

Abstract
To efficiently protect the integrity of genetic information, transcription is connected to nucleotide excision repair (NER), which allows preferential repair of the transcribed DNA strands (TS). As yet, the molecular basis of this connection remains elusive in eukaryotic cells. Here we show that, in haploids, the RAD26 gene is essential for the preferential repair of the TS during G1. However, in G2/M phase there is an additional RAD51-dependent process that enhances repair of TS. Importantly, the simultaneous deletion of both RAD26 and RAD51 led to complete abolishment of strand-specific repair during G2/M, indicating that these genes act through two independent but complementary subpathways. In diploids, however, RAD51 is involved in repair of the TS even in G1 phase, which unveils the implication of homologous recombination in the preferential repair of the TS. Importantly, the abolishment of NER, by abrogation of RAD1 or RAD14, completely stopped repair of UV damage even during G2/M phase. These results show the existence of functional cross-talk between transcription, homologous recombination and NER.
Keywords: RAD26, RAD51, S. cerevisiae, transcription-coupled repair, UV damage
Introduction
In most living cells, nucleotide excision repair (NER) represents the main repair mechanism for bulky adducts, including the highly mutagenic and carcinogenic UV-induced pyrimidine dimers (PDs). Several studies conducted in different experimental systems have indicated that NER efficiency is heterogeneous throughout the genome, mainly because of transcription and the compaction of genomic DNA into very complex chromatin structure (Thoma, 1999; Hanawalt, 2002). Indeed, the transcribed parts of the genome are more efficiently repaired than the genome overall, and DNA damage is removed faster from transcribed strands (TS) compared to the corresponding nontranscribed strands (NTS) of active genes (Hanawalt, 2002). This phenomenon, called transcription-coupled nucleotide excision repair (TCNER), constitutes an important branch of the NER pathway. The other NER subpathway, referred to as global genome nucleotide excision repair (GGNER), copes with lesions in the nontranscribed parts of the genome (Hanawalt et al, 2003). It is believed that the preferential repair of the TS is initiated by stalled RNA polymerase II (RNAPII) at a damaged site, followed by the displacement of the polymerase, hereby allowing access to the NER core complexes in order to remove the transcription-blocking lesion (Selby and Sancar, 1993; Svejstrup, 2002). To date, the eukaryotic protein or complex that insures this liaison between transcription and NER is still undefined and the TCNER mechanism is yet elusive. In humans, a defect in TCNER could lead to Cockayne syndrome, a genetic disease characterized by extreme photosensitivity and severe developmental defects (de Boer and Hoeijmakers, 2000). Cockayne syndrome group A (CSA) and Cockayne syndrome group B (CSB) are two genes specific for repair of the TS (Troelstra et al, 1992; Henning et al, 1995). The Saccharomyces cerevisiae homologues of CSA and CSB are RAD28 and RAD26, respectively (van Gool et al, 1994; Bhatia et al, 1996; Guzder et al, 1996). As CSB, RAD26 is solely involved in repair of the TS, while RAD28 seems not to be required. Unlike CSA and CSB mutants, rad26 and rad28 mutants are not sensitive to the killing effect of UV light (van Gool et al, 1994; Bhatia et al, 1996). Interestingly, Rad26 protein is required for the elongation by the RNAPII transcription machinery (Lee et al, 2001, 2002). It is noteworthy that RAD26 deletion does not have any effect on TCNER of the GAL7 gene (Verhage et al, 1996) and only reduces its extent in some other genes such as RPB2 (Bhatia et al, 1996; Verhage et al, 1996) and MFA2 (Teng and Waters, 2000), revealing the existence of a RAD26-independent TCNER mechanism. The elucidation of this mechanism will be of great importance, since it will shed light on the molecular basis of the TCNER process in eukaryotic cells.
In the present report, we show that homologous recombination is involved in the preferential repair of UV damage from transcribed strands of active genes.
Results
The efficiency of transcription-coupled NER is cell cycle modulated in haploid S. cerevisiae cells
To evaluate the efficiency of the NER process in different phases of the S. cerevisiae cell cycle, cells were synchronized in G1 and G2/M with α-factor. For G2/M, cells were first arrested at the G1/S boundary with α-factor, and then were released for cycling in α-factor-free medium until the majority of cells (∼70%) were large budded, with 2n DNA content, and less than 10% of cells were in the S phase (Figure 1A). This synchronization/release technique was performed in order to use the same synchronizing agent, and hence avoid any potential drug effect on the repair of UV damage. Subsequently, G1 and G2/M cells were UV-challenged (200 J m−2) and reincubated for 2 h of dark repair, during which cells remained in the same phase. Following different DNA repair periods, genomic DNA was purified and utilized to assess the removal of PDs from the ACT1 and GAL10 genes, with the aid of the very sensitive primer extension technique. Figure 1B shows the initial distribution pattern of PDs formed almost exclusively at adjacent pyrimidines (repair time 0), and the repair kinetics that occurred along the TS of the portion of the GAL10 gene, in both phases of the cell cycle. A close visual inspection of the representative autoradiograms shows a faster decrease in the intensities of the bands corresponding to the photolesions formed in G2/M cells, compared with those formed in cells arrested in G1, suggesting that the PD removal from the TS is more efficient during the G2/M phase of the cell cycle. Over 2 h of repair, while 80% of PDs were excised from the TS in G2/M cells, only about 60% were removed in G1 cells (Figure 1D). The repair rates obtained for the major PD cluster in this part of the GAL10 gene (CTTTTTTT) were 75 and 50% of PD removed over 2 h in G2/M and G1 cells, respectively. By contrast, the repair rate of the NTS is slightly lower in G2/M compared to the G1 phase; 30 and 40% of PDs were, respectively, removed over the 2 h of repair (Figure 1C and D). To show that this phenomenon is not gene-dependent, similar experiments were performed using the constitutively expressed ACT1 gene for repair assessment. Figure 1E shows that, as seen for the GAL10 gene, strand bias for repair remains more pronounced in G2/M as compared to the G1 phase. Importantly, similar results were obtained when cells were synchronized in G1 (α-factor) or in G2/M (nocodazole), UV-treated and kept arrested with the same agent during the whole repair period (Figure 1F). These results indicate that, in haploid S. cerevisiae cells, the strand bias for repair is more pronounced in G2/M than in G1.
Figure 1.
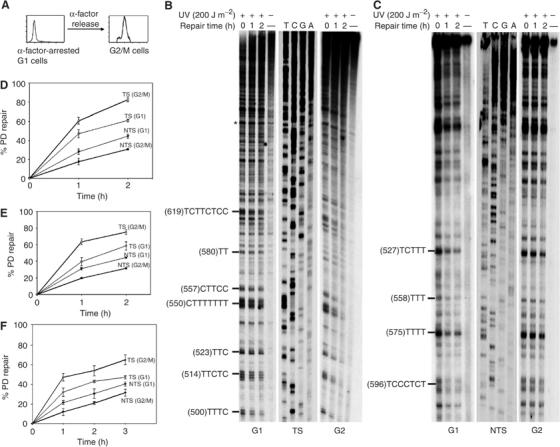
Strand bias for NER is more pronounced in G2/M phase of haploid cells. S. cerevisiae cells synchronized in G1 or G2/M were UV-irradiated and allowed to repair for the indicated periods of time, and then NER was analyzed by primer extension. (A) FACScan analysis showing the synchronization procedure. (B, C) Autoradiograms showing repair of the PDs formed along the GAL10 TS and NTS, respectively. Asterisks, nonspecific Taq polymerase arrests; sequencing reactions (T, C, G, A). The pyrimidine tracks on the left represent the main PD clusters, with the accompanying number in parentheses referring to the 5′ nucleotide of the cluster. (D–F) Quantitative analysis of PD removal from the GAL10 and ACT1 genes, respectively. Illustrated are the fractions (%) of PD removed at each repair time. Each data point corresponds to an average value for the repair of several PD clusters. Inter-lane loading differences were corrected as described in Materials and methods. Each error bar represents the standard deviation of at least three experiments.
