

Summary
GlycoRNAs are glycosylated RNAs that can be detected in many cell types and often partly reside on the outer cell surface, with a recently demonstrated role in mediating neutrophil-endothelium interaction. Here, we present a protocol for glycoRNA detection based on metabolic tracing and northwestern blot. We describe steps for metabolic labeling of cells, extraction and purification of RNA, biotin labeling of RNA, and northwestern blot for glycoRNA detection. We also incorporate optimized conditions for biotin labeling, RNA dye, and membrane blocking.
For complete details on the use and execution of this protocol, please refer to Zhang et al.1
Subject areas: Cell Biology, Cell culture, Molecular Biology, Molecular/Chemical Probes, Chemistry
Graphical abstract
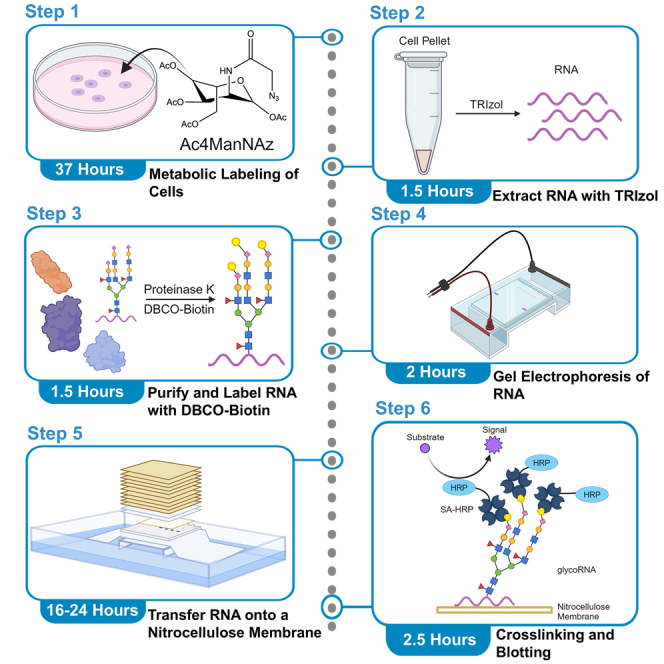
Highlights
-
•
Metabolic labeling of glycans, RNA extraction, and DBCO-biotin labeling
-
•
Denaturing electrophoresis and RNA transfer onto a nitrocellulose membrane
-
•
Crosslinking and blotting to visualize glycoRNA signals
-
•
Optimized DBCO-biotin reaction, RNA dye, and blocking
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
GlycoRNAs are glycosylated RNAs that can be detected in many cell types and often partly reside on the outer cell surface, with a recently demonstrated role in mediating neutrophil-endothelium interaction. Here, we present a protocol for glycoRNA detection based on metabolic tracing and northwestern blot. We describe steps for metabolic labeling of cells, extraction and purification of RNA, biotin labeling of RNA, and northwestern blot for glycoRNA detection. We also incorporate optimized conditions for biotin labeling, RNA dye, and membrane blocking.
Before you begin
GlycoRNAs are a group of glycosylated RNAs that can be detected in many cell types.1,2,3 The presence of RNAs with glycan modification2,4 raises substantial interest. It expands the realm of glycosylated molecular species beyond proteins and lipids. In addition, in many cell types, a fraction of glycoRNAs has been shown, with direct or indirect evidence, to be localized on the outer cell surface.1,2,3,5 The cell surface localization of RNAs, such as in human circulating monocytes,6 has raised interesting questions because the location is topologically different from the nucleus and cytoplasm where most cellular RNAs reside. The functional importance of glycoRNAs has recently been shown in the setting of neutrophil recruitment where the cell surface fraction of neutrophil glycoRNAs regulates neutrophil-endothelial-cell interaction.1 It is likely that many more functions of glycoRNAs will be discovered in the years ahead.
The most well-established method for the detection of glycoRNAs is based on metabolic labeling of cells with clickable sugar precursors,1,2,3 such as N-azidoacetylmannosamine-tetraacylated (Ac4ManNAz) that can be added to cell culture and incorporated into glycans as a sialic acid mimetic. With copper-free click chemistry, these RNAs can be analyzed by northwestern blot for detection. There are several other methods recently reported to detect glycoRNAs.3,4,5 One of such methods, RNA optimized periodate oxidation and aldehyde labeling (rPAL),4 oxidizes the chemical diol structures on sialic acid moieties in glycoRNAs into aldehydes, tags such aldehyde groups with biotin, and analyses RNAs by northwestern blots. While there are strengths and weaknesses for each of these two methods when cross-compared, a substantial part of the northwestern protocol described here can also be applied to the detection of glycoRNAs labeled by the rPAL method.
This protocol is modified based on the original procedures for glycoRNA labeling and detection described by Flynn et al.2 We have performed optimization experiments on three aspects of the protocol, including the click reaction condition with Dibenzocyclooctyne-(Polyethylene Glycol)4-Biotin (DBCO-PEG4-biotin, or abbreviated as DBCO-Biotin, Figure S1), the RNA dyes and their dilution for quantitative detection of RNA loading (Figure S2), and the blocking condition for streptavidin-based detection (Figure S3). The experiments and the results of our optimization experiments are detailed in Supplemental Materials. We also want to highlight two key considerations here. First, it is critical to avoid glycan contamination from protein or lipid sources when preparing RNAs. Therefore, having high-purity RNA preparations coupled with subsequent proteinase K digestion is important. By the same consideration, care needs to be put into the choice of the initial RNA preparation method both to minimize such contaminations and to avoid the loss of glycoRNAs which are mostly small RNAs.1,2 We routinely use TRIzol for RNA preparation which removes proteins and hydrophobic contaminants such as lipids and preserve small RNAs during precipitation. Second, we found that it is critical that the appropriate amount and type of RNA dye are used when running RNAs on gels. We found that the frequently used RNA dye, SYBR Gold, when used at the manufacturer suggested dilution of 1:10,000 (often referred to as 1×), is extremely poor to distinguish different RNA loading amounts, therefore such conditions can be prone to erroneous conclusions about sample equal loading.
