

Abstract
Several metabolites serve as substrates for histone modifications and communicate changes in the metabolic environment to the epigenome. Technologies such as metabolomics and proteomics have allowed us to reconstruct the interactions between metabolic pathways and histones. These technologies have shed light on how nutrient availability can have a dramatic effect on various histone modifications. This metabolism-epigenome cross talk plays a fundamental role in development, immune function, and diseases like cancer. Yet, major challenges remain in understanding the interactions between cellular metabolism and the epigenome. How the levels and fluxes of various metabolites impact epigenetic marks is still unclear. Here we discuss recent applications, and the potential of systems biology methods such as flux tracing and metabolic modeling to address these challenges and to uncover new metabolic-epigenetic interactions. These systems approaches can ultimately help elucidate how nutrients shape the epigenome of microbes and mammalian cells.
Graphical Abstract
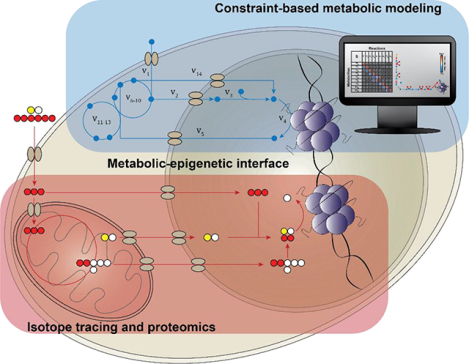
Histone post-translational modifications (PTMs) sense cellular metabolic state and regulate gene expression, thereby influencing normal physiology and disease progression. While histone PTMs rely on metabolic substrates, how nutrients impact the histone PTM code is unclear. Here we review systems biology technologies that can be used to study metabolic-epigenetic interactions.
1. Introduction
Histones are susceptible to a variety of post-translational modifications (PTMs). These include acetylation, methylation, phosphorylation, ubiquitination, sumoylation, glycosylation, and many others. These histone PTMs in combination serve as an ‘epigenetic code’ for transcriptional activation, repression and coordination of higher order chromatin structure.[1–3]
Histone PTMs are highly sensitive to cellular metabolism.[2,4,5] Several metabolites influence gene expression by serving as substrates for modification of histones or as regulators of epigenetic enzymes.[4] The metabolite S-Adenosyl Methionine (SAM) donates a methyl group to methyltransferase enzymes for histone methylation. Histone demethylating enzymes are dependent on intracellular levels of Flavin adenine dinucleotide (FAD), α-ketoglutarate, iron and oxygen. Acetyl-CoA, an important biomolecule produced from glucose, amino-acid and fatty acid catabolism, is the substrate for histone acetyltransferase enzymes, while NAD+ is the substrate for sirtuins, a class of deacetylase enzymes.
Histone PTMs thus represent an innate cellular mechanism that links metabolic status to gene expression. By sensing the levels of intracellular metabolites, cells can alter the expression of genes that are important for biological processes such as cell growth and differentiation. Furthermore, as metabolism provides the building blocks for histone PTMs, in many cases, these building blocks limit the creation of histone marks. For instance, lack of dietary folate can lead to impaired histone methylation during development [4]. Dysregulation of both metabolism and the epigenome are important hallmarks of cancers and metabolic disorders such as diabetes, obesity and hypertension.[2,4–6]
How histone PTMs sense and integrate various metabolic inputs is still unclear.[5] This has been challenging to investigate because histone PTMs sense several central metabolites that are in turn involved in numerous metabolic reactions.[5] Here we highlight recent technologies that can help us tackle the highly interconnected and compartmentalized nature of cellular metabolism and its myriad impacts on the epigenome.
2. Metabolism-histone interactions regulate normal and disease physiology
Metabolic-epigenetic interactions play a central role in development and normal physiology of various organisms.[2] For example, the interactions between histones and cellular metabolites are important for controlling gene expression during the cell cycle. The temporal peak in abundance of acetyl-CoA during the yeast cell cycle correlates with histone acetylation of growth genes.[7,8] Levels of histone glycosylation by acetylglucosamine (GlcNAc) also changes during the cell cycle. Histone GlcNAcylation depends on the activity of the hexosamine biosynthesis pathway and is sensitive to the availability of glucose, fatty acids, uridine and glutamine; thus it may act as a nutrient sensor of diverse metabolic pathways.[9,10] Metabolism and histone acetylation also play an important role in DNA repair in mammalian cells. Upon DNA damage, nuclear ATP-citrate lyase (ACLY) promotes acetyl-CoA production, facilitating histone acetylation at the sites of double-strand breaks and stimulates DNA repair. [11]
Some cellular metabolites directly regulate the expression of metabolic genes via histone modification. The FAD-dependent enzyme LSD1 has been shown to demethylate histones and regulate cellular energy levels by repressing genes involved in mitochondrial respiration and energy expenditure.[12,13] Another example is found in brown adipose tissue development. The master metabolic regulator – AMP-activated protein kinase (AMPK), causes increased production of α-ketoglutarate, the substrate for demethylases; α-ketoglutarate accumulation results in epigenetic activation of adipogenesis regulators by demethylation of their promoters.[14] In response to stress, AMPK also phosphorylates histone H2B serine residues (H2BS36) in mammalian cells and regulates the activity of histone acetyltransferases and deacetylases through phosphorylation.[15]
Metabolism driven epigenetic changes can influence cancer risk.[16] In mammalian cells, the set of genes activated by the c-Myc oncoprotein[17] through metabolic rewiring and chromatin remodeling[18] resembles the set of growth genes that are acetylated during acetyl-CoA peak abundance in the yeast cell cycle.[2,7,8] Metabolic gene mutations in diverse cancers cause an accumulation of succinate, fumarate and R-2-hydroxyglutarate. The accumulation of these metabolites is believed to contribute to tumorigenesis by inhibiting α-ketoglutarate-dependent demethylase enzymes including the tumor suppressor TET2.[2,19] Other studies have shown the NAD-dependent deacetylase enzymes – sirtuins, to be tumor suppressors, as they limit reactive oxygen species (ROS) synthesis.[20,21] Low NAD levels results in decreased sirtuin activity and increased risk for many cancers, likely due to DNA damage by ROS.[21,22] The enzyme Nicotinamide N-methyltransferase (NNMT) is overexpressed in numerous cancers. Increase in NNMT activity consumes SAM, impairing histone methylation and leading to altered expression of cancer-associated genes.[23]
Besides cancer, multiple studies have been conducted to better understand the effect of epigenetic changes on disease pathology. The level of H3K4 trimethylation, a histone mark associated with active transcription, in the promoters of genes involved in lipid metabolism, adipogenesis and inflammation, correlates with increasing BMI of individuals.[24] The availability of folate and other one-carbon donors during conception and pregnancy influences the epigenome and various phenotypes in offspring.[25] SAM and methionine availability also plays an important role in maintenance of pluripotency in stem cells. The depletion of SAM in stem cells diminishes H3K4 trimethylation levels and leads to enhanced differentiation.[26,27] In summary, these examples provide a connection between intracellular metabolites, histone marks and their effect on gene transcription, which may contribute to the progression of diseases.
3. Metabolic-epigenetic cross talk is complex and context-specific
The numerous metabolic pathways that intersect with histone PTMs make it highly challenging to understand their interdependencies. For example, acetylation is sensitive to acetyl-CoA and NAD+,[2,4] which are involved in hundreds of metabolic reactions. Methylation depends on highly connected metabolic intermediates (α-ketoglutarate, SAM) and redox factors (FAD), as well.[2,4]
As with any other process in biology, the metabolic impact on histone PTMs is context specific. For example, inhibiting the synthesis of SAM, the substrate for methylation, reduces histone methylation in primed murine embryonic stem cells.[28] However, the same inhibition does not alter bulk methylation in naïve embryonic stem cells.[28] Another layer of complexity results from extensive cross-talk between different histone PTMs with some PTMs stimulating or repressing others.[29,30] For example, H3K4 methylation can stimulate an increase in H3K9 acetylation. In contrast, butyrylation can preclude acetylation of the same histone.[30,31] Thus, understanding how cellular metabolism influences histone PTMs is a significant challenge.
