

Abstract
Recent studies indicate that the LKB1 tumour suppressor protein kinase is the major ‘upstream' activator of the energy sensor AMP-activated protein kinase (AMPK). We have used mice in which LKB1 is expressed at only ∼10% of the normal levels in muscle and most other tissues, or that lack LKB1 entirely in skeletal muscle. Muscle expressing only 10% of the normal level of LKB1 had significantly reduced phosphorylation and activation of AMPKα2. In LKB1-lacking muscle, the basal activity of the AMPKα2 isoform was greatly reduced and was not increased by the AMP-mimetic agent, 5-aminoimidazole-4-carboxamide riboside (AICAR), by the antidiabetic drug phenformin, or by muscle contraction. Moreover, phosphorylation of acetyl CoA carboxylase-2, a downstream target of AMPK, was profoundly reduced. Glucose uptake stimulated by AICAR or muscle contraction, but not by insulin, was inhibited in the absence of LKB1. Contraction increased the AMP:ATP ratio to a greater extent in LKB1-deficient muscles than in LKB1-expressing muscles. These studies establish the importance of LKB1 in regulating AMPK activity and cellular energy levels in response to contraction and phenformin.
Keywords: AMP-activated protein kinase, glucose transport, LKB1, phenformin, skeletal muscle
Introduction
The AMP-activated protein kinase (AMPK) is a major sensor of cellular energy levels and is activated by increased levels of 5′-AMP resulting from reduced energy availability (reviewed in Carling, 2004; Hardie, 2004). AMPK is a heterotrimeric complex formed from a catalytic α subunit and two regulatory subunits termed AMPKβ and AMPKγ. AMP activates the AMPK complex by binding to two sites formed by the CBS motifs located on the γ subunit (Scott et al, 2004). Once activated, AMPK phosphorylates substrates that inhibit anabolic processes and promote catabolic processes to restore cellular energy levels. One of the most studied physiological events that activates AMPK is exercise/contraction in skeletal muscle, where AMPK has been proposed to play a key role in stimulation of glucose uptake. Thus, treatment of muscle with an AMPK activator, 5-aminoimidazole-4-carboxamide riboside (AICAR), which is converted to an AMP mimetic within the cell, stimulated glucose uptake (Merrill et al, 1997; Hayashi et al, 1998). Moreover, overexpression of a constitutively active AMPK mutant in muscle cell lines increased glucose uptake (Fryer et al, 2002), while overexpression of a dominant-negative mutant of AMPK in mouse skeletal muscle blocked AICAR-induced glucose uptake (Mu et al, 2001). However, glucose uptake induced by contraction was only inhibited partially in mouse muscle expressing dominant-negative AMPK (Mu et al, 2001), suggesting that AMPK-independent pathway(s) may regulate glucose transport in contracting muscle. AMPK is also activated by metformin, the most widely utilised drug for reducing blood glucose levels in Type II diabetics (Zhou et al, 2001). The mechanism by which metformin, or its closely related analogue phenformin, activates AMPK has not yet been fully established, but may involve inhibition of ATP production via effects on complex I of the mitochondrial respiratory chain.
Activation of AMPK by AICAR, metformin and contraction requires phosphorylation of a threonine residue in the T-loop of the AMPKα subunit kinase domain (corresponding to Thr172 in both the AMPKα1 and AMPKα2 isoforms), by upstream kinase(s) (Carling, 2004; Hardie, 2004). Work performed initially in Saccharomyces cerevisiae (Hong et al, 2003; Nath et al, 2003; Sutherland et al, 2003), and subsequently in mammalian cells (Hawley et al, 2003; Woods et al, 2003a; Shaw et al, 2004), demonstrated that the LKB1 protein kinase can mediate Thr172 phosphorylation of AMPK both in vitro and in intact cells. LKB1 is a 50 kDa serine/threonine kinase that was originally identified as the product of the gene mutated in the autosomal dominantly-inherited Peutz–Jeghers cancer syndrome (Hemminki et al, 1998; Jenne et al, 1998). Like AMPK, LKB1 forms a heterotrimeric complex, in this case with regulatory proteins termed STRAD and MO25, which are required for its activation and cytosolic localisation (Baas et al, 2003; Boudeau et al, 2003, 2004). The LKB1 complex is not itself stimulated by AMP, and is constitutively active in cell lines (Woods et al, 2003a; Lizcano et al, 2004), as well as in skeletal muscle (Sakamoto et al, 2004). In vitro studies have suggested that binding of AMP to AMPK is likely to be the principle regulatory mechanism stimulating phosphorylation of AMPK by LKB1 (Hawley et al, 2003). The LKB1 complex also phosphorylates and activates a number of other protein kinases related to AMPK, whose functions are poorly understood (Lizcano et al, 2004; Jaleel et al, 2005). However, unlike AMPK, the other AMPK-related kinases phosphorylated by LKB1 are not activated by AICAR, phenformin or muscle contraction (Lizcano et al, 2004; Sakamoto et al, 2004).
Recent studies have suggested that activation of AMPK might be mediated independently of LKB1 in IGF1-stimulated fibroblasts and HeLa cells (Suzuki et al, 2004) or in cardiac muscle in response to ischaemia (Altarejos et al, 2005; Baron et al, 2005). Furthermore, in LKB1-deficient HeLa cells or mouse embryo fibroblasts, although AMPK is not activated by AICAR or other stimuli tested, it did possess significant basal activity and phosphorylation at Thr172 (Hawley et al, 2003; Shaw et al, 2004). This suggested that AMPK could be phosphorylated at Thr172 in vivo independently of LKB1. In this study, we wished to address the role that LKB1 plays in activating AMPK in a mammalian tissue in vivo, rather than in cultured cells in vitro. We have generated mice either lacking LKB1 in muscle, or in which the expression of LKB1 is reduced 10-fold, to define the role that LKB1 plays in regulating AMPK activity and glucose uptake in contracting skeletal muscle.
Results
Strategy employed to generate muscle-specific LKB1 knockout mice
We have generated mice conditional for expression of LKB1 and the structure of the floxed (fl) allele is shown in Figure 1A. Exons 5–7 of the LKB1 gene were replaced by a cDNA cassette encoding the remainder of the LKB1 sequence, and exons 4 of the LKB1 gene encoding residues 156–203 and the cDNA cassette are flanked by the loxP Cre excision sequence. This strategy was employed to ensure that in the LKB1fl/fl mice, full-length wild-type LKB1 would be expressed after exon 4 from the LKB1 cDNA cassette. The LKB1fl/fl mice were crossed with transgenic mice expressing the Cre recombinase under the muscle creatine kinase promoter, which induces expression of the Cre recombinase specifically in skeletal muscle and heart, just prior to birth (Bruning et al, 1998). In the resulting LKB1fl/flCre+/− mice, exon 4 as well as the LKB1 cDNA cassette would be specifically excised in skeletal and cardiac muscle, ablating functional LKB1 expression, but potentially resulting in expression of an N-terminal fragment of LKB1 encompassing exons 1–3 (encoding residues 1–155).
Figure 1.
