

Abstract
The traditional view of chronic obstructive pulmonary disease (COPD) as a self-inflicted disease caused by tobacco smoking in genetically susceptible individuals has been challenged by recent research findings. COPD can instead be understood as the potential end result of the accumulation of gene–environment interactions encountered by an individual over the life course. Integration of a time axis in pathogenic models of COPD is necessary because the biological responses to and clinical consequences of different exposures might vary according to both the age of an individual at which a given gene–environment interaction occurs and the cumulative history of previous gene–environment interactions. Future research should aim to understand the effects of dynamic interactions between genes (G) and the environment (E) by integrating information from basic omics (eg, genomics, epigenomics, proteomics) and clinical omics (eg, phenomics, physiomics, radiomics) with exposures (the exposome) over time (T)—an approach that we refer to as GETomics. In the context of this approach, we argue that COPD should be viewed not as a single disease, but as a clinical syndrome characterised by a recognisable pattern of chronic symptoms and structural or functional impairments due to gene–environment interactions across the lifespan that influence normal lung development and ageing.
Introduction
Chronic obstructive pulmonary disease (COPD) has traditionally been considered to be paradigmatic of a disease caused by gene–environment interactions, being viewed as occurring almost exclusively (with the exception of patients with α1-antitrypsin deficiency)1 in genetically susceptible smokers.2 However, several observations have challenged this conventional model.3 First, the central dogma of the gene–environment hypothesis is that a combination of gene(s) and environmental factor(s) determine disease risk.4 But few convincing examples of clear gene–environment interactions that have been replicated across studies have been identified for COPD,5,6 and the genetic effects of identified loci are quite similar in smokers and never-smokers.7,8 A recent, large genome-wide association analysis of 179 689 controls and 21 077 individuals with COPD of European ancestry from the UK Biobank5 confirmed the presence of interaction effects with smoking at previously reported COPD loci, but did not identify novel susceptibility loci. Two potential, non-mutually exclusive explanations can be considered here. On the one hand, molecular associations between smoking status and COPD might be detectable through other omics approaches, such as epigenomics, transcriptomics, or proteomics;6 as discussed below, the effect of smoking is well captured by epigenetic modifications.9–11 On the other hand, the toxic effects caused by the thousands of different compounds and metabolites in cigarette smoke might be too complex to be captured by a reductionist approach (ie, identification of a single gene or variant),12,13 so multilevel integrative approaches might be needed.6,14,15 Polygenic risk scores, which are based on information about all established risk variants from well powered genome-wide association studies, might help to identify individuals with a high genetic risk who are therefore more susceptible to developing COPD when exposed to environmental factors.16
Second, a substantial proportion of patients with COPD are never-smokers.17,18 Furthermore, epidemiological studies have shown that many different environmental exposures and host conditions other than smoking (eg, sex) are significantly associated with airflow limitation (the traditional diagnostic functional abnormality of COPD) over the lifespan.19 Of note, in a European population-based study, sex differences were found according to smoking status: 27·2% of females and 7·3% of males with COPD were never-smokers.20 Finally, several studies have now shown that there is a range of lung function trajectories over the life course (ie, a trajectome; panel 1 and figure 1A), some of which can lead to COPD in late adulthood through two different but non-mutually exclusive mechanisms: abnormal lung development and accelerated lung function decline.3,21–23
Figure 1: Potential lung function trajectories, opportunities for intervention, and research questions.

(A) Normal, below normal, and supranormal lung function trajectories are shown, with the possibility of catch-up during development and accelerated decline leading to premature death in adulthood, at least in those with reduced peak lung function. Abnormal lung development and accelerated lung function decline can lead to COPD. Reproduced from Agustí and Faner,22 by permission of Elsevier. (B) Risk or protective factors associated with lung function trajectories and the development of COPD could be targeted to improve lung function and reduce the risk of COPD. Strategies for risk modification during childhood and adolescence, adulthood, and later life are shown, with questions that need to be addressed to identify new therapeutic targets and develop new strategies for improved lung health and the prevention of COPD. COPD=chronic obstructive pulmonary disease.
In this Series paper, we argue that to advance understanding of the contributions of gene–environment interactions to the pathogenesis of COPD, we need to consider host and environmental factors other than smoking and their biological interactions in the context of a time dimension. We review evidence for this proposed approach to COPD, with a focus on its three main components: environmental and host factors, genetics and epigenetics, and the time axis. Furthermore, we consider the clinical implications of this lifespan perspective and key questions for future, multidisciplinary research.
Gene–environment interactions: a lifespan perspective
To understand fully the roles of factors that contribute to the pathogenesis of COPD, the following must be considered. First, the series of dynamic and cumulative gene–environment interactions beyond tobacco smoking that contribute to COPD risk, including many other environmental (eg, infections, air pollutants) and host (eg, prematurity, immune fitness, senesence24,25) factors. Second, a time dimension that takes into account the age of the individual at which a given gene–environment interaction occurs—because at different ages, the biological response(s) and clinical consequence(s) are likely to be influenced by unique sets of genes and epigenetic mechanisms that modulate the capacity of the system to respond (eg, immune function) and repair26,27—and the cumulative history of gene–environment interactions encountered by the individual throughout life (from before birth or even before conception28) that might render the lungs more fragile and less resilient to respond to new gene–environment interactions.29
We propose that future research should take an integrative approach that uses multiple omics platforms to improve understanding of the pathogenesis of COPD in the context of the complex interactions between genes (G) and the environment (E) over time (T); this approach, which we refer to as GETomics (panel 1 and figure 2),30–34 has also been proposed for other medical conditions35,36 and could be relevant for a range of chronic human diseases. Specifically, this term refers to the integration of information from basic omics approaches (eg, genomics, epigenomics, proteomics) with that from clinical omics approaches (eg, phenomics, physiomics, radiomics) and exposures (the exposome; panel 1) over time. The time axis is crucial because both the age of the individual at which a given gene–environment interaction occurs and the previous, cumulative gene–environment interactions that an individual has encountered through life can influence the final biological and clinical outcome.37
Figure 2: A GETomics approach to understanding COPD and other chronic human diseases.
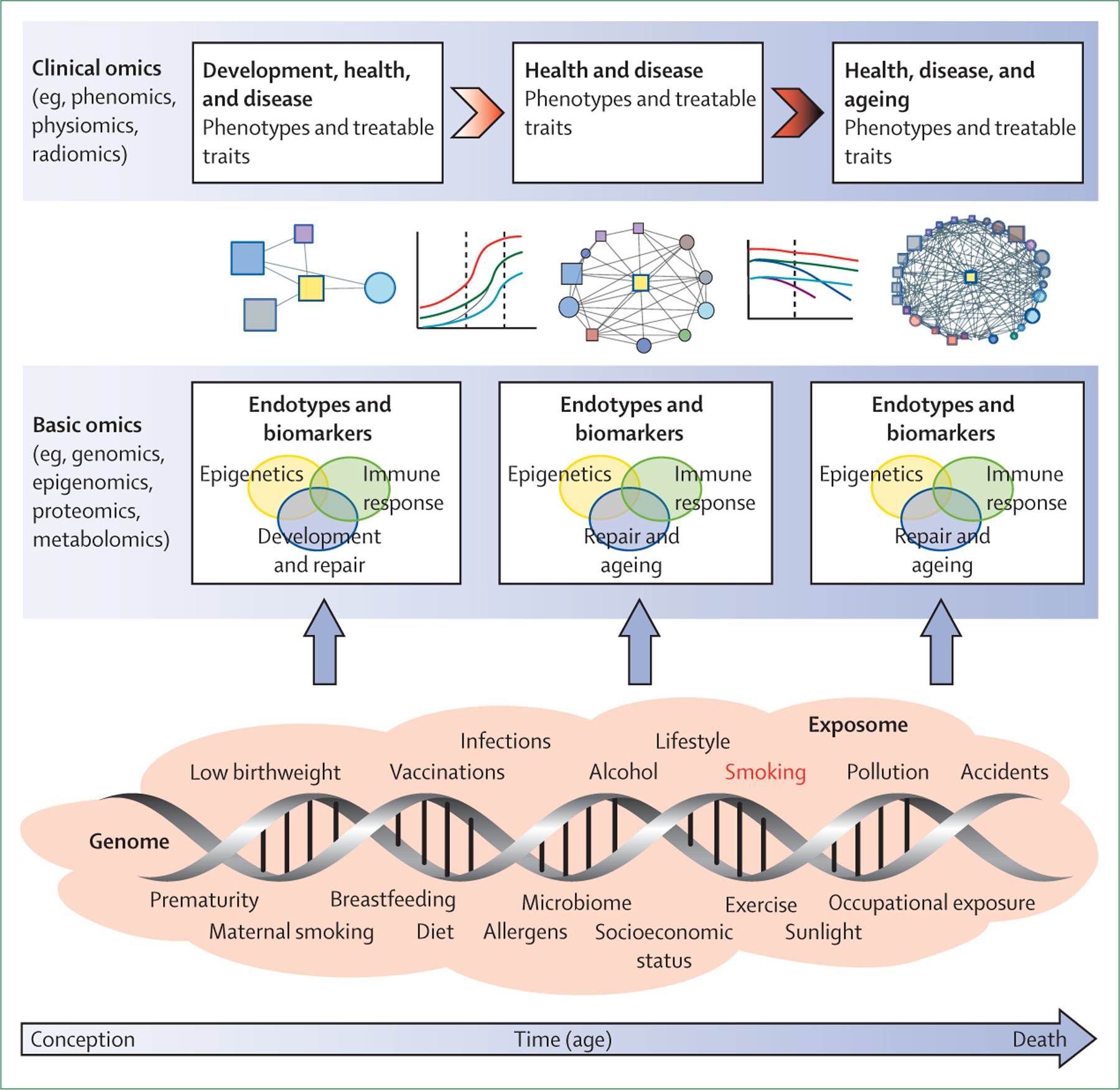
The biological effects and clinical outcomes of different gene–environment interactions depend not only on their specific characteristics, but also on a time dimension—ie, the age of the individual at which the interaction occurs and the cumulative history of the individual’s previously encountered gene–environment interactions. We propose that future research should take a holistic approach that considers the range of interactions between genes (G) and the environment (E) that occur over an individual’s lifespan (time, T) in the context of integrated omics approaches (ie, GETomics) to better understand the pathogenesis of COPD (and probably other chronic human diseases). Examples of environmental factors (the exposome30–33), from conception to death, are represented by orange shading. The positions of different exposures included in the shaded area are not necessarily related to the time axis (arrow) and might occur several times during the lifespan. At different timepoints, these environmental factors interact with the genomic background of the individual through epigenetic and other mechanisms that might be identified through various basic omics approaches. These interactions induce biological responses (endotypes34)—such as innate or acquired immune responses—that modulate organ structure (development, maintenance and repair, ageing) and function. Biomarkers for these endotypes are needed to be able to characterise objectively the pathogenic mechanisms linked to altered lung structure and function. Modulation of organ structure and function, represented here by different lung function trajectories associated with development and ageing, determines long-term phenotypes associated with health and disease, which can be explored through clinical omics approaches. COPD=chronic obstructive pulmonary disease.
