

Abstract
Simple Summary
Conventional cancer treatments, based on chemotherapy and radiotherapy, are often effective but suffer from serious side effects and a potential risk of resistance. Dual therapies, combining DNA alkylating agents and antimicrobial peptides, are generating great interest. Within chemotherapies, a frequently used mechanism is DNA alkylation, inducing DNA damage and subsequent cell death. Antimicrobial peptides, in turn, have demonstrated their efficacy as anticancer agents due to their ability to selectively alter cancer cell membranes. In this review, our aim has been to explore the synergistic potential of these two therapeutic modalities when used together.
Abstract
Cancer remains one of the most difficult diseases to treat, requiring continuous research into innovative therapeutic strategies. Conventional treatments such as chemotherapy and radiotherapy are effective to a certain extent but often have significant side effects and carry the risk of resistance. In recent years, the concept of dual-acting therapeutics has attracted considerable attention, particularly the combination of DNA alkylating agents and antimicrobial peptides. DNA alkylation, a well-known mechanism in cancer therapy, involves the attachment of alkyl groups to DNA, leading to DNA damage and subsequent cell death. Antimicrobial peptides, on the other hand, have been shown to be effective anticancer agents due to their ability to selectively disrupt cancer cell membranes and modulate immune responses. This review aims to explore the synergistic potential of these two therapeutic modalities. It examines their mechanisms of action, current research findings, and the promise they offer to improve the efficacy and specificity of cancer treatments. By combining the cytotoxic power of DNA alkylation with the unique properties of antimicrobial peptides, dual-action therapeutics may offer a new and more effective approach to fighting cancer.
Keywords: chemotherapy, alkylating agents, DNA alkylation, cancer, DNA damage and mutations
1. Introduction
Chemotherapy uses drugs to kill cancer cells. Chemical compounds are used to selectively shrink or destroy a tumor or at least limit its growth. Compared to surgery and radiotherapy, chemotherapy has the advantage that the drugs are distributed in most tissues of the body after oral or intravenous administration and can, therefore, also destroy tumor cells that metastasize or are located in protected areas such as the brain [1].
Today, many types of chemotherapy or chemotherapeutic drugs are used to fight cancer, which can be administered as combination treatments or individually or together with other treatments. These drugs vary greatly in their chemical composition, how they are prescribed and administered, how effective they are in treating certain types of cancer, and what side effects they may have. Cancer encompasses a large group of diseases characterized by the development of abnormal cells that divide and grow uncontrollably in any part of the body [2].
The chemical compounds used in chemotherapy are cytotoxic, meaning they are designed to kill cancer cells, which have the property of multiplying faster than normal cells, but they also act on normal tissue, meaning they are able to stop the replication or growth of cancer cells and cause their death. A specific type of drug is used, with the number of treatments and the length of treatment depending on the type of cancer and the patient’s particular circumstances [3].
Cancer drugs do not all work in the same way, and it is also important to consider the side effects they can cause depending on the dose being administered. This leads to side effects that vary from patient to patient. The most common are bone marrow suppression, hair loss, fatigue, nausea, and vomiting. These side effects reflect the fact that the cells of the bone marrow, gastrointestinal tract, and hair follicles divide faster than most normal tissues [4].
Cancer treatment usually involves more than one approach, and the strategy chosen depends on the type of cancer, the presence of a biomarker, and the stage of tumor progression. Some cancer patients require only one type of treatment, while others may need several, administered at different times and in specific combinations [5].
A comparison between targeted therapy and chemotherapy shows that both use drugs to attack and destroy cancer cells. However, targeted therapy is designed to act specifically on proteins with mutations that are unique to cancer cells, reducing the potential damage to normal tissue. Targeted therapy is often used in combination with other treatments. Currently, it is not an ideal method for eliminating cancer, as it still requires the use of strong chemicals that can also cause side effects such as hair and skin problems or high blood pressure [6].
2. Types of Chemotherapy Drugs
The anticancer drugs currently in use and under development act via different mechanisms to interfere with a number of processes, such as tumor cell growth, motility, survival, or angiogenesis. Chemotherapeutic drugs can be categorized into groups according to their mode of action, chemical structure, and interactions with other drugs. Some drugs act in more than one way and may belong to more than one group.
DNA-damaging agents, such as alkylating agents, platinum derivatives, anthracyclines, and camptothecins, are some of the most commonly used drugs in chemoattractants. Knowledge of ligand–DNA interactions is a key step in the development of new drugs. It is necessary to determine their DNA sequence specificity, the mode of binding to DNA, and the kinetic, dynamic, and structural parameters of binding [7].
The drugs used to fight cancer are called antineoplastic or cytotoxic. They have different mechanisms of action based on agents that are able to mimic properties of biomolecules to disrupt growth or destroy cells and are characterized by being non-selective, i.e., they act on both cancer cells and healthy cells [2].
To classify anticancer drugs, the following characteristics can be considered: (i) chemical structure, (ii) function, and (iii) interaction with other drugs [8]. The treatments used are based on the action of the pharmacological groups shown in Figure 1.
Figure 1.

Classification of oncological drugs and mechanism of action of each drug type.
Different antineoplastic drugs can act on one or more phases of the cell cycle or on the mechanisms controlling cell proliferation. The same drug may have more than one mode of action on the tumor cell, although usually one of them predominates. The classification currently used is based on the target or site of action of the antineoplastic [9,10] and is shown in Table 1.
Table 1.
Classification of antineoplastic drugs according to the target and mechanisms of action.
| Category | Mechanism of Action | Examples |
|---|---|---|
| Alkylating Agents | Act on the entire reproduction process; most effective during the DNA synthesis phase (S). | Derivatives of Nitrogen Mustards: cyclophosphamide, chlorambucil, ifosfamide. Tetrazenes–Triazenes: dacarbazine. Derivatives of Platinum: cisplatin, carboplatin, oxaliplatin. |
| Medications that Affect the Entire Process of Reproduction | Interfere with various stages of the cell cycle and reproduction process. | Nitrosureas: carmustine (bcnu), lomustine (ccnu), estramustine. |
| Antitumor Antibiotics | Interfere with DNA duplication and alter the membrane surrounding the cells. | Anthracyclines: doxorubicin, daunorubicin, epirrubicin, idarubicin, mitoxantrone. Others: bleomycin, mitomycin c, dactinomycin. |
| Antimetabolites | Act on the S phase (DNA synthesis), interfere with DNA and RNA. | Folic Acid Analogues: methotrexate. Pyrimidine Analogues: fluorouracil, cytarabine, gemcitabine, capecitabine. Purine Analogues: fludarabine, mercaptopurine, cladribine. |
| Campotecin Derivatives | Inhibit topoisomerase I, leading to DNA damage. | Irinotecan, topotecan. |
| Mitotic Inhibitors | Act during the M phase (mitosis), inhibit or stop mitosis, or inhibit enzymes needed for cell reproduction. | Vinca Alkaloids: vincristine, vinblastine, vindesine, vinorelbine. Taxoids: paclitaxel, docetaxel. Epipodophyllotoxins: etoposide, teniposide. |
| Other Mechanisms of Action | Affect tumors with hormone dependence or stimulate the immune system to attack cancer cells. | Hormones: diethylbestrol. Antiandrogens: flutamide, nilutamide, cyproterone. Antiestrogens: tamoxifen, anastrazole. Gonadotrophin Agonists: leuprolide, goserelin, buserelin, triptorelin. Progestagens: megestrol, medroxyprogesterone. |
| Immunotherapy | Stimulates the immune system’s natural defenses to destroy abnormal cells. | Interferons: interferon alpha 2a, alpha 2b. Monoclonal Antibodies: rituximab, cetuximab, trastuzumab. |
Within the antineoplastic drugs, alkylating agents are commonly used. These treatments are able to intervene in one or more phases of the cell cycle or in the mechanisms that control cell proliferation, and at least two drugs are used to act on different therapeutic targets and avoid the emergence of resistance.
Unfortunately, due to the lack of specificity of cytotoxic treatments, there are a variety of adverse effects, the most common of which are rapid death of healthy cells, bone marrow depression, anemia, nausea, vomiting, alopecia, impaired wound healing, growth failure in children, necrosis, sterility, and possible long-term carcinogenesis [11].
Anthracycline derivatives such as doxorubicin (DOX) are another of the most widely used compounds and are the main component of cancer treatment regimens currently used in clinical practice. However, the precise mechanisms of their action are not fully understood [12].
The anticancer activity of DOX may be attributed to the drug’s ability to intercalate into DNA, inhibit topoisomerase II, alter mitochondrial function, and enhance free radical generation and oxidative damage. The precise mechanisms of action of DOX are complex and still relatively unknown [13,14,15].
Like other anthracyclines, DOX intercalates DNA through the formation of hydrogen bonds with guanines in adjacent GC base pairs [16,17].
DOX induces DNA damage through three main mechanisms: DNA adduct formation, single-strand break (SSB) induction, and double-strand break (DSB) induction, in which DNA strands remain attached to trapped topoisomerase enzymes through DNA–protein cross-linking (DPC) and DOX intercalation into the DNA molecule. One possible mechanism postulated to explain the effect of DOX in malignant cells is that DOX intercalation into DNA unwinds the molecule and results in positive supercoiling of the DNA helix [18,19]. There is also evidence that DOX increases the turnover of nucleosomes surrounding active gene promoters due to its intercalation and induces changes in DNA topology [20]. It is possible that the DNA unwinding that occurs as a result of DOX intercalation provides a significant amount of positive torsional stress that destabilizes nucleosomes [21].
Topoisomerase II inhibitors are among the most effective antitumor drugs used in the treatment of human tumors [22].
Lemke K. et al., 2005, have described the antitumor drug 5-dimethylaminopropylamino-8-hydroxytriazoloacridinone, C-1305, which belongs to the group of acridine derivatives, binds to DNA by intercalation, and is an inhibitor of DNA topoisomerase II [23]. C-1305 induces structural perturbations in regions of DNA with three adjacent guanine residues. C-1305 binds to DNA by intercalation and induces unusual structural changes in regions of DNA with guanine triplets.
A study of direct C-1305-mediated microtubule stabilization as the central mediator leading to apoptosis was performed in 2020. Furthermore, C-1305 was found to promote G2 cell cycle arrest by modulating gene expression, indicating that C-1305 is the first microtubule-stabilizing agent identified together with topoisomerase II inhibitory activity [24].
3. Binding of a Ligand to DNA
The binding of a ligand to DNA can be classified into covalent and non-covalent binding (Figure 2).
Figure 2.

Modes of ligand binding to DNA. DNA can bind to small molecules or drugs through covalent or non-covalent interactions. Covalent binding in DNA can be irreversible and can lead to inhibition of all DNA processes that subsequently lead to cell death. Covalent interactions lead to permanent changes in the structure of nucleic acids. Non-covalent interaction of molecules with DNA can be due to electrostatic interaction, intercalation, and groove binding.
3.1. Non-Covalent Bond
Non-covalent bonds are hydrogen bonds, electrostatic interactions, or non-covalent bonds with ethidium bromide and quinacrine. Non-covalent binding can occur in different ways, e.g., groove binding and intercalation. Agents that interact with non-covalent DNA (groove, intercalators, and external binders) are generally considered less cytotoxic than agents that produce covalent DNA adducts and other types of DNA damage. The non-covalent mode of binding is reversible and is generally preferred over covalent adduct formation in terms of drug metabolism and toxic side effects. In addition, drugs that interact with non-covalent DNA can alter DNA conformation, torsionally stress DNA, disrupt protein–DNA interactions, and potentially lead to DNA strand breaks [25].
3.2. Covalent Bonding
The first cancer chemotherapeutics were those that formed a covalent bond by alkylating the nitrogenous bases of DNA. This binding is irreversible and leads to a complete inhibition of DNA processes and subsequent cell death [26].
The alkylation of adenine and guanine at the nitrogen atom and of guanine at the oxygen atom can easily be caused by alkylating chemicals. If these errors are not corrected by DNA repair processes, cell mutations can occur. In medicine, DNA alkylation is used beneficially in the treatment of cancer. Alkylating chemicals impair DNA replication and can, therefore, cause cell death. This effect is particularly pronounced in rapidly dividing cells, such as cancer cells [27].
4. DNA Alkylating Agents
Alkylating agents prevent the proliferation of cells by damaging their DNA. These drugs work at all stages of the cell cycle and are used to treat many different types of cancer, including lung, breast, and ovarian cancer, as well as leukemia, lymphoma, Hodgkin’s disease, multiple myeloma, and sarcoma.
As these drugs damage DNA, they can affect the bone marrow cells that form new blood cells. The risk of leukemia from alkylating agents is dose-dependent, i.e., the risk is lower at lower doses but increases as the total amount of the drug is increased. The risk of leukemia after administration of alkylating agents is highest about 5 to 10 years after treatment.
The characteristic toxicities of alkylating agents are (i) hematopoietic toxicity, (ii) gastrointestinal toxicity, and (iii) gonadal and central nervous system (CNS) toxicity. However, each of these agents has a characteristic set of toxicities that are determined by the reactivity, metabolism, and distribution of the drug.
Despite their toxicity, DNA alkylating drugs remain the cornerstone of cancer therapy. DNA-targeting alkylating agents were among the first anticancer drugs and are still the most commonly used in chemotherapy today. Alkylating agents used as anticancer drugs are able to react with DNA and proteins and disrupt cell function at several stages of the cell cycle, either killing the cell or preventing it from growing. The National Cancer Institute defines an alkylating agent as a type of drug that is used to treat cancer by interfering with the cell’s deoxyribonucleic acid (DNA) and inhibiting cancer growth. Although alkylating agents can be used to treat most types of cancer, they are usually most useful in treating slow-growing cancers [28]. Alkylating agents are used to treat the types of cancer shown in Figure 3.
Figure 3.
Types of cancers treated with alkylating agents.
Based on the number of alkyl groups in their molecule, alkylating agents are classified as monofunctional, bifunctional, and polyfunctional; the last two are the most effective antitumor agents. Alkylating compounds cause mono-alkylation or can intercalate into DNA. In addition, bis-alkylating compounds can form bridges within a single DNA strand or between two complementary DNA strands (Figure 4), as well as cross-links between DNA and associated proteins [29].
Figure 4.

Different types of links produced on DNA by bis-alkylating agents.
The structure and dynamics of DNA are strongly affected by the alkylation of its bases, leading to different types of effects: (i) prevention of DNA replication, (II) DNA fragmentation, (iii) induces nucleotide mismatches by disrupting the normal hydrogen bonds between bases, (iv) bis-alkylation causes intra-catenary cross-linking, (v) cross-linking between two complementary DNA strands preventing their separation during DNA replication or transcription, (vi) cross-linking between DNA and associated proteins. Bifunctional alkylating compounds are more cytotoxic than monofunctional ones. Many anticancer drugs interact with DNA by intercalation, whereby compounds containing aromatic or heteroaromatic rings are inserted between neighboring base pairs perpendicular to the helix axis without altering the overall stacking pattern due to Watson–Crick hydrogen bonding [30,31].
Based on the chemical structure of the reactive groups in antineoplastic drugs, they are categorized into the following groups: nitrogen mustards, nitrosoureas, alkyl sulphonates, triazines, and ethylenimines, and this group of covalent bonds also includes platinum compounds (Figure 5). Alkylating chemotherapies have been used for years and can be effective against many types of cancer. However, because they attack all cells, the side effects can be severe. The goal in using these older alkylating neoplastic agents is to carefully titrate the dose to ensure that the cancer cells are attacked the most while minimizing damage to non-cancerous cells.
Figure 5.

Classification of antineoplastics–alkylating agents according to the reactive groups. They may act as monofunctional agents or may form bifunctional derivatives by forming cross-links (inter- or intra-chain) in DNA or between DNA and proteins.
An alkylating agent is defined as a compound capable of covalently bonding an alkyl group to a biomolecule under physiological conditions (aqueous solution, 37 °C, pH 7.4); DNA alkylating agents interact with resting and proliferating cells at any stage of the cell cycle but are most cytotoxic in late G1 and S phase, as there is insufficient time to repair DNA damage. Alkylating agents, especially nitrogen-based alkylating agents, are commonly used in the treatment of hematologic and solid malignancies. They exert their antineoplastic effect in all phases of the cell cycle and prevent tumor cells from proliferating.
All nitrogen and oxygen atoms of these bases are nucleophilic, with the exception of the nitrogen atoms involved in nucleoside binding (N9 or N1 in purines or pyrimidines), therapeutically useful drugs always behaving as carbon electrophiles. Alkylating agents are electrophilic and bind covalently to electron-rich functional groups, such as the N-7, N-1, and O-6 positions of guanine [32]. Other nucleobases that can be alkylated and the atomic positions where alkylation preferentially occurs are the N-1, N-3, and N-7 positions of adenine, the N-3 position of cytosine, and the O-4 position of thymidine (Figure 6).
Figure 6.

