

Abstract
Myeloid leukemia factor 1 (MLF1) was first identified as the leukemic fusion protein NPM-MLF1 generated by the t(3;5)(q25.1;q34) chromosomal translocation. Although MLF1 expresses normally in a variety of tissues including hematopoietic stem cells and the overexpression of MLF1 correlates with malignant transformation in human cancer, little is known about how MLF1 is involved in the regulation of cell growth. Here we show that MLF1 is a negative regulator of cell cycle progression functioning upstream of the tumor suppressor p53. MLF1 induces p53-dependent cell cycle arrest in murine embryonic fibroblasts. This action requires a novel binding partner, subunit 3 of the COP9 signalosome (CSN3). A reduction in the level of CSN3 protein with small interfering RNA abrogated MLF1-induced G1 arrest and impaired the activation of p53 by genotoxic stress. Furthermore, ectopic MLF1 expression and CSN3 knockdown inversely affect the endogenous level of COP1, a ubiquitin ligase for p53. Exogenous expression of COP1 overcomes MLF1-induced growth arrest. These results indicate that MLF1 is a critical regulator of p53 and suggest its involvement in leukemogenesis through a novel CSN3–COP1 pathway.
Keywords: COP1, CSN3, MLF1, oncogenesis, p53
Introduction
Chromosomal translocations in human leukemia often create aberrant fusion proteins, which act as an initiating event in tumorigenesis by disrupting the normal hematopoietic control of proliferation, differentiation and cell death. Molecular functional analysis of the fusion proteins frequently leads to the discovery of novel regulatory pathways essential for both normal cell growth and tumorigenesis (for reviews, see Rabbitts, 1994; Look, 1997). The t(3;5)(q25.1;q34) chromosomal translocation is associated with myelodysplastic syndrome (MDS) often prior to acute myeloid leukemia (AML) (Raimondi et al, 1989) and generates a fusion protein consisting of nucleophosmin (NPM)/B23 and cytoplasmic protein, myeloid leukemia factor 1 (MLF1) (Yoneda-Kato et al, 1996). NPM is a ubiquitously expressed multifunctional nucleolar phosphoprotein, which plays a part in nuclear–cytoplasmic shuttling of ribonucleoproteins during the ribosomal assembly (Yung et al, 1985; Borer et al, 1989; Olson et al, 2000). NPM is a direct regulator of the Arf–Mdm2–p53 tumor suppressor protein pathway (Colombo et al, 2002; Itahana et al, 2003; Bertwistle et al, 2004; Kurki et al, 2004). NPM binds to Arf and recruits it to the nucleoli, which affects the regulation of the cell cycle. NPM also interacts with Mdm2 and prevents the degradation of p53 mediated by Mdm2.
By contrast, the biochemical activity of MLF1 remains unknown because of a lack of significant homology between MLF1 and any previously identified protein, but clues as to the biological function have been obtained. In clinical studies, MLF1 expression was preferentially detected in CD34+ primitive cells and declined in more lineage-committed cells during normal hematopoiesis (Matsumoto et al, 2000). MLF1 is aberrantly overexpressed in over 25% of patients with MDS-associated AML and the malignant transformation phase of MDS (Matsumoto et al, 2000). In addition, enforced expression of murine MLF1 disturbed the development of erythroid colonies in normal hematopoietic precursors and differentiation to erythrocytes in an erythropoietin-dependent cell line (Williams et al, 1999). These findings suggest that MLF1 normally functions as a regulatory factor in the development of primitive hematopoietic cells and its deregulation contributes to hematopoietic dysplasia and leukemogenesis. However, endogenous expression of MLF1 is not restricted in hematopoietic cells (Yoneda-Kato et al, 1996) and the aberrant overexpression of MLF1 is also reported in lung squamous cell carcinoma (Sun et al, 2004), suggesting that MLF1 is involved in a common regulatory pathway related to tumorigenesis.
We show here that MLF1 inhibits cell cycle progression through a p53-dependent mechanism. MLF1 binds directly to the third component of the COP9 signalosome complex (CSN3) and activates the p53/p21 pathway via a novel CSN3–COP1-mediated pathway. The COP9 signalosome (CSN) and COP1 were initially defined as repressors of photomorphogenesis in plants. CSN is placed upstream of COP1, which promotes the degradation of the transcription factor HY5, a positive regulator of photomorphogenesis, in the dark (Schwechheimer and Deng, 2001). Although COP1 was recently discovered to exhibit E3 ubiquitin ligase activity toward p53 (Dornan et al, 2004), the existence of the CSN–COP1–p53 pathway and its upstream regulator in mammalian cells has never been demonstrated before. The expression of MLF1 results in the downregulation of COP1 through CSN3 and consequent stabilization of p53. Importantly, knockdown of CSN3 protein by the small interfering RNA (siRNA) method not only impairs MLF1-induced cell cycle arrest but also interferes with the activation of p53 by genotoxic stress despite the presence of MLF1, implying that CSN3 is a critical factor mediating genotoxic stress leading to p53 activation. Identification of the novel pathway of p53 activation mediated by CSN3–COP1 in response to MLF1 signaling and DNA damage will provide new insight into the regulation of mammalian cell proliferation and tumorigenesis.
Results
MLF1 induces cell cycle arrest in G1 phase
To elucidate the function of MLF1, we ectopically expressed MLF1 protein in murine fibroblasts and examined its effect on cell proliferation. Human MLF1 has a single nucleotide polymorphism at the 226th amino-acid codon, which produces proline and threonine at a ratio of 3:1 in normal genomic alleles (Figure 1A). We designated the proteins P-MLF1 and T-MLF1, respectively, and introduced expression vectors encoding P-MLF1, T-MLF1 and NPM-MLF1 fused with green fluorescent protein (GFP) and a puromycin resistance gene into mouse NIH3T3 fibroblasts. The number of colonies that appeared after selection in puromycin was much smaller for MLF1-transfected cells (ca. 10%) than for the cells transfected with control GFP and NPM-MLF1. The colonies expressing P-MLF1 and T-MLF1 were smaller, consisted of fewer cells and exhibited lower cellular density (Figure 1B). T-MLF1 seemed to have a stronger suppressive effect on cell growth than did P-MLF1 because the number of colonies and the cell density were significantly lower on transfection of T-MLF1 than P-MLF1. In addition, cells expressing T-MLF1 ceased to proliferate much earlier than those expressing P-MLF1. Therefore, we performed the analyses mainly with the P-MLF1 transfectants. As expected, the cells expressing P-MLF1 grew slower than those expressing GFP alone and NPM-MLF1 (Figure 1C). A similar retardation of growth was observed in several independent single cell-derived clones as well as in mixed populations of GFP-positive cells, and in cells transfected with intact MLF1 and HA-tagged MLF1 under the control of several different promoters (data not shown), indicating that the phenotype seen in this experiment was not due to the clonal variation of the cells or to the fusion with the GFP molecule. Examination of the cell cycle distribution of these cell clones revealed that P-MLF1-expressing cells exhibited a markedly increased G1-phase population (77%) at the expense of a decrease in the S and G2/M phases (14 and 9%, respectively), while the control cell population contained 45% G1-phase cells, 35% S-phase cells and 19% G2/M-phase cells (Figure 1D). Importantly, these cell clones expressed comparable amounts of MLF1 and NPM-MLF1 proteins (Figure 1E). Taken together, these results indicate that ectopically expressed MLF1 induced cell cycle arrest at G1 phase and the fusion protein comprising NPM and MLF1 generated in leukemia cells lost this activity.
Figure 1.
