

Abstract
We have recently identified an intronic polymorphic CA-repeat region in the human endothelial nitric oxide synthase (eNOS) gene as an important determinant of the splicing efficiency, requiring specific binding of hnRNP L. Here, we analyzed the position requirements of this CA-repeat element, which revealed its potential role in alternative splicing. In addition, we defined the RNA binding specificity of hnRNP L by SELEX: not only regular CA repeats are recognized with high affinity but also certain CA-rich clusters. Therefore, we have systematically searched the human genome databases for CA-repeat and CA-rich elements associated with alternative 5′ splice sites (5′ss), followed by minigene transfection assays. Surprisingly, in several specific human genes that we tested, intronic CA RNA elements could function either as splicing enhancers or silencers, depending on their proximity to the alternative 5′ss. HnRNP L was detected specifically bound to these diverse CA elements. These data demonstrated that intronic CA sequences constitute novel and widespread regulatory elements of alternative splicing.
Keywords: alternative splicing, hnRNP L, repetitive sequence, splicing enhancer, splicing silencer
Introduction
Messenger RNA splicing is essential for generating and regulating highly complex proteomes from a relatively small number of human genes. Tissue- and development-specific regulation of mRNA splicing can occur either on the level of splice site (ss) choice (alternative splicing) or splicing efficiency. Considerable efforts have been undertaken to elucidate the cis-acting elements on the pre-mRNA that are responsible for these regulatory processes; as a result, in particular in the exon regions both positively and negatively acting regulatory elements have been described, called exonic splicing enhancers (ESE) and silencers (ESS), respectively. Among the exonic elements, several classes can be distinguished that function as potent splicing enhancers, including both purine-rich and AC-rich sequences (Coulter et al, 1997; Fairbrother et al, 2002; reviewed by Black, 2003).
In contrast to exonic elements, we know much less about characteristics and mechanisms of intronic splicing enhancers and -silencers (ISE and ISS elements); there are only few and diverse examples, such as uridine-rich enhancer elements downstream of alternative 5′ss, the UGCAUG hexanucleotide enhancer, or CUCUCU as an intronic splicing repression element (reviewed by Ladd and Cooper, 2002; Black, 2003). Finally, both positive and negative splicing-regulatory elements often occur clustered, assembling a regulatory complex.
In general, regulatory proteins can function as activators or repressors, and mediate—by their tissue-specific expression and by interacting with the general splicing machinery—regulation of alternative splicing pathways. Among the trans-acting protein factors, which recognize either exonic or intronic elements, are members of the SR (serine-arginine-rich) protein family, members of the hnRNP (heterogenous nuclear ribonucleoprotein) family, but also other specific proteins such as TIA-1, CUGBP, PTB, ETR-like factors (CELF) proteins, and the U1 snRNP (see references in Black, 2003). In particular, RNA recognition and function of the SR proteins have been well-characterized (reviewed by Graveley, 2000); less is known about other splicing regulators.
Alternative splicing is in many cases essential for understanding human genetic diseases: Many mutations have been found to result in splicing defects and map to splicing-relevant sequences, most often to the splice site regions (Krawczak et al, 1992; Cooper and Mattox, 1997). Moreover, mutations affecting ESE or ESS have been suggested as a novel disease mechanism on the level of pre-mRNA splicing (Blencowe, 2000; Cartegni et al, 2002). Examples include alternative splicing of the BRCA1 gene (Liu et al, 2001) and the human SMN locus, where mutations cause spinal muscular atrophy (SMA, Cartegni and Krainer, 2002; Kashima and Manley, 2003).
We have recently studied an unusual case of splicing regulation, involving the human gene for the endothelial NO synthase (eNOS); its intron 13 contains a polymorphic CA-repeat region, (CA)14–44, proximal to the 5′ss. eNOS plays a key role in vascular homeostasis, and there is an interesting correlation between the number of CA-repeat units and the risk of coronary artery disease (Stangl et al, 2000). We have established that this intronic CA-repeat element acts as an important sequence determinant for the efficiency of intron 13 splicing. Interestingly, the activity of this CA-repeat element correlates with the CA-repeat length and depends on the trans-acting factor hnRNP L (Hui et al, 2003; reviewed by Bilbao and Valcarcel, 2003).
In the human genome, 19.4 CA repeats occur per Mb, representing by far the most common simple-sequence repeat (SSR) motif (Waterston et al, 2002). The functional significance of these polymorphic sequence elements has remained largely unclear. Together with our initial findings on the CA-repeat element in the human eNOS gene, important questions are raised: Do CA repeats function as widespread regulatory elements, and is there a general role of the abundant hnRNP L protein as a splicing regulator? Although identified already 15 years ago (Piñol-Roma et al, 1989), it was not clear so far, what sequence determinant in the pre-mRNA is recognized by hnRNP L.
To address these issues, we have initially used mutational analysis of the intronic CA-repeats in the human eNOS gene; a SELEX approach was applied to define the RNA binding specificity of hnRNP L; sets of candidate genes were selected from human sequence databases as potential hnRNP L targets; and alternative splicing of such CA-element containing genes was analyzed by minigene transfections. In sum, we demonstrate here for the first time that (1) hnRNP L recognizes—in addition to regular CA-repetitive sequences—certain CA-rich clusters with high affinity; (2) such CA elements can act—depending on their 5′ss proximity—as either enhancers or silencers in alternative splicing regulation; and (3) hnRNP L is involved in mediating these regulatory processes. Thus, CA-repeat and -rich sequences have a high potential to function as important splicing-regulatory elements in the intronic regions of many human genes.
Results and discussion
Position requirements of the CA-repeat element: role in 5′ss activation
In the human eNOS gene, the polymorphic CA-repeat region that functions as a length-dependent sequence determinant of splicing efficiency is located close to the 5′ss of intron 13, starting after position +80 (Hui et al, 2003). To obtain more insight into the mechanism of this unusual regulatory element, we tested the position requirements of the enhancer function (Figure 1). Our normal minigene derivative, CA32(+80), contains eNOS exons 13 and 14 interrupted by a shortened version of intron 13 with (CA)32. RT–PCR assays of in vitro splicing reactions showed that splicing activity of the normal eNOS minigene construct could be detected, as previously described (panel A); no activity could be observed with pre-mRNAs, where the CA repeats had been deleted or replaced by a 64-nucleotide nonspecific sequence (data not shown; see Hui et al, 2003).
Figure 1.