Arrows indicate DNA base alkylation sites.
Nitrogen-based alkylating agents, which are frequently used to treat hematological and solid malignancies, often in combination with other chemotherapeutic agents, exert their antineoplastic effect at all stages of the cell cycle and prevent the replication of tumor cells. During this alkylation process, an alkyl residue is introduced into the DNA bases, a change that leads to fragmentation of the DNA by DNA repair enzymes that attempt to replace the alkylated base [33].
5. Nitrogen Mustards
One of the largest and most diverse groups of drugs are those that affect the structure and function of DNA. Part of the reason that DNA-interfering drugs are the largest group of agents in clinical use today is that nitrogen mustard was the first type of chemotherapeutic agent to be discovered. They have been used for more than 60 years [34]. Nitrogen mustard acts on the kidneys, heart, bladder, central nervous system, and gonads.
After ingestion, the alkylating agent is metabolized to a highly reactive aziridinium derivative that alkylates DNA and inhibits DNA reduplication. There are several generations of nitrogen-based alkylating drugs. The mustard compounds generated from the nitro group contain the bis(2-chloroethyl) group, which is the major chemical constituent, and all have a common mechanism of action, namely the in situ generation of a highly electrophilic aziridinium cation (Figure 7) by an intramolecular nucleophilic substitution reaction [35].
Figure 7.

In situ generation of an aziridinium cation.
5.1. Aliphatic Mustard
Mechlorethamine (Figure 8) was the first antitumor agent developed during the First World War as part of the mustard gas study. Due to its short half-life, it must be prepared and administered immediately. It can only be administered intravenously. At physiological pH, mechlorethamine is converted via the free base into a relatively stable aziridinium ion, which in turn reacts with available nucleophilic centers. Mechlorethamine has been replaced by new derivatives to improve selectivity and specificity in the fight against cancer cells [36].
Figure 8.
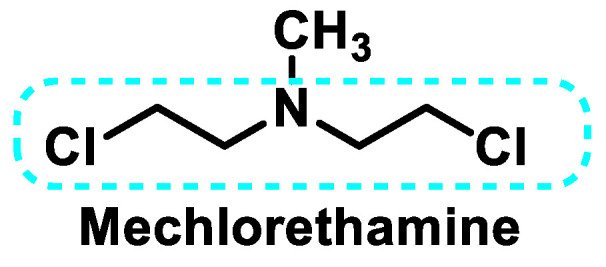
Mechlorethamine structure.
5.2. Phosphamide Nitrogen Mustard
The oxazaphosphorins have complex pharmacokinetics, and metabolism is crucial for their cytotoxic activity. There are several classes, such as cyclophosphamide, iphosphamide, maphosphamide, trophosphamide, bromophosphamide, and gluphosphamide (Figure 9).
Figure 9.

Phosphamide nitrogen mustard.
Cyclophosphamide is the most commonly used alkylating agent. It is a prodrug that is converted to its active form after metabolism by cytochrome P-450. Hydroxylation of the carbon at position 4 is the first metabolic reaction of cyclophosphamide and iphosphamide. This reaction occurs in the liver microsomes via the enzyme systems of the 2A6, 2B6, 2C, 3A4, and 3A5 subfamilies of the cytochrome P450 (CYP) enzyme complex. The main metabolite of this reaction is 4-hydroxycyclophosphamide or 4-hydroxyphosphamide, which is in tautomeric equilibrium with its keto form, aldophosphamide. From aldophosphamide, a β-elimination reaction produces phosphoramide mustard with an alkylating effect and acrolein, a urotoxic compound responsible for hemorrhagic cystitis [37] (Figure 10).
Figure 10.

Biotransformation mechanism of cyclophosphamide.
Its main toxic effects are myelosuppression for 10–14 days, hair loss, nausea, and vomiting. It can also cause hemorrhagic cystitis due to the effect of some of its metabolites, such as acrolein, on the bladder epithelium; high fluid intake for 24 to 48 h and Sodium-2-mercapto-sulphonate (mesna) to chelate acrolein and avoid this complication. It is a drug widely used in oncology and is part of polychemotherapy regimens, as its efficacy has been demonstrated in various neoplasms (leukemias, lymphomas, breast cancer, ovarian cancer, and sarcomas). It is also part of the main induction regimens prior to bone marrow transplantation.
Isophosphamide is a drug similar to cyclophosphamide but requires higher doses to achieve the same antitumor effect. It is only administered intravenously, always with sufficient fluid intake and mesna as a prophylactic measure [38]. Iphosphamide is a prodrug that must be converted into a cytotoxic metabolite (4-hydroxy derivatives) by the action of CYP3A4 isoforms [39]. Iphosphamide generates seven-atom DNA cross-links (compared to five-atom cross-links for cyclophosphamide).
Acrolein, a metabolite of cyclophosphamide and iphosphamide, is a highly toxic aldehyde. As a bifunctional electrophile, acrolein reacts with protein nucleophiles and DNA via Michael addition and Schiff base formation [40] (Figure 11).
Figure 11.

Mechanism of acrolein toxicity.
Maphosphamide is a cyclophosphamide analog, an alkylating agent, and an immunomodulator that induces a pro-inflammatory response and increased endothelial cell permeability in vitro [41]. Maphosphamide breaks down spontaneously into 4-OH-CP and mesna (Figure 12). The released 4-OH-CP is in equilibrium with its tautomer aldophosphamide, which can be degraded by β-elimination to generate cytotoxic phosphoramide mustard and acrolein. Thus, maphosphamide is directly active in cells and intracellular fluids. Although the drug is similar to CP, it does not need to be activated by the hepatic enzyme system [42].
Figure 12.

Spontaneous decomposition of Maphosphamide into 4-HO-CP and mesna.
Maphosphamide has significant in vitro activity against leukemia and solid tumor cell lines such as U-937, ML-1, MOLT-4, rhabdomyosarcoma, and MCF-7, and also in vivo against several transplantable mouse tumors, including P388 and L1210 leukemia, B16 melanoma, Lewis lung carcinoma, colon tumor 38, and cyclophosphamide-resistant P388 leukemia.
Trophosphamide was approved as one of the anticancer drugs in 1973. Like phosphamide and cyclophosphamide, they are non-specific cell cycle alkylating agents that are bioactivated by hepatic cytochrome P450 enzymes through either 4-hydroxylation or N-dechloroalkylation; both are NADPH-dependent [43] (Figure 13).
Figure 13.

Activation of trophosphamide by cytochrome P450 into oxazaphosphorine mustards. N-dealkylation by cytochrome P450s results in the formation of inactive metabolites and chloracetaldehyde.
Bromophosphamide, an analog of iphosphamide with a bromine substituent, causes less urotoxicity. It acts by cross-linking DNA strands or deoxyribose–protein cross-links and alkali-labile sites in a concentration-dependent manner in HeLa cells [44].
(S)-(-)-bromophosphamide (Figure 14) is an alkylating anticancer drug introduced into clinical practice in the 1980s and used to treat soft tissue sarcomas and a variety of pediatric tumors [45,46].
Figure 14.

Chemical structure of (S)-(-)-Bromophosphamide Gluphosphamide (β-D-glucoseisophosphoramide-mustard).
Gluphosphamide, also known as glucophosphamide (β-D-glucose isophosphoramide mustard), is a glycosidic conjugate between β-D-glucose and the active alkylating moiety of the antineoplastic drug iphosphamide. In gluphosphamide, the active part of iphosphamide is linked to a glucose molecule. Inside the cells, the bond between the glucose and the alkylating agent must be broken in order to release the active substance. Phase III trials are currently underway with this drug, comparing gluphosphamide with 5-FU, both in combination with gemcitabine, for the second-line treatment of metastatic pancreatic cancer [47].
5.3. Aromatic Nitrogen Mustards
The main aromatic nitrogen mustards are shown in Figure 15.
Figure 15.

Aromatic nitrogen mustards.
Melphalan is a phenylalanine derivative of nitrogen mustard and was approved by the FDA in 1964 [48]. It can be administered orally or intravenously. Melphalan is 90% bound to plasma proteins and is actively transported into cells primarily via the L-type amino acid transporter system. The oral bioavailability is 50–80%, and the half-life can be up to 8 h. It is partially excreted in the feces and partially in the urine. Melphalan was developed for the treatment of melanoma because it selectively targets tumor cells that actively use tyrosine. However, it has shown little effect in this neoplasm. Currently, its main indication is multiple myeloma. It has been shown to be effective at conventional doses in ovarian cancer and lymphoma and at high doses in breast cancer and acute myeloid leukemia.
Chlorambucil was approved by the FDA in 1957 [48]. It is stable in aqueous solution so that it is almost completely absorbed after administration and is rapidly and completely excreted from the blood. It is metabolized in the liver and has the alkylating metabolite PAAM (phenylacetic acid mustard). Adverse effects are uncommon, with the exception of bone marrow suppression. However, it can cause a severe generalized rash, which may develop into Stevens–Johnson syndrome or toxic epidermal necrolysis. In the event of a rash, further treatment with chlorambucil is contraindicated. This drug is indicated for the palliative treatment of chronic lymphocytic leukemia, malignant lymphoma, lymphosarcoma, follicular giant lymphoma, and Hodgkin’s disease [49].
Bendamustine: N-methylbenzimidazole replaced the benzene ring of chlorambucil to form bendamustine. The drug was approved by the FDA in 2008, although it was discovered in 1960. The incorporation of the benzimidazole ring gives the molecule the properties of a purine analog [50].
5.4. Steroid-Coupled Nitrogen Mustards
Alkylating agents have high toxicity and low specificity for alkylating agents, but when coupled with a steroid, they are more effective in improving the therapeutic profile. Steroid-coupled nitrogen mustards utilize the action of both the alkylating agent and a steroid for selective uptake or a combined anticancer effect. The steroid acts as a biological carrier, allowing the nitrogen mustard component to be easily transported and increasing the uptake of the mustard through the lipid bilayer of cells [51]. The result is a more effective, more selective, and less toxic antineoplastic treatment. Saha et al., 2018, have shown that steroid-bound mustard enhances the damaging effect on a specific DNA sequence and achieves better selectivity and lower toxicity compared to nitrogen mustard itself [51,52]. Some of the most important drugs in this class are listed below:
Estramustine phosphate sodium: Estramustine has a β-estradiol moiety attached to a nitrogen mustard moiety (mechlorethamine) via a carbamate bridge. Chemically, it is estra-1,3,5(10)-triene-3,17β-diol-3-bis(2-chloroethyl)-carbamate. Due to the stability of the carbamate bridge, which is not cleaved to release the nitrogen mustard, the compound has no alkylating activity.
In 1986, estramustine sodium phosphate was synthesized by condensation of estradiol with bis(2-chloroethyl) carbamyl chloride, followed by phosphatidylation with phosphorus oxychloride and finally treatment with NaOH in EtOH [53] (Figure 16).
Figure 16.

Chemical synthesis of Estramustine phosphate sodium.
The compound is dephosphorylated during absorption and broken down into estradiol, estrone, and estramustine as the main metabolites in the blood. The estradiol component increases the uptake of the alkylating agent into the target cells. Estramustine phosphate is a prodrug marketed for the treatment of advanced prostate cancer.
Prednimustine: The drug was produced from prednisolone ester (corticosteroid) and chlorambucil (Figure 17). It has both alkylating and corticosteroid properties. Acute leukemia, breast cancer, and lymphocytic leukemia are diseases for which this drug is commonly used [54,55].
Figure 17.

Chemical synthesis of Prednimustine.
Homoazasteroid (lactam steroids): This type of drug is formed by attaching a lactam residue to the esterified form of the alkylating steroid drug. They are effective in various neoplastic tumors and leukemias both in vivo and in vitro. Lactandrate and lactestoxate (Figure 18) showed excellent anticancer results in colon carcinomas. However, the latter showed better results than lactandrate [56]. In 2016, Trafalis et al. reported positive results with four modified forms of lactamsteroids [57].
Figure 18.

Chemical structure of Lactandrate and Lactestoxate.
5.5. Peptide-Coupled Nitrogen Mustards
Melphalan flufenamide, also known as melphalan flufenamide, is a third-generation drug consisting of the ethyl ester of melphalan and para-fluoro-L-phenylalanine (Figure 19).
Figure 19.

Chemical structure of Melflufen.
Being highly lipophilic, it is readily taken up by rare cells (such as myeloma cells) across the membrane barrier, leading to activation by hydrolytic cleavage of the peptide bond using peptidases and esterases overexpressed in multiple myeloma cells to release a toxic cargo into the cells, damaging the DNA and killing the cancer cells [58]. The alkylating component produced during hydrolysis is melphalan, which enters the cell nucleus and causes DNA alteration (Figure 20).
Figure 20.

Metabolism of Melflufen hydrochloride.
It has a high affinity for solid tumors and multiple myeloma, and phase I/II trials have led to its successful use over time [51]. The drug is effective in third-line treatment in patients who have not previously received alkylating chemotherapeutic agents and in patients who have received melphalan in previous treatments and who are also unsuitable for transplantation [59].
In comparison between melphalan and melflufen, melflufen has a superior cytotoxic effect, higher efficacy at a lower dose, and is effective in treating multiple myeloma cells in which melphalan was not actively involved [60].
Melflufen received accelerated approval in the HORIZON trial on 26 February 2021 [61]. In March 2021, the FDA approved melflufen in combination with dexamethasone for the treatment of adult patients with multiple myeloma. In October 2021, it was withdrawn from the market in the USA after too many deaths occurred in a confirmatory phase 3 study [61].
5.6. Mode of Action of Nitrogen Mustards
Nitrogen mustard reacts with DNA nucleobases preferentially at the N-7 position of guanine. Their alkylation leads to important changes in the chemical properties of guanine. Since a positive charge is generated on the imidazole ring, the formation of the enolic tautomer is favored, which leads to guanine preferentially pairing with thymine and not with cytosine. In purification processes, hydrolysis leads to local destabilization of the DNA, which would favor strand cleavage. Another consequence is the opening of the imidazole ring, which would also trigger purification (Figure 21). The mechanism of action involves the formation of DNA cross-links, which leads to an inhibition of DNA synthesis and function.
Figure 21.

Action of alkylating agents on guanine.
The reason for the high reactivity of nitrogen mustard as an alkylating agent under mild conditions is the formation of an aziridinium cation, which acts as an alkylating agent and is formed by intramolecular nucleophilic substitution. The aziridinium cation is highly reactive due to the positive charge of the N and the tension of the three-membered ring. Since nitrogen mustards are bifunctional alkylating agents, one of their cytotoxicity mechanisms is related to the ability of monoalkylated DNA species to form a covalent intercatalytic DNA strand and intracatalytic cross-links or DNA–protein complexes that lead to disruption of replication or transcription (Figure 22).
Figure 22.

(A) DNA monoalkylation and dialkylation mechanism. (B) DNA–protein complexes.
During alkylation, the normal pairing of the DNA bases between adenine–thymine and guanine–cytosine (Watson–Crick base pairs) is altered. The three hydrogen bonds connecting guanine and cytosine, for example, require the existence of a carbonyl at the C-6 position of the purine derivative. Alkylation at the N-7 position creates a positive charge in this center adjacent to the partial positive charge at C-6, which means that the tautomeric equilibrium shifts to the more stable 6-hydroxy form [62]. This change in the tautomeric form turns the hydrogen bond acceptor groups into hydrogen bond donors and vice versa. This leads to a weakening of the hydrogen bond with cytosine, as only two bonds can be formed, whereas the pairing of the 6-hydroxy species with thymine leads to a more stable complex (three hydrogen bonds). The normal pairing is changed to guanine–non-thymine, which leads to mutations (Figure 23).
Figure 23.

Hydrogen bonding interactions in guanine–cytosine and guanine–thymine pairs.
Another consequence of guanine alkylation is an increase in the electrophilicity of the positions adjacent to or conjugated with the positive charge on N-7, which leads to various hydrolytic reactions that alter the DNA structure. This increased electrophilicity allows cleavage of the heteroside bond (Figure 24) and leads to purification of the DNA.
Figure 24.

