

Abstract
Allogeneic natural killer (NK) cell therapies are a valuable treatment option for cancer, given their remarkable safety and favorable efficacy profile. Although the use of allogeneic donors allows for off-the-shelf and timely patient treatment, intrinsic interindividual differences put clinical efficacy at risk. The identification of donors with superior anti-tumor activity is essential to ensure the success of adoptive NK cell therapies. Here, we investigated the heterogeneity of 10 umbilical cord blood stem cell-derived NK cell batches. First, we evaluated the donors’ cytotoxic potential against tumor cell lines from solid and hematological cancer indications, to distinguish a group of superior, “excellent” killers (4/10), compared with “good” killers (6/10). Next, bulk and single-cell RNA sequencing, performed at different stages of NK differentiation, revealed distinct transcriptomic features of the two groups. Excellent donors showed an enrichment in cytotoxicity pathways and a depletion of myeloid traits, linked to the presence of a larger population of effector-like NK cells early on during differentiation. Consequently, we defined a multi-factorial gene expression signature able to predict the donors’ cytotoxic potential. Our study contributes to the identification of key traits of superior NK cell batches, supporting the development of efficacious NK therapeutics and the achievement of durable anti-tumor responses.
Keywords: MT: Regular Issue, NK cells, allogeneic NK cells, cell therapy, umbilical cord blood, hematopoietic stem cells, NK functionality, NK cytotoxicity, donor selection, omics, scRNA-seq
Graphical abstract
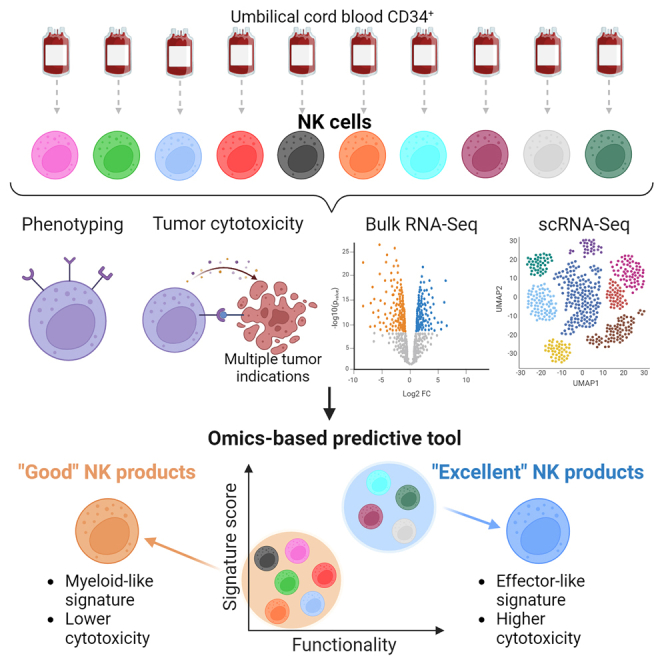
In this study, van Vliet and colleagues unravel key distinctive traits of superior allogeneic, stem cell-derived, natural killer cell therapeutics by integrating functional anti-tumor cytotoxicity, phenotypical, and molecular bulk and single-cell RNA-seq analysis. The identified multi-factorial gene expression signature supports the development of efficacious NK therapies for cancer treatment.
Introduction
Natural killer (NK) cells are innate lymphocytes that lyse cancer or infected cells without prior sensitization by actively secreting cytolytic granules or by initiating apoptosis via death receptors. In addition, NK cells secrete chemokines and cytokines to attract other innate and adaptive immune cells to sites of inflammation, triggering an enhanced immune response.1 The activation status of NK cells is dependent on a balance between multiple activating and inhibiting signals that are mediated via engagement of ligands expressed by encountered cells and activating or inhibiting receptors on the NK cell surface. The cumulative signal resulting from receptor engagement determine the “kill” or “not kill” signal.1 Recognition of self-major histocompatibility complex class I (MHC class I) molecules on the surface of healthy cells by inhibitory killer immunoglobulin-like receptors (KIRs) or by other receptors such as the NK group 2 member A (NKG2A) protects normal cells from NK-mediated killing. Both KIRs and NKG2A are associated with an immunoreceptor tyrosine-based inhibition motif.2,3 Conversely, in abnormal cells, such as tumor cells, the downregulation of MHC class I to avoid T cell responses and the upregulation of stress-related activating ligands unleash NK cell cytotoxicity. Activating receptors include natural killer group 2D (NKG2D), DNAX accessory molecule (DNAM-1), CD16, 2B4, and the natural cytotoxicity receptors (NCRs) NKp30, NKp44, and NKp46.4 The NCRs and CD16 signal via immunoreceptor tyrosine-based activation motifs,5,6,7,8 while 2B4 contains immunoreceptor tyrosine-based switch motifs.9 NKG2D is associated with the adaptor protein DAP10, which contains a YINM tyrosine-based signaling motif.10 Killing occurs via secretion of lytic granules containing perforin and granzymes or by death receptor-mediated apoptosis. Downstream of activating receptors, activation of the mitogen-activated protein kinase (MAPK) pathway leads to calcium flux, degranulation, and cytokine secretion.11 Death receptor-mediated cytotoxicity involves the engagement of tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) or Fas ligand on the surface of NK cells. While this pathway is classically described as slower than the release of granzymes and perforin,12 recent work showed how rapid lysis of tumor cells occurs via TRAIL/TRAILR receptor-mediated interactions.13
NK cells originate from hematopoietic stem cells (HSCs) primarily in the bone marrow, but also in secondary lymphoid tissues like the spleen and lymph nodes. A stepwise linear model for development and maturation of human NK cells was proposed in 2005, where HSCs differentiate into common lymphoid progenitors (CLPs) via lymphoid-primed multipotential progenitors (LMPPs). These CLPs still have the potential to commit to B, T, or NK cell lineages. NK cell lineage commitment occurs in the next stage, as NK precursors mature first into CD56brightCD94+ cells, then into CD56dim, CD16+, and KIR+ cells.14 Later, NK heterogeneity within and across individuals, and the advent of more sophisticated technologies, challenged the idea that NK cells develop exclusively from CLPs. In the branched model, LMPPs can generate both CLPs and common myeloid progenitors, either of which can further differentiate into NK cells.15 Such knowledge has guided the development of protocols for the ex vivo differentiation of CD34+ stem cells and induced pluripotent stem cells (iPSCs) into NK cells, for application in adoptive immunotherapy.
The innate cytotoxic capacity against tumor cells endows NK cells with unique advantages for therapeutic applications. Off-the-shelf availability and remarkable safety, with no risk of graft-vs.-host disease, accelerated the progression of allogeneic NK cell therapies toward the clinical stage in the past years.16 Multiple NK sources are available, such as peripheral blood NK cells (PBNKs), NK cell lines, cord blood NK cells, iPSC-derived NKs, or bone marrow or umbilical cord blood (UCB)-derived HSCs, the latter two employing ex vivo differentiation.17 When using donor-derived sources, the interindividual variability might affect product quality and attributes and thereby puts the success of the treatment at risk.18 Despite the urgency, limited effort has been made to evaluate donor heterogeneity or to standardize selection criteria for improved clinical efficacy.19
Glycostem Therapeutics leverages cord blood-derived HSCs to generate off-the-shelf therapeutic GTA002 NK cells. During the ex vivo culture process, CD34+ HSCs are first expanded and directed toward the NK cell progenitor stage, to be then differentiated into highly active CD56+ NK cells. Preclinical in vitro data showed high cytotoxic potential of GTA002 against melanoma13 and hematological cancer cell lines20 as well as in vivo efficacy against metastatic colorectal cancer.21 GTA002 is currently under clinical investigation in a phase I/IIa trial to evaluate safety and efficacy in patients with minimal residual disease in acute myeloid leukemia (NCT04632316). To advance product understanding toward standardization and improved clinical outcome, we aimed to investigate the qualities pertaining to the NK cell batches most effective at eliminating cancer cells. In this study, we used unsupervised clustering to compare the cytotoxic potential of 10 NK batches against tumor cell lines from multiple cancer indications at low effector-to-target (E:T) ratios. We identified a group of donors exhibiting superior cytotoxic capacity, representing the most favorable source for therapeutic NK products. We then used transcriptomics, i.e., bulk and single-cell RNA sequencing (scRNA-seq), and surface proteomics, i.e., multi-color flow cytometry and scRNA-seq techniques, to investigate the molecular features of such donors. We found that excellent, most potent donors are defined by a multi-factorial gene expression signature, composed of cytotoxicity- and lineage-related traits, which can be found early on during NK differentiation. In this article, we establish new criteria and novel methods for identifying qualities of superior donors, which can be leveraged to ensure consistent generation of optimal NK therapeutics with high anti-tumor potential for efficacious cancer treatment.
Results
Under challenging conditions, excellent NK cell donors show superior cytolysis of tumor cell lines from different cancer indications
To characterize interindividual variability of donors, we generated a dataset of 10 different NK batches with heterogeneous cytotoxic capacity, as recently published.13 A summary of the experimental setup and of the assessments performed is shown in Figure 1A. First, the cytotoxic potential of day 35 late differentiated NK cells from all donors (Figure S1A) was evaluated in a flow cytometry-based assay against the bone marrow cancer, chronic myeloid leukemia (CML), cell line K562 and in an impedance-based assay against three target cell lines of diverse solid tumor origin, namely melanoma A375, colon adenocarcinoma LoVo, and glioblastoma (GBM) LN-18 (Figure 1B). These indications were previously identified as suitable candidates for investigation in NK cell therapy based on promising in vitro and preclinical mouse model studies.13,21,22,23 The kinetic results of the impedance-based assay are reported in Figure S1B. After 20 h and at an E:T ratio of 10:1, all targets were efficiently lysed by all donors. Similarly, at 3:1, cytotoxic efficiency was still high, and only small differences could be observed between donors. Challenging E:T ratios of 1:1 and 1:3, conversely, demonstrated donor heterogeneity, as some maintained very high functionality (>75% cytolysis in 1:3 for LN-18 for donors 5, 6, 7, 8, and 10), while others were strongly impaired (<5% against K562 in 1:3 for donors 1, 2, and 4). Having shown that donor differences could be reliably detected only at lower E:T ratios, while overall high functionality was maintained, we co-cultured the 10 donors with the 4 target cell lines at a 1:1 E:T for 20 h and analyzed cytotoxicity via flow cytometry (Figure 1C). When ranked by mean cytotoxicity value, representing the donors’ anti-tumor efficiency across all 4 cell lines, the 10 donors showed a distinct, intrinsic behavior, independent of the targeted indication. In addition, some donors were more potent than others (Figure 1C). We then inquired if we could formally distinguish donors in groups, according to potency. Hierarchical clustering based on the mean cytotoxicity value defined 2 groups, separating the best performers (donors 7–10) from the others (donors 1–6) (Figure 1D). We therefore designated the 4 superior effectors as “excellent” donors, and the remainder as “good” donors. The presence of intrinsic donor features leading to superior cytotoxic capacity was also confirmed by extensive characterization of receptors on the donor cells’ surface. Clustering analysis of 30 surface antigens involved in NK activation, inhibition, adhesion, and maturation separated the excellent donors from the good donors, although less strongly than cytotoxicity (donor 9 was not included in the analysis due to lack of cell availability) (Figure 1E). If considered individually, none of the 30 antigens was able to discriminate excellent effectors, indicating that the excellent phenotype is defined by a combination of factors and not by expression of a specific receptor. To further investigate the distinctive features of excellent donors, we pursued whole transcriptome analysis.
