

Abstract
Abdominal aortic aneurysm (AAA) formation is a chronic vascular pathology characterized by inflammation, leukocyte infiltration and vascular remodeling. The aim of this study was to delineate the protective role of Resolvin D2 (RvD2), a bioactive isoform of specialized proresolving lipid mediators, via G-protein coupled receptor 18 (GPR18) receptor signaling in attenuating AAAs. Importantly, RvD2 and GPR18 levels were significantly decreased in aortic tissue of AAA patients compared with controls. Furthermore, using an established murine model of AAA in C57BL/6 (WT) mice, we observed that treatment with RvD2 significantly attenuated aortic diameter, pro-inflammatory cytokine production, immune cell infiltration (neutrophils and macrophages), elastic fiber disruption and increased smooth muscle cell α-actin expression as well as increased TGF-β2 and IL-10 expressions compared to untreated mice. Moreover, the RvD2-mediated protection from vascular remodeling and AAA formation was blocked when mice were previously treated with siRNA for GPR18 signifying the importance of RvD2/GPR18 signaling in vascular inflammation. Mechanistically, RvD2-mediated protection significantly enhanced infiltration and activation of monocytic myeloid-derived suppressor cells (M-MDSCs) by increasing TGF-β2 and IL-10 secretions in a GPR18-dependent manner to attenuate aortic inflammation and vascular remodeling. Collectively, this study demonstrates that RvD2 treatment induces an expansion of myeloid-lineage committed progenitors, such as M-MDSCs, activates GPR18-dependent signaling to enhance TGF-β2 and IL-10 secretion, and mitigates SMC activation that contributes to resolution of aortic inflammation and remodeling during AAA formation.
Graphical Abstract

Resolvin D2 acts via GPR18 receptors on M-MDSCs to upregulate TGF-β2 and IL-10 secretion leading to inhibition of smooth muscle cell (SMC) activation and matrix metalloproteinase (MMP)-2 expression during abdominal aortic aneurysm (AAA) formation. The potential crosstalk between infiltrating RvD2-induced M-MDSCs and aortic SMCs suggests modulation of subsequent aortic remodeling to cause attenuation of AAA formation.
INTRODUCTION
Abdominal aortic aneurysms (AAA) are a devastating clinical disease and a leading cause of mortality, especially among the elderly population in the US, with no specific medical therapies available(1–4). The pathological and immunological process of AAAs is characterized by a multi-faceted crosstalk between leukocyte infiltration into the aortic wall, such as neutrophils and macrophages, with subsequent destruction of elastin and collagen in the medial and adventitial layers of the aorta due to excessive production of pro-inflammatory cytokines and matrix degrading enzymes(1, 5–7). This chronic non-resolving inflammatory process during aging, termed as ‘inflammaging’, culminates in aortic smooth muscle cell (SMC) activation and remodeling of the aorta that can result in aortic wall thinning with eventual rupture and sudden death(8, 9).
The resolution of aortic inflammation is an active process that is dependent on the balance of pro-inflammatory (leukotrienes, prostaglandins, cytokines) and pro-resolving (specialized proresolving lipid mediators; SPMs) paracrine mediators(10). An important bioactive isoform of SPMs is Resolvin D2 (RvD2) which is derived from omega-3 fatty acids, such as docosahexaenoic acid (DHA)(11). RvD2 promotes a pro-resolving and pro-reparative phenotype via G-protein coupled receptor 18 (GPR18) to regulate phagocytosis and cytokine production(12). Our previous study demonstrated that treatment with RvD2 significantly influences macrophage polarization towards an anti-inflammatory phenotype to prevent AAA formation(13). However, the role of monocytes to reprogram towards a more pro-resolving and pro-reparative phenotype in chronic vascular inflammation remains to be deciphered. Furthermore, the cell-specific mechanism of RvD2-mediated resolution via GPR18 signaling also remains to be investigated in the pathogenesis of AAAs.