For the purposes of optimization and demonstrating the steps of the protocol, we used Ba/F3 cells.7 a murine hematopoietic progenitor cell line, due to the abundance of glycoRNAs in this cell line, the ease to culture, and fast proliferation rates. For other cell types, the levels of glycoRNAs are likely different from those in Ba/F3 cells and their tolerance of Ac4ManNAz treatment could be different. We recommend optimizing the duration of Ac4ManNAz treatment and the amount of RNAs to be loaded.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Agarose GPG/LE | American Bioanalytical | Cat # AB00972-00500 |
| MOPS (3-(N-morpholino) propanesulfonic acid) | American Bioanalytical | Cat # AB01270-01000 |
| Non-fat dry milk Omniblok | American Bioanalytical | Cat # AB10109-01000 |
| Tris, 2 M solution, pH 7.5 | American Bioanalytical | Cat # AB14116-01000 |
| EDTA, 0.5 M solution, pH 8.0 | American Bioanalytical | Cat # AB00502 |
| SDS, 20%, liquid solution | Apex Bioresearch Products | Cat # 18-234B |
| TBST (TBS-Tween 20), 10×, pH 7.6 | Apex Bioresearch Products | Cat # 18-235B |
| EveryBlot blocking buffer | Bio-Rad | Cat # 12010020 |
| N-azidoacetyl-mannosamine (Ac4ManNAz) | Click Chemistry Tools | Cat # 1084-100 |
| Dulbecco’s phosphate-buffered saline, 1× without calcium and magnesium | Corning | Cat # 21-031-CV |
| Formamide (certified ACS) | Fisher Chemical | Cat # F84-1 |
| Fetal bovine serum, heat inactivated | Gibco | Cat # 10438-026 |
| Penicillin-streptomycin-glutamine (100×) | Gibco | Cat # 10378-016 |
| RPMI 1640 medium | Gibco | Cat # 11875-093 |
| Trypan blue solution, 0.4% | Gibco | Cat # 15250061 |
| SYBR Gold nucleic acid gel stain | Invitrogen | Cat # S11494 |
| SYBR Safe DNA gel stain | Invitrogen | Cat # S33102 |
| TRIzol | Invitrogen | Cat # 15596-018 |
| UltraPure DNase/RNase-free distilled water | Invitrogen | Cat # 10977015 |
| EDTA disodium salt dihydrate, crystals | J.T.Baker | Cat # 8993-01 |
| Sodium acetate, anhydrous | J.T.Baker | Cat # JT3470-1 |
| Intercept (PBS) blocking buffer | LI-COR | Cat # 927-70003 |
| GelStar nucleic acid gel stain | Lonza | Cat # 50535 |
| Immobilon Crescendo Western HRP substrate, 500 mL | Millipore | Cat # WBLUR0500 |
| Gel loading dye, purple (6×) | New England Biolabs | Cat # B7024S |
| Recombinant murine IL-3 | PeproTech | Cat # 213-13 |
| High sensitivity streptavidin-HRP (SA-HRP) | Pierce | Cat # 21130 |
| Diamond nucleic acid dye | Promega | Cat # H1181 |
| Milli-Q Water | Provided by your institute | N/A |
| Recombinant proteinase K powder | Roche | Cat # 3115836001 |
| 2-propanol | Sigma-Aldrich | Cat # I9516-500ML |
| Chloroform | Sigma-Aldrich | Cat # 319988-500ML |
| D-(+)-galactose | Sigma-Aldrich | Cat # G0750-50G |
| DEPC, diethyl pyrocarbonate | Sigma-Aldrich | Cat # D5758-50ML |
| Dibenzocyclooctyne-PEG4-biotin conjugate | Sigma-Aldrich | Cat # 760749 |
| Ethyl alcohol, pure | Sigma-Aldrich | Cat # E7023-500ML |
| Formaldehyde solution | Sigma-Aldrich | Cat # 252549-500ML |
| N-acetylgalactosamine (GalNAc) | Sigma-Aldrich | Cat # A2795 |
| Sodium hydroxide solution, 10 M | Sigma-Aldrich | Cat # 72068-100ML |
| SSC buffer 20× concentrate | Sigma-Aldrich | Cat # S6639-1L |
| RNase AWAY surface decontaminant | Thermo Scientific | Cat # 7002PK |
| Critical commercial assays | ||
| RNA Clean & Concentrator-25 kits | Zymo Research Corporation | Cat # 1159U74 (R1018) |
| Experimental models: Cell lines | ||
| Ba/F3 cells | Lab stock | N/A |
| Other | ||
| Transparency film | Apollo | Cat # VWO100C-B |
| Nitrocellulose membrane, 0.2 μm pore | Bio-Rad | Cat # 1620112 |
| Corning 100 mm TC-treated culture dish | Corning | Cat # 430167 |
| 15 mL and 50 mL conical centrifuge tubes | Falcon | Cat # 14-959-49D and 14-432-22 |
| Syngene G:BOX Chemi XRQ | Fisher Scientific | Cat # 01-257-162 |
| Kleenex premiere paper towel, 11 × 10.4 in | Kleenex | Cat # 13964 |
| 1.7 mL microcentrifuge tubes | Posi-Lock | Cat # 280175 |
| Owl D2 wide-gel electrophoresis system | Thermo Scientific | Cat # D2-BP |
| Whatman filter paper | VWR | Cat # 28298020 |
Materials and equipment
Ba/F3 cell culture media
| Reagent | Final concentration | Amount |
|---|---|---|
| RPMI 1640 Medium | N/A | add to 50 mL |
| Fetal Bovine Serum, Heat Inactivated | 10% | 5 mL |
| Penicillin-Streptomycin-Glutamine (100×) | 1% | 0.5 mL |
| Interleukin-3 (IL-3) (20 μg/mL stock) | 3 ng/mL | 7.5 μL |
| Total | N/A | 50 mL |
Can be stored at 4°C.