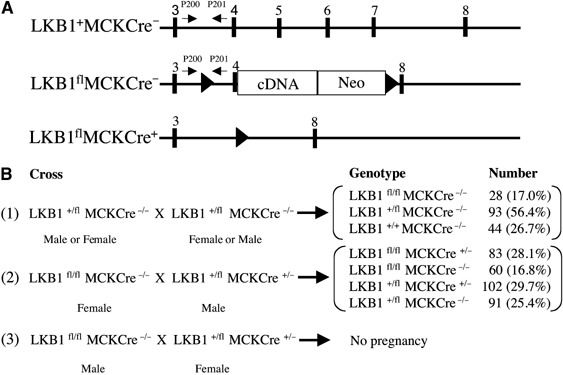
Generation of LKB1-deficient mice. (A) Diagram illustrating the positions of exons 3–8 (▪) in the wild-type LKB1+Cre− allele. In the LKB1flCre− allele, exon 4 of the LKB1 gene is flanked with loxP Cre recombinase excision sites (▴), and exons 5–7 encoding the catalytic domain of LKB1 are replaced with a cDNA construct encoding the remainder of the LKB1 sequence, as well as a neomycin (Neo) selection gene. The expression of the neomycin gene is driven by the LKB1 promoter and it is made as fusion mRNA with LKB1. Its translation is directed by an internal ribosome entry site. In the LKB1flCre+ allele, exons 4–7 of the LKB1 gene are deleted through action of the Cre recombinase, thereby ablating functional expression of LKB1. The positions of the PCR primers used to genotype the mice are indicated with arrows. (B) Breeding strategy employed to generate LKB1fl/fl and LKB1-muscle-deficient (LKB1fl/flCre+/−) mice, where MCKCre denotes transgenic mice expressing the Cre recombinase under the muscle creatine kinase promoter. The number and percentage of each genotype obtained are indicated.
Hypomorphic phenotype of LKB1fl/fl mice
LKB1fl/fl mice were bred as described in Cross 1 in Figure 1B and we observed that these were born at ∼30% lower than expected Mendelian frequency. Female LKB1fl/fl mice were fertile (Cross 2, Figure 1B) but, unexpectedly, male LKB1fl/fl mice were infertile (Cross 3, Figure 1B). To investigate whether this might result from reduced expression of LKB1, we quantified the level of LKB1 protein and activity in various tissues including testis, from 7–10-week-old LKB1fl/fl animals (Figure 2). This revealed that in testis, as well as skeletal muscle, heart, liver and kidney, LKB1 protein and activity was five- to 10-fold lower in LKB1fl/fl mice compared to littermate LKB1+/+ mice. The heterozygous LKB1+/fl mice expressed two-fold lower levels of LKB1 compared to LKB1+/+ mice. In testis, in addition to the 50 kDa protein corresponding to full-length LKB1, a major faster migrating ∼48 kDa species was also recognised by the LKB1 antibody in an immunoblot analysis (Figure 2F). Interestingly, the faster migrating species was not detected in the LKB1fl/fl mice, while the 50 kDa species was reduced ∼10-fold. In contrast to other tissues, the levels of LKB1 in the brain of LKB1fl/fl mice were only reduced slightly (Figure 2G).
Figure 2.
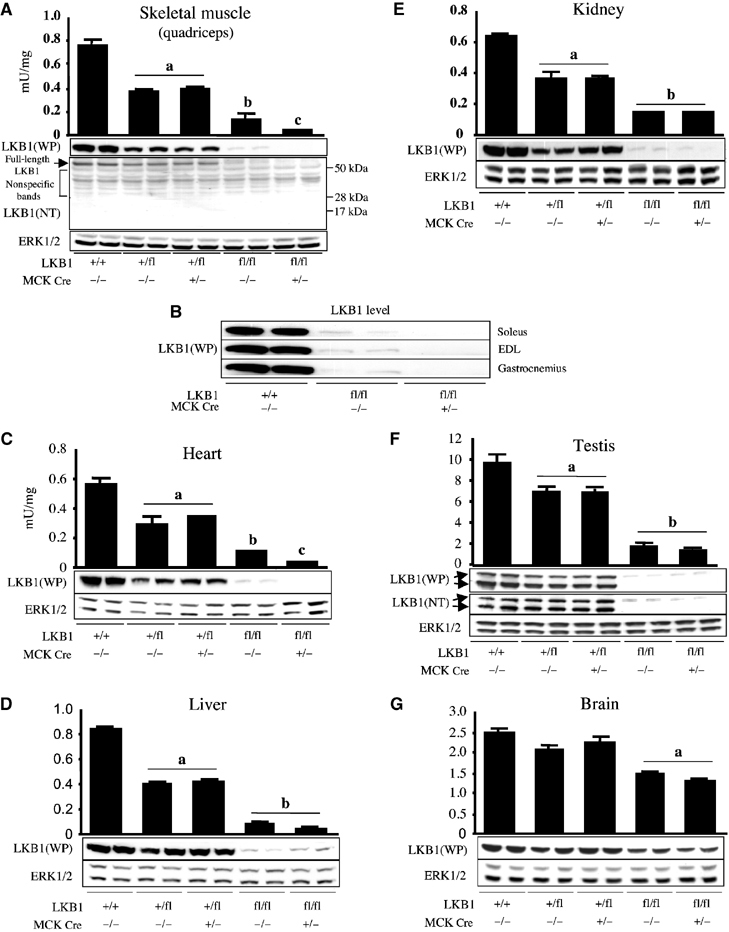
Expression and activity of LKB1 in mouse tissues. (A–G) Extracts of the indicated muscles and other tissues derived from wild-type and mutant mice (7–10 weeks of age) were generated. LKB1 activity was assessed following its immunoprecipitation and assay with the LKBtide peptide. Assays were performed in duplicate from tissues derived from three mice and results presented as the average±s.e.m. The level of LKB1 protein was assessed by immunoblot analysis of 30 μg of protein from tissue extracts using either anti-LKB1 antibody raised against the whole mouse LKB1 protein (WP antibody) or an anti-LKB1 N-terminal antibody raised against the peptide encompassing residues 24–39 of human LKB1 (NT antibody). The ERK1 and ERK2 kinases were immunoblotted as a loading control. The immunoblotting results are representative of independent experiments performed with tissues from two mice. (a) P<0.05 versus LKB1+/+; (b) P<0.05 versus LKB1+/flCre−/−; (c) P<0.05 versus LKB1fl/flCre−/−. Statistical analysis performed using unpaired Student's t test.
LKB1 expression is abolished in the muscle of LKB1fl/flCre+/− mice
We next analysed LKB1 in tissues of the LKB1fl/fl mice expressing the Cre recombinase, generated as described in Cross 2 in Figure 1B. We employed antibodies raised against the full-length LKB1 protein as well as an antibody raised against an N-terminal peptide to enable us to detect any expression of the N-terminal exon 1–3 fragment of LKB1, which would be expected to have a molecular mass of ∼17 kDa. We found that, in the LKB1fl/flCre+/− mice, no LKB1 expression or activity was detected using either antibody in quadriceps muscle (Figure 2A), indicating that Cre ablated expression of full-length LKB1 and that the exon 1–3 fragment of LKB1 is unstable. In three other skeletal muscle types or in cardiac muscle, expression of LKB1 was abolished in LKB1fl/flCre+/− animals (Figure 2B and C). As expected, in nonmuscle tissues of LKB1fl/flCre+/− mice, LKB1 was expressed at levels similar to those found in LKB1fl/flCre−/− littermate mice (Figure 2D–G). The LKB1fl/flCre+/− mice, as well as their hypomorphic littermate LKB1fl/flCre−/− mice, displayed no overt phenotype up to the age of 10 weeks. They possessed normal fasted and fed blood glucose levels, and growth curves from 4 to 10 weeks of age indicated that these animals were of normal size and weight compared to control littermates (data not shown).