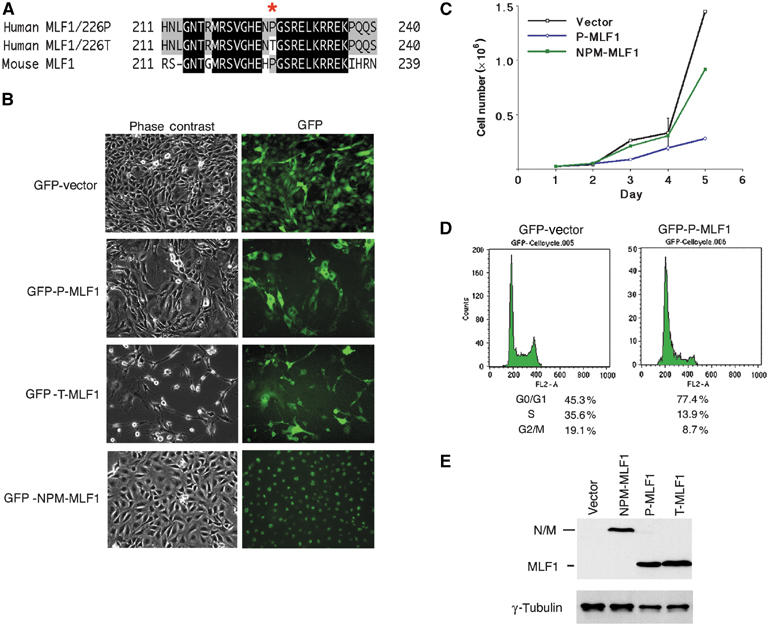
MLF1 induces cell cycle arrest in G1 phase. (A) A single nucleotide polymorphism at the 226th amino-acid sequence of human MLF1 produces proline (P) and threonine (T) at a ratio of 3:1 in normal genomic alleles. (B, C) MLF1 suppresses cell growth. NIH3T3 (p53 wild type, Arf null) mouse fibroblasts were transfected with GFP empty vector and GFP-fused P-MLF1(226 Proline), T-MLF1(226 Threonine) and NPM-MLF1. After selection with puromycin, GFP-positive single-cell clones expressing P-MLF1 and T-MLF1 exhibited a lower cell density and growth rate than the clones expressing GFP alone or NPM-MLF1. (D) The cell cycle distributions of the GFP-positive cells were analyzed using a flow cytometer after staining with propidium iodide. (E) Cell lysates from GFP-positive single-cell clones were immunoblotted with antibodies to MLF1 and anti-γ-tubulin.
To test whether the MLF1-associated growth suppressing phenotype is not restricted to fibroblasts, we introduced MLF1 expression vectors into several hematopoietic and adherent cell lines. As a result, we found that a stable expression of MLF1 in cells depends on the status of p53: we failed to establish clones expressing either type of MLF1 protein from 32D immature myeloid (p53 wild type) cells and U2OS (p53 wild type) cells, whereas MLF1 expression was stable in M1 (p53 null), K562 (p53 mutation) and 293T (p53 inactivated) cells. In addition, the NIH3T3 cells (provided by Drs CJ Sherr and MF Roussel) used in Figure 1 lack the ARF locus but retain the intact p53 allele.
MLF1-induced growth arrest is dependent on p53
To assess the molecular mechanism involved in MLF1-induced cell cycle arrest, we examined the effect of ectopic expression of MLF1 on the growth of genetically altered murine primary embryonic fibroblasts (MEFs). We infected MEF cells (wild type, p53−/− and p27−/−) with retroviruses that express GFP alone and GFP-(P-type and T-type)-MLF1 fusion proteins and selected them in puromycin. Growth curves show that wild-type MEF cells expressing either type of MLF1 protein grew markedly slower than those expressing GFP alone (Figure 2A), consistent with the results obtained in NIH3T3 cells (Figure 1C). In this experiment, the proliferation rate of T-MLF1-expressing cells was only marginally lower than that of cells expressing P-MLF1, but Western blot analysis revealed that expression levels of the protein were significantly lower in T-MLF1-expressing cells (Figure 2D, lanes 1–3), confirming the result that T-MLF1 was a more potent growth suppressor in murine fibroblasts. Protein analysis also shows that, in these growth-retarded cells, the expression of Cdk inhibitors, p21 and p27, and the tumor suppressor p53 was upregulated (Figure 2D, lanes 1–3). The growth curves of p53- or p27-deficient MEF cells infected with MLF1 retroviruses show that p53−/− MEF cells were refractory to the MLF1 action (Figure 2B), while ectopic MLF1 induced growth arrest in p27−/− MEF cells as effectively as in wild-type cells (Figure 2C). Protein analysis shows that MLF1 expression resulted in induction of p53 and its target protein p21 expression in p27−/−, but not p53−/−, MEF cells as expected. However, we did not detect upregulated p27 expression in p53−/− cells for unknown reasons (Figure 2D). Thus, consistent with the findings obtained from various cell lines with the different p53 status, these results demonstrate that MLF1-induced growth arrest depended on the integrity of the p53 allele.
Figure 2.
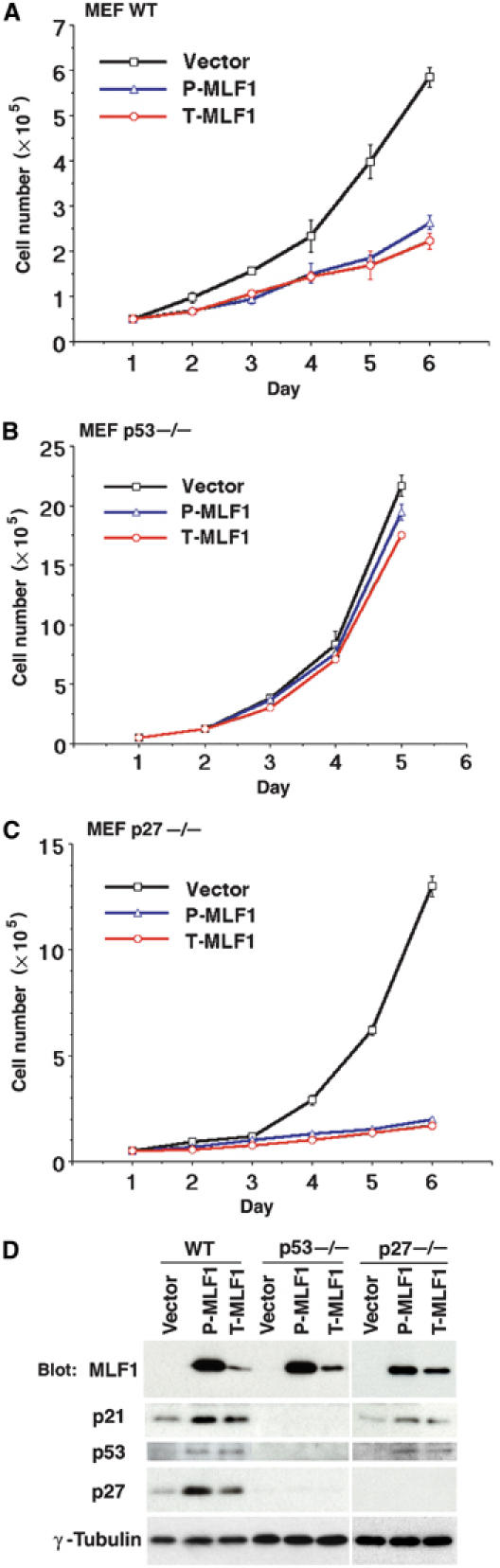
MLF1 induces G1 arrest in a p53-dependent manner. Growth curves of wild-type (A), p53−/− (B) and p27−/− (C) MEFs infected with retroviruses that expressed GFP alone, GFP-P-MLF1 and GFP-T-MLF1. (D) Cell lysates from MEFs were analyzed by immunoblotting using antibodies against MLF1, p21, p53, p27 and γ-tubulin.
MLF1 specifically interacts with CSN3
To search for factors involved in the MLF1-mediated signaling pathway, we utilized the yeast two-hybrid system to screen murine T-cell lymphoma and human K562 erythroleukemia cDNA libraries to identify genes whose products directly interact with MLF1. We eventually isolated four types of cDNA inserts as classified by sequence, one of which contained sequences 100% identical to those of the mouse and human COP9 signalosome subunit 3 (CSN3). We tested the specificity of the interaction between MLF1 and CSN3 in living cells. In Cos7 cells transiently transfected with HA-tagged CSN3 and MLF1, a substantial amount of MLF1 protein was detected in an anti-CSN3 immunoprecipitate (Figure 3A, middle panel). In a reciprocal experiment, we found a significant portion of CSN3 in an anti-MLF1 immunoprecipitate (Figure 3A, bottom panel). Furthermore, we successfully detected the interaction between endogenous CSN3 and MLF1 proteins in human leukemia K562 cells (Figure 3B). The majority of endogenous and ectopically expressed MLF1 protein is located in the cytoplasm, concentrated around the nucleus, with a small fraction in the nucleus in cultured mammalian cells (Yoneda-Kato et al, 1996; Matsumoto et al, 2000). By contrast, the endogenous CSN3 protein was detected in both the nucleus and cytoplasm, with the highest level in the nucleoplasm in murine fibroblasts (Figure 3C, the upper right panel). Ectopic MLF1 was colocalized with endogenous CSN3 in the perinuclear region of the cytoplasm with minimal changes in the subcellular distribution of CSN3 (Figure 3C, the lower panels). Taken together, these results indicate that MLF1 and CSN3 interact with each other in vivo.