Cleavage of the heteroside bond.
When attacked by water, a cyclic hemiketal forms in equilibrium with the open form, which has a good leaving group (oxygen phosphate) at the β-position with respect to the carbonyl group, facilitating its elimination and leading to DNA fragmentation.
C-8 of the guanine ring becomes more electrophilic as it is close to the positive charge generated in the N-7 alkylation reaction. The addition of water to C-8 provides the hydroxylated intermediate, which opens the purine ring, which then evolves into the imine that develops after hydrolysis to cleave the DNA strand [63] (Figure 25).
Figure 25.

DNA fragmentation that takes place following guanine monoalkylation.
6. Aziridines
Several aziridine derivatives have been described as antitumor agents [64]. It has been found that two aziridine units are required for these compounds to show good activity. It was also found that the presence of a third or fourth aziridine is not required, suggesting that the cytotoxicity is due to a cross-linking mechanism, as observed with nitrogen mustard. Aziridine alkylating agents are characterized by an aziridine ring that is structurally similar to the iminium found in nitrogen mustard. Since they have no charge on the nitrogen, they are less reactive than the nitrogen mustards. It should be noted that the reactivity of the aziridine group increases with protonation and is, therefore, enhanced at low pH. It is assumed that the ring opening of the aziridine ring is responsible for the alkylating activity of the molecule. The mechanism of action of these drugs is less understood. The first compounds of this family to be used are shown in Figure 26.
Figure 26.

The first compounds derived from aziridines introduced into therapeutics.
Thiotepa (tris(1-aziridinyl) phosphine sulfide) is an alkylating agent approved for the treatment of breast cancer, ovarian cancer, and bladder cancer [65,66,67]. Thiotepa is oxidatively desulfurized by hepatic cytochrome P450 to produce TEPA, a less cytotoxic form of the molecule [68,69]. Both compounds have been found in the plasma of patients undergoing this treatment. Thiotepa is currently still used for the treatment of bladder carcinomas. Position 7 of the guanine is the preferred alkylation site of thiotepa (Figure 27).
Figure 27.

Monoalkylation of guanine by thiotepa.
In vivo and in vitro studies show that DNA alkylation by ThioTEPA can occur via two pathways, but it is not yet clear which pathway is the exact mechanism of action [70] (Figure 28).
Figure 28.

(A) alkylation and cross-linking by sequential reaction of a single aziridine group. (B) Cross-linking produced by sequential alkylating reactions of two aziridine groups of thiotepa.
6.1. Aziridines Linked to a Benzoquinone System
Alkylating agents containing a quinone moiety require reduction of the quinone structure to activate their alkylating substituents, which are used in the clinic for cancer treatment [71,72].
6.1.1. Natural Aziridinyylbenzoquinone
Mitomycin C (mutamycin) [73] is a natural antitumor quinone from Streptomyces caespitosus [74] that contains a quinone unit and an aziridine (Figure 29). It has been used since the 1960s for the treatment of breast cancer and tumors of the gastrointestinal tract and is considered a cytotoxic agent [75]. Mitomycin C was approved by the FDA in 2020 for adult patients with low-grade urothelial carcinoma of the upper tract.
Figure 29.

Chemical structure of mitomycins.
Porfyromycin is the N-methyl derivative of mitomycin C and is also a natural product. Porfyromycin has been tested in phase III clinical trials for the treatment of head and neck cancer in combination with radiotherapy with acceptable toxicity and stimulating effect [76].
Mitomycin C and porfyromycin can be considered as prototypes of reductively activated alkylating agents leading to cytotoxic species [77]. Figure 30 includes two reduction steps with one electron transfer to semiquinone and subsequently to hydroquinone. Both species can initiate the reaction cascade leading to DNA alkylation, but the available evidence points to hydroquinone as the active species [78]. The next step would be the formation of the imine derivative by methanol elimination of the hydroquinone. The next step is the formation of an indole derivative, protonation of the aziridine nitrogen of this derivative, and subsequent elimination with simultaneous opening of the aziridine ring yielding methylquinone methylide. A Michael reaction with nucleophilic DNA groups takes place on this intermediate product, in which the N-2 amino group of the guanine or the N-7 position is involved [79]. With mitomycin, removal of the carbamate group generates an electrophilic iminium, which undergoes a second alkylation by attacking a 2-amino group of guanine, leading to DNA cross-linking [80,81] (Figure 30).
Figure 30.

Bioreductive alkylation of DNA by mitomycin C.
Both inter- and intra-strand cross-linking by mitomycin C have been observed, with the former predominating.
6.1.2. Synthetic Aziridinyylbenzoquinone
Antitumor compounds with two or three aziridine rings linked to a benzoquinone system can act as DNA bisalkylators and cross-linking agents and cross the blood–brain barrier due to their high ionization and reductively activated lipophilicity (Figure 31).
Figure 31.

Chemical structure of Aziridinylquinones.
Diaziquone (AZQ) Diaziquone (AZQ) [82] was granted orphan drug status by the FDA in the early 1980s but showed no improvement over existing drugs. AZQ is a synthetic, lipophilic aziridine that can cross cell membranes and the blood–brain barrier. This drug has been used in CNS cancer and has shown therapeutic effect in these tumors. AZQ has also been used in the treatment of other solid tumors and leukemia.
Diazichon (AZQ) and carboquinone (carbazilquinone) have been tested in the treatment of various cancers but show low efficacy and toxicity. Triazichon (Oncovedex) was used clinically for the treatment of cancer in 1960. It is highly toxic to the walls of blood vessels and bone marrow, so it was replaced by other, more important and effective agents [83].
Apaziquone (EO9), a bioreductive drug with only one azidirine moiety in its structure, showed no activity in phase II clinical trials when administered intravenously, but in subsequent studies, it proved to be active and well tolerated in patients with superficial transitional cell carcinoma of the bladder after intravesical administration [84]. Approval was denied in 2016 after poor results in phase III clinical trials.
Aziridinylquinone BZQ was also the subject of several clinical trials in humans [72]. Mitomycin C and diaziquone (AZQ) are currently used in chemotherapy.
6.1.3. Aziridines β-D-Galactopyranosides
Calderón-Montaño et al., 2021, investigated various β-D-galactopyranoside-aziridine derivatives as anticancer agents. The best performing was 2-methyl-2,3-[N-(4-methylbenzenesulphonyl)imino] propyl 2,3-di-O-benzyl-4,6-O-(S)-benzylidene-β-D-galactopyranoside (AzGalp) (Figure 32), which has been shown to induce DNA damage [64].
Figure 32.

Chemical structure of AzGalp.
6.2. Mechanism of DNA Alkylation by Aziridinylbenzoquinones
The mechanism of DNA alkylation by aziridinylbenzoquinone derivatives involves bioreductive processes [64,85,86,87] due to the action of one- or two-electron reductases. Aziridinylbenzoquinones enter the cell in an inactive, oxidized form and are reduced to hydroquinone by enzymes in the cell, increasing the basicity of the aziridine groups and enhancing protonation until they become cytotoxic (Figure 33). This DNA damage increases when electron-donating groups are present on the benzoquinone ring, while the presence of electron-withdrawing functions or bulky groups at the C-6 position results in lower potency or inactive compounds [88].
Figure 33.

Reduction mechanism and structure of cross-links in DNA by aziridinylbenzoquinone derivatives.
One of these enzymes is DT-diaphorase (DTD, NQO1), a two-electron reductase that is located in the cell nucleus and whose level is elevated in many tumors [89]. The reduction is mainly due to the hypoxic conditions that often prevail in tumor cells [90]. Research is mainly aimed at finding new drugs that target this enzyme and have lower toxicity in normal tissue.
6.3. Inactivation of Aziridinylbenzoquinones
In addition to alkylation, other reactions can also occur with aziridinylquinone derivatives. Other possible transformations that lead to the inactivation of these derivatives are shown in Figure 34—(A) Formation of quinone derivatives by loss of aziridine; (B) 1,5-sigmatropic hydrogen shift [91], which is then converted to ethylaminoquinone by tautomerism or to aminoquinone by a second 1,5-sigmatropic shift, followed by hydrolysis of the imine derivative in which acetaldehyde is lost; (C) formation of a quinone derivative by loss of aziridine; (D) formation of a quinone derivative by loss of aziridine, which then undergoes a second 1,5-sigmatropic hydrogen shift, followed by hydrolysis of the imine derivative, which undergoes loss of acetaldehyde.
Figure 34.

Inactivation of aziridinium benzoquinones (A) with loss of the aziridine ring and (B) initiated by a 1,5-sigmatropic shift.
The metabolic one-electron reduction of aziridinylquinones yields semiquinones. Their protonated derivatives undergo a 1,5-sigmatropic shift leading to inactive ethylaminoquinones and aminoquinones (Figure 35), as in the two-electron reduction [92].
Figure 35.

Inactivation of hydroquinone forms of aziridinylbenzoquinones by one-electron reduction.
7. Epoxides
The high reactivity of the epoxide ring towards nucleophilic groups in biomolecules is the basis for the use of ethylene oxide in side chains of compounds intended for the alkylation of DNA.
Treosulphan and mitobronitol are two prodrugs that undergo intramolecular double nucleophilic displacement to yield the diepoxide derivative. This compound is able to crosslink DNA by alkylating the guanine bases of DNA [93] (Figure 36).
Figure 36.

(A) DNA alkylation by mitobronitol and (B) DNA alkylation by treosulphan.
Treosulphan is mainly used for the treatment of ovarian cancer. In addition, it has shown preclinical and clinical activity in some other solid tumors and hematological malignancies and is used for bone marrow ablation prior to stem cell transplantation and for the treatment of malignant melanoma and breast cancer. Mitobronitol is the biomolecular analog of mannitol and is used for myelosuppression prior to allogeneic bone marrow transplantation in accelerated chronic granulocytic leukemia [94].
8. Nitrosoureas
Antineoplastic agents derived from nitrosoureas are alkylating agents. The most important property is their ability to reach the brain, and they are used specifically to treat brain tumors [95]. All other alkylating agents lack this property and cannot reach the brain. Nitrosoureas can penetrate the brain because they are able to cross the so-called blood–brain barrier, a special area that prevents most drugs from reaching the brain. Nitrosourea derivatives such as carmustine and lomustine are effective in brain tumors, Hodgkin’s disease, and other lymphomas. Side effects of nitrosourea derivatives include nausea, vomiting, phlebitis, and suppression of hematopoiesis. Some of the nitrosourea-derived drugs that can treat certain brain tumors are shown in Figure 37.
Figure 37.

Chemical structure of the main nitrosoureas.
1-Methyl-1-nitrosourea, the most important compound of the nitrosourea group, was found to be effective against intracerebrally implanted murine leukemia. However, it was found that the introduction of a 2-chloroethyl chain at the nitrogen-containing nitroso group led to a significant increase in the activity of the nitrosoureas. These 2-chloroethyl derivatives are lipophilic and can cross the blood–brain barrier and enable the treatment of brain tumors. In view of this activity, the nitrosoureas lomustine (CCNU), semustine, carmustine (BCNU), nimustine (ACNU), tauromustine, and fotemustine were synthesized, all of which are water-soluble but have significant toxicity problems.
Carmustine was tested in clinical trials in 1964 and approved by the FDA in 1977 for the treatment of various types of brain tumors (including glioma, glioblastoma multiforme, medulloblastoma, and astrocytoma), multiple myeloma, and lymphomas (Hodgkin’s and non-Hodgkin’s lymphoma).
A new formulation of carmustine with reduced systemic toxicity has been developed for the local treatment of brain tumors. The formulation, in the form of a slow-release “wafer” (Gliadel Wafer®; Polifeprosan 20 with carmustine), is implanted into the resection cavity remaining after surgical removal of the tumor. It was approved by the FDA in 1997 as an adjunct to surgery to prolong the survival of patients with recurrent GBM for whom surgical resection is indicated.
BCNU is a prodrug that breaks down to produce chloroethylalkylation chloroethyl residues that can form cross-links between DNA strands [96]. Carbamoylation of lysine residues in nucleoproteins via isocyanate intermediates may also play a role in BCNU’s anticancer mode of action [97].
Nimustine is used in combination with teniposide as second- or third-line chemotherapy for recurrent glioblastoma.
Streptozotocin (a glucosamine nitrosourea), a natural hydrophilic nitrosourea, was isolated from Streptomyces achromogenes (1967). This compound proved to be more effective and less toxic, was approved by the FDA in 1982 for the treatment of pancreatic islet cell cancer and marketed as Zanosar®, although its use is generally limited to patients whose cancer cannot be removed by surgery, and several analogs have been produced, such as chlorozotocin and ranimustine.
Streptozotocin differs from other nitrosoureas in that it does not cross the blood–brain barrier due to its high hydrophilicity and also has relatively low myelosuppression, as it penetrates less into the cells of the bone marrow.
Chlorozotocin has been shown in a phase II study to be as active against metastatic melanoma as other clinically used chloroethylnitrosoureas without causing bone marrow toxicity [98]. Chlorozotocin is probably carcinogenic to humans.
Mechanism of Action of Nitrosoureas
The mechanism of action of nitrosoureas has been extensively studied. Nitrosoureas decompose spontaneously to form two electrophiles: an isocyanate and a diazene hydroxide. The diazene hydroxide produces a diazonium salt that alkylates the DNA, and this appears to be the main reaction responsible for the antitumor activity [99]. The isocyanate reacts with the amino groups of proteins to cause carbamoylation of proteins, which leads to inhibition of various DNA repair mechanisms. This fragmentation pathway was proposed based on studies of thermal decomposition of nitrosoureas under anhydrous conditions, but in aqueous solution the reaction proved to be more complex [71] (Figure 38).
Figure 38.

(A) Decomposition of nitrosoureas under anhydrous conditions. (B) In aqueous solution with the products of the alkylation of DNA and proteins.
Almost all nitrosoureas contain a chloroethyl chain in the nitroside, which enables them to act as DNA cross-linkers. This monoalkylated product forms a 5-linked cyclic intermediate and then reacts with the N-3 atom of the cytosine unit on the complementary DNA strand to form the cross-linked end product (Figure 39).
Figure 39.

DNA cross-linking by nitrosoureas.
An alternative mechanism would be the direct alkylation of DNA by the nitrosourea itself and not by the diazonium salt. Most drugs from the nitrosourea group are no longer in use and have been replaced by other alkylating agents.
9. Methanesulphonates
The mesylate is a good leaving group due to the delocalization of the negative charge between its three oxygen atoms (mesylate anion) (Figure 40).
Figure 40.

Mesylate ion resonant forms and average structure.
Several compounds containing two methanesulphonate groups separated by a hydrocarbon–carbon chain was tested as antitumor agents, and the compound with four carbons (busulphan) was found to have the optimal activity. The other methanesulphonate derivatives tested include piposulphan, improsulphan, hepsulphan, and treosulphan, a di-epoxide prodrug, which are shown in Figure 41.
Figure 41.

Methanesulphonate used as antitumor agents.
Busulphan, 4-methylsulphonyloxybutylmethanesulphonate, is an antineoplastic agent from the class of bifunctional alkylating agents that has been used to treat various types of cancer since 1959. Busulphan is used to treat chronic myeloid leukemia and certain blood disorders such as polycythemia vera and myeloid metaplasia and is also used in some conditioning programs prior to bone marrow transplants [100,101,102,103]. In contrast to nitrogen mustard and nitrosoureas, busulphan mainly affects myeloid cells. In high doses, busulphan is used in combination with cyclophosphamide as part of a preparatory regimen for bone marrow transplants to treat childhood leukemias, lymphomas, and some solid tumors. Busulphan is toxic to lung tissue and this toxicity may limit the dose that can be used.
Close monitoring of blood counts is required in patients treated with busulphan. Hematologic toxicity, including severe neutropenia, leukopenia, thrombocytopenia, and anemia, is the dose-limiting factor for busulphan.
In aqueous solutions, busulphan hydrolyzes to release methanesulphate groups and generates carbonium cations, which are reactive and can alkylate DNA and other proteins, leading to cytotoxicity. Busulphan reacts more readily with the thiol groups of amino acids, with cysteine, which involves a double nucleophilic attack of busulphan on the amino acid (Figure 42), and also with proteins as nitrogen mustard. In the final step of this reaction, elimination of the tetrahydrothiophene takes place and a C-C double bond is formed. The alkylation reaction depends on the concentrations of both busulphan and the target compound. Busulphan binds to DNA at the N-7 position of guanine. The alkylation of guanine with busulphan occurs via an SN2 mechanism [104]. The mechanism involves the attack of the nucleophile on the carbon containing the leaving group (mesylate). This carbon has a clearly positive polarity due to the electro-negativity of the sulphonate residue. Simultaneously with the attack on the nucleophile (N-7), the carbon-mesylate bond is cleaved, forming the monoalkylation product. The formation of cross-links between DNA strands has been demonstrated for busulphan cross-links within the DNA strand, mainly in the guanine–adenine sequence [102,105] (Figure 42).
Figure 42.