In our view, the integration of a time axis in our understanding of disease pathogenesis could uncover novel opportunities for prevention or early treatment of COPD, and possibly also for prevention and treatment of the multimorbid conditions that frequently accompany COPD because they share similar risk factors and biological mechanisms.38
Environmental and host factors
Tobacco smoking remains a key, preventable environmental risk factor for COPD,2 and its impact on lung function can begin even before conception—eg, with exposures of future parents during adolescence.28,39 The effects of maternal tobacco smoking during pregnancy have been well described, with an estimated lower birthweight of about 19 g for each cigarette smoked per day by the mother.36 Stopping smoking during pregnancy at any time up to 30 weeks results in increased birthweight, although the greatest effects are seen when the mother quits before 16 weeks of pregnancy.40 Needless to say, the effects on lung function of passive smoking, both during childhood and in adulthood, as well as those of active smoking, are well established.2
However, smoking exposure is not the only risk factor for low lung function through life, as discussed in the second paper in this Series.18 A cross-sectional study of 11 423 participants in the LEAD (Lung, hEart, sociAl, body) cohort, a general population study in Austria that enrolled individuals with an age range of 6–82 years,41 showed that conditions associated with low lung function are different at different ages, increase in number with time and, importantly, interact with each other,19 so that the network of interactions becomes much more complex with advancing age (figure 3). In the younger age bins, an asthma signature (including the presence of an asthma diagnosis, wheezing, and elevated eosinophils) emerges as a risk factor for low lung function, in combination with other early-life factors, such as prematurity, no breastfeeding, hospital admission in infancy due to respiratory problems, systemic inflammation, poor nutrition, low fruit and vegetable intake, alcohol consumption in early life, low physical activity, and heavy smoking exposure at an age at which the lungs are still growing (figure 3).19 Importantly, some of these factors are preventable. In adulthood, the association network includes respiratory symptoms, allergies, exposure to environmental pollutants other than smoking, low socioeconomic status, and multimorbidity. The increasing complexity (density) of the network with increasing age suggests that any preventive or therapeutic intervention might be more effective in younger individuals.38 The relative individual effect of each of these factors is difficult to quantify, but their co-occurrence in the same individual significantly increases the risk of low lung function at any age.19 Importantly, reduced lung function might not always translate into clinical COPD.3,19
Figure 3: Network of interactions between risk factors for reduced lung function in different age bins.
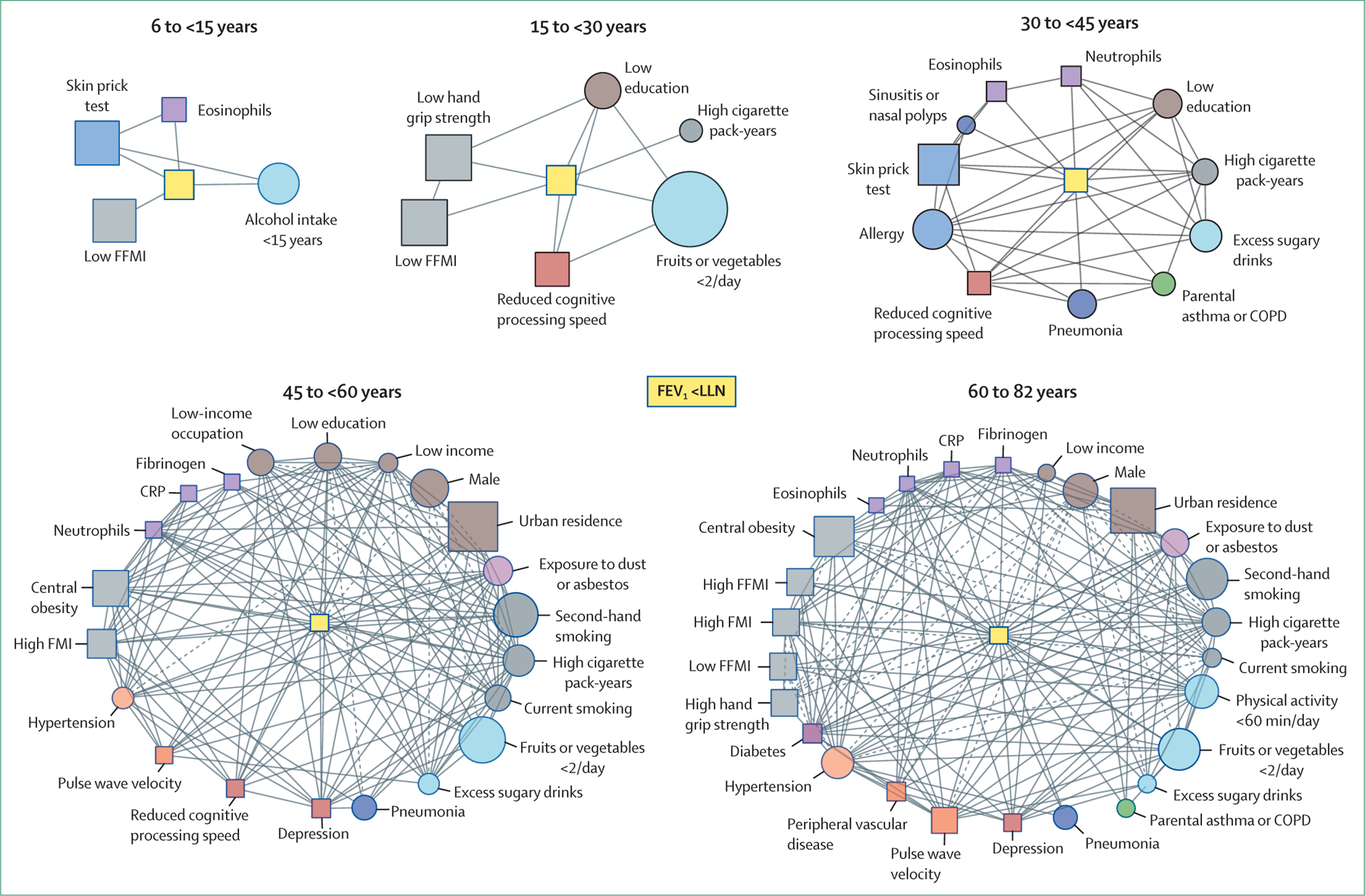
Network of interactions between risk factors for reduced lung function—FEV1 less than LLN (central yellow nodes)—in different age bins. Each node represents one variable (squares indicate that the variable was directly quantified, circles indicate that the variable was determined using questionnaires), node size is proportional to the prevalence of that variable in the specific age group, and node colour corresponds to the variable category (eg, biomarkers in purple, nutrition and activity in light blue). Links between nodes indicate the existence of a significant (p<0·05) relationship between variables and the line type indicates whether the odds ratio is greater than 1 (continuous; positive association) or less than 1 (dashed; negative association).19 The complexity of the network of interactions increases with advancing age. Modified from Breyer-Kohansal and colleagues,19 by permission of the American Thoracic Society. COPD=chronic obstructive pulmonary disease. CRP=C-reactive protein. FFMI=fat-free mass index. FMI=fat mass index. LLN=lower limit of normal.
Prematurity as a host risk factor for COPD deserves specific discussion. Premature birth (birth at <37 weeks of gestation), especially if it occurs before 32 weeks of gestation, results in smaller airways, fewer alveoli, and an impaired vascular system.42,43 As a result, children born prematurely do not often reach expected peak lung function in early adulthood and typically follow a low lung function trajectory that can potentially lead to a COPD-like phenotype later in life (figure 1A).44–46 The term dysanapsis, originally described by Mead in 1980,47 refers to the mismatch of airway tree calibre to lung size. Once believed to be a physiological curiosity,48 it is now considered to be clinically relevant in several conditions, including prematurity,49 obesity and asthma in children,50 and smoking in older adults.51 Recent studies showed that adults with evidence of dysanapsis on CT had more severe airflow limitation52 and a differential spirometric response to a standardised dose of inhaled bronchodilator when compared with those with no dysanapsis.53 Furthermore, microCT studies have shown that the number of patent terminal and transitional bronchioles is significantly reduced in adult patients with COPD.54,55 However, whether this is due to smoking-induced injury56 or abnormal lung development in early life (dysanapsis52) is unclear, although these mechanisms can coexist.3 Nevertheless, given that the incidence of prematurity has increased, and mortality of babies born prematurely has decreased significantly over the past three decades,57 a marked increase in adult survivors of premature birth with potential lung development limitations is expected in the near future.
