

Abstract
Adenoviruses infect a wide range of vertebrates including humans. Their icosahedral capsids are composed of three major proteins: the trimeric hexon forms the facets and the penton, a noncovalent complex of the pentameric penton base and trimeric fibre proteins, is located at the 12 capsid vertices. Several proteins (IIIa, VI, VIII and IX) stabilise the capsid. We have obtained a 10 Å resolution map of the human adenovirus 5 by image analysis from cryo-electron micrographs (cryoEMs). This map, in combination with the X-ray structures of the penton base and hexon, was used to build a quasi-atomic model of the arrangement of the two major capsid components and to analyse the hexon–hexon and hexon–penton interactions. The secondary proteins, notably VIII, were located by comparing cryoEM maps of native and pIX deletion mutant virions. Minor proteins IX and IIIa are located on the outside of the capsid, whereas protein VIII is organised with a T=2 lattice on the inner face of the capsid. The capsid organisation is compared with the known X-ray structure of bacteriophage PRD1.
Keywords: cryo-electron microscopy, minor capsid protein, structure
Introduction
Adenoviruses are double-stranded DNA viruses that are found in all vertebrates (Davison et al, 2003). Over 50 human serotypes cause a variety of rather mild diseases such as colds, eye infections and diarrhoea (Horwitz, 2001). Adenoviruses have an icosahedral capsid with a pseudo T number of 25 (Stewart et al, 1991) and a total protein mass of around 125 MDa (van Oostrum and Burnett, 1985). The major capsid components are the hexon, a trimeric protein with a hexagonal shape at its base, and the penton that is a noncovalent complex between the pentameric penton base and the trimeric fibre protein. In all, 240 hexons form the 20 facets of the icosahedron, whereas the pentons form and project from the 12 vertices. The fibre binds to host cell receptors with its C-terminal knob domain, whereas protein loops that extend from the penton base and that contain an Arg–Gly–Asp (RGD) sequence (except for the enteric adenovirus 40 and 41; Albinsson and Kidd, 1999) are required to bind to a secondary cell receptor (integrin αvβ3/5) to trigger endocytosis of the virus (Greber et al, 1993; Wickham et al, 1993). The capsid also contains minor components, proteins IIIa, VI, VIII and IX, that glue the hexons and pentons together at specific positions in the capsid.
Adenoviruses are also widely used as vectors for gene transfer and in anticancer protocols (Russell, 2000). However, their use as therapeutic agents for humans is problematic due to the massive immune response against the capsid proteins, due to the limited period of expression of trans-genes owing to nonintegration of viral DNA into the host genome and due to the difficulty in targeting specific cell types (Russell, 2000; Horwitz, 2001). Several of these problems may be overcome by modifying one or several of the viral capsid proteins, but for this a detailed knowledge of the capsid structure must be available.
Adenoviruses produce para-crystalline inclusions in the nuclei of infected cells (Boulanger et al, 1970), but it has not yet been possible to grow crystals of purified virus suitable for high-resolution structure determination. The atomic structure of the hexon of human adenovirus type 5 (Ad5) (Rux and Burnett, 2000) is available, as is that of the human adenovirus type 2 (Ad2) penton base (Zubieta et al, 2005). Thus, the shape and fold of these capsomers are known, as well as the interaction of the monomers in the hexon trimer and penton base pentamer. The penton base structure also gives information regarding the contacts between the bases in the penton base dodecahedron, a subviral particle consisting of 12 penton bases (Schoehn et al, 1996; Fender et al, 1997; Zubieta et al, 2005), and a view of how the trimeric fibre interacts with the penton base (Zubieta et al, 2005). Information about the architecture of the intact Ad capsid has come from three-dimensional (3D) reconstructions of virus particles imaged by cryo-electron microscopy (cryoEM). The first reconstruction, at 35 Å resolution, showed the complex organisation of the capsid with the overall positions of the major capsomers and the stabilising role of some of the minor capsid proteins (Stewart et al, 1991). Subsequently, a 25 Å reconstruction of the capsid allowed the crystal structure of the hexon to be placed into the EM map, giving additional information on the capsid organisation (Stewart et al, 1993). However, the atomic details on the capsomer interfaces at the different positions in the triangular facets and the interactions with the minor proteins were not revealed.
Here we describe the structure of the entire human Ad5 virion determined at a resolution of 10 Å by cryoEM. We also determined the structures of a mutant virus lacking proteins IIIa and IX, and the same virus after several cycles of freezing/thawing, which led to the expulsion of the pentons and the viral DNA. The difference maps between these three structures allow the densities corresponding to some of the minor proteins and the penton to be clearly delimited. We have generated a quasi-atomic model of the entire capsid by fitting the atomic structures of the hexon (Ad5) and the penton base (Ad2) into the 10 Å cryoEM map. Exposed protein loops disordered in the crystal structures of hexon and penton base are clearly visible in the cryoEM map, as are the loops involved in contacts between the capsomers. It is thus possible to describe, at the atomic level, these different contacts that nicely illustrate the quasi-equivalence principle: hexon loops interact differently with each other according to the position of the hexon in the capsid. We also present an image of the contacts between the fibre and the penton base as well as new details of the minor proteins that stabilise the internal face of the capsid. Finally, we compare the 10 Å structure of Ad with the recently reported atomic structure of the bacteriophage PRD1 capsid (Abrescia et al, 2004).