LKB1 is required for AICAR-induced AMPKα2 activation
To determine whether lack of LKB1 in skeletal muscle affected AMPK activation, we treated isolated extensor digitorum longus (EDL) muscle isolated from 7–10-week-old mice, in the presence or absence of 2 mM AICAR, which is converted by adenosine kinase into AICAR monophosphate (ZMP), a cellular mimetic of AMP which activates AMPK (Corton et al, 1995). We first measured the activity of the AMPKα2 isoform as well as its phosphorylation at Thr172, the site of LKB1 phosphorylation. In LKB1+/+ and LKB1+/fl EDL muscles, AICAR stimulated AMPKα2 activity ∼2-fold (Figure 3A) and robustly increased phosphorylation at Thr172 (Figure 3B). In contrast, in the EDL muscle of LKB1fl/flCre+/− lacking LKB1 activity, the basal AMPKα2 activity (1.8 mU/mg) was much lower than that observed (∼40 mU/mg) in LKB1-expressing LKB1+/+ or LKB1+/fl muscle, although AMPKα2 was expressed normally. Moreover, in LKB1-deficient muscle, AICAR barely increased AMPKα2 activity (to 2.3 mU/mg) compared to ∼80 mU/mg in LKB1-expressing LKB1+/+ or LKB1+/fl muscle. Consistent with the lack of AMPKα2 activation in LKB1fl/flCre+/− muscle, AICAR also failed to induce detectable phosphorylation of Thr172 (Figure 3B). AMPK activity in vivo was also assessed by analysing the phosphorylation of a downstream target of AMPK, namely the muscle isoform of acetyl-CoA carboxylase-2/β (ACC-2/β) isoform, at the primary site phosphorylated by AMPK (Ser212, equivalent to Ser79 phosphorylated on ACC-1/α; Davies et al, 1990). In LKB1+/+ or LKB1+/fl muscle, significant basal levels of ACC phosphorylation were observed, which were profoundly increased by AICAR (Figure 3B). In contrast, in both unstimulated and AICAR-treated LKB1fl/flCre+/− muscle, phosphorylation of ACC was undetectable.
Figure 3.
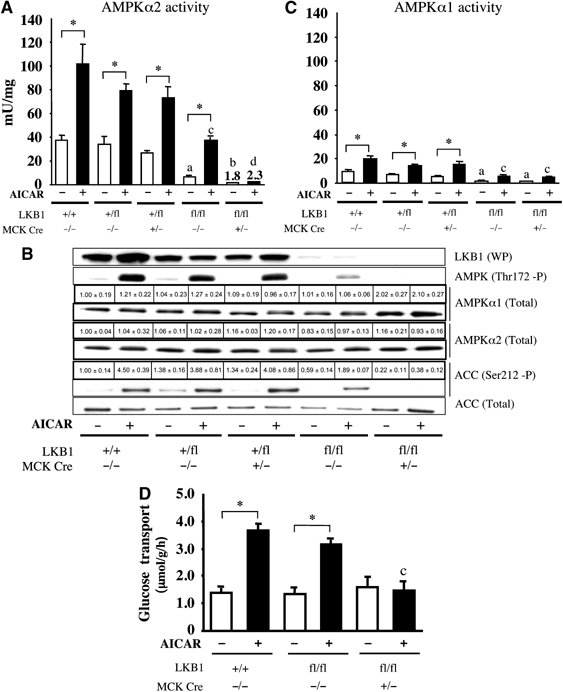
Role of LKB1 in regulating AICAR-induced AMPK activation and glucose transport. (A) Isolated mouse EDL muscles derived from wild-type and mutant mice (7–10 weeks of age) were incubated in the presence (+) or absence (−) of 2 mM AICAR for 60 min. AMPKα2 was immunoprecipitated and assayed with the AMARA peptide. Assays were performed in duplicate from tissues derived from 4–5 mice and results are presented as the average±s.e.m. (B) Equal amounts of protein (20–30 μg) from muscle extracts were immunoblotted with the indicated antibodies. The immunoblotting results are representative of independent experiments performed with tissues from at least three mice. Immunoblot analysis of AMPKα1 and AMPKα2 levels as well as ACC phosphorylation was also assessed by quantitative Li-Cor analysis. The data presented are the mean±s.e.m. Expression relative to expression in LKB1+/+ muscle derived from 3–4 mice. (C) As in (A), except that AMPKα1 was immunoprecipitated and assayed. (D) Glucose transport in isolated EDL muscle derived from the indicated mice was determined without (−) or with (+) 2 mM AICAR, as described in Materials and methods. The data are presented as the mean±s.e.m. for muscle isolated from 4–5 mice for each genotype. *P<0.05 basal versus AICAR within each genotype; (a), P<0.05 versus LKB1+/+ (basal); (b) P<0.05 versus LKB1fl/flCre−/− (basal); (c) P<0.05 versus LKB1+/+ (AICAR); (d) P<0.05 versus LKB1fl/flCre−/− (AICAR). Statistical analysis performed using one-way ANOVA and Tukey's post hoc test.
Further evidence that LKB1 regulated AMPKα2 activity came from the finding that, in the LKB1fl/fl hypomorphic mice, which express 10-fold lower levels of LKB1 in muscle (Figure 2A), the basal and AICAR-stimulated levels of AMPKα2 activity and Thr172 phosphorylation were reduced from four- to two-fold, compared to LKB1+/+ and LKB1+/fl muscle (Figure 3A and B). The phosphorylation of ACC in response to AICAR was reduced by 60% in the hypomorphic mice, as judged by quantitative immunoblot analysis using the Li-Cor Odyssey system (Figure 3B).
We next examined AMPKα1 activity in the EDL muscle of wild-type LKB1+/+ mice and found that its activity was markedly lower than that of AMPKα2 (compare Figure 3A and C), although AICAR still stimulated AMPKα1 activity two-fold. In the hypomorphic LKB1fl/flCre−/− mice, AMPKα1 was expressed normally, but its basal and AICAR-stimulated activities were reduced ∼4-fold compared to LKB1+/+ and LKB1+/fl muscle, suggesting that LKB1 does regulate AMPKα1 activity. In LKB1-deficient LKB1fl/flCre+/− muscle, we noticed that AMPKα1 protein levels were increased ∼2-fold, as judged by quantitative immunoblot analysis using the Li-Cor Odyssey system (Figure 3B). Interestingly however, the basal and stimulated AMPKα1 activities were not further reduced in LKB1-deficient LKB1fl/flCre+/− compared with LKB1fl/flCre−/− muscle, and AMPKα1 was still modestly activated by AICAR. This result might be explained if the AMPKα1 activity was derived from a minor amount of nonmuscle cells in the preparation, which would not express the Cre recombinase.
LKB1 is required for AICAR-induced glucose transport in skeletal muscle
A major physiological role of AMPK in muscle is to stimulate glucose transport (Winder and Hardie, 1999). We therefore measured 2-deoxyglucose uptake in isolated EDL muscle incubated for 60 min in the presence or absence of 2 mM AICAR, followed by 10 min in 3H-2-deoxyglucose. These studies revealed that the basal levels of glucose uptake were similar in LKB1+/+ or hypomorphic LKB1fl/flCre−/− muscle, and that glucose uptake was stimulated two- to three-fold by AICAR (Figure 3D). In contrast, in the LKB1-deficient LKB1fl/flCre+/− muscle, the basal uptake of 3H-2-deoxyglucose was similar to that observed in LKB1-expressing muscle, but AICAR-stimulated glucose uptake was blocked completely (Figure 3D).
