

Abstract
The E7 proteins of human papillomaviruses (HPVs) contribute to oncogenesis by associating with Rb family members as well class I histone deacetylases (HDACs). The binding of HDACs is also important for the maintenance of viral episomes during the differentiation-dependent productive life cycle. The effects of E7 and other viral proteins on E2F family members were examined in differentiating keratinocytes. E7 was found to specifically activate E2F2 transcription in suprabasal keratinocytes through its ability to bind HDACs. Chromatin immunoprecipitation assays demonstrated that, in differentiating cells, E7 acts to inhibit HDAC binding to the E2F2 promoter resulting in activation of expression. Reduction of E2F2 levels through the use of siRNA confirmed that E2F2 expression facilitated HPV replication but its loss did not affect cell proliferation. Our study demonstrates a mechanism by which binding of HDACs to E7 directly modulates viral replication and identifies E2F2 as a possible target for antiviral therapies.
Keywords: E2F2, E7, histone deacetylase, human papillomavirus
Introduction
The high-risk human papillomavirus (HPV) types are the etiological agents of cervical cancer as they are present in over 95% of tumors (Walboomers et al, 1999). The infectious papillomavirus life cycle is dependent on cellular replication machinery and epithelial differentiation. HPVs infect basal epithelial cells presumably through microwounds. Following entry and migration to the nucleus, genomes replicate as episomes largely through the action of cellular enzymes. After replication and division of an infected basal cell, one daughter cell migrates away from the basal layer and undergoes differentiation. The expression of viral genes allows differentiating suprabasal cells to remain active in the cell cycle and to induce DNA synthesis, resulting in amplification of viral genomes to high-copy numbers (Hummel et al, 1992; Cheng et al, 1995). Coincident with amplification, late viral genes are expressed and mature virion particles are assembled.
HPV genomes are approximately 8 kb in size and encode, on average, eight open reading frames. HPV E7 proteins of high-risk types act as oncoproteins, and, together with the E6 oncoprotein, are able to efficiently immortalize human foreskin keratinocytes (HFKs) (Munger et al, 1989; Halbert et al, 1991). E7 proteins contain three conserved regions consisting of a domain at the amino terminus, a central region containing an Rb binding site, and a third domain at the carboxyl terminus, which contains two zinc-finger-like motifs. Previous studies have suggested that the ability of high-risk E7 proteins to bind and mediate the degradation of the Rb family of proteins facilitates immortalization (Ciccolini et al, 1994). The Rb family of proteins control transcription of many genes involved in DNA synthesis as well as cell cycle control by interfering with the function of the E2F transcription factors. Recent studies, however, demonstrate that mutations to the zinc-finger-like region of E7 result in the retention of the ability to bind and degrade Rb but abrogate the ability to immortalize cells, suggesting that there exist other important activities of E7 (Helt and Galloway, 2001).
High-risk E7 proteins have been shown to bind to class I histone deacetylases (HDACs) through sequences in the zinc-finger-like region of the protein (Brehm et al, 1999; Longworth and Laimins, 2004). This binding, which is independent of E7's interaction with Rb, is indirect and is thought to be mediated through an association with the Mi2β protein, a component of the mammalian NuRD chromatin remodeling complex. HDACs act to remove acetyl groups from the amino-terminal tails of the histones present in nucleosomes bound to DNA. Removal of acetyl groups results in the tight binding of histone proteins to DNA, which leads to the formation of heterochromatin and inhibition of transcription factor binding. Class I HDACs, which include HDACs 1, 2, 3, and 8, are found in complexes with other transcriptional corepressors such as mSin3 and SMRT/N-CoR (Hassig et al, 1997; Guenther et al, 2001). HDACs regulate the activity of numerous promoters including those that are E2F dependent. In addition, HDACs can directly deacetylate E2F proteins, although the function of this modification is not well understood (de Ruijter et al, 2003). Previous experiments have shown that mutation of the amino acid L67 in HPV31 E7 abrogates binding to HDACs and results in viral genomes that are unable to be maintained as episomes or to complete the later stages of the viral life cycle (Longworth and Laimins, 2004). However, the mechanism by which viral replication is dependent on the binding of E7 to HDACs has not been described.
The E2F transcription factor family consists of seven members, and, of these E2Fs, 1–5 bind to DP cofactor proteins in order to effect transcription from gene promoters. E2Fs 1–3 are characterized as ‘activator E2Fs,' as their binding to promoters results in increased transcription, while E2Fs 4 and 5 are ‘repressor E2Fs' since they form complexes with p130, HDACs, and other factors to block transcription (Ren et al, 2002). Repressor E2Fs are present on promoters primarily in G0 under conditions that favor quiescence, while activator E2Fs increase transcription from promoters in late G1 prior to S phase (Takahashi et al, 2000). Since HDACs regulate transcription of activator E2F genes, it is possible that E7 binding to HDACs also modulates E2F levels.
In this study, we have examined the expression of E2F family members in HPV-positive cells and found that E2F2 levels were specifically increased in these cells during differentiation. This increased expression was found to be dependent on the ability of E7 to associate with HDACs and prevent their binding to E2F2 promoters. Furthermore, we observed significant decreases in viral DNA replication in HPV-positive cells transfected with siRNAs to E2F2, without any effect on cell proliferation. This study identifies E2F2 as an important cellular target of E7 association with HDACs.
Results
The E7 proteins of HPVs have been shown to play an important role in the productive life cycle by allowing differentiating epithelial cells to remain active in the cell cycle. This allows for differentiating suprabasal cells to re-enter S phase leading to viral DNA amplification and late gene expression. In order to investigate how E7 facilitates these activities, we examined the state of important mediators of S-phase progression, the family of E2F transcription factors, in HPV-positive differentiating keratinocytes.
E2F2 is the only E2F transcription factor that is upregulated in differentiated HPV-positive HFKs
It was first important to determine if the expression of any of the E2F family members was altered by the presence of HPV gene products. For these studies, we used Western blot analyses to determine E2F protein levels in HPV31-positive HFKs and compared those to that seen in untransfected HFKs. We examined levels in undifferentiated cells as well as in cells induced to differentiate by resuspension in methylcellulose for 24 or 48 h. Previous studies have shown that suspension of keratinocytes in methylcellulose for 24 h is sufficient to induce HPV late functions (Ruesch and Laimins, 1998). No significant changes were observed in protein levels for any of the E2F transcription factors in undifferentiated cells; however, following 24 h of differentiation, we observed substantial increases in E2F2 protein levels (Figure 1). This observation was confirmed using a different primary antibody to E2F2 (data not shown). In contrast, only minimal changes in protein levels of the other E2F transcription factors were observed in undifferentiated or differentiated cell lines. The modest difference in E2F4 levels seen in undifferentiated cells was not consistently seen in other experiments. To confirm that these changes were not specific to the methylcellulose system, we also investigated if E7 altered E2F2 levels in keratinocyte raft cultures. In rafts of normal, untransfected HFKs, E2F2 was found to be present primarily in the undifferentiated basal layer cells (Figure 2A) and to be localized in both the cytoplasm and the nucleus (data not shown). In contrast, in HPV31-positive rafts, strong staining for E2F2 protein was detected in differentiated cells in the suprabasal layers (Figure 2B). In these cells, E2F2 protein was observed to localize to intranuclear bodies, as well as to the cytoplasm (Figure 2C).
