drug release. Eur. J. Pharm. Biopharm 2018, 133, 285–308. 10.1016/j.ejpb.2018.10.017. [DOI] [PubMed] [Google Scholar]
- Jayaraman M.; Ansell S. M.; Mui B. L.; Tam Y. K.; Chen J.; Du X.; Butler D.; Eltepu L.; Matsuda S.; Narayanannair J. K.; et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem., Int. Ed. Engl. 2012, 124 (34), 8657–8661. 10.1002/ange.201203263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinc A.; Maier M. A.; Manoharan M.; Fitzgerald K.; Jayaraman M.; Barros S.; Ansell S.; Du X.; Hope M. J.; Madden T. D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol 2019, 14 (12), 1084–1087. 10.1038/s41565-019-0591-y. [DOI] [PubMed] [Google Scholar]
- Adams D.; Gonzalez-Duarte A.; O’Riordan W. D.; Yang C.-C.; Ueda M.; Kristen A. V.; Tournev I.; Schmidt H. H.; Coelho T.; Berk J. L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J. Med. 2018, 379 (1), 11–21. 10.1056/NEJMoa1716153. [DOI] [PubMed] [Google Scholar]
- Hald Albertsen C.; Kulkarni J. A.; Witzigmann D.; Lind M.; Petersson K.; Simonsen J. B. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv. Drug Deliv Rev. 2022, 188, 114416 10.1016/j.addr.2022.114416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X.; Lee R. J. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Deliv Rev. 2016, 99, 129–137. 10.1016/j.addr.2016.01.022. [DOI] [PubMed] [Google Scholar]
- Tenchov R.; Sasso J. M.; Zhou Q. A. PEGylated Lipid Nanoparticle Formulations: Immunological Safety and Efficiency Perspective. Bioconjug Chem. 2023, 34 (6), 941–960. 10.1021/acs.bioconjchem.3c00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell B. C.; Nabel E. M.; Murdock M. H.; Lao-Peregrin C.; Tsoulfas P.; Blackmore M. G.; Lee F. S.; Liston C.; Morishita H.; Petsko G. A. mGreenLantern: a bright monomeric fluorescent protein with rapid expression and cell filling properties for neuronal imaging. Proc. Natl. Acad. Sci. U. S. A. 2020, 117 (48), 30710–30721. 10.1073/pnas.2000942117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J. M.; Martin N. E.; Modi M. Pegylation: a novel process for modifying pharmacokinetics. Clin Pharmacokinet 2001, 40, 539–551. 10.2165/00003088-200140070-00005. [DOI] [PubMed] [Google Scholar]
- Kulkarni J. A.; Darjuan M. M.; Mercer J. E.; Chen S.; Van Der Meel R.; Thewalt J. L.; Tam Y. Y. C.; Cullis P. R. On the formation and morphology of lipid nanoparticles containing ionizable cationic lipids and siRNA. ACS Nano 2018, 12 (5), 4787–4795. 10.1021/acsnano.8b01516. [DOI] [PubMed] [Google Scholar]
- Whitfield R.; Anastasaki A.; Truong N. P.; Cook A. B.; Omedes-Pujol M.; Loczenski Rose V.; Nguyen T. A.; Burns J. A.; Perrier S.; Davis T. P.; et al. Efficient binding, protection, and self-release of dsrna in soil by linear and star cationic polymers. ACS Macro Lett. 2018, 7 (8), 909–915. 10.1021/acsmacrolett.8b00420. [DOI] [PubMed] [Google Scholar]
- Crawford R.; Dogdas B.; Keough E.; Haas R. M.; Wepukhulu W.; Krotzer S.; Burke P. A.; Sepp-Lorenzino L.; Bagchi A.; Howell B. J. Analysis of lipid nanoparticles by Cryo-EM for characterizing siRNA delivery vehicles. Int. J. Pharm. 2011, 403 (1–2), 237–244. 10.1016/j.ijpharm.2010.10.025. [DOI] [PubMed] [Google Scholar]
- Li Z.; Carter J.; Santos L.; Webster C.; van der Walle C. F.; Li P.; Rogers S. E.; Lu J. R. Acidification-Induced Structure Evolution of Lipid Nanoparticles Correlates with Their In Vitro Gene Transfections. ACS Nano 2023, 17 (2), 979–990. 10.1021/acsnano.2c06213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam M.; Jozic A.; Su G. L.; Herrera-Barrera M.; Curtis A.; Arrizabalaga S.; Tschetter W.; Ryals R. C.; Sahay G. Lipid nanoparticles with PEG-variant surface modifications mediate genome editing in the mouse retina. Nat. Commun. 2023, 14 (1), 6468. 10.1038/s41467-023-42189-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards K. A.; Baeumner A. J. Analysis of liposomes. Talanta 2006, 68 (5), 1432–1441. 10.1016/j.talanta.2005.08.031. [DOI] [PubMed] [Google Scholar]
- Ingebrigtsen L.; Brandl M. Determination of the size distribution of liposomes by SEC fractionation, and PCS analysis and enzymatic assay of lipid content. AAPS PharmSciTech 2002, 3, 9–15. 10.1208/pt030207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chazotte B. Labeling nuclear DNA with hoechst 33342. Cold Spring Harb Protoc 2011, 2011 (1), prot5557. 10.1101/pdb.prot5557. [DOI] [PubMed] [Google Scholar]
- Lai T.-Y.; Cao J.; Ou-Yang P.; Tsai C.-Y.; Lin C.-W.; Chen C.-C.; Tsai M.-K.; Lee C.-Y. Different methods of detaching adherent cells and their effects on the cell surface expression of Fas receptor and Fas ligand. Sci. Rep 2022, 12 (1), 5713. 10.1038/s41598-022-09605-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui L.; Renzi S.; Quagliarini E.; Digiacomo L.; Amenitsch H.; Masuelli L.; Bei R.; Ferri G.; Cardarelli F.; Wang J.; et al. Efficient delivery of DNA using lipid nanoparticles. Pharmaceutics 2022, 14 (8), 1698. 10.3390/pharmaceutics14081698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prazeres P. H. D. M.; Ferreira H.; Costa P. A. C.; da Silva W.; Alves M. T.; Padilla M.; Thatte A.; Santos A. K.; Lobo A. O.; Sabino A.; et al. Delivery of Plasmid DNA by Ionizable Lipid Nanoparticles to Induce CAR Expression in T Cells. Int. J. Nanomedicine 2023, Volume 18, 5891–5904. 10.2147/IJN.S424723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R.-Q.; Qu Y.-H.; Ke W.-L.; Zhu J.-H.; Pei Y.-Y.; Jiang C. Efficient gene delivery targeted to the brain using a transferrin-conjugated polyethyleneglycol-modified polyamidoamine dendrimer. FASEB J. 2007, 21 (4), 1117–1125. 10.1096/fj.06-7380com. [DOI] [PubMed] [Google Scholar]
- Yu M.; Su D.; Yang Y.; Qin L.; Hu C.; Liu R.; Zhou Y.; Yang C.; Yang X.; Wang G. D-T7 peptide-modified PEGylated bilirubin nanoparticles loaded with cediranib and paclitaxel for antiangiogenesis and chemotherapy of glioma. ACS Appl. Mater. Interfaces 2019, 11 (1), 176–186. 10.1021/acsami.8b16219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.