Media for metabolic labeling
| Reagent | Final concentration | Amount |
|---|---|---|
| Ba/F3 Cell Culture Media | N/A | add to 50 mL |
| N-Azidoacetyl-mannosamine (Ac4ManNAz) (500 mM) | 0.1 mM | 10 μL |
| D-(+)-Galactose (50 mM) | 10 μM | 10 μL |
| N-acetylgalactosamine (GalNAc) (500 mM) | 0.1 mM | 10 μL |
| Total | N/A | 50 mL |
Make fresh.
RNA formaldehyde denaturing del (1%)
| Reagent | Final concentration | Amount |
|---|---|---|
| Agarose GPG/LE | 1% | 1 g |
| Formaldehyde Solution (37%) | 6.66% | 18 mL |
| MOPS Buffer (10×) | 1× | 10 mL |
| DEPC-Treated Water | N/A | 72 mL |
| Total | N/A | 100 mL |
Make fresh.
Dye-free gel loading buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| Formamide | 95% | 950 μL |
| EDTA pH 8.0 (0.5 M) | 18 mM | 36 μL |
| SDS (20%) | 0.025% | 1.25 μL |
| UltraPure DNase/RNase-Free Distilled Water | N/A | 12.75 μL |
| Total | N/A | 1 mL |
Can be stored at 25°C.
Alternatives: SDS can be removed from the recipe and supplemented with water instead.
MOPS buffer (10×)
| Reagent | Final concentration | Amount |
|---|---|---|
| MOPS (3-(N-Morpholino) Propanesulfonic Acid) | 0.2 M | 41.86 g |
| Sodium Acetate | 0.05 M | 4.1 g |
| EDTA Disodium Salt Dihydrate | 0.01 M | 3.72 g |
| Sodium Hydroxide Solution(10 M) | N/A | adjust pH to 7.0 |
| DEPC-Treated Water | N/A | add to 1 L |
| Total | N/A | 1 L |
Can be stored at 25°C.
DEPC-treated water: In the fume hood, add 2 mL of Diethyl Pyrocarbonate (DEPC) into 2 L of MilliQ water. Stir with a stir bar for at least 2 h or vigorously mix by shaking the container at 25°C. Then autoclave to degrade excessive DEPC.
20 mg/mL Proteinase K Solution: Dissolve 25 mg of proteinase K with 1.25 mL DEPC-treated water. Once it is fully dissolved, incubate at 37°C for 5 min to inactivate any potential RNase contamination in the proteinase K product. This inactivation step only needs to be performed once. Proteinase K solution can be stored at −20°C. Routine tests of proteinase K activity are recommended, especially after proteinase K has been frozen and thawed multiple times. We use bovine serum albumin (BSA) as a substrate in these tests (an example of the test: We treat 0.5 mg/mL BSA with or without 1 mg/mL proteinase K in a total volume of 20 μL at 37°C for 30 min, and run the samples on a 10% SDS-PAGE (polyacrylamide gel electrophoresis) gel. We then stain the gel with the Coomassie blue stain. If the proteinase K is sufficiently active, all the BSA bands, especially the main band at ∼66 kDa, will disappear in the sample with proteinase K treatment.1
75% ethanol: To make 15 mL of 75% ethanol, add 11.25 mL 100% ethanol and then add DEPC-treated water to 15 mL.
Step-by-step method details
Metabolic labeling of Ba/F3 cells
Timing: ∼37 h
In this section, we describe how to introduce the metabolic tracer Ac4ManNAz into Ba/F3 cells. For other cell types, similar treatment can be used in the corresponding cell culture media.
-
1.
Prepare Ba/F3 culture media for maintenance as described in materials and equipment.
-
2.
Prepare Ba/F3 cell culture media for metabolic labeling as described in materials and equipment. Mix well and set aside at 25°C.
-
3.Prepare the cells:
-
a.Seed ∼2.25×106 Ba/F3 cells into each new 10 cm dish.Note: Use the labeling media so that for each 10 cm plate, there is 10 mL of labeling media.Note: The exact concentration of cells is not crucial. The number of cells to be plated should allow cells to be healthy during the metabolic labeling period and yield enough RNA for detection.Note: For any other cell types, a starting point of optimization can involve adding media containing the metabolic reporter 1.5 days before the cells become confluent. For some cells, a shorter labeling period may be necessary to avoid toxicity.
-
b.Gently move the plates so that cells are evenly distributed.
-
c.Put the cells back into the incubator.
-
d.Make note of the time when the labeling media was added.Note: We normally label cells for ∼36 h but shorter durations often yield acceptable results too.
-
a.
Harvest cells and extract RNA
In this section, we describe how to harvest Ba/F3 cells and extract RNA using TRIzol.
-
4.Harvest cells:
-
a.Harvest cells and transfer cells into a 15 mL or 50 mL conical tube.
-
b.Centrifuge to pellet cells (e.g., 201 g for 3 min for Ba/F3 cells).Note: For adherent cells, try to avoid using proteinase-based (e.g. trypsin) reagents to harvest cells, due to the possibility that proteinase digestion will disrupt the protein anchors/protectors of the cell surface fraction of glycoRNAs. Instead, use ice-cold PBS to wash cells on the plate and then scrape cells off using a cell scraper.Note: Count cells, when possible, to help estimate RNA yield per cell and gauge the amount of TRIzol to be used.
-
c.There will be a pellet at the bottom of the conical tube after centrifugation. Discard supernatant.
-
d.Resuspend cells in 1 mL of PBS.
-
e.Transfer cells to an Eppendorf tube.
-
f.Centrifuge to pellet cells.Note: Wash cells with PBS once or twice before this transfer step can help to further remove contaminants in the cell culture media that contain fetal bovine serum (FBS).