Figure 1.
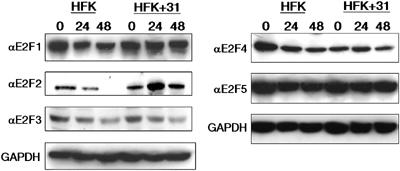
HPV31-positive HFKs exhibit changes in protein levels of E2F2. Western blot analyses were performed on cell lysates from untransfected HFKs (HFK) and HPV31 stably transfected HFKs (HFK+31). Cell lysates were harvested from undifferentiated cells (0 h time point) and cells induced to differentiate by suspension in methylcellulose (24 and 48 h time points). Primary antibodies were used to detect expression of E2F1, E2F2, E2F3, E2F4, and E2F5. Western blots were stripped and blotted with a primary antibody to GAPDH as a control for equal loading.
Figure 2.
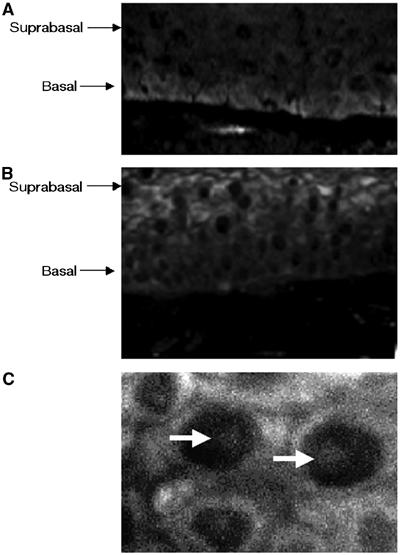
E2F2 localizes to suprabasal, differentiated layers of organotypic rafts of HPV31-positive HFKs. Immunofluorescence analysis with primary antibody to E2F2 was performed on (A) untransfected HFK raft sections and (B) HPV31-positive HFK raft sections. (C) Magnification (× 100) of the suprabasal cells in an HPV31-positive HFK raft section. Arrows depict intranuclear concentrations of E2F2.
The E7 HDAC binding site is necessary for the increase in E2F2 protein and transcript levels seen in HPV31-positive HFKs
We next sought to determine if the increase in E2F2 levels seen in differentiating cells was due to the action of the E7 protein. The L67R mutation in E7 specifically abrogates binding to HDACs, while the ΔLHCYE mutation abrogates binding of the Rb family of proteins. In previous studies, we demonstrated that neither mutation altered the stability of E7 (Longworth and Laimins, 2004). Normal keratinocytes were stably transfected with the wild-type HPV31 genomes, L67R mutant HPV31 genomes, or ΔLHCYE mutant HPV31 genomes and cell extracts were analyzed by Western blot analysis for E2F2 levels. The levels of E2F2 protein in undifferentiated cells transfected with either of the mutant genomes were, on average, found to be similar to that seen in untransfected HFKs. Undifferentiated cells harboring wild-type HPV31 genomes exhibited a small increase in E2F2 protein levels as compared to untransfected HFKs (Figure 3A). Upon differentiation for 24 h in methylcellulose, we observed a 2.5-fold increase in E2F2 protein levels in wild-type HPV31-positive cells as compared to untransfected HFKs (Figure 3B). The E2F2 protein levels in differentiated cells transfected with the HDAC binding mutant genome were decreased from that seen in cells with wild-type genomes. Similar results were seen in three independent experiments. We also observed that cells transfected with the Rb binding mutant genome exhibited E2F2 protein levels that were similar to those seen in both undifferentiated and differentiated, untransfected cells. This suggests that E7's ability to bind HDACs as well as Rb is necessary for the upregulation of E2F2 protein levels in HFKs. As expected, expression of other E2Fs did not change in cells transfected with HDAC or Rb binding mutant genomes (data not shown).
Figure 3.
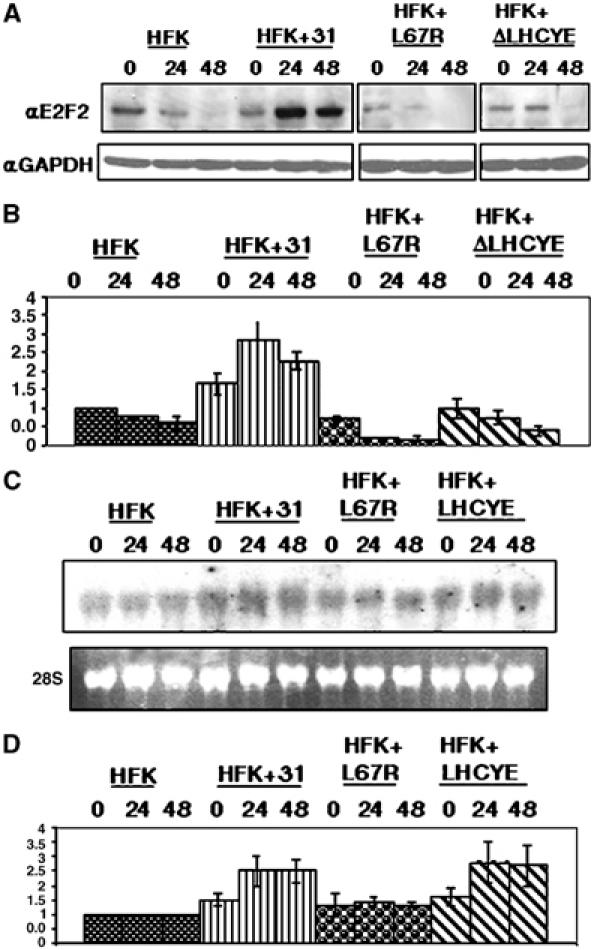
E2F2 transcripts and protein are upregulated in HPV31-positive HFKs and this is dependent on the presence of an intact HDAC binding site in E7. (A) Lysates from untransfected, wild-type, and mutant transfected HFKs either grown as undifferentiated cultures or induced to differentiate for 24 and 48 h in methylcellulose were examined by Western blot analyses using a primary antibody to E2F2. Western blots were stripped and blotted with a primary antibody to GAPDH as a control for equal loading. (B) Protein levels from three separate experiments were quantitated by Fluorchem computer analysis, normalized to levels observed in untransfected cells, and depicted in the bar graph. (C) Northern blot analysis of E2F2 transcription. RNA was isolated from untransfected HFKs, HFKs transfected with wild-type HPV31, and HFKs transfected with either HDAC or RB mutant genomes. Cells were grown either as undifferentiated monolayer cultures or induced to differentiate for 24 and 48 h in methylcellulose. Ethidium bromide staining of 28S ribosomal RNA levels was used to control for equal loading of samples. (D) RNA levels were quantitated by phosphoimager analysis and are depicted in the bar graph normalized to levels observed in untransfected cells. All results are the average of three experiments performed using two different primary cell lines.