Figure 3.
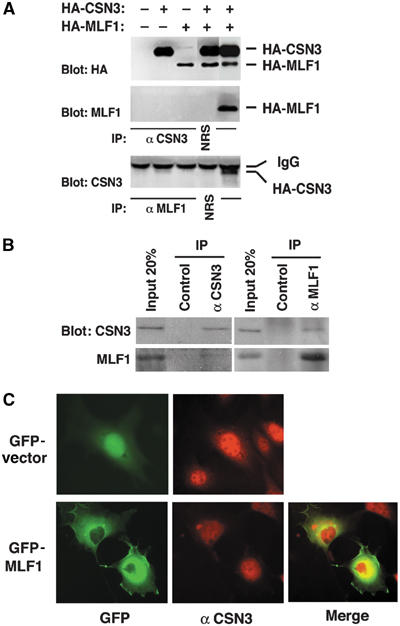
CSN3 specifically interacts with MLF1 in vivo. (A) CSN3 specifically interacts with MLF1 in ectopically overexpressed cells. Cos7 cells were transfected with the expression vectors shown at the top. Total expression was analyzed by anti-HA immunoblotting (upper panel). Cell lysates were analyzed by sequential immunoprecipitation and immunoblotting with antibodies to CSN3 and MLF1 as shown at the left of the panels (middle and lower panels). NRS, preimmune normal rabbit serum. (B) Specific interaction between endogenous CSN3 and MLF1 proteins. Endogenous CSN3 and MLF1 proteins were immunoprecipitated from the K562 cell lysate shown at the top, and analyzed by immunoblotting with antibodies to CSN3 (upper panel) and MLF1 (lower panel). (C) Subcellular localization of MLF1 and CSN3. NIH3T3 cells were transfected with the GFP-control (upper panel) and GFP-MLF1 (lower panel) expression vectors, stained with an antibody to CSN3 and viewed using fluorescence (GFP for GFP-MLF1 and Texas red for endogenous CSN3) microscopy. The merged panel is also shown.
CSN3 is required for the MLF1-induced growth arrest
To investigate whether CSN3 is involved in MLF1-mediated growth arrest and activation of the p53 pathway, we used an RNA interference (RNAi) technique to reduce the amount of endogenous CSN3 protein in mammalian cells, and examined its effect on the MLF1 action. NIH3T3 cells were transfected with expression vectors containing GFP alone and GFP-P-MLF1 fusion cDNA together with an siRNA vector specific for mouse CSN3 (CSN3 siRNA) and a luciferase (Luc siRNA, used as a negative control). The cells were selected in puromycin for 5 days and then allowed to form colonies for an additional 9 days. The flat morphology and low cell density induced by MLF1 was markedly recovered in cells cotransfected with CSN3 siRNA, but not with Luc siRNA (Figure 4A). We chose several independent colonies, which uniformly expressed GFP proteins, expanded them, and used them for further analyses. The results from two representative clones that express different levels of CSN3 are shown in Figure 4B and C, but similar results were obtained in the experiment using different clones. The introduction of MLF1 into mouse fibroblasts activated the p53 pathway and induced the expression of p21 as in Figure 2D, but a decrease in the intracellular level of CSN3 protein prevented activation of p53 and resultant induction of p21 (Figure 4B). Surprisingly, a moderate reduction of CSN3 was sufficient to prevent the full induction of p21 and suppression of cell proliferation (Figure 4B and C, clone #2). Importantly, reduction of the intracellular level of CSN3 did not affect the expression level or the intracellular localization of MLF1 protein. The growth curve of each clone shows that a reduction of endogenous CSN3 protein resulted in a marked increase in the rate of proliferation regardless of the ectopic MLF1 expression (Figure 4C). As expected from the above result (Figure 4B), even a moderate reduction of CSN3 protein was sufficient to abrogate MLF1-induced growth suppression (Figure 4C). Thus, maintenance of CSN3 expression is required for the growth suppressive function of MLF1, suggesting that CSN3 is one of the key downstream effectors of the MLF1-mediated signaling pathway.
Figure 4.
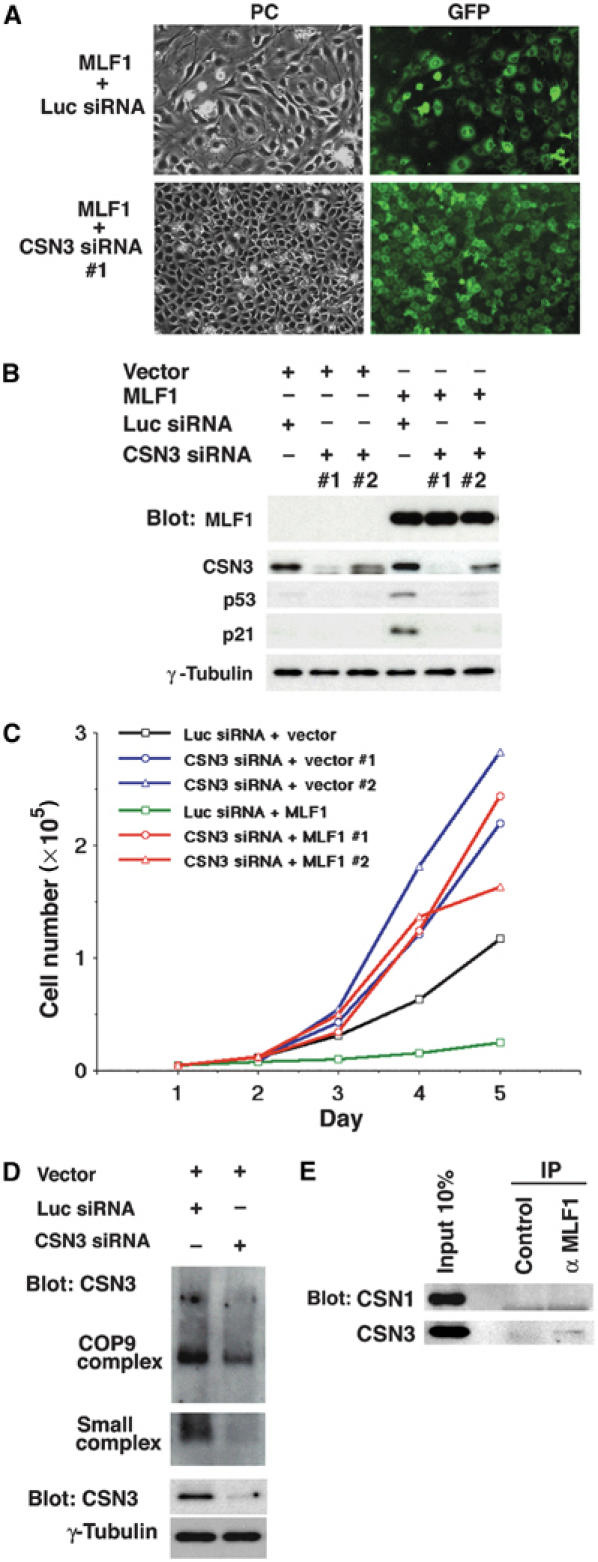
CSN3 is required for the MLF1-induced G1 arrest. NIH3T3 cells were transfected with control luciferase (Luc) siRNA and CSN3 siRNA expression vectors together with the GFP-control and GFP-MLF1 expression vectors and selected in puromycin for 5 days. (A) At 14 days post-transfection, representative GFP-positive single clones were photographed. PC, phase-contrast. (B) Two representative clones with extensive and moderate reductions of CSN3 were chosen from each transfectant and analyzed by Western blotting using antibodies to CSN3, p53, p21 and γ-tubulin. (C) Growth curves of each clone. (D) Cell lysates were separated by native-PAGE (COP9 complex and small complex) and by SDS–PAGE (total CSN3 and γ-tubulin) and analyzed by immunoblotting using an antibody to CSN3. (E) Endogenous MLF1 was immunoprecipitated from the K562 cell lysate and analyzed by immunoblotting with antibodies to CSN1 (upper panel) and CSN3 (lower panel).