-
a.
-
5.Extract RNA with TRIzol (follow TRIzol protocol):Note: Unless specified otherwise, the sample is kept on ice. We also use filter-tips to prevent contaminations.
-
a.Before extracting RNA, spray the benchtop and wipe the pipettes with RNase AWAY Surface Decontaminant.Note: This step helps to reduce the probability of RNase contamination.
-
b.Loosen the cell pellet by vertexing the tube briefly.Note: This step is highly recommended if the cell pellet is large. Loosen the cell pellet will help to avoid incomplete lysis of cells by TRIzol.
-
c.In a fume hood, add 1 mL of TRIzol into the cell pellet.
 CRITICAL: 1 mL of TRIzol can maximally take 100 μL of cell pellet. If cell pellet is larger, use more TRIzol or use the TRIzol LS reagent.Note: Alternatively, for Ba/F3 cells, 1 mL of TRIzol per ∼6 × 106 cells can be used.
CRITICAL: 1 mL of TRIzol can maximally take 100 μL of cell pellet. If cell pellet is larger, use more TRIzol or use the TRIzol LS reagent.Note: Alternatively, for Ba/F3 cells, 1 mL of TRIzol per ∼6 × 106 cells can be used. -
d.Incubate at room temperature for 15 min.
-
e.Add 0.2 mL of chloroform per 1 mL of TRIzol.
-
f.Vortex vigorously for 15 s and incubate on ice for 3 min.
-
g.Centrifuge the sample at 12,000–22,000 g for 10 min at 4°C.
-
h.Transfer the aqueous phase to a new 1.5 mL Eppendorf tube.Note: Make sure the pipette tip does not touch the interphase or the organic layer.CRITICAL: Pipette tip touching any layers other than the top layer could lead to contamination of the RNA sample with protein, lipid, or DNA.
-
i.Measure the volume of liquid taken from the aqueous phase with a 1 mL pipette.
-
j.Add the same volume of isopropanol, mix well, and incubate at −20°C for 20 min.
-
k.Centrifuge at 12,000–22,000 g for 15 min at 4°C.Note: There would be a white pellet at the bottom of the Eppendorf tube.
-
l.Discard the supernatant by pipetting with a 1 mL tip.Note: It is fine to leave ∼20–30 μL of supernatant to avoid touching pellet with tip.
-
m.Add 1 mL of 75% ethanol to wash the pellet.Note: Avoid disrupting the pellet by adding the wash solution against the tube wall.
-
n.Gently invert the tube for ∼5 to 10 times to wash the RNA pellet.
-
o.Centrifuge at 12,000–22,000 g for 5 min at 4°C.
-
p.Discard the supernatant by pipetting with a 1 mL tip.Note: It is fine to leave ∼20–30 μL of supernatant to avoid touching pellet with the tip.
-
q.Briefly centrifuge to collect all remaining supernatant (and RNA pellet) at the bottom of the tube
-
r.Remove the remaining supernatant with a 200 μL pipette.Note: Be careful to not touch the RNA pellet with the pipette tip.
-
s.Air-dry the RNA pellet for 1–5 min.Note: Over drying the pellet may make RNAs more difficult to dissolve later. If it does happen, the optional heating step below can facilitate the dissolution.
-
t.Resuspend the pellet in an appropriate amount of RNase-free water, with the final concentration of RNA around 1–2 mg/mL.Note: One can use estimates of RNA yield per cell coupled with the number of cells to estimate the amount of water to add.
-
u.Pipette up and down until the pellet completely dissolves.Note: As an optional step, incubate on a heat block at 55°C for 3 min to help dissolve the RNA. After the incubation, return the samples to ice immediately.Optional: Use another round of TRIzol preparation on the purified RNA to further clean up the RNA sample and reduce protein and lipid contaminations. This could be particularly helpful when the starting cell number is high.
-
v.Use a Nanodrop spectrophotometer to measure RNA concentration.Note: Use RNase-free water as a control. The A260/280 value should be ∼2 for RNA.
 Pause point: RNA samples can be stored at −80°C for months.
Pause point: RNA samples can be stored at −80°C for months.
-
a.
Remove protein contamination from isolated RNA and label RNA with DBCO-biotin
In this section, we describe how to use protein K digestion to remove trace-amounts of protein contamination and how to label the resultant RNA with DBCO-Biotin. There are two options for running these steps.
Option 1: Two-step protocol for Proteinase K treatment and DBCO labeling
-
6.Proteinase K treatment of RNA:
-
a.Add Tris pH 7.5, proteinase K, and RNase-free water into isolated RNA (listed below in Table 1).Note: The amount of RNA and the total amount of sample can be adjusted as long as the final concentrations of reagents remain unchanged.
-
b.Mix and incubate in a water bath at 37°C for 30 min.
-
c.After the incubation, immediately put the sample on ice.
-
a.
-
7.Purify RNA with the ZYMO RNA Clean & Concentrator kit:
-
a.Add RNase-Free Water to the RNA to make a final volume of 50 μL.
-
b.Add 100 μL of the RNA Binding Buffer to each sample.
-
c.Pipette up and down several times to mix.
-
d.Add 150 μL of 100% ethanol.
-
e.Pipette up and down several times to mix.
-
f.Transfer the sample to a Zymo-Spin IICR Column with a Collection Tube.
-
g.Centrifuge at 7,000 g for 30 s at 4°C. Discard the flow through.
-
h.Add 400 μL RNA Prep Buffer to the column.
-
i.Centrifuge at 13,000 g for 30 s at 4°C. Discard the flow through.
-
j.Add 700 μL RNA Wash Buffer to the column.
-
k.Centrifuge at 13,000 g for 30 s at 4°C.
-
l.Discard the flow through.
-
m.Add 400 μL RNA Wash Buffer to the column.