Results and discussion
EM models for wild-type (wt) Ad5 and Ad5 pIX° mutant viruses
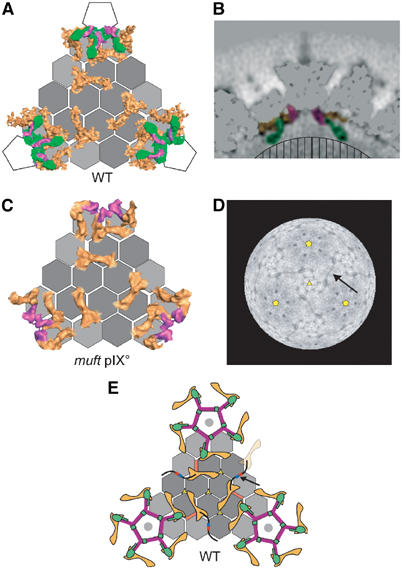
We used two Ad5 viruses: a GFP expressing Ad5 with a wt capsid (wt Ad5) and an Ad5 pIX° mutant virus with a deletion in the pIX coding sequence. The wt Ad5 was prepared for EM after a single cycle of freezing/thawing (necessary for transport between the laboratory of production and the site of the EM) that did not have any apparent influence on the structure of the virus. The Ad5 pIX° mutant virus was also prepared after a single freezing/thawing step (sift) or after multiple cycles of freezing/thawing (muft). Image reconstruction from cryoEM images for the three samples gave models with resolutions of about 10 Å for wt Ad5 (Fourier shell correlation criterion of 0.3, see Supplementary data), 16 Å for sift pIX° virus and 22 Å for muft pIX° virus. CryoEM images showed that the interior of the muft pIX° particles was much less dark than that of wt Ad5, indicating that these viruses had lost their DNA (not shown). Figure 1A shows a surface rendering of the EM density for wt Ad5 with the pentons (penton base plus fibre) coloured in blue. A slice of the density map cutting through the middle of the virus particle and four pentons shows the densities of the major and minor capsid proteins and the particle interior filled with DNA (Figure 1B). However, no detailed information on the DNA organisation is visible. The size of the capsid between the tips of the hexons on opposite facets is 890 Å. The fibre is only visible up to the third repeat in the shaft at which point it is flexible, thus smearing out the density (Ruigrok et al, 1990; Stewart et al, 1991; Wu et al, 2003). As the entire map contains so much information, for clarity, Figures 1C–E show only a single facet of wt Ad5, sift pIX° and muft pIX° viruses. When Ad is dissociated in vitro under mild conditions, one of the structures that remains intact is the ‘Groups Of Nine hexons' or GON (Laver et al, 1969; Furcinitti et al, 1989). GONs are indicated on four adjacent facets in Figure 1A and on the facet in Figure 1D. The capsid is composed of the pentons, the hexons around the penton (peripentonal hexons) and the GONs, plus minor capsid proteins. The structure of sift pIX° virus can be super-imposed on that of the wt virus and the difference density reveals two features: four triskelion-like (yellow) and three rod-like (red) structures are visible on each facet exterior (Figure 1C). The triangular complexes correspond to four trimers of protein IX as previously suggested by Furcinitti et al (1989), who performed image analysis on GONs. The red molecules correspond to three distinct monomers of protein IIIa (Stewart et al, 1993). As pointed out before (Furcinitti et al, 1989; Stewart et al, 1993), IX links the hexons within a GON, whereas IIIa stabilises the contacts between neighbouring GONs. The EM reconstructions show that in the sift pIX° sample both IX and IIIa are absent. Whereas the absence of IX was expected, we find that IIIa, although present in freshly purified pIX° virus (see Materials and methods), is released from the capsid after freezing/thawing. The most likely explanation for this is that, in the absence of IX, the GONs are less rigid, allowing expulsion of IIIa upon freezing. The structure of muft pIX° (Figure 1E) is the same as that of sift Ad5 pIX°, despite the fact that it also lacks all its pentons and viral DNA, although the peripentonal hexons remain in place.
Figure 1.
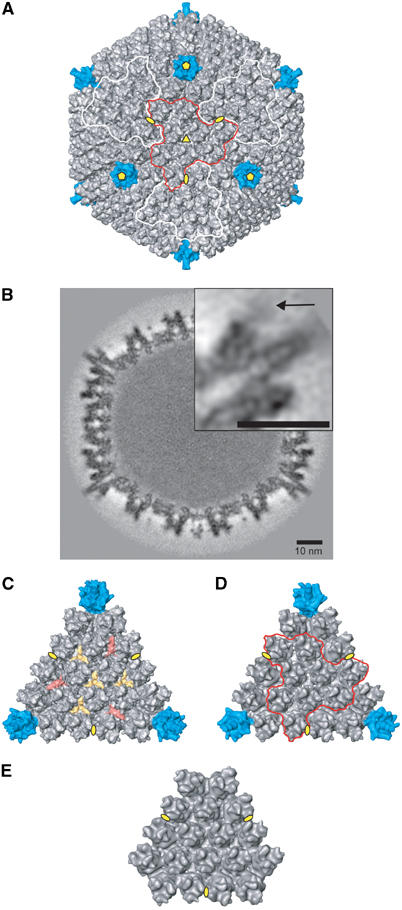
CryoEM reconstructions of adenovirus. (A) Isosurface representation of the 3D reconstruction of human Ad5 by cryoEM at 10 Å resolution viewed down a three-fold axis. The pentons are coloured in blue and the different symmetry axes are indicated in yellow (pentagons for five-fold, a triangle for three-fold and ellipses for two-fold). The GONs are also outlined on four adjacent facets (in red and white). (B) Central section through the density of the Ad5 seen in a 222 orientation. The protein and DNA densities are dark grey. The upper right inset is a magnification of the section through one penton and one protuberance. The arrow indicates the distal limit of the protuberance extension. Scale bars represent 10 nm. (C) Isosurface representation of one facet of the wt Ad5 capsid in the same orientation as that in (B). Red rods and yellow triskelions represent, respectively, monomers of minor protein IIIa and trimers of minor protein IX. (D) Isosurface representation of one facet of the pIX° sift Ad5 capsid at 16 Å resolution. One GON is outlined in red. (E) Isosurface representation of one facet of the pIX° muft Ad5 capsid at 22 Å.
Fitting the atomic structure of the penton base into the EM model
The isolated EM density for the penton is shown in Figure 2A. The visible part of the fibre and its connection with the penton base are shown in pink. The two protuberances of the penton base are indicated by red and yellow dots. The atomic structure of the Ad2 penton base was fitted into the EM envelope of wt Ad5 (Figure 2B). The penton base sequences from Ad2 and Ad5 are 98.6% identical, so it can be assumed that they have the same structure. The atomic structure shows only one protuberance at the top of the base (the variable loop, Zubieta et al, 2005) made up by two antiparallel β-strands, residues 142–169 (yellow dots). The second extension visible in the EM model (red dots) corresponds to residues 296–372, the loop that contains the RGD sequence (RGD loop). The part of the RGD loop shown in Figure 2A has the same density as the rest of the penton. However, in the magnified view of the density section in Figure 1B, it can be seen that diffuse density extends further out. Such weak density, probably resulting from flexibility, has been reported before in the Ad3 dodecahedron cryoEM structure (Schoehn et al, 1996) and in previous reconstructions of the virus (Chiu et al, 1999). Similarly, in the penton base crystal structure, most of the RGD loop is missing due to flexibility (Zubieta et al, 2005). Only 18 amino-acid residues are observed, which rise to about 20 Å above the level where the fibre attaches to the base. In the EM reconstruction the RGD loop is about 30 Å long with the same density as the penton base. Depending on the secondary structure in the rest of this loop (the crystal structure gives evidence for at least one helical region; Zubieta et al, 2005), the total length of the flexible RGD loop is predicted to be between 60 and 70 Å for Ad5. The smeared-out low density visible for the penton base (Figure 1B, arrow) extends to at least 50 Å, indicating that the RGD loop is not totally disordered.
Figure 2.