Although Arabidopsis CSN3 is exclusively found in the CSN complex (Peng et al, 2001), mammalian CSN3 forms a unique smaller subcomplex besides CSN in murine fibroblasts (Fukumoto et al, 2005). To investigate the effect of CSN3 knockdown on the formation of the CSN3-containing complexes, we performed the nondenaturing polyacrylamide gel electrophoresis (native-PAGE) analysis (Seeger et al, 1998; Fukumoto et al, 2005; Tomoda et al, 2005), and found that the small CSN3 subcomplex, rather than CSN, was predominantly reduced (Figure 4D). Furthermore, to know the specificity of interaction in vivo, we immunoprecipitated endogenous MLF1 protein from the K562 cell lysate and found that MLF1 forms a complex with CSN3 but not with CSN1 (Figure 4E). Because CSN1 is found only in CSN in Arabidopsis (Wei et al, 1994; Staub et al, 1996) and in murine fibroblasts (Fukumoto et al, 2005), the target of MLF1 seems to be specific to CSN3 rather than CSN. In addition, we used the Jab1/CSN5 siRNA in an attempt to rescue the growth suppression mediated by MLF1, but were not successful (data not shown), supporting the notion that MLF1 is involved in the CSN3-specific function, such as the small CSN3 subcomplex. However, we do see some reduction of CSN in CSN3 siRNA-treated cells (Figure 4D), and do not know the exact relationship between the small CSN3 subcomplex and CSN yet. Therefore, we do not exclude the possibility that CSN plays an important role in the MLF1-mediated signaling pathway, and it will be most adequate to say that both CSN and the small CSN3 subcomplex are involved in the regulation of p53 by MLF1.
Residues 50–125 of MLF1 are required for both CSN3 binding and growth inhibition
To map the region of MLF1 required for association with CSN3, a series of MLF1 deletion mutants were generated (Figure 5A). Glutathione S-transferase (GST)-tagged recombinant MLF1 proteins were tested for binding to CSN3 in the lysate prepared from Cos7 cells transfected with CSN3 cDNA. An MLF1 mutant lacking the N-terminal 49 amino acids (MLF1/50–268), but not the one lacking 125 amino acids (MLF1/126–268), bound to CSN3 in vitro (Figure 5B), suggesting that residues 50–125 are required for interaction with CSN3. To determine the domain of MLF1 critical for growth suppression, we transfected NIH3T3 cells with expression vectors containing GFP-fused MLF1 mutants and enumerated the number of GFP-positive colonies formed after selection in puromycin. Again, the mutant lacking the N-terminal 125 amino acids (MLF1/126–268) lost the ability to suppress cell growth (Figure 5C). Thus, as summarized in Figure 5A, only the mutant unable to bind to CSN3 lost the growth inhibitory activity, indicating that the interaction with CSN3 is essential for the MLF1-induced growth arrest.
Figure 5.
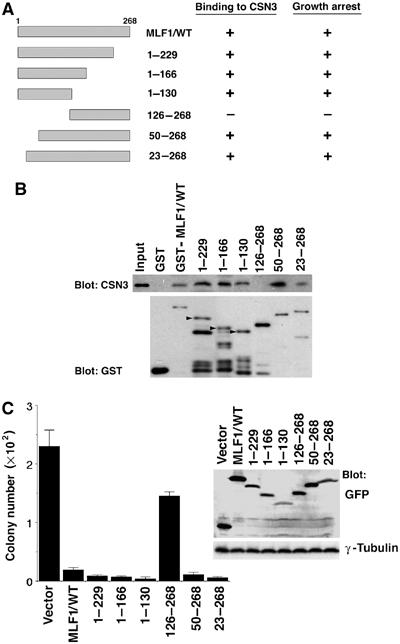
Mapping the domain of MLF1 required for CSN3 binding and growth inhibition. (A) Schematic representation of MLF1 deletion mutants. The results of CSN3 binding and growth inhibition are summarized on the right. (B) The region of MLF1 involved in binding to CSN3 was determined by the GST pull-down assay. Beads coated with GST-control or the GST-MLF1 mutant fusion proteins shown at the top of the panel were incubated with Cos7 cell lysates containing HA-tagged CSN3 proteins. Bound proteins were detected by immunoblotting with antibody to CSN3. The amounts of GST proteins absorbed on the beads were evaluated by anti-GST immunoblotting. The positions of full-length GST-MLF1 mutant fusion proteins are indicated with arrowheads in lanes 3–5. (C) NIH3T3 cells were transfected with GFP-control and GFP-MLF1 mutant expression vectors and selected in puromycin for 5 days. At 14 days post-transfection, GFP-positive colonies were enumerated. Upper right: Expression levels of GFP-MLF1 mutant proteins before puromycin selection are shown by Western blotting with antibodies to GFP and γ-tubulin.
CSN3 mediates MLF1 and genotoxic stress signaling in p53 activation via COP1
In plants, it was demonstrated that the COP9 signalosome complex is required for the proper functioning of the RING-finger-type E3 ubiquitin ligase COP1, which promotes the degradation of the transcription factor HY5, a positive regulator of photomorphogenesis, in the dark (Schwechheimer and Deng, 2001). Recently, COP1 was reported to serve as an E3 ubiquitin ligase for p53 in mammalian cells (Dornan et al, 2004). Therefore, we addressed whether COP1 is a downstream target leading to p53 activation in the MLF1 signaling. To avoid clonal deviation of the transfected cells, we isolated the GFP-positive population of cells by cell sorting from the mixed population of NIH3T3 cells transfected with GFP and siRNA vectors as shown in Figure 6A. We found that ectopic MLF1 expression resulted in a marked decrease in the endogenous COP1 protein level, whereas a reduction of CSN3 led to an increase of COP1 regardless of the MLF1 expression (Figure 6A), suggesting that MLF1 modulates COP1 expression via CSN3 to activate the p53 pathway. Quantitative RT–PCR analysis revealed that the relative levels of p53 and COP1 mRNA in cells transfected with MLF1 or CSN3 siRNA remained unchanged (Figure 6B), whereas the half-life of p53 in MLF1-transfected cells (ca. 30 min) determined by the pulse-chase analysis was significantly longer than that in control cells (ca. 18 min) (Figure 6C). Thus, MLF1 upregulates p53 through inhibition of protein degradation.
Figure 6.
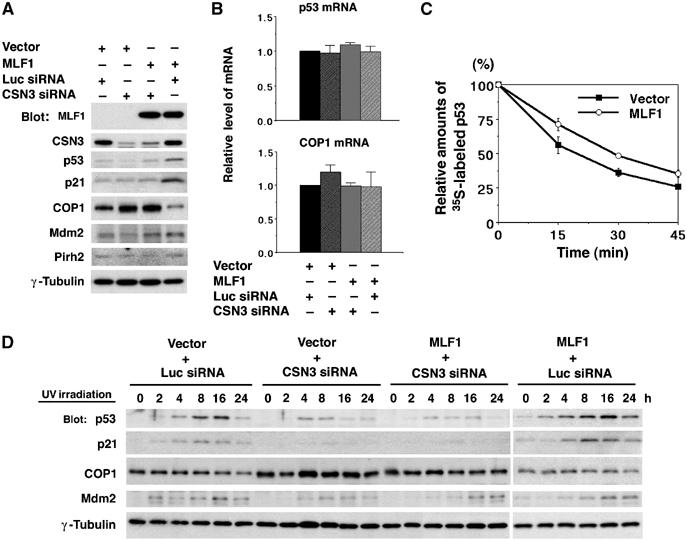
Effects of CSN3 siRNA on the MLF1-induced G1 arrest and p53 stability. NIH3T3 cells were transfected with control luciferase (Luc) siRNA and CSN3 siRNA expression vectors together with the GFP-control and GFP-MLF1 expression vectors. Multiclonal GFP-positive cells were isolated by cell sorting. (A) Cells were lysed and analyzed by Western blotting using antibodies against MLF1, CSN3, p53, p21, COP1, Mdm2 and Pirh2 and γ-tubulin. (B) Total RNAs were extracted from cells and used for the quantitative RT–PCR analysis to determine the relative amounts of p53 and COP1 transcripts. (C) GFP-control and GFP-MLF1 cells were metabolically labeled with [35S]methionine, and chased for the indicated periods of time. Labeled p53 protein was immunoprecipitated with an antibody to p53 and quantified using a phosphorimager. (D) Cells were treated with 25 J/m2 of UV irradiation for the indicated period of time before harvest. Cells were lysed and analyzed by Western blotting using antibodies against p53, p21, COP1, Mdm2 and γ-tubulin. Results shown in (A, D) are representative of three independent experiments. Results in (B, C) represent the average of triplicate experiments.