Cell cycle-dependent variability of strand bias for NER is transcription-related
After showing that strand bias for excision repair is more pronounced in G2/M phase, it was important to check whether or not this difference is transcription-related. To this end, DNA repair assessment was performed in G1 and G2/M phases using the repressed GAL10 gene as a target. The synchronization was performed as described in the previous paragraph (Figure 1A). Cells were grown and allowed to repair UV-damaged DNA in glucose-containing medium. The evaluation of DNA repair ability in both strands indicated that under these conditions the differential repair was completely abolished in both phases, as expected. Importantly, both strands were repaired at the same rate (∼40% of PD removed over the 2 h of repair) in both G1 and G2/M phases (Figure 2A and B). This clearly shows that the cell cycle-dependent strand bias for repair is transcription-related.
Figure 2.
Cell cycle-dependent TCNER is transcription-related. Cells were grown in glucose, synchronized either in G1 or in G2/M and then reincubated for dark repair. PD removal was assessed on both strands of the repressed GAL10 gene. (A) Autoradiograms, the legends are as in Figure 1. (B) Quantitative analysis, performed as described in Figure 1.
RAD26 deletion abolishes strand-specific repair in G1, but only reduces its extent in G2/M
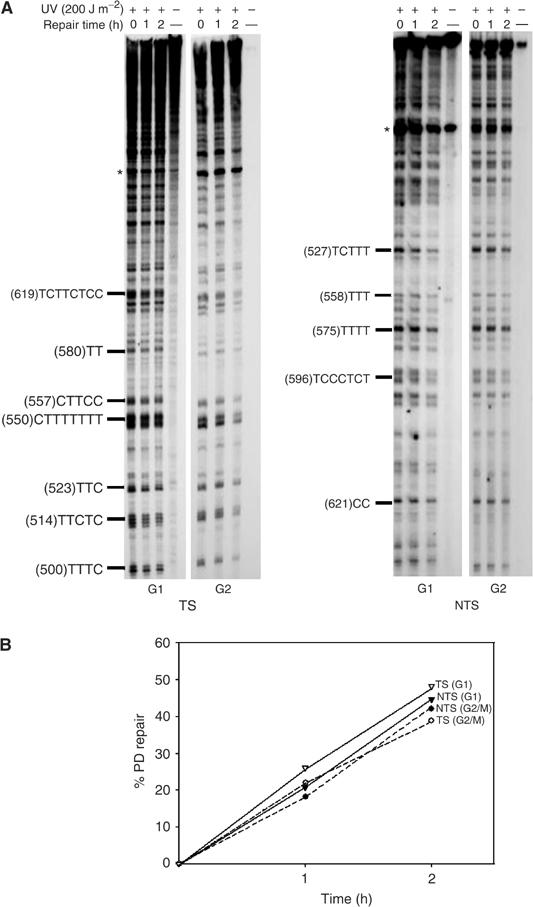
To check whether RAD26 is responsible for the cell cycle-dependent variability of strand bias for repair, excision repair along the TS and the NTS of the GAL10 gene was assessed in RAD26-deleted (rad26Δ) strain, in both G1 and G2/M phases obtained as described in Figure 1A. Figure 3A shows that the efficiency of PD removal from both the TS and the NTS is similar in the G1-rad26Δ cells. For both, about 30% of PDs were removed over the 2 h of repair (Figure 3B). However, as expected, the repair of the NTS was not affected by the RAD26 deletion (Figure 3B), indicating that the absence of RAD26 abolished the TS-preferential repair in G1 phase.
Figure 3.
RAD26 inactivation abolishes strand-specific repair and sensitizes cells in G1. rad26Δ cells were synchronized in G1 or G2/M, UV-challenged and then reincubated for repair. (A) Autoradiograms, the legends are as in Figure 1. (B) Quantitative analysis, performed as described in Figure 1D. Dashed lines represent quantification results from Figure 1D (G2/M) that were included for comparison. (C) Survival curves. Synchronized WT and rad26Δ cells were UV-irradiated with increasing UV fluences, and the percentage of surviving cells corresponding to each UV fluence was determined. Each error bar represents the standard deviation of three different experiments.
The assessment of excision repair in G2/M cells indicated that the RAD26 deletion reduced the extent of strand bias for repair by reducing the repair efficiency of the TS. Within the 2 h of repair, only 55% of PDs were removed in the rad26Δ cells, whereas 80% were excised in the WT cells (Figure 3C). In striking contrast, the repair rate of the NTS turned out to be slightly higher in the rad26Δ cells (35% in 2 h) than in the WT cells (25% in 2 h) (Figure 3B). This indicates that, in G2/M phase, the preferential repair of the TS is still taking place in spite of RAD26 deletion, but to a lower extent as compared to WT cells (Figure 3B). The comparison of DNA repair efficiencies of the TS in both stages showed that this strand is still better repaired in G2/M than in G1, as it is the case in the WT cells (Figures 1D and 3B). The repair assessment at the pyrimidine cluster (CTTTTTTT) indicated that G2/M cells removed 60% of the photolesions within 2 h, whereas G1 cells repaired only 38%. On the other hand, the NTS seems to be repaired with similar efficiencies in both phases of the cell cycle, in contrast to what was found in the RAD26-proficient cells (Figures 1D and 3B). At the cluster (TCCCTCT), G2/M and G1 cells removed 38 and 35% of the photolesions, respectively.
Using the same DNA preparations, the role of RAD26 in TCNER at different cell cycle stages was also assessed in the lowly transcribed ACT1 gene. Interestingly, the results corroborate the findings with the highly transcribed GAL10 gene. RAD26 deletion impaired the repair of the TS in both G1 and G2/M phases, but as for GAL10 the repair in G2/M is still higher than that in G1 (data not shown). These results show that while the differential repair is only partially dependent on the RAD26 gene during G2/M stage, it is completely RAD26-dependent during G1 phase.
RAD26 deletion sensitizes G1 but not G2/M cells to UV damage
To investigate the effect of RAD26 disruption on the cellular resistance to the killing effect of UV light, WT and rad26Δ cells were first synchronized in either G1 or G2/M phases, as described above, and then challenged with increasing UV fluences. The survival curves depicted in Figure 3C show that WT and rad26Δ cells irradiated in G2/M stage are more resistant than cells treated in late G1. The UV fluences resulting in 50% survival (D50 values) are 12 J m−2 for cells irradiated at the G1/S boundary and 35 J m−2 for G2/M cells, indicating that the latter are three-fold more resistant than the G1/S cells. Figure 3C also shows that WT- and rad26Δ-G2/M cells have similar resistance to UV damage. On the other hand, rad26Δ-G1 cells exhibited slight UV sensitivity as compared to their isogenic WT-G1 cells (Figure 3C). This parallels the abolition of TCNER during G1 phase in the rad26Δ cells.