It was next important to determine if the increase in E2F2 protein levels in HPV31-positive cells was due to altered transcript levels or protein stability. Northern blot analyses demonstrated increased E2F2 transcript levels in differentiated HPV31-positive cells, compared to untransfected HFKs (Figure 3C). An approximate 2.5-fold increase in transcripts was observed in the wild-type HPV31 transfected cells at 24 h of differentiation, as compared to untransfected HFKs at this time point. This mirrored the protein levels seen at this time point (Figure 3D). E2F2 transcript levels in cells that maintained Rb binding mutant genomes behaved identically to the wild-type transfected cells, while cells containing HDAC binding mutant genomes exhibited E2F2 transcript levels that closely resembled the levels seen in untransfected HFKs (Figure 3D). This indicates that the binding of E7 to HDACs is specifically required for the increase in E2F2 transcript levels in HPV31-positive HFKs. Interestingly, the E2F2 transcript levels in cells containing the Rb binding mutant genome were similar to those seen in wild-type HPV31 transfected cells, suggesting that effects on E2F2 protein levels in these cells were due to post-transcriptional regulatory mechanisms. We observed similar results in three individual experiments in independently derived cell lines. We conclude that E7's association with HDACs as well as Rb can independently modulate E2F2 protein levels.
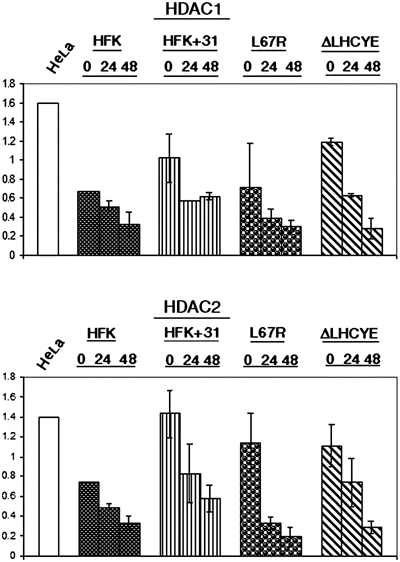
High levels of HDAC proteins are present in differentiated HPV-positive epithelial cells
In our previous studies, we demonstrated an association between HPV31 E7 and HDAC1 as well as HDAC2 (Longworth and Laimins, 2004). Using a GST pulldown assay, we first confirmed the ability of HPV31 E7 to bind to HDAC3 through this residue, as well (data not shown). Efforts to confirm binding between E7 and the fourth member of the class I HDAC family, HDAC8, were unsuccessful due to the lack of a functional antibody. The data described in the previous section suggested that E7's binding to HDACs played a role in specifically regulating the transcription of E2F2 in differentiating cells, and so we next examined if papillomavirus gene products significantly altered HDAC levels in undifferentiated and differentiated HFKs. For these studies, untransfected HFKs and wild-type or mutant HPV31 genome transfected HFKs were resuspended in methylcellulose and Western blots were performed using antibodies to HDAC1, 2, and 3. We did not observe any significant changes in HDAC expression levels for the wild-type or mutant HPV31 transfected cells until 48 h of differentiation (Figure 4). At this time point, HDAC levels were maintained at higher levels than those seen in untransfected cell lines. Interestingly, HDAC protein levels in HDAC binding mutant and Rb binding mutant transfected cells were almost identical to levels seen in wild-type HPV31 transfected cells, suggesting that these functions of E7 are not responsible for the maintenance of HDAC protein levels during differentiation. These experiments were repeated three times using two different sets of independently cloned cell lines derived from transfection of different donor keratinocytes. We conclude that HPV genomes do not significantly alter HDAC protein levels in undifferentiated cells or during early events of differentiation.
Figure 4.
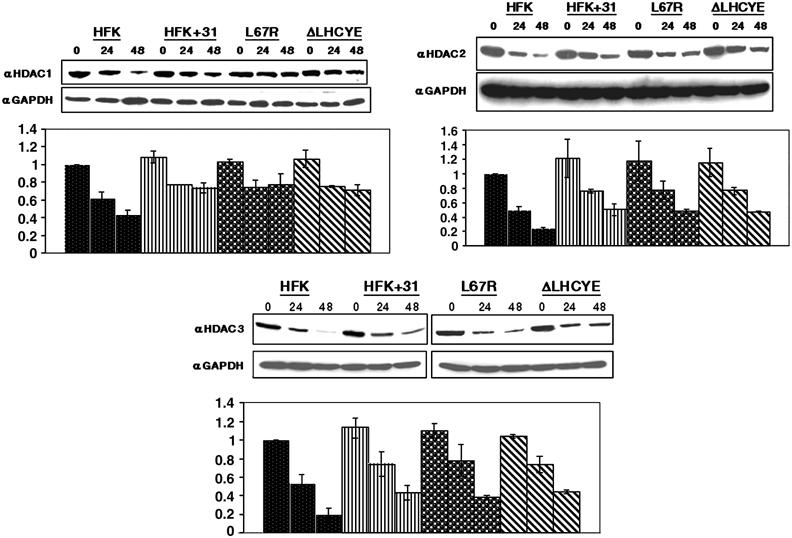
HDAC1, HDAC2, and HDAC3 proteins are maintained in differentiated HPV31-positive HFKs. Western blot analyses using antibodies to HDAC1, HDAC2, and HDAC3 were performed on cell lysates from untransfected HFKs, HFKs transfected with wild-type HPV31, and HFKs transfected with either HDAC or RB mutant genomes. Cell lysates were harvested from undifferentiated monolayer cultures or following differentiation induced by suspension in methylcellulose for 24 and 48 h. Results are representative of three experiments conducted on two separate clonal populations. Protein levels were quantitated by Fluorchem computer analysis and are depicted in bar graphs normalized to levels obtained from untransfected, undifferentiated cells. Western blots were stripped and blotted with a primary antibody to GAPDH as a control for equal loading.