To know whether CSN3 is more commonly involved in the regulation of p53, we examined the effect of the knockdown of CSN3 on a genotoxic stress-induced activation of p53. We treated the siRNA-transfected cells used in Figure 6A with ultraviolet light (UV, 25 J/m2) and analyzed the kinetics of endogenous p53, p21, COP1 and Mdm2 protein expression. Treatment with UV resulted in a marked increase of p53 and p21 and a subsequent decrease of COP1 in control cells and a more enhanced and prolonged increase of p53 and p21 in MLF1-transfected cells, but failed to induce full upregulation of p53 and p21 in cells transfected with a CSN3-specific siRNA vector regardless of the MLF1 expression. Importantly, a relatively large amount of COP1 was maintained in these cells after exposure to UV irradiation (Figure 6D). These results show that CSN3 is an essential mediator needed to suppress COP1 and activate p53 in the genotoxic stress-induced signaling pathway as well as in MLF1-mediated signaling.
COP1 interferes with the growth inhibitory activity of MLF1
To assess whether COP1 can counteract the action of MLF1 in p53 activation, we investigated the effect of ectopic COP1 expression on the growth arrest induced by MLF1. NIH3T3 cells were transfected with a constant amount of GFP-MLF1 cDNA together with an increasing amount of HA-tagged COP1 expression vector. First, transfected cells were harvested before selection and analyzed for the protein expression by immunoblotting with antibodies specifically recognizing MLF1 and COP1. Figure 7A shows that an equivalent amount of MLF1 protein was expressed in each transfectant regardless of the COP1 expression. Next, transfected cells were selected in puromycin for 5 days and allowed to form colonies for an additional 9 days in the absence of the drug. Introduction of the MLF1 cDNA alone did not affect survival much as expected. However, cotransfection with a COP1 expression vector substantially increased the number of GFP-positive colonies in a dose-dependent manner (Figure 7B). Importantly, the single cell-derived clones maintained an equivalent level of MLF1 and COP1 expression after colony formation, and the level of p53 was markedly lower in HA-COP1-transfected cells than in control cells (Figure 7C). Because COP1 elicits the E3 ligase activity toward p53 through its RING-finger domain (Dornan et al, 2004), we examined whether a COP1 mutant lacking the RING-finger domain, COP1ΔRING, could block the MLF1 action. As expected, COP1ΔRING failed to rescue cells from MLF1-mediated growth inhibition (Figure 7A and B, right). Thus, ectopic COP1 expression can block the MLF1-induced growth suppression via its specific E3 ligase activity, indicating that downregulation of COP1 is a critical event downstream of MLF1 signaling to control cell proliferation.
Figure 7.
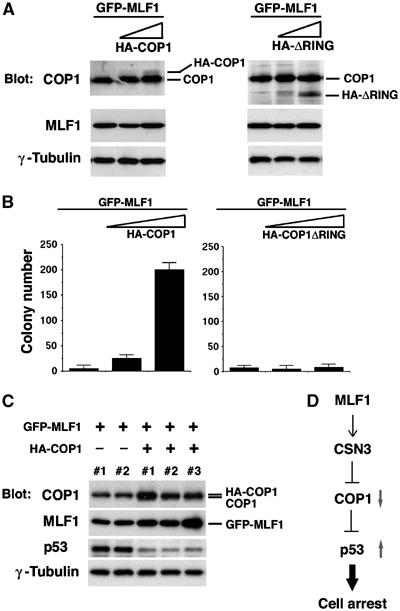
COP1 interferes with the growth inhibitory activity of MLF1. NIH3T3 cells were transfected with a constant amount (5 μg) of GFP-P-MLF1 cDNA together with various amounts (0, 2.5 and 5 μg) of HA-tagged COP1 and COP1ΔRING expression vectors. (A) Cells were harvested 5 days after transfection and analyzed by immunoblotting with antibodies to COP1, MLF1 and γ-tubulin. The positions of HA-tagged COP1 (HA-COP1), COP1ΔRING (HA-ΔRING) and endogenous COP1 (COP1) are shown. (B) Transfected cells were selected in puromycin for 5 days, and GFP-positive colonies were enumerated at 14 days post-transfection. (C) GFP-positive, single-cell-derived clones from (B) were isolated by a cylinder-cloning method and expanded. Total cell lysates were analyzed by immunoblotting with antibodies to COP1, MLF1, p53 and γ-tubulin. (D) Proposed model for the MLF1, CSN3, COP1 and p53 pathway.
Discussion
While analyzing the physiological function of MLF1, which is disturbed by formation of the leukemic fusion protein NPM-MLF1, we found a novel regulatory pathway of degradation of the tumor suppressor p53. As downstream elements of MLF1 leading to cell growth arrest due to p53 accumulation, we identified two factors, CSN3, the third component of the COP9 signalosome (CSN), and COP1, a recently characterized E3 ubiquitin ligase for p53 (Figure 7D). Genotoxic stress signals as well as the MLF1 signal are capable of initiating the CSN3–COP1-mediated accumulation of p53. Both signals induce a significant decrease in endogenous levels of COP1 and the resultant accumulation of p53 causes cell cycle arrest.
p53 is strictly maintained at a low level under normal conditions of cell growth by proteasome-mediated degradation. Once cells are exposed to cellular and genotoxic stress, p53 degradation is inhibited via multiple pathways (Vogelstein et al, 2000; Harper, 2004; Yang et al, 2004). While Mdm2 is the most characterized ubiquitin E3 ligase targeting p53 for degradation, recent studies revealed that additional RING-finger E3 ligases, Pirh2 and COP1, also promote the degradation of p53 by direct binding independently of Mdm2 (Leng et al, 2003; Dornan et al, 2004). However, little is known about the regulatory system of COP1-mediated p53 degradation. The functional relevance of CSN and COP1 is better defined in plants than in mammals because CSN and COP1 were first identified as COP/DET/FUS proteins showing the ability to repress photomorphogenesis, light-mediated development (Wei and Deng, 1999; Schwechheimer and Deng, 2001). Subsequent studies led to the characterization of COP1 and CSN as a RING-finger E3 ubiquitin ligase and putative essential modulator of the SCF (Skp1/Cul1/F-box protein) E3 ligase complex in ubiquitin-mediated proteolysis. Although mammalian cells do not undergo photomorphogenesis, basically similar functional properties should exist in these highly conserved proteins. Our findings have provided the first evidence that COP1 and a subunit of CSN function cooperatively in a crucial signaling pathway in mammals, whose deregulation is closely linked to leukemogenesis.