(A) Alkylation of cysteine residues by busulphan and (B) mechanism of DNA alkylation by busulphan.
Hepsulpham induced higher levels of DNA strand cross-linking than busulpham in three isolated samples from patients with chronic myeloid leukemia in blast crisis. However, busulphan caused a low number of DNA strand breaks in human cells.
Treosulphan in combination with fludarabine is a drug that is given to people before they undergo a bone marrow transplant from a donor, called an allogeneic hematopoietic stem cell transplant. It is used as a ‘conditioning treatment’ to cleanse the patient’s bone marrow and make room for the transplanted bone marrow cells, which can then produce healthy blood cells. It is used in both adult and pediatric patients older than one month with malignant neoplasms and benign diseases.
10. Triazenes
Triazenes are molecules that contain three contiguous nitrogen atoms in a linear arrangement. They have anticancer potential in many types of tumors such as leukemia, melanoma, and brain tumors as well as other therapeutic applications. The three triazenes used therapeutically are the anticancer drugs dacarbazine and temozolomide, and dia-minazole acetate, which is used in the treatment of trypanosomiasis in animals (Figure 43).
Figure 43.

Current therapeutic use triazenes.
Dacarbazine (DAC) (5-(3,3-dimethyl-1-triazenyl) imidazole-4-carboxamide) belongs to the triazene group of alkylating agents used in cancer therapy [106,107,108,109]. DAC has a triazene group linked to the imidazole ring, which, in turn, is linked to the carboxamide group. It is an electrophilic agent that acts specifically in the S phase of the cell cycle. Dacarbazine was approved in the 1970s in the USA and France for the treatment of metastatic malignant melanoma, Hodgkin’s lymphoma, sarcoma [110], and pancreatic islet cell carcinoma and is used in combination with other cancer drugs in more extreme cases. It has been classified in the group of antitumor agents for which there is no safe exposure range [111].
It is a prodrug that is activated in the liver and its cytotoxic activity is due to the formation of methyldiazonium during its metabolism, which methylates DNA [112]. Methyldiazonium has a very short half-life of about 0.4 s in aqueous solution. A mechanism for this process is summarized in Figure 44, where activation of dacarbazine by metabolic oxidative demethylation with formation of formaldehyde and (E)-5-(3-methyltriaz-1-en-1-yl)-1H-imidazole-4-carboxamide from the tautomer generates the reagent methyldiazonium. The formation of this cation is considered to be the main mechanism of its antitumor effect (Figure 44).
Figure 44.

Generation of methanediazonium from dacarbazine.
The most important methylation reaction takes place at the N-7 guanine atom and is relatively non-toxic. Methylation of the O-6 guanine also occurs and is considered to be the primary cytotoxic mechanism [113] (Figure 45).
Figure 45.

Mechanism of DNA methylation by Dacarbazine.
Disadvantages of dacarbazine are: (i) toxicity, (ii) excessive hydrophilicity leading to slow and incomplete oral absorption, necessitating intravenous administration, and (iii) its high photosensitivity with a very short half-life (about 30 min). This antineoplastic drug is excreted in the urine and its half-life in the body is 5 h. Dacarbazine is unstable in aqueous solution and is spontaneously degraded in the light, decomposing to 5-diazomidazole-4-carboxamide and di-methylamine and also undergoing a structural rearrangement to 2-azahipoxanthine [114,115] (Figure 46).
Figure 46.

Dacarbazine photodegradation products.
5-Diazomidazole-4-carboxamide can attack nucleophilic groups of DNA. However, the degradation products of dacarbazine probably do not contribute significantly to its cytotoxicity, although they may be involved in the painful local burning sensation on intravenous injection and in systemic problems associated with the drug.
Temozolamide (TMZ) was synthesized in 1980 as part of a series of new imidazo-tetrazinones to address the problems caused by the use of dacarbazine and has replaced dacarbazine in the clinic. It is a cytotoxic alkylating prodrug used for the treatment of glioblastoma multiforme [116,117] {Stevens, 1984 #12198}. Temozolomide, a methylating agent, was approved by the FDA in the USA in 1999 for the treatment of refractory anaplastic astrocytoma. The European Agency for the Evaluation of Medicinal Products approved the drug in 1998 for (i) progressive or recurrent glioblastoma multiforme after standard surgery and (ii) in 1999 for anaplastic astrocytoma, which is also resistant to treatment. Importantly, temozolomide is one of the few drugs approved specifically for a brain tumor and the first that can be administered orally.
The chemotherapeutic activity is achieved by converting temozolamide to the same intermediate (5-methyltriazenoimidazole-4-carboxamide) as dacarbazine, but in the case of temozolomide, the bioactivation process involves a non-enzymatic hydrolysis reaction followed by decarboxylation (Figure 47).
Figure 47.

Temozolomide hydrolysis.
The methyldiazonium ion formed during the degradation of MTIC mainly methylates the guanine residues in the DNA molecule, leading to the formation of O6- and N7-methylguanine. This aberrant DNA can no longer be corrected by the cellular machinery and ultimately leads to cell death through apoptosis. In addition, unlike dacarbazine, this drug can be administered both parenterally and orally. The main problem with this drug is bone marrow toxicity.
11. Fate and Effects of Anticancer Drugs in the Environment
Cytotoxic drugs, i.e., cancer drugs used in chemotherapy, are administered in European countries in the order of tons per year. After administration, these compounds are excreted by the chemotherapy patient either in their original form or as a product of the metabolization of the original compound into wastewater and subsequently discharged into rivers, seas, and oceans and can enter the environment via wastewater treatment plants, where they can have harmful effects. Due to their physicochemical properties, they can also be adsorbed in sewage sludge. The incidence of cancer has increased in recent years and is expected to continue to rise, as is the consumption of cytotoxic drugs. Therefore, their control in the environment is of utmost importance [118].
Assessing the risk posed by cytotoxic drugs is of particular interest as they are cytotoxic and, in many cases, carcinogenic. There is currently little data on the occurrence and effects of cytotoxic drugs in the environment.
The release of cytotoxic drugs used in chemotherapy has received less attention than other pharmaceuticals, although they may have cytotoxic, genotoxic, mutagenic, carcinogenic, endocrine, and/or teratogenic effects on various organisms.
The main sources of these effluents are hospital effluents, effluents from drugs excreted during outpatient treatment, and drug manufacturers. The immediate consequence is the release into the aquatic and terrestrial environment, even in minute quantities. The removal efficiency of wastewater treatment varies and depends on both the physicochemical properties of these by-products and the treatment process used [119].
In the 1990s, major efforts were made to investigate the occurrence, phase, and potential risks of human pharmaceuticals in the environment. Several large-scale collaborative programs have been conducted in Europe and the US, such as ‘Poseidon’ (EU, 2001–2004), Norman (EU, 2005–2008), PILLS (EU, 2007–2012), and Emerging Contaminants in the Environment (US Geological Survey, 2007–2011). The most recent PHARMAS (EU, 2011–2013) and CytoThreat (EU, 2011–2013) specifically target antibiotics and cancer drugs.
There is a need to sensitize both the scientific community and the general public to the potential of anticancer drugs as pollutants of increasing concern in aquatic and terrestrial ecosystems [120].
Antineoplastic drugs (ANPs) have raised concerns about their safe remediation. An excellent review paper by Abhilash Kumar Tripathi and co-authors provides an overview of the environmental sources of ANP agents, focusing on the remediation methods currently in use [121].
Other necessary considerations include the solid cytotoxic wastes generated during the preparation, reconstitution, and administration of these drugs to chemotherapy patients. There are no suitable treatment technologies for this waste in most parts of Spain, so it has to be transported to France and/or Germany for incineration, resulting in the emission of toxic and greenhouse gasses.
12. Antimicrobial Peptides
The search for more effective and less toxic cancer treatments has driven research into numerous innovative therapeutic strategies. Among these, antimicrobial peptides (AMPs) have emerged as a promising class of agents with potent anticancer properties [122]. Originally identified for their role in innate immunity and their ability to fight microbial infections, AMPs have shown a broad spectrum of biological activities that also have anticancer effects. These peptides exert their anticancer effects primarily through the selective destruction of cancer cell membranes, induction of apoptosis, and modulation of immune responses, making them highly versatile and potent candidates for cancer therapy [123]. Their unique mechanism of attacking cancer cells while sparing normal cells offers a significant advantage over conventional chemotherapeutic agents, which often result in significant collateral damage to healthy tissue [124,125].
Antimicrobial peptides (AMPs) are a critical component of the innate immune system and serve as one of the first lines of defense against a broad spectrum of pathogens, including bacteria, viruses, fungi, and parasites [126]. These small, naturally occurring peptides are ubiquitous in a variety of species, from microorganisms to humans, underscoring their evolutionary importance [126]. The discovery of AMPs can be traced back to the early 20th century, when scientists first discovered the antimicrobial properties of lysozyme, an enzyme found in egg white and human tears [127]. But it was not until the late 1980s that the field of AMPs gained prominence after magainin were isolated and characterized from the skin of the African clawed frog Xenopus laevis [128]. This groundbreaking work highlighted the strong antimicrobial activity of these peptides and initiated extensive research into their potential therapeutic applications.
AMPs are characterized by their amphipathic nature, which enables them to interact with and destroy microbial membranes [129]. Structurally, they typically consist of 12–50 amino acids and can adopt various conformations, including alpha helices, beta sheets, and extended or looped structures [129,130]. The ability of AMPs to attack and destroy the lipid bilayers of microbial membranes, while mammalian cells are generally spared, is attributed to differences in membrane composition [122]. This selectivity is crucial for their role in host defense and represents a significant advantage over conventional antibiotics, which often cause problems with resistance and collateral damage to host tissue [129].
Beyond their antimicrobial properties, AMPs have attracted attention for their diverse role in modulating the immune response, promoting wound healing and even their anticancer activity. In the context of cancer therapy, AMPs have been shown to selectively target cancer cells through mechanisms similar to those used to fight microbes [122]. Cancer cells often have particular membrane characteristics, such as a higher proportion of negatively charged phospholipids, which make them susceptible to AMP-induced membrane disruption. In addition, AMPs can induce apoptosis in cancer cells and modulate the tumor microenvironment to enhance antitumor immunity. These properties make AMPs promising candidates for novel cancer therapies, either as stand-alone treatments or in combination with other therapeutics [122].
The potential of AMPs in cancer therapy has generated considerable interest in the scientific community, particularly with regard to their use in combination with other therapeutic modalities [131]. One of these promising combinations is with DNA alkylating agents [132,133,134,135]. DNA alkylation involves the attachment of alkyl groups to DNA molecules, resulting in DNA damage and inhibition of cancer cell proliferation. Alkylating agents, such as cyclophosphamide and cisplatin, have been cornerstones of cancer treatment for decades [60]. However, their use is often limited by the development of resistance and severe side effects [136]. The combination of DNA alkylating agents with AMPs offers a novel approach to overcome these limitations by increasing the efficacy and specificity of the attack on cancer cells [137,138]. The reason for this combination lies in the complementary mechanisms of action of AMPs and alkylating agents. While alkylating agents have a direct genotoxic effect on cancer cells, AMPs can enhance this effect by compromising the integrity of cancer cell membranes, enabling better intracellular uptake of the drug and promoting apoptosis. In addition, AMPs can modulate the tumor microenvironment by enhancing the immune response against cancer cells, potentially attenuating the immunosuppressive effects often associated with alkylating chemotherapy. This synergistic interaction promises not only better therapeutic outcomes but also a reduction in the required doses of alkylating agents and thus a minimization of their undesirable side effects [2,139,140,141].
13. Mechanisms of Action of AMPs in Cancer Cells
Antimicrobial peptides (AMPs) exhibit a versatile mode of action against cancer cells by utilizing their innate properties to attack and destroy malignant cells. One of the most important mechanisms by which AMPs exert their anticancer effect is by destroying the membranes of cancer cells [122]. Cancer cells often have special membrane characteristics, including a higher content of negatively charged phospholipids and an altered lipid composition compared to normal cells. This makes them particularly susceptible to the amphipathic and cationic nature of AMPs. Upon interaction with the cancer cell membrane, AMPs can penetrate the lipid bilayer and form pores or degrade the membrane. This disruption impairs the integrity of the cell membrane and leads to cell lysis and cell death [142].
In addition to the direct disruption of the membrane, AMPs can also trigger apoptosis in cancer cells. Apoptosis, or programmed cell death, is a tightly regulated process that eliminates damaged or unwanted cells without causing inflammation. AMPs can trigger apoptosis via different pathways, including the mitochondrial pathway and the death receptor pathway. In the mitochondrial pathway, AMPs can cause the release of cytochrome c from the mitochondria into the cytosol, which subsequently activates caspases, the enzymes responsible for carrying out apoptosis. Alternatively, AMPs can interact with cell surface receptors to activate the death receptor pathway, leading to the formation of the death-inducing signaling complex (DISK) and activation of caspase-8. These pathways culminate in the cleavage of important cellular proteins and the eventual destruction of the cell.
AMPs can not only trigger apoptosis but also modulate the immune response, creating a hostile environment for cancer cells [143]. AMPs are known to have immunomodulatory properties. These include the ability to attract immune cells to the tumor site, promote the maturation and activation of dendritic cells, and enhance the presentation of tumor antigens. This immunomodulation can stimulate both the innate and adaptive immune response, leading to a stronger attack on the cancer cells [144]. By altering the tumor microenvironment, AMPs can help overcome the immunosuppressive barriers that often protect tumors from surveillance and destruction by the immune system [122,145].
In addition, some AMPs have been found to inhibit angiogenesis, the process by which tumors establish their own blood supply to sustain growth and metastasis [122,146]. Angiogenesis is a crucial step in tumor progression, and inhibiting it can supply the tumor with essential nutrients and oxygen [147]. AMPs can interfere with angiogenic signaling pathways by reducing the expression of pro-angiogenic factors such as vascular endothelial growth factor (VEGF) and disrupting the formation of new blood vessels [148,149]. This anti-angiogenic effect not only inhibits tumor growth but also limits the potential for metastasis [150].
14. Advantages of AMPs and Their Synergy with Alkylating Agents
One of the main problems with alkylating agents is their lack of selectivity, which results in damage to both cancer cells and healthy cells. This non-specificity leads to a number of serious side effects, including myelosuppression (bone marrow suppression), gastrointestinal toxicity, and an increased risk of secondary malignancies [151]. These side effects may limit the dose of alkylating agents that can be safely administered to patients, reducing their overall efficacy [136]. The membranes of cancer cells often contain higher levels of negatively charged phospholipids and other unique lipid components that attract the cationic and amphipathic nature of AMPs [122,152]. This selectivity minimizes collateral damage to healthy cells and reduces the side effects commonly associated with conventional chemotherapy [153] (Figure 48).
Figure 48.