RAD51 and RAD54 deletions impair the preferential repair of the TS during G2/M and eliminates the cell cycle-dependent differential repair
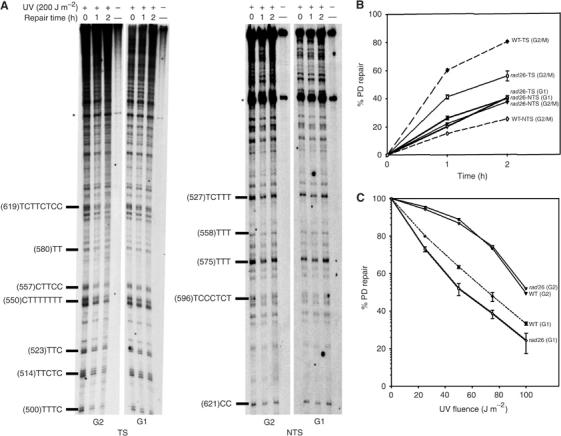
The existence of a RAD26-independent transcription–repair coupling process that operates solely during the post-replicative stages, during which recombination between homologues is possible in haploid cells, suggested a plausible role of recombination in the preferential repair of the TS during G2/M. To test this hypothesis, NER was assessed in RAD51-deleted (rad51Δ) cells synchronized either in G1 or in G2/M phases as described above (Figure 4A). Like for WT cells, 70% of cells were in G2/M phase, while less than 10% were in S phase. RAD51 is a RecA-like protein required for homologous recombination in S. cerevisiae (Aboussekhra et al, 1992; Basile et al, 1992; Shinohara et al, 1992). Following synchronization, WT and rad51Δ cells were UV-challenged (200 J m−2), and then reincubated for repair. Figure 4B shows that in the rad51Δ mutant the TS is repaired with similar rates in both G1 and G2/M. Up to 48% of PDs were removed during the 2 h of repair in both phases (Figure 4C). Likewise, the repair efficiency of the NTS was similar in G1 and G2/M, indicating that in the rad51 mutant the repair rates are equivalent in G1 and G2/M phases. This shows that the cell cycle-dependent preferential repair of the TS is RAD51-dependent, and hence could be homologous recombination-related.
Figure 4.
RAD51 and RAD54, but not RAD50, are required for the preferential repair of the TS in the haploid G2/M cells. WT, rad50Δ, rad51Δ, and rad54Δ cells were synchronized in G1 or G2/M, UV-challenged with 200 J m−2 (B, C) or 100 J m−2 (D) and then reincubated for repair. (A) FACScan analysis showing G2/M synchronized rad51Δ cells. (B) Autoradiograms, the legends are as in Figure 1. (C, D) Quantitative analysis, performed as described in Figure 1D. Each error bar represents the standard deviation of three experiments.
Importantly, during G2/M phase, the PD removal from the TS in rad51Δ cells is not as fast as in WT cells (Figures 1B and 4B). Over 2 h of repair, while about 80% of photolesions were excised from the TS in the WT cells, only 48% were repaired in rad51 mutant (Figure 4C). This shows a deficiency in the repair of the TS in the rad51Δ cells. Regarding the repair of the NTS, like in the rad26Δ mutant, the repair rates in the rad51Δ mutant were slightly higher than in WT cells (Figure 4C), indicating that RAD51 deletion impairs specifically the repair of the TS, without affecting that of the NTS. Therefore, RAD51 is required for the preferential repair of the TS during the post-replicative phases of the cell cycle. Indeed, the TS is repaired with similar rates in both rad51 and rad26 mutants (Figure 5D), suggesting that these genes are both involved in the transcription–repair coupling process during G2/M phase and they are both needed for optimal TCR process.
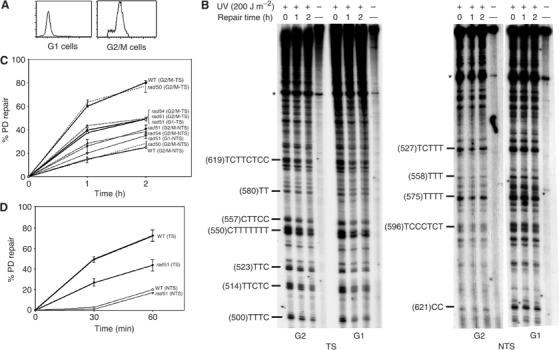
Figure 5.
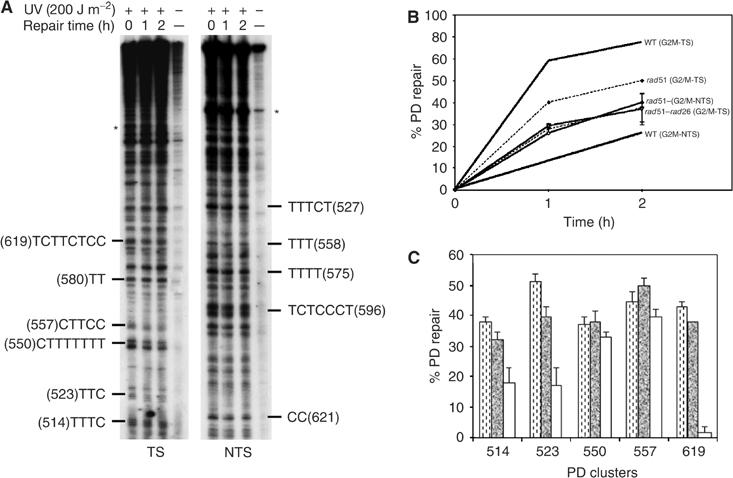
Simultaneous deletion of RAD51 and RAD26 abolishes strand-specific repair in G2/M. WT as well as the double mutant rad26Δrad51Δ cells were synchronized in G2/M, UV-challenged (200 J m−2) and then reincubated for repair. (A) Autoradiograms, the legends are as in Figure 1. (B) Quantitative analysis, performed as described in Figure 1D. Dashed lines represent quantification results from Figure 4C (rad51, G2/M) that were included for comparison. (C) Site-specific removal of PDs from the indicated PD clusters of the GAL10 TS, after 2 h of dark repair: rad51Δ (dashed bars), rad26Δ (solid bars), rad26Δrad51Δ (open bars). Each error bar represents the standard deviation of three experiments.
In G1 phase, the TS is repaired in both WT and rad51 mutant with similar rates, about 40% of PD were removed in 1 h. Similarly, the NTS was repaired with analogous rates in both backgrounds (Figures 1D and 4C). These results show that RAD51 deletion does not affect the excision repair of PDs during the G1 phase of the cell cycle and that, in contrast to RAD26, the role of RAD51 in this process is restricted to G2/M stage. This suggests that it is not RAD51 per se that is involved in the preferential repair, but probably the homologous recombination as a process that occurs exclusively during G2/M phase in haploids. To confirm this, we investigated the role of another homologous recombination gene, RAD54, in the preferential repair of the TS during G2/M stage. Importantly, the rad54Δ cells were found to be as deficient in the repair of the TS as the rad51Δ cells (Figure 4C), indicating that RAD54 is also involved in the repair of the TS during G2/M, which supports the possible role of homologous recombination in the preferential repair of the TS.
To show that this role is not related to the high UV dose used that introduce high number of UV lesions, the role of RAD51 in the preferential repair of the TS during G2/M phase was also analyzed in response to lower UV dose (100 J m−2). Figure 4D shows the excision repair kinetics over 1 h at the GAL10 gene, indicating that RAD51 deletion reduced the repair efficiency of the TS as compared to normal cells. After 1 h, 75% of lesions were removed in WT cells, whereas not more than 40% were excised in rad51Δ cells, corroborating the results described above. However, RAD51 disruption did not have a significant effect on the repair of the NTS (Figure 4D). Similar results were obtained when repair was assessed in the URA3 gene (data not shown), which shows that the role of RAD51 in the preferential repair of the TS is not gene-dependent and is sustained even at low UV fluence.