LKB1 is required for contraction-induced AMPKα2 activation
One of the major physiological activators of AMPK in skeletal muscle is contraction or exercise (Winder and Hardie, 1999). In order to investigate the role of LKB1 in enabling AMPK to be activated by muscle contraction, in situ contractions of hind limb muscle were induced in one leg via electrical stimulation of the sciatic nerve in anaesthetised mice, while the other leg served as the noncontracted control. Contraction was performed for 5 min, a time point that maximally activates AMPKα2 using the protocol employed (Sakamoto et al, 2004). We measured AMPKα2 activity (Figure 4A), and phosphorylation of AMPK at Thr172 and ACC at Ser212, in mixed tibialis anterior and EDL muscles (Figure 4B). In LKB1+/+ and LKB1+/fl muscle, contraction increased AMPKα2 activity six-fold and also induced marked phosphorylation of AMPK at Thr172 and ACC at Ser212. In contrast, in the LKB1-deficient LKB1fl/flCre+/− muscle, the basal and contraction-induced AMPKα2 activities were reduced ∼50-fold, compared to LKB1+/+ and LKB1+/fl muscles (Figure 4A). Furthermore, in LKB1-deficient muscle, contraction failed to stimulate phosphorylation of AMPK at Thr172, while phosphorylation of ACC was reduced by nearly 90% (Figure 4B).
Figure 4.
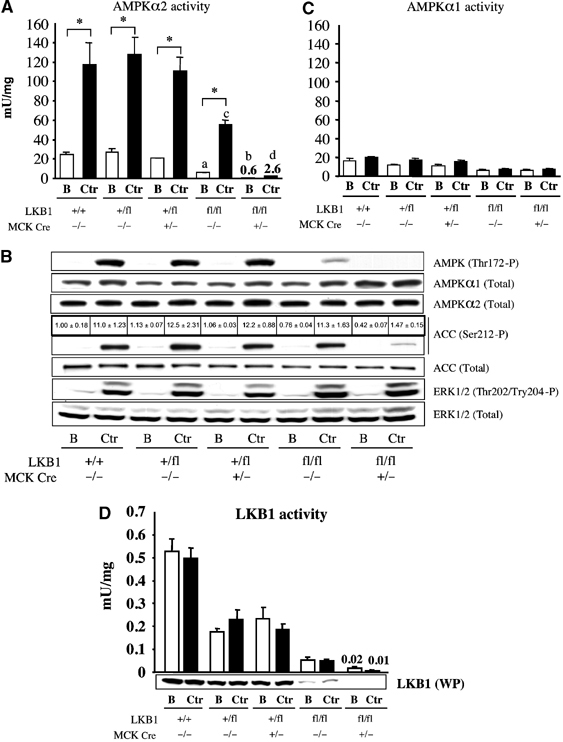
Role of LKB1 in regulating contraction-induced AMPK activation. (A) One leg from anaesthetised mice of the indicated genotype (7–10 weeks of age) was subjected to in situ hindlimb muscle contraction (Ctr, contraction) via sciatic nerve stimulation for 5 min, and the other leg served as noncontracted control (B, basal). Tibialis anterior and EDL muscles from both legs were rapidly extracted and snap frozen in liquid nitrogen. AMPKα2 was immunoprecipitated and assayed with the AMARA peptide. Assays were performed in duplicate from muscles derived from four to five mice and results are presented as the average±s.e.m. (B) Equal amounts of protein (20–30 μg) from the muscle extracts were immunoblotted with the indicated antibodies. Immunoblot analysis of ACC phosphorylation was also assessed by quantitative Li-Cor analysis. The immunoblotting results are representative of independent experiments performed with tissues from at least three mice. (C, D) As in (A), except that AMPKα1 (C) or LKB1 (D) was immunoprecipitated and assayed with AMARA or LKBtide. *P<0.05 basal versus contraction within each genotype; (a) P<0.05 versus LKB1+/+ (basal); (b) P<0.05 versus LKB1fl/flCre−/− (basal) (c) P<0.05 versus LKB1+/+ (ctr); (d) P<0.05 versus LKB1fl/flCre−/− (ctr). Statistical analysis performed using one-way ANOVA and Tukey's post hoc test.
In the hypomorphic LKB1fl/flCre−/− muscle, the basal AMPKα2 activity was 6 mU/mg (compared to 25 mU/mg in LKB1+/+ muscle) and was stimulated during contraction to 55 mU/mg (compared to 115 mU/mg in LKB1+/+ muscle). In the LKB1 hypomorphic mice, contraction-induced phosphorylation of AMPK at Thr172 was reduced significantly, but, in contrast to AICAR treatment (Figure 3B), the phosphorylation of ACC at Ser212 was similar to that observed in LKB1+/+ muscle (Figure 4B).
In contrast to AMPKα2, AMPKα1 activity was not increased significantly by muscle contraction (Figure 4C), consistent with the notion outlined above that AMPKα1 detected in mixed tibialis anterior and EDL muscle may largely be derived from nonmuscle cells. Previous studies have also found that the AMPKα1 is not activated in contracting rat or human muscle under conditions where AMPKα2 is stimulated (Fujii et al, 2000). AMPKα1 activity was ∼3-fold lower in hypomorphic LKB1fl/flCre−/− muscle compared to LKB1+/+ muscle, and its activity was not reduced further in the Cre-expressing muscles (Figure 4C; see also Figure 3C). In the hypomorphic and LKB1 knockout muscles, contraction induced normal T-loop phosphorylation of the ERK1/ERK2 kinases (Figure 4B), which are potently activated by contraction or exercise (Sakamoto and Goodyear, 2002). Consistent with previous reports in rat muscle (Sakamoto et al, 2004), LKB1 activity in mouse muscle was not affected by contraction (Figure 4D).
LKB1 is required for contraction-induced glucose transport in skeletal muscle
Muscle contraction stimulates glucose uptake, but the contribution that AMPK makes to this process relative to other signalling pathways has been controversial (Aschenbach et al, 2004). We therefore measured glucose uptake in EDL muscle using in situ (Figure 5A) as well in vitro (Figure 5B) methodologies to induce muscle contraction. For the in situ approach, contraction was induced through sciatic nerve stimulation for 5 min and then EDL muscles were isolated and glucose transport measured following immersion for 10 min in 3H-2-deoxyglucose. This revealed that, in LKB1+/+ or hypomorphic LKB1fl/flCre−/− muscle, basal levels of glucose transport were similar and contraction stimulated the uptake of 3H-2-deoxyglucose two-fold (Figure 5A). In contrast, in the LKB1-deficient LKB1fl/flCre+/− muscle, the basal uptake of 3H-2-deoxyglucose, although similar to that observed in LKB1-expressing muscle, was marginally stimulated, even though this was not statistically significant (Figure 5A). In the in vitro approach, isolated EDL muscles were induced to contract for 10 min by electrical stimulation and then glucose transport quantified following immersion of the muscle for 10 min in 3H-2-deoxyglucose. This revealed that the 1.7-fold stimulation of glucose uptake induced by contraction in wild-type and hypomorphic muscles was largely blocked in the LKB1-deficient LKB1fl/flCre+/− muscle (Figure 5B). We also monitored muscle force production during in vitro contraction and observed that, in the LKB1-lacking or LKB1-hypomorphic muscles, the force profiles were identical to those of wild-type muscle, indicating that the ability of LKB1-deficient muscles to contract is not impaired (Figure 5C). As a further control to ensure that the glucose transport apparatus was functional in the LKB1-deficient muscle, we also measured glucose transport in the presence or absence of insulin, which stimulates the process through an AMPK-independent pathway. This revealed that insulin stimulated 2-deoxyglucose uptake two-fold in the LKB1-lacking LKB1fl/flCre+/− muscle, similar to that observed in LKB1-expressing muscle (Figure 5D).
Figure 5.