Another potential host factor to consider is obesity. A recent mendelian randomisation analysis of the SAPALDIA (Swiss Cohort Study on Air Pollution and Lung Diseases in Adults) cohort,58 a general population study in Switzerland that included 9651 participants aged 18–60 years at baseline (51% female) in whom lung function and body-mass index were measured three times over 20 years of follow-up, showed that gene scores linked to childhood body-mass index were associated with lung function later in life. This finding highlights the importance of a life-course perspective in studies of the causal role of body-mass index (or other environmental and host factors) in respiratory health.
Finally, it is important to highlight that many previous studies have shown that childhood asthma can (in some individuals) track impaired lung function into adulthood, so an early-life diagnosis of asthma is often considered to be an important host risk factor for COPD later in life.59 For instance, early-life predictors of COPD in adulthood in the Tasmanian Longitudinal Health Study60 included childhood asthma, bronchitis, pneumonia, allergic rhinitis, eczema, parental asthma, and maternal smoking. However, what is often missed is that asthma is a clinical diagnosis based on a pattern of respiratory symptoms such as wheezing, dyspnoea, chest tightness or cough, and variable expiratory airflow limitation.61 This same pattern of symptoms can occur in children who, for whatever reason, have impaired lung development and who, most likely, will be diagnosed with (and treated for) asthma. A careful interpretation of the evidence indicates that it is a diagnosis of asthma in childhood that tracks with poor lung function in adulthood (and perhaps with clinical COPD), and this diagnosis might correspond to the disease that we call asthma or, alternatively, to deficient lung development, the symptoms of which might clinically resemble asthma and lead to COPD later in life.62 Of note, a recent report from the ECRHS (European Community Respiratory Health Survey) indicated that the asthma–COPD overlap syndrome in adults aged 40–68 years seems to have its origins earlier in life than does COPD alone.63
To advance understanding of the complex set of host risk factors and the exposome, future research will have to encompass the dynamic integration of multilevel basic, clinical, and epidemiological data through the lifespan of individuals in diverse populations. This approach might eventually allow the identification and validation of biomarkers representative of relevant endotypes as early as possible during the life course of an individual, facilitating prevention or early therapeutic intervention (figure 2 and panel 2).
Genetics and epigenetics
The genetic basis of COPD is reviewed in the first paper in this Series6 and will not be discussed in detail here. Many gene variants associated with risk of COPD, lung function (eg, FEV1, forced vital capacity), and other pulmonary and non-pulmonary traits of the disease have been identified (thus helping to explain the clinical heterogeneity of the disease), although the largest contribution to the phenotype (panel 1) arises from the combined effect of many common variants of small effect size and not from rare variants with large effect sizes.6 Of particular relevance, large-scale genome-wide association studies have found a clear and consistent overlap between genes related to lung function in children and in adults,64–66 and some of the genes associated with COPD are related to lung development.6 The use of polygenic risk scores supports findings of overlapping genetics for lung function traits in adults and children.67
Conversely, environmental exposures might interact with the genome via epigenetic modifications, which primarily refer to chemical changes to the DNA molecule that affect gene expression but do not alter the genetic sequence.26,68,69 DNA methylation, the addition of a methyl group to cytosines next to guanines (CpG sites), is the most widely studied epigenetic modification in humans, but other epigenetic mechanisms such as histone modifications (acetylation or methylation) and microRNA also modify gene expression. Epigenetic changes can be inherited across cell generations, and recent data indicate that these changes could be transmitted across human generations (ie, transgenerational changes).70 Of note, even in the absence of disease, age-associated epigenetic changes can occur. In fact, chronological age is one of the strongest determinants of DNA methylation levels.71
Central to the concept of epigenetics is that gene expression activity is affected. If detrimental exposures and associated epigenetic changes occur early in life, perhaps starting in utero (eg, through maternal smoking), lifelong cellular trajectories could be affected, which might be followed by tissue and organ effects that have pathophysiological consequences. For instance, maternal smoking during pregnancy is associated with widespread changes in offspring DNA methylation across the genome at birth; importantly, these changes appear to persist at least until adolescence.72 Changes in the DNA methylome as a consequence of maternal smoking are associated with diseases such as childhood asthma, but differential methylation as a causal mechanism in the relationship between maternal smoking and asthma has not been proven.13 Similarly, epigenetic marks identified in fetal lung tissue might signal risk for COPD in later life.73 Finally, the epigenetic effects of smoking relate to both the intensity and the duration of exposure and can have long-lasting effects, from years to decades, further emphasising the relevance of the time axis in this gene–environment interaction.11
Similar to maternal smoking during pregnancy, gestational age has been associated with DNA methylation levels in thousands of genes in newborns, some of which might persist through childhood.74 A similar effect on methylation profiles has been observed for birthweight.75 During the fetal period, epigenetic processes are important for controlling development and growth.76 The examples of the associations of maternal smoking during pregnancy and gestational age at birth with persistent epigenetic changes in newborns provide compelling evidence of early and long-lasting effects of certain environmental exposures and highlight the potential role of such early-life events and related mechanisms in development, growth, and health and disease in later life.
Extending the notion that trajectories of the epigenome can be set in utero, network-based approaches have been applied to capture the complexity of gene–environment interactions on a genome-wide scale. For example, a co-methylation analysis revealed network features that were highly preserved between fetal and adult lung data associated with in-utero smoke exposure, COPD, and reduced adult lung function, with enrichment in developmental and inflammatory pathways, including Hippo, Wnt, transforming growth factor-β, and PI3K/AKT (phosphatidylinositol 3-kinase/protein kinase B) pathways.73 A recent study has shown that lung methylation profiles are more closely related to smoking in patients with mild-to-moderate COPD than in patients with severe disease.77 In this study, however, patients with severe disease were significantly younger, suggesting that different epigenetic patterns are associated with different lung function trajectories, some of which can lead to severe COPD at younger ages.77
Finally, investigation of DNA methylation in lung tissue has shown differences between men and women.78 A recent study showed that sex-specific methylation patterns were associated with trajectories of lung function measured at ages 10, 18, and 26 years, and that pathways enriched for methylation were associated with poorer lung function trajectories.79 Sex-specific differences in COPD symptoms later in life might therefore begin in early life with methylation patterns that track with specific lung function trajectories. Indeed, sex-specific associations with COPD have been observed with different omics levels, including genetics, epigenetics, transcriptomics, metabolomics, and proteomics,80 with sex and gender emerging as important endogenous (eg, by way of hormones or sex chromosomes) and exogenous (eg, through smoking behaviours or occupational roles) determinants of the final outcomes of similar gene–environment interactions over the life course, with varying and complex effects on health and disease.
Future research will need to integrate the findings of a range of clinical and basic omics analyses to unravel the many gene–environment interactions, including a range of genetic and epigenetic factors, that contribute to different lung function trajectories and, eventually, to the development of COPD (panel 2). A better understanding of these factors could also help to identify those at risk of COPD.
The time axis
Normal lung development starts in utero during the embryonic phase (gestational weeks 4–7) with lung bud differentiation and trachea and bronchi formation.81 It continues throughout the fetal and postnatal periods, with lung growth and cellular proliferation occurring until maximally attained lung function peaks at around age 20–25 years for men (earlier in women).82,83 This developmental phase is followed by a relatively short plateau phase of about a decade, with mild lung function decline thereafter (about 20 mL/year for women, more in men82) due to physiological lung ageing22 (figure 1A).
Any insult to the airways or lung parenchyma during the very long (>20 years) lung developmental period could have long-term consequences for health and disease,27 a fact now recognised in the concept of the developmental origins of health and disease (DOHaD).84,85 Indeed, as we have discussed, several asthma, COPD, and lung function genes identified in studies of adults also seem to have relevant functional roles in fetal developmental processes.6,86 Conversely, adult lung diseases might be the consequence of an acceleration of physiological age-associated changes.56 Ageing-related DNA methylation pathways have also been linked to pathways of lung development;87 therefore, biological age, starting in early life, might reflect linear and non-linear cumulative environmental pressures on the epigenome that could increase the risk of COPD later in life. Given that lung ageing seems to recapitulate key developmental epigenetic signatures of transcription factors, and that differential methylation of transcription factors has been associated with COPD,88 close inspection of the molecular endotypes (ie, the biological mechanisms that underlie particular phenotypes) that might emerge from investigations of gene–environment interactions over time could reveal novel opportunities for the early prevention of COPD through targeting of gene regulation, among other potential molecular strategies.