Role of RAD51 in preferential repair of the TS during G2/M is transcription-dependent
To investigate the link between the role of RAD51 in the preferential repair of the TS and transcription, the removal of UV lesions was assessed in transcriptionally inactive GAL10 gene in rad51Δ cells grown in glucose-containing medium and synchronized in G2/M phase, as described above. As expected, both GAL10 strands were repaired with similar rates during the 2 h of incubation. Indeed, 50 and 47% of photolesions were excised from the NTS and the TS of the GAL10 gene, respectively. These values are similar to those obtained in WT cells (Figure 2B), indicating that the role of RAD51 in the removal of UV damage is transcription-dependent.
RAD50 is not involved in repair of UV damage
The RAD50 gene is involved in recombinational repair of double-strand DNA breaks. Rad50 forms a complex with two other proteins, Mre11 and Xrs2, and this hetero-complex is involved in double-strand break nucleolytic processing (Game, 2000). Based on the phenotype of their mutants and the functions of their products, RAD50, XRS2, and MRE11 genes form a separate subgroup into the RAD52 epistasis group (Game, 2000). To investigate the role of this nuclease in the preferential repair of UV damage in G2/M phase, rad50Δ and WT cells were synchronized in G2/M, treated with 200 J m−2, and then repair of UV lesions was assessed on both GAL10 strands. Figure 4C shows that RAD50 deletion did not affect repair of DNA photolesions from both the TS and the NTS as compared to WT cells. In both cell backgrounds, 25 and 80% of PDs were removed from the NTS and the TS, respectively. This shows that the Rad50 protein is not involved in homologous recombination-mediated excision repair of UV damage.
rad51 rad26 double mutant abolishes the preferential repair of the TS in G2/M phase
The rad26 mutant abolished the preferential repair of the TS in G1. However, in G2/M, the efficiency of TCNER was only reduced, but not abrogated in both rad51 and rad26 mutants. Therefore, we sought to analyze NER in both the WT and the double mutant rad26 rad51 in G2/M phase. Figure 5A shows that in the double mutant the removal of the PDs from the TS is very slow, similar to that occurring in the NTS, whose repair rate increased slightly as compared to that in WT cells. After 2 h of dark repair, an average of 33% of PDs were removed from both strands, showing the absence of transcription-coupled repair in this double mutant (Figure 5B). It is noteworthy that the PD removal from the TS became heterogeneous in the double mutant, while the repair of the same PD clusters was more homogeneous in the parental single mutants (Figure 5A and C). This heterogeneity in the repair rate is a feature of the NTS, while repair in the TS is, in principle, homogeneous (Wellinger and Thoma, 1997). Figure 5A and C clearly shows that the repair rate at many PD clusters on the TS in the double mutant is lower than that assessed in each single mutant and the WT. This reveals that in G2/M phase the preferential repair of the TS is under the control of both RAD26 and RAD51, which may act through independent but cooperative pathways.
Role of RAD51 in repair of the TS is cell cycle-independent in diploid cells
Since, in diploids, radiation-induced homologous recombination can take place even during G1 phase, it was important to investigate whether the RAD51 gene is involved in TCNER during this phase in diploid cells. The effect of RAD51 deletion on preferential repair was first analyzed in exponentially growing cells. WT (FF18735) and rad51Δ/rad51Δ (FF18960) cells were challenged with a UV fluence of 200 J m−2 and then were reincubated for 2 h of dark repair. While 85% of photolesions were in average excised from the TS in WT cells, not more than 50% were excised in the mutant (Figure 6A). For the PD cluster (557)CTTCC for example, while 80% of the PDs were excised in 2 h in WT cells, not more than 40% were repaired in the mutant (Figure 6A). These results confirm the role of RAD51 in the repair of the TS.
Figure 6.
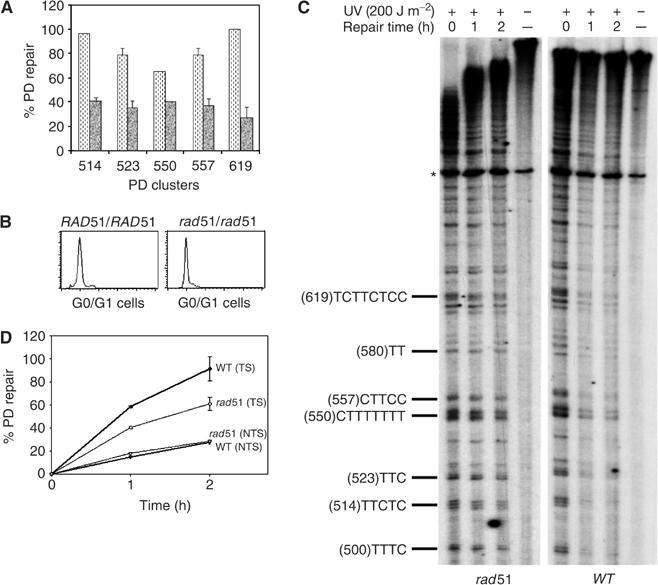
RAD51 is required for the preferential repair of the TS in diploid cells. WT and rad51Δ diploid cells were UV-challenged (200 J m−2) and then reincubated for repair. (A) Log phase cells. Illustrated is quantitative analysis of site-specific removal of PD from the indicated PD clusters of the GAL10 TS, after 2 h of dark repair: WT (dashed bars), rad51Δ (solid bars). (B) G1 cells, DNA profiles by FACScan analysis. (C) G1 cells, Autoradiograms, the legends are as in Figure 1. (D) G1 cells. Quantitative analysis, performed as described in Figure 1D. Each error bar represents the standard deviation of three experiments.
Next, the role of RAD51 in the repair of the TS was analyzed in G1 stage. WT and rad51Δ diploid cells were grown in YEPG for 2 days until 90% of cells were single. Subsequently, cells were diluted in the same medium and reincubated for 2 h, before being UV-treated (Figure 6B). The irradiated cells remained in G1 (single cells) during the whole repair period (2 h). As for haploid cells, the DNA repair assay was performed on both the GAL10 and URA3 genes. Figure 6C shows PD removal from the TS of the GAL10 gene, and clearly indicates that the excision of PDs from the rad51Δ diploid cells is slower than that in WT cells. In average, 90% of PDs were removed from the TS in the WT cells during the 2 h of repair, whereas in the rad51Δ mutant only about 50% were removed within the same period of time (Figure 6D). In the diploids as well, RAD51 deletion did not affect the repair of the NTS (Figure 6D). Interestingly, similar repair rates were obtained when cells were challenged in G2/M phase (obtained by treatment with nocodazole), indicating that in diploid cells TCNER efficiency is not cell cycle-dependent (data not shown). The PDs formed on the TS of the URA3 gene were also more slowly repaired in the mutant rad51Δ cells than in the WT cells (data not shown), which confirms the data obtained for GAL10. Together, these results indicate that, even in G1 phase, the diploid rad51Δ cells are partially deficient in the repair of UV-induced PDs, which was not the case for the haploid cells, suggesting the involvement of RAD51-dependent homologous recombination in the preferential repair of the TS.
RAD1 and RAD14 deletions abolish NER in G2/M cells
To test whether the role of homologous recombination in the preferential repair of UV damage is direct, or indirect through the NER process, repair of UV damage was assessed in NER-deficient rad1Δ and rad14Δ cells during G2/M phase of the cell cycle. RAD14 gene codes for a DNA damage-binding protein that plays key roles in the first steps of the excision repair of UV damage (Guzder et al, 1993). Cells were synchronized as described above and treated with a UV fluence of 200 J m−2. As expected, after 2 h, the repair of the PDs in the TS was undetectable (Figure 7). Indeed, the repair of PDs from the different clusters was between 0 and 10%, indicating that NER was abolished in these G2/M-rad14Δ cells. Similar results were obtained in the rad1Δ cells that are defective for the Rad1/Rad10 endonuclase activity (Tomkinson et al, 1993), which is essential for the NER process (data not shown).