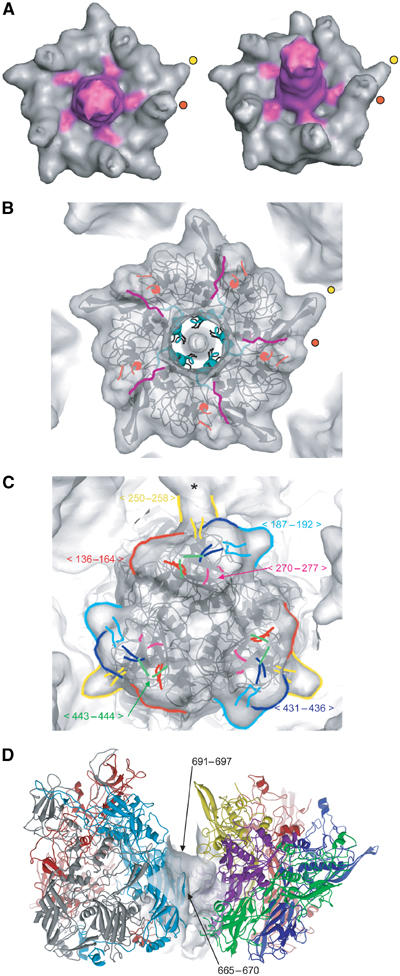
Fitting the atomic structure in the EM densities of pentons and hexons. (A) Isosurface view of two isolated pentons from the wt Ad5 capsid cryoEM map. The right penton is tilted by about 20° compared to the left one. The fibre and the connection between the fibre and the penton base are coloured in pink. The two protuberances are marked by yellow and red dots (yellow: variable loop; red: RGD loop). (B) Fitting of the penton base crystal structure in complex with amino acids 11–20 from the N-terminal part of the fibre into the cryoEM envelope at 10 Å resolution. The figure is labelled with yellow and red dots as in (A). The two strands of the RGD loop seen in the X-ray structure are coloured in orange. The fibre peptide is in pink and the part of the penton base interacting with the fibre is in cyan. (C) Fitting of the hexon crystal structure into hexon 2 (according to Figure 3B) of the cryoEM envelope at 10 Å resolution. Densities for missing loops in the crystal structure that are present in the EM map are labelled in different colours. * indicates the contact between Hexon 2 and protein IIIa through the yellow loop (250–258). (D) Detailed view of the peripentonal hexon–penton base atomic interactions. Hexon (grey, red, blue) and penton (in five colours) are in ribbons, whereas the cryoEM density of the connection is in grey. Interactions occur through a hexon B face (in blue). Arrows indicate two out of the three interacting hexon loops.
The crystal structure of the penton base without fibre peptide, when fitted into the EM map (not shown), does not account for the connection between the fibre and the penton base (coloured pink in the EM density in Figure 2A). However, fitting the penton base structure containing amino acids 11–20 from the N-terminus of the fibre protein (Zubieta et al, 2005) shows that these amino acids fall inside and fill up this connection (Figure 2B). Owing to the imposition of icosahedral symmetry during the image-processing procedure, the densities of the five connections were equal even though the fibre is a trimer. In reality, three connections should have a density 5/3 times higher and two should be absent. This five-fold averaging only affects the connections because the conformational change that occurs upon fibre binding is thought to be cooperative with all penton base monomers having the same structure even if only three tails are bound to them (Zubieta et al, 2005). The three tails of the individual trimeric fibres can probably associate randomly with the five binding sites on the penton base. In Figure 1B, it can be seen that the lowest part of the fibre shaft is hollow. The structure of the fibre shaft at the level of the regular fibre repeats is dense and rigid (van Raaij et al, 1999). The first regular shaft repeat starts at amino acid 47 (Chroboczek et al, 1995). The hollow structure at the base of the fibre, probably composed of the proline-rich region 21–46, appears to constitute a rigid link between the first 20 residues that are fixed onto the penton base and the first repeat in the shaft.
Fitting the atomic structure of the hexon into the Ad5 EM model
As the GON contains a three-fold axis, there are only three unique hexons in a GON (hexons 2, 3 and 4 in Figure 3B). The fourth structurally unique hexon in the capsid is the peripentonal hexon (hexon 1 in Figure 3B; Burnett, 1985). The crystal structure of the Ad5 hexon (Rux and Burnett, 2000) was fitted independently into the four unique hexon densities that make up the asymmetric unit (AU), with the assumption that all hexons have essentially the same structure. As for the penton, the hexon crystal structure fits very well into the EM map, with the addition that a number of loops at the top of the hexons and disordered in the crystal structure are resolved (Figure 2C). The good fit of the crystal structure to the EM density, particularly near the base of the hexon (not shown), indicates that the N- and C-termini of the hexon (of which only the four N-terminal and five C-terminal residues are not ordered in the crystal; Rux and Burnett, 2000) are unlikely to be available for extensive hexon–hexon and hexon–penton contacts, as seen in the capsid structure of bacteriophage PRD1 (Abrescia et al, 2004).
Figure 3.
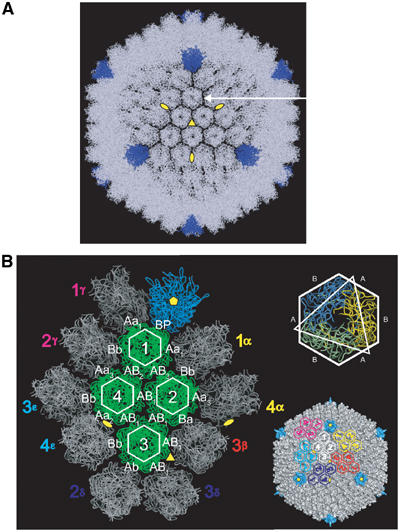
Quasi-atomic model of the Ad5 capsid. (A) View down a three-fold axis (similar to Figure 1A) of the quasi-atomic model of the Ad5 capsid made by fitting the hexon (light blue) and penton base (dark blue) crystal structures into the 10 Å resolution EM map. Three-fold (triangle) and two-fold (ellipses) symmetries axes are indicated in yellow. The arrow indicates one position of protein IIIa. (B) Detailed view of the different capsomer contacts. The penton base is in blue. The four hexons coming from one AU are coloured in green with a white hexagon. The hexons at the outside of the AU are in grey and are labelled uniquely. The positions of six AUs (including only the hexons) are also indicated in the complete virus on the right lower part of the figure. The top right shows that the hexon is a trimeric protein with a hexagonal base. Hexagon sides formed exclusively from one monomer are denoted as B; those formed from two monomers are denoted as A. At the left, the different contacts between the capsomers are labelled in white with the following convention: the labels are in capitals when the interaction side belongs to the same AU, and in small letters when coming from another AU; Aa1, Aa2 and Aa3 represent, respectively, contacts between peripentonal hexons, hexons connected across a two-fold symmetry axis, and hexon–hexon connected by protein IIIa. All the atomic contacts are listed in Table I.
After fitting all the unique positions of the major constituents of the capsid, we built a quasi-atomic model of the capsid using its 532 symmetry with the pentons in dark blue and the hexons in light blue (Figure 3A). In the quasi-atomic model, it is apparent that the spacing between the hexons depends on their position on the facet. The separation is particularly wide at the position of protein IIIa (see below, Figure 1C and arrow in Figure 3A).