-
n.Centrifuge at 13,000 g for 1 min at 4°C, and discard the flow through.
-
o.Centrifuge for 30 s at 4°C to remove any remaining Wash Buffer from the column.
-
p.Transfer the column into a 1.5 mL Eppendorf tube.
-
q.Add 25 μL RNase-Free Water into the column.
-
r.Wait for 1 min, and centrifuge at 13,000 g for 30 s at 4°C.
-
s.Put the eluted RNA on ice.
-
t.Use a Nanodrop spectrophotometer to measure RNA concentration.
-
a.
Note: Use RNase-Free Water as a control for Nanodrop.
-
8.DBCO-Biotin Label Purified RNA.
-
a.Add DBCO-PEG4-Biotin and UltraPure DNase/RNase-free distilled water into purified RNA (listed below in Table 2).Note: The amount of RNA and the total amount of the sample can be adjusted as long as the final concentrations of reagents remain unchanged.
-
b.Mix and incubate at 37°C for 30 min.
-
c.After incubation, put the sample immediately on ice.
-
a.
-
9.
Purify RNA with the ZYMO RNA Clean & Concentrator kit (Follow ZYMO RNA Clean & Concentrator protocol) as in step 7.
Table 1.
Proteinase K treatment mixture
| Reagent | Final concentration | Amount |
|---|---|---|
| RNA | N/A | variable |
| Tris pH 7.5 (200 mM, diluted with RNase-free Water from 2 M Tris Solution, pH 7.5) | 10 mM | 2 μL |
| Proteinase K Solution (20 mg/mL) | 1 mg/mL | 2 μL |
| UltraPure DNase/RNase-Free Distilled Water | N/A | add to 40 μL |
| Total | N/A | 40 μL |
Table 2.
DBCO-biotin label mixture
| Reagent | Final concentration | Amount |
|---|---|---|
| RNA | N/A | 5 μg to 15 μg |
| DBCO-PEG4-Biotin (10 mM) | 1 mM | 1.5 μL |
| UltraPure DNase/RNase-Free Distilled Water | N/A | add to 15 μL |
| Total | N/A | 15 μL |
Option 2: One-step protocol for Proteinase K treatment and DBCO labeling
This one-step protocol has slightly lower efficiency of DBCO labeling compared to the two-step protocol (Figure S1). But it saves time and a purification step.
-
10.Proteinase K treatment and DBCO labeling of RNA:
-
a.Add Tris pH 7.5, proteinase K, and RNase-free water into isolated RNA (listed below in Table 3).Note: The RNA amount and total sample amount can be adjusted as long as the final concentrations of reagents remain unchanged.
-
b.Mix and incubate in a water bath at 37°C for 15 min.
-
c.After incubation, add 1.5 μL of 10 mM DBCO-Biotin to reach a final concentration of 1 mM.
-
d.Mix and incubate in a water bath at 37°C for 30 min.
-
e.After incubation, put the sample immediately on ice.
-
a.
-
11.
Purify RNA with the ZYMO RNA Clean & Concentrator kit, as in step 7.
Table 3.
Proteinase K and DBCO-biotin mixture
| Reagent | Final concentration | Amount |
|---|---|---|
| RNA | N/A | 5 μg–15 μg |
| Tris pH 7.5(200 mM, diluted with RNase-free Water from 2 M Tris-HCl Solution, pH 7.5) | 11.1 mM | 0.75 μL |
| Proteinase K Solution (20 mg/mL) | 1.1 mg/mL | 0.75 μL |
| UltraPure DNase/RNase-Free Distilled Water | N/A | add to 13.5 μL |
| Total | N/A | 13.5 μL |
Run DBCO-biotin-labeled RNA on a denaturing formaldehyde gel
In this section, we describe how to run RNA on a denaturing agarose gel. We normally avoid using mini-size gel systems because it would be difficult to balance the weight during the transfer step.
-
12.Follow the recipe for RNA formaldehyde denaturing gel in materials and equipment. Prepare 1% agarose RNA formaldehyde gel:CRITICAL: The entire process of making the gel and waiting for the gel to solidify should be performed inside the fume hood.
-
a.Add 72 mL RNase-free water into a 100 mL autoclaved glass bottle.
-
b.Weigh 1 g of agarose and add it into the bottle.
-
c.Microwave the bottle to dissolve agarose.Note: Microwave in several segments and take the bottle out in between to swirl. This can help to make sure the agarose is dissolved thoroughly.
-
d.Leave bottle at room temperature for 5 min until the agarose solution cools down a little.Note: While waiting for the solution to cool down, wash the gel cast, gel comb, and gel tray and dry them.
-
e.Add 10 mL of 10× MOPS buffer, mix well, and then add 18 mL of 37% formaldehyde solution.
-
f.Swirl to mix.
-
g.Pour the gel mixture into the gel casting tray and insert the gel comb.
-
h.Leave the gel at room temperature for >40 min to solidify.Note: While waiting, proceed to the next step.Note: Leave the gel to solidify in the fume hood as formaldehyde still evaporates during the cooling.
-
a.
-
13.Prepare RNA samples for gel loading:
-
a.Add dye-free gel loading buffer, SYBR Gold nucleic acid dye, and RNase-free water into the RNA sample (listed below in Table 4).Note: SYBR Gold is added into RNA samples instead of the gel. When SYBR gold is added into the gel in a 1× (1:10,000 dilution of the commercial stock) final concentration, the dye will preferentially label the 5s/5.8s rRNAs, presumably due to the small RNAs taking up most of the dye first during electrophoresis. If our recommended 100× final concentration of SYBR Gold is used to make a gel, a large amount of SYBR Gold would be needed for each gel. Therefore, the recommended procedure is to add SYBR Gold directly into the RNA samples.
-
b.Mix and incubate at 60°C for 2 min.
-
c.Put the samples on ice.
-
a.
-
14.Running RNA gel:
-
a.Prepare 1× MOPS buffer by diluting 10× MOPS buffer with DEPC-treated water.