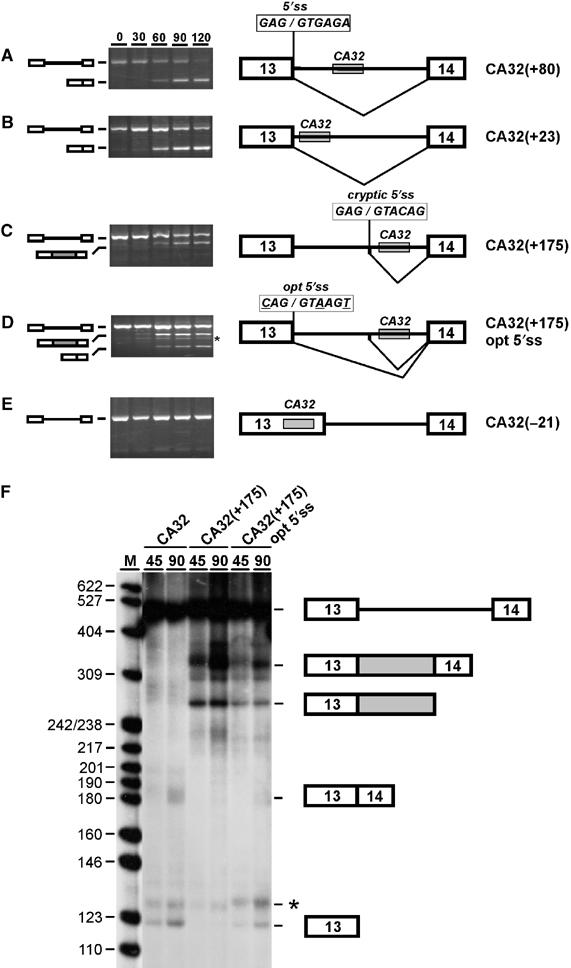
Position requirements of the CA-repeat element in eNOS intron 13. (A–E) Pre-mRNAs are schematically shown on the right and are derived from exons 13–14 of the human eNOS gene with a shortened version of intron 13 containing the (CA)32 splicing enhancer (gray box). The (CA)32 enhancer is located at its normal intronic position (+80; A), moved upstream (+23; panel B), downstream (+175; C and D), or into exon 13 (−21; E). In addition, for CA32(+175) opt 5′ss, the 5′ splice site (5′ss; see A) was converted to the consensus sequence (opt 5′ss; see D, sequence as shown with three mutated positions underlined). Each eNOS pre-mRNA derivative was spliced in vitro, and the reaction at time 0, and after 30, 60, 90, and 120 min was analyzed by RT–PCR. The positions corresponding to pre-mRNA and product mRNAs are schematically indicated on the left, predominant splicing patterns also on the right. Note that a cryptic 5′ss is activated with the (CA)32 enhancer at intron position +175 (C and D). The band marked by the asterisk (D) represents a nonspecific PCR artifact, as determined by sequence analysis. (F) Direct RNA analysis of in vitro splicing of 32P-labeled eNOS pre-mRNA CA32(+80) and derivatives CA32(+175) and CA32(+175) opt5′ss (compare with panels A, C, and D). Time points of 45 and 90 min have been analyzed, as indicated. The positions of pre-mRNAs, spliced products, and first-exon intermediates are indicated on the right. The gray box represents the additional exon sequence incorporated as a result of use of the cryptic 5′ss. The asterisk marks most likely a degradation product of the splicing substrate, since it occurred splicing-independently and varied in intensity when different preparations of nuclear extract were used. M, pBR322/HpaII markers (sizes in nucleotides).
We first moved the (CA)32 sequence to a position closer to the 5′ss, resulting in construct CA32(+23), which contains 32 CA repeats after position +23 in intron 13. Splicing of CA32(+23) pre-mRNA did not alter the splicing pattern compared with the normal substrate (compare panels A and B). Next, we moved the (CA)32 sequence further downstream, giving construct CA32(+175) (panel C). Surprisingly, the normal spliced product was only very weakly detectable, but instead, a larger RT–PCR product appeared during the time course of the splicing reaction. Cloning and sequence analysis of this additional product revealed that it reflects usage of a cryptic 5′ss mapping 37 nucleotides upstream of the CA repeats (5′-GAG/GTACAG-3′; the slash marking the 5′ss).
Why was the normal 5′ss used only weakly in comparison to the cryptic 5′ss? Since there are three deviations from the consensus (positions −3, +3, and +6) in the normally used 5′ss, we converted it to the consensus sequence by three point mutations (G−3 → C, G+3 → A, A+6 → T; opt 5′ss; see panel D). In vitro splicing analysis for this construct, CA32(+175) opt 5′ss, showed that the change to the consensus 5′ss did reactivate the normal 5′ss, but only partially; the cryptic 5′ss was still used at a similar efficiency (panel D). These RT–PCR based results were confirmed by direct RNA analysis with 32P-labeled RNA, using the most important constructs, CA32(+80), CA32(+175), and CA32(+175) opt5′ss (panel F). In an additional control construct, the nonspecific sequence was inserted at position +175: No significant in vitro splicing activity could be observed, confirming that the activation of the cryptic 5′ss depended on the specific CA-repeat sequence and was not caused by changes in the relative distances of other elements (data not shown).
We conclude that moving the CA repeats to a more downstream position in intron 13 resulted in activation of a novel cryptic 5′ss, mapping 37 nucleotides upstream of the CA repeats. This implies that the CA-repeat region functions in activating a 5′ss located directly upstream of the CA repeats. It further suggests that CA repeats can determine not only splicing efficiency but might also be involved in 5′ss selection during alternative splicing (see further below).
Position requirements of the CA-repeat element: intron specificity
To further study the position requirements, we also made two constructs with the CA-repeat enhancer in an exonic context. First, in CA32(−21) the (CA)32 sequence was inserted upstream of position −21 in the eNOS exon 13 (Figure 1E). In vitro splicing of this construct demonstrated that with CA repeats in an exonic context, no splicing stimulation could be detected. Second, this was confirmed in a different, heterologous construct consisting of exons 3 and 4 of the Drosophila dsx gene; as previously characterized, splicing of this reporter depends on an exonic splicing enhancer in exon 4 (Tanaka et al, 1994; Woerfel and Bindereif, 2001). In this context, both in the absence of an insert, with 32 CA repeats, or with a nonspecific sequence of identical length inserted, only very low background levels of in vitro splicing activity were detected. In contrast, a positive control, which carries a known strong exonic enhancer derived from the IgM exon M2, strongly enhanced splicing activity (data not shown). In conclusion, we have demonstrated that in two different exonic contexts no splicing stimulation could be detected. Therefore, the (CA)32 enhancer appears to function in an intron-specific manner.
Intronic CA repeats are sufficient for splicing enhancement
Next, the CA repeats were placed into a heterologous intronic context, to test whether they are sufficient for splicing activation. We used the T7 DUP4-1 construct, which is derived from the human β-globin gene and contains three exons and two introns (Figure 2A). The first and last exons are β-globin exons 1 and 2, respectively. In between, an additional 33-nucleotide hybrid exon containing the duplicated 3′ss of β-globin exon 2 and the duplicated 5′ss of β-globin exon 1 had been inserted. Thus, this clone contains two identical 5′ss, two identical 3′ss, and two identical introns (intron A and intron B). In vivo, the hybrid central exon is skipped due to its short length (Modafferi and Black, 1997), and the combination of introns A and B and the hybrid central exon was named ‘intron C'.
Figure 2.