Hexon–hexon and hexon–penton contacts in the quasi-atomic model
Hexons are trimeric complexes with a pseudo-hexagonal base, the six faces of which are of two types depending on whether the face is made up of only one monomer or of two monomers (respectively B and A in Figure 3B). There are eight different hexon–hexon contacts (Table I), depending on whether these are contacts between peripentonal hexons or hexons in the GON, unique positions in the GON or whether minor proteins are present between the hexons (Figure 3B). When the face comes from a hexon or penton base within the AU it is labelled with a capital A, B or P, and when it comes from a hexon outside the AU it is labelled with ‘a or b' (Figure 3B), as originally introduced by Burnett (1985). For the different contacts, all possible interactions at a distance ⩽5 Å between the surfaces of the hexons and the penton are given in Table I. All contacts concern the lower double jellyroll region of the hexon (eight-stranded antiparallel β sheets, regions V1, V2, see Figure 4A). The nine different hexons in the GON are all making four contacts (contacts AB1) with an angle of about 10°. This is also the case between hexons 1–4 and 1–2 in the same AU. Deviations from this angle are located at the IIIa interface, where the angle is about 30° (no hexon–hexon contact), between peripentonal hexons (30° and two contacts) and between hexons through the two-fold axis (12° and four contacts). The structure of the GON is further stabilised through the presence of protein IX at these AB1 contacts. It is of course possible that in the viral capsid the hexon loops are slightly re-arranged compared to their conformation in the crystal structure. This, combined with the limited resolution of the EM map, implies that residue-to-residue contacts cannot be given to better than a few residues. Although the loops are not specifically enriched in charged or hydrophobic residues, in the majority of cases two interacting loops contain residues with opposite charges (see Table I).
Table 1.
Hexon–hexon, hexon–penton base and hexon–minor protein contacts in the capsid
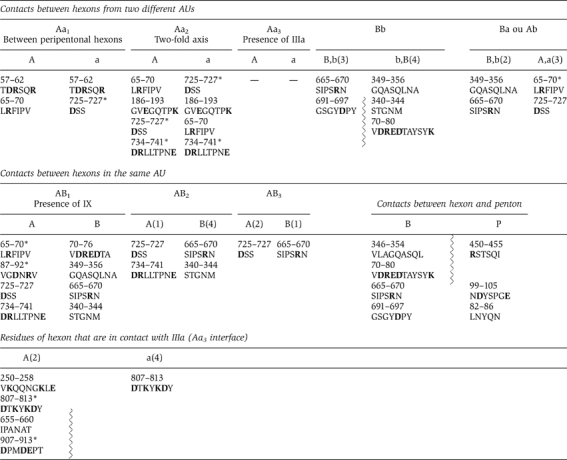 |
| Only contact regions (numbers that give the positions of the amino acid in the sequence) that are separated by less than 5 Å are listed. Hexons have two types of sides, B and A, depending on the fact whether they are made up of one or two monomers, respectively. Capitals are used for the hexons that make part of the AU and lower case for hexons of another AU. There are three types of Aa contact depending on their position in the capsid (i.e. between peripentonal hexon, when IIIa is present and between hexons on the two-fold axis). ‘*' indicates that the contacts are made by two different monomers in an A-side of the corresponding hexon. The amino acids involved in the interaction are listed and the charged amino acids are in bold. When a zigzag line is present, two loops from one capsomer are interacting with one loop from another capsomer. |
Figure 4.
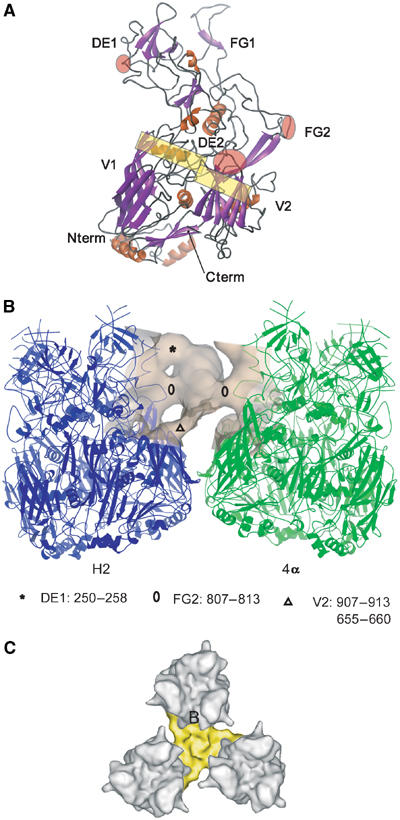
Connections between hexon and proteins IIIa/IX at the atomic level. (A) Ribbon view of one monomer of the hexon. The secondary structure elements are marked, as well as some of the interacting regions with proteins IIIa (red ellipses) and IX (yellow rectangles). The N and C termini are also marked. (B) Atomic interactions between protein IIIa (EM density) and its two neighbouring hexons H2 and 4α (ribbons). The asterisk, triangle and circles mark the interactions of IIIa with the hexons and the corresponding structural elements are indicated at the bottom of this figure and in part A. (C) Detailed top view of the hexon–protein IX interaction on the three-fold of the virus. The protein IX monomer wraps only around the hexon B face.
The peripentonal hexons interact with the penton through a B-face (Figures 2D and 3B). The penton base–hexon interaction is also just below the middle of the two structures and involves loops of the jellyroll motifs of both proteins (Figure 2D; Table I). Only one hexon loop implicated in hexon–hexon contacts (loop 665–670) is also involved in the hexon–penton base contact; the two others are different. Two of the penton base loops, 99–105 and 450–455, are also involved in inter-penton base contacts in the penton base dodecahedron where penton bases interact directly (Zubieta et al, 2005).
The interactions that a hexon makes with its neighbours depend on its position in the facet and this illustrates the principle of quasi-equivalence. For example, loop 725–727 interacts either with loop 65–70 (when the hexons are across the two-fold axis) or with loop 665–670 (hexons around the three-fold axis).
Position and contacts of minor proteins IIIa and IX on the outside of the capsid
Protein IIIa forms a rod with a length of about 60 Å and a maximum width of about 30 Å and links two neighbouring GONs at an Aa3 hexon–hexon contact (Figures 1C and 3B and Table I). It lies at an angle of 45° to the capsid surface and touches the side of two hexons near their middle at the level of structural elements FG2 (807–813, Figures 4A and B). As seen in Figure 4B, the top of protein IIIa is attached to one hexon by a loop that is missing in the X-ray structure (250–258, element DE1) and the bottom of protein IIIa is connected to two elements (655–660; 907–913) from the V2 domain of the same hexon (Rux and Burnett, 2000; Figures 4A and B and Table I). This position leads to a separation in space of the two neighbouring hexons that have no densities that approach within the 5 Å cutoff distance that was used to determine subunit contacts (see arrow in Figure 3A). This implies that at this position capsid integrity is maintained only through hexon–IIIa–hexon interactions.