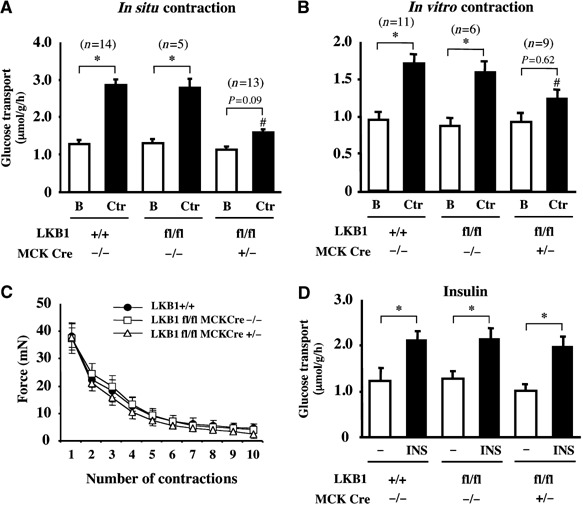
Role of LKB1 in regulating contraction-induced glucose transport. (A) One leg from anaesthetised mice of the indicated genotype (7–10 weeks of age) was subjected to in situ hindlimb muscle contraction (Ctr, contraction) via sciatic nerve stimulation for 5 min, and the other leg served as noncontracted control (B, basal). Following contraction, EDL muscle was isolated and glucose transport measured as described in Materials and methods. (B) Isolated EDL muscles were either subjected to in vitro contraction (Ctr, contraction) via electrical stimulation for 10 min or left unstimulated (B, basal). Glucose transport was then measured as described in Materials and methods. The data in (A) and (B) are presented as the mean±s.e.m. for muscle isolated from the indicated number (n) of mice of each genotype. (C) Force production during contraction protocol. Isolated EDL muscles were subjected to 10 sets of 10-s in vitro contraction and total force generated for each contraction was calculated as described in Supplementary data. The data are presented as the mean±s.e.m. for muscle isolated from three mice for each genotype. (D) Glucose transport activity in isolated EDL muscle from the indicated mice, determined without (−) or with 100 nM insulin (INS) as described in Materials and methods. The data are presented as the mean±s.e.m. for muscle isolated from three mice for each genotype. *P<0.05 basal versus contraction within each genotype; #P<0.05 versus LKB1+/+ (contraction). Statistical analysis performed using one-way ANOVA and Tukey's post hoc test.
LKB1 is required for phenformin-induced AMPKα2 activation
To study whether LKB1 was required for the activation of AMPKα2 by the antidiabetic drug phenformin, we treated isolated EDL muscle in the presence or absence of 2 mM phenformin for 60 min. Phenformin was employed rather than metformin, as this close relative of metformin activates AMPK much more potently in muscle than metformin (KS, unpublished results). Phenformin induced a three-fold stimulation of AMPKα2 in LKB1-expressing muscle (Figure 6A), which was accompanied by a marked increase in phosphorylation of AMPK at Thr172 and ACC at Ser212 (Figure 6B). In the LKB1 hypomorphic muscle, the basal activity of AMPKα2 was reduced, but phenformin still stimulated AMPKα2, albeit to a level lower than observed in wild-type muscle (Figure 6A). By contrast, in LKB1-deficient muscle, phenformin failed to significantly increase the low AMPKα2 activity or the phosphorylation of AMPK at Thr172 and ACC at Ser212. We attempted to measure phenformin-stimulated glucose uptake in EDL muscles. However, under the conditions employed, we were unable to measure any significant increase in glucose uptake in LKB1-expressing muscles (KS, data not shown). To our knowledge, no other group has reported that phenformin can stimulate glucose uptake in isolated muscle.
Figure 6.
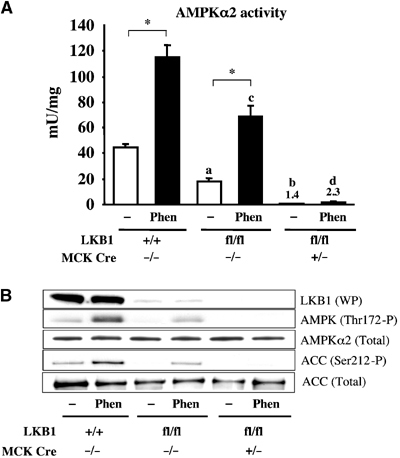
Role of LKB1 in regulating phenformin-induced AMPKα2 activation. (A) Isolated mouse EDL muscles derived from wild-type and mutant mice (7–10 weeks of age) were incubated in the presence (Phen) or absence (−) of 2 mM phenformin for 60 min. AMPKα2 was immunoprecipitated and assayed with the AMARA peptide. Assays were performed in duplicate from tissues derived from four mice and results are presented as the average±s.e.m. (B) Equal amounts of protein (20–30 μg) from the muscle extracts were immunoblotted with the indicated antibodies. The immunoblotting results are representative of independent experiments performed with tissues from at least two mice. *P<0.05 basal versus phenformin within each genotype; (a) P<0.05 versus LKB1+/+ (basal); (b) P<0.05 versus LKB1fl/flCre−/− (basal); (c) P<0.05 versus LKB1+/+ (Phen); (d) P<0.05 versus LKB1fl/flCre−/− (Phen). Statistical analysis performed using one-way ANOVA and Tukey's post hoc test.
Role of LKB1 in controlling cellular energy charge
Activation of AMPK during muscle contraction is thought to regulate cellular energy balance (Hardie et al, 2003). We therefore measured ADP:ATP and AMP:ATP ratios in control and contracted muscles (tibialis anterior and EDL) by quantitative capillary electrophoresis (Figure 7). As AMP can be converted to IMP by AMP deaminase, we also measured IMP:ATP ratios. In unstimulated LKB1-deficient LKB1fl/flCre+/− muscle, the ADP:ATP, AMP:ATP and IMP:ATP ratios were similar to those found in resting LKB1+/+ and LKB1fl/fl muscle (Figure 7). In wild-type LKB1+/+ muscle, contraction only moderately increased the ADP:ATP (Figure 7A) and AMP:ATP (Figure 7B) ratios. However, in LKB1 hypomorphic muscle, contraction increased ADP:ATP and AMP:ATP ratios to statistically significant higher levels than those observed in wild-type muscle. These levels were further increased in the LKB1fl/flCre+/− muscle. In LKB1+/+ muscle, IMP:ATP ratios were similar to those observed in LKB1fl/flCre+/− muscle, in which the basal ratios were very low, but increased ∼100-fold after contraction (Figure 7C). We found that in the LKB1 hypomorphic as well as the LKB1fl/flCre+/− muscles the IMP:ATP ratios were further increased by contraction.
Figure 7.
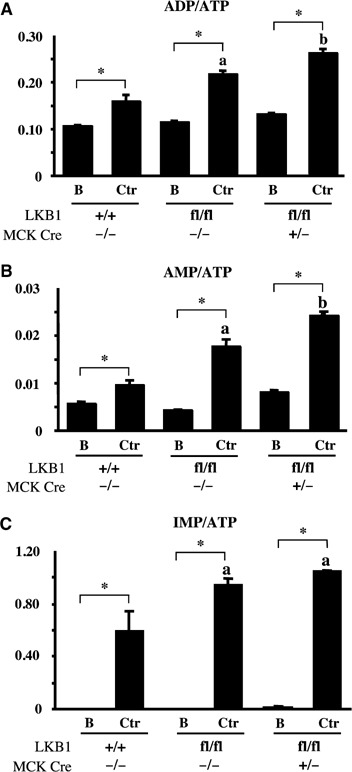
Measurement of ADP:ATP, AMP:ATP and IMP:ATP ratios in contracting muscle. One leg from anaesthetised mice of the indicated genotype (7–10 weeks of age) was subjected to in situ hindlimb muscle contraction (Ctr, contraction) via sciatic nerve stimulation for 5 min, and the other leg served as noncontracted control (B, basal). Following contraction, tibialis anterior and EDL muscle was isolated and nucleotides extracted and analysed by capillary electrophoresis as described in Supplementary data. The ratios of ADP:ATP (A), AMP:ATP (B) and IMP:ATP (C) derived from analysis of three independent muscle samples for each condition were measured. The results are shown as the average±s.e.m. *P<0.05 basal versus contraction within each genotype; (a) P<0.05 versus LKB1+/+ (basal); (b) P<0.05 versus LKB1fl/flCre−/− (ctr). Statistical analysis performed using one-way ANOVA and Tukey's post hoc test.