Figure 1.

Cytotoxicity analysis of 10 stem cell-derived NK batches against multiple tumor cell lines distinguishes donors with superior anti-tumor efficacy
(A) Overview of the experimental setup. (B) NK cytotoxicity, expressed as percentage of tumor cell cytolysis, analyzed after 20 h of co-culture with different cell lines at multiple E:T ratios (from 1:3 to 10:1). Cytolysis was assessed via flow cytometry for K562, and on the impedance-based platform xCELLigence for A375, LoVo, and LN-18. Each dot represents a donor and is the average of a technical triplicate. (C) Flow cytometry-based overnight cytotoxicity assay at a 1:1 E:T ratio; cytolysis was assessed as percentage of 7AAD+ cells. Data are shown as mean ± SD of technical triplicates. Donors are ordered from low to high cytotoxic capacity based on the mean cytotoxicity value across the four cell lines. (D–E) Heatmaps showing the hierarchical clustering of donors based on the mean cytotoxicity value across the four cell lines (D) or the expression of 30 surface antigens (E). Excellent and good donors are labeled with different colors. E:T, effector-to-target; 7AAD, 7-aminoactinomycin D; SD, standard deviation.
Excellent donors express distinctive cytotoxicity genes and pathways, while lacking myeloid traits
The transcriptome profiles of late differentiated day 35 cells from all 10 donors (Figure S1A) were analyzed by RNA-seq, each in single or biological duplicates, depending on cell availability. To identify similarities, samples were clustered based on their expression of the top 1,000 most variable genes (Figure 2A), accounting for the majority of the variation (Figure S2). Remarkably, this analysis distinguished the same 2 groups identified based on cytotoxicity (Figure 1D), suggesting that intrinsic gene expression is linked to cytotoxic capacity. Notably, biological replicates clustered together, indicating absence of batch effect. To further explore transcriptomics differences between excellent and good donors, differential gene expression (DGE) analysis was performed between the 2 groups (Figure 2B). A total of 130 genes were upregulated in the excellent group, while 549 genes were downregulated. The most upregulated gene in the excellent group was B3GAT1, encoding for the NK cell maturation marker CD57, the expression of which has been reported to correlate with higher granzyme B and perforin levels, as well as CD16-mediated cytotoxic potential.24 Similarly, several genes related to NK cell maturation and functionality were enriched in the excellent group, including GZMB (granzyme B) and GZMK (granzyme K), IFNG (IFN-γ), highly expressed in mature NK cells,25 CD247 (CD3ζ) required for NKp30, NKp46, and CD16 signaling,26 SELL (CD62L, or L-selectin), inversely correlating with maturation but also distinguishing an intermediate stage of maturation with high cytotoxic potential,27 and TNFRSF4 (OX40), important for NK cell cytotoxicity and IFN-γ expression in combination with FcγR crosslinking.28 In addition, we observed increased expression of the calcium-binding protein S100B, marker of cytotoxic lymphocytes in the peripheral blood,29 the mitochondrial peptide MTRNR2L8, protecting cells from oxidative stress and hypoxia30 and observed to be expressed in PBNKs,31 INSRR, involved in protein tyrosine kinase activity of transmembrane receptors,32 and SOX13, a transcription factor involved in innate lymphoid cell development.28 Notably, downregulated genes included several markers of macrophage and monocyte innate immune effectors, such as surface receptors CD14, TLR4, and CD4,33 anti-microbial enzyme lysozyme LYZ, calcium-binding proteins modulating the inflammatory response S100A8 and S100A9,34 complement components C1QA, C1QB, and C1QC,35 chemokines CCL1736 and CXCL8,37 and metalloproteinases MMP1238 and MMP9.39 Using gene set enrichment analysis with the Gene Ontology Biological Processes (GO BP) database, we identified pathways enriched in the excellent group as related to the cell cycle, signaling pathways, response to external stimuli and intracellular transport (Figure 2C). Pathways upregulated in the good donors included regulation of the innate immune response, defense response and response to bacteria and cytokines (Figure 2C), consistent to the observed monocyte/macrophage gene traits.28
Figure 2.

Bulk RNA-seq identifies the enrichment of cytotoxic pathways and the absence of myeloid traits in excellent donors
(A) Heatmap showing the hierarchical clustering based on the 1,000 most variable genes, analyzed from 10 donors on day 35. For each donor, single or two biological replicates were used (a and b). (B) Volcano plot of differentially expressed genes in excellent vs. good donors. (C and D) Ten most enriched pathways in excellent and good donors, using in (C) the differentially expressed genes and the GO Biological Process database and in (D) the normalized gene counts and the KEGG database. Significant enrichment threshold was considered as NES p < 0.05 (colored bars) and FDR < 0.25 (bars with diagonal pattern). (E) Heatmap showing the genes from the NK cell-mediated cytotoxicity KEGG pathway that contribute to pathway enrichment in excellent donors. Excellent and good donors are labeled with different colors. DE, differentially expressed; GO BP, Gene Ontology Biological Process; KEGG, Kyoto Encyclopedia of Genes and Genomes; NES, normalized enrichment score; FDR, false discovery rate.
Since DGE analysis only accounts for large differences in the expression of few genes, it could mask biologically relevant effects resulting from small changes occurring in groups of genes belonging to the same pathway. Therefore, we also performed gene set enrichment analysis (GSEA), using the normalized gene counts for all expressed genes and the Kyoto Encyclopedia of Genes and Genomes (KEGG) Canonical Pathways gene set database. Notably, we observed significant enrichment of the NK cell-mediated cytotoxicity pathway in the excellent group (Figure 2D), which is in line with our functional assessments. Among the genes contributing to the enrichment, we found granule molecules and cytokines (GZMB, PRF1, IFNG), receptors (CD247, NCR1), signal transducers (ITGB2, MAP2K2, MAPK3, ITGAL, ZAP70), and kinases (SYK, PIK3R3, PIK3CD, PIK3CG, LCK) (Figure 2E). Notably, GZMB, CD247, IFNG, CSF2, ULBP1, and ULBP2 were identified as pivotal markers of excellent donors from both DGE and NK cytotoxicity gene set analysis. Additional pathways enriched in the excellent group included the Notch signaling pathway, important for early NK cell development40 but also for enhanced NK functionality at later stages,41 the non-small cell lung cancer pathway, including cell-cycle-related members of the PI3 kinase family, MAP kinases, CCND1, AKT1 and 2, HRAS, GRB2 and E2F transcription factors, in accordance with the highly proliferative signature observed in excellent donors from the DGE analysis (Figure 2C). Interestingly, such signals are also activated downstream of NKG2D and other NK surface receptors, and lead to actin polarization and NK cell activation.11 Other pathways include the T cell receptor pathway, with genes that are also important mediators of NK cell functions such as CD247, IFNG, FYN, and ZAP70,11 and the endocytosis pathway, used by NK cells to regulate surface receptor expression and function,42,43 with genes such as ARF6, GRK, ARAP. Conversely, several pathways related to inflammation were enriched in the good group, including complement and coagulation cascades, NOD-like receptor signaling pathway, IgA production in the intestine, and cytokine-cytokine receptor interaction, involving several genes of the CC (CCL) and C-X-C motif (CXCL) chemokine ligands and of the CXCR chemokine receptor families, cytokine interleukin-6 (IL-6) and IL-6 receptor, C1Q complement factors. Of note, the KEGG cell cycle pathway was enriched in the good donors, in contrast to what was observed in DGE analysis (Figure 2D). However, the lists of cell cycle genes from KEGG and GO do not overlap, except for one gene, CCNA1, which was upregulated in the good donors. Such discrepancy between GO and KEGG can explain the disagreement, although makes the cell cycle analysis inconclusive.
Transcriptome analysis of late differentiated NK cells identified distinct gene expression the excellent donors, which is related to increased NK cell activation and cytotoxicity-related traits. We then questioned if these traits could already discriminate the most potent killers earlier during the NK differentiation process, as younger populations are more heterogeneous and not all cells have acquired expression of CD56 (Figure S1A). Therefore, we next analyzed the transcriptome of day 28 cells with scRNA-seq.
During NK differentiation, excellent donors show early enrichment of effector-like cells and lack of myeloid lineage populations
To investigate the presence of differences distinguishing superior cytotoxic capacity in day 28 cell populations, we analyzed gene expression (RNA) and surface protein expression (AbSeq) from 8 of the donors in the dataset, i.e., excellent donors 7, 8, and 10, and good donors 1–5, with scRNA-seq. First, we removed cells expressing too few or too many genes and cells with higher percentage of mitochondrial DNA (Figure S3A), leaving ∼7,500 cells for further analysis (Figure S3B). Then, we visualized all cells in two dimensions by Uniform Manifold Approximation and Projection (UMAP). Hierarchical clustering with Leiden algorithm identified 6 clusters, i.e., clusters 1–6 (Figures 3A and S3C). All clusters were present in both excellent and good donors (Figure 3B); nevertheless, we observed differences in their distribution. Notably, cluster 1 cells were overrepresented in excellent donors (55.4% vs. 39.3% in the good group), while clusters 2 and 6 cells were more abundant in the good donors (23.6% vs. 7.8% and 2.1% vs. 0.6%, respectively) (Figure 3C). Clusters 3, 4, and 5 were evenly distributed.
Figure 3.

scRNA-seq reveals early enrichment of NK effector-like cells and lack of myeloid lineage cells during differentiation of excellent donors
(A) UMAP of cluster distribution of cells, analyzed with Leiden algorithm on day 28 from 8 donors. (B) Relative distribution of clusters per donor, expressed as percentage of total. (C) Distribution of clusters across excellent and good donors (n = 3 and 5, respectively), expressed as percentage of total. Clusters are ranked by overall size. Data are shown as mean ± SD. Statistical analysis was performed using two-way ANOVA with Šídák correction for multiple comparisons. Significance is shown as ∗∗∗p <0.001. (D) Heatmap showing the top 10 markers of each cluster identified by differential expression analysis. (E) Selected GO Biological Process pathways enriched in each cluster. (F and G) Dot plots of (F) selected NK cell-related genes and (G) selected myeloid-related genes. The size of the dot is proportional to the percentage of cells expressing the gene, while the color scale indicates the average scaled gene expression. Excellent and good donors are labeled with different colors. UMAP, Uniform Manifold Approximation and Projection; GO, Gene Ontology; NES, normalized enrichment score; Padj, adjusted p value.