As the density of the internal minor proteins in the sift Ad5 pIX° reconstruction does not differ from those in wt virus (not shown but similar to Figure 5A), we conclude that IIIa is positioned only on the outside of the capsid. In a previous model of the Ad2 capsid structure, IIIa was proposed to traverse the width of the capsid with a rod-like density (corresponding to what we see) near the top of the hexon and another domain near the bottom of the hexon (Stewart et al, 1993). However, we assign the latter density on the inside of the capsid to a distinct minor capsid component, most likely protein VIII (see below and Figure 5). The reason for this different interpretation is probably due to the fact that the resolution of our model is better (10 Å versus 25 Å) and also because the atomic model for the hexon that was used in 1993 was corrected in 2000 (Rux and Burnett, 2000). At the resolution of our 3D reconstruction, the volume for IIIa corresponds only to about half the volume of a protein of 63 kDa. Most of the C-terminal 200 amino acids of IIIa are predicted to be disordered (FOLDINDEX; Uversky et al, 2000) and may occupy an extended conformation in the virus that is too diffuse to be resolved at the present resolution or may deviate from icosahedral symmetry. There is a nonattributed thin density running through the two-fold axis at the inside of the particle that starts just under the protein IIIa (see Discussion and Figure 5D). It is not possible to see if this feature is present or absent in the sift capsid that lacked IIIa, because it is only visible at higher resolution.
Figure 5.
Structural views of the inside of Ad5 capsid. (A) View of the inside of one facet of the wt Ad5. Hexons and penton base are represented diagrammatically with the GON in dark grey. The densities of the minor proteins coming from difference imaging of the cryoEM map with the wt 10 Å filtered quasi-atomic model exhibit three kinds of extra densities: a hammer-like orange shape (120 copies in the capsid), which probably corresponds to protein VIII (see text) and magenta and green densities that have not been attributed. The interactions between protein VIII and the four hexons are the same around the five-fold and around the three-fold symmetry axis. (B) Central slice through the density of one penton showing the internal organisation of the minor proteins under the five-fold symmetry axis. This view is generated from the difference map between the 10 Å filtered quasi-atomic model and the wt Ad5 cryoEM map. The missing density (coming from the subtraction of the quasi-atomic model) is in grey. The remaining different densities under the five-fold are coloured according to Figure 5A. The green density extends up to 40 Å under the hexon. Parallel lines indicate schematically the DNA. (C) Same view as in (A), but using the difference image from the muft pIX° adenovirus mutant reconstruction and the quasi-atomic model filtered to 18 Å. The colour codes are the same as in (A) and (B). (D) Spherical section view (one voxel thick) at a radius of 330 Å down the three-fold axis of the wt Ad5 cryoEM reconstruction that shows a thin connection (10 Å in diameter) over the two-fold symmetry axis (arrow). This connection is visible between radius 325 and 335 in Å (i.e. in four slices). In the map, the extremities of this connection start just below the big part of the orange hammer-like density, which is also vertically beneath the bottom of protein IIIa (see (E)). (E) Schematic distribution of the major and minor proteins in one facet of the Ad particle seen from the inside of the virus particle. Hexons are in grey (GON in dark grey) with a hexagonal shape; pentons are in white with a grey disk representing the fibre. Protein IIIa is in red, protein IX in yellow, VIII in orange. Un-attributed densities around the five-fold symmetry axis are colour coded according to (A)–(C). The connections across the two-fold axis (coming from VIII or IIIa) are in red and blue.
Protein IX is positioned at the AB1 hexon contacts (Figures 1C and 3B and Table I). This trimeric molecule has three arms with a length of about 40 Å (from the triskelion axis to the end of one arm) and a maximum diameter of 20 Å. The protein only wraps around the B face of the hexon, leaving the opposite A face free (Figure 4C). The interactions between IX and the hexon B face are too numerous to be listed in Table I, but they are clearly mediated via the top of jelly-roll V1 and V2 of the hexon (yellow rectangles in Figure 4A). As IX is relatively thin and sits on the top of the hexon–hexon interacting region, most of the normal interactions between the hexons are not perturbed. The fact that the hexon–hexon contacts can occur independently of the presence of IX probably explains why capsids can form in the absence of this protein although the virus is less stable at elevated temperatures (Colby and Shenk, 1981).
Positions of minor capsid proteins on the inside of the capsid
Adenoviruses have several minor proteins on the inner surface of the capsid. Polypeptide VI is thought to form a ring underneath the peripentonal hexons (Stewart et al, 1993). The position of protein VIII has not yet been assigned (Stewart et al, 1993), but was recently proposed as a possible size-determining or ‘tape-measure' protein similar to P30 of PRD1 (Abrescia et al, 2004; Rux and Burnett, 2004). Figure 5A and B show the EM density map of the inside of wt Ad5 capsid after subtraction of the crystallographic densities of hexon (virtually the entire molecule) and penton base (excluding residues 1–52 since their structure is not known) filtered to about 10 Å resolution (IIIa and IX have also been removed to clarify the figure). Figure 5C shows the inside of the muft pIX° map after subtraction of the hexon density filtered to 18 Å (this virus sample had already lost its pentons and DNA and probably all other proteins only attached to pentons and DNA). The difference map of the sift pIX° mutant is similar to Figure 5A, except for being at lower resolution (not shown).
The most conspicuous feature in Figure 5A and C (and also in the difference map of sift pIX°) is the orange hammer-like structure organised in a T=2 lattice (120 copies), with a volume corresponding to a protein of about 20 kDa (this is probably an overestimate due to the many fine details in the structure that could not be taken into account). Protein VIII is the only capsid protein likely to be associated with both DNA and hexons, and which is present with this stoichiometry. Stewart and Burnett (1995) estimated the presence of about 127±3 copies, assuming a molecular weight of processed VIII of 15.4 kDa (c.f. full-length precursor pVIII Ac2-227, 24.6 kDa). In previous, lower-resolution EM maps of Ad2 (Stewart et al, 1993), the hammer-like density was thought to be part of protein IIIa. However, due to the fact that the density is present in the muft pIX° capsid (which does not contain protein IIIa) and particularly its presence in a T=2 lattice, this assignment is excluded. Note that the exact state of protein VIII in mature viral capsids is not totally established, since it is uncertain which potential protease sites are actually cleaved and which fragments actually remain in the virus. A recent analysis suggests that whereas empty viral capsids contain full-length pVIII, mature viral particles contain pVIII-derived 7.6 and 12 kDa peptides with estimated copy numbers of 160 and 130 per particle (Vellekamp et al, 2001). Other analyses reveal the likely presence of only the so-called propeptide domain of pVIII (Ac2-111, MW 12 kDa, protease cleavage at LAGG/F) in the virion (Blanche et al, 2001; Liu et al, 2003). These results are generally consistent with the volume and stoichiometry of the hammer-like EM density we observe.
The T=2 lattice implies two nonequivalent positions in the capsid for protein VIII (Figure 5A). The first is around the five-fold axis where five copies of protein VIII generate a ring that connects the peripentonal hexons and links them to the GON. These molecules remain in place after the removal of the penton. The second localisation occurs around the three-fold axis, where three molecules of VIII form a second ring that most likely stabilises the GONs as well as makes links between different GONs, complementing, respectively, the binding function of proteins IX and IIIa on the external side of the capsid. Independent of their location around the three- or the five-fold axis, each protein VIII appears, at the resolution of the map, to interact in exactly the same manner with four hexons. Looking carefully at spherical slices of the difference map at a radius between 325 and 335 Å, another feature with a diameter of 10 Å is apparent. This makes a connection across the two-fold axis between the rings of protein VIII around the three-fold axis, linking the GONs of two facets (arrow in Figure 5D). It is not possible to be sure of the origin of this linker at the current resolution, but it does suggest that unfolded regions of minor capsid proteins (possibly VIII or IIIa) are indeed involved in forming extended arms, perhaps in a fashion reminiscent of P30 of PRD1 (Abrescia et al, 2004; Rux and Burnett, 2004).