CSN is a multifunctional, eight-subunit complex participating in signaling, regulation of the cell cycle and development in mammals (Wei and Deng, 2003). CSN has the ability to regulate protein kinases that phosphorylate p53, c-Jun and IκBα (Seeger et al, 1998; Sun et al, 2002; Uhle et al, 2003). As an additional activity, CSN removes a small ubiquitin-like protein Nedd8 from the cullin-1 subunit of the SCF ubiquitin ligase (Lyapina et al, 2001; Cope et al, 2002). This ‘deneddylation' activity of CSN maintains the SCF ligase activity at a low level, resulting in suppression of the ubiquitin-mediated proteolysis. Our results that CSN3 is a key mediator to facilitate COP1 downregulation and p53 accumulation seem to be associated with the function of the CSN complex. A recent report showed that a family of cullins is not the only target of neddylation: Mdm2 is also neddylated and promotes conjugation of NEDD8 to p53, which inhibits its transcription activity (Xirodimas et al, 2004). These results raised the important questions of whether CSN promotes deneddylation of p53 and its regulators Mdm2 and COP1 as well as that of cullin in the SCF complex, and whether CSN3 and COP1 are involved in CSN-mediated phosphorylation or deneddylation of p53. The existence of smaller complexes containing each subunit of CSN complicates the study of the regulatory mechanisms (Tomoda et al, 2002, 2004; Fukumoto et al, 2005). Jab1, the fifth subunit of CSN, forms a unique smaller subcomplex induced by normal growth signals and oncogenic signals such as IL-3 and Bcr-Abl kinase and promotes the degradation of the Cdk inhibitor p27 by its transportation from the nucleus to the cytoplasm (Tomoda et al, 1999, 2005), whereas CSN inhibits its degradation by promoting deneddylation of cullin-1 (Yang et al, 2002). Considering the case of Jab1, further experiments are required to clarify whether the CSN3 activity is part of the CSN complex or specific for the subunit. Nonetheless, the finding that the knocking down of CSN3 protein markedly decreased the sensitivity to UV-induced p53 accumulation leading to growth arrest indicates that CSN3 is essential for the activity.
As for the function of MLF1 in normal hematopoiesis, its strong ability to inhibit cell growth and expression in CD34+ hematopoietic stem cells (HSCs) (Matsumoto et al, 2000) suggests that MLF1 negatively regulates the proliferation of HSCs by affecting cell cycle inhibitors. In fact, most HSCs under normal conditions are quiescent and only a very limited number of cells enter cell cycling for self-renewal or commitment to become progenitor cells (Hodgson and Bradley, 1979; Lerner and Harrison, 1990). This regulation is crucial to protect stem cells from premature exhaustion and prolonged insult to genotoxic stress. Analyses using mice revealed that many cell cycle inhibitors are involved in this negative regulation (Cheng, 2004). For instance, p21 controls the cell cycle entry of HSCs, and a defect of p21 causes increased cell cycling and consequent exhaustion of the HSC pool (Cheng et al, 2000). However, the intrinsic factors regulating the cell cycle machinery responsible for the maintenance of HSCs are poorly understood. MLF1 is a putative candidate for one of the factors contributing to the maintenance of HSCs upstream of cell cycle inhibitors. This reminds us of Bmi-1 activity. Bmi-1 acts as a potent positive regulator of the self-renewal of HSCs by repressing the transcription of the Ink4a/Arf locus that encodes the tumor suppressors p16 and p19Arf (Jacobs et al, 1999; Lessard and Sauvageau, 2003; Park et al, 2003). However, further studies including targeted disruption of the MLF1 gene locus will be necessary to clarify the role of MLF1 in hematopoiesis and the physiological importance of MLF1 in other normal tissues.
Our findings suggest that inactivation of MLF1 will lead to tumorigenesis due to prevention of p53 accumulation. On the contrary, aberrant overexpression of MLF1 is reported in patients with myeloid leukemia and lung carcinoma (Matsumoto et al, 2000; Sun et al, 2004), which seems contradictory to the proposed action of MLF1. However, cancer cell lines overexpressing MLF1 exclusively harbor nonfunctional p53 such as missense mutations and deletions (our unpublished observation), suggesting that aberrant overexpression of MLF1 in cancer cells appears to result from physiological response to prevent deregulated cell proliferation during the multiprocess of tumorigenesis. This is luminescent to the case of the INK4a locus in human cancer, in which tumor suppressor p16INK4a is overexpressed in a variety of human cancers due to the loss of Rb protein during the early stage of tumorigenesis (Sherr and Roberts, 1995; Hall and Peters, 1996).
We clarified two important issues in this study. First, MLF1 is a strong inducer of cell growth arrest, which regulates a novel oncogenic pathway leading to a central ‘guardian of the genome', p53, independent of the Arf pathway. Second, CSN may function upstream of COP1 in mammalian signal transduction. Although numerous questions have been raised about this novel pathway, manipulations of the MLF1 pathway may be useful for clinical applications such as cancer therapy or ex vivo expansion of normal HSCs.
Materials and methods
Plasmid construction and retroviral production
We constructed a GFP fusion protein expression vector by modifying the retroviral vector pMSCV-IRES-puro (Clontech). cDNA fragments of P-MLF1, T-MLF1 and NPM-MLF1 were subcloned into the retroviral vector in-frame with GFP. For viral production, the plasmid was cotransfected into 293T cells (provided by Dr David Baltimore) together with a plasmid encoding an ecotropic helper virus containing a defective virion-packaging (ψ2) sequence. Culture supernatants containing retroviruses harvested 48–72 h after transfection were used to infect proliferating MEF cells.
Cell culture, transfection and infection
NIH3T3 (Arf null, p53 wild type) mouse fibroblasts (provided by Drs CJ Sherr and MF Roussel) and Cos7 monkey cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS, and transfected with expression vectors via the calcium phosphate–DNA precipitation method (Chen and Okayama, 1987; Yoneda-Kato et al, 1999). Single cell clones from transfected NIH3T3 cells after culture with puromycin (5 ng/ml; Clontech) were isolated by a cylinder-cloning method and expanded in six-well plates. Wild-type, p53−/− and p27−/− MEF cells were cultured in the same conditions and infected with pMSCV-GFP-P-MLF1, pMSCV-GFP-T-MLF1 and empty pMSCV-GFP retroviral supernatants. GFP-positive cells with puromycin resistance were used for further analysis.
Cell cycle analysis
For flow cytometric analysis of DNA content, cells were suspended in a 1 ml solution of 0.1% sodium citrate and 0.1% Triton X-100 containing 50 μg/ml of propidium iodide and treated with 1 μg/ml of RNase for 30 min at room temperature. GFP-positive cells were gated and fluorescence from the propidium iodide–DNA complex was measured with a FACScan flow cytometer (Becton Dickinson). The percentages of cells in the G1, S and G2/M phases of the cell cycle were determined using the Cell Fit cell cycle software.
Protein analyses
Cell lysis, immunoprecipitation, gel electrophoresis and immunoblotting were performed as described (Tomoda et al, 1999, 2004). MLF1 and NPM-MLF1 proteins were detected using mouse monoclonal antibodies generated with bacterially produced human MLF1 polypeptides, which effectively crossreacted with the mouse protein. Rabbit polyclonal antibodies to GST (Z-5) and p53 (FL-393), and a goat polyclonal antibody to p21 (C-19) were purchased from Santa Cruz Biotechnology. Mouse monoclonal antibodies to HA peptide epitopes (12CA5) and γ-tubulin (GTU-88) were obtained from Sigma. A mouse monoclonal antibody to p27, a sheep polyclonal antibody to p53 (Ab-7) and rabbit polyclonal antibodies to p53 (CM5), GFP (Living Colors) and Pirh2 (BL588) were obtained from Transduction Laboratories, Calbiochem, Nobocastra Laboratories, BD Biosciences and Bethyl Laboratories, respectively. Rabbit polyclonal antibodies to MLF1, CSN1, CSN3 and COP1 were generated using bacterially produced polypeptides in our laboratory. A mouse monoclonal antibody to MDM2 was provided by Dr Arnold J Levine.
Nondenaturing gel electrophoresis (native-PAGE) was performed as described previously (Fukumoto et al, 2005). In brief, cells were lysed in a digitonin lysis buffer (50 mM Tris, pH 8.0, 120 mM NaCl, 1 mM EDTA and 0.1% digitonin) at 1 × 105 cells/μl. The sample was supplemented with 40 mM Tris–HCl pH 6.8 and 10% glycerol and equal amounts (ca. 100 μg) of total protein were separated on precast native gradient gels (5–15%; Bio-Craft) at 4°C without SDS at 5 mA for 16 h followed by 10 mA for 6 h, and analyzed by anti-CSN3 immunoblotting.