The histone PTM levels in a cell depend on the activity of both PTM writers and erasers. The writers comprise enzymes such as histone methyltransferases and histone acetyltransferases, while histone demethylases and histone deacetylases are examples of “erasers”. The availability of substrates and cofactors, such as folates, acetyl-CoA, SAM, and α-Ketoglutarate, influences the activity of these epigenetic enzymes (i.e. writers and erasers).[2,4–6,28] For example, the levels of a single histone PTM - acetylation, depends on the levels of its substrate acetyl-coA, 17 distinct acetyltransferases, 18 different deacetylases and their substrates such as NAD, and the presence of other histone marks like methylation. Hence increased acetylation in a cell could occur due to either high acetyl-coA production or due to reduced deacetylase activity arising from a change in redox metabolism. Consequently, acetylation has been found to increase in both nutrient excess and starvation conditions! [32,33]
There are over 250 such epigenetic enzymes in humans with both distinct and overlapping substrates and targets (i.e. histone sites).[34] Some histone sites can even be non-enzymatically modified directly by metabolites.[4] Furthermore, the extent of sensitivity to metabolism for different PTMs changes with respect to their positions in the histone.[4,35–37] For example, acetylation at H3K9, H3K27 and H3K56, but not at H3K14, H3K18 and H3K23, have been found to be sensitive to acetate addition.[35] Similarly acetylation at H3K9, H3K14, H3K18, H4K8, H4K12, and H4K16 was most sensitive to glucose levels while other sites did not change significantly.[36] Our understanding of how histone PTMs are influenced by metabolism can benefit from systems biology approaches that account for numerous components.
4. How to trace metabolic signals to the nucleus
While there are numerous pathways that can theoretically synthesize specific epigenetic substrates, such as acetyl-CoA, stable isotope tracing analysis can tell us which pathway predominates in a given condition. Combining isotope-labeled metabolomics with proteomics is a powerful approach for uncovering metabolic-epigenetic interactions. For instance, treating cells with heavy isotope labeled glucose or acetate followed by proteomics measurement of histone PTMs can help identify which molecule and metabolic pathway contributes more to histone PTM synthesis. Further, measuring the incorporation of stable isotope labeled glucose or other metabolites over time can track the dynamics of PTMs and trace metabolic pathways that support the synthesis of PTM substrates (Figure 2).
Figure 2.
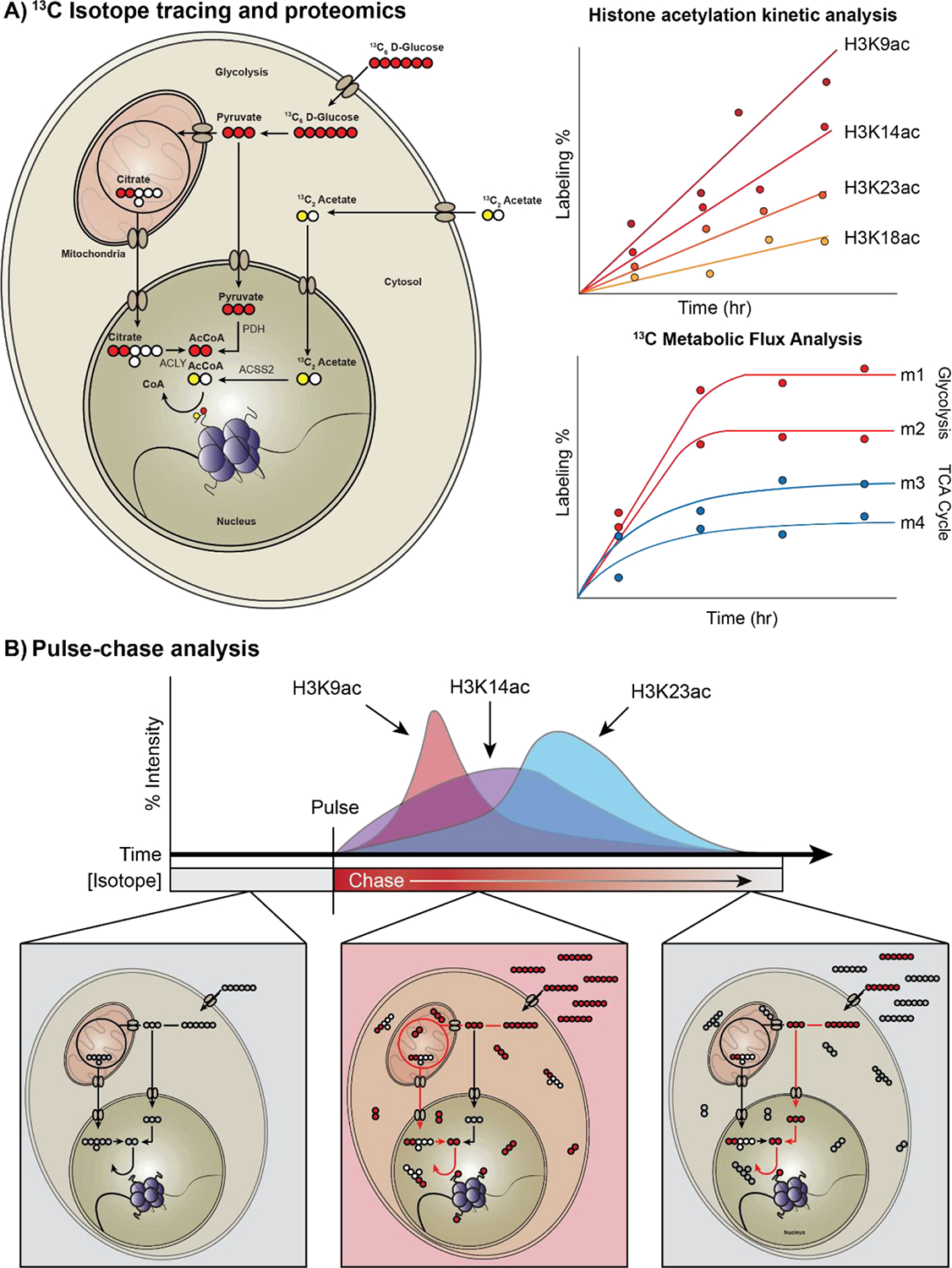
Flux tracing experiments and proteomic profiling under different conditions reveal the impact of metabolic rewiring on histone acetylation and other histone PTMs. A. Histone acetylation labeling using 13C-labeled glucose and acetate. Measuring incorporation of the labeled acetyl-group over time using mass spectrometry enables quantification of histone acetylation dynamics and kinetic profiles of different histone writers and erasers. To quantify the impact of metabolite concentrations on histone acetylation, Metabolic Flux Analysis (MFA) can be used to infer intracellular fluxes for small metabolic networks. The rate of labeling incorporation over time can be used to measure metabolic activity for various metabolic pathways. Combining both proteomic kinetic profiling and MFA under different nutrient- and genetic perturbation conditions can reveal unique metabolic dependencies of various histone PTMs. B. A schematic of a pulse-chase analysis to quantify histone acetylation/deacetylation rates from different nutrient sources. In a pulse-chase experiment, an isotope tracer is introduced (pulse), and an unlabeled form of the nutrient replaces the tracer over time (chase). The time it takes for the unlabeled acetyl-groups to replace the labeled acetylated histones is the deacetylation rate for a given acetylation species.