Advantages of antimicrobial peptides in combination with alkylating agents for cancer therapy.
AMPs also exhibit a broad spectrum of anticancer mechanisms, making it difficult for cancer cells to develop resistance. Their ability to disrupt cell membranes, induce apoptosis, modulate immune responses, and inhibit angiogenesis provides a versatile approach to destroying cancer cells. By attracting immune cells to the tumor site and promoting the activation and maturation of dendritic cells, AMPs can stimulate both the innate and adaptive immune systems. This immune modulation can create a more hostile environment for cancer cells, further enhancing the anticancer effects of AMPs and also helps to prevent the emergence of resistant cancer cell populations, which is a major problem with single-target chemotherapeutics [154].
The natural origin of many AMPs and their relatively simple structure also facilitate their synthesis and modification, enabling the development of a wide range of AMP derivatives with improved stability, potency, and selectivity. Advances in peptide engineering and delivery systems have further enhanced the therapeutic potential of AMPs, making them more suitable for clinical use [142]. These properties make AMPs promising candidates for the development of novel cancer therapies, either as stand-alone treatments or in combination with existing chemotherapeutics to increase efficacy and reduce toxicity.
The combination of AMPs with alkylating agents can improve the efficacy of cancer therapy by several mechanisms (Table 2): (i) by enhancing DNA damage, since alkylating agents act by attaching alkyl groups to DNA and AMPs can enhance this effect by interfering with DNA repair mechanisms, making cancer cells more susceptible to damage by alkylating agents; (ii) by modulating the tumor microenvironment, affecting angiogenesis (formation of new blood vessels) and immune responses, making tumor cells more susceptible to the cytotoxic effects of alkylating agents; (iii) overcoming drug resistance by disrupting cell signaling pathways that cancer cells use to escape the effects of DNA damage; and (iv) activating signaling pathways that lead to apoptosis (programmed cell death) and complement the cytotoxic effects of alkylating agents. This dual approach may lead to more effective elimination of cancer cells.
Table 2.
Synergistic mechanisms of AMPs and alkylating agents in cancer therapy.
| Mechanism | Description | Reference |
|---|---|---|
| Membrane Disruption | AMPs disrupt cancer cell membranes, leading to cell lysis, facilitating the entry of alkylating agents. | [155] |
| DNA Damage | Alkylating agents cause DNA strand breaks, facilitated by AMPs. | [156] |
| Immune Modulation–Induction of Apoptosis | AMPs trigger apoptosis, which can be amplified by alkylating agents. | [157,158] |
| Anti-Angiogenesis | AMPs inhibit angiogenesis, which complements the cytotoxic effects of alkylating agents. | [159] |
| Overcoming Drug Resistance | AMPs reduce the potential for drug resistance to alkylating agents. | [160] |
15. Technological Advances in the Administration of AMPs and Alkylating Drugs
The development of effective delivery systems is critical to the therapeutic success of antimicrobial peptides (AMPs) and alkylating agents in cancer treatment (Figure 49). One of the most important advances in AMP delivery is the use of nanoparticle-based systems [161]. Nanoparticles can be engineered to encapsulate AMPs, protect them from enzymatic degradation, and improve their stability in the bloodstream [162,163]. These nanoscale carriers can be functionalized with targeted ligands that recognize and bind to specific markers on the cancer cells to ensure that the AMPs are delivered precisely to the tumor. This targeted delivery not only increases the therapeutic efficacy of the AMP, but also minimizes off-target effects and reduces systemic toxicity. Various nanoparticle platforms, including liposomes, polymeric nanoparticles, and metallic nanoparticles, have been evaluated for their ability to effectively deliver AMPs to tumors [164].
Figure 49.