We then sought to understand the features of each cluster by DGE analysis, using both expressed genes and surface markers. We identified the top-defining RNA and AbSeq markers of each cluster and visualized the top 10 in heatmaps (Figures 3D and S3D, respectively), and all AbSeq markers and the top 5 RNA markers as feature plots in UMAP (Figures S4 and S5). Cluster 1, representing the largest population (46% of total cells), showed high surface expression of CD45RA, CD56, NKp44, GITR, and CD62L (Figures S3D and S4) and enrichment of 518 genes, including NK effector genes NKG7, PRF1, GNLY, GZMB, and CCL5 (Figures 3D and S5). Other gene markers of cluster 1 included IL2RB (IL-2β/IL-15 receptor), essential for NK cell differentiation,44 CTSW (Cathepsin W),45 HOPX (transcription co-factor),46 and CD7 (associated with active NK cells),47 all known to be expressed by NK cells. Analysis of GO BP associated to cluster 1 revealed enrichment of NK cell effector functions, i.e., NK cell cytotoxicity, immunity, and activation, and response to cytokines, including IFN-γ (Figure 3E). Cluster 2 (18%) showed low CD56 but enrichment of several myeloid markers on the cell surface, including CD4, CD14, CD11c, HLA-DR, CD25, and CD127 (Figures S3D and S4), and of 979 genes, the top ones being LYZ, S100A8, S100A9, CXCL8, and FCN1 (Figures 3D and S5). GO analysis identified several myeloid-related functions, such as macrophage and granulocyte activation and chemotaxis, innate immune response, as well as the inflammatory response (Figure 3E). Cluster 3 (15.5%) showed CD56 expression on the surface (Figures S3D and S4), and the 672 enriched marker genes included ALDOC, IL1R1, FURIN, LACC1, and KIT (Figures 3D and S5). Enriched immune processes were mostly related to regulation of lymphocyte proliferation, differentiation and activation, regulation of the immune response, and regulation of cytokine production and of molecular mediators of the immune response (Figure 3E). Cluster 4 (12%) showed a similar surface profile to cluster 1, with surface expression of CD56, NKp44, and GITR although at a lower level, and of CD62L (Figures 3D and S5). The 761 enriched genes, with top markers TOP2A, MKI67, TUBB, STMN1, and HMGB2 (Figures 3D and S5), were strongly associated with the cell-cycle process, such as mitotic spindle and cytoskeleton organization, chromosome separation, and cell division (Figure 3E), suggesting that this cluster consists of highly proliferating cells. Cluster 5 (7%) had surface expression of CD56, NKp44, and GITR (Figures S3D and S4). 913 genes were enriched, including ATP8B4, ATP10A, RABGAP1L, SMYD3, MAML2 and MAML3, KCNQ5, and CARD11 (Figures 3D and S5), associated to the regulation of developmental processes, response to stimulus, GTPase activity, signal transduction, and (immune) cell surface receptor signaling (Figure 3E). Cluster 6 (1.5%) showed common markers with cluster 2, i.e., CD11c, HLA-DR, and CD25, and unique markers TNFRSF9/CD137, BTLA/CD272, and CD196 (Figures S3D and S4). The 881 marker genes included CCL17, CCL22, HLA-DPA1, -DPB1, and -DQA1, FSCN1, and CD74 (Figures 3D and S5). Enrichment analysis showed functions related to cytokine production, response, and pathway, defense response, innate response, and granulocyte and neutrophil chemotaxis (Figure 3E).
We then looked in more detail at the expression of NK cell-related genes in the different clusters. As shown by GO analysis, cluster 1 cells showed a strong cytotoxic profile (Figure 3E), despite not being fully differentiated (Figure S1A). Cluster 1 cells showed a CD56bright phenotype based on both surface protein and RNA expression (Figure S6), and key markers included cytotoxicity molecules PRF1, GZMB, GNLY, GZMA, GZMK, and NKG7, receptors IL2RB, NCR1/NKp30, NCR3/NKp46, KLRC1/NKG2A, and CD226/DNAM-1, co-stimulatory receptors such as KLRD1/CD94, TNFRSF18/GITR, and CRTAM/CD355, ligands such as TNFSF10/TRAIL, IFN-γ response-related genes OAS2 and ISG15, chemokines such as XLC1, XCL2, CCL3, CCL4, and CCL5 (Figure 3F). Notably, expression of NCR2/NKp44 was high on the cell surface, but low at the RNA level (Figures 3F, S3D, and S4). Receptors KLRK1/NKG2D, FCGR3A/CD16a, B3GAT1/CD57, and KIR family members were low (not shown). Cluster 4 had a similar profile, enriched with PRF1, GZMB, GNLY, TRAIL, NKG2A, and CD94, albeit to a lower level than cluster 1 (Figure 3F). A similar trend was observed with surface markers, including a CD56 intermediate (CD56int) phenotype (Figure S6). Likely, the cytotoxic profile was diminished by the high proliferative potential of the cells, as shown from gene ontology analysis (Figure 3E) and cell cycle analysis (Figures S7A and S7B). Clusters 3 and 5, CD56int from surface analysis (Figure S3D), had lower expression of NK genes, especially cluster 5 (Figure 3F). Notably, cluster 5 showed the highest RNA expression of CD56, in contrast with surface levels (Figure S6). Clusters 2 and 6, characterized as CD56low/neg on the cell surface (Figure S6), had low expression of granule molecules and of NK receptors; on the contrary, both sub-populations were characterized by myeloid markers, namely CD4, LYZ, and HLA-DR (Figures 3G and S3D). In comparison, cluster 2 showed higher expression of CD14, CD68, CXCL8, S100A8, S100A9, ITGAM/CD11b, C5AR1/CD88, FCAR/CD89, and TLR4, suggesting that these cells were of monocytic origin,48 while cluster 6 expressed higher levels of CD40, CCL17, CCL22, LAMP3, CD80, CD86, CCR7, FSCN1, CD274/PD-L1, and IDO1, indicating a more dendritic cell (DC)-like phenotype.49 Notably, the immunosuppressive enzymes ARG1, NOS1, and NOS250 were absent in all clusters, except for a small percentage of cells from cluster 6 (Figure 3G). In conclusion, cluster analysis of differentiating day 28 cells identified different sub-populations, either with a differentiating NK phenotype (expressing CD56) or with a myeloid phenotype (CD56 negative). While the most cytotoxic population was enriched in the excellent donors, the myeloid populations were poorly present, in line with observations from bulk RNA-seq. Interestingly, CD56-expressing populations showed heterogeneity of NK genes and functions, suggesting different degrees of maturation between the NK clusters. We therefore investigated this aspect by analyzing the status of NK development of clusters 1, 3, 4, and 5.
Early effector-like cells from excellent and good donors are at a more advanced stage of NK differentiation
We pursued the analysis of the maturation of the CD56-expressing sub-populations by looking at the expression of surface and transcriptional markers of NK cell developmental stages during hematopoiesis, as reported by Abel et al. and by Wang and Malarkannan.14,51 As NK maturation is marked by the appearance of the neural cell adhesion molecule NCAM1/CD56,14 we compared its expression in the clusters, both at the surface protein (AbSeq analysis) and RNA level (gene expression) (Figure S6). Notably, CD56 expression varied greatly between clusters. Not surprisingly, we observed differences between protein and RNA levels, given the complex regulation of their expression, their half-life, and the heterogeneity of the detection methods.52 Cluster 3 cells, CD56int by surface analysis (Figure S6), showed consistent RNA levels of NCAM1/CD56, highest expression of KIT/CD117, IL1R1, IL7R/CD127, and KLRB1/CD161, low expression of KLRC1/NKG2A, NCR1/CD335, and NCR3/CD337, with high expression of early transcription factors RUNX3, ID2, and ETS1 (Figure 4A), suggesting compatibility with stage 3/stage 4a progenitors. Likely, the different environmental conditions and stimuli present during ex vivo culture, compared with NK maturation in the bone marrow and lymphoid organs, induced this “hybrid” profile between the two stages. Cluster 5 cells showed an increase in NCAM1/CD56, reaching the highest expression at the RNA level (Figures 4A and S6), a reduction of KIT/CD117 and of IL1R1, and an increase in NCR1/NKp46, compatible with stage 4a. However, other markers such as NCR3 or KLRC1 were still poorly expressed, and the overall gene expression of the cluster was low, making it difficult to assign cells to a specific stage (Figure 4A). Clusters 1 and 4 showed a bright CD56 phenotype, especially on the cell surface (Figures 4A and S6) and traits of stage 4 cells (loss of KIT and IL1R1, increase in KLRC1/NKG2A, NCRs). In addition, both clusters showed increased expression of KLRF1/NKp80 and of the late-stage transcription factor EOMES (Figure 4A), making them compatible with stage 4b. In addition to the Abel model, expression of KLRD1/CD94 is linked to NK cell lineage commitment, and its expression is increased on CD56bright cells.53 We observed KLRD1 expression in clusters 1, 4, and 5, with the maximum reached in cluster 1 (Figure 4A). Developmental stages 5 and 6 could not be identified in any cluster, likely as cells were still undergoing differentiation.
Figure 4.

NK sub-populations show differences consistent with developmental stages, while myeloid cells derive from a separate trajectory
(A) Dot plot of selected NK development genes. The size of the dot is proportional to the percentage of cells expressing the gene, while the color scale indicates the average scaled gene expression. (B) UMAP of pseudotime inference, computed selecting cluster 3 as the root. Each cluster is profiled in a different color. (C) Boxplots of cluster ordering and cell distribution based on pseudotime. UMAP, Uniform Manifold Approximation and Projection.
We then applied pseudotime analysis to reconstruct the cells’ trajectories through differentiation. Setting cluster 3 as the starting point, the trajectory divided over two main branches, one leading to myeloid-like cells in clusters 2 and 6, and one to NK cell clusters (Figure 4B). The highest NK cell maturity was observed for clusters 1 and 4 (Figure 4C), consistent with the observed effector-like gene expression signature (Figure 3F). Notably, the branched trajectory of clusters 2 and 6 was compatible with the expression of myeloid developmental markers CEBPB and SPI1/PU.1,54 which were nearly absent in the other clusters (Figure 3G). In summary, analysis of sub-populations during cell differentiation identified CD56+ cells in different stages of development; the most mature ones, although present in both groups, were more abundant in the excellent donors. CD56– myeloid-like cells, depleted in excellent donors, were likely branching from a separate lineage trajectory.
Gene expression markers identified with bulk and scRNA-seq predict the cytotoxic potential of ex vivo differentiated NK cell donors
From our donor analysis, both bulk and scRNA-seq identified specific transcriptomics features of excellent donors, which could be related to their superior cytotoxic potential. Aiming to find measurable markers of donor cytotoxicity that can be analyzed during ex vivo cell culture, we sought to integrate the two approaches. When visualized by UMAP, all donors showed large overlap, although segregation was visible, especially in clusters 1 and 2 (Figure 5A). As previously done with bulk RNA-seq, we performed DGE analysis of scRNA-seq between the two groups of excellent vs. good donors, including all clusters, and found 76 upregulated and 208 downregulated genes in the excellent donors. Notably, we observed a large overlap with the day 35 bulk analysis (Figure 2B), with 5 upregulated and 88 downregulated common genes (Figure 5B). No gene showed an opposite trend between the two lists. The 5 upregulated genes were granule molecules GZMB and GZMK, the adhesion receptor SELL/CD62L, the calcium-binding protein S100B, and the mitochondrial peptide MTRNR2L8. Interestingly, when performing DGE analysis between excellent and good donors including only cells from cluster 1, no significant genes were found, suggesting that the same potent NK effectors are present in both groups, and that the differences observed in cytotoxic capacity are truly due to the differences in abundance of this population within the total cell pool of a donor. As high expression of granzyme B was one of the strongest traits of the excellent phenotype, we validated this finding at the transcriptome level by qPCR, and at the protein level by intracellular staining (Figures 5C and 5D). Moreover, detection of granzyme B in the secretome upon 5 h of co-culture with K562 at a 1:1 E:T ratio showed significantly higher release by the excellent donors (Figure 5E). A trend of higher granule release and activation was also observed for other effector molecules such as PRF1/perforin, IFN-γ, TNF, granzyme A, and granulysin, although not always at significant levels (Figures 5C–5E). Of the 88 downregulated genes that were in common between bulk and single-cell analyses there were, among others, previously identified myeloid markers such as CD14, LYZ, S100A8, S100A9, CD4, TLR4, CCL17, CXCL8, and MMP9, which were also indicators of cluster 2.