As discussed, there is a range of lung function trajectories through life,3,22 both above and below the normal trajectory (figure 1A). Lung function trajectories below the expected normal range can be due to suboptimal growth and development in childhood and adolescence, leading to reduced peak lung function, or to shortened plateau or accelerated lung function decline caused by accelerated lung ageing or other factors that damage the lungs,89 and are known to be associated with increased risk of developing COPD in adulthood.21,60,90 Results from the population-based CARDIA (Coronary Artery Risk Development in Young Adults) study showed that both lower peak and accelerated decline in FEV1 are risk factors for future emphysema identified on CT scans, independent of smoking status.91 Moreover, low peak lung function in early adulthood is associated with increased prevalence and earlier incidence of cardiovascular and metabolic conditions, as well as premature death,92 suggesting that abnormal lung development could be associated with altered development of other organ systems. If this is the case, reduced lung function in infancy, adolescence, or early adulthood could be a marker of a more systemic deficit.3,22 Conversely, recent research has shown that supranormal trajectories (figure 1A) are associated with healthier ageing93 and reduced risk of developing COPD.94 Therefore, a lifespan approach to understanding the gene–environment interactions that underlie these different lung function trajectories is of paramount importance to prevent and treat the disease-associated conditions as early and efficiently as possible, and to promote healthier ageing (figure 1B).38
Finally, like any organ, the immune system develops, matures, and ages.95 As a result, there are marked differences in immune function and immune phenotypes between children and adults.95 Therefore, the immune responses elicited by gene–environment interactions might differ depending on the age (time) at which they occur.25,96 Studies of the asthma locus on chromosome 17q12–21 have taught us important lessons about the effects of gene–environment interactions over time, because single-nucleotide polymorphisms at this major locus have been repeatedly associated with childhood asthma.97 Data from seven US birth cohorts (n=3786) suggest that the association with this locus primarily reflects early-life susceptibility to respiratory viruses,98 which might have long-term consequences for lung health.99 Functional interactions between one of the key 17q12–21 genes, GSDMB (gasdermin B), and COPD-associated genes such as IL27 (interleukin 27) and HHIP (hedgehog-interacting protein) have now been identified, thus linking this asthma locus to COPD.100
In experimental models, neutrophilic inflammation during development disrupts the extracellular matrix and predisposes adult mice to a COPD-like phenotype,101 whereas aged T cells in young mice (with mitochondrial dysfunction) induce premature ageing and multimorbidity.102 Furthermore, infection with γ-herpesvirus in aged mice causes a more intense inflammatory response than that elicited in young animals, suggesting that the same environmental insult can have different consequences depending on the age at which it occurs.37 Finally, cell senescence, a hallmark of cellular ageing,103 also has a key role in organ development and disease, in which senescence cells accumulate and secrete a specific set of immune mediators collectively known as the senescence-associated secretory phenotype.104 Thus, an excessively aged immune system for a given chronological age might also increase the risk of COPD.
In summary, future research will have to address the role of the time axis by combining longitudinal studies with different omic platforms to better understand the determinants of lung health in adulthood, lung ageing, and COPD (panel 2).
Clinical implications of integrating the time axis
Understanding the pathogenesis of COPD in terms of the occurrence and accumulation of gene–environment interactions along the time axis has implications for disease prevention and management. We propose that COPD should no longer be viewed as a single disease caused almost exclusively by tobacco smoking, but as a complex clinical syndrome (panel 1).105 A syndrome is a recognisable pattern of symptoms and clinical signs that can be caused by different mechanisms (eg, febrile syndrome).106,107 According to this definition, we propose that COPD should be considered as a syndrome characterised by a recognisable pattern of chronic symptoms (one or more of dyspnoea, cough, or expectoration) and structural (one or more of bronchitis, bronchiolitis, or emphysema) or functional (airflow limitation or abnormal gas exchange) impairments, or both, due to mechanisms (endotypes34) that are the end result of different and cumulative gene–environment interactions through the lifetime of the individual. Currently, the presence of non-fully reversible airflow limitation is a mandatory requirement for the diagnosis of COPD,108 but there is increasing recognition of the existence of a pre-COPD stage, with symptoms, structural abnormalities, and functional abnormalities other than airflow limitation,109 although its natural history and response to treatment is still unclear.110
Regarding the prevention of COPD, there is no question that avoiding smoke or inhaled exposures at all times (in utero and during infancy through to adolescence and adulthood) and favouring early quitting of active smoking82 are key preventive measures that must continue to be fully endorsed. However, the study of gene–environment interactions in the context of a lifetime perspective opens opportunities for the prevention of COPD that go well beyond smoking (figure 1B).27 For instance, in a chronically undernourished population in Nepal, maternal supplementation of diet with vitamin A before, during, and after pregnancy improved lung function in the offspring in preadolescent years.111 Similarly, supplemental vitamin C taken by pregnant American smokers improved newborn lung function and decreased wheezing in the offspring up to the age of 1 year.112 Whether other preventive strategies can contribute to improved lung health in other populations requires further research.113 There is a knowledge gap regarding lung function during the transition from infancy to adulthood, sometimes referred to as a black box; however, this phase might hold promise for prevention and early intervention.114 For instance, studies have shown that the presence of chronic bronchitis in children115 and young adults116 is predictive of the development of airflow limitation later in life. In this context, spirometry should be viewed as a test with global application that, if used routinely for screening in young populations (eg, during school, university, army, driving licence testing), might help to identify individuals at risk of respiratory and non-respiratory multimorbidity and premature death who might benefit from intervention.38,92,117
Finally, regarding disease management, an understanding of lung function impairment and the development of COPD in terms of gene–environment interactions along the time axis requires translation of the concept of lifelong lung function (the trajectome) to clinical practice. Unfortunately, lung function is not currently measured routinely in infancy, adolescence, or early adulthood, so identification in the clinic of the lung function trajectory that an individual patient has followed so far (or will follow in the future) would be challenging. This strategy would clearly require that the attending physician specifically questions the patient on potentially relevant early-life factors through careful clinical history taking (eg, prematurity, maternal smoking, allergies and respiratory infections in infancy). Validation of biomarkers associated with different lung function trajectories is also needed to aid early detection of those at risk of disease and to ascertain which lung function trajectory a given patient might have followed before being seen in the clinic, because it is likely that different lung function trajectories are associated with different prognosis and might require different treatment strategies. Several examples of such biomarkers already exist. First, serum concentrations of the pneumoprotein CC16 (club cell secretory protein) appear to be consistently associated with low lung function in different age groups,118 have been associated with accelerated lung function decline,119,120 and are influenced by smoking.121–123 CC16 concentrations are also related to persistence of asthma and reduced lung function, both in childhood and adulthood.118 Furthermore, several genetic variants have been associated with serum CC16 concentrations.124 Second, CHI3L1 (chitinase-3-like protein 1, also known as YKL-40) is a secreted human glycoprotein involved in airway remodelling that has been associated with early-life inflammation in children with a history of prematurity, and is now considered to be a biomarker of altered lung development.125 Third, plasma concentrations of the microRNA miR-145-5p in children with asthma appear to be associated with abnormal lung growth leading to COPD.126 Fourth, eosinophil concentrations seem to be associated with the development of persistent airflow limitation in children with a diagnosis of asthma.127 More recent research has confirmed that higher circulating eosinophil concentrations were associated with increased risk of developing COPD,128 and that FEV1 decline was faster in patients with mild-to-moderate COPD and higher blood eosinophil counts.129 Finally, some biomarkers of ageing, such as telomere shortening, have also been associated with structural remodelling of the lung in children with bronchopulmonary dysplasia, and in young patients with COPD,130 supporting another potential link between altered lung development and ageing.125 Therefore, current and future therapeutic interventions need to be tested in younger individuals,110 in whom they might be more effective because of the reduced complexity of the network of interacting risk factors (figure 3).38,131 For instance, treatment with tiotropium in the UPLIFT (Understanding Potential Long-Term Impacts on Function with Tiotropium) study reduced the decline of lung function in young (≤50 years)132 but not in older patients with COPD.133
If the GETomics approach proposed here, in which COPD is viewed as a syndrome attributable to one or several mechanisms (endotypes), is adopted in research and applied in practice, it could allow us to develop novel preventive strategies and to consider much earlier intervention (even in the absence of poorly reversible airflow limitation, the current functional defining characteristic of COPD) to promote respiratory health. Moving upstream to younger individuals holds the promise of more effective prevention and treatment of COPD; a recent publication presents the rationale for this approach and discusses the design of appropriate clinical trials in young patients with COPD and those with pre-COPD.110
Conclusions and future directions
Life starts as a tabula rasa that begins to be written from conception (and even before39,134). As discussed here, integration of the time axis in our understanding of the origins of COPD—with improved understanding of the range of contributing gene–environment interactions and their biological effects—has the potential to enable more effective prevention and management of COPD and, potentially, other chronic non-communicable diseases of the elderly that often share genetic and environmental risk factors with COPD.114 Furthermore, taking a gene–environment–time approach to COPD research could provide relevant information not only on the determinants of lung function, but also on the timeframe for COPD development and timing of diagnosis, severity of disease, and heterogeneity of clinical presentation, including exacerbations, chronic bronchitis versus emphysema, systemic manifestations, associated multimorbidity, and response to therapy.