Figure 7.
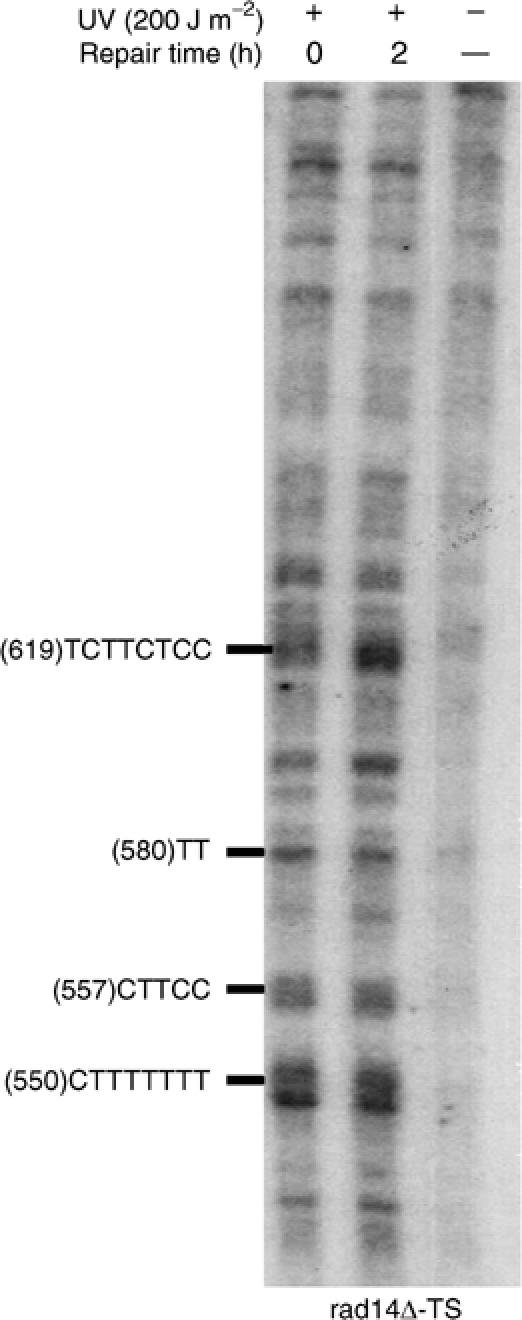
RAD14 deletion abolishes repair of the TS in G2/M phase. rad14Δ cells were synchronized in G2/M, irradiated with a UV fluence of 200 J m−2, and then reincubated for dark repair. The legends of the autoradiogram are as in Figure 1.
Discussion
Transcription-coupled NER efficiency is cell cycle modulated in haploid, but not diploid S. cerevisiae cells
It is shown in the present investigations that strand bias for NER is more pronounced in the post-replicative phases than in G1 phase of the haploid S. cerevisiae cells. This phenomenon is not locus-dependent, but it is transcription-related since it was abolished in transcriptionally inactive GAL10 gene (Figure 2). By contrast, no cell cycle-dependent variation in TCNER was observed in diploid cells, which parallels the results that were obtained in both human and rodent cells (Lommel et al, 1995; Petersen et al, 1995).
RAD26 is essential for TCNER in G1, but not in G2/M phase of haploid cells
It is shown here that RAD26 deletion abolishes the preferential repair of the TS in G1, but only reduces its extent in G2/M phase. This effect of RAD26 is not locus-dependent nor transcription rate-dependent, since it has been shown in the highly transcribed GAL10 gene (Figure 3) and the lowly transcribed ACT1 gene (data not shown). These results indicate that, while RAD26 is necessary for TCNER during G1 phase, there are both RAD26-dependent and -independent transcription–repair coupling processes in G2/M. Interestingly, rad26Δ exhibits slight UV sensitivity in G1 but not in G2/M, nor in Log phase. Together, these results indicate that the lack of UV sensitivity and the absence/slight effect on the preferential repair of the TS that has been previously found in the rad26Δ mutants (Bhatia et al, 1996; Verhage et al, 1996) may be due to the presence of a significant proportion of cells in G2/M in the irradiated population.
Homologous recombination couples transcription to the NER process
We have also shown that RAD51 deletion impairs the repair of the TS, but not that of the NTS, during the post-replicative phases. However, no effect was observed in G1 phase (Figure 4C). This indicates that, in haploid cells, RAD51 plays an important role in the repair of the TS, but only in G2/M phase, during which exchanges between sister chromatids can take place. This strongly suggests a role of recombination in this process. In keeping with this possibility, we have found that RAD54, another homologous recombination gene that belongs to the same epistasis group, is also involved in the preferential repair of the TS during G2/M phase of haploid cells (Figure 4C). This indicates that the RAD26-independent TCR process is related to a pathway, but not to a particular gene. Importantly, RAD51 is required for the preferential repair of the TS even during G1 phase of diploid cells, wherein homologous recombination is possible (Figure 6), indicating that the role of RAD51 in this process is not cell cycle-dependent, but is related to the presence of homologous molecules. Together, these results present the first evidence that homologous recombination is involved in the removal of PDs from the TS of active genes. Indeed, in the absence of transcription (repressed GAL10), RAD51 deletion did not have any effect on the repair of both the TS and the NTS in G2/M phase, showing that the role of RAD51 in the preferential repair of the TS during G2/M is transcription-dependent. More importantly, the effect of RAD51 deletion on the repair of the TS was observed in most PD clusters in both GAL10 and ACT1 genes, indicating that the effect is not limited to some PDs at specific sites, but is a general event. These data indicate that homologous recombination is coupled to transcription, in order to insure the most efficient repair of UV damage from the TS. Is the removal of PDs by HR direct or indirect through the NER process? In fact, the abrogation of the NER RAD1 or RAD14 genes abolished the repair of the TS of the GAL10 gene in G2/M, indicating that during this phase, wherein homologous recombination participates in the preferential repair of UV damage, NER is still essential for this repair process. This suggests that homologous recombination is not directly involved in the excision of UV damage, but only couples transcription to the NER process that insures PDs removal in all phases of the cell cycle. This indicates that, in addition to the role of homologous recombination in UV damage avoidance (bypass) during DNA replication, it also insures efficient repair of part of these lesions from TS of active genes. It is noteworthy that this novel function of HR in promoting the removal of DNA photolesions is unrelated to DNA replication, since (i) the cells used here were synchronized either in G1 or in G2/M; (ii) the role of RAD51 in the preferential repair of UV damage has been found to be strand-specific and transcription-dependent; (iii) while rad50 mutant is known to be defective for the post-replication repair process (Kadyk and Hartwell, 1993), it has been found here to be normal for the preferential repair of the TS during G2/M phase (Figure 4C); (iv) while HR insures only an adaptive role during S phase, it enables an efficient repair of UV damage during the post-replicative phases.
In human cells, CSA and CSB are specifically involved in repair of the TS. Unlike rad26 mutant, CSA and CSB mutants are defective in the preferential repair of the TS (Venema et al, 1990; van Hoffen et al, 1993), suggesting that either the homologous recombination is not involved in TCNER or CSA and CSB mutants are defective in all the TCNER pathways. The human hRad51 protein, a sequence and functional homologue of yeast Rad51, is also required for strand exchange during homologous recombination (Benson et al, 1994; Baumann et al, 1996). hRad51 protein binds directly with Brca2 and interacts with Brca1, two breast cancer susceptibility gene products that are also involved in the homologous recombination mechanism (Venkitaraman, 2002). Importantly, BRCA1 and BRCA2 are required for transcription-coupled repair of the oxidative 8-oxoguanine DNA damage (Abbott et al, 1999; Le Page et al, 2000). In fact, this constitutes another example where proteins required for homologous recombination are also involved in TCR. This suggests that homologous recombination could be involved in the coupling between transcription and repair of oxidative DNA damage as well as UV damage, in human cells.