-
b.Put the formaldehyde gel into the electrophoresis tank.
-
c.Fill the tank with 1× MOPS until it covers the gel.
-
d.Load the RNA samples into the wells.Note: The samples do not have running dyes to indicate the progress of electrophoresis. One can add 4 μL of Gel Loading Dye, Purple (6×) in lanes next to the RNA samples as an indicator.
-
e.Run gel at room temperature at 120 V for ∼ 1 h and 15 min.Note: While waiting, proceed to preparing transfer materials.Note: We use the Thermo Scientific Owl D2 Wide-Gel electrophoresis system. We run the gel until the indicator dye in the Purple Gel Loading Dye runs ∼5 centimeters down the gel.
-
f.Once the gel is done running, image it under blue light or UV light.Note: There should be at least two clear bands showing 28S and 18S ribosomal RNAs (rRNAs). The 5S rRNA bands can also be visible if sufficient RNAs are loaded (Figure S2).
-
a.
Table 4.
Gel loading mixture
| Reagent | Final concentration | Amount |
|---|---|---|
| RNA | N/A | 0.5–10 μg |
| Dye-Free Gel Loading Buffer | 50% | 10 μL |
| SYBR Gold Nucleic Acid Dye (Stock as 1:10 diluted with DMSO) | 1:100 | 2 μL |
| RNase-Free Water | N/A | add to 20 μL |
| Total | N/A | 20 μL |
Transferring RNAs onto a nitrocellulose membrane
This transfer method is the same as the bottom-up transfer for northern blotting.
-
15.Preparing transfer materials (Figure 1A).
-
a.Make 1× SSC buffer by diluting 20× SSC buffer with DEPC-treated water.Note: The amount of transfer buffer necessary to achieve a good transfer is dependent on the exact setup including tray size etc. and the amount of RNAs loaded. A good starting point for optimization is 500 mL of transfer buffer.
-
b.A roller about 10 cm long (can be made from a 10 mL plastic serological pipette by breaking it in the middle).
-
c.1 pair of clean plastic forceps.
-
d.1 large tray to serve as the transfer tank.
-
e.1 gel cast, cleaned with MilliQ water and dried.
-
f.Make a stack of dry paper towels to be put on top of the Whatman filter paper.Note: If the paper towels are of good quality and strong enough, they could be reused for multiple experiments as long as they are dried after each experiment.Note: The amount of paper towels needed is dependent on several factors, including the absorbency of the paper and the amount of RNAs to be transferred. A good starting point for optimization is a stack of 10–15 cm of paper towels.
-
g.Make a gel mask with a transparency film which can be used to prevent paper towels from touching the filter bridge and shortcutting the liquid flow.Note: This gel mask can be made by taping four pieces of transparency film strips with the central opening being slightly larger than the gel.
-
h.Cut 2 pieces of Whatman filter paper that are the same sizes as the RNA gel.
-
i.Cut 1 piece of Whatman filter paper with its width the same as the length of the RNA gel, and its length about the same as that of the large container. This will be the bridge.
-
j.Cut 1 piece of nitrocellulose membrane that can cover the area of the loaded RNA samples.
-
k.Plastic wrap to cover the transfer apparatus, preventing evaporation.
-
l.Weight, such as a protein gel tank with water in it, or a reagent bottle with water in it.
-
a.
-
16.Transfer:
-
a.Assemble the transfer as shown in Figures 1B and 1C.
-
i.Put a clean gel cast upside down into the empty transfer tank.
-
ii.Add 1× SSC buffer into the transfer tank.
-
iii.Put the filter paper bridge on top of the gel cast covering the entire area of the gel cast.
-
iv.Pour some 1× SSC buffer to wet the bridge.
-
v.Take the RNA gel out of its gel cast and put it onto the wetted bridge.
-
vi.Using a pair of forceps, take the nitrocellulose membrane and wet it in the 1× SSC buffer in the tank.Note: Cover the area with RNA lanes with the wetted nitrocellulose membrane.
-
vii.Use the roller to roll through the nitrocellulose membrane, making sure there are no bubbles between the gel and the membrane.
-
viii.Put the 2 pieces of Whatman filter papers (same sizes as the RNA gel) on top of the nitrocellulose membrane, covering the entire gel area.
-
ix.Add some 1× SSC buffer to wet the filter papers and roll out any bubbles.
-
x.Add the transparency gel mask around the gel.
-
xi.Put the stack of dry paper towels onto the setup, covering the entire gel area.
-
xii.Use plastic wraps to cover the setup, preventing evaporation.
-
xiii.Add weight at the top of the dry paper towel stack, with the weight ∼1 kg.Note: Use a small tray (labeled as weight 2 in Figure 1A) below the weight to create an even surface for even pressure on the paper towel stack. Other objects, such as a flat glass plate, can also be used as an alternative.
-
i.
-
b.Transfer at room temperature for 15–24 h.Note: The more RNA amounts are loaded, the longer the transfer is required to achieve a complete transfer.
-
a.
Figure 1.

Setup for RNA transfer
(A) Materials and equipment needed before starting. The 1× SSC buffer and gel are not shown. Weight 1 is a protein gel tank filled with water and used as a weight. Weight 2 is a tray that we put in between the plastic wrap and the protein gel tank so the weight can be evenly distributed across the setup.
(B) Step by step picture showing how to assemble the transfer setup (in sequence from 1–9).
(C) Cartoon figure illustrating the setup.
Crosslinking and blotting
In this section, we describe how to visualize biotin-labeled glycoRNAs on a nitrocellulose membrane.
-
17.Crosslinking
-
a.Take down the transfer setup and take the gel to imaging.Note: All RNAs should be transferred and there should not be visible RNA signals on the gel if the transfer is complete.
-
b.Using clean plastic forceps, put the nitrocellulose membrane, with sample-side up, onto a piece of clean Whatman filter paper.