To achieve this potential, however, we need to improve understanding of the paradigm proposed here. Key questions for future research are presented in panel 2. The ideal study would involve a birth cohort that is followed prospectively until death with repeated and detailed pheno-endotypic characterisation (including biomarker measurement), ample biological profiling assessment, and comprehensive monitoring of the exposome (perhaps through the blood exposome, which includes all biologically active chemicals135), as well as tissue probes, if possible. As this ideal study is not currently a realistic option, a potential alternative is to use different cohorts that span different age groups to understand the biological mechanisms and clinical effects of the range of lung function trajectories and their gene–environment determinants over the lifespan. This is precisely the goal of the CADSET (Chronic Airway DiSeases Early sTratification) network, a European Respiratory Society Clinical Research Collaboration.136 In this setting, investigations of the gene–environment interactions that underpin the range of lung function trajectories over time will have to consider not only the specific nature of a given gene–environment interaction but also the following: first, the phase of the lung function trajectory in which the specific gene–environment interaction occurs (development vs ageing); second, the specific lung function trajectory that the individual was following when the gene–environment interaction took place; third, the duration, intensity, and repetitiveness of the exposure; and fourth, the previous, cumulative exposome history of the individual (figure 2). Comprehensive analytical, non-reductionistic, and agnostic approaches (network analysis, systems biology) that integrate different omics levels and environmental and clinical data will be required to provide a more integrated view of gene–environment interactions over time (a GETomics approach) by showing both their relationships with the outcomes of interest and between the components of the network.30,137–140
Finally, we also need to deploy this gene–environment–time perspective in daily clinical practice when considering the potential need for preventive or therapeutic interventions. This would require a considerable educational effort directed to health-care professionals. For instance, asking about early-life events and exposures should become routine in clinics for adults with chronic airway diseases, as well as considering the role of potential biomarkers. It is time to implement a looking-back-to-look-forward concept in addressing COPD in particular and, importantly, respiratory and human health in general.
Key messages.
COPD has traditionally been understood as a self-inflicted disease caused by tobacco smoking in genetically susceptible individuals, but recent research has challenged this view; many genetic variants and a range of different environmental exposures and host factors are now known to be associated with risk for COPD over the lifespan
COPD might have its origins in early life, with impaired lung development (low peak lung function) or lung ageing (accelerated lung function decline) affecting an individual’s lung function trajectory and risk of developing COPD; improved understanding of gene–environment interactions over the lifespan in the pathogenesis of COPD is needed
To understand the origins and pathobiology of COPD, the time axis needs to be considered because the age of an individual at which a given gene–environment interaction occurs, as well as the cumulative history of previous gene–environment interactions, might affect the biological response(s) and clinical consequence(s) of a given exposure
Comprehensive investigation of the interactions between genes (G) and the environment (E) over time (T) is needed to attain a complete picture of COPD pathogenesis; to achieve this aim, we propose an approach that integrates information from basic omics and clinical omics approaches with exposures (the exposome) over the lifespan—a strategy that we refer to as GETomics
Rather than being viewed as a single disease caused mainly by tobacco smoking, we argue that COPD might instead be regarded as a clinical syndrome that is characterised by a recognisable pattern of chronic respiratory symptoms and structural lung abnormalities or functional lung impairments, or both, and due to one or more mechanisms (endotypes) that reflect different and cumulative gene–environment interactions over the life course
A GETomics approach—leading to improved understanding of underlying pathogenic mechanisms over the lifespan—could enable identification of novel targets for the prevention and early treatment of COPD
COPD=chronic obstructive pulmonary disease.
Panel 1: Key terms and definitions.
The following terms and definitions are used in this Series paper to describe and propose approaches to understanding the nature and origins of chronic obstructive pulmonary disease.
Basic omics
The range of methods that can be used to comprehensively characterise and quantify pools of biological molecules and potentially identify molecular mechanisms of disease (endotypes); examples include genomics, transcriptomics, epigenomics, proteomics, lipidomics, and metabolomics
Chronic obstructive pulmonary disease (COPD)
A clinical syndrome characterised by a recognisable pattern of chronic symptoms and structural or functional abnormalities (including, but not limited to, airflow limitation), or both, and attributable to one or several mechanisms (endotypes) that reflect different, cumulative gene–environment interactions occurring through the lifetime of the individual
Clinical omics
The range of methods that can be used to comprehensively characterise and quantify the clinical features of health and disease; examples include phenomics (study of the phenome, encompassing the phenotypes of cells, tissues, organs, and organisms), physiomics (study of the physiological function of cells, tissues, organs, and organisms), radiomics (study of the range of qualitative and quantitative features on medical imaging), and microbiomics (study of the microbiome of organs and systems, including the bacteriome, mycobiome, and virome)
Exposome
The full set of (qualitative and quantitative) environmental exposures of an individual in their lifetime
GETomics
A holistic strategy that considers all gene–environment interactions that an individual might encounter through the lifespan (the time axis, T) to better understand the roles of genes (G) and environmental exposures (E) in disease pathogenesis; the approach aims to integrate basic omics, clinical omics, and exposures (the exposome) over time, accounting for both the age of the individual at which a given gene–environment interaction occurs and the previous, cumulative gene–environment interactions that the individual might have encountered through life
Clinical phenotype
A single disease attribute or combination of attributes that describe differences between patients as they relate to clinically meaningful outcomes (eg, symptoms, exacerbations, response to therapy, rate of disease progression, or death)
Trajectome
The range of possible lung function trajectories across the lifespan; lung function trajectories below the expected normal range—due to suboptimal growth and development in childhood and adolescence, or early or accelerated lung function decline in adulthood—can be associated with an increased risk of COPD
Panel 2: Questions for future research.
How do genetic, clinical, epidemiological, and biological factors associated with lung function trajectories and airflow limitation differ across the lifespan?
How do responses to the same environmental exposure differ across the lifespan, and what can omics analyses teach us about detrimental responses after exposure?
Are differences in the response to specific environmental factors at a particular timepoint determined by regenerative capacity or ageing effects on different pathways, or by the history of previous exposures?
Which early-life exposures result in a lower threshold for developing lung disease, including COPD, later in life?
Which biological and clinical thresholds determine health and disease outcomes in different age groups, and how are these thresholds affected by exposures at different ages and by the accumulation of gene–environment interactions over time?
How can these thresholds be measured in a clinically meaningfully way (eg, through assessments of lung function, breathomics, blood biomarkers)?
How might a GETomics understanding of the origins of disease be harnessed to prevent or manage COPD?
COPD=chronic obstructive pulmonary disease.
Search strategy and selection criteria.
This narrative review is based mainly on the knowledge and judgement of the authors, supported by selected references from a PubMed search for papers published from database inception to Oct 27, 2021, using three sets of search terms: (“gene–environment” OR “early life”) AND (“lung function” OR “FEV1” OR “FVC” OR “COPD”); “lifelong” AND “exposure” AND “COPD”; and “genetic” AND (“environmental” OR “environment”) AND “exposure” AND “COPD”. For the three searches, we retrieved 188, 22, and 153 studies, respectively, reporting gene–environment interactions occurring in early life with different lung function indices both in childhood and early adulthood and evidence of risk factors associated with lung function trajectories. Key papers from the authors’ own files and online searches were also considered. Papers were selected, without language restrictions, on the basis of relevance to our review and proposal.
Acknowledgments
AA and RF are supported by the Instituto de Salud Carlos III (PI17/00379, PI18/00018, PI21/00735, PMP21/00090) and Sociedad Española Neumología Cirugia Toracica (PI948/2019). EM is supported by the European Research Council (TRIBAL, 757919), The Swedish Research Council, The Swedish Heart-Lung Foundation, and Region Stockholm (ALF). DLD is supported by the US National Institutes of Health (P01 HL132825, R21 HL156122), an Alpha-1 Foundation Award, and a Brigham and Women’s Hospital Connors Center IGNITE First in Women Precision Medicine Award. RF receives support from the European Research Council (PredictCOPD, 101044387), and the Serra Hunter Program.
Footnotes
Declaration of interests
AA has received research funds and honoraria as a speaker and consultant from AstraZeneca, GlaxoSmithKline, Chiesi, and Menarini for initiatives related to chronic obstructive pulmonary disease (COPD), outside of the submitted work. EM has received advisory board reimbursements and fees as a speaker from AstraZeneca, Chiesi, Novartis, and Sanofi, outside of the submitted work. DLD has received research funds from the National Institutes of Health, Alpha-1 Foundation, and Bayer, and honoraria from Novartis. RB-K has received honoraria as a speaker from AstraZeneca, GlaxoSmithKline, Menarini, and Novartis. RF has received research funds from AstraZeneca, GlaxoSmithKline, and Menarini, honoraria as a speaker from Chiesi, and consultancy fees from GlaxoSmithKline for COPD-related initiatives outside of the submitted work.
Contributor Information
Alvar Agustí, Càtedra Salut Respiratòria, Universitat Barcelona, Barcelona, Spain; Respiratory Institute, Hospital Clinic, Barcelona, Spain; Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain; Centro de Investigación Biomédica en Red de Enfermedades Respiratorias (CIBERES), Barcelona, Spain.
Erik Melén, Department of Clinical Science and Education, Södersjukhuset, Karolinska Institutet, Stockholm, Sweden; Sachs’ Children and Youth Hospital, Södersjukhuset, Stockholm, Sweden.
Dawn L DeMeo, Channing Division of Network Medicine, and Division of Pulmonary and Critical Care Medicine, Brigham and Women’s Hospital, Boston, MA, USA.
Robab Breyer-Kohansal, Ludwig Boltzmann Institute for Lung Health, Vienna, Austria; Department of Respiratory and Critical Care Medicine, Clinic Penzing, Vienna, Austria.
Rosa Faner, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain; Centro de Investigación Biomédica en Red de Enfermedades Respiratorias (CIBERES), Barcelona, Spain.