Homologous recombination and RAD26 control different TCNER subpathways
Both mutants rad26Δ and rad51Δ are defective, but not completely, in the preferential repair of the TS. Importantly, the double mutant displayed no strand-specific repair, indicating that RAD51 and RAD26 are both required for optimal TCNER during G2/M phase of haploid cells and belong to two additive and interactive TCNER subpathways. These subpathways act on the same photolesions, since the repair rate at each PD cluster on the TS was reduced in both rad51Δ and rad26Δ mutants (Figure 5C). This indicates that the same primary lesions are operated on in the WT cells by both the RAD26- and the HR-dependent processes, which complement each other to insure the best repair of the TS. It is noteworthy that when the repair of the TS decreased in the repressed GAL10 gene, in the double mutant as well as in the single mutants (rad51Δ and rad26Δ), the repair of the NTS increased slightly, which may suggest an inter-relationship between repair of the TS and its corresponding NTS of each particular gene, and that, during transcription, repair of the TS takes place at the cost of that of the NTS.
It has been recently reported that Rpb9, a subunit of the S. cerevisiae RNAPII, mediates a subpathway of TCNER (Li and Smerdon, 2002). In the light of the present results, the role of Rpb9 could be either indirect by affecting chromatin remodeling during transcription or by enabling the RNAPII to resume transcription after being arrested by DNA damage. Indeed, Rpb9 sequence shows 30% identity with that of TFIIS (Kaine et al, 1994), and are both normally involved in reactivating paused RNAPII during transcription, in the absence of DNA damage (Awrey et al, 1997; Hemming and Edwards, 2000; Hemming et al, 2000).
How homologous recombination could connect transcription to NER?
A wide range of DNA lesions can block the elongation of the RNAPII. Hence, fast removal of the lesion is crucial for resuming transcription and escaping the mutagenic or lethal effects of transcription inhibition. In an in vitro reaction with purified proteins, it has been shown that the arrested RNAPII occluded the lesion, making it inaccessible to repair enzymes (Tornaletti et al, 1999). This suggests that rapid removal of these lesions requires fast retraction/dissociation of the blocked RNAPII and immediate recruitment of repair complexes. In E. coli, these processes are carried out by the Mfd protein (Selby and Sancar, 1993), which is not a transcription nor a NER factor, but insures the coupling between these two DNA metabolism processes. As yet, no such factor has been identified in eukaryotic cells, where proteins that belong to the RNAPII-elongating machinery, such as Rad26/CSB, seem to facilitate repair of the transcription-blocking lesions (Svejstrup, 2003). However, unlike Mfd, CSB is not able to disrupt the ternary transcription complex of stalled RNA polymerase in vitro (Selby and Sancar, 1997). Thereby, it has become clear that, in eukaryotic cells, the coupling between transcription and NER is an extremely complex process, which requires different proteins. In the present report, we present evidence that the homologous recombination mechanism is also involved in this process. Therefore, how Rad26 and HR insure the preferential repair of UV damage from TS of active genes? Since Rad26 is an ATPase protein with similarities with the ATP-dependent chromatin-remodeling enzymes of the Swi/Snf family (Eisen et al, 1995), it is possible that this protein is involved in the remodeling of the ternary complex of the arrested RNAPII. This could take place either by exposing the lesion to the repair proteins or by retracting/dissociating the RNAPII complex. These reactions, especially the dissociation of the RNAPII from the damage site, may not be always possible or may lead to/require the introduction of a DNA strand break, which will necessitate the participation of HR. Indeed, several lines of evidence point to the role of transcription arrest or impairment in the stimulation of recombination. Recently, it has been shown that the impairment of transcription elongation and the presence of DNA:RNA hybrids, which are present at the RNAPII arrested site, can cause hyper-recombination (Chavez et al, 2000; Aguilera, 2002). In addition, several lines of evidence suggest that transcription elongation may contribute to the formation of DNA breaks. In a recent report, it has been shown that transcription and double-strand breaks induce similar mitotic recombination events (Gonzalez-Barrera et al, 2002). Rad51 and the other homologous recombination proteins, including Rpa and Rad54, can bind to the transcription arrest-dependent recombinogenic structure and initiate the recombination process by searching for homology. Next, the pairing and branch migration can take place, leading to the formation of a double Holliday junction. These events do not need the heterocomplex Rad50–Mre11–Xrs2, since RAD50 deletion did not affect the repair of UV damage (Figure 4C). This suggests that this transcription-related homologous recombination does not need strand break processing. Subsequently, NER proteins will be recruited, may be through the RPA protein, which is involved in both recombination and the first steps of the excision repair mechanism. Indeed, RPA interacts with Rad51 and stimulates presynaptic complex formation and strand exchange, and also associates with Rad14/XPA to insure NER (Sugiyama et al, 1997; Rodriguez et al, 1998; de Laat et al, 1999; Stauffer and Chazin, 2004). Indeed, we have shown here that Rad14 and the endonuclease Rad1/Rad10 are essential for the removal of UV damage in G2/M phase, wherein homologous recombination is also involved (Figure 7). This suggests that NER proteins will be recruited by the HR proteins to excise the damaged fragment and then fill in the resulting gap. Simultaneously or subsequently, the Holliday junctions will be resolved, allowing the resumption of transcription on a damage free DNA template.
Assuming that this process is initiated upon the arrest of the RNAPII at DNA damage site, it is plausible that recombination is involved in coupling transcription with repair of different RNAPII-blocking DNA lesions.
Materials and methods
Strains and media
The haploid S. cerevisiae strains used were FF181268 (MATa; bar1∷LEU2; leu2; ura3; trp1; his7; lys1) (Aboussekhra et al, 1996), AA5 (MATa; bar1∷LEU2; leu2; ura3; trp1; his7; lys1; rad26∷KAN), AA6 (MATa; bar1∷LEU2; leu2; ura3; trp1; his7; lys1; rad51∷URA3), AA7 (MATa; bar1∷LEU2; leu2; ura3; trp1; his7; lys1; rad26∷KAN; rad51∷URA3), FF18973 (MATa; leu2; ura3; trp1; his7; lys1; rad54∷LEU2), FF181655 (MATa; leu2; ura3; trp1; his7; lys1; rad14∷LEU2), and FF18964 (MATa; leu2; ura3; trp1; his7; lys1; rad50∷URA3). The isogenic diploid strains used were FF18735 (MATa/MATα; leu2/leu2; ura3/ura3; trp1/trp1; his7/his7; lys1/lys1) and FF18960 (MATa/MATα; leu2/leu2; ura3/ura3; trp1/trp1; his7/his7; lys1/lys1; rad51∷URA3/ rad51∷URA3) (Aboussekhra et al, 1992).
Cells were grown at 30°C in complete medium containing 2% glucose (YEPD) or galactose (YEPG) to a density of ∼107 cells/ml. α-Factor and nocodazole (Sigma) were prepared and used as described previously (Aboussekhra et al, 1996).