Yeast two-hybrid screen
The entire coding sequence of human MLF1 was fused in-frame to the GAL4 DNA-binding domain of the pAS2 vector (Harper et al, 1993). The resulting ‘bait' plasmid (pAS2-MLF1) was used to screen pACT-mouse T-cell lymphoma and human K562 erythroleukemia libraries (Clontech) by the yeast two-hybrid method in Y190 yeast cells (Durfee et al, 1993; Harper et al, 1993; Tomoda et al, 1999). Briefly, yeast transfectants were cultured on a Trp−Leu−His− selection medium and the resultant colonies were tested for β-galactosidase activity. After segregation, plasmids containing cDNA inserts were recovered from yeast cells and tested as to whether they could confer the His requirement to Y190 cells transformed with the plasmid, pAS2-MLF1, but not with an empty pAS2 vector.
Immunofluorescent staining
Cells grown on coverslips were fixed in 4% paraformaldehyde, permeabilized in methanol, stained with anti-CSN3 rabbit polyclonal antibodies and incubated with Texas red (TR)-linked anti-rabbit IgG (Amersham). The cell samples were viewed using phase-contrast or fluorescence microscopy.
RNA interference
The vectors for RNAi specific to mouse CSN3 were constructed based on the pSilencer expression vector system (Ambion) according to the manufacturer's instructions. The siRNA sequence targeting the CSN3 gene corresponded to nucleotides 61–79 from the first nucleotide of the start methionine codon (sense, 5′-ATG ACT CAG CTT TGT GAA CTT CAA GAG AGT TCA CAA AGC TGA GTC ATT TTT TT-3′; antisense, 5′-AAT TAA AAA AAT GAC TCA GCT TTG TGA ACT CTC TTG AAG TTC ACA AAG CTG AGT CAT GGC C-3′). As a control, we used the luciferase sequence (sense, 5′-CGT ACG CGG AAT ACT TCG ATT CAA GAG ATG GAA GTA TTC CGC GTA CGT TTT TT-3′; antisense, 5′-AAT TAA AAA ACG TAC GCG GAA TAC TTC GAT CTC TTG AAT CGA AGT ATT CCG CGT ACG GGC C-3′).
GST pull-down assay
cDNA fragments containing the wild-type and mutant MLF1 coding sequences were inserted into the pGEX vector (Pharmacia) in-frame with GST. GST fusion proteins were expressed in bacteria and purified as described (Tomoda et al, 1999). Crude cell extracts containing CSN3 protein were prepared from Cos7 cells transfected with the CSN3 expression vector in an EBC buffer (50 mM Tris–HCl, pH 7.5, 120 mM NaCl, 0.5% NP-40 and 1 mM EDTA) containing 5 μg/ml of aprotinin, 1 mM PMSF, 0.1 mM NaF, 0.1 mM NaVO4 and 1 mM DTT. Binding was performed in the EBC buffer and the protein complexes were washed in the same buffer and the bound protein was detected by immunoblotting. The same amounts of beads used for the binding assay were subjected to immunoblotting analysis with antibodies to GST to quantitatively examine the GST proteins.
Pulse-chase analysis
NIH3T3 cells were metabolically labeled with [35S]methionine for 15 min. Labeled cells were washed twice in PBS and incubated in medium containing excess cold methionine for 0, 15, 30 or 45 min before collecting. p53 was immunoprecipitated with FL393 polyclonal antibody and resolved by SDS–PAGE. 35S-labeled p53 proteins were visualized by autoradiography and quantified using a phosphorimager (Fuji BAS-2000).
Quantitative RT–PCR
Total RNA was isolated using the ISOGEN reagent (Nippon Gene) and reverse transcribed using RNase-free Superscript reverse transcriptase (Invitrogen) according to the manufacturer's instructions. Quantitative RT–PCR was performed within a linear range as described previously (Matsumoto et al, 2000) and the data were normalized by the expression level of β-actin for each sample. We confirmed that the reactions were quantitatively performed within a linear range by several control experiments. The following oligonucleotide primers specific to mouse p53, COP1 and β-actin were used: p53, 5′-ATG ACT GCC ATG GAG GAG TC-3′ (sense) and 5′-GTC AGT CTG AGT CAG GCC C-3′ (antisense); COP1, 5′-AGG TTT CAG TGG GAC CTC TC-3′ (sense) and 5′-GAC CTT TGA CCT CTG TCC TG-3′ (antisense); β-actin, 5′-CTT CTA CAA TGA GCT GCG TGT-3′ (sense) and 5′-CAA CGT CAC ACT TCA TGA TGG-3′ (antisense).
Acknowledgments
We thank Dr J Fujisawa for the plasmid, Drs CJ Sherr and MF Roussel for the NIH3T3 cell line and Ms I Nakamae for excellent technical assistance. This work was supported by Grants-in-Aid for Scientific Research and for Cancer Research from the Ministry of Education, Science, and Culture of Japan and by Hayashi Memorial Foundation for Female Natural Scientists.
References
- Bertwistle D, Sugimoto M, Sherr CJ (2004) Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol Cell Biol 24: 985–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borer RA, Lehner CF, Eppenberger HM, Nigg EA (1989) Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell 56: 379–390 [DOI] [PubMed] [Google Scholar]
- Chen C, Okayama H (1987) High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol 7: 2745–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T (2004) Cell cycle inhibitors in normal and tumor stem cells. Oncogene 23: 7256–7266 [DOI] [PubMed] [Google Scholar]
- Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT (2000) Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science 287: 1804–1808 [DOI] [PubMed] [Google Scholar]
- Colombo E, Marine JC, Danovi D, Falini B, Pelicci PG (2002) Nucleophosmin regulates the stability and transcriptional activity of p53. Nat Cell Biol 4: 529–533 [DOI] [PubMed] [Google Scholar]
- Cope GA, Suh GS, Aravind L, Schwarz SE, Zipursky SL, Koonin EV, Deshaies RJ (2002) Role of predicted metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1. Science 298: 608–611 [DOI] [PubMed] [Google Scholar]
- Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, O'Rourke K, Koeppen H, Dixit VM (2004) The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 429: 86–92 [DOI] [PubMed] [Google Scholar]
- Durfee T, Becherer K, Chen PL, Yeh SH, Yang Y, Kilburn AE, Lee WH, Elledge SJ (1993) The retinoblastoma protein associates with the protein phosphatase type 1 catalytic subunit. Genes Dev 7: 555–569 [DOI] [PubMed] [Google Scholar]
- Fukumoto A, Tomoda K, Kubota M, Kato JY, Yoneda-Kato N (2005) Small Jab1-containing subcomplex is regulated in an anchorage- and cell cycle-dependent manner, which is abrogated by ras transformation. FEBS Lett 579: 1047–1054 [DOI] [PubMed] [Google Scholar]
- Hall M, Peters G (1996) Genetic alterations of cyclins, cyclin-dependent kinases, and Cdk inhibitors in human cancer. Adv Cancer Res 68: 67–108 [DOI] [PubMed] [Google Scholar]
- Harper JW (2004) Neddylating the guardian; Mdm2 catalyzed conjugation of Nedd8 to p53. Cell 118: 2–4 [DOI] [PubMed] [Google Scholar]
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ (1993) The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75: 805–816 [DOI] [PubMed] [Google Scholar]
- Hodgson GS, Bradley TR (1979) Properties of haematopoietic stem cells surviving 5-fluorouracil treatment: evidence for a pre-CFU-S cell? Nature 281: 381–382 [DOI] [PubMed] [Google Scholar]