Recent evidence suggests that myeloid-derived suppressor cells (MDSCs) are an immunosuppressive cell population that are generated during acute and chronic pathologies secondary to infections, sterile inflammation or tumor biology (14–16). This heterogenous cell population, comprising of monocytic- (M-) and granulocytic (G- or PMN-)- MDSCs, are generated by enhanced myelopoiesis and characterized by myriad of immune modulating activities, including release of cytokines, exhaustion of pro-inflammatory cells, and promotion of pro-resolving cell phenotypes (17–20). These immature progenitor cells are notably distinguished for their ability to act in an immunosuppressive manner, ultimately modulating the survival, function, and proliferation of other immune cells(18, 21–31). Therefore, the enhancement of endogenous immunosuppression via M-MDSCs represents a significant signaling axis that could potentially be exacerbated by pharmacological modalities.
In this study, the immunomodulation of vascular inflammation and remodeling during AAA formation by RvD2 and GPR18 signaling was investigated using an established topical elastase-treatment model of experimental murine AAA. Aortic wall from human patients undergoing AAA repair demonstrated a marked decrease in RvD2 expression. In addition, treatment with RvD2 in the experimental murine models of AAA showed preserved aortic morphology and reduced inflammatory cytokine expression. RvD2 treatment correlated with increased M-MDSCs in the aortic tissue and in vitro studies demonstrated the ability of this monocytic subset in downregulation of aortic smooth muscle cell activation to attenuate vascular remodeling.
MATERIALS AND METHODS
Animals
Eight to 12-week old wild-type C57BL/6 male mice (Jackson Laboratory, Bar Harbor, ME) were housed and maintained at 70°F, 50% humidity, in 12-hour light-dark cycles as per institutional animal protocols. Mice were provided drinking water and standard chow diet ad libitum. All experiments were approved by and conducted in accordance to the Institutional Animal Care and Use Committee of the University of Florida (protocol # 201910902).
Human aortic tissue analysis
Collection of human aortic tissue was approved by the University of Florida’s Institutional Review Board (#IRB201902782). Consent was obtained from all patients before surgery. Aortic tissue from AAA patients was resected during open surgical repair as well as during organ transplant donor surgery (controls). Aortic tissue was homogenized in Trizol, and RNA was purified per manufacturer’s protocol (Qiagen, Valencia, CA). cDNA was synthesized using the iScript cDNA Synthesis Kit (BioRad, Hercules, CA). Quantitative Real-Time (RT-PCR) was performed with primer sets (MWG/Operon, Huntsville, AL) in conjunction with SsoFast EvaGreen Supermix (BioRad, Hercules, CA). Gene expression was calculated by using the relative quantification method according to the following equation: 2(−ΔCT), where ΔCT=(Average gene of interest)−(Average reference gene), where GAPDH was used as the reference gene.
Topical Elastase AAA Model
C57BL/6 (wild-type; WT) mice were anesthetized using isoflurane and underwent exposure of the infrarenal abdominal aorta, as previously described(32). The aorta was dissected circumferentially away from the surrounding tissues and subjected topically for 5 minutes to either 5 μL of elastase (0.4 U/mL type 1 porcine pancreatic elastase, Sigma Aldrich, St. Louis, MO) or heat-inactivated elastase (deactivated elastase was used as control). Treated aortic sections were harvested postoperatively and preserved in formalin for immunohistochemistry or snap-frozen in liquid nitrogen for protein and RNA extraction. Aortic diameter was measured using video micrometry (AmScope, Irvine, CA). Aortic dilation percentage was determined by [(maximal AAA diameter – self-control aortic diameter)/(self-control aortic diameter)] × 100. An aortic dilation of ≥100% was considered positive for AAA.
Treatment of Mice with RvD2
To evaluate the attenuative effects of RvD2 in AAA formation, mice received intraperitoneal injections (i.p.) with 0.3 mL vehicle (0.1% ethanol in 0.9% saline) or RvD2 (300ng/kg; Cayman Chemical, Ann Arbor, MI) on postoperative days 1 through 13 and subsequently aortas were harvested on post-operative day 14.