The second major feature of the difference map is the magenta coloured ring of density around the five-fold axis in Figure 5A–C. In the wt Ad5 map, two additional green blobs of density connected by a green arm are also present (Figure 5A and B), but not when the pentons and the DNA are removed (Figure 5C). The largest green blob is attached to protein VIII and to the magenta density, and appears to be a hollow sphere. The smaller green blob and the arm are only attached to the magenta ring. We do not have any positive way of identifying the protein nature of these densities that may consist of the amino-terminal 52 amino acids of the penton base, protein VI, other viral proteins or even viral DNA. However, the magenta ring cannot consist of the N-terminus of the penton base because it remains there when the penton is expelled. Stewart et al (1993) also observed density underneath the peripentonal hexons and attributed this to protein VI. Protein VI has nonspecific DNA-binding activity and also is an activator of the Ad protease (Russell and Precious, 1982). It is predicted to have 44% disordered residues (FOLDINDEX, residues 1–32; 65–126; 170–177). However, neither the green, the magenta, nor the green plus magenta regions together represent enough density for the 342 copies (7.6 MDa) of protein VI estimated to be present in the virion (van Oostrum and Burnett, 1985; Stewart and Burnett, 1995) unless a large portion of the protein is disordered. Whatever the identity of these densities, the largest green blob points radially inwards towards the middle of the virus particle (up to 40 Å under the base of the hexon protein, Figure 5B), away from the capsid and in contact with the DNA at exactly the same position as the trans-membrane protein P16 of PRD1 that binds the capsid to the viral DNA on the inner side of the viral lipid membrane (Abrescia et al, 2004).
Organisation of the DNA in the capsid
There have been a number of models proposing the organisation of the DNA in human adenoviruses (Newcomb et al, 1984). From our results, it is apparent, as from the previous work of Stewart et al (1991, 1993), that the DNA is not organised with any icosahedral symmetry, because, if it were, dsDNA strands would have been visible around the symmetry axes. Other viruses, such as Herpes virus and bacteriophages lambda and T4, have closely packed dsDNA that is readily visible in cryoEM images (Lepault et al, 1987; Booy et al, 1991). The absence of any ordered density directly on the micrographs or in the 3-D model indicates that there is neither icosahedral symmetry nor liquid crystalline organisation of the DNA of Ad5. Nevertheless, it is still possible to deduce some information about the DNA packing in the virus from the EM map. Only two types of connections seem to be present between the capsid proteins and the DNA (Figure 5B and not shown). The first one occurs through the spherical blob of green density that points to the inside of the particle. The arms of protein VIII, independent of their position, make the second type of connection.
Comparison between adenovirus and PRD1 capsid structures
Hypotheses on an evolutionary relationship between adenoviruses and bacteriophage PRD1 are based on the similarity in overall organisation of their protein capsids and the structures of the hexon (P3 in PRD1) and penton base (P31). However, for both hexons and pentons, whereas the bottom parts of the proteins have the same fold, the upper elaborations are not present in PRD1 (Benson et al, 1999, 2004; Rux and Burnett, 2000; Abrescia et al, 2004; Zubieta et al, 2005). The Ad capsid is heavily stabilised by a network of connections made by interactions with at least four types of minor protein both on the inside and the outside of the capsid (Figure 5E). On the outside of the Ad capsid, proteins IIIa and IX interact mainly with the upper part of the hexons, above the jelly-roll domain. There cannot be any equivalent proteins or interactions in the PRD1 structure because this top part of P3 is absent.
In PRD1, the contacts between P3–P3 and P3–P31 are stabilised by specific interactions of their respective N- and C-terminal peptides, resulting in specific conformations of these termini at specific positions in the AU. In Ad, all hexons are connected with each other by protein VIII which, therefore, has a role functionally equivalent to the N and C termini of P3 (PRD1 hexon). Furthermore, it seems that for PRD1, the P3–P31 connection is stronger than the P3–P3 connection, since when PRD1 empty particles or the sus1 mutant (a mutant that does not package DNA) are treated with SDS, P31 plus peripentonal P3 are released (Martin et al, 2001). For Ad empty capsids (the muft structure in this work and also natural empty capsids; Vellekamp et al, 2001), only the penton is ejected with the peripentonal hexons remaining in place, probably because of the stabilisation of the contact between the GON and the peripentonal hexons by protein VIII.
The only minor capsid protein of PRD1 that is organised with icosahedral symmetry is the ‘tape-measure protein' P30. Interestingly, two PRD1 P30 proteins meet over the two-fold axis just like the weak density seen in Ad (arrows in Figure 5D and E). Another apparent similarity is the presence of a protein element attached to the peripentonal hexons (the ‘green density' for Ad, P16 for PRD1), which extends inwards probably to interact with the viral DNA (Figure 5B). However, the major difference between the two viruses remains the presence of the lipid membrane in PRD1, needed for the infection mechanism, but which is absent in adenoviruses.
Materials and methods
Viruses
E1/E3-deleted Ad5 (the E1 region was replaced by the gene for GFP) that is wt for the capsid (wt Ad5) was obtained from the Gene Vector Production Network (http://www.gvpn.org/). For the recombinant Ad5 pIX° virus, viral genomes were constructed as infectious plasmids by homologous recombination in Escherichia coli, as described previously (Rosa-Calatrava et al, 2001). Ad5 pIX° contained an additional deletion that removed the entire coding sequence for minor capsid protein IX. Growing, purification, titration and storage were as described previously (Rosa-Calatrava et al, 2001). As checked by SDS–PAGE and immunoblotting using polyclonal antibody against the virus capsid (gift from P Boulanger), penton base, fibre and IIIa proteins were present both in infected cell extracts and after CsCl purification, whereas IX was absent (see Supplementary data and not shown).
Electron cryomicroscopy
Frozen hydrated specimens of wt virus were prepared on quantifoil grids as described previously (Schoehn et al, 1997) and with quantifoil grids covered with a thin layer of continuous carbon for Ad5 pIX° in order to increase the virus concentration (Schoehn et al, 1997). Specimens were imaged with less than 10 electrons/Å2 at a temperature close to −186°C by using a Gatan 686 cryo-holder in a LaB6 Philips CM200 electron microscope operating at 200 kV. Tobacco mosaic virus (TMV) was used to determine the absolute magnification of the microscope, which was found to be 28 600±100 times.