Recent studies have used isotope-labeled tracing to uncover how different acetylation sites exhibit unique sensitivity and dynamics for different substrates.[35,36,38,39] This approach can also help determine the quantitative relationship between histone marks and metabolite levels and fluxes. Cluntun et al created a tunable system to titrate glucose at various concentrations in a human colon cancer cell line.[36] They performed kinetic flux profiling experiments using 13C labeled glucose to manipulate glycolytic flux. They found that this glucose titration leads to different histone acylation (multiple PTMs) patterns, in which different sites show differing degrees of sensitivity to glycolytic flux. Similarly, Mentch et al found a quantitative link between methionine levels and histone H3 trimethylation.[37] They found that the SAM/SAH ratio was predictive of the levels of histone methylation in response to methionine restriction. While a severe reduction in histone di- and tri-methylation in response to depletion of SAM or methionine has been observed in many systems,[23,26–28,37,40] Haws et al found that mammalian cells mount a highly coordinated response to preserve H3K9 mono-methylation.[41]
Tracing has also provided novel insights on how metabolic pathways are rewired to impact histone PTMs in stem cells. Chandrasekaran et al traced 13C labeled glucose, glutamine and serine to show how carbon from glucose impacts histone methylation during the naïve to primed transition in pluripotent stem cells.[28] In these cells, glycolytic flux is routed towards the one-carbon metabolic pathway, which influences serine and folate metabolism, and leads to increased SAM synthesis and ultimately histone methylation. Similarly, Moussaieff et al used labeled glucose tracing to uncover the impact of glycolytic flux on acetyl-CoA synthesis and histone acetylation in naïve and primed pluripotent stem cells.[42]
While glucose is considered the most common carbon source for histone acetylation,[38,43,44] eukaryotic cells, especially cancer cells, also utilize alternate carbon sources.[45] McDonnell et al combined 13C carbon tracing with acetyl-proteomics in immortalized hepatocytes (AML12 cells) to show that up to 90% of acetylation on histone lysines can be derived from fatty acid carbon (octanoate), even in the presence of excess glucose.[46] A large proportion of tumors also utilize carbon from acetate for histone acetylation.[47] Using 13C labeling and multiple reactions monitoring mass spectrometry, Gao et al found that during hypoxia, acetate becomes a major carbon source for histone acetylation in tumors.[35] Furthermore, carbons from branched-chain amino acid oxidation is used to synthesize acetyl-CoA to support histone acetylation in pancreatic acinar cells and contributes to the development of pancreatic ductal adenocarcinoma.[48]
Flux tracing analyses have helped uncover how aberrant metabolic rewiring can influence histone PTMs in tumors. Morrish et al used 13C glucose tracing to demonstrate that Myc overexpression increases the mitochondrial synthesis of acetyl-CoA, and a 40% increase in H4K16ac.[18] Cancer cells can channel carbon flux into acetyl-CoA to sustain high levels of histone acetylation even when glucose is limiting. This is achieved by increasing the activity of ATP-Citrate Lyase (ACLY) or acetyl-CoA synthetase 2 (ACSS2) enzymes that synthesize acetyl-CoA and support histone acetylation.[47] In hypoxic tumors, acetate from histone deacetylation is recaptured by nuclear ACSS2 and channeled for histone acetylation. ACSS2 helps maintain adequate nuclear acetyl-CoA levels to support histone acetylation even when there is high cytosolic demand for acetyl-CoA to support lipogenesis.[44]
New histone marks have also been discovered using tracing and mass-spectrometry. For example, metabolic labelling using 13C L-lactate followed by mass-spectrometry analysis has demonstrated that a novel histone mark - lysine lactylation, can be derived from lactate.[49]
Furthermore, metabolic labelling experiments using isotopic glucose have demonstrated that lysine lactylation is endogenously derived from glucose.
A limitation of these isotope labeled tracing studies is that they are done using bulk cellular measurements and as a result, subcellular compartment information is lost. Acetyl-CoA and other histone PTM substrates exist in distinct pools in mitochondria, nucleus and other compartments.[50] Metabolite pools in the mitochondria may not have significant impact on histone modifications in the nucleus. Recent studies have begun to address this limitation through a variety of ways including fractionation to separate organelles, compartment-specific chemical probes, and via computational modeling. Lee et al measured fluxes in mitochondria and cytosol by combining isotope tracing with subcellular fractionation and metabolomics.[51] However, the subcellular fractionation process itself can lead to artifacts. Trefely et al have developed a post-labeling correction strategy to account for the disruption caused by the fractionation of compartments.[52] Computational models can also be used to deconvolute compartment-specific metabolism from bulk measurements. Chandrasekaran et al were able to differentiate mitochondrial and cytosolic folate metabolism from bulk metabolomics measurement using a computational model of metabolism in various compartments, and validated the model using chemical inhibitors that target folate metabolism in distinct compartments.[28].
An essential requirement for isotope labeling experiments is the steady-state labeling of metabolites, i.e. the isotopic labeling does not change over time. However, the exchange of intracellular and external metabolites can significantly reduce labeling rates and labeling may not reach steady state. For example, cytosolic acetate freely exchanges with both acetyl-CoA and extracellular acetate. This free exchange along with rapid protein acetylation-deacetylation cycles can lead to labeling of histones without a net carbon transfer. This makes it challenging to study histone acetylation labeling by acetate. To overcome this, Bulusu et al utilized a chemical derivative of acetate to determine net acetate exchange rate and quantify labeling of histone-bound acetate.[44]
Although isotope labeling patterns of metabolites can directly provide qualitative information on relative pathway activities, 13C metabolic flux analysis (MFA)[53,54] can provide a more quantitative estimate of fluxes at key branch points. In MFA, labeling patterns of metabolites are used to computationally estimate metabolic fluxes.[55] However, MFA is time-intensive and computational models with detailed atomic mapping are currently available only for a limited set of pathways in central metabolism.[53,54]
Finally, the interpretation of large numbers of metabolic changes observed in tracing and metabolomics measurements is a significant challenge.[56,57] While flux tracing is limited to a small set of well-studied pathways, extensive metabolic changes occur during differentiation or tumorigenesis resulting in altered epigenetic modifications. Mechanistic modeling tools have now been developed to interpret omics datasets. Recent studies have also applied metabolic modeling methods to understand the influence of diverse metabolic changes on histone modifications.
5. Constraint based modeling can predict and interpret metabolism-histone cross talk
All living cells contain numerous highly interconnected metabolic pathways with varying degrees of activity. Transcriptomics or metabolomics analysis can provide a snapshot of cellular metabolism; however, transcript or metabolite changes do not directly provide insights on the activity of various metabolic reactions. For instance, increased accumulation of TCA cycle metabolites may be due to increased activity of glycolysis and TCA cycle or decreased activity of oxidative phosphorylation pathway. Similarly, changes in mRNA levels of one pathway should be interpreted in the context of all other pathways that are linked to it. Interpreting metabolic changes through traditional informatics approaches such as grouping genes into pathways is also challenging.[56] Usually, individual proteins in a pathway do not change coherently as a whole. Given the highly inter-connected nature of the metabolic network, the underlying assumption behind pathway analysis that each pre-defined pathway is independent of each other does not hold for metabolism as adjacent pathways can influence each other’s activity. A systems-level model is needed to account for changes at both the individual protein level and the overall network level.
Metabolic network reconstructions address these challenges and provide a virtual map of all known metabolic reactions that happen in a human cell.[58] Metabolic network reconstructions represent the mechanistic relationships between genes, proteins, and metabolites in a cell. For example, the human metabolic model (Recon 2) contains 7,440 reactions, 1,789 genes, 2,194 transcripts, 2,657 proteins, 1,052 protein complexes, 8 cellular compartments and 5,063 metabolites.[59]
Several theoretical approaches that utilize metabolic reconstructions to interpret transcriptomics[60–63] and metabolomics[28,64,65] data have been developed. All these approaches build upon a fundamental theoretical concept called Constraint-Based Modeling (CBM). CBM is a powerful theoretical tool that is capable of simulating hundreds of enzymes in the metabolic network.[66,67] Using CBM, we can identify an optimal path through the network from nutrients to biomass components based on thermodynamic, stoichiometric and enzyme expression constraints. CBM does not require any kinetic parameters and can be used to simulate models with thousands of reactions. Flux balance analysis, the oldest and most commonly used CBM method, is formulated as an optimization problem, wherein fluxes are estimated by assuming cellular metabolism is optimized for the production of biomass components, subject to stoichiometric constraints resulting from mass balances for intracellular metabolites (Figure 3).[68] Further external nutrient levels and metabolic secretion rates provide boundary constraints on intracellular fluxes. Due to redundancies in the metabolic network, additional constraints from transcriptomics or metabolomics data are frequently used to limit the feasible space of possible fluxes through the network.
Figure 3.