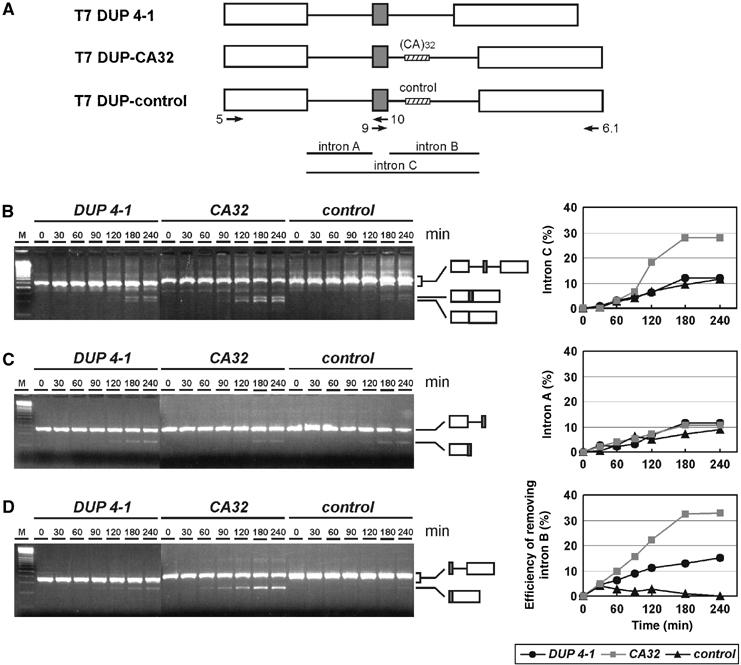
Intronic CA repeats are sufficient for splicing enhancement. (A) Schematic representation of heterologous constructs: T7 DUP 4-1 is derived from exons 1 and 2 (white boxes) of the human β-globin gene, with an artificial short exon (gray box) between two identical introns A and B. T7 DUP-CA32 contains in intron B the (CA)32 sequence, T7 DUP-control a nonspecific sequence (shaded boxes). The positions of the primers used for RT–PCR assays are shown by arrows with primer names labeled. (B–D) Each of these three pre-mRNAs was spliced in vitro, and aliquots were assayed by RT–PCR at time points between 0 and 240 min (as indicated above the lanes), using primer combinations indicative of splicing of the entire intron C unit (B), of intron A (C), and of intron B (D). Positions of products corresponding to pre-mRNAs and spliced mRNAs are schematically shown on the right. Splicing efficiencies for removing different introns were quantitated and diagrammed on the right. In panel B, the efficiency of removing intron C was quantitated as the ratio of the mRNA signal without the central exon to the sum of pre-mRNA and mRNA signals. In panels C and D, the efficiencies of removing intron A and B, respectively, were quantitated as the ratio of the mRNA signal to the sum of pre-mRNA and mRNA signals (expressed as percentages). M, 100-bp ladder (Fermentas).
Two new clones were derived from the DUP4-1 construct: T7 DUP-CA32 and T7 DUP-control, which contain 32 CA repeats or a 64-nucleotide control sequence, respectively, inserted in intron B after position +45. Splicing activities of these constructs were tested in vitro, using RT–PCR analysis and primer combinations that monitor separately the excision of the entire unit (intron C, primers 5/6.1; Figure 2B), of intron A (primers 5/10; Figure 2C), and of intron B (primers 9/6.1; Figure 2D).
In the original construct, T7 DUP 4-1, intron C was spliced out in vitro only inefficiently (to ∼10%; see Figure 2B, lanes DUP 4-1). Inserting the (CA)32 sequence into intron B greatly stimulated splicing activity (to almost 30%; see Figure 2B, lanes CA32). This effect was sequence-specific, since splicing activity of the control construct was similar to the starting construct (Figure 2B; compare lanes control and DUP4-1). Using an intron A-specific primer pair showed there were no significant differences in intron A removal between the three constructs (Figure 2C); in contrast, the CA-repeat sequence strongly and specifically stimulated intron B excision (Figure 2D; compare lanes DUP 4-1 and CA32). Moreover, insertion of the control sequence appears to suppress splicing activity of intron B, possibly because of the length increase of intron B in comparison to the construct without insert (Figure 2D, lanes control). The result that CA repeats are sufficient for specifying splicing enhancement of the intron they reside in was reproduced several times (for two repeats in different nuclear extract preparations, see Supplementary Figure S1).
Taken together, these results confirm that (CA)32 has the properties of a strong splicing-regulatory element. More importantly, the CA repeats are sufficient for activating splicing in a heterologous context, acting specifically on excision of the intron they reside in.
Defining the RNA binding specificity of hnRNP L by SELEX
We had previously established that hnRNP L represents the major CA-repeat RNA binding protein (Hui et al, 2003). To test whether repetitive CA sequences are the only target RNA of hnRNP L, we applied an in vitro SELEX approach (systematic evolution of ligands by exponential enrichment; Tuerk and Gold, 1990). Baculovirus-expressed GST-hnRNP L was immobilized on glutathione-Sepharose and presented with a large molar excess of RNA containing a randomized 20-nucleotide region flanked by two specific sequences. RNA was bound and—after extensive washing—recovered and amplified by RT–PCR. Following T7 transcription, this selection was repeated five times, monitoring the enrichment of hnRNP L-bound sequences by a GST pull-down assay. After the final amplification round, products were cloned, resulting in 108 distinct sequences (see Supplementary Materials and methods). From these sequences we extracted a consensus (see Figure 3A for a graphic representation of a 10-nucleotide consensus, and Supplementary Materials and methods for details): Clearly, there is a high enrichment of CA dinucleotides. To obtain a minimal binding motif, the occurrence of all possible 256 tetranucleotides in the 108 selected sequences was analyzed (see Figure 3B for the 20 most commonly occurring tetranucleotides): As a result, ACAC and CACA were found most frequently, representing together ∼16% of all tetranucleotides in the selected sequences (high-score motifs); these were followed by TACA and CACC, each representing ∼3% (low-score motifs; compared to 0.39% for any tetranucleotide in a random sequence). In sum, this indicates that not only regular CA repeats are among the winner sequences but also sequences with certain deviations, most of which maintain an alternating purine–pyrimidine pattern.
Figure 3.

Defining the RNA binding specificity of hnRNP L by SELEX. (A) The 10-nucleotide consensus sequence. The frequency of each of the four nucleotides at any position in the consensus is represented by the letter height. Boxes mark the two highly conserved core tetranucleotides. (B) Tetranucleotide frequency in selected sequences. The first 20, most common tetranucleotide sequences, are given, in the order of their frequencies in the 108 selected sequences (heavy line, SELEX) and in the 20 sequences taken from the initial pool (thin line, control; both diagrammed as percentage of the total). (C) Characteristics of 11 SELEX-derived (clone numbers on the left) and two control sequences (with asterisks; #20 and 15). Given are the individual sequences (with high-score motifs in red, low-score motifs underlined) and the KD values (in nM; with standard deviations, P<0.05).
Next, we picked 11 SELEX-derived specific RNA sequences for a more detailed analysis (Figure 3C); in addition, two sequences of the original starting pool were included (#20 and 15). These 13 RNAs were T7-transcribed and their binding affinities to hnRNP L determined by filter binding assays. There are four sequences (#51, 55, 1, and 63) with the highest apparent KD values (between 7 and 18 nM), three more with intermediate values (#33, 105, and 26; between 32 and 71 nM), and four others with relatively low KD values (#72, 49, 23, and 6; above 300 nM). The affinities of the two RNAs of the control group (#20 and 15) were close to the millimolar range.