Figure 5.

Identification of a multi-factorial gene expression signature predicting donor cytotoxic potential
(A) UMAP of excellent and good donor distribution. No major separation is distinguishable. (B) Overlap of differentially expressed genes between excellent and good donors identified with bulk and scRNA-seq. (C) Relative mRNA expression of GZMB and PRF1 of day 35 cells, analyzed by qPCR on the 10 donors. The good group is used as control. (D) Intracellular protein levels of granzyme B and perforin of day 35 cells, analyzed via flow cytometry on 9 donors. The good group is used as control. (E) Detection of effector molecules in the supernatant after 5 h co-culture with K562 target cells, analyzed via flow cytometry on 9 donors. In (C)–(E), data are shown as mean ± SD and statistical analysis was performed using unpaired t test, with Holm-Šídák correction for multiple comparisons. Significance is shown as ∗p < 0.05, ∗∗p < 0.01. (F and G) Linear correlation analysis between the mean cytotoxicity value, calculated across 4 target cell lines, and the signature score S, from (F) bulk RNA-seq (10 donors) and (G) scRNA-seq (8 donors). (H) Summary of the main findings from this study. Excellent and good NK cell-related genes, contributing to the predictive signature, are listed in the blue and orange regions, respectively. MFI, mean fluorescence intensity.
Having identified the presence of cytotoxic attributes and the absence of myeloid ones as distinct traits of excellent donors, we sought to explain them as measurable entities to potentially predict the anti-tumor potential of any donor. We therefore selected 22 predictor genes from the enrichment observed in either bulk or scRNA-seq. First, we identified markers of superior donors, including the 5 genes that were enriched in excellent donors from both bulk and scRNA-seq (e-genes: GZMB, GZMK, GZMA, MTRNR2L8, and SELL) and 8 additional genes enriched in excellent donors from bulk or scRNA-seq (e-genes: S100B, CD247, TNFRSF4, B3GAT1, IL2RB, NKG7, PRF1, and IFNG). Next, we added 9 myeloid lineage genes, enriched in the good donors from both bulk and scRNA-seq, which were also highly enriched in clusters 2 and/or 6 from scRNA-seq (g-genes: CD14, LYZ, S100A8, S100A9, TLR4, CCL17, CXCL8, MMP9, and CD4). We then calculated a signature score S for each donor from bulk RNA-seq read counts (day 35 cells) or from scRNA-seq average gene expression (day 28 cells), where e-genes and g-genes contributed positively and negatively to the overall score value, respectively. Excellent donors showed higher scores than good donors, and a linear correlation was observed between the S score and cytotoxicity (Figure 1D), with Pearson’s correlation coefficient r of 0.95 and 0.94 for day 35 and day 28, respectively (Figures 5F and 5G). Although not all 22 genes were enriched in both bulk and scRNA-seq, their expression reliably predicted the donors’ cytotoxic potential already on day 28 as well as on day 35, when assessed with either method.
In conclusion, our study has identified a multi-factorial gene expression signature, which enables prediction of the cytotoxic potential of mature CD34+ HSC-derived NK donors already from earlier stages of ex vivo differentiation (Figure 5H).
Discussion
Allogeneic NK cell therapies are rapidly emerging as a promising anti-tumor cellular immunotherapy, with proven efficacy and remarkable safety.16 Currently, clinical development of NK therapies employs different cell sources, mostly derived from healthy donors, which are expanded, activated, or differentiated ex vivo.16 Donor-derived therapeutic NK cells bring remarkable advantages toward sustainable manufacturing of cell therapies, as they allow for large-scale batch production and off-the-shelf availability, providing a timely treatment for patients in need while reducing costs.55 However, not all donor-derived NK cell batches are equipped with the same cytotoxic potential, thus influencing anti-tumor efficacy. The identification and use of highly functional NK cell products is therefore essential to ensure clinical success. So far, limited consideration has been given to the identification and selection of donors with superior anti-tumor activity, although attempts have been made,56,57 and currently there are no standardized criteria for donor selection.19 Clinical interest for donor selection is restricted to KIR/HLA match/mismatch, which is not predictive of the cells’ cytotoxic potential and is not relevant for some ex-vivo-expanded NK cell therapies, as they lack KIR expression. Thus, it is necessary to understand donor heterogeneity and to identify key intrinsic traits of donors with superior cytolytic efficacy to generate potent allogeneic NK cell therapies.
Historically, characterization based on phenotypic analysis has led the assessment of the quality, functionality, and safety of therapeutic NK cells. The benefit of phenotyping is its widespread application and accessibility, and it is still highly relevant for cell therapy in-process control and release assessment. However, phenotypic methods are inadequate at revealing donor heterogeneity, especially as consistent discrimination of highly cytotoxic NK subsets solely based on surface markers cannot be made,58,59 and additional aspects, such as splice variants, are involved.60 More recently, the advent of high-throughput technologies allowed for deeper investigation of donor differences. Foley et al. assessed the anti-leukemic potential of 49 PBNK donors through in vitro functional screening, and identified a pool of superior donors, which could represent a source of effective and functional therapeutic NK cells.18 As a follow-up, Barnes et al. proposed a paradigm shift away from traditional phenotypic characterization, toward a functional profile based on molecular and metabolic characteristics.19 Recently, Marin et al. demonstrated how donor cord blood characteristics and transcriptomics heterogeneity of cord blood NK cells determined clinical outcome in a phase 1/2 trial where chimeric antigen receptor-engineered NKs were used to treat CD19+ B cell tumors, while notable phenotypic differences of the infused NK cells were not observed.61 Leveraging high-resolution transcriptomics can provide unprecedented insight into the mechanisms driving NK cell development and function, enabling functional understanding of donor diversity and the selection of superior effectors.
Here, we showed that stem cell-derived, ex-vivo-expanded NK cells show intrinsic heterogeneity of cytotoxic capacity, appreciable in challenging assay conditions, and irrespective of the targeted tumor indication. With hierarchical clustering, we were able to distinguish a group of excellent donors, showing superior anti-tumor capacity, compared with good donors. Notably, transcriptome analysis of late differentiated day 35 cells, analyzed by bulk RNA-seq, defined the same groups, indicating that a donor’s cytotoxic capacity is inherently connected to its molecular ensemble. Looking at the genes distinguishing the two groups, we found that excellent donors were characterized by the concurrent enrichment of NK effector-like and depletion of myeloid-like traits. Investigation by scRNA-seq analysis on younger day 28 cells linked such traits to different sub-populations present during NK differentiation. In agreement with bulk RNA analysis, excellent donors showed a larger proportion of early cytotoxic CD56bright cells and a depletion of CD14+ myeloid cells. Our findings are in line with the observations from Lehmann et al.,62 who analyzed ex vivo expansion cultures of CD34+ cells to NK cells, finding significant upregulation of effector genes including GZMB, PRF1, and GZMA in CD56bright NK cells, comparable with PBNK CD56dim and similar to the early cytotoxic cell CD56bright population we found enriched in the excellent donors. Notably, high intensity of CD56 on the cell surface is mostly influenced by cytokine stimulation and is not a sign of poorly cytotoxic cells, showing how phenotypic criteria should be revised in view of the development of therapeutic NK cell products from ex vivo cultures. Besides early cytotoxic cells, our analysis distinguished several additional clusters within the NK population. Although our transcriptomics-based characterization did not completely overlap with in vivo developmental stages, likely due to the incomparability of ex vivo culture systems with the human body, we observed that the heterogeneity of differentiating cells is due to the coexistence of NK cells at different stages of development, rather than to the presence of different subtypes. As cells were still undergoing differentiation, we did not identify populations from final NK maturation stages; however, phenotypic characterization and in vivo evaluation show later maturation and upregulation of CD16 and KIR.63 Characterization of the myeloid cells, enriched in the good donors, distinguished a larger cluster expressing genes associated with the monocytic lineage, and a smaller cluster featuring a DC-like phenotype, likely reflective of in vivo lineage differentiation pathways. Importantly, these clusters lack immunosuppressive traits, suggesting that their impact on donor cytotoxicity is due to the reduced pool of active NK cells, rather than to a myeloid-mediated suppression of NK function. The presence of myeloid-like cells during NK differentiation was also reported by Lehmann et al. CD14+ monocytes accumulate transiently during ex vivo culture, and could have a functional role by stimulating NK cell differentiation.62 However, based on surface phenotypic assessment, almost all cells were fully differentiated into CD56+, and the myeloid signature was still observed in the good donors on day 35. It is not known if during the final week of differentiation the myeloid sub-populations differentiated into NK cells, in line with the branched NK development model,15 or if they mostly disappeared from the culture, possibly undergoing apoptosis induced by a lack of sustaining growth factors. Pseudotime analysis suggests that the myeloid-like cells branch separately from NK cells, supporting the hypothesis of myeloid cell death during the latest phase of the culture. Further investigation is needed to comprehend the fate of the myeloid cells, and to learn how to reduce expression of the myeloid signature, to make all donors excellent. Remarkably, we observed that the early, day 28, cytotoxic NK cells from cluster 1 were present in both excellent and good donors, although in different proportions, suggesting that each donor has the potential of being excellent if this effector population is sufficiently enriched.
Bulk RNA-seq and scRNA-seq, although performed at two different time points during NK differentiation, showed comparable outcomes, with a significant overlap of genes enriched in the two groups. By combining the two approaches, we defined a unique, multi-factorial gene expression signature that predicts a donor’s cytotoxic capacity, including both positive cytotoxicity-related, and negative myeloid lineage-related, traits. When applying this signature to bulk and scRNA-seq data, we found a comparable positive correlation with cytotoxicity, further demonstrating the agreement between the two methods, and indicating that the signature can be predictive both earlier and later during NK differentiation. Notably, the signature genes include different molecular types, namely effector molecules, receptors, co-stimulatory factors, signal peptides, and enzymes, which would be impossible to measure by phenotyping but could be amenable to measurement through a targeted qPCR panel. A customized qPCR kit would allow for simultaneous and rapid analysis of all signature genes, in association with intracellular staining of effector molecules. Continuous dataset expansion will strengthen our correlation model and allow us to define robust threshold criteria to assign a high or low signature score S to excellent and good donors. As the efforts in leveraging transcriptomics techniques to define NK signatures applicable to disease treatment and outcome are increasing,18,61,64,65,66,67,68,69,70 this is the one of first reports of measurable pivotal traits of NK cytotoxic efficacy that can support the establishment of improved criteria for donor selection to be applied during ex vivo manufacturing of off-the-shelf NK therapeutics. Remarkably, in vitro prediction methods can be easily scaled-up, allowing for matrix testing of multiple target cell lines, indications, and donors at once; the same would not be possible by using animal models, limited by ethical and practical reasons. Finally, combining transcriptomics-based gene signature analysis with genomics parameters would further improve the prediction of excellent donors and enable confirmation of superior product profiles before infusion.