Image analysis
Images were screened by optical diffraction, and the best were digitised on a Z/I Imaging scanner (Photoscan TD) with a pixel size of 7 μm that corresponds to a sample pixel size of 2.45 Å (see calibration above). The particles were selected interactively using x3d and boxed into 427 × 427 pixels with a circular mask applied of radius 209 pixels plus four fade pixels (Conway and Steven, 1999). The starting model for the image analysis procedure was calculated from images of negatively stained (2% ammonium molybdate) wt Ad5 using the classical common lines orientation search (Crowther et al, 1970) and performing single-particle reconstructions. The best single-particle reconstruction (showing clear T=25 triangulation) was then used as the starting model in a model-based approach using a modified version of the Polar Fourier Transform (PFT) algorithm (Baker and Cheng, 1996; Conway and Steven, 1999). From a total of 250 images of negatively stained particles, 120 were used in a final reconstruction at 25 Å resolution (Fourier Shell Correlation 0.5 criterion; Yang et al, 2003). This negative stain model was used to start analysis of the cryoEM images. Determination of particle origin and orientation were again performed using the model-based PFT programs after images had been corrected for contrast transfer function effects (CTFMIX, Conway and Steven, 1999).
For wt Ad5, we selected 6131 images of particles from nine focal pair micrographs plus three single negatives. After completing PFT refinement, one supplementary cycle was carried out using a new version of PFT: PFT2 (http://www.niams.nih.gov/rcn/labbranch/lsbr/software). Of the total selected particles, two-thirds (4100) were chosen for the final reconstruction according to a figure of merit calculated from a combination of the PFT coefficients ccPRJ and ccCMP. The reconstruction includes data to a spatial frequency limit of (9 Å−1) and the resolution was estimated at (10.3 Å−1), where the Fourier shell correlation between independent half data-set density maps crossed below 0.3 (see Supplementary data).
For the sift Ad5 pIX° virus, we selected 1202 images of particles from 10 focal pair micrographs plus two single negatives, of which 1062 were included in the final reconstruction, yielding a resolution of 15.5 Å. For the muft Ad5 pIX° virus, we selected 820 images of particles from five focal pair micrographs. For the final reconstruction we included 660 particles, obtaining a resolution of 22 Å.
The contour level of the isosurface views was chosen using three filtered X-ray structures of the hexon (10, 16 and 22 Å) so that the surface contours the right molecular weight of the protein with an average protein density of 0.84 Da/Å3.
Fitting the atomic structures of hexon and penton base into the EM map and contacts between capsomers
The atomic structures of the Ad5 hexon and that of the Ad2 penton base were fitted into the EM model for wt Ad5. The atomic structures were placed manually into the four hexons that make up the AU and into one penton base using the program ‘O' (Jones et al, 1991). The optimisation procedure was carried out using SITUS (Wriggers and Birmanns, 2001) for the penton base and using URO (Navaza et al, 2002) for the four hexons. The quality of the final fit is given by a correlation coefficient of 94.4% and crystallographic R factor of 26.7%. From the optimised positions of the four hexons in the AU plus the penton, the quasi-atomic model of the whole capsid was built using 532 symmetry.
Supplementary Material
Supplementary Data
Acknowledgments
We thank the Production and Control department of Genethon which is supported by the Association Française contre les Myopathies (AFM), for providing us with the Ad5GFP sample. We also thank Dr David Belnap for providing pft3dr2 software and Jorge Navaza for help with the URO software. Figures were made using AMIRA (TGS Europe, Merignac, France), Povscript (Avatar Software AB) and Chimera (Pettersen et al, 2004). JFC was supported in part by an Action Thématique et Incitative sur Programme (ATIP) from the CNRS. The EM map has been deposited in the EM database associated with the Macromolecular Structure Database. The accession numbers are, respectively, 1111, 1112 and 1113 for muft, sift and wild-type Ad. PDB accession number for the quasi-atomic model is 2bld.
References
- Abrescia NG, Cockburn JJ, Grimes JM, Sutton GC, Diprose JM, Butcher SJ, Fuller SD, San Martin C, Burnett RM, Stuart DI, Bamford DH, Bamford JK (2004) Insights into assembly from structural analysis of bacteriophage PRD1. Nature 432: 68–74 [DOI] [PubMed] [Google Scholar]
- Albinsson B, Kidd AH (1999) Adenovirus type 41 lacks an RGD alpha(v)-integrin binding motif on the penton base and undergoes delayed uptake in A549 cells. Virus Res 64: 125–136 [DOI] [PubMed] [Google Scholar]
- Baker TS, Cheng RH (1996) A model-based approach for determining orientations of biological macromolecules imaged by cryoelectron microscopy. J Struct Biol 116: 120–130 [DOI] [PubMed] [Google Scholar]
- Benson SD, Bamford JK, Bamford DH, Burnett RM (1999) Viral evolution revealed by bacteriophage PRD1 and human adenovirus coat protein structures. Cell 98: 825–833 [DOI] [PubMed] [Google Scholar]
- Benson SD, Bamford JK, Bamford DH, Burnett RM (2004) Does common architecture reveal a viral lineage spanning all three domains of life? Mol Cells 16: 673–685 [DOI] [PubMed] [Google Scholar]
- Blanche F, Monegier B, Faucher D, Duchesne M, Audhuy F, Barbot A, Bouvier S, Daude G, Dubois H, Guillemin T, Maton L (2001) Polypeptide composition of an adenovirus type 5 used in cancer gene therapy. J Chromatogr A 921: 39–48 [DOI] [PubMed] [Google Scholar]
- Booy FP, Newcomb WW, Trus BL, Brown JC, Baker TS, Steven AC (1991) Liquid-crystalline, phage-like packing of encapsidated DNA in herpes simplex virus. Cell 64: 1007–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulanger PA, Torpier G, Biserte G (1970) Investigation on intranuclear paracrystalline inclusions induced by adenovirus 5 in KB cells. J Gen Virol 6: 329–332 [DOI] [PubMed] [Google Scholar]
- Burnett RM (1985) The structure of the adenovirus capsid. II. The packing symmetry of hexon and its implications for viral architecture. J Mol Biol 185: 125–143 [DOI] [PubMed] [Google Scholar]
- Chiu CY, Mathias P, Nemerow GR, Stewart PL (1999) Structure of adenovirus complexed with its internalization receptor, alphavbeta5 integrin. J Virol 73: 6759–6768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chroboczek J, Ruigrok RW, Cusack S (1995) Adenovirus fiber. Curr Top Microbiol Immunol 199 (Part 1): 163–200 [DOI] [PubMed] [Google Scholar]