Discussion
Our results establish that LKB1 is a major regulator of AMPKα2 activity in skeletal muscle, and that lack of LKB1 is not compensated for by other kinases. Our results also support the notion that activation of AMPK plays a major role in maintaining cellular energy levels. This is based on the finding that, in LKB1-deficient muscles in which AMPK is not activated, muscle contraction resulted in an abnormal elevation of the ADP:ATP, AMP:ATP and IMP:ATP ratios (Figure 7). We believe that this is the first genetic evidence directly demonstrating the important role that LKB1 pathway plays in maintaining cellular energy balance in vivo. Moreover, our data clarify the mechanism by which muscle contraction activates AMPK. This is likely to be through elevation of AMP levels via the action of adenylate kinase, subsequent to depletion of ATP. AMP then interacts with the AMPKγ subunit, inducing conformational changes in the AMPK complex that stimulate the rate of phosphorylation of the AMPKα catalytic subunit by the LKB1 complex.
In contrast to the results with AMPKα2, we observed that AICAR (Figure 3) as well as phenformin (data not shown) still induced substantial activation of AMPKα1, in LKB1-deficient muscle. However, the finding that, in LKB1 hypomorphic muscle expressing 10-fold lower levels of LKB1, both the basal and AICAR-stimulated AMPKα1 activity were reduced three- to four-fold (Figure 3) provides genetic evidence that LKB1 does regulate the activity of AMPKα1. A likely reason as to why AMPKα1 activity was not further reduced by expression of Cre recombinase could be that AMPKα1 activity was derived mainly from nonmuscle cell types, such as fibroblasts, endothelial, fat or blood cells, which are known to be present in the muscle tissue and which would not express the Cre recombinase. This could also explain why AMPKα1 is not activated in contracting muscle under conditions where AMPKα2 was stimulated (Figure 4). However, our data do not rule out the possibility that AMPKα1 is regulated by an LKB1-independent mechanism that is not triggered by contraction. LKB1 does activate AMPK complexes containing either AMPKα1 or AMPKα2 in cell-free assays (Hawley et al, 2003). Most studies in LKB1-deficient cell lines have been performed using approaches that immunoprecipitate both AMPKα1 and AMPKα2 activities (Hawley et al, 2003; Woods et al, 2003b; Shaw et al, 2004). However, in one study in which AMPKα1 activity was assayed specifically, AICAR and phenformin were unable to stimulate AMPKα1 activity in LKB1-deficient HeLa cells and mouse embryo fibroblasts (Lizcano et al, 2004), demonstrating that LKB1 can regulate AMPKα1 activity in intact cultured cell lines.
Our finding that male LKB1fl/fl mice are infertile suggests a role for LKB1 in regulating spermatogenesis and/or sperm motility. In testis, a faster migrating 48 kDa species of LKB1 is detected by antibodies raised against an N-terminal peptide of LKB1, which is absent in the LKB1fl/fl mice (Figure 2F). In the LKB1fl/fl mice, LKB1 is expressed after exon 4 from a cDNA fragment, therefore suggesting that the 48 kDa species is formed as a C-terminal splice variant of LKB1. In future work, it will be of interest to explore whether the faster migrating form of LKB1 plays a specific role in testis, and whether AMPK or one of the other AMPK-related kinases activated by LKB1 play a role in spermatogenesis and/or sperm motility. The SNRK kinase that is activated by LKB1 is specifically expressed in testis (Jaleel et al, 2005).
Our data demonstrate that a 10-fold reduction in LKB1 expression in the muscle of LKB1fl/fl mice reduced basal AMPKα2 activity as well as its activation by AICAR and contraction, although by much less than 10-fold. This suggests that, in muscle tissues, the normal level of LKB1 is not rate-limiting and exerts only a small degree of control over the activation of AMPKα2. This may be a common feature of protein kinase signalling networks, as we have previously shown that, in hypomorphic mice that express only 10% of the normal level of PDK1, two of its downstream targets, that is, protein kinase B and ribosomal protein S6 kinase, are activated normally (Lawlor et al, 2002). Our results also illustrate the sensitivity and spare capacity that exists in the LKB1–AMPK signalling pathway. For example, AMPKα2 activation induced by contraction was reduced ∼2-fold in the hypomorphic LKB1fl/fl mice, but this had no significant effect on the downstream actions of AMPK that we have investigated, namely phosphorylation of ACC (Figure 4B) or stimulation of glucose uptake (Figure 5A and B). Thus, only very small increases in AMPK activity may be required to stimulate these downstream processes, and above that level the system becomes saturated. We are unable to rule out the possibility that the LKB1 hypomorphic mice may have adapted in order to compensate for the reduction in expression of LKB1. Humans with Peutz Jeghers syndrome lacking one allele of the LKB1 gene have not been reported to suffer from an inability to exercise or to possess any metabolic disorders. Our findings that AMPK is significantly activated in LKB1 hypomorphic mice may suggest that a single copy of the LKB1 allele in humans with Peutz–Jeghers syndrome is sufficient to normally regulate most processes downstream of AMPK.
In LKB1-deficient muscle, we found that phosphorylation of ACC, although greatly reduced, was not abolished. The residual phosphorylation of ACC that is observed could be mediated by trace residual activation of AMPKα2 in the LKB1-deficient muscle, or by a kinase other than AMPK that is also stimulated by contraction. We also noticed that, in the hypomorphic mice, AICAR-induced, but not contraction-induced, phosphorylation of ACC was inhibited. This may provide further evidence that exercise can stimulate ACC phosphorylation independently of AMPK, and moreover it is also possible that exercise might promote ACC phosphorylation through the inhibition of a protein phosphatase. Interestingly, in LKB1-deficent fibroblasts, AICAR- and hydrogen-peroxide-induced phosphorylation of ACC was reduced, but not abolished (Shaw et al, 2004). Our findings suggest that AMPK is the major kinase phosphorylating the regulatory site (Ser212) on muscle ACC, but indicate that there may be other kinases that phosphorylate this site in vivo.
A complication that arises when generating muscle-specific knockouts using the Cre/loxP system is that it is sometimes difficult to obtain a complete ablation of gene expression throughout a muscle fibre, resulting from muscles being formed from fused myoblasts. Thus, if the Cre recombinase fails to delete the loxP-targeted gene in a small percentage of nuclei within a muscle fibre, this would result (in contrast to other tissues) in the targeted gene being expressed at low levels throughout the fibre. We have previously attempted to ablate PDK1 expression in skeletal muscle using the Cre/loxP system and found that, despite a 95% reduction in PDK1 expression in muscle tissues, the remaining PDK1 that is expressed throughout the muscle fibre is sufficient to normally activate its downstream substrates (Mora et al, 2003). In this study, we may have overcome the inherent problem of creating conditional knockouts in mouse muscle, by ablating LKB1 expression in a hypomorphic background. Had the LKB1fl/fl mice not been hypomorphic for LKB1 expression, it is possible that a 95% reduction in LKB1 expression in the muscle of these animals would have only partially reduced AMPKα2 activation and would not have affected contraction-induced ACC phosphorylation or glucose uptake.