To test whether the RNA binding specificity of hnRNP L might be based on this minimal tetranucleotide consensus, we counted how often the high- (ACAC, CACA) and low-score motifs (TACA, CACC) occur in these sequences (Figure 3C). Significantly, the number of tetranucleotide motifs correlates well with the apparent affinities: Each of the four sequences within the top group (KD below 18 nM) carries between two and four motifs, mostly of the high-score type; the three sequences in the intermediate group (KD between 32 and 71 nM) contain only one or two high-score motifs each, and the members of the low-affinity group only a single, low-score motif. In contrast, the two control RNAs contain none of these tetranucleotide motifs (#15 or 20). In addition, we note that C/A-richness does not suffice for high hnRNP L binding affinity: For example, sequence #23 consists entirely of cytidine and adenosine nucleotides, yet shows only a single low-score motif and binds hnRNP L with very low affinity.
Finally, this newly defined RNA-binding specificity of hnRNP L also explains older data in the literature, where hnRNP L had been crosslinked to specific RNAs, but where the binding motif had not yet been recognized (see Table I). First, Liu and Mertz (1995) had identified an RNA element in the HSV-TK gene, called pre-mRNA processing enhancer (PPE), to which hnRNP L could be crosslinked. The wild-type sequence contains three closely spaced high-score motifs (see double-underlining). In contrast, three mutant derivatives, LS0, LS1, and LS2, which are defective or reduced in hnRNP L binding (relative crosslinking efficiencies given in parentheses), carry only one (mutants LS0 and LS1) or two high-score motifs (mutant LS2). Therefore, there is a clear correlation between the number of motifs and the hnRNP L crosslinking efficiency.
Table 1.
Correlation of hnRNP L crosslinking data and binding motifs
| Genes | Sequences | Reference | |
|---|---|---|---|
| HSV-TK | Wild type (1X) | 5′-TCGCGAACATCTACACCACACAACACCGCCTCGA-3′ | Liu and Mertz (1995) |
| LS0 (0.05X) | TAG G T TAGA | ||
| LS1 (0.45X) | G TCT | ||
| LS2 (0.6X) | AGATCT | ||
| VEGF | 5′-AGACACACCCACCCACATACATACAT-3′ | Shih and Claffey (1999) | |
| HCV | 5′-AGCACAAATCCT/AAACCTCAAAGAAAAACC/AAAAGAAACACAAACCGCCGCCCACAGG-3′ | Hahm et al (1998) | |
| High- and low-score motifs are indicated by double- and single-underlining, respectively. The relative crosslinking efficiencies for wild type and mutant sequences of the HSV-TK gene are given in parentheses. The sequence for HCV IRES element corresponds to the 5′ end of HCV mRNA (nucleotides 345–402; Hahm et al, 1998). The slashes indicate the boundaries of different 3′ truncations (after positions 356 and 374). | |||
Second, hnRNP L had been identified as a factor binding to the 3′ UTR region of the vascular endothelial growth factor (VEGF) gene (Shih and Claffey, 1999). The relevant region contains a striking cluster of two high- and three low-score motifs (indicated by double- and single-underlining).
Third, Hahm et al (1998) identified hnRNP L as a protein binding to the 5′ end of HCV IRES element. Progressive deletions from the 3′ end (boundaries indicated by the slashes) gradually abolished hnRNP L binding, concomitantly with the loss of binding motifs.
In conclusion, hnRNP L recognizes not only regular CA repeats with high affinity but also certain CA-rich clusters that can be described as high- (ACAC, CACA) or low-score motifs (TACA, CACC); those can be organized as direct tandem repeats, or closely spaced within a short RNA region. The number and strength of these SELEX-derived motifs appear to be of high predictive value for hnRNP L binding. Further experimental evidence for this was derived from analyzing hnRNP L binding to our candidate genes (see below).
Selection of candidate genes
Using the NCBI Human Genome Maps and UniGene cluster data, we built a data set of 31 188 gene-based clusters, which have at least one mRNA/complete cds sequence. Transcribed sequences within each cluster were selected only if, first, all splice sites had GT–AG, GC–AG, or AT–AC boundaries, and second, if the quality of the alignment with the genomic sequence (NCBI contig annotation) was greater than 95%.
Based on our SELEX analysis of the hnRNP L recognition sequence (see above), we searched within the data set for CA-repeat regions that contained a minimum of two CACA or ACAC high-score motifs, allowing a spacer in between of maximally eight nucleotides. Based on the UniGene Build #172, a total of 155 325 CA-repeat regions were found, of which 142 542 (92%) were located exclusively in introns, 9067 (6%) exclusively in exons, the remaining rest of 3716 (2%) in alternatively spliced regions, that is in intron or exon regions, depending on the splicing pattern. Of the total 155 325 CA-repeat regions, 1955 (1.3%) are located within 100 bp downstream of a 5′ss. Among these, there are 431 elements with evidence of alternative usage of the upstream 5′ss. After further restricting the analysis to exon skipping cases, we identified 147 candidate genes, for which there is evidence of alternative inclusion or skipping of the upstream exon, but where the distal 5′ss and the downstream 3′ss are constitutive (data not shown).
Intronic CA repeats and CA-rich sequences: role in alternative splicing
Based on our mutational studies on the CA repeats of the human eNOS gene (see above), we tested whether CA repeats and CA-rich intronic elements can determine alternative splicing patterns in vivo. Therefore, we selected from the data sets described above four human genes, MAPK10, GSTZ1, SLC2A2, and RFXANK, representing different lengths of the elements and different distances to the alternatively used 5′ss, all cases of exon skipping, where CA-repeat or CA-rich elements occur within a 100-nucleotide distance to the upstream, alternatively used 5′ss. Each of these three-exon minigene constructs was made, using the pcDNA3 vector under CMV promoter control. The minigene units are comprised of the alternatively used exon and the upstream and downstream flanking exons (Figure 4). Only in the case of MAPK10 and SLC2A2, the two introns had to be shortened for experimental reasons. After transient transfection into HeLa cells, alternative splicing patterns were analyzed by RT–PCR from total RNA, using primers specific for the two flanking exons. Mock controls carried out in the absence of transfected DNA demonstrated that the splicing patterns observed in each case were due to the transfected constructs, and that there was no interference from endogenous mRNA.
Figure 4.