References
- 1.Barnes PJ. Endo-phenotyping of COPD patients. Expert Rev Respir Med 2021; 15: 27–37. [DOI] [PubMed] [Google Scholar]
- 2.Fletcher C, Peto R. The natural history of chronic airflow obstruction. BMJ 1977; 1: 1645–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agustí A, Hogg JC. Update on the pathogenesis of chronic obstructive pulmonary disease. N Engl J Med 2019; 381: 1248–56. [DOI] [PubMed] [Google Scholar]
- 4.Hunter DJ. Gene–environment interactions in human diseases. Nat Rev Genet 2005; 6: 287–98. [DOI] [PubMed] [Google Scholar]
- 5.Kim W, Prokopenko D, Sakornsakolpat P, et al. Genome-wide gene-by-smoking interaction study of chronic obstructive pulmonary disease. Am J Epidemiol 2021; 190: 875–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cho MH, Hobbs BD, Silverman EK. Genetics of chronic obstructive pulmonary disease: understanding the pathobiology and heterogeneity of a complex disorder. Lancet Respir Med 2022; published online April 12. 10.1016/S2213-2600(21)00510-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wain LV, Shrine N, Miller S, et al. Novel insights into the genetics of smoking behaviour, lung function, and chronic obstructive pulmonary disease (UK BiLEVE): a genetic association study in UK Biobank. Lancet Respir Med 2015; 3: 769–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shrine N, Guyatt AL, Erzurumluoglu AM, et al. New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat Genet 2019; 51: 481–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guida F, Sandanger TM, Castagné R, et al. Dynamics of smoking-induced genome-wide methylation changes with time since smoking cessation. Hum Mol Genet 2015; 24: 2349–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilson R, Wahl S, Pfeiffer L, et al. The dynamics of smoking-related disturbed methylation: a two time-point study of methylation change in smokers, non-smokers and former smokers. BMC Genomics 2017; 18: 805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McCartney DL, Stevenson AJ, Hillary RF, et al. Epigenetic signatures of starting and stopping smoking. EBioMedicine 2018; 37: 214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gref A, Merid SK, Gruzieva O, et al. Genome-wide interaction analysis of air pollution exposure and childhood asthma with functional follow-up. Am J Respir Crit Care Med 2017; 195: 1373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.London SJ, Melén E. Genomic interactions with exposure to inhaled pollutants. J Allergy Clin Immunol 2019; 143: 2011–13.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Faner R, Cruz T, Casserras T, et al. Network analysis of lung transcriptomics reveals a distinct B-cell signature in emphysema. Am J Respir Crit Care Med 2016; 193: 1242–53. [DOI] [PubMed] [Google Scholar]
- 15.Cruz T, López-Giraldo A, Noell G, et al. Multi-level immune response network in mild-moderate chronic obstructive pulmonary disease (COPD). Respir Res 2019; 20: 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang PD, Zhang XR, Zhang A, et al. Associations of genetic risk and smoking with incident chronic obstructive pulmonary disease. Eur Respir J 2021; published online June 25. 10.1183/13993003.01320-2021. [DOI] [PubMed] [Google Scholar]
- 17.Salvi SS, Barnes PJ. Chronic obstructive pulmonary disease in non-smokers. Lancet 2009; 374: 733–43. [DOI] [PubMed] [Google Scholar]
- 18.Yang IA, Jenkins CR, Salvi SS. Chronic obstructive pulmonary disease in never-smokers: risk factors, pathogenesis, and implications for prevention and treatment. Lancet Respir Med 2022; published online April 12. 10.1016/S2213-2600(21)00506-3. [DOI] [PubMed] [Google Scholar]
- 19.Breyer-Kohansal R, Faner R, Breyer M-K, et al. Factors associated with low lung function in different age bins in the general population. Am J Respir Crit Care Med 2020; 202: 292–96. [DOI] [PubMed] [Google Scholar]
- 20.Terzikhan N, Verhamme KM, Hofman A, Stricker BH, Brusselle GG, Lahousse L. Prevalence and incidence of COPD in smokers and non-smokers: the Rotterdam Study. Eur J Epidemiol 2016; 31: 785–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lange P, Celli B, Agustí A, et al. Lung-function trajectories leading to chronic obstructive pulmonary disease. N Engl J Med 2015; 373: 111–22. [DOI] [PubMed] [Google Scholar]
- 22.Agusti A, Faner R. Lung function trajectories in health and disease. Lancet Respir Med 2019; 7: 358–64. [DOI] [PubMed] [Google Scholar]
- 23.Marott JL, Ingebrigtsen TS, Çolak Y, Vestbo J, Lange P. Lung function trajectories leading to chronic obstructive pulmonary disease as predictors of exacerbations and mortality. Am J Respir Crit Care Med 2020; 202: 210–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Filgueira TO, Castoldi A, Santos LER, et al. The relevance of a physical active lifestyle and physical fitness on immune defense: mitigating disease burden, with focus on COVID-19 consequences. Front Immunol 2021; 12: 587146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Faner R, Rojas M, Macnee W, Agustí A. Abnormal lung aging in chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2012; 186: 306–13. [DOI] [PubMed] [Google Scholar]
- 26.Boyce WT, Sokolowski MB, Robinson GE. Genes and environments, development and time. Proc Natl Acad Sci USA 2020; 117: 23235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agustí A, Faner R. COPD beyond smoking: new paradigm, novel opportunities. Lancet Respir Med 2018; 6: 324–26. [DOI] [PubMed] [Google Scholar]
- 28.Accordini S, Calciano L, Johannessen A, et al. Prenatal and prepubertal exposures to tobacco smoke in men may cause lower lung function in future offspring: a three-generation study using a causal modelling approach. Eur Respir J 2021; 58: 2002791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crispi F, Crovetto F, Larroya M, et al. Low birth weight as a potential risk factor for severe COVID-19 in adults. Sci Rep 2021; 11: 2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rappaport SM. Biomarkers intersect with the exposome. Biomarkers 2012; 17: 483–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wild CP. Complementing the genome with an “exposome”: the outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol Biomarkers Prev 2005; 14: 1847–50. [DOI] [PubMed] [Google Scholar]
- 32.Wild CP. The exposome: from concept to utility. Int J Epidemiol 2012; 41: 24–32. [DOI] [PubMed] [Google Scholar]
- 33.Vrijheid M The exposome: a new paradigm to study the impact of environment on health. Thorax 2014; 69: 876–78. [DOI] [PubMed] [Google Scholar]
- 34.Woodruff PG, Agusti A, Roche N, Singh D, Martinez FJ. Current concepts in targeting chronic obstructive pulmonary disease pharmacotherapy: making progress towards personalised management. Lancet 2015; 385: 1789–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bradley WG, Andrew AS, Traynor BJ, Chiò A, Butt TH, Stommel EW. Gene-environment-time interactions in neurodegenerative diseases: hypotheses and research approaches. Ann Neurosci 2018; 25: 261–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boyce WT, Levitt P, Martinez FD, McEwen BS, Shonkoff JP. Genes, environments, and time: the biology of adversity and resilience. Pediatrics 2021; 147: e20201651. [DOI] [PubMed] [Google Scholar]
- 37.Naik PN, Horowitz JC, Moore TA, Wilke CA, Toews GB, Moore BB. Pulmonary fibrosis induced by γ-herpesvirus in aged mice is associated with increased fibroblast responsiveness to transforming growth factor-β. J Gerontol A Biol Sci Med Sci 2012; 67: 714–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Agusti A, Alcazar B, Cosio B, et al. Time for a change: anticipating the diagnosis and treatment of chronic obstructive pulmonary disease. Eur Respir J 2020; 56: 2002104. [DOI] [PubMed] [Google Scholar]
- 39.Svanes C, Koplin J, Skulstad SM, et al. Father’s environment before conception and asthma risk in his children: a multi-generation analysis of the Respiratory Health In Northern Europe study. Int J Epidemiol 2017; 46: 235–45. [DOI] [PubMed] [Google Scholar]
- 40.MacArthur C, Knox EG. Smoking in pregnancy: effects of stopping at different stages. Br J Obstet Gynaecol 1988; 95: 551–55. [DOI] [PubMed] [Google Scholar]
- 41.Breyer-Kohansal R, Hartl S, Burghuber OC, et al. The LEAD (Lung, Heart, Social, Body) Study: objectives, methodology, and external validity of the population-based cohort study. J Epidemiol 2019; 29: 315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martinez FD. Early-life origins of chronic obstructive pulmonary disease. N Engl J Med 2016; 375: 871–78. [DOI] [PubMed] [Google Scholar]
- 43.Baraldi E, Filippone M. Chronic lung disease after premature birth. N Engl J Med 2007; 357: 1946–55. [DOI] [PubMed] [Google Scholar]
- 44.Doyle LW, Andersson S, Bush A, et al. Expiratory airflow in late adolescence and early adulthood in individuals born very preterm or with very low birthweight compared with controls born at term or with normal birthweight: a meta-analysis of individual participant data. Lancet Respir Med 2019; 7: 677–86. [DOI] [PubMed] [Google Scholar]
- 45.Simpson SJ, Turkovic L, Wilson AC, et al. Lung function trajectories throughout childhood in survivors of very preterm birth: a longitudinal cohort study. Lancet Child Adolesc Health 2018; 2: 350–59. [DOI] [PubMed] [Google Scholar]
- 46.Bui DS, Perret JL, Haydn Walters E, et al. Association between very to moderate preterm births, lung function deficits, and COPD at age 53 years: analysis of a prospective cohort study. Lancet Respir Med 2022; published online Feb 18. 10.1016/S2213-2600(21)00508-7. [DOI] [PubMed] [Google Scholar]
- 47.Mead J Dysanapsis in normal lungs assessed by the relationship between maximal flow, static recoil, and vital capacity. Am Rev Respir Dis 1980; 121: 339–42. [DOI] [PubMed] [Google Scholar]
- 48.Thompson BR. Dysanapsis—once believed to be a physiological curiosity—is now clinically important. Am J Respir Crit Care Med 2017; 195: 277–78. [DOI] [PubMed] [Google Scholar]
- 49.Duke JW, Gladstone IM, Sheel AW, Lovering AT. Premature birth affects the degree of airway dysanapsis and mechanical ventilatory constraints. Exp Physiol 2018; 103: 261–75. [DOI] [PubMed] [Google Scholar]
- 50.Forno E, Weiner DJ, Mullen J, et al. Obesity and airway dysanapsis in children with and without asthma. Am J Respir Crit Care Med 2017; 195: 314–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheel AW, Guenette JA, Yuan R, et al. Evidence for dysanapsis using computed tomographic imaging of the airways in older ex-smokers. J Appl Physiol 2009; 107: 1622–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Smith BM, Kirby M, Hoffman EA, et al. Association of dysanapsis with chronic obstructive pulmonary disease among older adults. JAMA 2020; 323: 2268–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vameghestahbanati M, Kirby M, Maltais F, et al. Dysanapsis and the spirometric response to inhaled bronchodilators. Am J Respir Crit Care Med 2021; 204: 997–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McDonough JE, Yuan R, Suzuki M, et al. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med 2011; 365: 1567–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koo H-K, Vasilescu DM, Booth S, et al. Small airways disease in mild and moderate chronic obstructive pulmonary disease: a cross-sectional study. Lancet Respir Med 2018; 6: 591–602. [DOI] [PubMed] [Google Scholar]