A key finding of this study is that contraction-induced glucose uptake is largely inhibited in LKB1-deficient EDL muscle (Figure 5). This demonstrates that LKB1 is an important regulator of muscle contraction-induced glucose uptake. A previous study overexpressing dominant-negative AMPK from a muscle creatine kinase promoter (Mu et al, 2001) reported only a partial inhibition of glucose uptake, suggesting that AMPK-independent pathway(s) also regulate contraction-induced glucose uptake. Recently, it was also reported that global knockouts of either AMPKα1 or AMPKα2 in mice had no effect on the ability of muscle contraction to induce glucose uptake (Jorgensen et al, 2004). By contrast, AICAR-induced glucose uptake was entirely blocked in the mice expressing the dominant-negative AMPK mutant in muscle (Mu et al, 2001) and in the AMPKα2 knockout mice (Jorgensen et al, 2004). Our results with the LKB1 hypomorphic muscles, in which AMPK activation induced by contraction is considerably reduced, but glucose uptake is normal, indicate that a small activation of AMPK may be sufficient to induce normal glucose uptake. It is therefore possible that, in the muscles expressing the dominant-negative mutant, a small residual activation of endogenous AMPK is sufficient to cause a partial stimulation of glucose uptake. Similarly, in the individual AMPKα2 knockout mice, there is a compensatory increase in the expression of AMPKα1 (Viollet et al, 2003), and this might be sufficient to stimulate glucose uptake in the muscle. Interestingly, we also observed a two-fold increase in AMPKα1 levels in the LKB1-deficient muscle, but presumably this would not be activated due to the lack of LKB1 and thus could not stimulate glucose uptake (Figure 3B). These data suggest that there is a negative feedback pathway by which expression of AMPKα1 protein is upregulated following downregulation of AMPK activity, via an as yet undetermined mechanism. In contrast to the AMPKα2 knockout mice (Viollet et al, 2003), the LKB1-deficient animals are not glucose intolerant. A likely explanation for this difference is that glucose intolerance in the AMPKα2-deficient mice arises from defects in the AMPK kinase signalling pathways in tissues such as the autonomic nervous system, rather than in muscle (Viollet et al, 2003).
Our results do not exclude the possibility that LKB1 may be able to stimulate contraction-induced glucose uptake through an AMPK-independent mechanism. The most likely candidates for downstream targets of LKB1 that might mediate this effect would be the AMPK-related kinases that are also phosphorylated and activated by LKB1 (Lizcano et al, 2004). Although we were previously unable to demonstrate that any of these enzymes that were detectable in skeletal muscle (QSK, QIK, MARK2, MARK3 and MARK4) were stimulated by muscle contraction, AICAR or phenformin (Sakamoto et al, 2004), we cannot rule out the possibility that these enzymes might play a permissive role in enabling contraction to induce glucose uptake.
In conclusion, we provide here the first genetic evidence of the crucial role that LKB1 plays in regulating AMPK activation in muscle. Our data suggest that contraction/exercise-induced activation of AMPK and glucose uptake requires LKB1. We also demonstrate for the first time that contraction in muscles lacking AMPK activity results in abnormal increases in cellular AMP:ATP ratio, supporting the notion that LKB1 network functions as a sensor and regulator of cellular energy charge that protects muscle cells against the metabolic stress of contraction.
Materials and methods
Materials and details of antibodies and procedures used to prepare tissue lysates, perform immunoblotting, assay of LKB1/AMPK as well as glucose transport, contraction force, nucleotide measurements and statistics employed are described in Supplementary data.
Mice breeding and genotype analysis
All animal studies and breeding were approved by the University of Dundee ethical committee and performed under a UK Home Office project license. LKB1fl/fl mice were crossed to transgenic mice expressing Cre recombinase under muscular creatine kinase promoter (Cre) (Bruning et al, 1998), which had been backcrossed for seven generations to the C57BL/6J strain. Genotyping was performed by PCR using genomic DNA isolated from tails. The presence of a wild-type or floxed LKB1 allele was detected using two primers, p200, 5′-CCAGCCTTCTGACTCTCAGG-3′ and p201, 5′-GTAGGTATTCCAGGCCGTCA-3′. For the detection of CRE, the following primers were employed: Cre1, 5′-AAATGGTTTCCCGCAGAACC-3′ and Cre10, 5′-TAGCTGGCTGGTGGCAGATG-3′.
Incubation of isolated muscle with AICAR or phenformin
Mice (7–10 weeks of age) were fasted overnight (16 h) prior to experiment and killed by cervical dislocation, and EDL muscles were rapidly and carefully removed. Tendons from both ends of each muscle were tied with suture (silk 4–0) and mounted on an incubation apparatus. The muscles were incubated as described previously (Sakamoto et al, 2004). Muscles were incubated in 8 ml of Krebs-Ringer bicarbonate (KRB) buffer (117 mM NaCl, 2.5 mM CaCl2, 1.2 mM KH2PO4, 1.2 mM MgSO4, 24.6 mM NaHCO3, pH 7.4) containing 2 mM pyruvate for 60 min at 37°C, in the presence or absence of 2 mM AICAR or 2 mM phenformin. At the end of the incubation period, muscles were quickly frozen in liquid nitrogen. Muscles were stored at −80°C.
In situ muscle contraction
Mice (7–10 weeks of age) were fasted overnight (16 h) and were anaesthetised with sodium pentobarbital (90 mg/kg of body weight, administered intraperitoneally); the sciatic nerves to both legs were surgically exposed, and electrodes were attached. One leg was subjected to electrical stimulation for 5 min (train rate, 1/s; train duration, 500 ms; pulse rate, 100 Hz; duration, 0.1 ms at 2–5 V), and the other leg served as sham-operated (noncontracted) control as described previously (Sakamoto et al, 2002). Immediately after nerve stimulation, mice were killed by cervical dislocation, and tibialis anterior and EDL muscles were rapidly removed and then frozen in liquid nitrogen. Muscle tissues were stored at −80°C.
In vitro muscle contraction
Mice (7–10 weeks of age) were fasted overnight (16 h) prior to experiment and killed by cervical dislocation, EDL muscles were rapidly removed. Tendons from both ends of each muscle were tied with suture and mounted on an incubation apparatus with resting length. The muscles were incubated in 8 ml of KRB buffer containing 2 mM pyruvate for 50 min at 37°C. When contracted, muscles were electrically stimulated during the last 10 min of this period (train rate, 2/min; train duration, 10 s; pulse rate, 100 Hz; duration, 0.1 ms at 100 V).
Supplementary Material
Supplementary Data
Acknowledgments
We thank Ronald Kahn (Joslin Diabetes Center) for providing the Cre expressing mice, Gail Fraser, Victoria Murray-Tait and Laura Armit for technical assistance, Jose Bayascas and Kirsty Mustard for helpful advice, Greg Stewart and the protein production and antibody purification teams (Division of Signal Transduction Therapy (DSTT), University of Dundee) co-ordinated by Hilary McLauchlan and James Hastie for affinity purification of antibodies, the Sequencing Service (School of Life Sciences, University of Dundee, Scotland, www.dnaseq.co.uk) for DNA sequencing. We thank the Association for International Cancer Research (DRA), Cancer Research UK and Breakthrough Breast Cancer (AA), Diabetes UK (DRA+DG), the Medical Research Council UK (DRA+DG), the Moffat Charitable Trust, the European Commission (DGH, QLG1-CT-2001-0148800 RTD contract), the Wellcome Trust (DGH), as well as the pharmaceutical companies that support the Division of Signal Transduction Therapy (AstraZeneca, Boehringer-Ingelheim, GlaxoSmithKline, Merck & Co. Inc., Merck KGaA and Pfizer) for financial support.