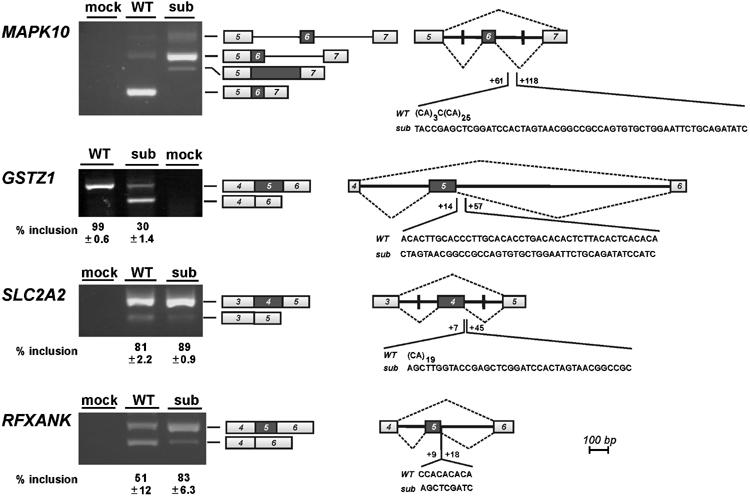
CA-repeat and CA-rich sequences function as regulatory elements of alternative splicing. Alternative splicing of four representative candidate genes that contain intronic CA-repeat elements (MAPK10, SLC2A2, RFXANK) or a CA-rich sequence (GSTZ1) has been characterized, using minigenes diagrammed on the right. The alternatively used exon is indicated by the dark box, and intron and exon parts are shown in scale. Wild type (WT) and substituted (sub) sequence elements are given, with boundaries relatively to the 5′ss of the second intron. Vertical lines within the introns mark the positions where introns have been shortened. Minigenes were transfected into HeLa cells, and alternative splicing tested by RT–PCR (lanes WT, sub); the control transfections (lanes mock) were carried out in the absence of DNA. The positions corresponding to pre-mRNA and alternative mRNA products are indicated to the right of the gel pictures. For the GSTZ1, SLC2A2, and RFXANK minigenes, the percentages of exon inclusion with standard deviations (n=3) are given below the corresponding lanes.
The mitogen-activated protein kinase 10 (MAPK10) gene of approximately 343 kb is responsible for phosphorylation of transcription factor Jun. Alternative splicing through skipping or inclusion of exon 6 (NM_002753) is documented, and our minigene covers exons 5–7, with shortened introns in between (Figure 4). There is an extended CA-repeat region, (CA)3C(CA)25, starting 61 nucleotides downstream from the 5′ss of exon 6, which in the substitution was replaced by a nonspecific sequence of identical length.
Substituting the long CA-repeat element resulted in complete suppression of the normal splice form (exons 5–6–7); instead, the complete intron 6 was retained, indicating that the CA repeats are necessary for efficient splicing of intron 6. In addition, we observed with the altered minigene a minor splice variant, which originated from activation of a cryptic 5′ss within intron 6, downstream of the substituted sequence (as determined by cloning and sequence analysis; data not shown). In sum, this provides evidence for an intronic CA-repeat element functioning as a splicing enhancer.
GSTZ1 encodes a multifunctional enzyme with the glutathione S-transferase (Zeta-1 type) activity, conjugating glutathione to a wide variety of substrates, as well as with maleylacetoacetate isomerase activity, which is important in the catabolism of phenylanaline and tyrosine. The 10.6-kb gene of nine exons (NM_145870) carries in its intron 5 a CA-rich region downstream of the alternatively used exon 5 (positions +14 to +57; Figure 4). To analyze a potential role of the CA-rich region in alternative splicing, we constructed a minigene with exons 4–6; in the substitution construct, the CA-rich sequence was replaced by a nonspecific sequence of corresponding length.
Transfection of the GSTZ1 minigene gave almost exclusively the constitutive splicing pattern (exons 4–5–6; 99±0.6%). However, after substitution the skipped form (exons 4–6) became the predominant form, and only 30±1.4% were included. Therefore, we conclude that a CA-rich element in 5′ss proximity can also act as a splicing enhancer.
Next we analyzed two cases, SLC2A2 and RFXANK, where CA-repeat elements reside in the immediate neighbourhood of 5′ss. First, the SLC2A2 gene of approximately 30 kb contains 11 exons (NM_000340), coding for a specialized glucose transporter molecule, GLUT2, which is expressed in the β-cells of pancreatic islets and likely functions as a glucose sensor. Its exon 4 is included or skipped, and the 5′ss of exon 4 is followed by a (CA)19 sequence starting at position +7 of intron 4 (Figure 4). In this minigene the two introns had to be shortened, and in the substitution derivative, (CA)19 was replaced by a nonspecific sequence.
For the wild-type version of this minigene, we detected mostly the mRNA with exons 3–4–5 joined with each other (81±2.2%), and only a small fraction of skipped mRNA (exons 3–5). In case of the altered minigene, inclusion of exon 4 increased to 89±0.9%, and the skipped form was hardly detectable, indicating a negative effect of the CA-repeat region on splicing (silencer effect).
Second, the RFXANK (regulatory factor X-associated ankyrin-containing) gene, which codes for a transactivator of MHC class II genes, is organized in 10 exons spanning 9.7 kb (NM_003721). Exon 5 is alternative, and a short CA-repeat region [C(CA)4] starts at position +9 of the downstream intron (Figure 4). Our minigene construct comprises exons 4–6 and the full-length introns. In the substitution derivative the wild-type CA-repeat sequence, [C(CA)4], was replaced by 5′-AGCTCGATC-3′.
For the RFXANK minigene, a similar effect was detected as for SLC2A2: The distribution of the included (exons 4–5–6) versus the skipped form (exons 4–6) was clearly shifted towards exon inclusion by the substitution (percentage of exon inclusion: 51±12% for the wild type, 83±6.3% for the substitution); this indicates a splicing silencer role of the short CA-repeat region, C(CA)4.
HnRNP L binds specifically to CA-rich elements of candidate genes
As previously demonstrated by UV-crosslinking experiments (Hui et al, 2003), hnRNP L binds directly and with high affinity to CA-repeat RNAs ((CA)32, (CA)20, and (CA)10). Since we have shown here for the first time, that CA-repeat and CA-rich RNA elements can regulate alternative splicing, we next determined whether hnRNP L directly interacts also with CA-rich (GSTZ1) and with very short CA-repeat sequences (RFXANK).
First, we tested this by GST pull-down assays with recombinant GST-hnRNP L protein (Figure 5A). 32P-labeled RNAs of 44 nucleotides each were transcribed, covering the CA-elements of GSTZ1 and RFXANK, as well as their corresponding substituted sequences of identical length; (CA)20 served as positive control. These RNAs were incubated with baculovirus-expressed GST-hnRNP L, which had been immobilized on glutathione-Sepharose. After extensive washing, bound RNAs were recovered and analyzed by denaturing gel electrophoresis. As expected, more than 50% of the input (CA)20 RNA bound to hnRNP L (lanes CA20). The CA-rich element of the wild-type GSTZ1 sequence bound very efficiently as well, whereas no significant bound material could be recovered from the corresponding substituted sequence (compare lanes wild type (WT) and substitution (S) for GSTZ1). This can be nicely explained by the presence of five high- and one low-score motifs (underlined) in the GSTZ1 wild-type element; in contrast, the GSTZ1 substitution sequence carries none of these motifs:
Figure 5.