- Itahana K, Bhat KP, Jin A, Itahana Y, Hawke D, Kobayashi R, Zhang Y (2003) Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol Cell 12: 1151–1164 [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, Kieboom K, Marino S, DePinho RA, van Lohuizen M (1999) The oncogene and Polycomb-group gene bmi-1 regulates cell proliferation and senescence through the ink4a locus. Nature 397: 164–168 [DOI] [PubMed] [Google Scholar]
- Kurki S, Peltonen K, Latonen L, Kiviharju TM, Ojala PM, Meek D, Laiho M (2004) Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell 5: 465–475 [DOI] [PubMed] [Google Scholar]
- Leng RP, Lin Y, Ma W, Wu H, Lemmers B, Chung S, Parant JM, Lozano G, Hakem R, Benchimol S (2003) Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 112: 779–791 [DOI] [PubMed] [Google Scholar]
- Lerner C, Harrison DE (1990) 5-Fluorouracil spares hemopoietic stem cells responsible for long-term repopulation. Exp Hematol 18: 114–118 [PubMed] [Google Scholar]
- Lessard J, Sauvageau G (2003) Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 423: 255–260 [DOI] [PubMed] [Google Scholar]
- Look AT (1997) Oncogenic transcription factors in the human acute leukemias. Science 278: 1059–1064 [DOI] [PubMed] [Google Scholar]
- Lyapina S, Cope G, Shevchenko A, Serino G, Tsuge T, Zhou C, Wolf DA, Wei N, Deshaies RJ (2001) Promotion of NEDD–CUL1 conjugate cleavage by COP9 signalosome. Science 292: 1382–1385 [DOI] [PubMed] [Google Scholar]
- Matsumoto N, Yoneda-Kato N, Iguchi T, Kishimoto Y, Kyo T, Sawada H, Tatsumi E, Fukuhara S (2000) Elevated MLF1 expression correlates with malignant progression from myelodysplastic syndrome. Leukemia 14: 1757–1765 [DOI] [PubMed] [Google Scholar]
- Olson MO, Dundr M, Szebeni A (2000) The nucleolus: an old factory with unexpected capabilities. Trends Cell Biol 10: 189–196 [DOI] [PubMed] [Google Scholar]
- Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, Clarke MF (2003) Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 423: 302–305 [DOI] [PubMed] [Google Scholar]
- Peng Z, Serino G, Deng XW (2001) A role of Arabidopsis COP9 signalosome in multifaceted developmental processes revealed by the characterization of its subunit 3. Development 128: 4277–4288 [DOI] [PubMed] [Google Scholar]
- Rabbitts TH (1994) Chromosomal translocations in human cancer. Nature 372: 143–149 [DOI] [PubMed] [Google Scholar]
- Raimondi SC, Dube ID, Valentine MB, Mirro J Jr, Watt HJ, Larson RA, Bitter MA, Le Beau MM, Rowley JD (1989) Clinicopathologic manifestations and breakpoints of the t(3;5) in patients with acute nonlymphocytic leukemia. Leukemia 3: 42–47 [PubMed] [Google Scholar]
- Schwechheimer C, Deng XW (2001) COP9 signalosome revisited: a novel mediator of protein degradation. Trends Cell Biol 11: 420–426 [DOI] [PubMed] [Google Scholar]
- Seeger M, Kraft R, Ferrell K, Bech-Otschir D, Dumdey R, Schade R, Gordon C, Naumann M, Dubiel W (1998) A novel protein complex involved in signal transduction possessing similarities to 26S proteasome subunits. FASEB J 12: 469–478 [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM (1995) Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev 9: 1149–1163 [DOI] [PubMed] [Google Scholar]
- Staub JM, Wei N, Deng XW (1996) Evidence for FUS6 as a component of the nuclear-localized COP9 complex in Arabidopsis. Plant Cell 8: 2047–2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Zhang K, Zhang X, Lei W, Xiao T, Ma J, Guo S, Shao S, Zhang H, Liu Y, Yuan J, Hu Z, Ma Y, Feng X, Hu S, Zhou J, Cheng S, Gao Y (2004) Identification of differentially expressed genes in human lung squamous cell carcinoma using suppression subtractive hybridization. Cancer Lett 212: 83–93 [DOI] [PubMed] [Google Scholar]
- Sun Y, Wilson MP, Majerus PW (2002) Inositol 1,3,4-trisphosphate 5/6-kinase associates with the COP9 signalosome by binding to CSN1. J Biol Chem 277: 45759–45764 [DOI] [PubMed] [Google Scholar]
- Tomoda K, Kato JY, Tatsumi E, Takahashi T, Matsuo Y, Yoneda-Kato N (2005) The Jab1/COP9 signalosome subcomplex is a downstream mediator of Bcr-Abl kinase activity and facilitates cell-cycle progression. Blood 105: 775–783 [DOI] [PubMed] [Google Scholar]
- Tomoda K, Kubota Y, Arata Y, Mori S, Maeda M, Tanaka T, Yoshida M, Yoneda-Kato N, Kato JY (2002) The cytoplasmic shuttling and subsequent degradation of p27Kip1 mediated by Jab1/CSN5 and the COP9 signalosome complex. J Biol Chem 277: 2302–2310 [DOI] [PubMed] [Google Scholar]
- Tomoda K, Kubota Y, Kato J (1999) Degradation of the cyclin-dependent-kinase inhibitor p27Kip1 is instigated by Jab1. Nature 398: 160–165 [DOI] [PubMed] [Google Scholar]
- Tomoda K, Yoneda-Kato N, Fukumoto A, Yamanaka S, Kato JY (2004) Multiple functions of Jab1 are required for early embryonic development and growth potential in mice. J Biol Chem 279: 43013–43018 [DOI] [PubMed] [Google Scholar]
- Uhle S, Medalia O, Waldron R, Dumdey R, Henklein P, Bech-Otschir D, Huang X, Berse M, Sperling J, Schade R, Dubiel W (2003) Protein kinase CK2 and protein kinase D are associated with the COP9 signalosome. EMBO J 22: 1302–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B, Lane D, Levine AJ (2000) Surfing the p53 network. Nature 408: 307–310 [DOI] [PubMed] [Google Scholar]
- Wei N, Chamovitz DA, Deng XW (1994) Arabidopsis COP9 is a component of a novel signaling complex mediating light control of development. Cell 78: 117–124 [DOI] [PubMed] [Google Scholar]
- Wei N, Deng XW (1999) Making sense of the COP9 signalosome. A regulatory protein complex conserved from Arabidopsis to human. Trends Genet 15: 98–103 [DOI] [PubMed] [Google Scholar]
- Wei N, Deng XW (2003) The COP9 signalosome. Annu Rev Cell Dev Biol 19: 261–286 [DOI] [PubMed] [Google Scholar]
- Williams JH, Daly LN, Ingley E, Beaumont JG, Tilbrook PA, Lalonde JP, Stillitano JP, Klinken SP (1999) HLS7, a hemopoietic lineage switch gene homologous to the leukemia-inducing gene MLF1. EMBO J 18: 5559–5566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xirodimas DP, Saville MK, Bourdon JC, Hay RT, Lane DP (2004) Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell 118: 83–97 [DOI] [PubMed] [Google Scholar]
- Yang X, Menon S, Lykke-Andersen K, Tsuge T, Di X, Wang X, Rodriguez-Suarez RJ, Zhang H, Wei N (2002) The COP9 signalosome inhibits p27(kip1) degradation and impedes G1–S phase progression via deneddylation of SCF Cul1. Curr Biol 12: 667–672 [DOI] [PubMed] [Google Scholar]
- Yang Y, Li CC, Weissman AM (2004) Regulating the p53 system through ubiquitination. Oncogene 23: 2096–2106 [DOI] [PubMed] [Google Scholar]
- Yoneda-Kato N, Fukuhara S, Kato J (1999) Apoptosis induced by the myelodysplastic syndrome-associated NPM-MLF1 chimeric protein. Oncogene 18: 3716–3724 [DOI] [PubMed] [Google Scholar]
- Yoneda-Kato N, Look AT, Kirstein MN, Valentine MB, Raimondi SC, Cohen KJ, Carroll AJ, Morris SW (1996) The t(3;5)(q25.1;q34) of myelodysplastic syndrome and acute myeloid leukemia produces a novel fusion gene, NPM-MLF1. Oncogene 12: 265–275 [PubMed] [Google Scholar]
- Yung BY, Busch H, Chan PK (1985) Translocation of nucleolar phosphoprotein B23 (37 kDa/pI 5.1) induced by selective inhibitors of ribosome synthesis. Biochim Biophys Acta 826: 167–173 [DOI] [PubMed] [Google Scholar]