Cell cycle blocks and UV irradiation
Exponentially growing cells were arrested at the G1/S boundary or in G2/M with α-factor. The synchronization at G2/M was performed with the synchronization/release technique. Cells were first synchronized at the G1/S border, and then harvested and washed out from α-factor. Next, cells were reincubated in α-factor-free medium containing 10 μg/ml of pronase for recycling for 120 min. When a significant number of cells were in the desired phase (large budded, with 2n DNA content), cells were collected by centrifugation and resuspended in water. UV irradiation was performed using a germicidal UV lamp (predominantly 254 nm), with a UV-fluence rate of 1 J m−2 s−1. The UV dosimetry was performed using an ultraviolet meter (Spectronics Corporation, NY, USA).
Flow cytometry
2 × 106 cells were fixed in 1 ml ethanol (70%) for 60 min at room temperature. Harvested cells were resuspended in 1 ml Na citrate (50 mM) containing 250 μg RNase A and incubated at 50°C for 60 min. Next, cells were resuspended in the same buffer containing 4 μg propidium iodine (PI) and incubated for 30 min before being analyzed by a Becton-Dickinson FACScan.
DNA repair
Once treated with a UV fluence of either 100 or 200 J m−2, cells were reincubated in the dark. Cells were then collected at various repair times and chilled immediately on ice to stop DNA repair. The percentage of cells in G1 or in G2/M phases was monitored microscopically during the entire repair period. In all cases, cells did not escape the phase in which they were irradiated. A nonirradiated sample served as a sham control.
DNA preparation and enzyme digestion
Genomic DNA was prepared from samples corresponding to different repair times using QIAGEN columns and protocols, whereupon the DNA was incised with the EcoRI restriction enzyme.
Primer extension analysis
Primer labeling and primer extension were carried out as described previously (Aboussekhra and Thoma, 1998), using the GAL10, ACT1, and URA3 genes as templates and the following primers: GAL10 gene bottom strand: 5′-TCTTCTGCTACTGCTTATGGTGATG-3′; top strand: 5′-CTAGATCAACTACGTGGATATAATC-3′. URA3 gene bottom strand: 5′-GCAGGAAACGAAGATAAATCATGTC-3′; top strand: 5′-GCAATTTGACTGTATTACCAATGTC-3′. ACT1 gene bottom strand: 5′-GTAACATCGTTATGTCCGGTGGTA-3′; top strand: 5′-CTTGTGGTGAACGATAGATGGACC-3′.
Primer extension was achieved by 30 cycles of repeated denaturation (95°C for 1 min), annealing (60°C for 4 min) and extension (72°C for 3 min) reaction, with 0.2 U of Taq polymerase (QIAGEN). The reaction products were ethanol precipitated and analyzed on a 5% polyacrylamide, urea (w/v; 40%) gel. DNA sequencing reactions were performed in parallel, using the Sanger chain termination technique with the same primer. The gels were subsequently dried on 3MM paper and then exposed to X-ray films (Kodak) and to a phosphorImager screen (Molecular Dynamics, Typhoon 8600) in order to be analyzed.
Quantification
The sequencing gels were used for the quantification of the relative repair of UV-induced DNA damage. Briefly, a volume box was made around each band obtained from a UV-induced DNA lesion and the corresponding gel background was subtracted using a volume box of the same size outside of the loaded lanes. This value was then divided by that obtained for a volume box which covered the whole lane, so as to correct for inter-lane loading differences. The values obtained for the nonirradiated DNA were then subtracted in order to correct for unspecific background signal due to DNA nicking or nonspecific Taq DNA polymerase blockage. For standardization, the corrected values obtained at time 0 (no repair) were defined as 100% damage.
Acknowledgments
We are grateful to Dr K Al-Hussein, PS Manogaran and Z Al-Mukhalafi for their help with the DNA flow cytometry. We thank Dr Charles I White for helpful discussions and critical reading of the manuscript and Dr F Fabre for providing us with strains and for valuable comments on the manuscript. We also thank S Alanti for her help with statistical analysis and curves. This work was supported by the King Faisal Specialist Hospital and Research Center, under the RAC proposal #990025.
References
- Abbott DW, Thompson ME, Robinson-Benion C, Tomlinson G, Jensen RA, Holt JT (1999) BRCA1 expression restores radiation resistance in BRCA1-defective cancer cells through enhancement of transcription-coupled DNA repair. J Biol Chem 274: 18808–18812 [DOI] [PubMed] [Google Scholar]
- Aboussekhra A, Chanet R, Adjiri A, Fabre F (1992) Semidominant suppressors of Srs2 helicase mutations of Saccharomyces cerevisiae map in the RAD51 gene, whose sequence predicts a protein with similarities to procaryotic RecA proteins. Mol Cell Biol 12: 3224–3234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboussekhra A, Thoma F (1998) Nucleotide excision repair and photolyase preferentially repair the nontranscribed strand of RNA polymerase III-transcribed genes in Saccharomyces cerevisiae. Genes Dev 12: 411–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aboussekhra A, Vialard JE, Morrison DE, de la Torre-Ruiz MA, Cernakova L, Fabre F, Lowndes NF (1996) A novel role for the budding yeast RAD9 checkpoint gene in DNA damage-dependent transcription. EMBO J 15: 3912–3922 [PMC free article] [PubMed] [Google Scholar]
- Aguilera A (2002) The connection between transcription and genomic instability. EMBO J 21: 195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awrey DE, Weilbaecher RG, Hemming SA, Orlicky SM, Kane CM, Edwards AM (1997) Transcription elongation through DNA arrest sites. A multistep process involving both RNA polymerase II subunit RPB9 and TFIIS. J Biol Chem 272: 14747–14754 [DOI] [PubMed] [Google Scholar]
- Basile G, Aker M, Mortimer RK (1992) Nucleotide sequence and transcriptional regulation of the yeast recombinational repair gene RAD51. Mol Cell Biol 12: 3235–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann P, Benson FE, West SC (1996) Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell 87: 757–766 [DOI] [PubMed] [Google Scholar]
- Benson FE, Stasiak A, West SC (1994) Purification and characterization of the human Rad51 protein, an analogue of E. coli RecA. EMBO J 13: 5764–5771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia PK, Verhage RA, Brouwer J, Friedberg EC (1996) Molecular cloning and characterization of Saccharomyces cerevisiae RAD28, the yeast homolog of the human Cockayne syndrome A (CSA) gene. J Bacteriol 178: 5977–5988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez S, Beilharz T, Rondon AG, Erdjument-Bromage H, Tempst P, Svejstrup JQ, Lithgow T, Aguilera A (2000) A protein complex containing Tho2, Hpr1, Mft1 and a novel protein, Thp2, connects transcription elongation with mitotic recombination in Saccharomyces cerevisiae. EMBO J 19: 5824–5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer J, Hoeijmakers JH (2000) Nucleotide excision repair and human syndromes. Carcinogenesis 21: 453–460 [DOI] [PubMed] [Google Scholar]
- de Laat WL, Jaspers NG, Hoeijmakers JH (1999) Molecular mechanism of nucleotide excision repair. Genes Dev 13: 768–785 [DOI] [PubMed] [Google Scholar]