-
c.Crosslink the membrane at 0.18 Jules twice.Note: After crosslinking, the crosslinking chamber will emit a mild ozone smell.
-
d.Put the membrane under a blue light and use a pencil to mark the 28s, 18s, and if visible, the 5s/5.8s RNA locations.
-
e.Also mark the location of the lanes with samples loaded.Note: If needed, the membrane can be cut smaller.Note: 5s/5.8s ribosomal can be hard to visualize when a small amount of RNAs is loaded (e.g. less than 2.5 μg)
-
f.Using clean plastic forceps, move the nitrocellulose membrane with samples into a container larger than the membrane.Note: We found that clean plastic forceps coupled with larger containers give lower background. Rusty iron forceps leave dark spots on the membrane when exposing and imaging the membrane.
-
a.
-
18.Blocking.
-
a.Block the membrane in Bio-Rad EveryBlot blocking buffer for 1 h at room temperature, on a rocking shaker.Note: Cover the container with plastic wrap to prevent evaporation.
-
b.After blocking, remove the old blocking buffer, and add new Bio-Rad EveryBlot blocking buffer with SA-HRP added in a 1:5000 ratio.Note: Cover the container with plastic wrap to prevent evaporation.
-
c.Stain the membrane for 1 h at room temperature, on a rocking shaker.
-
d.After staining, wash with 1× TBST for 4 times on a rocking shaker, 10 min each at room temperature.Note: Remove as much liquid as possible between the washes.
-
a.
-
19.Imaging the blot.
-
a.After washing, take the membrane out with a pair of clean plastic forceps and dry the excessive liquid off by picking up the membrane with the forceps and letting the sides of the membrane slightly touching a piece of kimwipe.
-
b.Lay the membrane flat on a plastic wrap and apply Immobilon Crescendo Western HRP substrate dropwise, enough to cover the membrane.
-
c.Incubate the membrane in HRP substrate for 1 min and use clean plastic forceps to take the membrane out.
-
d.Dry the excessive liquid off by picking up the membrane with the forceps and letting the sides of the membrane slightly touching a piece of kimwipe and lay the membrane flat onto a piece of transparency film.
-
e.Image with a membrane imager.Note: We use a Syngene G:BOX Chemi XRQ imager and use the chemiluminescence channel for imaging. We routinely perform a series of exposure times ranging from 5 s to 20 min.
-
a.
Expected outcomes
This protocol is designed for the visualization of glycoRNAs in cultured cells. Upon completion of this protocol, we expect to (1) detect the existence of glycoRNAs in samples and (2) quantitatively compare glycoRNA expression levels between different samples of the same cell type under various experimental conditions.
Limitations
One limitation of this protocol lies in its requirement of the metabolic tracer, Ac4ManNAz or equivalent. Ac4ManNAz is an azide-containing sialic acid mimetic that can be incorporated into glycans, and in this protocol, it is crucial for the visualization of glycans after northwestern blotting. This strategy of metabolic labeling is most suited for cells that can be cultured. In addition, not all cell types take up Ac4ManNAz equally, making it more challenging to compare glycoRNA levels across different cell types. As noted earlier, the rPAL method,4 which relies on the preferential peroxidation of sialic acids, can utilize much of the northwestern protocol described here after the glycan labeling step. The rPAL method can utilize RNA samples without the need for metabolic labeling and is reported to be more sensitive for glycoRNA detection,4 which are its advantages. On the other hand, the rPAL method may yield some background labeling of RNAs without glycans, and therefore needs attention in the procedure and data interpretation.
Another limitation is that this protocol for the detection of glycoRNAs requires micrograms of total RNAs, and for some cells, even more. This makes the detection of glycoRNAs in small populations of cells difficult.
A third limitation is that this protocol does not cover the detailed validation steps to verify whether the signals are from glycoRNAs, which could involve the digestion of RNA samples by RNase or glycosidases such as PNGase F followed by a purification step. But having an additional round of TRIzol extraction or column-based purification at the initial RNA preparation stage can help reduce contaminating signals from protein/lipid sources.
Troubleshooting
Problem 1
There are smears of signals in the RNA lane after running the gel (related to step 14f).
Potential solution
-
•
For most cell types, this suggests that RNA is partially degraded.
-
•
Run a gel to determine which step(s) the degradation likely occurred, whether it is the initial RNA preparation, or the follow up processing steps including proteinase K digestion and DBCO labeling. Load samples after the initial RNA preparation and those after the other processing steps.
-
•
Depending on the result of the diagnosis step above, tailor troubleshooting experiments accordingly. There are several potential aspects to investigate. If the RNA is degraded as a result of the initial RNA preparation, it might be because the cell pellet is not immediately dissolved in TRIzol. TRIzol contains chemicals that inhibit RNases. If some cells are not completely lysed, they may be prone to RNA degradation. It is also possible that some cell types contain higher levels of endogenous RNases. If so, alternative methods of RNA preparation, such as using commercial kits that retain small RNAs, or changing the incubation temperature (on ice) after TRIzol lysis, could be attempted. If the degradation is mostly occurring as a consequence of the processing steps after the initial RNA preparation, examine the reagents, plasticware and pipetting habit as potential sources of contamination, because pure RNA is stable at 37°C for 30 min in most cases.
Problem 2
Transfer is not complete: there are still much RNA signals left in the gel, but the transfer buffer has been mostly used up or is completely dried up (related to step 17a).
Potential solution
-
•
When setting up the transfer, make sure the two layers of Whatman paper on top of the membrane is not larger than the gel itself.
-
•
Make sure to use plastic wraps to cover the transfer setup, before putting on the weight, to prevent the evaporation of the transfer buffer.
-
•
Many times, inefficient transfer is due to liquid flow “short circuits” during the transfer, e.g., with the paper towel unintentionally touching the transfer bridge. We found that using a gel mask to surround the gel can help prevent such liquid shortcuts.