HnRNP L binds specifically to intronic CA-repeat and CA-rich elements of candidate genes. (A) GST-hnRNP L pull-down assays. 32P-labeled short RNAs containing the CA-repeat and CA-rich elements of candidate genes GSTZ1 and RFXANK (wild type, lanes WT, and substitution version, lanes S) as well as the positive control (CA)20 (lanes CA20) were incubated with GST-hnRNP L, which had been immobilized on glutathione-Sepharose. Bound RNAs were recovered and analyzed by denaturing gel electrophoresis. For each pull-down assay, 10% of the input was loaded (left half of figure) and the total amount of recovered material (right half). M, markers (36, 67, and 76 nucleotides). (B) Anti-hnRNP L immunoprecipitation assays from HeLa nuclear extract. 32P-labeled short RNAs containing the CA-repeat and CA-rich elements of candidate genes GSTZ1 and SLC2A2 (wild type, lanes WT, and substitution version, lanes S) as well as the positive control (CA)20 (lanes CA20) were incubated in HeLa nuclear extract. Following immunoprecipitation with anti-hnRNP L antibodies, coprecipitated RNAs were analyzed by denaturing gel electrophoresis. For each assay, 10% of the input was loaded (left half of figure) and the total of immunoprecipitated material (right half). M, markers (36, 67, and 76 nucleotides). (C) Coupled UV-crosslinking/anti-hnRNP L immunoprecipitation assays. Incubations as described in panel B were subjected to UV-crosslinking, followed by anti-hnRNP L immunoprecipitation. For each assay, 10% of the reaction after crosslinking was loaded (left half of figure) and the total of crosslinked/immunoprecipitated material (right half). The arrow indicates the mobility of crosslinked hnRNP L (protein markers in kDa).
GSTZ1, wild type: ACACTTGCACCCTTGCACACCTGACACACTCTTAC ACTCACACA
GSTZ1, substitution: CTAGTAACGGCCGCCAGTGTGCTGGAATTCTGCAG ATATCCATC
The RFXANK element also bound hnRNP L in a sequence-specific manner (compare lanes wild type (WT) and substitution (S) for RFXANK), although much less efficiently than the GSTZ1 element and (CA)20 did. Consistent with the high predictive value of the binding motifs described (see above under our SELEX analysis), the RFXANK wild-type sequence contains only two high-score motifs in direct tandem orientation (CCACACACA); the RFXANK substitution derivative (AGCTCGATC), however, carries no motif.
Second, we did in vitro binding assays in nuclear extract, using immunoprecipitations of the endogenous hnRNP L protein (Figure 5B). Short 32P-labeled RNAs of between 44 and 50 nucleotides in length were incubated in nuclear extract, followed by anti-hnRNP L immunoprecipitation. In addition to GSTZ1 wild type and substituted sequences, we included the CA-repeat element of SLC2A2 (wild type versus substitution), as well as CA20 as a control. The MAPK10 element was not tested, since its long CA-repeat stretches are very similar to the SLC2A2 element. These more stringent assays clearly showed that endogenous hnRNP L associates very efficiently with the wild-type sequences of both GSTZ1 and SLC2A2 elements, as well as with CA20; in contrast, binding to the substitution derivatives was undetectable. Binding of hnRNP L to the RFXANK element under these conditions was not significant and similar for wild type and altered versions (data not shown).
Third, we studied whether hnRNP L contacts the RNA elements directly, using a combined crosslinking/immunoprecipitation protocol (Figure 5C): Again, both GSTZ1 and SLC2A2 elements were incubated in nuclear extract, resulting in crosslinking of an hnRNP L-sized protein band for both the GSTZ1 and SLC2A2 element, as well for CA20; the identity of hnRNP L was subsequently confirmed by anti-hnRNP L immunoprecipitation from the crosslinking reactions.
In sum, hnRNP L directly binds in extract both the CA-rich element of GSTZ1, which functions as an enhancer, and the CA-repeat silencer element of SLC2A2. Thus, the same protein can recognize both CA-repeat and CA-rich RNA elements and may participate in both enhancer and silencer functions. Although the short CA element in RFXANK has a clear silencer role (see Figure 4), hnRNP L binding could be detected only with purified recombinant protein (see Figure 5A), and only at a very low level. However, this is not surprising, since in our earlier study we had found that an RNA containing only 10 CA repeats crosslinked only very weakly to hnRNP L (Hui et al, 2003). Therefore, in this case hnRNP L binding is probably functionally not significant, and for such short CA elements proteins in addition to or alternatively to hnRNP L may assemble to enhancer/silencer complexes.
We conclude that CA-rich or CA-repeat elements can regulate alternative splice site choice either positively or negatively, and hnRNP L may participate in both processes. Based on the selection of genes analyzed, it appears that short distances of the CA-sequence to the alternative 5′ss correlate with a silencer role (SLC2A2, RFXANK), longer distances with enhancer function (MAPK10, GSTZ1, eNOS). A likely explanation would be that hnRNP L (or alternative proteins) can work as an activator of the 5′ss only from a certain distance (as demonstrated for the eNOS CA-repeat enhancer; Hui et al, 2003); in contrast, bound at a 5′ss-proximal position, proteins may sterically block recogition of the 5′ss by the general machinery. What specifies positive or negative regulation, whether additional proteins or interactions are responsible and sensitive to the 5′ss proximity, is of particular interest and remains to be investigated by detailed mechanistic studies.
Materials and methods
Oligonucleotides, constructs
The sequences of all oligonucleotides are listed in Supplementary Materials and methods. All constructions were confirmed by sequence analysis.
pT7 DUP-CA32 and pT7 DUP-control. The parent plasmid from which these two clones were made was pT7 DUP4-1, which is a T7 version of pDUP4-1 (Modafferi and Black, 1997). It contains three exons and two introns derived from human β-globin sequence. A fragment containing 32 CA repeats was generated by annealing two oligonucleotides, DUP1 and DUP2, and cloned into the BglII site in the second intron of pT7 DUP4-1, resulting in pDUP-CA32. Clone pDUP-control was constructed by insertion of a 64-nucleotide nonspecific sequence into pT7 DUP4-1 at the BglII site. The 64-nucleotide sequence was amplified from the polylinker region of pGEM5Zf(+) (Promega, USA), using oligonucleotides DUP3 and DUP4.
pGEM-CA32(−21), pGEM-CA32(+23), pGEM-CA32(+175), and pGEM-opt 5′ss. A fragment containing 32 CA repeats was generated by annealing two oligonucleotides, DUP1 and DUP2, and cloned into the BamHI site in the eNOS exon 13 of pGEM-CA32 (Hui et al, 2003), resulting in pGEM-CA32(−21). To construct pGEM-CA32(+23) and pGEM-CA32(+175), a BglII site was introduced in pGEM-CA32 at positions +23 and +175 of intron 13, using a two-step PCR mutagenesis method and oligonucleotides, EN1, EN2, +23up, +23dw, +175up, and +175dw. The annealed DUP1 and DUP2 oligonucleotides containing 32 CA repeats were subsequently inserted into the introduced BglII site. To make pGEM-opt 5′ss, a PCR fragment containing three point mutations at the 5′ss of intron 13 was amplified using oligonucleotides css and EN2 with pGEM-CA32(+175) as template, and cloned between the BamHI and XbaI sites of pGEM-CA32.
pDSX-CA32. A fragment containing 32 CA repeats was generated by annealing two oligonucleotides, DSX1 and DSX2, and cloned into the XbaI and HindIII sites of pDSX-XH (Woerfel and Bindereif, 2001), resulting in pDSX-CA32.