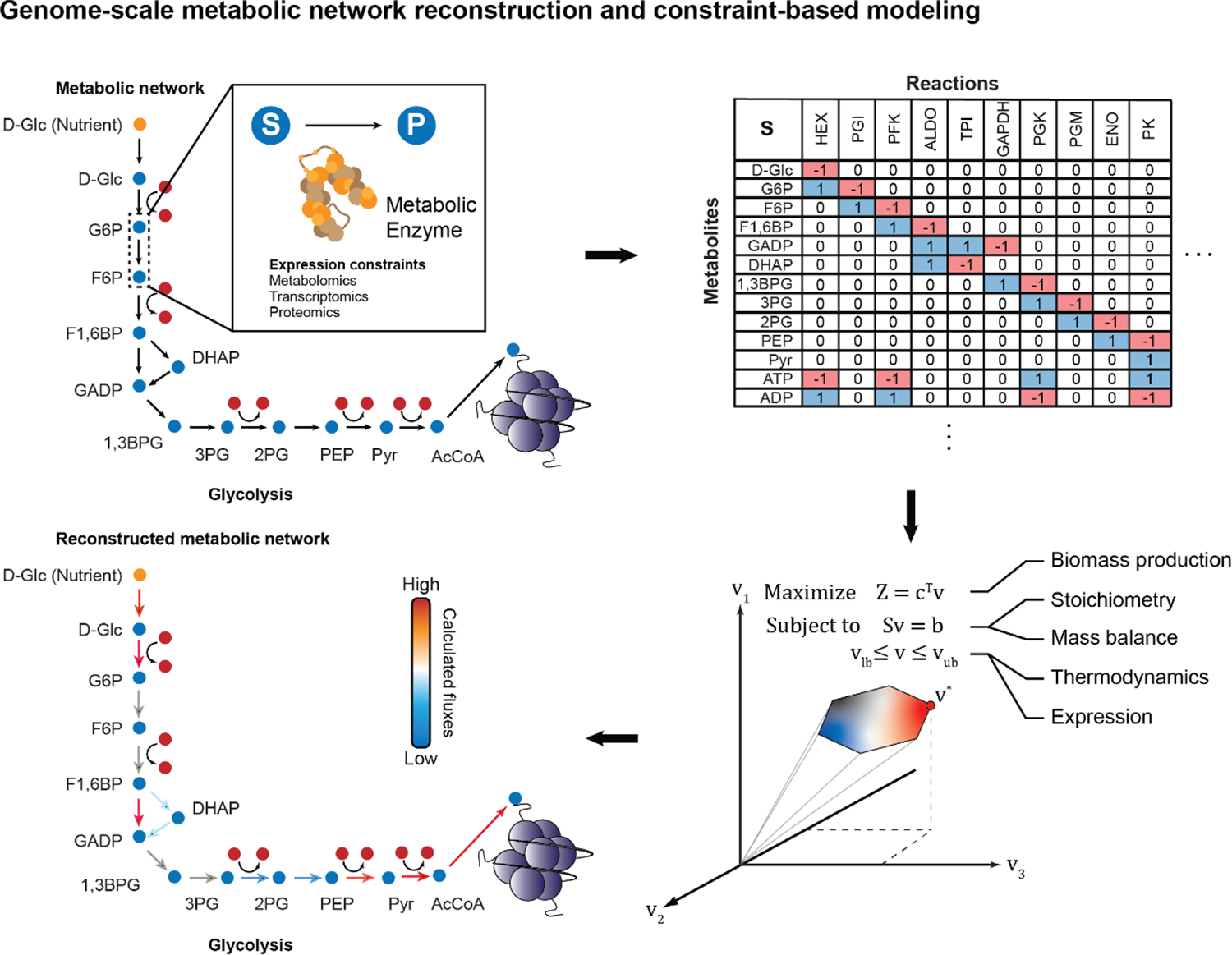
Using Constraint-based Modeling (CBM) to compute metabolic fluxes through the metabolic network. Genome-scale metabolic network reconstructions map all known gene-protein-reaction associations using an iterative process of literature curation, database mining, and model refinement. The resulting metabolic network is converted to a mathematical form as a matrix of the stoichiometries (S) for every single reaction-metabolite pair. The product of the stoichiometric matrix (S) and the desired vector of metabolic fluxes going through each reaction (v) is equal to the rate of change of metabolites (b). If b is set to 0, this represents quasi-steady state conditions. Solving for v provides steady-state fluxes from the metabolic reconstruction. To get a unique biologically feasible flux distribution, several constraints must be imposed on the model. A cellular objective is set, where a cell is assumed to fulfill a specific metabolic task such as maximizing biomass production. Further, the structure of the metabolic network itself (S), given that each reaction is mass- and charge-balanced, provides another constraint.
CBM has been successfully used to predict the metabolic state of various mammalian systems, including cancer cells and stem cells.[28,60,69]. CBM models have led to the discovery of biomarkers, metabolic vulnerabilities and drug targets.[58,70] For example, CBM identified dysregulation of mannose metabolism in obese patients, which was validated by quantifying plasma mannose levels in lean and obese individuals.[71] Similarly, modelling of hepatocyte metabolism revealed serine deficiency in patients with non-alcoholic fatty liver disease.[72]
CBM is highly effective in predicting lethality of single and combinatorial gene knockouts as this approach is good at predicting infeasible metabolic states. For example, using a metabolic model of renal-cell cancer cells, Frezza et al discovered a synthetic lethal interaction between the enzymes fumarate hydratase (FH) and haem oxygenase.[73] Since FH mutations are common in these cancers, haem oxidation could be targeted in tumors with FH inactivating mutations, while sparing normal cells with wild-type FH. A metabolic model of hepatocellular carcinoma (HCC) revealed that de novo lipogenesis is substantially upregulated in patients with HCC and identified mitochondrial acetate as the substrate for lipogenesis through the upregulation of mitochondrial acetyl-CoA synthetase (ACSS1).[74]
Like all models, the accuracy of CBM depends on the availability of high-quality datasets to build and curate the metabolic models. Further, the underlying assumptions of metabolic steady state and optimization of biomass are not applicable for all systems. Nevertheless, these assumptions can be adjusted based on experimental evidence. For example, time-course metabolomics measurements can be used to identify metabolites that are not at steady-state.[28,64] Similarly complex objectives based on biochemical tasks performed by a cell type such as neuron or hepatocyte can be used as an alternate optimization goal instead of optimizing biomass.[59]
CBM methods have also been recently applied to gain insight on how metabolic changes can influence histone modifications. For example, a variation of CBM called Dynamic Flux Activity (DFA) uses snapshots of metabolite levels taken at different time points and subsequently overlays this onto a metabolic network model.[28] DFA was used to compare the metabolism of embryonic- and induced-pluripotent stem cells using time-course metabolomics of each cell state. DFA revealed the activation of the one-carbon metabolic pathway in mouse embryonic stem cells transitioning from naïve to primed pluripotent state. This activation enhances the synthesis of SAM and supports extensive histone methylation in primed stem cells. Inhibiting this pathway reduced histone methylation in primed cells but not in naïve cells, as predicted by the model. DFA also uncovered differences in folate metabolism between mitochondria and cytosol, which is usually lost during bulk metabolomics measurement. These predictions were then experimentally confirmed using inhibition of folate enzymes in different compartments.
Notably, a new computational model based on CBM for directly simulating the dynamics of histone acetylation was recently developed by Shen et al.[32] This enabled them to predict the impact of metabolic alterations on histone acetylation (Figure 3). To simulate acetylation using the metabolic network model, the authors added biochemical reactions corresponding to histone acetylation and synthesis of acetyl-CoA in the nucleus.[43] This model enabled them to correctly predict the histone acetylation levels of various cell lines based on their metabolic activity, suggesting a quantitative relationship between the two processes. This model suggests that excess carbon that is not used for biomass synthesis supports acetylation. It also explains why acetylation can increase in certain nutrient stress conditions such as nitrogen starvation that result in excess carbon levels. Finally, it also revealed that the diversion of carbon flux for histone acetylation will have limited impact on overall metabolism in an actively dividing cell. This is significant given that histone acetylation accounts for 74% of all acetylated lysines in mammalian cells.[75]
CBM of metabolism-epigenome interactions is still in its infancy. Existing models cannot yet differentiate between specific histone sites (e.g. H3K9 or H3K27). Further, CBM in general cannot model the feedback regulation of metabolism by transcriptional regulation induced by metabolic changes. New approaches are being developed to model this feedback in microbes,[62,76] and may soon be able to tackle the regulatory complexity in mammalian cells.
Thermodynamic parameters such as Gibbs Free Energy can be used to set reaction directions. Finally, datasets such as transcriptomics, proteomics, and metabolomics can be used to limit enzyme activity, flux bounds and substrate uptake rates. Together, these constraints can produce condition-specific metabolic profiles. Extending the metabolic network to include reactions required for histone PTMs can simulate metabolomic-epigenomic interactions at the genome-scale.