Technological advances in the administration of AMPs and alkylating drugs for cancer therapy.
Similarly, advances in the delivery of alkylating agents have focused on improving their selectivity and reducing their toxicity. Liposomal encapsulation is one of these promising strategies. Liposomes are spherical vesicles consisting of lipid bilayers that can encapsulate hydrophilic and hydrophobic drugs. Encapsulation of alkylating agents in liposomes may help to protect them from premature degradation and reduce their systemic toxicity by enhancing their accumulation in tumor tissue through the effect of enhanced permeability and retention (EPR). This effect takes advantage of the leaky vasculature and poor lymphatic drainage of tumors to preferentially deliver liposomal encapsulated drugs to the tumor [165,166,167].
Another significant advance is the development of antibody–drug conjugates (ADCs), which combine the targeting ability of monoclonal antibodies with the potent cytotoxic activity of alkylating agents. ADCs are designed to bind specifically to antigens expressed on the surface of cancer cells, delivering the alkylating agent directly to the tumor and sparing normal tissue. This targeted approach maximizes the therapeutic index of alkylating agents and increases their efficacy while minimizing adverse effects [168,169].
Polymeric micelles have also been used to enhance the delivery of AMP and alkylating agents. These self-assembling nanocarriers can encapsulate hydrophobic actives in their core and form a hydrophilic shell that improves the solubility and stability of the encapsulated actives. Polymeric micelles can be engineered to release their payload in response to specific stimuli, such as changes in pH or temperature, ensuring controlled and sustained drug release at the tumor site [164,170,171,172].
In addition to nanoparticle-based systems, peptide conjugation strategies have been developed to improve the delivery of AMPs. Conjugation of AMPs with cell-penetrating peptides (CPPs) can facilitate their translocation across cell membranes, improving their intracellular release and therapeutic efficacy. CPPs can be designed to recognize and bind to specific receptors on cancer cells, ensuring targeted delivery of AMPs and enhancing their anticancer effects [173,174]. For example, a combination of the cell-penetrating peptide octoarginine–oxaliplatin was developed to efficiently and rapidly transport oxaliplatin into colon cancer cells. The in vivo study in mice showed that the compound successfully suppressed tumor development and had a significantly strong anticancer effect [175]. Similarly, the complex of the cell-penetrating peptide Tansportan-10 and cisplatin produced a stronger effect on the cancer cell lines (HeLa, OS143B) compared to that observed after separate treatment [176]. Conjugation of cell-penetrating peptides with cobalt complexes also showed superior efficacy against the HepG2 liver cancer cell line compared to the original Co(III) complex [177]. The combination of cell-penetrating peptides with nanotechnology is a promising strategy. Encapsulation of the cell-penetrating peptide EIP103, which specifically targets the nucleus, in PEG-PE micelles showed significant inhibitory effects in H446 and A549 cells and demonstrated a promising therapy to improve the clinical treatment of lung cancer [178].
Advances in 3D printing technology have opened new avenues for the local delivery of AMPs and alkylating agents; 3D-printed scaffolds and hydrogels can be designed to release these agents in a controlled manner and ensure sustained therapeutic concentration at the tumor site. This local delivery approach can minimize systemic exposure and reduce the risk of adverse effects [179,180,181]. These advances demonstrate the importance of continuing drug delivery research and development to fully realize the therapeutic potential of AMPs and alkylating agents.
16. Can AI Tools Accelerate Current Research into Alkylating Agents?
The question of whether artificial intelligence (AI) can accelerate research into alkylating agents in cancer treatment is a hot topic that is increasingly being answered in the affirmative.
Alkylating agents are a class of chemotherapeutic agents that damage the DNA of cancer cells. However, their use is limited by significant side effects and the emergence of tumor resistance. This is where AI can make a decisive difference.
AI can accelerate research by discovering new drugs by analyzing large amounts of data. AI can analyze huge databases of molecules to identify new chemical structures with potential antitumor activity and through molecular modeling that simulates the interaction between molecules and proteins [182]. AI can predict the efficacy and toxicity of new drugs, reducing the time and cost of preclinical trials [183].
It can also help optimize treatments, such as personalizing therapies by analyzing each patient’s genomic data and identifying the most appropriate treatments that minimize side effects. It can also be used to predict response to treatment by analyzing medical imaging and clinical data so that therapy can be better tailored [184]. Another potential area of application is understanding resistance mechanisms and identifying potential biomarkers that predict the occurrence of resistance to alkylating agents [185].
In the negative perception, AI relies on large amounts of high-quality data. Therefore, more comprehensive and standardized databases need to be developed, and tools need to be developed to understand how the models arrive at their conclusions [186].
AI therefore has the potential to revolutionize alkylating agent research and ultimately improve outcomes for cancer patients. However, it is important to address the existing challenges and encourage collaboration between researchers from different disciplines to achieve the most out of this technology [187].
17. Conclusions
In the last thirty years, more and more drugs have been used to treat tumors, as genetic and environmental factors have led to an increased incidence of cancer in the human population. The structure and dynamics of DNA are strongly influenced by the alkylation of its bases. Alkylation prevents DNA replication and RNA transcription of the affected DNA molecule. It also leads to DNA fragmentation through hydrolytic reactions and through the action of repair enzymes that attempt to remove the alkylated bases. Alkylation also leads to mismatching of nucleotides by disrupting the normal hydrogen bonds between the bases.
Current research on alkylating agents should focus on the development of more selective alkylating agents with fewer side effects and adequate bioavailability. New derivatives of nitrogen mustard continue to be investigated for improved efficacy and reduced toxicity with combinations of nitrogen mustard with monoclonal antibodies and small molecule targeted agents.
The increasing use of these drugs and their degradation products has resulted in their appearance in water bodies around the world, impacting ecosystems, polluting the environment, and having negative effects on biota.
The combination of antimicrobial peptides (AMPs) and alkylating agents is a promising strategy towards more effective and targeted cancer therapies. AMPs offer significant advantages over conventional chemotherapeutics with their versatile mechanisms of action, including membrane disruption, apoptosis induction, immune modulation, and anti-angiogenic properties. These peptides selectively target cancer cells while sparing healthy tissue, minimizing systemic toxicity and reducing the risk of resistance development. Meanwhile, alkylating agents remain central to cancer treatment due to their ability to cause DNA damage and inhibit cell proliferation, albeit with significant limitations such as non-specific toxicity and the potential for resistance.
The combination of AMPs with alkylating agents capitalizes on the complementary strengths of these agents and may improve therapeutic efficacy through synergistic interactions. Technological advances in drug delivery, such as nanoparticle-based systems, antibody–drug conjugates, and 3D printing technologies, have further optimized the delivery of AMPs and alkylating agents to tumor sites, improving their bioavailability and therapeutic outcomes.
Combining AMPs with alkylating agents can also improve the efficacy of cancer therapy by enhancing DNA damage, modulating the tumor microenvironment, overcoming drug resistance, and activating signaling pathways that lead to apoptosis. These efforts may yield a more effective elimination of malignant cells.
While challenges such as resistance mechanisms, off-target effects, and optimal dosing strategies remain unresolved, ongoing research and clinical trials continue to explore the full potential of this dual mode of action. Future directions include the development of novel AMP derivatives, personalized medicine approaches, and combination therapies tailored to the individual patient profile. Ultimately, the convergence of AMPs and alkylating agents represents a promising paradigm shift in cancer therapy, offering hope for more effective treatments and better outcomes for patients with this difficult disease.
Author Contributions
Conceptualization, C.M.C.A. and C.A.J.; investigation, E.B.M., C.M.C.A. and C.A.J.; writing—review and editing, E.B.M., C.M.C.A., C.A.J., J.M.P.d.l.L. and E.P.-L.; supervision, E.B.M., C.M.C.A., C.A.J. and J.M.P.d.l.L. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Funding Statement
This research was funded by the State Plan for Scientific, Technical Research and Innovation 2021–2023 from the Spanish Ministry of Science and Innovation (project PLEC2022-009507) and by the COCREA innovation project of the Spanish National Research Council (CSIC) to promote solutions to global challenges.
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Amjad M.T., Chidharla A., Kasi A. Cancer Chemotherapy. StatPearls Publishing; Treasure Island, FL, USA: 2023. [PubMed] [Google Scholar]
- 2.Anand U., Dey A., Chandel A.K.S., Sanyal R., Mishra A., Pandey D.K., De Falco V., Upadhyay A., Kandimalla R., Chaudhary A., et al. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2023;10:1367–1401. doi: 10.1016/j.gendis.2022.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wiltse J., Dellarco V.L. US Environmental Protection Agency guidelines for carcinogen risk assessment: Past and future. Mutat. Res./Rev. Genet. Toxicol. 1996;365:3–15. doi: 10.1016/S0165-1110(96)90009-3. [DOI] [PubMed] [Google Scholar]
- 4.De, Surya Kanta . Fundamentals of Cancer Detection, Treatment, and Prevention. John Wiley & Sons; Hoboken, NJ, USA: 2022. An Overview of Cancer; pp. 1–20. [Google Scholar]
- 5.Passaro A., Al Bakir M., Hamilton E.G., Diehn M., André F., Roy-Chowdhuri S., Mountzios G., Wistuba I.I., Swanton C., Peters S. Cancer biomarkers: Emerging trends and clinical implications for personalized treatment. Cell. 2024;187:1617–1635. doi: 10.1016/j.cell.2024.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee Y.T., Tan Y.J., Oon C.E. Molecular targeted therapy: Treating cancer with specificity. Eur. J. Pharmacol. 2018;834:188–196. doi: 10.1016/j.ejphar.2018.07.034. [DOI] [PubMed] [Google Scholar]
- 7.Misiak M., Mantegazza F., Beretta L.G. Methods for Elucidation of DNA-Anticancer Drug Interactions and their Applications in the Development of New Drugs. Curr. Pharm. Des. 2016;22:6596–6611. doi: 10.2174/1381612822666160831114622. [DOI] [PubMed] [Google Scholar]
- 8.Thurston D.E., Pysz I. Chemistry and Pharmacology of Anticancer Drugs. CRC Press; Boca Raton, FL, USA: 2021. [Google Scholar]
- 9.Lagunas-Rangel F.A., Liu W., Schiöth H.B. Can Exposure to Environmental Pollutants Be Associated with Less Effective Chemotherapy in Cancer Patients? Int. J. Environ. Res. Public Health. 2022;19:2064. doi: 10.3390/ijerph19042064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoofnagle J.H., Serrano J., Knoben J.E., Navarro V.J. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury. National Institute of Diabetes and Digestive and Kidney Diseases; Bethesda, MD, USA: 2012. [Google Scholar]
- 11.Holland J.F. Holland-Frei Cancer Medicine 8. Volume 8 PMPH-USA; Shelton, CT, USA: 2010. [Google Scholar]
- 12.Kciuk M., Gielecińska A., Mujwar S., Kołat D., Kałuzińska-Kołat Ż., Celik I., Kontek R. Doxorubicin—An Agent with Multiple Mechanisms of Anticancer Activity. Cells. 2023;12:659. doi: 10.3390/cells12040659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Zanden S.Y., Qiao X., Neefjes J. New insights into the activities and toxicities of the old anticancer drug doxorubicin. FEBS J. 2021;288:6095–6111. doi: 10.1111/febs.15583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carvalho C., Santos R.X., Cardoso S., Correia S., Oliveira P.J., Santos M.S., Moreira P.I. Doxorubicin: The good, the bad and the ugly effect. Curr. Med. Chem. 2009;16:3267–3285. doi: 10.2174/092986709788803312. [DOI] [PubMed] [Google Scholar]
- 15.Tacar O., Sriamornsak P., Dass C.R. Doxorubicin: An update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol. 2013;65:157–170. doi: 10.1111/j.2042-7158.2012.01567.x. [DOI] [PubMed] [Google Scholar]
- 16.Lipscomb L.A., Peek M.E., Zhou F.X., Bertrand J.A., VanDerveer D., Williams L.D. Water ring structure at DNA interfaces: Hydration and dynamics of DNA-anthracycline complexes. Biochemistry. 1994;33:3649–3659. doi: 10.1021/bi00178a023. [DOI] [PubMed] [Google Scholar]
- 17.Howerton S.B., Nagpal A., Dean Williams L. Surprising roles of electrostatic interactions in DNA–ligand complexes. Biopolymers. 2003;69:87–99. doi: 10.1002/bip.10319. [DOI] [PubMed] [Google Scholar]
- 18.Zlatanova J., Victor J.M. How are nucleosomes disrupted during transcription elongation? HFSP J. 2009;3:373–378. doi: 10.2976/1.3249971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang F., Kemp C.J., Henikoff S. Doxorubicin enhances nucleosome turnover around promoters. Curr. Biol. 2013;23:782–787. doi: 10.1016/j.cub.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salerno D., Brogioli D., Cassina V., Turchi D., Beretta G.L., Seruggia D., Ziano R., Zunino F., Mantegazza F. Magnetic tweezers measurements of the nanomechanical properties of DNA in the presence of drugs. Nucleic Acids Res. 2010;38:7089–7099. doi: 10.1093/nar/gkq597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang F., Teves S.S., Kemp C.J., Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta. 2014;1845:84–89. doi: 10.1016/j.bbcan.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hande K.R. Clinical applications of anticancer drugs targeted to topoisomerase II. Biochim. Biophys. Acta. 1998;1400:173–184. doi: 10.1016/S0167-4781(98)00134-1. [DOI] [PubMed] [Google Scholar]
- 23.Lemke K., Wojciechowski M., Laine W., Bailly C., Colson P., Baginski M., Larsen A.K., Skladanowski A. Induction of unique structural changes in guanine-rich DNA regions by the triazoloacridone C-1305, a topoisomerase II inhibitor with antitumor activities. Nucleic Acids Res. 2005;33:6034–6047. doi: 10.1093/nar/gki904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Króliczewski J., Bartoszewska S., Dudkowska M., Janiszewska D., Biernatowska A., Crossman D.K., Krzymiński K., Wysocka M., Romanowska A., Baginski M., et al. Utilizing Genome-Wide mRNA Profiling to Identify the Cytotoxic Chemotherapeutic Mechanism of Triazoloacridone C-1305 as Direct Microtubule Stabilization. Cancers. 2020;12:864. doi: 10.3390/cancers12040864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kellett A., Molphy Z., Slator C., McKee V., Farrell N.P. Molecular methods for assessment of non-covalent metallodrug-DNA interactions. Chem. Soc. Rev. 2019;48:971–988. doi: 10.1039/C8CS00157J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bauer G.B., Povirk L.F. Specificity and kinetics of interstrand and intrastrand bifunctional alkylation by nitrogen mustards at a G-G-C sequence. Nucleic Acids Res. 1997;25:1211–1218. doi: 10.1093/nar/25.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Andrés C.M.C., Lastra J.M.P.d.l., Juan C.A., Plou F.J., Pérez-Lebeña E. Chemical Insights into Oxidative and Nitrative Modifications of DNA. Int. J. Mol. Sci. 2023;24:15240. doi: 10.3390/ijms242015240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.David E.G., Luminita M.V. Intercalative Binding of Small Molecules to Nucleic Acids. Curr. Org. Chem. 2000;4:915–929. doi: 10.2174/1385272003375978. [DOI] [Google Scholar]
- 29.Ralhan R., Kaur J. Alkylating agents and cancer therapy. Expert Opin. Ther. Pat. 2007;17:1061–1075. doi: 10.1517/13543776.17.9.1061. [DOI] [Google Scholar]
- 30.Guerra C.F., Bickelhaupt F.M. CHAPTER 4-Watson–Crick hydrogen bonds: Nature and role in DNA replication. In: Starikov E.B., Lewis J.P., Tanaka S., editors. Modern Methods for Theoretical Physical Chemistry of Biopolymers. Elsevier Science; Amsterdam, The Netherlands: 2006. pp. 79–97. [Google Scholar]
- 31.Langkjær N., Wengel J., Pasternak A. Watson-Crick hydrogen bonding of unlocked nucleic acids. Bioorg. Med. Chem. Lett. 2015;25:5064–5066. doi: 10.1016/j.bmcl.2015.10.024. [DOI] [PubMed] [Google Scholar]
- 32.Nelson S.M., Ferguson L.R., Denny W.A. DNA and the chromosome-varied targets for chemotherapy. Cell Chromosome. 2004;3:2. doi: 10.1186/1475-9268-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Paul A., Bhattacharya S. Chemistry and biology of DNA-binding small molecules. Curr. Sci. 2012;102:212–231. [Google Scholar]
- 34.Highley M.S., Landuyt B., Prenen H., Harper P.G., De Bruijn E.A. The Nitrogen Mustards. Pharmacol. Rev. 2022;74:552–599. doi: 10.1124/pharmrev.120.000121. [DOI] [PubMed] [Google Scholar]
- 35.Chen Y., Jia Y., Song W., Zhang L. Therapeutic Potential of Nitrogen Mustard Based Hybrid Molecules. Front. Pharmacol. 2018;9:1453. doi: 10.3389/fphar.2018.01453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J.J., Li J.J. Laughing Gas, Viagra, and Lipitor: The Human Stories Behind the Drugs We Use. Oxford University Press; Oxford, UK: 2006. Cancer Drugs: From Nitrogen Mustards to Gleevec. [Google Scholar]
- 37.Ogino M.H., Tadi P. StatPearls. StatPearls Publishing LLC.; Treasure Island, FL, USA: 2024. Cyclophosphamide. [PubMed] [Google Scholar]
- 38.Idle J.R., Beyoğlu D. Ifosfamide-History, efficacy, toxicity and encephalopathy. Pharmacol. Ther. 2023;243:108366. doi: 10.1016/j.pharmthera.2023.108366. [DOI] [PubMed] [Google Scholar]
- 39.Scripture C.D., Sparreboom A., Figg W.D. Modulation of cytochrome P450 activity: Implications for cancer therapy. Lancet Oncol. 2005;6:780–789. doi: 10.1016/S1470-2045(05)70388-0. [DOI] [PubMed] [Google Scholar]
- 40.Burcham P.C., Fontaine F.R., Kaminskas L.M., Petersen D.R., Pyke S.M. Protein adduct-trapping by hydrazinophthalazine drugs: Mechanisms of cytoprotection against acrolein-mediated toxicity. Mol. Pharmacol. 2004;65:655–664. doi: 10.1124/mol.65.3.655. [DOI] [PubMed] [Google Scholar]