References
- Altarejos JY, Taniguchi M, Clanachan AS, Lopaschuk GD (2005) Myocardial ischemia differentially regulates LKB1 and an alternate 5′AMP-activated protein kinase kinase. J Biol Chem 280: 183–190 [DOI] [PubMed] [Google Scholar]
- Aschenbach WG, Sakamoto K, Goodyear LJ (2004) 5′ adenosine monophosphate-activated protein kinase, metabolism and exercise. Sports Med 34: 91–103 [DOI] [PubMed] [Google Scholar]
- Baas AF, Boudeau J, Sapkota GP, Smit L, Medema R, Morrice NA, Alessi DR, Clevers HC (2003) Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. EMBO J 22: 3062–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron SJ, Li J, Russell RR III, Neumann D, Miller EJ, Tuerk R, Wallimann T, Hurley RL, Witters LA, Young LH (2005) Dual mechanisms regulating AMPK kinase action in the ischemic heart. Circ Res 96: 337–345 [DOI] [PubMed] [Google Scholar]
- Boudeau J, Baas AF, Deak M, Morrice NA, Kieloch A, Schutkowski M, Prescott AR, Clevers HC, Alessi DR (2003) MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. EMBO J 22: 5102–5114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudeau J, Scott JW, Resta N, Deak M, Kieloch A, Komander D, Hardie DG, Prescott AR, van Aalten DM, Alessi DR (2004) Analysis of the LKB1–STRAD–MO25 complex. J Cell Sci 117: 6365–6375 [DOI] [PubMed] [Google Scholar]
- Bruning JC, Michael MD, Winnay JN, Hayashi T, Horsch D, Accili D, Goodyear LJ, Kahn CR (1998) A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol Cell 2: 559–569 [DOI] [PubMed] [Google Scholar]
- Carling D (2004) The AMP-activated protein kinase cascade—a unifying system for energy control. Trends Biochem Sci 29: 18–24 [DOI] [PubMed] [Google Scholar]
- Corton JM, Gillespie JG, Hawley SA, Hardie DG (1995) 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem 229: 558–565 [DOI] [PubMed] [Google Scholar]
- Davies SP, Sim AT, Hardie DG (1990) Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur J Biochem 187: 183–190 [DOI] [PubMed] [Google Scholar]
- Fryer LG, Foufelle F, Barnes K, Baldwin SA, Woods A, Carling D (2002) Characterization of the role of the AMP-activated protein kinase in the stimulation of glucose transport in skeletal muscle cells. Biochem J 363: 167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii N, Hayashi T, Hirshman MF, Smith JT, Habinowski SA, Kaijser L, Mu J, Ljungqvist O, Birnbaum MJ, Witters LA, Thorell A, Goodyear LJ (2000) Exercise induces isoform-specific increase in 5′AMP-activated protein kinase activity in human skeletal muscle. Biochem Biophys Res Commun 273: 1150–1155 [DOI] [PubMed] [Google Scholar]
- Hardie DG (2004) The AMP-activated protein kinase pathway—new players upstream and downstream. J Cell Sci 117: 5479–5487 [DOI] [PubMed] [Google Scholar]
- Hardie DG, Scott JW, Pan DA, Hudson ER (2003) Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett 546: 113–120 [DOI] [PubMed] [Google Scholar]
- Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG (2003) Complexes between the LKB1 tumor suppressor, STRADalpha/beta and MO25alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Hirshman MF, Kurth EJ, Winder WW, Goodyear LJ (1998) Evidence for 5′ AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes 47: 1369–1373 [DOI] [PubMed] [Google Scholar]
- Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Hoglund P, Jarvinen H, Kristo P, Pelin K, Ridanpaa M, Salovaara R, Toro T, Bodmer W, Olschwang S, Olsen AS, Stratton MR, de la Chapelle A, Aaltonen LA (1998) A serine/threonine kinase gene defective in Peutz–Jeghers syndrome. Nature 391: 184–187 [DOI] [PubMed] [Google Scholar]
- Hong SP, Leiper FC, Woods A, Carling D, Carlson M (2003) Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc Natl Acad Sci USA 100: 8839–8843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaleel M, McBride A, Lizcano JM, Deak M, Toth R, Morrice NA, Alessi DR (2005) Identification of the sucrose non-fermenting related kinase SNRK, as a novel LKB1 substrate. FEBS Lett 579: 1417–1423 [DOI] [PubMed] [Google Scholar]
- Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, Muller O, Back W, Zimmer M (1998) Peutz–Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 18: 38–43 [DOI] [PubMed] [Google Scholar]
- Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, Schjerling P, Vaulont S, Richter EA, Wojtaszewski JF (2004) Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem 279: 1070–1079 [DOI] [PubMed] [Google Scholar]
- Lawlor MA, Mora A, Ashby PR, Williams MR, Murray-Tait V, Malone L, Prescott AR, Lucocq JM, Alessi DR (2002) Essential role of PDK1 in regulating cell size and development in mice. EMBO J 21: 3728–3738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, Alessi DR (2004) LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J 23: 833–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill GF, Kurth EJ, Hardie DG, Winder WW (1997) AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol 273: E1107–E1112 [DOI] [PubMed] [Google Scholar]
- Mora A, Davies AM, Bertrand L, Sharif I, Budas GR, Jovanovic S, Mouton V, Kahn CR, Lucocq JM, Gray GA, Jovanovic A, Alessi DR (2003) Deficiency of PDK1 in cardiac muscle results in heart failure and increased sensitivity to hypoxia. EMBO J 22: 4666–4676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu J, Brozinick JT Jr, Valladares O, Bucan M, Birnbaum MJ (2001) A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cells 7: 1085–1094 [DOI] [PubMed] [Google Scholar]
- Nath N, McCartney RR, Schmidt MC (2003) Yeast Pak1 kinase associates with and activates Snf1. Mol Cell Biol 23: 3909–3917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto K, Goodyear LJ (2002) Invited review: intracellular signaling in contracting skeletal muscle. J Appl Physiol 93: 369–383 [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Goransson O, Hardie DG, Alessi DR (2004) Activity of LKB1 and AMPK-related kinases in skeletal muscle; effects of contraction, phenformin and AICAR. Am J Physiol Endocrinol Metab 287: E310–E317 [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Hirshman MF, Aschenbach WG, Goodyear LJ (2002) Contraction regulation of Akt in rat skeletal muscle. J Biol Chem 277: 11910–11917 [DOI] [PubMed] [Google Scholar]
- Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, Norman DG, Hardie DG (2004) CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest 113: 274–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC (2004) The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA 101: 3329–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland CM, Hawley SA, McCartney RR, Leech A, Stark MJ, Schmidt MC, Hardie DG (2003) Elm1p is one of three upstream kinases for the Saccharomyces cerevisiae SNF1 complex. Curr Biol 13: 1299–1305 [DOI] [PubMed] [Google Scholar]
- Suzuki A, Kusakai G, Kishimoto A, Shimojo Y, Ogura T, Lavin MF, Esumi H (2004) IGF-1 phosphorylates AMPK-alpha subunit in ATM-dependent and LKB1-independent manner. Biochem Biophys Res Commun 324: 986–992 [DOI] [PubMed] [Google Scholar]
- Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, Vaulont S (2003) The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest 111: 91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winder WW, Hardie DG (1999) AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol 277: E1–E10 [DOI] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M, Carling D (2003a) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 13: 2004–2008 [DOI] [PubMed] [Google Scholar]
- Woods A, Vertommen D, Neumann D, Turk R, Bayliss J, Schlattner U, Wallimann T, Carling D, Rider MH (2003b) Identification of phosphorylation sites in AMP-activated protein kinase (AMPK) for upstream AMPK kinases and study of their roles by site-directed mutagenesis. J Biol Chem 278: 28434–28442 [DOI] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE (2001) Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108: 1167–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data