- 56.Verleden SE, Kirby M, Everaerts S, et al. Small airway loss in the physiologically ageing lung: a cross-sectional study in unused donor lungs. Lancet Respir Med 2021; 9: 167–74. [DOI] [PubMed] [Google Scholar]
- 57.Chawanpaiboon S, Vogel JP, Moller AB, et al. Global, regional, and national estimates of levels of preterm birth in 2014: a systematic review and modelling analysis. Lancet Glob Health 2019; 7: e37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Probst-Hensch N, Jeong A, Stolz D, et al. Causal effects of body mass index on airflow obstruction and forced mid-expiratory flow: a Mendelian randomization study taking interactions and age-specific instruments into consideration toward a life course perspective. Front Public Health 2021; 9: 584955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Melén E, Guerra S, Hallberg J, Jarvis D, Stanojevic S. Linking COPD epidemiology with pediatric asthma care: implications for the patient and the physician. Pediatr Allergy Immunol 2019; 30: 589–97. [DOI] [PubMed] [Google Scholar]
- 60.Bui DS, Lodge CJ, Burgess JA, et al. Childhood predictors of lung function trajectories and future COPD risk: a prospective cohort study from the first to the sixth decade of life. Lancet Respir Med 2018; 6: 535–44. [DOI] [PubMed] [Google Scholar]
- 61.Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention (2020 update). 2020. https://ginasthma.org/wp-content/uploads/2020/04/GINA-2020-full-report_-final-_wms.pdf (accessed April 2, 2022).
- 62.Pavord ID, Beasley R, Agusti A, et al. After asthma: redefining airways diseases. Lancet 2018; 391: 350–400. [DOI] [PubMed] [Google Scholar]
- 63.Marcon A, Locatelli F, Dharmage SC, et al. The coexistence of asthma and COPD: risk factors, clinical history and lung function trajectories. Eur Respir J 2021; 58: 2004656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Soler Artigas M, Wain LV, Miller S, et al. Sixteen new lung function signals identified through 1000 Genomes Project reference panel imputation. Nat Commun 2015; 6: 8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ranjan A, Singh A, Walia GK, Sachdeva MP, Gupta V. Genetic underpinnings of lung function and COPD. J Genet 2019; 98: 98. [PubMed] [Google Scholar]
- 66.Liao SY, Lin X, Christiani DC. Gene-environment interaction effects on lung function- a genome-wide association study within the Framingham heart study. Environ Health 2013; 12: 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moll M, Sakornsakolpat P, Shrine N, et al. Chronic obstructive pulmonary disease and related phenotypes: polygenic risk scores in population-based and case-control cohorts. Lancet Respir Med 2020; 8: 696–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Feinberg AP. The key role of epigenetics in human disease prevention and mitigation. N Engl J Med 2018; 378: 1323–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sharp GC, Relton CL. Epigenetics and noncommunicable diseases. Epigenomics 2017; 9: 789–91. [DOI] [PubMed] [Google Scholar]
- 70.Jawaid A, Jehle KL, Mansuy IM. Impact of parental exposure on offspring health in humans. Trends Genet 2021; 37: 373–88. [DOI] [PubMed] [Google Scholar]
- 71.Ryan CP. “Epigenetic clocks”: theory and applications in human biology. Am J Hum Biol 2021; 33: e23488. [DOI] [PubMed] [Google Scholar]
- 72.Joubert BR, Felix JF, Yousefi P, et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am J Hum Genet 2016; 98: 680–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kachroo P, Morrow JD, Kho AT, et al. Co-methylation analysis in lung tissue identifies pathways for fetal origins of COPD. Eur Respir J 2020; 56: 1902347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Merid SK, Novoloaca A, Sharp GC, et al. Epigenome-wide meta-analysis of blood DNA methylation in newborns and children identifies numerous loci related to gestational age. Genome Med 2020; 12: 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Küpers LK, Monnereau C, Sharp GC, et al. Meta-analysis of epigenome-wide association studies in neonates reveals widespread differential DNA methylation associated with birthweight. Nat Commun 2019; 10: 1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hussain N Epigenetic influences that modulate infant growth, development, and disease. Antioxid Redox Signal 2012; 17: 224–36. [DOI] [PubMed] [Google Scholar]
- 77.Casas-Recasens S, Noell G, Mendoza N, et al. Lung DNA methylation in COPD: relationship with smoking status and airflow limitation severity. Am J Respir Crit Care Med 2021; 231: 129–34. [DOI] [PubMed] [Google Scholar]
- 78.Koo HK, Morrow J, Kachroo P, et al. Sex-specific associations with DNA methylation in lung tissue demonstrate smoking interactions. Epigenetics 2021; 16: 692–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sunny SK, Zhang H, Mzayek F, et al. Pre-adolescence DNA methylation is associated with lung function trajectories from pre-adolescence to adulthood. Clin Epigenetics 2021; 13: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.DeMeo DL. Sex and gender omic biomarkers in men and women with COPD: considerations for precision medicine. Chest 2021; 160: 104–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kajekar R Environmental factors and developmental outcomes in the lung. Pharmacol Ther 2007; 114: 129–45. [DOI] [PubMed] [Google Scholar]
- 82.Kohansal R, Martinez-Camblor P, Agustí A, Buist AS, Mannino DM, Soriano JB. The natural history of chronic airflow obstruction revisited: an analysis of the Framingham offspring cohort. Am J Respir Crit Care Med 2009; 180: 3–10. [DOI] [PubMed] [Google Scholar]
- 83.Melén E, Guerra S. Recent advances in understanding lung function development. F1000 Res 2017; 6: 726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bousquet J, Anto JM, Berkouk K, et al. Developmental determinants in non-communicable chronic diseases and ageing. Thorax 2015; 70: 595–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gluckman PD, Hanson MA, Beedle AS. Early life events and their consequences for later disease: a life history and evolutionary perspective. Am J Hum Biol 2007; 19: 1–19. [DOI] [PubMed] [Google Scholar]
- 86.Melén E, Koppelman GH, Guerra S. On genetics, lung developmental biology, and adult lung function. Am J Respir Crit Care Med 2020; 202: 791–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kachroo P, Morrow JD, Vyhlidal CA, et al. DNA methylation perturbations may link altered development and aging in the lung. Aging (Albany NY) 2021; 13: 1742–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morrow JD, Cho MH, Hersh CP, et al. DNA methylation profiling in human lung tissue identifies genes associated with COPD. Epigenetics 2016; 11: 730–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Agustí A, Celli B. Natural history of COPD: gaps and opportunities. ERJ Open Res 2017; 3: 00117–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Belgrave DCM, Granell R, Turner SW, et al. Lung function trajectories from pre-school age to adulthood and their associations with early life factors: a retrospective analysis of three population-based birth cohort studies. Lancet Respir Med 2018; 6: 526–34. [DOI] [PubMed] [Google Scholar]
- 91.Washko GR, Colangelo LA, Estépar RSJ, et al. Adult life-course trajectories of lung function and the development of emphysema: the CARDIA Lung Study. Am J Med 2020; 133: 222–230.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Agustí A, Noell G, Brugada J, Faner R. Lung function in early adulthood and health in later life: a transgenerational cohort analysis. Lancet Respir Med 2017; 5: 935–45. [DOI] [PubMed] [Google Scholar]
- 93.Çolak Y, Nordestgaard BG, Vestbo J, Lange P, Afzal S. Relationship between supernormal lung function and long-term risk of hospitalisations and mortality: a population-based cohort study. Eur Respir J 2021; 57: 2004055. [DOI] [PubMed] [Google Scholar]
- 94.Çolak Y, Nordestgaard BG, Lange P, Vestbo J, Afzal S. Supernormal lung function and risk of COPD: a contemporary population-based cohort study. EClinicalMedicine 2021; 37: 100974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Müller L, Di Benedetto S, Pawelec G. The immune system and its dysregulation with aging. Subcell Biochem 2019; 91: 21–43. [DOI] [PubMed] [Google Scholar]
- 96.Faner R, Cruz T, Agusti A. Immune response in chronic obstructive pulmonary disease. Expert Rev Clin Immunol 2013; 9: 821–33. [DOI] [PubMed] [Google Scholar]
- 97.Melén E Asthma genetics revisited: understanding disease mechanisms by studying ethnically diverse groups. Lancet Respir Med 2020; 8: 427–29. [DOI] [PubMed] [Google Scholar]
- 98.Hallmark B, Wegienka G, Havstad S, et al. Chromosome 17q12–21 variants are associated with multiple wheezing phenotypes in childhood. Am J Respir Crit Care Med 2021; 203: 864–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zar HJ, Nduru P, Stadler JAM, et al. Early-life respiratory syncytial virus lower respiratory tract infection in a South African birth cohort: epidemiology and effect on lung health. Lancet Glob Health 2020; 8: e1316–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maiorino E, Baek SH, Guo F, et al. Discovering the genes mediating the interactions between chronic respiratory diseases in the human interactome. Nat Commun 2020; 11: 811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Benjamin JT, Plosa EJ, Sucre JM, et al. Neutrophilic inflammation during lung development disrupts elastin assembly and predisposes adult mice to COPD. J Clin Invest 2021; 131: 139481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Desdín-Micó G, Soto-Heredero G, Aranda JF, et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 2020; 368: 1371–76. [DOI] [PubMed] [Google Scholar]
- 103.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013; 153: 1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Muñoz-Espín D, Cañamero M, Maraver A, et al. Programmed cell senescence during mammalian embryonic development. Cell 2013; 155: 1104–18. [DOI] [PubMed] [Google Scholar]
- 105.Celli BR, Agustí A. COPD: time to improve its taxonomy? ERJ Open Res 2018; 4: 00132–02017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Scadding JG. Health and disease: what can medicine do for philosophy? J Med Ethics 1988; 14: 118–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pearce JM. Disease, diagnosis or syndrome? Pract Neurol 2011; 11: 91–97. [DOI] [PubMed] [Google Scholar]
- 108.Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease (2022 report). 2022. https://goldcopd.org/wp-content/uploads/2021/12/GOLD-REPORT-2022-v1.1-22Nov2021_WMV.pdf (accessed April 2, 2022).