- Eisen JA, Sweder KS, Hanawalt PC (1995) Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acids Res 23: 2715–2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Game JC (2000) The Saccharomyces repair genes at the end of the century. Mutat Res 451: 277–293 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Barrera S, Garcia-Rubio M, Aguilera A (2002) Transcription and double-strand breaks induce similar mitotic recombination events in Saccharomyces cerevisiae. Genetics 162: 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzder SN, Habraken Y, Sung P, Prakash L, Prakash S (1996) RAD26, the yeast homolog of human Cockayne's syndrome group B gene, encodes a DNA-dependent ATPase. J Biol Chem 271: 18314–18317 [DOI] [PubMed] [Google Scholar]
- Guzder SN, Sung P, Prakash L, Prakash S (1993) Yeast DNA-repair gene RAD14 encodes a zinc metalloprotein with affinity for ultraviolet-damaged DNA. Proc Natl Acad Sci USA 90: 5433–5437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt PC (2002) Subpathways of nucleotide excision repair and their regulation. Oncogene 21: 8949–8956 [DOI] [PubMed] [Google Scholar]
- Hanawalt PC, Ford JM, Lloyd DR (2003) Functional characterization of global genomic DNA repair and its implications for cancer. Mutat Res 544: 107–114 [DOI] [PubMed] [Google Scholar]
- Hemming SA, Edwards AM (2000) Yeast RNA polymerase II subunit RPB9. Mapping of domains required for transcription elongation. J Biol Chem 275: 2288–2294 [DOI] [PubMed] [Google Scholar]
- Hemming SA, Jansma DB, Macgregor PF, Goryachev A, Friesen JD, Edwards AM (2000) RNA polymerase II subunit Rpb9 regulates transcription elongation in vivo. J Biol Chem 275: 35506–35511 [DOI] [PubMed] [Google Scholar]
- Henning KA, Li L, Iyer N, McDaniel LD, Reagan MS, Legerski R, Schultz RA, Stefanini M, Lehmann AR, Mayne LV, Friedberg EC (1995) The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell 82: 555–564 [DOI] [PubMed] [Google Scholar]
- Kadyk LC, Hartwell LH (1993) Replication-dependent sister chromatid recombination in rad1 mutants of Saccharomyces cerevisiae. Genetics 133: 469–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaine BP, Mehr IJ, Woese CR (1994) The sequence, and its evolutionary implications, of a Thermococcus celer protein associated with transcription. Proc Natl Acad Sci USA 91: 3854–3856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Page F, Randrianarison V, Marot D, Cabannes J, Perricaudet M, Feunteun J, Sarasin A (2000) BRCA1 and BRCA2 are necessary for the transcription-coupled repair of the oxidative 8-oxoguanine lesion in human cells. Cancer Res 60: 5548–5552 [PubMed] [Google Scholar]
- Lee SK, Yu SL, Prakash L, Prakash S (2001) Requirement for yeast RAD26, a homolog of the human CSB gene, in elongation by RNA polymerase II. Mol Cell Biol 21: 8651–8656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Yu SL, Prakash L, Prakash S (2002) Requirement of yeast RAD2, a homolog of human XPG gene, for efficient RNA polymerase II transcription. implications for Cockayne syndrome. Cell 109: 823–834 [DOI] [PubMed] [Google Scholar]
- Li S, Smerdon MJ (2002) Rpb4 and Rpb9 mediate subpathways of transcription-coupled DNA repair in Saccharomyces cerevisiae. EMBO J 21: 5921–5929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lommel L, Carswell-Crumpton C, Hanawalt PC (1995) Preferential repair of the transcribed DNA strand in the dihydrofolate reductase gene throughout the cell cycle in UV-irradiated human cells. Mutat Res 336: 181–192 [DOI] [PubMed] [Google Scholar]
- Petersen LN, Orren DK, Bohr VA (1995) Gene-specific and strand-specific DNA repair in the G1 and G2 phases of the cell cycle. Mol Cell Biol 15: 3731–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez K, Talamantez J, Huang W, Reed SH, Wang Z, Chen L, Feaver WJ, Friedberg EC, Tomkinson AE (1998) Affinity purification and partial characterization of a yeast multiprotein complex for nucleotide excision repair using histidine-tagged Rad14 protein. J Biol Chem 273: 34180–34189 [DOI] [PubMed] [Google Scholar]
- Selby CP, Sancar A (1993) Molecular mechanism of transcription–repair coupling. Science 260: 53–58 [DOI] [PubMed] [Google Scholar]
- Selby CP, Sancar A (1997) Human transcription–repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J Biol Chem 272: 1885–1890 [DOI] [PubMed] [Google Scholar]
- Shinohara A, Ogawa H, Ogawa T (1992) Rad51 protein involved in repair and recombination in S. cerevisiae is a RecA-like protein. Cell 69: 457–470 [DOI] [PubMed] [Google Scholar]
- Stauffer ME, Chazin WJ (2004) Physical interaction between replication protein A and Rad51 promotes exchange on single-stranded DNA. J Biol Chem 279: 25638–25645 [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Zaitseva EM, Kowalczykowski SC (1997) A single-stranded DNA-binding protein is needed for efficient presynaptic complex formation by the Saccharomyces cerevisiae Rad51 protein. J Biol Chem 272: 7940–7945 [DOI] [PubMed] [Google Scholar]
- Svejstrup JQ (2002) Mechanisms of transcription-coupled DNA repair. Nat Rev Mol Cell Biol 3: 21–29 [DOI] [PubMed] [Google Scholar]
- Svejstrup JQ (2003) Rescue of arrested RNA polymerase II complexes. J Cell Sci 116: 447–451 [DOI] [PubMed] [Google Scholar]
- Teng Y, Waters R (2000) Excision repair at the level of the nucleotide in the upstream control region, the coding sequence and in the region where transcription terminates of the Saccharomyces cerevisiae MFA2 gene and the role of RAD26. Nucleic Acids Res 28: 1114–1119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoma F (1999) Light and dark in chromatin repair: repair of UV-induced DNA lesions by photolyase and nucleotide excision repair. EMBO J 18: 6585–6598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkinson AE, Bardwell AJ, Bardwell L, Tappe NJ, Friedberg EC (1993) Yeast DNA repair and recombination proteins Rad1 and Rad10 constitute a single-stranded-DNA endonuclease. Nature 362: 860–862 [DOI] [PubMed] [Google Scholar]
- Tornaletti S, Reines D, Hanawalt PC (1999) Structural characterization of RNA polymerase II complexes arrested by a cyclobutane pyrimidine dimer in the transcribed strand of template DNA. J Biol Chem 274: 24124–24130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troelstra C, van Gool A, de Wit J, Vermeulen W, Bootsma D, Hoeijmakers JH (1992) ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne's syndrome and preferential repair of active genes. Cell 71: 939–953 [DOI] [PubMed] [Google Scholar]
- van Gool AJ, Verhage R, Swagemakers SM, van de Putte P, Brouwer J, Troelstra C, Bootsma D, Hoeijmakers JH (1994) RAD26, the functional S. cerevisiae homolog of the Cockayne syndrome B gene ERCC6. EMBO J 13: 5361–5369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Hoffen A, Natarajan AT, Mayne LV, van Zeeland AA, Mullenders LH, Venema J (1993) Deficient repair of the transcribed strand of active genes in Cockayne's syndrome cells. Nucleic Acids Res 21: 5890–5895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venema J, Mullenders LH, Natarajan AT, van Zeeland AA, Mayne LV (1990) The genetic defect in Cockayne syndrome is associated with a defect in repair of UV-induced DNA damage in transcriptionally active DNA. Proc Natl Acad Sci USA 87: 4707–4711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkitaraman AR (2002) Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 108: 171–182 [DOI] [PubMed] [Google Scholar]
- Verhage RA, van Gool AJ, de Groot N, Hoeijmakers JH, van de Putte P, Brouwer J (1996) Double mutants of Saccharomyces cerevisiae with alterations in global genome and transcription-coupled repair. Mol Cell Biol 16: 496–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellinger RE, Thoma F (1997) Nucleosome structure and positioning modulate nucleotide excision repair in the non-transcribed strand of an active gene. EMBO J 16: 5046–5056 [DOI] [PMC free article] [PubMed] [Google Scholar]