6. Next-generation technologies for discovering new metabolic-epigenetic interactions
The development of new imaging, omics and modeling technologies can help discover new interactions between these two central cellular processes in the future. Ultimately these technologies together may make it possible to track a labelled metabolite in a live cell, watch its transition between cellular compartments and ultimately identify which histone modification it ends up in at which gene.
A major influence in this area of research was the surprising discovery that key mitochondrial energy metabolism enzymes are present in the nucleus and provide metabolites for histone modification.[77,78] These observations support the possibility that other metabolic enzymes, which are primarily thought to function in the mitochondria or the cytoplasm, may directly facilitate epigenetic change in the nucleus. Imaging techniques and sensors to locate enzymes and metabolites in space are likely to be powerful tools in this hunt.
A limitation of metabolomics and tracing approaches is that they lack spatial resolution within a compartment like nucleus. Many metabolic substrates are synthesized in the nucleus and metabolism driven epigenetic alterations may happen in specific regions of the chromatin. Histone modifications may change spatially at different loci even though the bulk levels may remain the same. Next-generation sequencing approaches such as chromatin-immunoprecipitation and sequencing (ChIP-seq) allow identification of gene-specific epigenetic effects of metabolism. Notably, Aranda et al recently developed a DNA-mediated chromatin pull-down technology to identify chromatin-bound proteins in pluripotent stem cells.[79] Using this approach, they discovered that the enzyme adenosyl-homocysteinase (AHCY) influences SAM/SAH ratio, thereby affecting methylation in chromatin sub-compartments. Linking spatial metabolite levels coupled with genome-wide and spatial measurements of histone marks using ChIP-seq or chromatin capture technologies[80] will enable the characterization of these local effects.
The advent of machine learning algorithms has revolutionized many areas of biology.[81] Unlike traditional biochemical modeling approaches, machine learning algorithms can learn patterns in data without relying on prior knowledge. In contrast, the mechanistic models (CBMs) employed in studies highlighted above were built using biochemical data on enzymes and substrates curated from literature.[32] Using advanced machine learning algorithms like Deep learning may soon make it possible to directly predict histone modifications and ramifications of metabolic alterations on the epigenome without knowledge of underlying mechanism. While mechanistic modeling is limited to known reactions or interactions in literature, machine learning algorithms are ideally suited for uncovering novel interactions. However, machine learning algorithms are data-driven. Hybrid approaches that integrate machine-learning and mechanistic modeling[82,83] can enable us to effectively harness large-scale metabolic and epigenomic datasets in the future.
7. Future directions for metabolism-epigenetics research
Technological advances described above will reveal important metabolic-epigenetic interactions in diverse areas of biology, as well as opening up new research areas, and breathing life into old biological puzzles. They can provide insights on the impact of the tissue micro-environment on the epigenome during tumorigenesis, development, or ageing. Identification of unique growth requirements of stem cells based on their epigenetic state to improve their viability in culture can transform regenerative medicine applications such as disease modeling and stem-cell therapy.[84] Recent discoveries in neuroscience on the importance of metabolism and epigenetics for memory and behavior[85–89] and altered brain energy metabolism in conditions such as obesity and aging[90,91] could underlie known associations between diet and the brain.[92] Another area that links metabolism and epigenetics is transgenerational epigenetic inheritance. Research has now confirmed that environmental exposures (e.g. diet, stress, toxins) can alter the phenotypes of future generations without altering DNA sequences.[93,94] In some cases, these changes appear to provide a mechanism of short-term adaption to the exposure, which contrasts with the slower evolutionary process of natural selection.[95,96] Numerous cases of transgenerational epigenetic inheritance in rodents, C. elegans and Drosophila stimulated by nutritional exposures suggests that metabolism-epigenetic interactions may be part of these inheritance mechanisms. Future work will reveal how widespread and important these mechanisms are for adaptation in all species, including our own.
The growth of metabolism-epigenetic research will bring translational benefits through modulation of physiology for therapy and agriculture. Manipulation of metabolism-epigenetic mechanisms is a consequence of the ketogenic diet, a treatment for epilepsy and some current cancer therapies.[97,98] Considering the recently discovered importance of metabolism-epigenetic regulation of immune cell development[99] it is likely that therapies targeting those mechanisms will be developed for a variety of inflammatory and infectious diseases. Understanding the interdependencies of metabolic epigenetic processes can identify synergistic and antagonistic combinations of epigenetic and metabolic inhibitors. Epigenetic drugs such as histone deacetylase inhibitors are being explored for treating immunological, oncological, and neurological disorders.[100] Similarly, anti-metabolites such as methotrexate, gemcitabine and nucleotide analogs are widely used for cancer therapy.[101] Thus, identifying synergistic combinations of antimetabolites with epigenetic inhibitors can enhance the efficacy of current therapies.[102] Finally, as more is learned about how metabolism-epigenetic interactions influence growth, health and inheritance, there could be improvements in animal production and crop yields through nutritional supplementation at multiple stages of the life cycle or even multi-generationally.[103]
8. Conclusion
Regulation of gene expression through epigenetic chemical modifications is highly responsive to various metabolic cues. Mass spectrometry-based proteomics and metabolomics technologies are helping us uncover these interactions between metabolism and the epigenome. Yet interpreting these vast datasets to understand the interdependencies between these processes is challenging. Building virtual biochemical models represents an important and timely opportunity to harness the vast amounts of omics data and gain a better understanding of interaction mechanisms. Further, it is likely that we have only scratched the surface on the interplay between these two central cellular processes. Combining cutting-edge tools from systems biology, imaging, mass spectrometry and artificial intelligence can ultimately uncover interactions between metabolism, histone marks, and gene regulation that underlie numerous biological processes and diseases.
Figure 1.
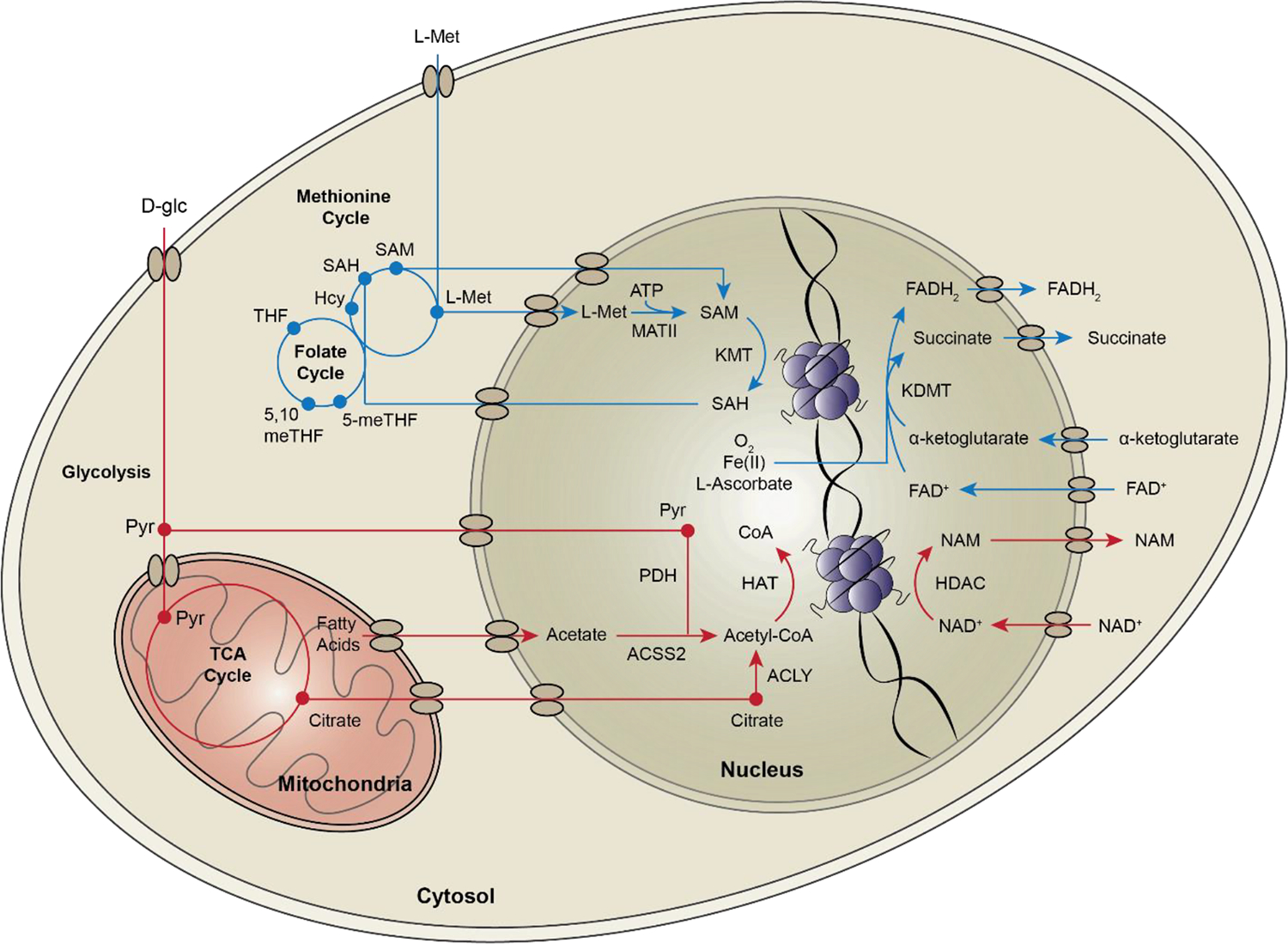
Interactions between metabolites and epigenetic enzymes impact the histone code. Cells normally utilize glucose (red) to synthesize the acetylation substrate acetyl-CoA. There are several nuclear metabolic enzymes that supply a local source of acetyl-CoA for histone acetylation, including PDH, ACLY, and ACSS2. Acetyl-CoA in the nucleus is used by histone acetyltransferases (HAT) to modify lysine groups on histone tails. Sirtuins are histone deacetylases (HDACs) that depend on local NAD+ to deacetylate histone tails. Histone methylation (blue) depends on one-carbon donors - methionine and folate. The metabolic enzyme MAT in the cytosol and the nucleus, converts L-methionine to S-adenosylmethionine, the substrate for histone methylation. Histone lysine methyltransferases (KMT) use SAM to methylate lysine groups on histone tails. Folate allows recycling of homocysteine (Hcy) back to L-methionine to continue the production of SAM. Histone demethylases (KDMT) remove methyl groups using two distinct mechanisms. LSD family demethylases act using a FAD-dependent amine oxidase reaction to demethylate histone lysine residues. JmjC domain family demethylases use an α-ketoglutarate-Fe(II)-dependent dioxygenase reaction for demethylation.