Localization of HDACs does not change in the presence of the viral genome
While E7 has been shown to be a predominantly nuclear protein, it also associates with proteins that shuttle from the nucleus to the cytoplasm (Greenfield et al, 1991). We therefore investigated if binding of E7 to the class I HDACs could act to relocalize the proteins to the cytoplasm. HDAC1 and HDAC2 are known to localize predominantly to the nuclei of cells, while HDAC3 has been reported to localize to both the nucleus and the cytoplasm (de Ruijter et al, 2003). Immunofluorescence analyses were performed on untransfected and HPV31 stably transfected HFKs to determine if there was a difference in localization of the class I HDACs in keratinocyte raft cultures. We observed HDAC1 and HDAC2 staining in the cell nuclei of untransfected keratinocyte raft cultures and their localization did not change in HPV31-positive keratinocyte raft cultures (Figure 5). Furthermore, E7 did not alter the cellular localization of HDAC3 proteins (data not shown).
Figure 5.
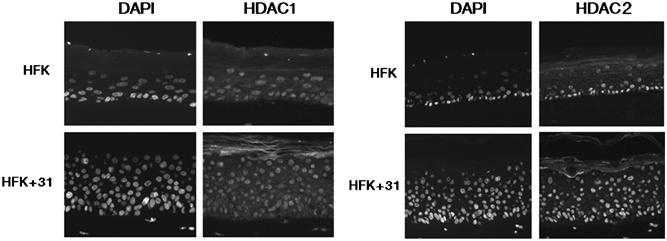
The localization of HDACs in HFKs does not change in the presence of the HPV31 genome in undifferentiated or differentiated cells. Immunofluorescence analysis using primary antibodies to HDAC1 and HDAC2 was performed on cross-sections of raft cultures of untransfected HFKs or HPV31 stably transfected HFKs (HFK+31). Nuclei were visualized using DAPI stain.
HPV31 induces an E7-independent increase in HDAC activity in undifferentiated HFKs
Since HDACs are enzymes that deacetylate histones, it was important to determine if viral genes affected enzyme activity. HDAC activity assays were performed using HDAC1, HDAC2, and HDAC3 immunoprecipitates that were isolated from untransfected HFKs or HFKs that had been stably transfected with wild-type HPV31 genome or the E7-HDAC binding or E7-Rb binding mutant genomes. We observed that HDAC1 and HDAC2 activity levels were moderately higher in undifferentiated HPV31-positive HFKs than in undifferentiated, untransfected HFKs (Figure 6). Mutation of either the HDAC binding site or the Rb binding sites present in the E7 protein did not affect the HPV31-induced increase in HDAC activity. We also observed that HDAC activity decreased with differentiation in all cell lines tested in a manner proportionately similar to that seen in HFKs. HDAC3 activity levels were also measured in untransfected and stably transfected HFKs; however, the signals were minimal and definitive conclusions could not be made (data not shown). We conclude that E7 only moderately alters HDAC activity levels. These data and the data presented in the previous paragraph suggest that E7's effects on the E2F2 promoter are specific to this regulatory region and are not the result of global changes in HDAC protein or activity levels.
Figure 6.
HDAC activity levels are increased in HPV31-positive HFKs. The Fluor de Lys kit (Biomol) was used to measure HDAC activity from HDAC1 and HDAC2 immunoprecipitates taken from untransfected HFKs, HFKs transfected with wild-type HPV31, and HFKs transfected with either HDAC or RB mutant genomes. Activity was measured from cells present in undifferentiated cultures and from cells suspended in methylcellulose for 24 or 48 h. HeLa nuclear extracts were used as a positive control. Results were normalized to the values obtained for the untransfected cell lines and corrected for background levels of activity. The results are the average of three experiments performed in two separate clonal cell populations.
HDAC binding by E7 is necessary for removal of HDACs from the E2F2 promoter
To investigate how the binding of E7 to HDACs contributed to the specific upregulation of E2F2 transcription, chromatin immunoprecipitation (ChIP) assays were performed on the E2F2 promoter. In these studies, untransfected HFKs as well as keratinocytes transfected with wild-type, HDAC binding mutant, or Rb binding mutant HPV31 genomes were examined. Using lysates from cells induced to differentiate in methylcellulose for 24 h, we investigated if p130, Rb, all three class I HDACs, and E2F4 were bound to the E2F2 promoter, as all of these proteins have been previously shown to play a role in the regulation of E2F transcription (Marzio et al, 2000; Takahashi et al, 2000; Ren et al, 2002; Narita et al, 2003). In HPV31-positive cells, no binding of HDAC1 and HDAC3 was observed on the E2F2 promoter (Figure 7A). In contrast, in both untransfected and HDAC binding mutant transfected HFKs, HDAC1 and HDAC3 were detected to be bound to the E2F2 promoter. The lack of HDAC binding correlates with the increase in E2F2 transcription seen in HPV-positive HFKs following 24 h of differentiation (Figure 3B). Screening of cells for the presence of Rb on the E2F2 promoter confirmed its presence on the E2F2 promoter in untransfected keratinocytes as well as cells that maintained HDAC binding or Rb binding mutant genomes after 24 h of differentiation. In contrast, Rb binding was not seen in cells that maintained wild-type HPV31 genomes (Figure 7A). Interestingly, p130 was found to be bound to the E2F2 promoter, regardless of whether wild-type or mutant HPV genomes were present, suggesting that it does not have an effect on E2F2 transcription. A similar spectrum of factors was also found bound to the E2F2 promoter at 48 h of differentiation (data not shown). To determine if E7 had a similar effect on the binding of factors to another E2F-dependent promoter, we next performed ChIP on the cdc6 promoter, but found no difference in HDAC binding to this promoter in any of the cell lines tested (Figure 7B). These data indicate that E7 acts to prevent HDAC binding to the E2F2 promoter in differentiating keratinocytes, which contributes to the upregulation of E2F2 transcription.
Figure 7.