- 109.Han MK, Agusti A, Celli BR, et al. From GOLD 0 to pre-COPD. Am J Respir Crit Care Med 2021; 203: 414–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Martinez FJ, Agusti A, Celli BR, et al. Treatment trials in young patients with COPD and pre-COPD patients: time to move forward. Am J Respir Crit Care Med 2022; 205: 275–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Checkley W, West KP Jr, Wise RA, et al. Maternal vitamin A supplementation and lung function in offspring. N Engl J Med 2010; 362: 1784–94. [DOI] [PubMed] [Google Scholar]
- 112.McEvoy CT, Schilling D, Clay N, et al. Vitamin C supplementation for pregnant smoking women and pulmonary function in their newborn infants: a randomized clinical trial. JAMA 2014; 311: 2074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lim SS, Vos T, Flaxman AD, et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012; 380: 2224–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Agusti A, Breyer-Kohansal R, Faner R. Transitioning from infancy to adulthood: a black box full of opportunities. Eur Respir J 2021; 57: 2003997. [DOI] [PubMed] [Google Scholar]
- 115.Wang G, Kull I, Bergström A, et al. Early-life risk factors for reversible and irreversible airflow limitation in young adults: findings from the BAMSE birth cohort. Thorax 2021; 76: 503–07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Allinson JP, Hardy R, Donaldson GC, Shaheen SO, Kuh D, Wedzicha JA. The presence of chronic mucus hypersecretion across adult life in relation to chronic obstructive pulmonary disease development. Am J Respir Crit Care Med 2016; 193: 662–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Agusti A, Fabbri LM, Baraldi E, et al. Spirometry: a practical lifespan predictor of global health and chronic respiratory and non-respiratory diseases. Eur J Intern Med 2021; 89: 3–9. [DOI] [PubMed] [Google Scholar]
- 118.Guerra S, Halonen M, Vasquez MM, et al. Relation between circulating CC16 concentrations, lung function, and development of chronic obstructive pulmonary disease across the lifespan: a prospective study. Lancet Respir Med 2015; 3: 613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Faner R, Tal-Singer R, Riley JH, et al. Lessons from ECLIPSE: a review of COPD biomarkers. Thorax 2014; 69: 666–72. [DOI] [PubMed] [Google Scholar]
- 120.Vestbo J, Edwards LD, Scanlon PD, et al. Changes in forced expiratory volume in 1 second over time in COPD. N Engl J Med 2011; 365: 1184–92. [DOI] [PubMed] [Google Scholar]
- 121.Robin M, Dong P, Hermans C, Bernard A, Bersten AD, Doyle IR. Serum levels of CC16, SP-A and SP-B reflect tobacco-smoke exposure in asymptomatic subjects. Eur Respir J 2002; 20: 1152–61. [DOI] [PubMed] [Google Scholar]
- 122.Lam DC-L, Kwok H-H, Yu W-C, et al. CC16 levels correlate with cigarette smoke exposure in bronchial epithelial cells and with lung function decline in smokers. BMC Pulm Med 2018; 18: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhu L, Di PY, Wu R, Pinkerton KE, Chen Y. Repression of CC16 by cigarette smoke (CS) exposure. PLoS One 2015; 10: e0116159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Milne S, Li X, Hernandez Cordero AI, et al. Protective effect of club cell secretory protein (CC-16) on COPD risk and progression: a Mendelian randomisation study. Thorax 2020; 75: 934–43. [DOI] [PubMed] [Google Scholar]
- 125.Henckel E, James A, Konradsen JR, et al. A novel association between YKL-40, a marker of structural lung disease, and short telomere length in 10-year-old children with bronchopulmonary dysplasia. Children (Basel) 2021; 8: 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tiwari A, Li J, Kho AT, et al. COPD-associated miR-145–5p is downregulated in early-decline FEV1 trajectories in childhood asthma. J Allergy Clin Immunol 2021; 147: 2181–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Caudri D, Wijga AH, Hoekstra MO, et al. Prediction of asthma in symptomatic preschool children using exhaled nitric oxide, Rint and specific IgE. Thorax 2010; 65: 801–07. [DOI] [PubMed] [Google Scholar]
- 128.Park HY, Chang Y, Kang D, et al. Blood eosinophil counts and the development of obstructive lung disease: the Kangbuk Samsung Health Study. Eur Respir J 2021; 58: 2003823. [DOI] [PubMed] [Google Scholar]
- 129.Tan WC, Bourbeau J, Nadeau G, et al. High eosinophil counts predict decline in FEV1: results from the CanCOLD study. Eur Respir J 2021; 57: 2000838. [DOI] [PubMed] [Google Scholar]
- 130.Casas-Recasens S, Mendoza N, Lopez-Giraldo A, et al. Telomere length but not mitochondrial DNA copy number is altered in both young and old COPD. Front Med (Lausanne) 2021; 8: 761767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Holgate S, Agusti A, Strieter RM, et al. Drug development for airway diseases: looking forward. Nat Rev Drug Discov 2015; 14: 367–68. [DOI] [PubMed] [Google Scholar]
- 132.Morice AH, Celli B, Kesten S, Lystig T, Tashkin D, Decramer M. COPD in young patients: a pre-specified analysis of the four-year trial of tiotropium (UPLIFT). Respir Med 2010; 104: 1659–67. [DOI] [PubMed] [Google Scholar]
- 133.Tashkin DP, Celli B, Senn S, et al. A 4-year trial of tiotropium in chronic obstructive pulmonary disease. N Engl J Med 2008; 359: 1543–54. [DOI] [PubMed] [Google Scholar]
- 134.Chang RC, Wang H, Bedi Y, Golding MC. Preconception paternal alcohol exposure exerts sex-specific effects on offspring growth and long-term metabolic programming. Epigenetics Chromatin 2019; 12: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rappaport SM, Barupal DK, Wishart D, Vineis P, Scalbert A. The blood exposome and its role in discovering causes of disease. Environ Health Perspect 2014; 122: 769–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Agusti A, Faner R, Donaldson G, et al. Chronic Airway Diseases Early Stratification (CADSET): a new ERS Clinical Research Collaboration. Eur Respir J 2019; 53: 1900217. [DOI] [PubMed] [Google Scholar]
- 137.Agusti A, Sobradillo P, Celli B. Addressing the complexity of chronic obstructive pulmonary disease: from phenotypes and biomarkers to scale-free networks, systems biology, and P4 medicine. Am J Respir Crit Care Med 2011; 183: 1129–37. [DOI] [PubMed] [Google Scholar]
- 138.Vermeulen R, Schymanski EL, Barabási AL, Miller GW. The exposome and health: where chemistry meets biology. Science 2020; 367: 392–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Diez D, Agustí A, Wheelock CE. Network analysis in the investigation of chronic respiratory diseases. From basics to application. Am J Respir Crit Care Med 2014; 190: 981–88. [DOI] [PubMed] [Google Scholar]
- 140.Faner R, Agustí A. Network analysis: a way forward for understanding COPD multimorbidity. Eur Respir J 2015; 46: 591–92. [DOI] [PubMed] [Google Scholar]