Materials and methods
Cell line culture
The CML cell line K562 (ATCC-CCL-243) was cultured in Iscove’s modified Dulbecco’s medium (Lonza). The melanoma cell line A375 (ATCC-CRL-1619) and the GBM cell line LN-18 (ATCC-CRL-2610) were cultured in Dulbecco’s modified Eagle’s medium (Lonza). The colon adenocarcinoma cell line LoVo (ACC-350, DSMZ) was cultured in Roswell Park Memorial Institute 1640 Medium (Lonza). All media were supplemented with 10% fetal bovine serum (Gibco). All cell lines were cultured at 37°C and 5% CO2. Cell line authentication was confirmed by short tandem repeat profiling and cells were regularly tested for mycoplasma.
NK cell culture and phenotypic characterization
Fresh and frozen UCB units were purchased from Anthony Nolan (UK) or NHS Blood and Transplant (UK), and mononuclear cells (MNCs) were isolated by Ficoll Paque Plus (1.077 g/mL, GE Healthcare) density gradient centrifugation. Hematopoietic CD34+ stem cells were selected from the MNCs using the CD34+ magnetic microbead kit (Miltenyi Biotech) according to the manufacturer’s protocol. CD34+ cells from 10 UCB donors (purity 67.6% ± 15.2%) were seeded in six-well tissue culture-treated plates (Corning) for expansion culture in Glycostem Basal Growth Medium (Fertipro) supplemented with human serum (Sanquin), thrombopoietin (TPO), IL-7, FMS-like tyrosine kinase 3 ligand (Flt-3L), granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-6, stem cell factor (all from Cellgenix), and Neupogen (G-CSF, Amgen) as described previously.71 After 9 days, TPO was replaced by IL-15 (Cellgenix) and, after 14 days of NK progenitor expansion, Flt-3L was replaced by Proleukin (IL-2, Novartis) for the NK cell differentiation phase. Cells were cultured until day 28 and cryopreserved. Upon thawing, cells were directly used for assays, or cultured for an additional 7 days in differentiation medium before use. In this article, when referring to donors, we allude to the 10 different UCB donors from which we originated the 10 different NK cell batches. To characterize the receptor expression profile on NK cells, the antibodies listed in Table 1 were used; 5 × 104 cells were stained for 30 min at 4°C, followed by washing with flow cytometry buffer and data acquisition using the Cytoflex LX flow cytometer (Beckman Coulter).
Table 1.
Overview of antibodies/stains used for phenotypic characterization of NK cells and their progenitors during ex vivo cell culture
| Antibody | Fluorochrome | Clone | Cat. no. | Supplier |
|---|---|---|---|---|
| Brilliant staining buffer plus (BSBP) | – | – | 566385 | BD Biosciences |
| ViaKrome | IR885 | – | C36628 | Beckman Coulter |
| CD56 | APC-Fire750 | QA17A16 | 392408 | BioLegend |
| CD3 | PerCPCy5.5 | SK7 | 344808 | BioLegend |
| CD14 | PerCPCy5.5 | HCD14 | 325622 | BioLegend |
| CD15 | PerCPCy5.5 | HI98 | 301922 | BioLegend |
| CD19 | PerCPCy5.5 | HIB19 | 302230 | BioLegend |
| CD16 | BV421 | 3G8 | 302038 | BioLegend |
| NKp30 | BV605 | P30-15 | 325234 | BioLegend |
| NKp46 | BV785 | 9E2 | 331946 | BioLegend |
| DNAM-1 | PE | DX11 | 559789 | BD Biosciences |
| NKp44 | PE-Cy7 | P44-8 | 325116 | BioLegend |
| NKG2D CD159D | APC | 1D11 | 320808 | BioLegend |
| LFA-1 | BV421 | M24 | 363408 | BioLegend |
| CRACC | BV605 | 235614 | 235614 | BD Biosciences |
| CD94 | BV786 | HP-3D9 | 743953 | BD Biosciences |
| NKG2A CD159A | PE | Z199 | IM3291U | Beckman Coulter |
| NKG2C CD159C | PE-Vio770 | REA205 | 130-120-449 | Miltenyi |
| SLAMF6 CD352/NTBA | APC | REA339 | 130-105-543 | Miltenyi |
| CD25 | BV421 | BC96 | 302630 | BioLegend |
| PD-1 | BV605 | NAT105 | 367426 | BioLegend |
| CD69 | BV785 | FN50 | 310932 | BioLegend |
| TIM-3 | PE | F38-2E2 | 345006 | BioLegend |
| FAS-L | PE-Cy7 | Nok-1 | 306418 | BioLegend |
| TRAIL | APC | RIK-2 | 308210 | BioLegend |
| CD96 | BV421 | 6F9 | 742794 | BD biosciences |
| 2B4 | BV605 | C1.7 | 329536 | BioLegend |
| CD2 | BV785 | RPA-2.10 | 300234 | BioLegend |
| SIGLEC7 | PE | 6–434 | 339204 | BioLegend |
| KIR2D | PE-Vio770 | REA1042 | 130-117-483 | Miltenyi |
| KIR3DL1/L2 | PE-Vio770 | REA970 | 130-116-180 | Miltenyi |
| CD161 | APC | HP-3G10 | 339912 | BioLegend |
| CXCR4 | BV421 | 12G5 | 306518 | BioLegend |
| CCR6 | BV605 | G034E3 | 353420 | BioLegend |
| CD62L | BV785 | DREG-56 | 304830 | BioLegend |
| CD57 | PE | QA17A04 | 393308 | BioLegend |
| CCR7 | PE-Cy7 | G043H7 | 353226 | BioLegend |
| CXCR3 | APC | G025H7 | 353708 | BioLegend |
Flow cytometry-based cytotoxicity assay
Cell lines were trypsinized when necessary and washed with phosphate-buffered saline before labeling with 5 μM Pacific blue succinimidyl ester (Thermo Fisher Scientific). NK cells were cultured for 7 days after thawing, then (on day 35) co-cultured with target tumor cells at an E:T ratio of 1:1 in a 96-well plate. As controls, target cells and NK cells alone were plated. After 20 h incubation at 37°C, cells were trypsinized and transferred to a V-bottom plate and stained with 7-aminoactinomycin D (7AAD) (Sigma) and anti-CD56 APC-A750 (N901, Beckman Coulter). Samples were acquired in technical triplicates with Cytoflex LX (Beckman Coulter). Data analysis was performed with Kaluza software (Beckman Coulter). Target cell killing, expressed as “percentage cytotoxicity,” was determined as follows (Equation 1):
| (Equation 1) |
Impedance-based cytotoxicity assay
Impedance-based cytotoxicity assays were performed on the xCELLigence Real-Time Analysis platform (Agilent). After baseline measurement with medium, cell lines A375, LN-18, and LoVo were seeded in a 96-well PET E-plate (Agilent) at 25,000, 30,000, and 30,000 cells/well, respectively, 1 day before effector cell addition to allow target cells to adhere. The next day, a few extra wells of the seeded cells were trypsinized and counted to match E:T ratios. NK cells were added in the following E:T ratios: 10:1, 3:1, 1:1, and 1:3. Impedance during the co-culture was measured every 15 min for a total of 24 h. Data analysis was performed with the RTCA software Pro (Agilent) using the immunotherapy module.
Bulk RNA-seq and data analysis
To preserve RNA expression patterns, 1 × 106 cells were collected from all 10 donors on day 35, then immediately diluted in RNAprotect Cell Reagent (QIAGEN) and stored at −80°C until mRNA isolation. The RNAeasy Plus Mini kit (QIAGEN) was used to isolate RNA according to manufacturer’s instructions. RNA samples were stored at −80°C until sequencing. RNA quality, measured as RNA integrity number, was >8 for all samples.
RNA-seq libraries were prepared according to the Illumina TruSeq stranded mRNA protocol. In brief, 200 ng of total RNA was purified using poly(T) oligo-attached magnetic beads to end up with poly(A)-containing mRNA. The poly(A)-tailed mRNA was fragmented and cDNA was synthesized using SuperScript II and random primers in the presence of actinomycin D. cDNA fragments were end repaired, purified with AMPure XP beads, and A-tailed using Klenow exo-enzyme in the presence of dATP. Paired end adapters with dual index (Illumina) were ligated to the A-tailed cDNA fragments and purified using AMPure XP beads. The resulting adapter-modified cDNA fragments were enriched by PCR using Phusion polymerase as follows: 30 s at 98°C, 15 cycles of 10 s at 98°C, 30 s at 60°C, and 30 s at 72°C, and 5 min at 72°C. PCR products were purified using AMPure XP beads and eluted in 30 μL of resuspension buffer. 1 μL was loaded on an Agilent Technologies 2100 Bioanalyzer using a DNA 1000 assay to determine the library concentration and for quality check. The resulting libraries were sequenced according to the Illumina TruSeq Rapid v.2 protocol on an Illumina HiSeq2500 sequencer. Paired-end reads of 100 base pairs in length were generated. Illumina adapter and poly(A) sequences were trimmed off with an in-house adapter-trimming script and the trimmed reads were mapped against the human GRCh38 reference using HiSat2 (v.2.1.0).72 Gene expression values were determined using htseq-count (v. 0.11.2)73 and Ensembl release-91 gene and transcript annotation. Sample QC, differential expression analysis, and data visualization were performed in the R environment for statistical computing v.3.6.3, using the packages DESeq2 v.1.26.0,74 tidyverse v.1.3.0, and ggplot2.75
Genes with >10 average counts were considered significantly expressed with a log2 fold change (log2FC) >1 or <−1 and an adjusted p value (Padj) < 0.05. GSEA was performed using GSEA v.4.1.0.76 The following gene sets were used: KEGG c2.cp.kegg.v2022.1.Hs.symbols.gmt and GO BP c5.go.bp.v2023.1.Hs.symbols. Classic enrichment statistics were used to calculate the statistics of the enrichment score and normalized enrichment score. A false discovery rate < 0.25, concomitant with a nominal p < 0.05, was considered significant.
scRNA-seq and data analysis
Immediately after thawing, 1 × 105 day 28 cells from 8 donors (1–5, 7, 8, and 10) were processed through the BD Rhapsody workflow (BD Biosciences). First, cells were resuspended in stain buffer, and each donor was labeled with a Sample Tag from the Human Single-Cell Multiplexing Kit. After washing, donors were pooled and stained with the BD AbSeq Immune Discovery Panel (BD Biosciences). Next, cells were loaded into a BD Rhapsody cartridge and checked for viability by Calcein AM (Thermo Fisher Scientific) and DRAQ7 (BD Biosciences) in the BD Rhapsody Scanner. A total of 33,774 living cells (72.4% viable) were loaded in the cartridge. Cells were captured in the microwells of the cartridge and cDNA and exonuclease treatment was performed using the Rhapsody cDNA kit according to manufacturer’s instruction. Libraries were prepared using the BD Rhapsody WTA Amplification kit according to manufacturer’s instructions. In brief, cDNA of the BD AbSeq and Sample Tags were denatured from the beads and supernatant was collected. A total of three libraries were generated: RNA library, BD AbSeq library, and Sample Tag library. To generate the RNA library, random priming and extension was performed, followed by a clean-up to remove primer dimers and other small-molecular weight by-products, and amplification by PCR. The amplification product was purified, and quality control and quantification were performed using the Qubit dsDNA HS Assay (Thermo Fisher Scientific) and Agilent High Sensitivity D1000 Screen Tape Assay (Agilent). The AbSeq/Sample Tag libraries were generated using the supernatant after denaturation by amplification through PCR. The PCR1 products were purified and quantified, followed by a PCR2 amplification for the Sample Tag library. To make the libraries compatible with the Illumina sequencing platform, full-length Illumina sequencing adapters and indexes were added through PCR. Sequencing was performed on an Illumina NovaSeq 6000 using a Novaseq 2 × 150 bp kit with a preferred number of reads per cell of 50,000 for the RNA library, 16,000 for the AbSeq library, and 1,000 for the Sample Tag library.