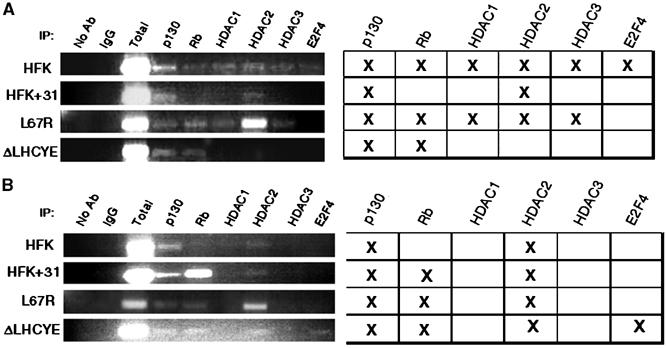
Mutation of the E7 HDAC binding site results in increased HDAC binding to the E2F2 promoter in differentiated HFKs. ChIP assays were performed on untransfected HFKs, HFKs transfected with wild-type HPV31, and HFKs transfected with either L67R or ΔLHCYE mutant genomes following differentiation in methylcellulose for 24 h. PCR was performed on DNA that had been crosslinked and then immunoprecipitated with antibodies to p130, Rb, HDAC1, HDAC2, HDAC3, and E2F4. The PCR primers used amplified a region upstream of the E2F2 promoter TATA box that contains multiple E2F binding sites (A) or to a region upstream of the cdc6 promoter that also contains E2F binding sites (B). No antibody samples and samples immunoprecipitated with an antibody to IgG were used as negative controls, while a total chromatin sample was used as a positive control. PCR experiments were performed three times on each of the different cell lines. The tables are representative of the bands depicted in the gels to the left and similar results were seen in all experiments.
E2F2 is necessary for HPV31 replication
Previously, it has been shown that the HDAC binding site in HPV31 E7 is required for stable maintenance of viral episomes (Longworth and Laimins, 2004). Since our studies identified the E2F2 promoter as a primary target of E7-HDAC activity, we investigated if the high levels of E2F2 seen in our analyses were necessary for viral replication. For technical reasons, we are unable to transfect cells suspended in methylcellulose with siRNAs. As an alternate approach, we took advantage of the observation that when cells that maintain HPV31 episomes are grown to high densities in monolayer cultures, viral genome copy number amplifies in a manner similar to differentiation-dependent viral amplification. In this system, we also observed a similar increase in E2F2 levels as seen in cells differentiated in methylcellulose or in raft cultures that correlated with increased HPV copy number. As shown in Figure 8, HPV31-positive cells transfected with control siRNAs grew to high densities in monolayer cultures, amplified HPV copy number by approximately four-fold at confluency, and increased E2F2 protein levels about five-fold. In contrast, HPV31-positive cells transfected with siRNAs specific to E2F2 exhibited a 60% reduction in HPV copy number at similar cellular density (Figure 8C–E). There was no change in cellular proliferation ability or any significant cell death induced by transfection of these cells with siRNAs to E2F2 (Figure 8E). In these E2F2 siRNA transfected cells, we observed a 50% reduction in E2F2 protein levels compared to that seen in control transfected cells, and this correlated with an inhibition of HPV DNA amplification (Figure 8A and B). Similar results were observed in two additional experiments. We conclude that a reduction in E2F2 levels leads to a significant reduction in HPV genome replication.
Figure 8.
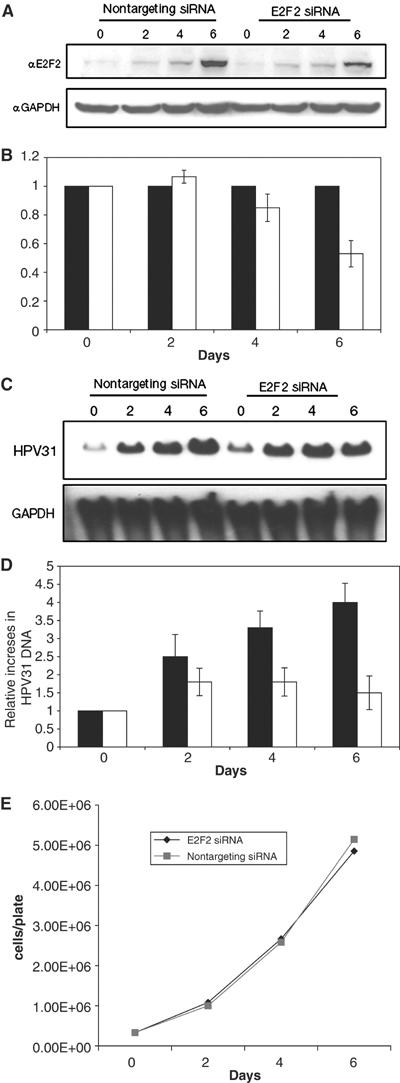
Transfection of E2F2 siRNA inhibits viral DNA replication in HPV31-positive HFKs. HPV31-positive HFKs grown in monolayer culture were transiently transfected with either nontargeting control siRNAs or E2F2 siRNAs (Dharmacon) on days 0 and 3. (A) Western blot analyses using a primary antibody to E2F2 were performed on cell lysates isolated from control siRNA transfected and E2F2 siRNA transfected cells at 2-day intervals. Western blots were stripped and blotted with a primary antibody to GAPDH expression as a measurement of equal loading. (B) E2F2 protein was quantified by Fluorchem computer analysis and levels from nontargeting control siRNA transfected cells were set to 1. E2F2 protein levels from E2F2 siRNA transfected cells were then normalized to those in control transfected cells. Results presented in the bar graph are the average of three different experiments in two separate clonal cell populations. Black bars represent cells transfected with nontargeting control siRNA and white bars represent cells transfected with E2F2 siRNAs. (C) Southern blot analyses using the linearized HPV31 genome as a probe were performed on DNA harvested from HPV31-positive HFKs transfected with nontargeting control siRNAs or E2F2 siRNAs. The DNA was digested prior to Southern analysis in order to study total HPV31 DNA present in cells. (D) Total HPV31 DNA was quantified by phosphoimager analysis and results presented in the bar graph are the average of three different experiments in two separate clonal cell populations. Black bars represent nontargeting control siRNA transfected cells and white bars represent E2F2 siRNA transfected cells. (E) Growth of HPV31-positive HFKs transfected with nontargeting control siRNAs or E2F2 siRNAs. Cells were transfected at day 0 and counted every 2 days. The line graph represents the average of three cell counts for each time point.
Discussion
In this study, we investigated the effects of viral gene products on the expression of E2F family members during the differentiation-dependent HPV life cycle. We found that a major target of E7 activation was the E2F2 gene, and that this activity was important for viral replication. In contrast, neither E7 nor any other viral proteins had any significant effects on other E2F family members. Importantly, the transcriptional activation of E2F2 was found to be mediated through E7's association with HDACs. E7 was found to prevent class I HDAC binding to the E2F2 promoter to facilitate high-level expression in differentiating cells. Our previous studies demonstrated a role for E7 association with HDACs as important for the stable maintenance of viral episomes in cells. The current study suggests a potential mechanism for this activity and identifies additional important activities in differentiated cells resulting from E7's association with HDACs. We cannot exclude the possibility that other factors bind to E7 through similar domains as those required for HDAC association, but our study identifies the interaction with HDACs as being of high importance.