- Colby WW, Shenk T (1981) Adenovirus type 5 virions can be assembled in vivo in the absence of detectable polypeptide IX. J Virol 39: 977–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JF, Steven AC (1999) Methods for reconstructing density maps of ‘single' particles from cryoelectron micrographs to subnanometer resolution. J Struct Biol 128: 106–118 [DOI] [PubMed] [Google Scholar]
- Crowther RA, Amos LA, Finch JT, De Rosier DJ, Klug A (1970) Three dimensional reconstructions of spherical viruses by Fourier synthesis from electron micrographs. Nature 226: 421–425 [DOI] [PubMed] [Google Scholar]
- Davison AJ, Benko M, Harrach B (2003) Genetic content and evolution of adenoviruses. J Gen Virol 84: 2895–2908 [DOI] [PubMed] [Google Scholar]
- Fender P, Ruigrok RW, Gout E, Buffet S, Chroboczek J (1997) Adenovirus dodecahedron, a new vector for human gene transfer. Nat Biotechnol 15: 52–56 [DOI] [PubMed] [Google Scholar]
- Furcinitti PS, van Oostrum J, Burnett RM (1989) Adenovirus polypeptide IX revealed as capsid cement by difference images from electron microscopy and crystallography. EMBO J 8: 3563–3570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber UF, Willetts M, Webster P, Helenius A (1993) Stepwise dismantling of adenovirus 2 during entry into cells. Cell 75: 477–486 [DOI] [PubMed] [Google Scholar]
- Horwitz MS (2001) Adenoviruses. In Fields' Virology, Knipe DM, Howley PM (eds), pp 2301–2326. Philadelphia, USA: Lippincott Williams and Wilkins [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47 (Part 2): 110–119 [DOI] [PubMed] [Google Scholar]
- Laver WG, Wrigley NG, Pereira HG (1969) Removal of pentons from particles of adenovirus type 2. Virology 39: 599–604 [DOI] [PubMed] [Google Scholar]
- Lepault J, Dubochet J, Baschong W, Kellenberger E (1987) Organization of double-stranded DNA in bacteriophages: a study by cryo-electron microscopy of vitrified samples. EMBO J 6: 1507–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YH, vellekamp G, Chen G, Mirza UA, Wylie D, Twarowska B, Tang JT, Porter FW, Wang S, Nagabhushan TL, Pramanik BN (2003) Proteomic study of recombinant adenovirus 5 encoding human p53 by matrix-assisted laser desorption/ionization mass spectrometry in combination with database search. Int J Mass Spectrom 226: 55–69 [Google Scholar]
- Martin CS, Burnett RM, de Haas F, Heinkel R, Rutten T, Fuller SD, Butcher SJ, Bamford DH (2001) Combined EM/X-ray imaging yields a quasi-atomic model of the adenovirus-related bacteriophage PRD1 and shows key capsid and membrane interactions. Structure (Camb) 9: 917–930 [DOI] [PubMed] [Google Scholar]
- Navaza J, Lepault J, Rey FA, Alvarez-Rua C, Borge J (2002) On the fitting of model electron densities into EM reconstructions: a reciprocal-space formulation. Acta Crystallogr D Biol Crystallogr 58: 1820–1825 [DOI] [PubMed] [Google Scholar]
- Newcomb WW, Boring JW, Brown JC (1984) Ion etching of human adenovirus 2: structure of the core. J Virol 51: 52–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Rosa-Calatrava M, Grave L, Puvion-Dutilleul F, Chatton B, Kedinger C (2001) Functional analysis of adenovirus protein IX identifies domains involved in capsid stability, transcriptional activity, and nuclear reorganization. J Virol 75: 7131–7141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruigrok RW, Barge A, Albiges-Rizo C, Dayan S (1990) Structure of adenovirus fibre. II. Morphology of single fibres. J Mol Biol 215: 589–596 [DOI] [PubMed] [Google Scholar]
- Russell WC (2000) Update on adenovirus and its vectors. J Gen Virol 81: 2573–2604 [DOI] [PubMed] [Google Scholar]
- Russell WC, Precious B (1982) Nucleic acid-binding properties of adenovirus structural polypeptides. J Gen Virol 63 (Part 1): 69–79 [DOI] [PubMed] [Google Scholar]
- Rux JJ, Burnett RM (2000) Type-specific epitope locations revealed by X-ray crystallographic study of adenovirus type 5 hexon. Mol Ther 1: 18–30 [DOI] [PubMed] [Google Scholar]
- Rux JJ, Burnett RM (2004) Adenovirus structure. Hum Gene Ther 15: 1167–1176 [DOI] [PubMed] [Google Scholar]
- Schoehn G, Fender P, Chroboczek J, Hewat EA (1996) Adenovirus 3 penton dodecahedron exhibits structural changes of the base on fibre binding. EMBO J 15: 6841–6846 [PMC free article] [PubMed] [Google Scholar]
- Schoehn G, Moss SR, Nuttall PA, Hewat EA (1997) Structure of Broadhaven virus by cryoelectron microscopy: correlation of structural and antigenic properties of Broadhaven virus and bluetongue virus outer capsid proteins. Virology 235: 191–200 [DOI] [PubMed] [Google Scholar]
- Stewart PL, Burnett RM (1995) Adenovirus structure by X-ray crystallography and electron microscopy. Curr Top Microbiol Immunol 199 (Part 1): 25–38 [DOI] [PubMed] [Google Scholar]
- Stewart PL, Burnett RM, Cyrklaff M, Fuller SD (1991) Image reconstruction reveals the complex molecular organization of adenovirus. Cell 67: 145–154 [DOI] [PubMed] [Google Scholar]
- Stewart PL, Fuller SD, Burnett RM (1993) Difference imaging of adenovirus: bridging the resolution gap between X-ray crystallography and electron microscopy. EMBO J 12: 2589–2599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN, Gillespie JR, Fink AL (2000) Why are ‘natively unfolded' proteins unstructured under physiologic conditions? Proteins 41: 415–427 [DOI] [PubMed] [Google Scholar]
- van Oostrum J, Burnett RM (1985) Molecular composition of the adenovirus type 2 virion. J Virol 56: 439–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Raaij MJ, Mitraki A, Lavigne G, Cusack S (1999) A triple beta-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature 401: 935–938 [DOI] [PubMed] [Google Scholar]
- Vellekamp G, Porter FW, Sutjipto S, Cutler C, Bondoc L, Liu YH, Wylie D, Cannon-Carlson S, Tang JT, Frei A, Voloch M, Zhuang S (2001) Empty capsids in column-purified recombinant adenovirus preparations. Hum Gene Ther 12: 1923–1936 [DOI] [PubMed] [Google Scholar]
- Wickham TJ, Mathias P, Cheresh DA, Nemerow GR (1993) Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell 73: 309–319 [DOI] [PubMed] [Google Scholar]
- Wriggers W, Birmanns S (2001) Using situs for flexible and rigid-body fitting of multiresolution single-molecule data. J Struct Biol 133: 193–202 [DOI] [PubMed] [Google Scholar]
- Wu E, Pache L, Von Seggern DJ, Mullen TM, Mikyas Y, Stewart PL, Nemerow GR (2003) Flexibility of the adenovirus fiber is required for efficient receptor interaction. J Virol 77: 7225–7235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Yu X, Galkin VE, Egelman EH (2003) Issues of resolution and polymorphism in single-particle reconstruction. J Struct Biol 144: 162–171 [DOI] [PubMed] [Google Scholar]
- Zubieta C, Schoehn G, Chroboczek J, Cusack S (2005) The structure of the human adenovirus 2 penton. Mol Cell 17: 121–135 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data