Fastq files were processed using the BD Rhapsody WTA Analysis Pipeline 2.0 in SevenBridges. Reads with low quality were filtered out based on read length (R1 < 60 bp or R2 < 40), Phred score (mean base quality score <20), and single-nucleotide frequency (R1 ≥ 0.55 or R2 ≥ 0.80). High-quality R1 reads were aligned to the pre-defined cell label sequences and the unique molecular identifiers (UMIs). High-quality R2 reads were aligned to human reference genome for mRNA and a reference panel of sequences for AbSeq information. Next, reads with the same cell label, UMI sequence, and same gene were collapsed into a single raw molecule to determine gene expression. Recursive substitution error correction and distribution-based error correction (DBEC) algorithms were used to correct for UMI errors and sequencing errors. Putative cells were determined by comparing the number of unique cell labels vs. number of reads on a log-transformed cumulative curve, where the inflection point separates putative cells from noise. Lastly, sample of origin was determined based on Sample Tag reads count.
The DBEC files with molecules per cell were imported in the R package Seurat v.4.3.077 and a Seurat object was created for further analysis. A total of 17,397 cells were sequenced and initial quality control analysis excluded cells with more than 20% mitochondrial gene expression or expressing fewer than 200 or more than 7,000 bioproducts (mRNA/AbSeq). Untagged cells were excluded. Further analysis was performed with the remaining 7,518 cells. Log normalization was performed for RNA data and centered log-ratio normalization for AbSeq data. Data dimensionality was reduced by PCA on both RNA and AbSeq data and elbow plots were used to retain relevant components. To integrate the multimodal mRNA and AbSeq datasets, the FindMultiModalNeighbors function was used, selecting 30 dimensions for RNA and 18 dimensions for AbSeq. Integrated data were visualized using UMAP78 after applying the RunUMAP function. Clusters were determined with the FindClusters function, using the Leiden algorithm79 with a resolution of 0.05, resulting in 7 clusters. Only clusters present in at least 5/8 samples were retained. Differentially expressed genes between clusters or groups were identified using FindAllMarkers or FindMarkers function, to obtain only upregulated genes with the following parameters: log2FC > 0.25 and Padj < 0.05. GSEA was performed with the clusterprofiler v.4.8.180 package. The GO Biological Process and KEGG databases were used. Significance was calculated using Benjamini-Hochberg correction and gene sets with Padj < 0.05 were considered. Pseudotime analysis was performed using Monocle 3,81 selecting cluster 3 as the root cluster. Cell-cycle analysis was performed using the CellCycleScoring function from Seurat.
Validation of gene expression by qPCR
Genes of interest for validation were selected based on genes that contributed to the core enrichment of the KEGG NK cell-mediated cytotoxicity pathway. For all tested genes, primers were designed with the Primer-BLAST tool from NCBI (Table 2). The iTaq Universal SYBR Green One-Step Kit (Bio-Rad) was used according to the manufacturer’s instructions. In brief, each reaction contained 5 μL iTaq reaction mix, 0.125 μL reverse transcriptase, 2 ng RNA, and 500 nM of forward and reverse primers. The reaction was performed at 50°C for 10 min, 95°C for 1 min, and for 40 cycles of 10 s at 95°C and 30 s at 60°C using a CFX Connect Real-Time PCR Detection System (Bio-Rad). After the amplification, a melt curve analysis was performed by increasing the temperature from 65°C to 95°C (0.5°C increment per step) for 5 s per step. No primer dimer peaks were observed. Fold change was calculated using the ΔΔCt method.82 Reference genes PPIA and YWHAZ were used for normalization and the good group was considered as control.
Table 2.
Forward and reverse primer sequences used for qPCR
| Gene | Forward primer | Reverse primer |
|---|---|---|
| GZMB | ACCATTGAGTTGTGCGTGGG | AGGCATGCCATTGTTTCGTC |
| PRF1 | CACCAGGACCAGTACAGCTT | GTGGAGGCGTTGAAGTGGT |
| PPIA | TCATCTGCACTGCCAAGACTG | CATGCCTTCTTTCACTTTGCC |
| YWHAZ | ACTTTTGGTACATTGTGGCTTCAA | CCGCCAGGACAAACCAGTAT |
Intracellular staining
Intracellular stainings were performed on 9 donors (1–8 and 10) on day 35 of culture. After count, 1 × 105 cells were stained with anti-CD56 BV421 (NCAM16.2, BD Biosciences) and live/dead fixable aqua (Thermo Fisher Scientific). After washing, cells were fixed and permeabilized using the cytofix/cytoperm kit (BD Biosciences) according to the manufacturer’s instructions. Cells were stained with anti-perforin PerCP-Cy5.5 (B-D48, BioLegend), anti-granzyme B AF647 (GB11, BioLegend), and mouse IgG1 isotype control in PerCP-Cy5.5 or AF647 (MOPC-21, BioLegend). All antibodies were used at the recommended concentration, and the good group was considered as control.
Secretome analysis
For secretome analysis, 1:1 co-cultures of 9 day 35 donors (1–8, 10) and K562 targets were incubated similarly to the flow cytometry-based cytotoxicity assay. Supernatant was collected after 5 h. The CD8/NK LEGENDplex (BioLegend) kit was used according to the manufacturer’s protocol to detect the following analytes: TNF, IFN-γ, granzyme A, granzyme B, perforin, and granulysin.
Statistical analysis
Hierarchical clustering based on cytotoxicity was performed first by calculating the mean cytotoxicity value, i.e., the geometric mean of the Δ 7AAD+ cells (%) values (Equation 1) across the four target cell lines for each donor, followed by calculating the distance matrix using the Euclidean method and computing the dendrogram with the single agglomeration method, and finally by visualizing distances as a heatmap. For surface receptors, the percentage of signal-positive cells per donor was expressed as Z scores, then the distance was calculated with the Euclidean method and the dendrogram was computed with the Ward algorithm. Two-way ANOVA with Šídák correction for multiple comparison or unpaired t-test with Holm-Šídák correction for multiple comparison was performed where indicated.
In scRNA-seq cluster analysis, qPCR and intracellular staining analysis, the statistical test was considered significant with (adjusted) p < 0.05. Significance is shown as ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
For the gene expression signature, a signature score was defined, based on 13 cytotoxicity genes from the excellent (e) group and 9 myeloid genes from the good (g) group, with a higher value determined by concomitant high expression of e-genes and low expression of g-genes. For each e- and g-genes, normalized gene counts (bulk RNA-seq) or average gene expression values calculated with the AverageExpression function in Seurat77 (scRNA-seq) were expressed as Z scores (, where and , where ). was calculated for each donor as the average of the combined Z scores (Equation 2),
| (Equation 2) |
where the sign of the Z scores of the g-genes was reversed. To improve visualization, the minimum value was shifted to 1 (). With biological replicates, the final score per donor was calculated as the average of the replicates. Linear regression analysis between cytotoxicity (geometric mean of four cell lines per donor) and the signature score was performed; Pearson’s correlation coefficient r was calculated between the two variables.
Data and code availability
Processed and raw RNA-seq data are available at the Gene Expression Omnibus Database (accession no. GEO: GSE249992). Additional data that support the findings of this study are available from the corresponding authors upon request.
Acknowledgments
The authors gratefully acknowledge Hasi Patel and Dr. Soudabeh Rad Pour from BD Biosciences for their support with designing the scRNA-seq experiments, the team at GalSeq S.R.L. for performing library preparation and sequencing for the scRNA-seq experiments, and Dr. Alessandro Corbetta for his helpful suggestions on data analysis and visualization.
Author contributions
A.A.v.V. designed the study, performed the majority of the experimental work, analyzed the data, analyzed the scRNA-seq data, prepared the figures, and drafted and wrote the manuscript. M.G.C.N.v.d.H. and W.F.J.v.I. performed bulk sequencing and analyzed the results. D.S. performed NK cell cultures and qPCR experiments. A.D.D. and A-M.G. supported with experimental design and reviewed the manuscript. T.D.d.G. interpreted data and reviewed and edited the manuscript. J.S. conceived the project idea, secured funding, and reviewed the manuscript. M.R. conceived the project idea, designed the study, performed experiments, analyzed the scRNA-seq data, prepared figures, and wrote and reviewed the manuscript. All authors reviewed and approved the final draft for submission.
Declaration of interests
A.A.v.V., D.S., A.D.D., A.-M.G., J.S., and M.R. are employees of Glycostem Therapeutics. All authors declare no competing interests. Data and results generated, shared, and reported in conjunction with this publication have been filed in a patent application by Glycostem Therapeutics.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omton.2024.200870.
Contributor Information
Amanda A. van Vliet, Email: amanda@glycostem.com.
Monica Raimo, Email: monica@glycostem.com.