- 41.Martinez-Sanchez J., Pascual-Diaz R., Palomo M., Moreno-Castaño A.B., Ventosa H., Salas M.Q., Rovira M., Escolar G., Carreras E., Diaz-Ricart M. Mafosfamide, a cyclophosphamide analog, causes a proinflammatory response and increased permeability on endothelial cells in vitro. Bone Marrow Transplant. 2023;58:407–413. doi: 10.1038/s41409-023-01912-w. [DOI] [PubMed] [Google Scholar]
- 42.Moon K.-Y., Kwon C.-H. N3-methyl-mafosfamide as a chemically stable, alternative prodrug of mafosfamide. Bioorganic Med. Chem. Lett. 1998;8:1673–1678. doi: 10.1016/S0960-894X(98)00287-X. [DOI] [PubMed] [Google Scholar]
- 43.May-Manke A., Kroemer H., Hempel G., Bohnenstengel F., Hohenlöchter B., Blaschke G., Boos J. Investigation of the major human hepatic cytochrome P450 involved in 4-hydroxylation and N-dechloroethylation of trofosfamide. Cancer Chemother. Pharmacol. 1999;44:327–334. doi: 10.1007/s002800050985. [DOI] [PubMed] [Google Scholar]
- 44.Liang J., Huang M., Duan W., Yu X.Q., Zhou S. Design of new oxazaphosphorine anticancer drugs. Curr. Pharm. Des. 2007;13:963–978. doi: 10.2174/138161207780414296. [DOI] [PubMed] [Google Scholar]
- 45.O’Byrne K., Steward W.P. The role of chemotherapy in the treatment of adult soft tissue sarcomas. Oncology. 1999;56:13–23. doi: 10.1159/000011924. [DOI] [PubMed] [Google Scholar]
- 46.Advani S.H. The role of ifosfamide in paediatric cancer. Aust. N. Z. J. Med. 1998;28:410–413. doi: 10.1111/j.1445-5994.1998.tb01976.x. [DOI] [PubMed] [Google Scholar]
- 47.Dayyani F., Macarulla T., Johnson A., Wainberg Z.A. Second-line treatment options for patients with metastatic pancreatic ductal adenocarcinoma: A systematic literature review. Cancer Treat. Rev. 2023;113:102502. doi: 10.1016/j.ctrv.2022.102502. [DOI] [PubMed] [Google Scholar]
- 48.Diethelm-Varela B., Ai Y., Liang D., Xue F. Nitrogen Mustards as Anticancer Chemotherapies: Historic Perspective, Current Developments and Future Trends. Curr. Top. Med. Chem. 2019;19:691–712. doi: 10.2174/1568026619666190401100519. [DOI] [PubMed] [Google Scholar]
- 49.Summerfield G.P., Taylor P.R., Mounter P.J., Proctor S.J. High-dose chlorambucil for the treatment of chronic lymphocytic leukaemia and low-grade non-Hodgkin’s lymphoma. Br. J. Haematol. 2002;116:781–786. doi: 10.1046/j.0007-1048.2002.03362.x. [DOI] [PubMed] [Google Scholar]
- 50.Lalic H., Aurer I., Batinic D., Visnjic D., Smoljo T., Babic A. Bendamustine: A review of pharmacology, clinical use and immunological effects (Review) Oncol. Rep. 2022;47:114. doi: 10.3892/or.2022.8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh R.K., Kumar S., Prasad D.N., Bhardwaj T.R. Therapeutic journery of nitrogen mustard as alkylating anticancer agents: Historic to future perspectives. Eur. J. Med. Chem. 2018;151:401–433. doi: 10.1016/j.ejmech.2018.04.001. [DOI] [PubMed] [Google Scholar]
- 52.Saha P., Debnath C., Bérubé G. Steroid-linked nitrogen mustards as potential anticancer therapeutics: A review. J. Steroid Biochem. Mol. Biol. 2013;137:271–300. doi: 10.1016/j.jsbmb.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 53.Scholar E. Estramustine. In: Enna S.J., Bylund D.B., editors. xPharm: The Comprehensive Pharmacology Reference. Elsevier; New York, NY, USA: 2007. pp. 1–5. [Google Scholar]
- 54.Bastholt L., Johansson C.J., Pfeiffer P., Svensson L., Johansson S.A., Gunnarsson P.O., Mouridsen H. A pharmacokinetic study of prednimustine as compared with prednisolone plus chlorambucil in cancer patients. Cancer Chemother. Pharmacol. 1991;28:205–210. doi: 10.1007/BF00685510. [DOI] [PubMed] [Google Scholar]
- 55.Zolottsev V.A., Latysheva A.S., Pokrovsky V.S., Khan I.I., Misharin A.Y. Promising applications of steroid conjugates for cancer research and treatment. Eur. J. Med. Chem. 2021;210:113089. doi: 10.1016/j.ejmech.2020.113089. [DOI] [PubMed] [Google Scholar]
- 56.Trafalis D.T.P. Hybrid aza-steroid alkylators in the treatment of colon cancer. Cancer Lett. 2006;243:202–210. doi: 10.1016/j.canlet.2005.11.034. [DOI] [PubMed] [Google Scholar]
- 57.Trafalis D., Geromichalou E., Dalezis P., Nikoleousakos N., Sarli V. Synthesis and evaluation of new steroidal lactam conjugates with aniline mustards as potential antileukemic therapeutics. Steroids. 2016;115:1–8. doi: 10.1016/j.steroids.2016.07.009. [DOI] [PubMed] [Google Scholar]
- 58.Wickström M., Nygren P., Larsson R., Harmenberg J., Lindberg J., Sjöberg P., Jerling M., Lehmann F., Richardson P., Anderson K., et al. Melflufen—A peptidase-potentiated alkylating agent in clinical trials. Oncotarget. 2017;8:66641–66655. doi: 10.18632/oncotarget.18420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mateos M.V., Bladé J., Bringhen S., Ocio E.M., Efebera Y., Pour L., Gay F., Sonneveld P., Gullbo J., Richardson P.G. Melflufen: A Peptide-Drug Conjugate for the Treatment of Multiple Myeloma. J. Clin. Med. 2020;9:3120. doi: 10.3390/jcm9103120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lehmann F., Wennerberg J. Evolution of Nitrogen-Based Alkylating Anticancer Agents. Processes. 2021;9:377. doi: 10.3390/pr9020377. [DOI] [Google Scholar]
- 61.Olivier T., Prasad V. The approval and withdrawal of melphalan flufenamide (melflufen): Implications for the state of the FDA. Transl. Oncol. 2022;18:101374. doi: 10.1016/j.tranon.2022.101374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Persmark M., Guengerich F.P. Spectroscopic and thermodynamic characterization of the interaction of N7-guanyl thioether derivatives of d(TGCTG*CAAG) with potential complements. Biochemistry. 1994;33:14368. doi: 10.1021/bi00251a601. [DOI] [PubMed] [Google Scholar]
- 63.Ryan B.J., Yang H., Bacurio J.H.T., Smith M.R., Basu A.K., Greenberg M.M., Freudenthal B.D. Structural Dynamics of a Common Mutagenic Oxidative DNA Lesion in Duplex DNA and during DNA Replication. J. Am. Chem. Soc. 2022;144:8054–8065. doi: 10.1021/jacs.2c00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burgos-Morón E., Pastor N., Orta M.L., Jiménez-Alonso J.J., Palo-Nieto C., Vega-Holm M., Vega-Pérez J.M., Iglesias-Guerra F., Mateos S., López-Lázaro M., et al. In Vitro Anticancer Activity and Mechanism of Action of an Aziridinyl Galactopyranoside. Biomedicines. 2022;10:41. doi: 10.3390/biomedicines10010041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eder J.P., Elias A., Shea T.C., Schryber S.M., Teicher B.A., Hunt M., Burke J., Siegel R., Schnipper L., Frei E., 3rd A phase I-II study of cyclophosphamide, thiotepa, and carboplatin with autologous bone marrow transplantation in solid tumor patients. J. Clin. Oncol. 1990;8:1239–1245. doi: 10.1200/JCO.1990.8.7.1239. [DOI] [PubMed] [Google Scholar]
- 66.Langer C.J., Nash S., Catalano R., Rosenblum N.G., Hogan W.M., Comis R.L., O’Dwyer P.J. Phase II trial of thio-TEPA in relapsed and refractory ovarian carcinoma. Gynecol. Oncol. 1991;43:242–246. doi: 10.1016/0090-8258(91)90028-4. [DOI] [PubMed] [Google Scholar]
- 67.Teicher B.A., Herman T.S., Holden S.A., Epelbaum R., Liu S.-d., Frei E., III. Lonidamine as a modulator of alkylating agent activity in vitro and in vivo. Cancer Res. 1991;51:780–784. [PubMed] [Google Scholar]
- 68.Miller B., Tenenholz T., Egorin M.J., Sosnovsky G., Rao N.U.M., Gutierrez P.L. Cellular pharmacology of N, N′, N ″-triethylene thiophosphoramide. Cancer Lett. 1988;41:157–168. doi: 10.1016/0304-3835(88)90112-7. [DOI] [PubMed] [Google Scholar]
- 69.MILLER D.G. ALKYLATING AGENTS AND HUMAN SPERMATOGENESIS. Obstet. Gynecol. Surv. 1972;27:150–151. doi: 10.1097/00006254-197203000-00003. [DOI] [Google Scholar]
- 70.Van Maanen M., Smeets C., Beijnen J. Chemistry, pharmacology and pharmacokinetics of N, N′, N′′-triethylenethiophosphoramide (ThioTEPA) Cancer Treat. Rev. 2000;26:257–268. doi: 10.1053/ctrv.2000.0170. [DOI] [PubMed] [Google Scholar]
- 71.Beall H.D., Winski S. Mechanisms of action of quinone-containing alkylating agents. I: NQO1-directed drug development. Front. Biosci. 2000;5:D639–D648. doi: 10.2741/beall. [DOI] [PubMed] [Google Scholar]
- 72.Begleiter A. Clinical applications of quinone-containing alkylating agents. Front. Biosci. 2000;5:153–171. doi: 10.2741/begleit. [DOI] [PubMed] [Google Scholar]
- 73.Bass P.D., Gubler D.A., Judd T.C., Williams R.M. Mitomycinoid alkaloids: Mechanism of action, biosynthesis, total syntheses, and synthetic approaches. Chem. Rev. 2013;113:6816–6863. doi: 10.1021/cr3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Puyo S., Montaudon D., Pourquier P. From old alkylating agents to new minor groove binders. Crit. Rev. Oncol./Hematol. 2014;89:43–61. doi: 10.1016/j.critrevonc.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 75.DeVita V.T., Lawrence T.S., Rosenberg S.A. DeVita, Hellman, and Rosenberg’s Cancer: Principles & Practice of Oncology. Volume 2 Lippincott Williams & Wilkins; Philadelphia, PA, USA: 2008. [Google Scholar]
- 76.Haffty B.G., Son Y.H., Wilson L.D., Papac R., Fischer D., Rockwell S., Sartorelli A.C., Ross D., Sasaki C.T., Fischer J.J. Bioreductive alkylating agent porfiromycin in combination with radiation therapy for the management of squamous cell carcinoma of the head and neck. Radiat. Oncol. Investig. Clin. Basic Res. 1997;5:235–245. doi: 10.1002/(SICI)1520-6823(1997)5:5<235::AID-ROI4>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 77.Wolkenberg S.E., Boger D.L. Mechanisms of in situ activation for DNA-targeting antitumor agents. Chem. Rev. 2002;102:2477–2496. doi: 10.1021/cr010046q. [DOI] [PubMed] [Google Scholar]
- 78.Paz M.M., Pritsos C.A. Chapter Seven-The Molecular Toxicology of Mitomycin C. In: Fishbein J.C., editor. Advances in Molecular Toxicology. Volume 6. Elsevier; Amsterdam, The Netherlands: 2012. pp. 243–299. [Google Scholar]
- 79.Andrés C.M.C., Pérez de la Lastra J.M., Bustamante Munguira E., Andrés Juan C., Pérez-Lebeña E. Michael Acceptors as Anti-Cancer Compounds: Coincidence or Causality? Int. J. Mol. Sci. 2024;25:6099. doi: 10.3390/ijms25116099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tomasz M., Palom Y. The mitomycin bioreductive antitumor agents: Cross-linking and alkylation of DNA as the molecular basis of their activity. Pharmacol. Ther. 1997;76:73–87. doi: 10.1016/S0163-7258(97)00088-0. [DOI] [PubMed] [Google Scholar]
- 81.Tomasz M., Chawla A.K., Lipman R. Mechanism of monofunctional and bifunctional alkylation of DNA by mitomycin C. Biochemistry. 1988;27:3182–3187. doi: 10.1021/bi00409a009. [DOI] [PubMed] [Google Scholar]
- 82.Eagan R.T., Dinapoli R.P., Cascino T.L., Scheithauer B., O’Neill B.P., O’Fallon J.R. Comprehensive phase II evaluation of aziridinylbenzoquinone (AZQ, diaziquone) in recurrent human primary brain tumors. J. Neuro-Oncol. 1987;5:309–314. doi: 10.1007/BF00148387. [DOI] [PubMed] [Google Scholar]
- 83.Obe G., Beek B. Trenimon: Biochemical, physiological and genetic effects on cells and organisms. Mutat. Res. 1979;65:21–70. doi: 10.1016/0165-1110(79)90012-5. [DOI] [PubMed] [Google Scholar]
- 84.Phillips R.M., Hendriks H.R., Peters G.J. EO9 (Apaziquone): From the clinic to the laboratory and back again. Br. J. Pharmacol. 2013;168:11–18. doi: 10.1111/j.1476-5381.2012.01996.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pierce S.E., Guziec L.J., Guziec F.S., Jr., Brodbelt J.S. Characterization of Aziridinylbenzoquinone DNA Cross-Links by Liquid Chromatography− Infrared Multiphoton Dissociation− Mass Spectrometry. Chem. Res. Toxicol. 2010;23:1097–1104. doi: 10.1021/tx1000738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hargreaves R., Hartley J.A., Butler J. Mechanisms of action of quinone-containing alkylating agents: DNA alkylation by aziridinylquinones. Front. Biosci. 2000;5:E172–E180. doi: 10.2741/hargreav. [DOI] [PubMed] [Google Scholar]
- 87.Naylor M., Thomson P. Recent advances in bioreductive drug targeting. Mini Rev. Med. Chem. 2001;1:17–29. doi: 10.2174/1389557013407241. [DOI] [PubMed] [Google Scholar]
- 88.Fourie J., Guziec F., Jr., Guziec L., Monterrosa C., Fiterman D.J., Begleiter A. Structure-activity study with bioreductive benzoquinone alkylating agents: Effects on DT-diaphorase-mediated DNA crosslink and strand break formation in relation to mechanisms of cytotoxicity. Cancer Chemother. Pharmacol. 2004;53:191–203. doi: 10.1007/s00280-003-0718-5. [DOI] [PubMed] [Google Scholar]
- 89.Danson S., Ward T.H., Butler J., Ranson M. DT-diaphorase: A target for new anticancer drugs. Cancer Treat. Rev. 2004;30:437–449. doi: 10.1016/j.ctrv.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 90.Denny W.A. Prodrug strategies in cancer therapy. Eur. J. Med. Chem. 2001;36:577–595. doi: 10.1016/S0223-5234(01)01253-3. [DOI] [PubMed] [Google Scholar]
- 91.Zhou R., Skibo E.B. Chemistry of the pyrrolo [1, 2-a] benzimidazole antitumor agents: Influence of the 7-substituent on the ability to alkylate DNA and inhibit topoisomerase II. J. Med. Chem. 1996;39:4321–4331. doi: 10.1021/jm960064d. [DOI] [PubMed] [Google Scholar]
- 92.Li B., Gutierrez P.L., Amstad P., Blough N.V. Hydroxyl radical production by mouse epidermal cell lines in the presence of quinone anti-cancer compounds. Chem. Res. Toxicol. 1999;12:1042–1049. doi: 10.1021/tx990095m. [DOI] [PubMed] [Google Scholar]
- 93.Hartley J., O’hare C., Baumgart J. DNA alkylation and interstrand cross-linking by treosulfan. Br. J. Cancer. 1999;79:264–266. doi: 10.1038/sj.bjc.6690043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Szebeni J., Barna K., Uher F., Milosevits J., Pálóczi K., Gaál D., Petrányi G., Kelemen E. Comparison of the lymphoid toxicities of mitobronitol and busulphan in mice: Reduced B cell toxicity and improved thymic recovery as possible contributors to the reduced risk for complications following BMT with mitobronitol preconditioning. Leukemia. 1997;11:1769–1774. doi: 10.1038/sj.leu.2400741. [DOI] [PubMed] [Google Scholar]
- 95.Schepartz S.A. Early history and development of the nitrosoureas. Cancer Treat. Rep. 1976;60:647. [PubMed] [Google Scholar]
- 96.Kohn K.W. Interstrand cross-linking of DNA by 1, 3-bis (2-chloroethyl)-1-nitrosourea and other 1-(2-haloethyl)-1-nitrosoureas. Cancer Res. 1977;37:1450–1454. [PubMed] [Google Scholar]
- 97.Panasci L.C., Green D., Nagourney R., Fox P., Schein P.S. A structure-activity analysis of chemical and biological parameters of chloroethylnitrosoureas in mice. Cancer Res. 1977;37:2615–2618. [PubMed] [Google Scholar]
- 98.Samson M., Baker L., Cummings G., Talley R., McDonald B., Bhathena D. Clinical trial of chlorozotocin, DTIC, and dactinomycin in metastatic malignant melanoma. Cancer Treat. Rep. 1982;66:371–373. [PubMed] [Google Scholar]
- 99.Montgomery J.A., James R., McCaleb G.S., Johnston T.P. The modes of decomposition of 1, 3-bis (2-chloroethyl)-1-nitrosourea and related compounds. J. Med. Chem. 1967;10:668–674. doi: 10.1021/jm00316a033. [DOI] [PubMed] [Google Scholar]
- 100.Buggia I., Locatelli F., Regazzi M.B., Zecca M. Busulfan. Ann. Pharmacother. 1994;28:1055–1062. doi: 10.1177/106002809402800911. [DOI] [PubMed] [Google Scholar]
- 101.Krivoy N., Hoffer E., Lurie Y., Bentur Y., Rowe J.M. Busulfan use in hematopoietic stem cell transplantation: Pharmacology, dose adjustment, safety and efficacy in adults and children. Curr. Drug Saf. 2008;3:60–66. doi: 10.2174/157488608783333899. [DOI] [PubMed] [Google Scholar]
- 102.Iwamoto T., Hiraku Y., Oikawa S., Mizutani H., Kojima M., Kawanishi S. DNA intrastrand cross-link at the 5′-GA-3′ sequence formed by busulfan and its role in the cytotoxic effect. Cancer Sci. 2004;95:454–458. doi: 10.1111/j.1349-7006.2004.tb03231.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Myers A.L., Kawedia J.D., Champlin R.E., Kramer M.A., Nieto Y., Ghose R., Andersson B.S. Clarifying busulfan metabolism and drug interactions to support new therapeutic drug monitoring strategies: A comprehensive review. Expert Opin. Drug Metab. Toxicol. 2017;13:901–923. doi: 10.1080/17425255.2017.1360277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Farmer P. Metabolism and reactions of alkylating agents. Pharmacol. Ther. 1987;35:301–358. doi: 10.1016/0163-7258(87)90099-4. [DOI] [PubMed] [Google Scholar]
- 105.Bedford P., Fox B.W. DNA-DNA interstrand crosslinking by dimethanesulphonic acid esters: Correlation with cytotoxicity and antitumour activity in the yoshida lymphosarcoma model and relationship to chain length. Biochem. Pharmacol. 1983;32:2297–2301. doi: 10.1016/0006-2952(83)90176-4. [DOI] [PubMed] [Google Scholar]
- 106.Serrone L., Zeuli M., Sega F.M., Cognetti F. Dacarbazine-based chemotherapy for metastatic melanoma: Thirty-year experience overview. J. Exp. Clin. Cancer Res. 2000;19:21–34. [PubMed] [Google Scholar]
- 107.Reid J.M., Kuffel M.J., Miller J.K., Rios R., Ames M.M. Metabolic activation of dacarbazine by human cytochromes P450: The role of CYP1A1, CYP1A2, and CYP2E1. Clin. Cancer Res. 1999;5:2192–2197. [PubMed] [Google Scholar]
- 108.Nussbaumer S., Bonnabry P., Veuthey J.L., Fleury-Souverain S. Analysis of anticancer drugs: A review. Talanta. 2011;85:2265–2289. doi: 10.1016/j.talanta.2011.08.034. [DOI] [PubMed] [Google Scholar]
- 109.Moody C.L., Wheelhouse R.T. The medicinal chemistry of imidazotetrazine prodrugs. Pharmaceuticals. 2014;7:797–838. doi: 10.3390/ph7070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.García-del-Muro X., López-Pousa A., Maurel J., Martín J., Martínez-Trufero J., Casado A., Gómez-España A., Fra J., Cruz J., Poveda A. Randomized phase II study comparing gemcitabine plus dacarbazine versus dacarbazine alone in patients with previously treated soft tissue sarcoma: A Spanish Group for Research on Sarcomas study. J. Clin. Oncol. 2011;29:2528–2533. doi: 10.1200/JCO.2010.33.6107. [DOI] [PubMed] [Google Scholar]
- 111.Bouraoui S., Brahem A., Tabka F., Mrizek N., Saad A., Elghezal H. Assessment of chromosomal aberrations, micronuclei and proliferation rate index in peripheral lymphocytes from Tunisian nurses handling cytotoxic drugs. Environ. Toxicol. Pharmacol. 2011;31:250–257. doi: 10.1016/j.etap.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 112.Meer L., Janzer R.C., Kleihues P., Kolar G.F. In vivo metabolism and reaction with DNA of the cytostatic agent, 5-(3,3-dimethyl-1-triazeno)imidazole-4-carboxamide (DTIC) Biochem. Pharmacol. 1986;35:3243–3247. doi: 10.1016/0006-2952(86)90419-3. [DOI] [PubMed] [Google Scholar]
- 113.Kyrtopoulos S.A., Souliotis V.L., Valavanis C., Boussiotis V.A., Pangalis G.A. Accumulation of O6-methylguanine in human DNA after therapeutic exposure to methylating agents and its relationship with biological effects. Environ. Health Perspect. 1993;99:143–147. doi: 10.1289/ehp.9399143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Horton J.K., Stevens M.F. A new light on the photo-decomposition of the antitumour drug DTIC. J. Pharm. Pharmacol. 1981;33:808–811. doi: 10.1111/j.2042-7158.1981.tb13944.x. [DOI] [PubMed] [Google Scholar]
- 115.Iwamoto T., Hiraku Y., Okuda M., Kawanishi S. Mechanism of UVA-dependent DNA damage induced by an antitumor drug dacarbazine in relation to its photogenotoxicity. Pharm. Res. 2008;25:598–604. doi: 10.1007/s11095-007-9413-2. [DOI] [PubMed] [Google Scholar]
- 116.Newlands E.S., Stevens M.F., Wedge S.R., Wheelhouse R.T., Brock C. Temozolomide: A review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat. Rev. 1997;23:35–61. doi: 10.1016/S0305-7372(97)90019-0. [DOI] [PubMed] [Google Scholar]