Our studies indicate that the binding of E7 to class I HDACs results in increased E2F2 transcription in differentiating epithelial cells. E2F2 transcripts were significantly increased in HPV31-positive HFKs following 24 h of differentiation in methylcellulose and were specifically dependent on the ability of E7 to bind to HDACs. No significant changes were seen in the expression of other E2F family members including E2F4, which has been shown to repress transcription from the E2F2 promoter (Ren et al, 2002). In addition, we believe that the upregulation of E2F2 levels is not due to nonspecific activation of cell cycle genes by E7, since other E2F family members are not similarly increased in expression. ChIP analyses were also performed on the cdc6 promoter, which directs expression of a protein involved in the loading of the pre-replication complex onto DNA, but found that the E7–HDAC interaction does not play a role in the removal of HDACs from this promoter. This demonstrates that the effects of E7 binding to HDACs do not induce identical effects on all E2F-dependent promoters.
While HDAC activities were modestly increased by HPV gene products in undifferentiated cells, the levels of HDAC proteins were not significantly altered by E7. Upon differentiation of normal HFKs, HDAC protein levels rapidly decline, while in HPV-positive cells, levels are maintained as seen in cells grown in rafts and suspended in methylcellulose. However, this global increase in HDAC levels in differentiated HPV-positive cells cannot explain the effects that we see that are specific to the E2F2 promoter. E7 has also been reported to specifically increase HDAC activity at the interferon-β (IFN-β) promoter in undifferentiated cells. The E7 protein can recruit HDACs to the IFN-β promoter and prevent its transactivation by the interferon regulatory factor-1 (IRF-1) transcription factor (Park et al, 2000). Interestingly, we observed that the binding of E7 to HDACs led to removal of HDACs from the E2F2 promoter at 24 h of differentiation leading to activation of its expression. These observations together with the results of our studies indicate that E7 has the ability to recruit HDACs to a specific set of promoters to induce transcriptional repression and facilitate removal of HDACs from other promoters to increase transcription. This differential regulation is likely to be influenced by the state of cellular differentiation and may be orchestrated by additional transcription factors that are also bound to E7–HDAC complexes, since in vitro and in vivo studies have shown that class I HDACs are unable to regulate transcription on their own (de Ruijter et al, 2003).
One surprising observation from our study was that E7 proteins that retain the ability to associate with HDACs but are unable to bind to Rb act to reduce E2F2 protein levels but not transcription. Cells harboring Rb binding mutant genomes maintained increased E2F2 transcript levels, while cells containing E7 proteins unable to associate with HDACs (E7 L67R) exhibited low levels of E2F2 transcripts. These data are consistent with our ChIP analyses, which demonstrated the presence of HDACs on the E2F2 promoter in cells that maintain L67R genomes. This suggests that the binding of HDACs by E7 is more important for the increase in E2F2 transcript levels since its abrogation directly correlates with downregulation of transcription, while binding of E7 to Rb does not. Similar to our observations, an increase in free p107, another Rb family member, has been shown to alter protein translation and to prolong the transition from G0 to S phase in rat fibroblasts (Makris et al, 2002). Since abrogation of Rb binding by E7 results in an increase in free Rb family members, it is possible that this could lead to a decrease in the translation of proteins involved in the G0–S transition, including E2F2. The post-transcriptional regulation of E2F2 proteins by Rb may be due to a direct effect of binding E7 or an indirect effect due to failure of differentiating cells to remain active in the cell cycle, which has been attributed to the association of E7 with the Rb protein (Dyson et al, 1989; Edmonds and Vousden, 1989).
Our studies indicate that E2F2 plays an important role in viral pathogenesis, as reductions in E2F2 levels were found to reduce HPV replication without modulating cell proliferation. More specifically, transfection of HPV31-positive HFKs with E2F2 siRNA resulted in a >2.5-fold reduction in total viral DNA present in the cells with no effect on proliferation. In our studies using monolayer cultures, the E2F2 protein levels increased at confluence. Transfection of HPV-positive cells with E2F2 siRNAs resulted in significant decreases in E2F2 protein levels at 6 days, when cells were confluent. Our studies also indicate that HPV31 DNA levels increase at confluence and we believe that this induces cells to begin to differentiate and amplify viral DNA. Transfection of cells with E2F2 siRNAs had no effect on cell viability or proliferation rates. The ability to specifically decrease viral DNA replication by transfection of E2F2 siRNA suggests that the targeting of E2F2 in infected cells by antivirals could lead to effective therapeutics.
Studies in E2F2 knockout mice show that while they are viable, they are susceptible to certain types of tumors (Li et al, 2003). This confirms that E2F2 expression is not essential for cell viability. Our studies demonstrate that E2F2 expression is increased approximately 2.5-fold upon differentiation of HPV31 wild-type cells in methylcellulose as compared to differentiated normal keratinocytes. Unlike other E2F family members, E2F2 levels decrease upon differentiation of normal keratinocytes and E7 proteins act to maintain high levels of this protein. This suggests that E2F2 does not play a significant positive role in controlling normal keratinocyte differentiation. One reason why papillomaviruses may specifically activate E2F2 and not the other activator E2Fs could be that E2F1 and E2F3 are both involved in regulation of apoptotic pathways. High-level expression of these two proteins could induce aberrant apoptosis in differentiating cells (Ginsberg, 2004). Recent studies suggest a role for both of these proteins in specific regulation of the Arf promoter and subsequent modulation of cell cycle kinetics (Aslanian et al, 2004). We also observed E2F2 protein to be localized to intranuclear bodies that may coincide with ND10 bodies, which are sites for replication of many DNA tumor viruses, including HPV (Swindle et al, 1999). In support of this idea, E2F2 has been shown to associate with proteins involved in recombinational DNA repair, Mre11 and Nbs1, at both viral and cellular origins of replication (Maser et al, 2001). Alternatively, E2F2 proteins may be directly involved in the transcription of viral DNAs, since degenerate E2F sites can be found in HPV genes. Finally, it is possible that E2F2 may have a novel role in viral pathogenesis that is independent of its action as an ‘activator E2F.' In particular, it may associate with complexes of other proteins to target specific E2F-dependent promoters that are not activated by other E2F family members.