Minigene constructs. All minigene constructs are composed of three exons and two introns. The genomic sequences of the minigene unit were amplified by using genomic DNA isolated from primary HUVEC cells as template (kindly provided by Dr Karl Stangl, Charité, Berlin). To construct the wild-type GSTZ1 and RFXANK minigenes, PCR fragments carrying the complete minigene unit were generated by primer pairs GSTZ-C1/-C2, and RFXANK-C1/-C2, and inserted into pcDNA3 vector (Invitrogen, USA). In the case of MAPK10 and SLC2A2, most of the intron sequences was deleted from 100 nt downstream of the 5′ss to 100 nt upstream of the 3′ss, except in the second intron of MAPK10 minigene, where the deleted sequence starts 150 nt downstream of the 5′ss to 100 nt upstream of the 3′ss. A series of PCRs were performed, using oligonucleotides MAPK-C1 to -C6 and SLC2A2-C1 to -C6 as primers. A complete fragment was assembled by a second-step PCR, using the previous PCR products as templates, and inserted into the corresponding sites of pcDNA3 vector. In the substituted minigene constructs, the CA repeats or C/A-rich sequences in the wild-type minigene constructs were replaced by nonspecific control sequences, using a similar two-step PCR mutagenesis method and oligonucleotides MAPK-C1, -C2, and -C7 to -C10; GSTZ-C1, -C2, and -C5 to -C8; SLC2A2-C1, -C2, -C7, and -C8; RFXANK-C1 to -C4. The nonspecific sequences in SLC2A2, GSTZ1, and MAPK10 substitution constructs were amplified from the polylinker region of pcDNA3.
In vitro transcription and splicing
pDUP and pGEM derivatives were cut with XbaI and transcribed in the presence of m7GpppG cap analog; 10 ng of pre-mRNA per 25-μl reaction was spliced in HeLa nuclear extract (Bindereif and Green, 1987). Splicing activity was detected by denaturing polyacrylamide gel electrophoresis or by RT–PCR analysis, using oligonucleotides EN2 and DUP6.1 as primers for RT, and oligonucleotides EN1, EN2, DUP5, DUP6.1, DUP9, and DUP10 as primers for PCR. 32P-labeled short RNAs for GST pull-down assays were in vitro transcribed as above, except that [32P]CTP was used. DNA templates for transcription were obtained by PCR, using primers GSTZ-U1 to -U4, and RFXANK-U1 to -U3, and the corresponding minigene constructs as templates, or alternatively, by annealing oligonucleotides SLC2A2-U1/-U2 and -U3/U4.
Recombinant hnRNP L-GST fusion protein
To construct pFASTBAC HTb-hnRNP L-GST, the KpnI–HindIII cleavage product, which contains the 3′ end and 3′-UTR of hnRNP L in pFASTBAC HTb-hnRNP L (Hui et al, 2003), was substituted by a fragment obtained by a two-step PCR method. For the first set of PCRs, the DNA fragments encoding the C-terminus of hnRNP L and GST protein were amplified using oligonucleotide pairs LGST-1/-2, LGST-3/-4, respectively. A second-step PCR was performed with primers LGST-1 and -4, and the above fragments as templates. The resulting PCR product was subsequently digested by KpnI and HindIII, and cloned into pFASTBAC HTb-hnRNP L. Recombinant baculoviruses were generated to express C-terminally GST-tagged hnRNP L in Sf21 cells according to the protocols of Invitrogen, followed by purification through glutathione-Sepharose (Amersham Biosciences, Sweden) and dialysis against buffer D (20 mM HEPES, pH 8.0, 20% glycerol, 100 mM KCl, 0.2 mM EDTA, 1 mM DTT, and 0.5 mM PMSF) at 4°C for 3 h.
SELEX
The SELEX procedure and the analysis of the consensus sequence are described in the Supplementary Materials and methods.
Filter binding assay
The filter-binding assay was carried out on a 96-well vacuum filtration apparatus (BioRad, USA). Approximately 50 fmol in vitro transcribed, [32P]CTP-labeled RNA was incubated with different amounts of baculovirus-expressed hnRNP L-GST protein in a final volume of 50 μl of binding buffer (10 mM Tris–Cl pH 8.0, 100 mM KCl, 2.5 mM MgCl2, 0.01% NP40, 10% glycerol, and 0.2 mg/ml BSA) at room temperature for 20 min. The binding reaction was applied onto the nitrocellulose membrane (0.45 μm, Macherey-Nagel, Germany), which had been preincubated with washing buffer (10 mM Tris–Cl pH 8.0, 100 mM KCl, and 2.5 mM MgCl2) at room temperature for 30 min. After each well was washed five times with washing buffer, the filter was subsequently dried and quantitated by a phosphorimager system (Biorad, USA).
Cell transfection, RNA isolation, RT–PCR analysis
The day before transfection, 5 × 105 HeLa cells were seeded on a 6-cm tissue culture dish. Minigene constructs (8 μg) was transfected by the calcium phosphate method (Sambrook et al, 1989). Cells were harvested 2 days after transfection. Total RNA was isolated using guanidinium thiocyanate (Xie and Rothblum, 1991). Total RNA (1 μg) was annealed to 2 pmol vector-specific primer bGH Rev (Invitrogen, USA) and reverse-transcribed by SuperScript™ III RNase-H-minus reverse transcriptase (Invitrogen, USA), according to the manufacturer's instruction. The resulting first-strand cDNA was further amplified by PCR, using primers against the flanking exons. The PCR conditions were optimized for each minigene.
GST pull-down assay
Baculovirus-expressed hnRNP L-GST fusion protein (5 pmol) immobilized on glutathione-Sepharose beads was incubated with 120 fmol in vitro transcribed, [32P]CTP-labeled short RNAs in a final volume of 200 μl of binding buffer (20 mM HEPES pH 7.5, 100 mM KCl, 10 mM MgCl2, 1 mM DTT, and 0.01% NP40). The short RNAs carry the CA sequences or the corresponding substituted sequences derived from the GSTZ1 and RFXANK minigene constructs. After binding at room temperature for 20 min, the beads were washed four times with 1 ml each of washing buffer (20 mM HEPES pH 7.5, 300 mM KCl, 10 mM MgCl2, 1 mM DTT, and 0.01% NP40), followed by treatment with 80 μg of proteinase K in 200 μl PK buffer (100 mM Tris–Cl pH 7.5, 12.5 mM EDTA, 150 mM NaCl, and 1% SDS) at 37°C for 30 min. Bound RNA was recovered by extraction with phenol/chloroform and ethanol precipitation, and separated on a 15% denaturing polyacrylamide gel.