Acknowledgements
This work was supported by faculty start-up funds from the University of Michigan to SC and Advanced Proteome Informatics of Cancer Training Program T32 CA140044 to SEC.
List of abbreviations
- ACCS1
Acyl-CoA Synthetase 1
- ACLY
ATP-Citrate Lyase
- ACSS2
Acyl-CoA Synthetase 2
- AHCY
Adenosyl-homocysteinase
- AMPK
AMP-activated protein kinase
- CBM
Constraint-Based Modeling
- ChIP-seq
Chromatin-immunoprecipitation and sequencing
- DFA
Dynamic Flux Analysis
- FAD
Flavin Adenine Dinucleotide
- FH
Fumarate Hydratase
- GlcNAc
N-acetylglucosamine
- HAT
Histone acetyltransferase
- HCC
Hepatocellular Carcinoma
- Hcy
Homocysteine
- HDAC
Histone deacetylases
- KDMT
Lysine demethylases
- KMT
Lysine methyltransferases
- MFA
Metabolic Flux Analysis
- NAD
Nicotinamide Adenine Dinucleotide
- NNMT
Nicotinamide N-methyltransferase
- PDH
Pyruvate Dehydrogenase
- PTMs
Post-Translational Modifications
- ROS
Reactive Oxygen Species
- SAH
S-Adenosyl Homocysteine
- SAM
S-Adenosyl Methionine
- TCA Cycle
Tricarboxylic Acid Cycle
References
- [1].Margueron R, Trojer P, Reinberg D, Curr. Opin. Genet. Dev 2005, DOI 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- [2].Kaelin WG, McKnight SL, Cell 2013, 153, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Fischle W, Mootz HD, Schwarzer D, Curr. Opin. Chem. Biol 2015, DOI 10.1016/j.cbpa.2015.07.005. [DOI] [PubMed] [Google Scholar]
- [4].Su X, Wellen KE, Rabinowitz JD, Curr. Opin. Chem. Biol 2016, 30, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Reid MA, Dai Z, Locasale JW, Nat. Cell Biol 2017, DOI 10.1038/ncb3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kinnaird A, Zhao S, Wellen KE, Michelakis ED, Nat. Rev. Cancer 2016. [DOI] [PubMed] [Google Scholar]
- [7].Cai L, Sutter BM, Li B, Tu BP, Mol Cell 2011, 42, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tu BP, Kudlicki A, Rowicka M, McKnight SL, Science (80-.). 2005, DOI 10.1126/science.1120499. [DOI] [PubMed] [Google Scholar]
- [9].Singh JP, Zhang K, Wu J, Yang X, Cancer Lett. 2015, DOI 10.1016/j.canlet.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wellen KE, Thompson CB, Nat. Rev. Mol. Cell Biol 2012, DOI 10.1038/nrm3305. [DOI] [PubMed] [Google Scholar]
- [11].Sivanand S, Rhoades S, Jiang Q, Lee JV, Benci J, Zhang J, Yuan S, Viney I, Zhao S, Carrer A, Bennett MJ, Minn AJ, Weljie AM, Greenberg RA, Wellen KE, Mol. Cell 2017, DOI 10.1016/j.molcel.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hino S, Sakamoto A, Nagaoka K, Anan K, Wang Y, Mimasu S, Umehara T, Yokoyama S, Kosai KI, Nakao M, Nat. Commun 2012, DOI 10.1038/ncomms1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y, Cell 2004, DOI 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- [14].Yang Q, Liang X, Sun X, Zhang L, Fu X, Rogers CJ, Berim A, Zhang S, Wang S, Wang B, Foretz M, Viollet B, Gang DR, Rodgers BD, Zhu M-J, Du M, Cell Metab. 2016, DOI 10.1016/j.cmet.2016.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bungard D, Fuerth BJ, Zeng PY, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, Berger SL, Science (80-.). 2010, DOI 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Tran TQ, Lowman XH, Kong M, Clin. Cancer Res 2017, DOI 10.1158/1078-0432.CCR-16-2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Ji H, Wu G, Zhan X, Nolan A, Koh C, de Marzo A, Doan HM, Fan J, Cheadle C, Fallahi M, Cleveland JL, Dang CV, Zeller KI, PLoS One 2011, DOI 10.1371/journal.pone.0026057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Morrish F, Noonan J, Perez-Olsen C, Gafken PR, Fitzgibbon M, Kelleher J, VanGilst M, Hockenbery D, J. Biol. Chem 2010, DOI 10.1074/jbc.M110.141606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Solary E, Bernard OA, Tefferi A, Fuks F, Vainchenker W, Leukemia 2014, DOI 10.1038/leu.2013.337. [DOI] [PubMed] [Google Scholar]
- [20].Sebastián C, Zwaans BMM, Silberman DM, Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber D, Cosentino C, Greenson JK, MacDonald AI, McGlynn L, Maxwell F, Edwards J, Giacosa S, Guccione E, Weissleder R, Bernstein BE, Regev A, Shiels PG, Lombard DB, Mostoslavsky R, Cell 2012, DOI 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, Aykin-Burns N, Pennington JD, van der Meer R, Nguyen P, Savage J, Owens KM, Vassilopoulos A, Ozden O, Park SH, Singh KK, Abdulkadir SA, Spitz DR, Deng CX, Gius D, Cancer Cell 2010, DOI 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].German NJ, Haigis MC, Curr. Biol 2015, DOI 10.1016/j.cub.2015.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ulanovskaya OA, Zuhl AM, Cravatt BF, Nat. Chem. Biol 2013, DOI 10.1038/nchembio.1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Castellano-Castillo D, Denechaud PD, Fajas L, Moreno-Indias I, Oliva-Olivera W, Tinahones F, Queipo-Ortuño MI, Cardona F, PLoS One 2019, DOI 10.1371/journal.pone.0215083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wolff GL, Kodell RL, Moore SR, Cooney CA, FASEB J. 1998, DOI 10.1096/fasebj.12.11.949. [DOI] [PubMed] [Google Scholar]
- [26].Shiraki N, Shiraki Y, Tsuyama T, Obata F, Miura M, Nagae G, Aburatani H, Kume K, Endo F, Kume S, Cell Metab. 2014, 19, 780. [DOI] [PubMed] [Google Scholar]
- [27].Shyh-Chang N, Locasale JW, Lyssiotis CA, Zheng Y, Teo RY, Ratanasirintrawoot S, Zhang J, Onder T, Unternaehrer JJ, Zhu H, Asara JM, Daley GQ, Cantley LC, Science 2013, 339, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chandrasekaran S, Zhang J, Sun Z, Zhang L, Ross CA, Huang Y-C, Asara JM, Li H, Daley GQ, Collins JJ, Cell Rep. 2017, 21, DOI 10.1016/j.celrep.2017.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Margueron R, Reinberg D, Nat. Rev. Genet 2010, DOI 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lee JS, Smith E, Shilatifard A, Cell 2010, DOI 10.1016/j.cell.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dutta A, Abmayr SM, Workman JL, Mol. Cell 2016, DOI 10.1016/j.molcel.2016.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shen F, Boccuto L, Pauly R, Srikanth S, Chandrasekaran S, Genome Biol. 2019, 20, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].McBrian MA, Behbahan IS, Ferrari R, Su T, Huang TW, Li K, Hong CS, Christofk HR, Vogelauer M, Seligson DB, Kurdistani SK, Mol. Cell 2013, DOI 10.1016/j.molcel.