- 117.Baker S.D., Wirth M., Statkevich P., Reidenberg P., Alton K., Sartorius S.E., Dugan M., Cutler D., Batra V., Grochow L.B., et al. Absorption, metabolism, and excretion of 14C-temozolomide following oral administration to patients with advanced cancer. Clin. Cancer Res. 1999;5:309–317. [PubMed] [Google Scholar]
- 118.Turci R., Sottani C., Spagnoli G., Minoia C. Biological and environmental monitoring of hospital personnel exposed to antineoplastic agents: A review of analytical methods. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2003;789:169–209. doi: 10.1016/S1570-0232(03)00100-4. [DOI] [PubMed] [Google Scholar]
- 119.Ioannou-Ttofa L., Fatta-Kassinos D. Fate and Effects of Anticancer Drugs in the Environment. Springer; Cham, Switzerland: 2020. Cytostatic drug residues in wastewater treatment plants: Sources, removal efficiencies and current challenges; pp. 103–138. [Google Scholar]
- 120.Li D., Chen H., Liu H., Schlenk D., Mu J., Lacorte S., Ying G.-G., Xie L. Anticancer drugs in the aquatic ecosystem: Environmental occurrence, ecotoxicological effect and risk assessment. Environ. Int. 2021;153:106543. doi: 10.1016/j.envint.2021.106543. [DOI] [PubMed] [Google Scholar]
- 121.Tripathi A.K., David A., Govil T., Rauniyar S., Rathinam N.K., Goh K.M., Sani R.K. Environmental Remediation of Antineoplastic Drugs: Present Status, Challenges, and Future Directions. Processes. 2020;8:747. doi: 10.3390/pr8070747. [DOI] [Google Scholar]
- 122.Tornesello A.L., Borrelli A., Buonaguro L., Buonaguro F.M., Tornesello M.L. Antimicrobial peptides as anticancer agents: Functional properties and biological activities. Molecules. 2020;25:2850. doi: 10.3390/molecules25122850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mader J.S., Hoskin D.W. Cationic antimicrobial peptides as novel cytotoxic agents for cancer treatment. Expert Opin. Investig. Drugs. 2006;15:933–946. doi: 10.1517/13543784.15.8.933. [DOI] [PubMed] [Google Scholar]
- 124.Roudi R., Syn N.L., Roudbary M. Antimicrobial peptides as biologic and immunotherapeutic agents against cancer: A comprehensive overview. Front. Immunol. 2017;8:1320. doi: 10.3389/fimmu.2017.01320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Deslouches B., Di Y.P. Antimicrobial peptides with selective antitumor mechanisms: Prospect for anticancer applications. Oncotarget. 2017;8:46635. doi: 10.18632/oncotarget.16743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Erdem Büyükkiraz M., Kesmen Z. Antimicrobial peptides (AMPs): A promising class of antimicrobial compounds. J. Appl. Microbiol. 2022;132:1573–1596. doi: 10.1111/jam.15314. [DOI] [PubMed] [Google Scholar]
- 127.Wang G.S. Antimicrobial Peptides: Discovery, Design and Novel Therapeutic Strategies. Cabi; Wallingford, UK: 2017. [Google Scholar]
- 128.Narayana J.L., Chen J.-Y. Antimicrobial peptides: Possible anti-infective agents. Peptides. 2015;72:88–94. doi: 10.1016/j.peptides.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 129.Li S., Wang Y., Xue Z., Jia Y., Li R., He C., Chen H. The structure-mechanism relationship and mode of actions of antimicrobial peptides: A review. Trends Food Sci. Technol. 2021;109:103–115. [Google Scholar]
- 130.Decker A.P., Mechesso A.F., Wang G. Expanding the landscape of amino acid-rich antimicrobial peptides: Definition, deployment in nature, implications for peptide design and therapeutic potential. Int. J. Mol. Sci. 2022;23:12874. doi: 10.3390/ijms232112874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Luong H.X., Thanh T.T., Tran T.H. Antimicrobial peptides–Advances in development of therapeutic applications. Life Sci. 2020;260:118407. doi: 10.1016/j.lfs.2020.118407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ramos-Martín F., Herrera-León C., d’Amelio N. Molecular basis of the anticancer, apoptotic and antibacterial activities of Bombyx mori Cecropin A. Arch. Biochem. Biophys. 2022;715:109095. doi: 10.1016/j.abb.2021.109095. [DOI] [PubMed] [Google Scholar]
- 133.de Almeida Gomes I., da Lima A.B., da Silva Brito D.M., Almeida Lima A., de Oliveira F.L., Espino Zelaya E.A., Magalhães Rebello Alencar L., Castelo Branco de Souza Collares Maia D., Amaral de Moraes M.E., Pantoja Mesquita F. Recalculating the Route: Repositioning Antimicrobial Peptides for Cancer Treatment. Chem. Biodivers. 2024;21:e202301840. doi: 10.1002/cbdv.202301840. [DOI] [PubMed] [Google Scholar]
- 134.Tolos A.M., Moisa C., Dochia M., Popa C., Copolovici L., Copolovici D.M. Anticancer potential of antimicrobial peptides: Focus on buforins. Polymers. 2024;16:728. doi: 10.3390/polym16060728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kanaujia K.A., Mishra N., Rajinikanth P., Saraf S.A. Antimicrobial peptides as antimicrobials for wound care management: A comprehensive review. J. Drug Deliv. Sci. Technol. 2024;95:105570. doi: 10.1016/j.jddst.2024.105570. [DOI] [Google Scholar]
- 136.van den Boogaard W.M., Komninos D.S., Vermeij W.P. Chemotherapy side-effects: Not all DNA damage is equal. Cancers. 2022;14:627. doi: 10.3390/cancers14030627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Toribio M., Campos E.L., Monteiro G.J., Henriques J.C.G., Gonzaga W.J.C., Lorenzon E.N., Miranda C.S.S. The combination of peptides and cisplatin for the treatment of oral cancer. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2024;137:e308. doi: 10.1016/j.oooo.2023.12.740. [DOI] [Google Scholar]
- 138.Khine H.E.E., Ecoy G.A.U., Roytrakul S., Phaonakrop N., Pornputtapong N., Prompetchara E., Chanvorachote P., Chaotham C. Chemosensitizing activity of peptide from Lentinus squarrosulus (Mont.) on cisplatin-induced apoptosis in human lung cancer cells. Sci. Rep. 2021;11:4060. doi: 10.1038/s41598-021-83606-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bhattacharjee R., Dey T., Kumar L., Kar S., Sarkar R., Ghorai M., Malik S., Jha N.K., Vellingiri B., Kesari K.K. Cellular landscaping of cisplatin resistance in cervical cancer. Biomed. Pharmacother. 2022;153:113345. doi: 10.1016/j.biopha.2022.113345. [DOI] [PubMed] [Google Scholar]
- 140.Aghamiri S., Zandsalimi F., Raee P., Abdollahifar M.-A., Tan S.C., Low T.Y., Najafi S., Ashrafizadeh M., Zarrabi A., Ghanbarian H. Antimicrobial peptides as potential therapeutics for breast cancer. Pharmacol. Res. 2021;171:105777. doi: 10.1016/j.phrs.2021.105777. [DOI] [PubMed] [Google Scholar]
- 141.Teng Q.-X., Luo X., Lei Z.-N., Wang J.-Q., Wurpel J., Qin Z., Yang D.-H. The multidrug resistance-reversing activity of a novel antimicrobial peptide. Cancers. 2020;12:1963. doi: 10.3390/cancers12071963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jafari A., Babajani A., Sarrami Forooshani R., Yazdani M., Rezaei-Tavirani M. Clinical applications and anticancer effects of antimicrobial peptides: From bench to bedside. Front. Oncol. 2022;12:819563. doi: 10.3389/fonc.2022.819563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Seyfi R., Kahaki F.A., Ebrahimi T., Montazersaheb S., Eyvazi S., Babaeipour V., Tarhriz V. Antimicrobial peptides (AMPs): Roles, functions and mechanism of action. Int. J. Pept. Res. Ther. 2020;26:1451–1463. doi: 10.1007/s10989-019-09946-9. [DOI] [Google Scholar]
- 144.Aria H., Rezaei M. Immunogenic cell death inducer peptides: A new approach for cancer therapy, current status and future perspectives. Biomed. Pharmacother. 2023;161:114503. doi: 10.1016/j.biopha.2023.114503. [DOI] [PubMed] [Google Scholar]
- 145.Tripathi A.K., Vishwanatha J.K. Role of anti-cancer peptides as immunomodulatory agents: Potential and design strategy. Pharmaceutics. 2022;14:2686. doi: 10.3390/pharmaceutics14122686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kardani K., Bolhassani A. Antimicrobial/anticancer peptides: Bioactive molecules and therapeutic agents. Immunotherapy. 2021;13:669–684. doi: 10.2217/imt-2020-0312. [DOI] [PubMed] [Google Scholar]
- 147.Al-Ostoot F.H., Salah S., Khamees H.A., Khanum S.A. Tumor angiogenesis: Current challenges and therapeutic opportunities. Cancer Treat. Res. Commun. 2021;28:100422. doi: 10.1016/j.ctarc.2021.100422. [DOI] [PubMed] [Google Scholar]
- 148.Hu Q., Chen C., Lin Z., Zhang L., Guan S., Zhuang X., Dong G., Shen J. The antimicrobial peptide Esculentin-1a (1–21) NH2 stimulates wound healing by promoting angiogenesis through the PI3K/AKT pathway. Biol. Pharm. Bull. 2023;46:382–393. doi: 10.1248/bpb.b22-00098. [DOI] [PubMed] [Google Scholar]
- 149.Kang M.J., Roh K.-H., Lee J.S., Lee J.H., Park S., Lim D.W. Vascular Endothelial Growth Factor Receptor 1 Targeting Fusion Polypeptides with Stimuli-Responsiveness for Anti-angiogenesis. ACS Appl. Mater. Interfaces. 2023;15:32201–32214. doi: 10.1021/acsami.3c03989. [DOI] [PubMed] [Google Scholar]
- 150.Del Genio V., Bellavita R., Falanga A., Hervé-Aubert K., Chourpa I., Galdiero S. Peptides to overcome the limitations of current anticancer and antimicrobial nanotherapies. Pharmaceutics. 2022;14:1235. doi: 10.3390/pharmaceutics14061235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Basak D., Arrighi S., Darwiche Y., Deb S. Comparison of anticancer drug toxicities: Paradigm shift in adverse effect profile. Life. 2021;12:48. doi: 10.3390/life12010048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Szlasa W., Zendran I., Zalesińska A., Tarek M., Kulbacka J. Lipid composition of the cancer cell membrane. J. Bioenerg. Biomembr. 2020;52:321–342. doi: 10.1007/s10863-020-09846-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Vedadghavami A., Zhang C., Bajpayee A.G. Overcoming negatively charged tissue barriers: Drug delivery using cationic peptides and proteins. Nano Today. 2020;34:100898. doi: 10.1016/j.nantod.2020.100898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Divyashree M., Mani M.K., Reddy D., Kumavath R., Ghosh P., Azevedo V., Barh D. Clinical applications of antimicrobial peptides (AMPs): Where do we stand now? Protein Pept. Lett. 2020;27:120–134. doi: 10.2174/0929866526666190925152957. [DOI] [PubMed] [Google Scholar]
- 155.Chernov A.N., Kim A.V., Skliar S.S., Fedorov E.V., Tsapieva A.N., Filatenkova T.A., Chutko A.L., Matsko M.V., Galimova E.S., Shamova O.V. Expression of molecular markers and synergistic anticancer effects of chemotherapy with antimicrobial peptides on glioblastoma cells. Cancer Chemother. Pharmacol. 2024;93:455–469. doi: 10.1007/s00280-023-04622-8. [DOI] [PubMed] [Google Scholar]
- 156.Jana A., Narula P., Chugh A., Kulshreshtha R. Efficient delivery of anti-miR-210 using Tachyplesin, a cell penetrating peptide, for glioblastoma treatment. Int. J. Pharm. 2019;572:118789. doi: 10.1016/j.ijpharm.2019.118789. [DOI] [PubMed] [Google Scholar]
- 157.Wu J., Chen X., Zhang J., Chen J., Wang Y., Wei T., Ma J., Li Y., Mo T., He Z., et al. Tachyplesin induces apoptosis in non-small cell lung cancer cells and enhances the chemosensitivity of A549/DDP cells to cisplatin by activating Fas and necroptosis pathway. Chem. Biol. Drug Des. 2021;97:809–820. doi: 10.1111/cbdd.13810. [DOI] [PubMed] [Google Scholar]
- 158.Lin M.-C., Lin S.-B., Chen J.-C., Hui C.-F., Chen J.-Y. Shrimp anti-lipopolysaccharide factor peptide enhances the antitumor activity of cisplatin in vitro and inhibits HeLa cells growth in nude mice. Peptides. 2010;31:1019–1025. doi: 10.1016/j.peptides.2010.02.023. [DOI] [PubMed] [Google Scholar]
- 159.Chen L., Wang Q., Jiang Y., Xu L., Wei N., Lu C., Chang C., Song D., Wang Y., Wu L., et al. A novel anti-angiogenesis peptide in combination with cisplatin self-assembling into tube-like nanomedicine for oral treatment of gastric cancer. Chem. Eng. J. 2024;496:154169. doi: 10.1016/j.cej.2024.154169. [DOI] [Google Scholar]
- 160.Luo X., Teng Q.-X., Dong J.-Y., Yang D.-H., Wang M., Dessie W., Qin J.-J., Lei Z.-N., Wang J.-Q., Qin Z. Antimicrobial peptide reverses ABCB1-mediated chemotherapeutic drug resistance. Front. Pharmacol. 2020;11:1208. doi: 10.3389/fphar.2020.01208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Teixeira M.C., Carbone C., Sousa M.C., Espina M., Garcia M.L., Sanchez-Lopez E., Souto E.B. Nanomedicines for the delivery of antimicrobial peptides (AMPs) Nanomaterials. 2020;10:560. doi: 10.3390/nano10030560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Parchebafi A., Tamanaee F., Ehteram H., Ahmad E., Nikzad H., Haddad Kashani H. The dual interaction of antimicrobial peptides on bacteria and cancer cells; mechanism of action and therapeutic strategies of nanostructures. Microb. Cell Factories. 2022;21:118. doi: 10.1186/s12934-022-01848-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gera S., Kankuri E., Kogermann K. Antimicrobial peptides–unleashing their therapeutic potential using nanotechnology. Pharmacol. Ther. 2022;232:107990. doi: 10.1016/j.pharmthera.2021.107990. [DOI] [PubMed] [Google Scholar]
- 164.Tang Z., Ma Q., Chen X., Chen T., Ying Y., Xi X., Wang L., Ma C., Shaw C., Zhou M. Recent advances and challenges in nanodelivery systems for antimicrobial peptides (AMPs) Antibiotics. 2021;10:990. doi: 10.3390/antibiotics10080990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ghafari M., Haghiralsadat F., Khanamani Falahati-pour S., Zavar Reza J. Development of a novel liposomal nanoparticle formulation of cisplatin to breast cancer therapy. J. Cell. Biochem. 2020;121:3584–3592. doi: 10.1002/jcb.29651. [DOI] [PubMed] [Google Scholar]
- 166.Tsvetkova D., Ivanova S. Application of approved cisplatin derivatives in combination therapy against different cancer diseases. Molecules. 2022;27:2466. doi: 10.3390/molecules27082466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Renault-Mahieux M., Vieillard V., Seguin J., Espeau P., Le D.T., Lai-Kuen R., Mignet N., Paul M., Andrieux K. Co-encapsulation of fisetin and cisplatin into liposomes for glioma therapy: From formulation to cell evaluation. Pharmaceutics. 2021;13:970. doi: 10.3390/pharmaceutics13070970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Tan X., Wang Y., Wu Z., Zhou Q., Tang Y., Liu Z., Yuan G., Luo S., Zou Y., Guo S. The role of Her-2 in penile squamous cell carcinoma progression and cisplatin chemoresistance and potential for antibody-drug conjugate-based therapy. Eur. J. Cancer. 2023;194:113360. doi: 10.1016/j.ejca.2023.113360. [DOI] [PubMed] [Google Scholar]
- 169.Fuentes-Antrás J., Genta S., Vijenthira A., Siu L.L. Antibody–drug conjugates: In search of partners of choice. Trends Cancer. 2023;9:339–354. doi: 10.1016/j.trecan.2023.01.003. [DOI] [PubMed] [Google Scholar]
- 170.Işık G. Ph.D. Thesis. Middle East Technical University; Ankara, Turkey: 2022. Development and Characterization of PEG-B-PCL Micelles Carrying Anticancer Agents. [Google Scholar]
- 171.Xia W., Tao Z., Zhu B., Zhang W., Liu C., Chen S., Song M. Targeted delivery of drugs and genes using polymer nanocarriers for cancer therapy. Int. J. Mol. Sci. 2021;22:9118. doi: 10.3390/ijms22179118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Eskandari R., Asoodeh A., Mousavi S.-D., Firouzi Z. The effect of a novel drug delivery system using encapsulated antimicrobial peptide protonectin (IL-12) into nano micelle PEG-PCL on A549 adenocarcinoma lung cell line. J. Polym. Res. 2021;28:341. doi: 10.1007/s10965-021-02699-4. [DOI] [Google Scholar]
- 173.Gessner I., Neundorf I. Nanoparticles modified with cell-penetrating peptides: Conjugation mechanisms, physicochemical properties, and application in cancer diagnosis and therapy. Int. J. Mol. Sci. 2020;21:2536. doi: 10.3390/ijms21072536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Zhou M., Zou X., Cheng K., Zhong S., Su Y., Wu T., Tao Y., Cong L., Yan B., Jiang Y. The role of cell-penetrating peptides in potential anti-cancer therapy. Clin. Transl. Med. 2022;12:e822. doi: 10.1002/ctm2.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Singh T., Kang D.H., Kim T.W., Kong H.J., Ryu J.S., Jeon S., Ahn T.S., Jeong D., Baek M.J., Im J. Intracellular delivery of oxaliplatin conjugate via cell penetrating peptide for the treatment of colorectal carcinoma in vitro and in vivo. Int. J. Pharm. 2021;606:120904. doi: 10.1016/j.ijpharm.2021.120904. [DOI] [PubMed] [Google Scholar]
- 176.Izabela R., Jarosław R., Magdalena A., Piotr R., Ivan K. Transportan 10 improves the anticancer activity of cisplatin. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2016;389:485–497. doi: 10.1007/s00210-016-1219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Śmiłowicz D., Metzler-Nolte N. Bioconjugates of Co(III) complexes with Schiff base ligands and cell penetrating peptides: Solid phase synthesis, characterization and antiproliferative activity. J. Inorg. Biochem. 2020;206:111041. doi: 10.1016/j.jinorgbio.2020.111041. [DOI] [PubMed] [Google Scholar]
- 178.Jiang M., Fang X., Ma L., Liu M., Chen M., Liu J., Yang Y., Wang C. A nucleus-targeting peptide antagonist towards EZH2 displays therapeutic efficacy for lung cancer. Int. J. Pharm. 2022;622:121894. doi: 10.1016/j.ijpharm.2022.121894. [DOI] [PubMed] [Google Scholar]
- 179.Safhi A.Y. Three-dimensional (3D) printing in cancer therapy and diagnostics: Current status and future perspectives. Pharmaceuticals. 2022;15:678. doi: 10.3390/ph15060678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zhu H., Zheng J., Oh X.Y., Chan C.Y., Low B.Q.L., Tor J.Q., Jiang W., Ye E., Loh X.J., Li Z. Nanoarchitecture-integrated hydrogel systems toward therapeutic applications. ACS Nano. 2023;17:7953–7978. doi: 10.1021/acsnano.2c12448. [DOI] [PubMed] [Google Scholar]
- 181.Binaymotlagh R., Chronopoulou L., Haghighi F.H., Fratoddi I., Palocci C. Peptide-based hydrogels: New materials for biosensing and biomedical applications. Materials. 2022;15:5871. doi: 10.3390/ma15175871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chinnaiyan K., Mugundhan Laakshmi S., Narayanasamy D., Mohan M. Revolutionizing Healthcare and Drug Discovery: The Impact of Artificial Intelligence on Pharmaceutical Development. Curr. Drug Ther. 2024;19:1–16. doi: 10.2174/0115748855313948240711043701. [DOI] [Google Scholar]
- 183.Saraf S., De A., Tripathy B. Computational Intelligence for Oncology and Neurological Disorders. CRC Press; Boca Raton, FL, USA: 2024. Effective Use of Computational Biology and Artificial Intelligence in the Domain of Medical Oncology; pp. 228–252. [Google Scholar]
- 184.Dlamini Z., Francies F.Z., Hull R., Marima R. Artificial intelligence (AI) and big data in cancer and precision oncology. Comput. Struct. Biotechnol. J. 2020;18:2300–2311. doi: 10.1016/j.csbj.2020.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Riaz I.B., Khan M.A., Haddad T.C. Potential application of artificial intelligence in cancer therapy. Curr. Opin. Oncol. 2024;36:437–448. doi: 10.1097/CCO.0000000000001068. [DOI] [PubMed] [Google Scholar]
- 186.Kim J., Kusko R., Zeskind B., Zhang J., Escalante-Chong R. A primer on applying AI synergistically with domain expertise to oncology. Biochim. Biophys. Acta (BBA)-Rev. Cancer. 2021;1876:188548. doi: 10.1016/j.bbcan.2021.188548. [DOI] [PubMed] [Google Scholar]
- 187.Mukherjee D., Roy D., Thakur S. Transforming Cancer Care: The Impact of AI-Driven Strategies. Curr. Cancer Drug Targets. 2024;24:1–4. doi: 10.2174/0115680096323564240703102748. [DOI] [PubMed] [Google Scholar]