In vitro RNA binding/immunoprecipitation
In total, 4 ng of in vitro transcribed, [32P]CTP-labeled short RNAs was incubated at 30°C for 15 min in HeLa nuclear extract under standard splicing conditions (Bindereif and Green, 1987). For immunoprecipitation experiments, 100 μl packed volume of protein A Sepharose CL-4B (Amersham Pharmacia Biotech) in N100 buffer (100 mM NaCl, 50 mM Tris–Cl pH 8.0, 0.05% NP-40) were first incubated overnight with 10 μl of anti-hnRNP L antibody 4D11 at 4°C, and washed five times with 1 ml of N100. In total, 20 μl packed volume of beads in 175 μl of N100 were then incubated together with 25 μl of splicing reaction at 4°C for 2 h with rotation. The beads were washed three times with 1 ml of N100 buffer, and heated at 80°C for 10 min in 200 μl of PK buffer. Immunoprecipitated RNA was extracted by phenol/chloroform followed by ethanol precipitation, and separated on a 12% denaturing polyacrylamide gel.
UV-crosslinking/immunoprecipitation
UV-crosslinking was performed as described (Hui et al, 2003). Briefly, 8 ng of in vitro transcribed, [32P]CTP-labeled short RNAs was incubated at 30°C for 15 min in HeLa nuclear extract (with tRNA added to a final concentration of 0.2 mg/ml). UV-crosslinking was carried out on ice for 20 min with 254 nm UV light at an intensity of 2700 μW/cm2. Unprotected RNA was digested with RNase A (1 mg/ml) at 37°C for 20 min. For immunoprecipitation, 20 μl of protein A Sepharose beads (prepared as above) in 175 μl of N100 were incubated with the UV-crosslinking reaction at 4°C for 2 h with rotation. The beads were then washed three times with 1 ml of N100 buffer. Immunoprecipitated proteins were fractionated on a 12% SDS–polyacrylamide gel.
Note added in proof:
We note that hnRNP L has recently been found to repress exon splicing via an exonic splicing silencer (CR Rothrock, AE House, KW Lynch, manuscript submitted).
Supplementary Material
Supplementary Figure
Supplementary Materials and methods
Acknowledgments
We thank Karl Stangl (Charité, Berlin) for providing genomic DNA samples, Doug Black for construct pDUP4-1, and Gideon Dreyfuss for anti-hnRNP L monoclonal antibody 4D11. We also thank Uwe Kühn (University of Halle) and Peter Friedhoff (University of Giessen) for help with filter binding assay, Martin Vingron (MPI for Molecular Genetics, Berlin) for discussions, and Andrey Damianov (University of Giessen) for comments on the manuscript. This work was supported by the Deutsche Forschungsgemeinschaft (Grants Bi 316/10-1, 10-2, and 10-3 to AB).
References
- Bilbao D, Valcarcel J (2003) Getting to the heart of a splicing enhancer. Nat Struct Biol 10: 6–7 [DOI] [PubMed] [Google Scholar]
- Bindereif A, Green MR (1987) An ordered pathway of snRNP binding during mammalian pre-mRNA splicing complex assembly. EMBO J 6: 2415–2424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DL (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72: 291–336 [DOI] [PubMed] [Google Scholar]
- Blencowe BJ (2000) Exonic splicing enhancers: mechanism of action, diversity and role in human genetic diseases. Trends Biochem Sci 25: 106–110 [DOI] [PubMed] [Google Scholar]
- Cartegni L, Chew SL, Krainer AR (2002) Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 3: 285–298 [DOI] [PubMed] [Google Scholar]
- Cartegni L, Krainer AR (2002) Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet 30: 377–384 [DOI] [PubMed] [Google Scholar]
- Cooper TA, Mattox W (1997) The regulation of splice-site selection, and its role in human disease. Am J Hum Genet 61: 259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter LR, Landree MA, Cooper TA (1997) Identification of a new class of exonic splicing enhancers by in vivo selection. Mol Cell Biol 17: 2143–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbrother WG, Yeh RF, Sharp PA, Burge CB (2002) Predictive identification of exonic splicing enhancers in human genes. Science 297: 1007–1013 [DOI] [PubMed] [Google Scholar]
- Graveley BR (2000) Sorting out the complexity of SR protein functions. RNA 6: 1197–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahm B, Kim YK, Kim JH, Kim TY, Jang SK (1998) Heterogeneous nuclear ribonucleoprotein L interacts with the 3′ border of the internal ribosomal entry site of hepatitis C virus. J Virol 72: 8782–8788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui J, Stangl K, Lane WL, Bindereif A (2003) HnRNP L stimulates splicing of the eNOS gene by binding to variable-length CA repeats. Nat Struct Biol 10: 33–37 [DOI] [PubMed] [Google Scholar]
- Kashima T, Manley JL (2003) A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet 34: 460–463 [DOI] [PubMed] [Google Scholar]
- Krawczak M, Reiss J, Cooper DN (1992) The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet 90: 41–54 [DOI] [PubMed] [Google Scholar]
- Ladd AN, Cooper TA (2002) Finding signals that regulate alternative splicing in the post-genomic era. Genome Biol 3, reviews 0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HX, Cartegni L, Zhang MQ, Krainer AR (2001) A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat Genet 27: 55–58 [DOI] [PubMed] [Google Scholar]
- Liu X, Mertz JE (1995) HnRNP L binds a cis-acting RNA sequence element that enables intron-independent gene expression. Genes Dev 9: 1766–1780 [DOI] [PubMed] [Google Scholar]
- Modafferi EF, Black DL (1997) A complex intronic splicing enhancer from the c-src pre-mRNA activates inclusion of a heterologous exon. Mol Cell Biol 17: 6537–6545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñol-Roma S, Swanson MS, Gall JG, Dreyfuss G (1989) A novel heterogeneous nuclear RNP protein with a unique distribution on nascent transcripts. J Cell Biol 109: 2575–2587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbour, New York, USA: Cold Spring Harbour Laboratory Press [Google Scholar]
- Shih SC, Claffey KP (1999) Regulation of human vascular endothelial growth factor mRNA stability in hypoxia by heterogeneous nuclear ribonucleoprotein L. J Biol Chem 274: 1359–1365 [DOI] [PubMed] [Google Scholar]
- Stangl K, Cascorbi I, Laule M, Klein T, Stangl V, Rost S, Wernecke KD, Felix SB, Bindereif A, Baumann G, Roots I (2000) High CA repeat numbers in intron 13 of the endothelial nitric oxide synthase gene and increased risk of coronary artery disease. Pharmacogenetics 10: 133–140 [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watakabe A, Shimura Y (1994) Polypurine sequences within a downstream exon function as a splicing enhancer. Mol Cell Biol 14: 1347–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuerk C, Gold L (1990) Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249: 505–510 [DOI] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigo R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, McCarthy M, McCombie WR, McLaren S, McLay K, McPherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O'Connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Von Niederhausern AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang SP, Zdobnov EM, Zody MC, Lander ES, Mouse Genome Sequencing Consortium (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562 [DOI] [PubMed] [Google Scholar]
- Woerfel G, Bindereif A (2001) In vitro selection of exonic splicing enhancer sequences: identification of novel CD44 enhancers. Nucleic Acids Res 29: 3204–3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie WQ, Rothblum LI (1991) Rapid, small-scale RNA isolation from tissue culture cells. BioTechniques 11: 326–327 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure
Supplementary Materials and methods