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Huang Z, Jiang H, Liu X, Chen Y, Wong J, Wang Q, Huang W, Shi T, Zhang J, PLoS One 2012, DOI 10.1371/journal.pone.0039917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Gao X, Lin SH, Ren F, Li JT, Chen JJ, Yao CB, Bin Yang H, Jiang SX, Yan GQ, Wang D, Wang Y, Liu Y, Cai Z, Xu YY, Chen J, Yu W, Yang PY, Lei QY, Nat. Commun 2016, DOI 10.1038/ncomms11960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cluntun AA, Huang H, Dai L, Liu X, Zhao Y, Locasale JW, Cancer Metab. 2015, 3, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mentch SJ, Mehrmohamadi M, Huang L, Liu X, Gupta D, Mattocks D, Gómez Padilla P, Ables G, Bamman MM, Thalacker-Mercer AE, Nichenametla SN, Locasale JW, Cell Metab. 2015, 22, 861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Evertts AG, Zee BM, Dimaggio PA, Gonzales-Cope M, Coller HA, Garcia BA, J. Biol. Chem 2013, DOI 10.1074/jbc.M112.428318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, Jackson E, Aiello NM, Haas NB, Rebbeck TR, Judkins A, Won KJ, Chodosh LA, Garcia BA, Stanger BZ, Feldman MD, Blair IA, Wellen KE, Cell Metab. 2014, DOI 10.1016/j.cmet.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sperber H, Mathieu J, Wang Y, Ferreccio A, Hesson J, Xu Z, Fischer KA, Devi A, Detraux D, Gu H, Battle SL, Showalter M, Valensisi C, Bielas JH, Ericson NG, Margaretha L, Robitaille AM, Margineantu D, Fiehn O, Hockenbery D, Blau CA, Raftery D, Margolin AA, Hawkins RD, Moon RT, Ware CB, Ruohola-Baker H, Nat. Cell Biol 2015, 17, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Haws SA, Yu D, Ye C, Wille CK, Nguyen LC, Krautkramer KA, Tomasiewicz JL, Yang SE, Miller BR, Liu WH, Igarashi K, Sridharan R, Tu BP, Cryns VL, Lamming DW, Denu JM, Mol. Cell 2020, DOI 10.1016/j.molcel.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Moussaieff A, Rouleau M, Kitsberg D, Cohen M, Levy G, Barasch D, Nemirovski A, Shen-Orr S, Laevsky I, Amit M, Bomze D, Elena-Herrmann B, Scherf T, Nissim-Rafinia M, Kempa S, Itskovitz-Eldor J, Meshorer E, Aberdam D, Nahmias Y, Cell Metab. 2015, DOI 10.1016/j.cmet.2015.02.002. [DOI] [PubMed] [Google Scholar]
- [43].Wellen KE, Hatzivassiliou G, Sachdeva UM, V Bui T, Cross JR, Thompson CB, Science (80-.). 2009, 324, 1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bulusu V, Tumanov S, Michalopoulou E, van den Broek NJ, MacKay G, Nixon C, Dhayade S, Schug ZT, Vande Voorde J, Blyth K, Gottlieb E, Vazquez A, Kamphorst JJ, Cell Rep. 2017, DOI 10.1016/j.celrep.2016.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Sebastián C, Mostoslavsky R, Trends Endocrinol. Metab 2017, DOI 10.1016/j.tem.2016.11.001. [DOI] [PubMed] [Google Scholar]
- [46].McDonnell E, Crown SB, Fox DB, Kitir B, Ilkayeva OR, Olsen CA, Grimsrud PA, Hirschey MD, Cell Rep. 2016, DOI 10.1016/j.celrep.2016.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, Horton JD, Hammer RE, Mcknight SL, Tu BP, Cell 2014, DOI 10.1016/j.cell.2014.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Li JT, Yin M, Wang D, Wang J, Lei MZ, Zhang Y, Liu Y, Zhang L, Zou SW, Hu LP, Zhang ZG, Wang YP, Wen WY, Lu HJ, Chen ZJ, Su D, Lei QY, Nat. Cell Biol 2020, DOI 10.1038/s41556-019-0455-6. [DOI] [PubMed] [Google Scholar]
- [49].Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, Ding J, Czyz D, Hu R, Ye Z, He M, Zheng YG, Shuman HA, Dai L, Ren B, Roeder RG, Becker L, Zhao Y, Nature 2019, DOI 10.1038/s41586-019-1678-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wellen KE, Snyder NW, Curr. Opin. Clin. Nutr. Metab. Care 2019, DOI 10.1097/MCO.0000000000000580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Lee WD, Mukha D, Aizenshtein E, Shlomi T, Nat. Commun 2019, DOI 10.1038/s41467-019-09352-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Trefely S, Liu J, Huber K, Doan MT, Jiang H, Singh J, von Krusenstiern E, Bostwick A, Xu P, Bogner-Strauss JG, Wellen KE, Snyder NW, Mol. Metab 2019, DOI 10.1016/j.molmet.2019.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Buescher JM, Antoniewicz MR, Boros LG, Burgess SC, Brunengraber H, Clish CB, DeBerardinis RJ, Feron O, Frezza C, Ghesquiere B, Gottlieb E, Hiller K, Jones RG, Kamphorst JJ, Kibbey RG, Kimmelman AC, Locasale JW, Lunt SY, Maddocks ODK, Malloy C, Metallo CM, Meuillet EJ, Munger J, N??h K, Rabinowitz JD, Ralser M, Sauer U, Stephanopoulos G, St-Pierre J, Tennant DA, Wittmann C, Vander Heiden MG, Vazquez A, Vousden K, Young JD, Zamboni N, Fendt SM, Curr. Opin. Biotechnol 2015, 34, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Antoniewicz MR, Exp. Mol. Med 2018, DOI 10.1038/s12276-018-0060-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Long CP, Antoniewicz MR, Nat. Protoc 2019, DOI 10.1038/s41596-019-0204-0. [DOI] [PubMed] [Google Scholar]
- [56].Shen F, Cheek C, Chandrasekaran S, Humana, New York, NY, 2019, pp. 305–320. [DOI] [PubMed] [Google Scholar]
- [57].Campit S, Chandrasekaran S, Inferring Metabolic Flux from Time-Course Metabolomics, 2020. [DOI] [PubMed] [Google Scholar]
- [58].O’Brien EJ, Monk JM, Palsson BO, Cell 2015, 161, 971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Thiele I, Swainston N, Fleming RMT, Hoppe A, Sahoo S, Aurich MK, Haraldsdottir H, Mo ML, Rolfsson O, Stobbe MD, Thorleifsson SG, Agren R, Bölling C, Bordel S, Chavali AK, Dobson P, Dunn WB, Endler L, Hala D, Hucka M, Hull D, Jameson D, Jamshidi N, Jonsson JJ, Juty N, Keating S, Nookaew I, Le Novère N, Malys N, Mazein A, Papin JA, Price ND, Selkov E, Sigurdsson MI, Simeonidis E, Sonnenschein N, Smallbone K, Sorokin A, Van Beek JHGM, Weichart D, Goryanin I, Nielsen J, Westerhoff HV, Kell DB, Mendes P, Palsson BO, Nat. Biotechnol 2013, 31, 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Shlomi T, Cabili MN, Herrgård MJ, Palsson BØ, Ruppin E, Nat. Biotechnol 2008, 26, 1003. [DOI] [PubMed] [Google Scholar]