Supplemental information
References
- 1.Vivier E., Raulet D.H., Moretta A., Caligiuri M.A., Zitvogel L., Lanier L.L., Yokoyama W.M., Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–49. doi: 10.1126/science.1198687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Binstadt B.A., Billadeau D.D., Jevremović D., Williams B.L., Fang N., Yi T., Koretzky G.A., Abraham R.T., Leibson P.J. SLP-76 is a direct substrate of SHP-1 recruited to killer cell inhibitory receptors. J. Biol. Chem. 1998;273:27518–27523. doi: 10.1074/jbc.273.42.27518. [DOI] [PubMed] [Google Scholar]
- 3.Stebbins C.C., Watzl C., Billadeau D.D., Leibson P.J., Burshtyn D.N., Long E.O. Vav1 dephosphorylation by the tyrosine phosphatase SHP-1 as a mechanism for inhibition of cellular cytotoxicity. Mol. Cell Biol. 2003;23:6291–6299. doi: 10.1128/mcb.23.17.6291-6299.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pegram H.J., Andrews D.M., Smyth M.J., Darcy P.K., Kershaw M.H. Activating and inhibitory receptors of natural killer cells. Immunol. Cell Biol. 2011;89:216–224. doi: 10.1038/icb.2010.78. [DOI] [PubMed] [Google Scholar]
- 5.Pende D., Parolini S., Pessino A., Sivori S., Augugliaro R., Morelli L., Marcenaro E., Accame L., Malaspina A., Biassoni R., et al. Identification and molecular characterization of NKp30, a novel triggering receptor involved in natural cytotoxicity mediated by human natural killer cells. J. Exp. Med. 1999;190:1505–1516. doi: 10.1084/jem.190.10.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sivori S., Vitale M., Morelli L., Sanseverino L., Augugliaro R., Bottino C., Moretta L., Moretta A. p46, a novel natural killer cell-specific surface molecule that mediates cell activation. J. Exp. Med. 1997;186:1129–1136. doi: 10.1084/jem.186.7.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vitale M., Bottino C., Sivori S., Sanseverino L., Castriconi R., Marcenaro E., Augugliaro R., Moretta L., Moretta A. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non-major histocompatibility complex-restricted tumor cell lysis. J. Exp. Med. 1998;187:2065–2072. doi: 10.1084/jem.187.12.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lanier L.L., Yu G., Phillips J.H. Analysis of Fc gamma RIII (CD16) membrane expression and association with CD3 zeta and Fc epsilon RI-gamma by site-directed mutation. J. Immunol. 1991;146:1571–1576. [PubMed] [Google Scholar]
- 9.Eissmann P., Beauchamp L., Wooters J., Tilton J.C., Long E.O., Watzl C. Molecular basis for positive and negative signaling by the natural killer cell receptor 2B4 (CD244) Blood. 2005;105:4722–4729. doi: 10.1182/blood-2004-09-3796. [DOI] [PubMed] [Google Scholar]
- 10.Upshaw J.L., Arneson L.N., Schoon R.A., Dick C.J., Billadeau D.D., Leibson P.J. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat. Immunol. 2006;7:524–532. doi: 10.1038/ni1325. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y., Lu D., Churov A., Fu R. Research Progress on NK Cell Receptors and Their Signaling Pathways. Mediators Inflamm. 2020;2020:6437057. doi: 10.1155/2020/6437057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J., Figueira S.K., Vrazo A.C.A., Binkowski B.F., Butler B.L., Tabata Y., Filipovich A., Jordan M.B., Risma K.A. Real-time detection of CTL function reveals distinct patterns of caspase activation mediated by Fas versus granzyme B. J. Immunol. 2014;193:519–528. doi: 10.4049/jimmunol.1301668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Vliet A.A., Peters E., Vodegel D., Steenmans D., Raimo M., Gibbs S., de Gruijl T.D., Duru A.D., Spanholtz J., Georgoudaki A.M. Early TRAIL-engagement elicits potent multimodal targeting of melanoma by CD34(+) progenitor cell-derived NK cells. iScience. 2023;26:107078. doi: 10.1016/j.isci.2023.107078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abel A.M., Yang C., Thakar M.S., Malarkannan S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018;9:1869. doi: 10.3389/fimmu.2018.01869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cichocki F., Grzywacz B., Miller J.S. Human NK Cell Development: One Road or Many? Front. Immunol. 2019;10:2078. doi: 10.3389/fimmu.2019.02078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamers-Kok N., Panella D., Georgoudaki A.M., Liu H., Özkazanc D., Kučerová L., Duru A.D., Spanholtz J., Raimo M. Natural killer cells in clinical development as non-engineered, engineered, and combination therapies. J. Hematol. Oncol. 2022;15:164. doi: 10.1186/s13045-022-01382-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Myers J.A., Miller J.S. Exploring the NK cell platform for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021;18:85–100. doi: 10.1038/s41571-020-0426-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foley B., Ta C., Barnes S., de Jong E., Nguyen M., Cheung L.C., Buzzai A., Wagner T., Wylie B., Fernandez S., et al. Identifying the optimal donor for natural killer cell adoptive therapy to treat paediatric B- and T-cell acute lymphoblastic leukaemia. Clin. Transl. Immunol. 2020;9:e1151. doi: 10.1002/cti2.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barnes S.A., Trew I., de Jong E., Foley B. Making a Killer: Selecting the Optimal Natural Killer Cells for Improved Immunotherapies. Front. Immunol. 2021;12:765705. doi: 10.3389/fimmu.2021.765705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spanholtz J., Tordoir M., Eissens D., Preijers F., van der Meer A., Joosten I., Schaap N., de Witte T.M., Dolstra H. High log-scale expansion of functional human natural killer cells from umbilical cord blood CD34-positive cells for adoptive cancer immunotherapy. PLoS One. 2010;5:e9221. doi: 10.1371/journal.pone.0009221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Veluchamy J.P., Lopez-Lastra S., Spanholtz J., Bohme F., Kok N., Heideman D.A.M., Verheul H.M.W., Di Santo J.P., de Gruijl T.D., van der Vliet H.J. In Vivo Efficacy of Umbilical Cord Blood Stem Cell-Derived NK Cells in the Treatment of Metastatic Colorectal Cancer. Front. Immunol. 2017;8:87. doi: 10.3389/fimmu.2017.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saha T., van Vliet A.A., Cui C., Macias J.J., Kulkarni A., Pham L.N., Lawler S., Spanholtz J., Georgoudaki A.M., Duru A.D., Goldman A. Boosting Natural Killer Cell Therapies in Glioblastoma Multiforme Using Supramolecular Cationic Inhibitors of Heat Shock Protein 90. Front. Mol. Biosci. 2021;8:754443. doi: 10.3389/fmolb.2021.754443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toffoli E.C., van Vliet A.A., Verheul H.W.M., van der Vliet H.J., Tuynman J., Spanholtz J., de Gruijl T.D. Allogeneic NK cells induce monocyte-to-dendritic cell conversion, control tumor growth, and trigger a pro-inflammatory shift in patient-derived cultures of primary and metastatic colorectal cancer. J. Immunother. Cancer. 2023;11:e007554. doi: 10.1136/jitc-2023-007554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez-Vergès S., Milush J.M., Pandey S., York V.A., Arakawa-Hoyt J., Pircher H., Norris P.J., Nixon D.F., Lanier L.L. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood. 2010;116:3865–3874. doi: 10.1182/blood-2010-04-282301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luetke-Eversloh M., Cicek B.B., Siracusa F., Thom J.T., Hamann A., Frischbutter S., Baumgrass R., Chang H.D., Thiel A., Dong J., Romagnani C. NK cells gain higher IFN-γ competence during terminal differentiation. Eur. J. Immunol. 2014;44:2074–2084. doi: 10.1002/eji.201344072. [DOI] [PubMed] [Google Scholar]
- 26.Lanier L.L. Natural killer cell receptor signaling. Curr. Opin. Immunol. 2003;15:308–314. doi: 10.1016/s0952-7915(03)00039-6. [DOI] [PubMed] [Google Scholar]
- 27.Juelke K., Killig M., Luetke-Eversloh M., Parente E., Gruen J., Morandi B., Ferlazzo G., Thiel A., Schmitt-Knosalla I., Romagnani C. CD62L expression identifies a unique subset of polyfunctional CD56dim NK cells. Blood. 2010;116:1299–1307. doi: 10.1182/blood-2009-11-253286. [DOI] [PubMed] [Google Scholar]
- 28.Xu Y., Zhang J., Hu Y., Li X., Sun L., Peng Y., Sun Y., Liu B., Bian Z., Rong Z. Single-cell transcriptome analysis reveals the dynamics of human immune cells during early fetal skin development. Cell Rep. 2021;36:109524. doi: 10.1016/j.celrep.2021.109524. [DOI] [PubMed] [Google Scholar]
- 29.Steiner J., Marquardt N., Pauls I., Schiltz K., Rahmoune H., Bahn S., Bogerts B., Schmidt R.E., Jacobs R. Human CD8(+) T cells and NK cells express and secrete S100B upon stimulation. Brain Behav. Immun. 2011;25:1233–1241. doi: 10.1016/j.bbi.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 30.Yen K., Lee C., Mehta H., Cohen P. The emerging role of the mitochondrial-derived peptide humanin in stress resistance. J. Mol. Endocrinol. 2013;50:R11–R19. doi: 10.1530/jme-12-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu H., Zhao R., Qin R., Sun H., Huang Q., Liu L., Tian Z., Nashan B., Sun C., Sun R. Panoramic comparison between NK cells in healthy and cancerous liver through single-cell RNA sequencing. Cancer Biol. Med. 2022;19:1334–1351. doi: 10.20892/j.issn.2095-3941.2022.0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Makhijani P., Basso P.J., Chan Y.T., Chen N., Baechle J., Khan S., Furman D., Tsai S., Winer D.A. Regulation of the immune system by the insulin receptor in health and disease. Front. Endocrinol. 2023;14:1128622. doi: 10.3389/fendo.2023.1128622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Crocker P.R., Jefferies W.A., Clark S.J., Chung L.P., Gordon S. Species heterogeneity in macrophage expression of the CD4 antigen. J. Exp. Med. 1987;166:613–618. doi: 10.1084/jem.166.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang S., Song R., Wang Z., Jing Z., Wang S., Ma J. S100A8/A9 in Inflammation. Front. Immunol. 2018;9:1298. doi: 10.3389/fimmu.2018.01298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sontheimer R.D., Racila E., Racila D.M. C1q: its functions within the innate and adaptive immune responses and its role in lupus autoimmunity. J. Invest. Dermatol. 2005;125:14–23. doi: 10.1111/j.0022-202X.2005.23673.x. [DOI] [PubMed] [Google Scholar]
- 36.Lupancu T.J., Eivazitork M., Hamilton J.A., Achuthan A.A., Lee K.M.C. CCL17/TARC in autoimmunity and inflammation-not just a T-cell chemokine. Immunol. Cell Biol. 2023;101:600–609. doi: 10.1111/imcb.12644. [DOI] [PubMed] [Google Scholar]
- 37.Xiong X., Liao X., Qiu S., Xu H., Zhang S., Wang S., Ai J., Yang L. CXCL8 in Tumor Biology and Its Implications for Clinical Translation. Front. Mol. Biosci. 2022;9:723846. doi: 10.3389/fmolb.2022.723846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aristorena M., Gallardo-Vara E., Vicen M., de Las Casas-Engel M., Ojeda-Fernandez L., Nieto C., Blanco F.J., Valbuena-Diez A.C., Botella L.M., Nachtigal P., et al. MMP-12, Secreted by Pro-Inflammatory Macrophages, Targets Endoglin in Human Macrophages and Endothelial Cells. Int. J. Mol. Sci. 2019;20:3107. doi: 10.3390/ijms20123107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yabluchanskiy A., Ma Y., Iyer R.P., Hall M.E., Lindsey M.L. Matrix metalloproteinase-9: Many shades of function in cardiovascular disease. Physiology. 2013;28:391–403. doi: 10.1152/physiol.00029.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haraguchi K., Suzuki T., Koyama N., Kumano K., Nakahara F., Matsumoto A., Yokoyama Y., Sakata-Yanagimoto M., Masuda S., Takahashi T., et al. Notch activation induces the generation of functional NK cells from human cord blood CD34-positive cells devoid of IL-15. J. Immunol. 2009;182:6168–6178. doi: 10.4049/jimmunol.0803036. [DOI] [PubMed] [Google Scholar]