-
•
Make sure the transfer buffer is not too much and is lower than the height of the gel tray during the bridge setup.
Problem 3
Transfer is not complete: there are still some RNA signals left in the gel and there is still quite some transfer buffer left (related to step 17a).
Potential solution
-
•
If the top layer of the paper towel is damp after transfer, one can increase the amount of paper towel to achieve a better transfer.
-
•
If the top layer of the paper towel is still dry, it suggests that more time is needed. Efficient transfer takes time, especially when the amount of loaded RNAs is large. Extend the time of transfer often helps.
-
•
It has been reported that changing the pH of the transfer buffer4 may enhance the transfer efficiency or speed. We have not tested this strategy.
Problem 4
There are “bubbles” in the RNA lane(s) after transfer (related to step 17d).
Potential solution
-
•
This is likely due to the presence of air bubbles during the assembly of the transfer setup. After putting the membrane onto the gel, and after putting the two Whatman papers on top of the membrane, make sure to use the roller to squeeze out any air bubbles.
Problem 5
No or weak signals of glycoRNAs (related to step 19c).
Potential solution
-
•
Use a positive control cell type, if possible, to diagnose whether this is due to the biology of the cell type used or due to technical issues. We often use Ba/F3, K562, or 293T cells as positive controls.
-
•
If the issue is biological, try to examine whether cells can incorporate Ac4ManNAz efficiently. We often do this by directly reacting live cells (after Ac4ManNAz culture) with DBCO-Biotin, and then run flow cytometry using fluorescently labeled streptavidin to examine cell surface presence of Ac4ManNAz-labeled glycans. This is because Ac4ManNAz can be incorporated into protein glycans in addition to glycoRNAs.
-
•
If the issue is technical, trouble shoot to see if reagents are good, by devising positive controls along the procedure.
Problem 6
Background is high or uneven at the glycoRNA detection (related to step 19e).
Potential solution
-
•
The background is always relative to the signal. When signal is low, background will be higher with longer exposures. If signal is low (i.e., needs long exposures), increase RNA amount or optimize the Ac4ManNAz treatment step.
-
•
Make sure the rocking shaker can make sufficient liquid movements.
-
•
Make sure the membrane is sufficiently shaken during the blocking, washing, and detection steps. We found using rocking shakers give lower background signal than using orbital shakers. We use an Ultra Cruz 2D rocker with speed ∼20 rpm.
-
•
Larger containers for blocking, staining, and washing tend to generate lower backgrounds.
-
•
This blocking condition is optimized based on the SA-HRP as a detector. If using other detectors, such as anti-biotin antibodies, different blocking conditions should be tested and optimized.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jun Lu (jun.lu@yale.edu).
Technical contact
Questions about the technical specifics of performing the protocol should be directed to and will be answered by the technical contact, Luoyi Li (luoyi.li@yale.edu).
Materials availability
-
•
This study did not generate new unique reagents.
Data and code availability
-
•
This study did not generate genomic datasets or analysis codes.
Acknowledgments
This work was supported in part by the NIH grant R01AI181308 (to J.L.).
Figure 1 and the graphical abstract were created with BioRender.com.
Author contributions
L.L. and J.L. performed the optimization experiments and wrote the manuscript. N.Z. initiated the northwestern blot system in our lab and helped to design/interpret the experiments. C.P. and Y.W. helped to optimize the crosslinking and blotting section of the protocol. J.L. supervised the study.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2024.103321.
Contributor Information
Luoyi Li, Email: luoyi.li@yale.edu.
Jun Lu, Email: jun.lu@yale.edu.
Supplemental information
References
- 1.Zhang N., Tang W., Torres L., Wang X., Ajaj Y., Zhu L., Luan Y., Zhou H., Wang Y., Zhang D., et al. Cell surface RNAs control neutrophil recruitment. Cell. 2024;187:846–860.e17. doi: 10.1016/j.cell.2023.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flynn R.A., Pedram K., Malaker S.A., Batista P.J., Smith B.A.H., Johnson A.G., George B.M., Majzoub K., Villalta P.W., Carette J.E., Bertozzi C.R. Small RNAs are modified with N-glycans and displayed on the surface of living cells. Cell. 2021;184:3109–3124.e22. doi: 10.1016/j.cell.2021.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma Y., Guo W., Mou Q., Shao X., Lyu M., Garcia V., Kong L., Lewis W., Ward C., Yang Z., et al. Spatial imaging of glycoRNA in single cells with ARPLA. Nat. Biotechnol. 2024;42:608–616. doi: 10.1038/s41587-023-01801-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xie Y., Hemberger H., Till N.A., Chai P., Watkins C.P., Lebedenko C.G., Caldwell R.M., George B.M., Bertozzi C.R., Garcia B.A., Flynn R.A. The modified RNA base acp3U is an attachment site for N-glycans in glycoRNA. bioRxiv. 2023 doi: 10.1101/2023.11.06.565735. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu H., Li X., Ren Y., Yang Y., Chen Y., Ju H. In Situ Visualization of RNA-Specific Sialylation on Living Cell Membranes to Explore N-Glycosylation Sites. J. Am. Chem. Soc. 2024;146:8780–8786. doi: 10.1021/jacs.4c01826. [DOI] [PubMed] [Google Scholar]
- 6.Huang N., Fan X., Zaleta-Rivera K., Nguyen T.C., Zhou J., Luo Y., Gao J., Fang R.H., Yan Z., Chen Z.B., et al. Natural display of nuclear-encoded RNA on the cell surface and its impact on cell interaction. Genome Biol. 2020;21:225. doi: 10.1186/s13059-020-02145-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guo S., Bai H., Megyola C.M., Halene S., Krause D.S., Scadden D.T., Lu J. Complex oncogene dependence in microRNA-125a-induced myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA. 2012;109:16636–16641. doi: 10.1073/pnas.1213196109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This study did not generate genomic datasets or analysis codes.