To our knowledge, no studies to date have described specific and essential roles for the human E2F2 protein in pathogenesis. Our studies have shown E2F2 to be predominantly located in the suprabasal layers of HPV31-positive raft cultures and we suspect that E2F2 is also a primary regulator of viral amplification, since amplification occurs in the differentiating layers of the epithelium. Our siRNA experiments indicated that E2F2 plays an important role in regulating HPV31 amplification. While our siRNA studies were performed in monolayer cultures, we are confident that reduction in E2F2 levels would similarly impact viral amplification in methylcellulose-treated cells or cells grown in organotypic rafts. Our studies have focused on the HPV high-risk type 31, but similar HDAC binding sites are also present in HPV16 and 18, and we suspect in low-risk HPV E7 proteins, as well (Brehm et al, 1999). This suggests that activation of E2F2 by E7 in differentiating epithelia may be a common mechanism necessary for replication of many papillomavirus types.
Materials and methods
Cell culture
HFKs were isolated from neonatal human foreskin epithelium and maintained in E-medium as described previously (Fehrmann and Laimins, 2004). Stable transfections of HFKs, drug selection, and expansion for further analyses including resuspension in 1.5% methylcellulose were performed as previously described (Longworth and Laimins, 2004).
Plasmids
The pBR322min plasmid contains the complete HPV31 genome, and all E7 mutations were generated in this plasmid using the Quikchange-XL site-directed mutagenesis kit (Stratagene, La Jolla, CA). The presence of mutations was confirmed by sequencing the entire HPV genome. The mutant genome plasmids were previously characterized by Longworth and Laimins (2004) and are as follows: pΔLHCYE contains an in-frame deletion of the Rb binding domain and pL67R contains a point mutation to the HDAC binding site, which converts a leucine to an arginine.
Immunoblot analysis
Whole-cell extracts were prepared with NP-40 lysis buffer and immunoblotting was performed using 100 μg of protein per sample, as previously described (Longworth and Laimins, 2004). Primary antibodies used included αHDAC1, αHDAC3, αRb-4H1 (Cell Signaling Technologies, Beverly, MA), αHDAC-2 (Calbiochem, San Diego, CA), αGAPDH (Abcam, Cambridge, MA), and αE2F1, αE2F2, αE2F3, αE2F4, αE2F5 (Santa Cruz Biotechnology, Santa Cruz, CA). Secondary antibodies used included horseradish peroxidase (HRP)-linked α-rabbit (Cell Signaling Technologies) and HRP-linked α-mouse (Amersham, Piscataway, NJ). Protein bands were quantitated using the Fluorchem computer program and a multi-image light cabinet (Alpha Innotech Corporation, San Leandro, CA).
Immunohistochemistry
Sections from normal keratinocyte raft cultures or wild-type HPV31 transfected keratinocyte raft cultures were examined by immunofluorescence as described previously (Fehrmann et al, 2003). Primary antibodies used are described above and dilutions used were as follows: HDAC1 at 1:500, HDAC2 at 1:50, HDAC3 at 1:50, and E2F2 at 1:50.
Northern blot analysis
Total RNA was isolated from cells that were resuspended into 1.5% methylcellulose by using Trizol reagent (Invitrogen, Carlsbad, CA) as instructed by the manufacturer, and examined by Northern analyses as previously described (Fehrmann et al, 2003). Equal loading of RNA was monitored through quantitation by spectrophotometry and through quantitation of 28S ribosomal RNA.
Histone deacetylase activity assays
Stable cell lines were grown to 80% confluency and either passaged or resuspended in 1.5% methylcellulose for 24 or 48 h. Protein was harvested from these cells as described by Longworth and Laimins (2004). A 1 mg portion of total cellular protein was suspended in 500 μl of NP-40 lysis buffer and precleared with 50 μl of protein A agarose bead slurry (Roche Diagnostics, Mannheim, Germany) for 5 h at 4°C on a rotating platform. Samples were then centrifuged for 20 s at 12 000 g to precipitate beads and the supernatant was transferred to a fresh eppendorf tube. A 10 μg portion of antibody was added to each precleared sample and tubes were incubated overnight at 4°C on a rotating platform. Antibody-bound beads were washed twice in 1 ml of wash buffer #1 (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1 Complete Mini protease tablet (Roche Diagnostics)), twice with 1 ml of wash buffer #2 (50 mM Tris–HCl, pH 7.5, 500 mM NaCl, 0.1% NP-40, 0.05% sodium deoxycholate) and once with 1 ml of buffer #3 (10 mM Tris–HCl, pH 7.5, 0.1% NP-40, 0.05% sodium deoxycholate) at 4°C for 20 min per wash. The beads were then resuspended in 25 μl of cold assay buffer (BIOMOL International, Plymouth Meeting, PA), and protein was quantified against a BSA curve and a control of protein A agarose bead slurry. A 10 μg portion of immunoprecipitated HDAC proteins was used with the Fluor de Lys kit (BIOMOL International) for the assay, which was performed as per the manufacturer's protocol.
Chromatin immunoprecipitation assays
HFKs were treated with Versene as previously described (Fehrmann and Laimins, 2004), trypsinized, and counted using a hemacytometer. A total of 4 × 106 cells were resuspended in 1.5% methylcellulose for 24 h. Methylcellulose was removed by washes with PBS as previously described (Fehrmann and Laimins, 2004) and resuspended in 270 μl of 37% formaldehyde. Samples were incubated for 10 min at 37°C and were then washed with PBS, centrifuged at 100 g for 4 min at room temperature, and resuspended in 500 μl of SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris–HCl, pH 8.1, 1 Complete Mini protease tablet (Roche Diagnostics)). Using a hand-held sonicator, samples were sonicated on ice for 15, 5 s pulses, each with a 3 s interval in between. Samples were centrifuged at 15 700 g at 4°C for 10 min and the supernatant was collected and added to 4050 μl of cold ChIP dilution buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris–HCl, pH 8.1, 167 mM NaCl, 1 Complete Mini protease tablet). A measure of 250 μl was removed and stored at 4°C for use as the total chromatin sample. The remaining cell extract/ChIP buffer solution was divided into 1 ml aliquots in eppendorf tubes and the assay was completed according to the manufacturer's protocol (Upstate, Lake Placid, NY). DNA was harvested through phenol–chloroform extraction as described by Longworth and Laimins (2004) and was resuspended in 50 μl of deionized water. PCR was performed on the cells using Taq DNA polymerase (New England Biolabs, Beverly, MA) and primers for the regions from −186 to −161 and −72 to −47 on the E2F2 promoter and for the regions from −84 to −62 and +76 to +103 on the cdc6 promoter. Antibodies used included HDAC1, HDAC3, Rb-4H1 (Cell Signaling Technologies), HDAC2 (Calbiochem), p130, and E2F4 (Santa Cruz Biotechnologies).