- 41.Felices M., Ankarlo D.E.M., Lenvik T.R., Nelson H.H., Blazar B.R., Verneris M.R., Miller J.S. Notch signaling at later stages of NK cell development enhances KIR expression and functional maturation. J. Immunol. 2014;193:3344–3354. doi: 10.4049/jimmunol.1400534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peruzzi G., Masilamani M., Borrego F., Coligan J.E. Endocytosis as a mechanism of regulating natural killer cell function: unique endocytic and trafficking pathway for CD94/NKG2A. Immunol. Res. 2009;43:210–222. doi: 10.1007/s12026-008-8072-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Molfetta R., Quatrini L., Zitti B., Capuano C., Galandrini R., Santoni A., Paolini R. Regulation of NKG2D Expression and Signaling by Endocytosis. Trends Immunol. 2016;37:790–802. doi: 10.1016/j.it.2016.08.015. [DOI] [PubMed] [Google Scholar]
- 44.Huntington N.D., Legrand N., Alves N.L., Jaron B., Weijer K., Plet A., Corcuff E., Mortier E., Jacques Y., Spits H., Di Santo J.P. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J. Exp. Med. 2009;206:25–34. doi: 10.1084/jem.20082013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stoeckle C., Gouttefangeas C., Hammer M., Weber E., Melms A., Tolosa E. Cathepsin W expressed exclusively in CD8+ T cells and NK cells, is secreted during target cell killing but is not essential for cytotoxicity in human CTLs. Exp. Hematol. 2009;37:266–275. doi: 10.1016/j.exphem.2008.10.011. [DOI] [PubMed] [Google Scholar]
- 46.Bourque J., Opejin A., Surnov A., Iberg C.A., Gross C., Jain R., Epstein J.A., Hawiger D. Landscape of Hopx expression in cells of the immune system. Heliyon. 2021;7:e08311. doi: 10.1016/j.heliyon.2021.e08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Milush J.M., Long B.R., Snyder-Cappione J.E., Cappione A.J., 3rd, York V.A., Ndhlovu L.C., Lanier L.L., Michaëlsson J., Nixon D.F. Functionally distinct subsets of human NK cells and monocyte/DC-like cells identified by coexpression of CD56, CD7, and CD4. Blood. 2009;114:4823–4831. doi: 10.1182/blood-2009-04-216374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dutertre C.-A., Becht E., Irac S.E., Khalilnezhad A., Narang V., Khalilnezhad S., Ng P.Y., Van Den Hoogen L.L., Leong J.Y., Lee B., et al. Single-Cell Analysis of Human Mononuclear Phagocytes Reveals Subset-Defining Markers and Identifies Circulating Inflammatory Dendritic Cells. Immunity. 2019;51:573–589.e8. doi: 10.1016/j.immuni.2019.08.008. [DOI] [PubMed] [Google Scholar]
- 49.Li J., Zhou J., Huang H., Jiang J., Zhang T., Ni C. Mature dendritic cells enriched in immunoregulatory molecules (mregDCs): A novel population in the tumour microenvironment and immunotherapy target. Clin. Transl. Med. 2023;13:e1199. doi: 10.1002/ctm2.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Groth C., Hu X., Weber R., Fleming V., Altevogt P., Utikal J., Umansky V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer. 2019;120:16–25. doi: 10.1038/s41416-018-0333-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang D., Malarkannan S. Transcriptional Regulation of Natural Killer Cell Development and Functions. Cancers. 2020;12:1591. doi: 10.3390/cancers12061591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.de Sousa Abreu R., Penalva L.O., Marcotte E.M., Vogel C. Global signatures of protein and mRNA expression levels. Mol. Biosyst. 2009;5:1512–1526. doi: 10.1039/b908315d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eissens D.N., Spanholtz J., van der Meer A., van Cranenbroek B., Dolstra H., Kwekkeboom J., Preijers F.W.M.B., Joosten I. Defining early human NK cell developmental stages in primary and secondary lymphoid tissues. PLoS One. 2012;7:e30930. doi: 10.1371/journal.pone.0030930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosenbauer F., Tenen D.G. Transcription factors in myeloid development: balancing differentiation with transformation. Nat. Rev. Immunol. 2007;7:105–117. doi: 10.1038/nri2024. [DOI] [PubMed] [Google Scholar]
- 55.Laskowski T.J., Biederstädt A., Rezvani K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat. Rev. Cancer. 2022;22:557–575. doi: 10.1038/s41568-022-00491-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meazza R., Ruggeri L., Guolo F., Minetto P., Canevali P., Loiacono F., Ciardelli S., Bo A., Luchetti S., Serio A., et al. Donor selection for adoptive immunotherapy with NK cells in AML patients: Comparison between analysis of lytic NK cell clones and phenotypical identification of alloreactive NK cell repertoire. Front. Immunol. 2023;14:1111419. doi: 10.3389/fimmu.2023.1111419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haroun-Izquierdo A., Vincenti M., Netskar H., van Ooijen H., Zhang B., Bendzick L., Kanaya M., Momayyezi P., Li S., Wiiger M.T., et al. Adaptive single-KIR+NKG2C+ NK cells expanded from select superdonors show potent missing-self reactivity and efficiently control HLA-mismatched acute myeloid leukemia. J. Immunother. Cancer. 2022;10:e005577. doi: 10.1136/jitc-2022-005577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Poznanski S.M., Nham T., Chew M.V., Lee A.J., Hammill J.A., Fan I.Y., Butcher M., Bramson J.L., Lee D.A., Hirte H.W., Ashkar A.A. Expanded CD56(superbright)CD16(+) NK Cells from Ovarian Cancer Patients Are Cytotoxic against Autologous Tumor in a Patient-Derived Xenograft Murine Model. Cancer Immunol. Res. 2018;6:1174–1185. doi: 10.1158/2326-6066.Cir-18-0144. [DOI] [PubMed] [Google Scholar]
- 59.Wagner J.A., Rosario M., Romee R., Berrien-Elliott M.M., Schneider S.E., Leong J.W., Sullivan R.P., Jewell B.A., Becker-Hapak M., Schappe T., et al. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J. Clin. Invest. 2017;127:4042–4058. doi: 10.1172/jci90387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Siewiera J., Gouilly J., Hocine H.R., Cartron G., Levy C., Al-Daccak R., Jabrane-Ferrat N. Natural cytotoxicity receptor splice variants orchestrate the distinct functions of human natural killer cell subtypes. Nat. Commun. 2015;6:10183. doi: 10.1038/ncomms10183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marin D., Li Y., Basar R., Rafei H., Daher M., Dou J., Mohanty V., Dede M., Nieto Y., Uprety N., et al. Safety, efficacy and determinants of response of allogeneic CD19-specific CAR-NK cells in CD19(+) B cell tumors: a phase 1/2 trial. Nat. Med. 2024;30:772–784. doi: 10.1038/s41591-023-02785-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lehmann D., Spanholtz J., Osl M., Tordoir M., Lipnik K., Bilban M., Schlechta B., Dolstra H., Hofer E. Ex Vivo Generated Natural Killer Cells Acquire Typical Natural Killer Receptors and Display a Cytotoxic Gene Expression Profile Similar to Peripheral Blood Natural Killer Cells. Stem Cells Dev. 2012;21:2926–2938. doi: 10.1089/scd.2011.0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dolstra H., Roeven M.W.H., Spanholtz J., Hangalapura B.N., Tordoir M., Maas F., Leenders M., Bohme F., Kok N., Trilsbeek C., et al. Successful Transfer of Umbilical Cord Blood CD34(+) Hematopoietic Stem and Progenitor-derived NK Cells in Older Acute Myeloid Leukemia Patients. Clin. Cancer Res. 2017;23:4107–4118. doi: 10.1158/1078-0432.Ccr-16-2981. [DOI] [PubMed] [Google Scholar]
- 64.Cursons J., Souza-Fonseca-Guimaraes F., Foroutan M., Anderson A., Hollande F., Hediyeh-Zadeh S., Behren A., Huntington N.D., Davis M.J. A Gene Signature Predicting Natural Killer Cell Infiltration and Improved Survival in Melanoma Patients. Cancer Immunol. Res. 2019;7:1162–1174. doi: 10.1158/2326-6066.Cir-18-0500. [DOI] [PubMed] [Google Scholar]
- 65.Crinier A., Dumas P.Y., Escalière B., Piperoglou C., Gil L., Villacreces A., Vély F., Ivanovic Z., Milpied P., Narni-Mancinelli É., Vivier É. Single-cell profiling reveals the trajectories of natural killer cell differentiation in bone marrow and a stress signature induced by acute myeloid leukemia. Cell. Mol. Immunol. 2021;18:1290–1304. doi: 10.1038/s41423-020-00574-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dufva O., Gandolfi S., Huuhtanen J., Dashevsky O., Saeed K., Klievink J., Nygren P., Bouhlal J., Lahtela J., Näätänen A., et al. Cold Spring Harbor Laboratory; 2022. Single-cell Functional Genomics of Natural Killer Cell Evasion in Blood Cancers. [DOI] [Google Scholar]
- 67.Guo C., Wu M., Huang B., Zhao R., Jin L., Fu B., Wang P., Wang D., Zheng M., Fang J., et al. Single-cell transcriptomics reveal a unique memory-like NK cell subset that accumulates with ageing and correlates with disease severity in COVID-19. Genome Med. 2022;14:46. doi: 10.1186/s13073-022-01049-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ren C., Li M., Zheng Y., Cai B., Du W., Zhang H., Wu F., Tong M., Lin F., Wang J., Quan R. Single-cell RNA-seq reveals altered NK cell subsets and reduced levels of cytotoxic molecules in patients with ankylosing spondylitis. J. Cell Mol. Med. 2022;26:1071–1082. doi: 10.1111/jcmm.17159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li L., Mohanty V., Dou J., Huang Y., Banerjee P.P., Miao Q., Lohr J.G., Vijaykumar T., Frede J., Knoechel B., et al. Loss of metabolic fitness drives tumor resistance after CAR-NK cell therapy and can be overcome by cytokine engineering. Sci. Adv. 2023;9:eadd6997. doi: 10.1126/sciadv.add6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li S., Du H., Gan D., Li X., Zao X., Ye Y. Integrated analysis of single-cell and bulk RNA-sequencing reveals tumor heterogeneity and a signature based on NK cell marker genes for predicting prognosis in hepatocellular carcinoma. Front. Pharmacol. 2023;14:1200114. doi: 10.3389/fphar.2023.1200114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Spanholtz J., Preijers F., Tordoir M., Trilsbeek C., Paardekooper J., de Witte T., Schaap N., Dolstra H. Clinical-grade generation of active NK cells from cord blood hematopoietic progenitor cells for immunotherapy using a closed-system culture process. PLoS One. 2011;6:e20740. doi: 10.1371/journal.pone.0020740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim D., Langmead B., Salzberg S.L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods. 2015;12:357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Anders S., Pyl P.T., Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wickham H. Springer International Publishing; 2016. ggplot2 Elegant Graphics for Data Analysis. [Google Scholar]
- 76.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Becht E., McInnes L., Healy J., Dutertre C.A., Kwok I.W.H., Ng L.G., Ginhoux F., Newell E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018;37:38–44. doi: 10.1038/nbt.4314. [DOI] [PubMed] [Google Scholar]
- 79.Traag V.A., Waltman L., Van Eck N.J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 2019;9:5233. doi: 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation. 2021;2:100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bookout A.L., Mangelsdorf D.J. Quantitative real-time PCR protocol for analysis of nuclear receptor signaling pathways. Nucl. Recept. Signal. 2003;1:e012. doi: 10.1621/nrs.01012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Processed and raw RNA-seq data are available at the Gene Expression Omnibus Database (accession no. GEO: GSE249992). Additional data that support the findings of this study are available from the corresponding authors upon request.