- [61].Jensen PA, Papin JA, Bioinformatics 2011, 27, 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Chandrasekaran S, Price ND, Proc. Natl. Acad. Sci 2010, 107, 17845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Machado D, Herrgård M, PLoS Comput Biol 2014, 10, e1003580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bordbar A, Yurkovich JT, Paglia G, Rolfsson O, Sigurjónsson ÓE, Palsson BO, Sci. Rep 2017, DOI 10.1038/srep46249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yizhak K, Benyamini T, Liebermeister W, Ruppin E, Shlomi T, Bioinformatics 2010, DOI 10.1093/bioinformatics/btq183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].O’brien EJ, Lerman JA, Chang RL, Hyduke DR, Palsson BØ, Mol. Syst. Biol 2013, 9, 693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Price ND, Papin JA, Schilling CH, Palsson BO, Trends Biotechnol 2003, 21, 162. [DOI] [PubMed] [Google Scholar]
- [68].Orth JD, Thiele I, Palsson BØ, Nat. Biotechnol 2010, 28, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Bordbar A, Monk JM, King ZA, Palsson BO, Nat. Rev. Genet 2014, 15, 107. [DOI] [PubMed] [Google Scholar]
- [70].Yizhak K, Chaneton B, Gottlieb E, Ruppin E, Mol. Syst. Biol 2015, 11, 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lee S, Zhang C, Kilicarslan M, Piening BD, Bjornson E, Hallström BM, Groen AK, Ferrannini E, Laakso M, Snyder M, Cell Metab 2016, 24, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Mardinoglu A, Agren R, Kampf C, Asplund A, Uhlen M, Nielsen J, Nat. Commun 2014, DOI 10.1038/ncomms4083. [DOI] [PubMed] [Google Scholar]
- [73].Frezza C, Zheng L, Folger O, Rajagopalan KN, MacKenzie ED, Jerby L, Micaroni M, Chaneton B, Adam J, Hedley A, Kalna G, Tomlinson IPM, Pollard PJ, Watson DG, Deberardinis RJ, Shlomi T, Ruppin E, Gottlieb E, Nature 2011, 477, 225. [DOI] [PubMed] [Google Scholar]
- [74].Björnson E, Mukhopadhyay B, Asplund A, Pristovsek N, Cinar R, Romeo S, Uhlen M, Kunos G, Nielsen J, Mardinoglu A, Cell Rep. 2015, DOI 10.1016/j.celrep.2015.10.045. [DOI] [PubMed] [Google Scholar]
- [75].Hansen BK, Gupta R, Baldus L, Lyon D, Narita T, Lammers M, Choudhary C, Weinert BT, Nat. Commun 2019, DOI 10.1038/s41467-019-09024-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Covert MW, Knight EM, Reed JL, Herrgard MJ, Palsson BO, Nature 2004, 429, 92. [DOI] [PubMed] [Google Scholar]
- [77].Sutendra G, Kinnaird A, Dromparis P, Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E, Michelakis ED, Cell 2014, DOI 10.1016/j.cell.2014.04.046. [DOI] [PubMed] [Google Scholar]
- [78].Sivanand S, Viney I, Wellen KE, Trends Biochem. Sci 2018, DOI 10.1016/j.tibs.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Aranda S, Alcaine-Colet A, Blanco E, Borràs E, Caillot C, Sabidó E, Di Croce L, Sci. Adv 2019, DOI 10.1126/sciadv.aav2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJM, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M, Nature 2015, DOI 10.1038/nature14248. [DOI] [Google Scholar]
- [81].Libbrecht MW, Noble WS, Nat. Rev. Genet 2015, DOI 10.1038/nrg3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Oruganty K, Campit SE, Mamde S, Lyssiotis CA, Chandrasekaran S, Cancer Metab. 2020, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Zampieri G, Vijayakumar S, Yaneske E, Angione C, PLoS Comput. Biol 2019, DOI 10.1371/journal.pcbi.1007084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Xu J, Du Y, Deng H, Cell Stem Cell 2015, 16, 119. [DOI] [PubMed] [Google Scholar]
- [85].Williams-Spooner MJ, Westbrook RF, Holmes NM, J. Neurosci 2019, DOI 10.1523/JNEUROSCI.0768-19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].David Sweatt J, Tamminga CA, Dialogues Clin. Neurosci 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Mews P, Egervari G, Nativio R, Sidoli S, Donahue G, Lombroso SI, Alexander DC, Riesche SL, Heller EA, Nestler EJ, Garcia BA, Berger SL, Nature 2019, DOI 10.1038/s41586-019-1700-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Chandrasekaran S, Rittschof CC, Djukovic D, Gu H, Raftery D, Price ND, Robinson GE, Genes, Brain Behav. 2015, 14, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Shpigler HY, Saul MC, Murdoch EE, Cash-Ahmed AC, Seward CH, Sloofman L, Chandrasekaran S, Sinha S, Stubbs LJ, Robinson GE, Genes, Brain Behav. 2017. [DOI] [PubMed] [Google Scholar]
- [90].Goyal MS, Vlassenko AG, Blazey TM, Su Y, Couture LE, Durbin TJ, Bateman RJ, Benzinger TLS, Morris JC, Raichle ME, Cell Metab. 2017, DOI 10.1016/j.cmet.2017.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Wardzinski EK, Kistenmacher A, Melchert UH, Jauch-Chara K, Oltmanns KM, Metabolism. 2018, DOI 10.1016/j.metabol.2018.02.013. [DOI] [PubMed] [Google Scholar]
- [92].Marx W, Moseley G, Berk M, Jacka F, Proc. Nutr. Soc 2017, DOI 10.1017/S0029665117002026. [DOI] [PubMed] [Google Scholar]
- [93].Bodden C, Hannan AJ, Reichelt AC, Trends Endocrinol. Metab 2020, DOI 10.1016/j.tem.2019.10.005. [DOI] [PubMed] [Google Scholar]
- [94].Perez MF, Lehner B, Nat. Cell Biol 2019, DOI 10.1038/s41556-018-0242-9. [DOI] [PubMed] [Google Scholar]
- [95].Jablonka E, Interface Focus 2017, DOI 10.1098/rsfs.2016.0135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Sarkies P, Semin. Cell Dev. Biol 2020, DOI 10.1016/j.semcdb.2019.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kaur J, Daoud A, Eblen ST, Curr. Mol. Pharmacol 2019, DOI 10.2174/1874467212666190215112915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Youngson NA, Morris MJ, Ballard B, Seizure 2017, DOI 10.1016/j.seizure.2017.09.005. [DOI] [PubMed] [Google Scholar]
- [99].Domínguez-Andrés J, Fanucchi S, Joosten LAB, Mhlanga MM, Netea MG, Curr. Opin. Cell Biol 2020, DOI 10.1016/j.ceb.2019.12.006. [DOI] [PubMed] [Google Scholar]
- [100].Falkenberg KJ, Johnstone RW, Nat. Rev. Drug Discov 2014, DOI 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- [101].Luengo A, Gui DY, Vander Heiden MG, Cell Chem. Biol 2017, DOI 10.1016/j.chembiol.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Sdelci S, Rendeiro AF, Rathert P, You W, Lin JMG, Ringler A, Hofstätter G, Moll HP, Gürtl B, Farlik M, Schick S, Klepsch F, Oldach M, Buphamalai P, Schischlik F, Májek P, Parapatics K, Schmidl C, Schuster M, Penz T, Buckley DL, Hudecz O, Imre R, Wang SY, Maric HM, Kralovics R, Bennett KL, Müller AC, Mechtler K, Menche J, Bradner JE, Winter GE, Klavins K, Casanova E, Bock C, Zuber J, Kubicek S, Nat. Genet 2019, DOI 10.1038/s41588-019-0413-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Thompson RP, Nilsson E, Skinner MK, Anim. Reprod. Sci 2020, DOI 10.1016/j.anireprosci.2020.106316. [DOI] [PubMed] [Google Scholar]