siRNA transfection of HPV31-positive HFKs
E2F2 smartpool siRNA and siControl nontargeting smartpool siRNA (Dharmacon, Lafayette, CO) were each resuspended in 1 × siRNA buffer (Dharmacon) and transfected onto stably transfected HPV31-positive HFKs on day 0, using 25 μl of Fugene added to 75 μl of serum-free DMEM (Invitrogen). The final concentration of transfected siRNA was 50 nM. Protein was extracted using NP-40 and DNA was harvested at 2-day intervals, through day 6. Cells were retransfected with E2F2 or nontargeting siRNA on day 3. Southern analysis and the stable replication assay were performed as described by Longworth and Laimins (2004). A probe to GAPDH DNA was used as a loading control. The total number of cells per plate was counted at 2-day intervals using a hemacytometer.
Acknowledgments
We thank the members of the Laimins laboratory for advice and comments on protocols. MSL was supported by National Cancer Institute Carcinogenesis Training grant no. T32CA09560. This work was supported by a Sexually Transmitted Diseases Cooperative Research Center grant funded through the National Institute of Allergy and Infectious Diseases.
References
- Aslanian A, Iaquinta PJ, Verona R, Lees JA (2004) Repression of the Arf tumor suppressor by E2F3 is required for normal cell cycle kinetics. Genes Dev 18: 1413–1422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm A, Nielsen SJ, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T (1999) The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J 18: 2449–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S, Schmidt-Grimminger D, Murant T, Broker T, Chow L (1995) Differentiation-dependent up-regulation of the human papillomavirus E7 gene reactivates cellular DNA replication in suprabasal differentiated keratinocytes. Genes Dev 9: 2335–2349 [DOI] [PubMed] [Google Scholar]
- Ciccolini F, Di Pasquale G, Carlotti F, Crawford L, Tommasino M (1994) Functional studies of E7 proteins from different HPV types. Oncogene 9: 2633–2638 [PubMed] [Google Scholar]
- de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB (2003) Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J 370: 737–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson N, Howley PM, Munger K, Harlow E (1989) The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 243: 934–937 [DOI] [PubMed] [Google Scholar]
- Edmonds C, Vousden KH (1989) A point mutational analysis of human papillomavirus type 16 E7 protein. J Virol 63: 2650–2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrmann F, Klumpp DJ, Laimins LA (2003) Human papillomavirus type 31 E5 protein supports cell cycle progression and activates late viral functions upon epithelial differentiation. J Virol 77: 2819–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrmann F, Laimins LA (2004) Human papillomavirus type 31 life cycle: methods for study using tissue culture models. Methods Mol Biol 292: 317–330 [DOI] [PubMed] [Google Scholar]
- Ginsberg D (2004) E2F3—a novel repressor of the ARF/p53 pathway. Dev Cell 6: 742–743 [DOI] [PubMed] [Google Scholar]
- Greenfield I, Nickerson J, Penman S, Stanley M (1991) Human papillomavirus 16 E7 protein is associated with the nuclear matrix. Proc Natl Acad Sci USA 88: 11217–11221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Barak O, Lazar MA (2001) The SMRT and N-CoR corepressors are activating cofactors for histone deacetylase 3. Mol Cell Biol 21: 6091–6101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert CL, Demers GW, Galloway DA (1991) The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol 65: 473–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassig CA, Fleischer TC, Billin AN, Schreiber SL, Ayer DE (1997) Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell 89: 341–347 [DOI] [PubMed] [Google Scholar]
- Helt AM, Galloway DA (2001) Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J Virol 75: 6737–6747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel M, Hudson JB, Laimins LA (1992) Differentiation-induced and constitutive transcription of human papillomavirus type 31b in cell lines containing viral episomes. J Virol 66: 6070–6080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FX, Zhu JW, Tessem JS, Beilke J, Varella-Garcia M, Jensen J, Hogan CJ, DeGregori J (2003) The development of diabetes in E2f1/E2f2 mutant mice reveals important roles for bone marrow-derived cells in preventing islet cell loss. Proc Natl Acad Sci USA 100: 12935–12940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longworth MS, Laimins LA (2004) The binding of histone deacetylases and the integrity of zinc finger-like motifs of the E7 protein are essential for the life cycle of human papillomavirus type 31. J Virol 78: 3533–3541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makris C, Voisin L, Giasson E, Tudan C, Kaplan DR, Meloche S (2002) The Rb-family protein p107 inhibits translation by a PDK1-dependent mechanism. Oncogene 21: 7891–7896 [DOI] [PubMed] [Google Scholar]
- Marzio G, Wagener C, Gutierrez MI, Cartwright P, Helin K, Giacca M (2000) E2F family members are differentially regulated by reversible acetylation. J Biol Chem 275: 10887–10892 [DOI] [PubMed] [Google Scholar]
- Maser RS, Mirzoeva OK, Wells J, Olivares H, Williams BR, Zinkel RA, Farnham PJ, Petrini JH (2001) Mre11 complex and DNA replication: linkage to E2F and sites of DNA synthesis. Mol Cell Biol 21: 6006–6016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger K, Phelps WC, Bubb V, Howley PM, Schlegel R (1989) The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol 63: 4417–4421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113: 703–716 [DOI] [PubMed] [Google Scholar]
- Park JS, Kim EJ, Kwon HJ, Hwang ES, Namkoong SE, Um SJ (2000) Inactivation of interferon regulatory factor-1 tumor suppressor protein by HPV E7 oncoprotein. Implication for the E7-mediated immune evasion mechanism in cervical carcinogenesis. J Biol Chem 275: 6764–6769 [DOI] [PubMed] [Google Scholar]
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD (2002) E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev 16: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruesch MN, Laimins LA (1998) Human papillomavirus oncoproteins alter differentiation-dependent cell cycle exit on suspension in semisolid medium. Virology 250: 19–29 [DOI] [PubMed] [Google Scholar]
- Swindle CS, Zou N, Van Tine BA, Shaw GM, Engler JA, Chow LT (1999) Human papillomavirus DNA replication compartments in a transient DNA replication system. J Virol 73: 1001–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Rayman JB, Dynlacht BD (2000) Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev 14: 804–816 [PMC free article] [PubMed] [Google Scholar]
- Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, Shah KV, Snijders PJ, Peto J, Meijer CJ, Munoz N (1999) Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol 189: 12–19 [DOI] [PubMed] [Google Scholar]