GPR18 Expression Quantification
To quantify expression of the GPR18 receptors, RNA was isolated from the aortic samples using TRIzol Reagent (Ambion, Carlsbad, CA). cDNA was synthesized from the isolated RNA via the iScript Reverse Transcription supermix (BioRad, Hercules, CA) and quantitative (real-time) RT-PCR was performed with primer sets (MWG/Operon, Huntsville, AL) in conjunction with SsoFast EvaGreen Supermix (BioRad, Hercules, CA), as previously described(33). To evaluate expression of GPR18 receptors, the following primers were used for evaluating human aortic tissue: GPR18, Forward: GCCAAGCGTTACACTGGAAA; Reverse: TGATACTTAGAAACTCCTGTCCATC, and the following primers were used for murine aortic tissue: GPR18 Forward: TCATGATCGGGTGCTACGTG; GPR18 Reverse: CTTGTAGCATCAGGACGGCA, GAPDH Forward: TTGATGGCAACAATCTCCAC; GAPDH Reverse: CGTCCCGTAGACAAAATGGT. Gene expression was calculated by using the relative quantification method according to the following equation: 2(−ΔCT), where ΔCT = (average gene of interest)−(average reference gene), where GAPDH was used as the reference gene. Each PCR reaction was carried out in triplicate, and the relative quantification of gene expression was quantified as fold change.
In vivo Suppression of GPR18
Mice were injected i.p. with siRNA targeting murine GPR18 (10nmol; ON-TARGETplus SMARTPool siRNA, Dharmacon, Lafayette, CO) or a non-targeting control siRNA (5nmol; ON-TARGETplus Non-targeting Control siRNA, Dharmacon, Lafayette, CO). The day after injection, mice underwent surgery using the topical elastase model to induce AAAs, as previously described(34). Aortas were harvested on day 14 and expression of GPR18 was measured by RT-qPCR from isolated RNA, as described above. RvD2 was injected i.p. from days 1 to 13 postoperatively in mice that received siRNA for GPR18 and non-targeting controls. Aortic tissue was harvested on postoperative day 14 for further analysis and aortic dilation percentage was measured.
Histology
Harvested aortic tissue was fixed in 10% zinc-buffered formalin overnight, processed with ethanol, and embedded into paraffin. Antibodies for immunohistochemical staining were anti-mouse Mac2 for macrophages (1:10,000, Cedarlane Laboratories, Burlington, ON, Canada; catalog no. CL8942AP), anti-mouse neutrophils for polymorphonuclear neutrophils (PMNs) (1:10,000, AbD Serotec, Oxford, United Kingdom; catalog no. MCA771GA), anti-mouse α-smooth-muscle-actin (α-SMA, 1:1000, Sigma, St. Louis, MO; catalog no. A5691), anti-mouse Calponin (1:1000; Abcam, Cambridge, UK; catalog no. ab46794, or anti-mouse smooth muscle myosin (1:2000; Sigma Aldrich, St. Louis, MO; catalog no. M7786). Aortic sections were also stained with hematoxylin and eosin, and Verhoeff-Van Gieson for elastin (Polysciences, Inc., Warrington, PA; catalog no. 25089–1). Elastin degradation was quantified by counting the number of breaks per vessel, then was averaged and graphed. Histological analysis was performed on three different sections of aortic tissue from each animal and quantified using two independent observers. Representative images from different animals from each group are depicted for accuracy. Images were taken with 20X magnification by a Nikon microscope equipped with a digital camera and Elements BR software. Staining was quantified by measuring the percent intensity of positive sections relative to the whole aortic cross-section using ImagePro software (Media Cybernetics, Rockville, MD, USA).
Cytokine measurements
Cytokine expression in murine aortic tissue homogenates and cell culture supernatants was quantified using the Luminex Bead Array technique using a multiplex cytokine-panel assay (Bio-Rad Laboratories, Hercules, CA).
Enzyme-linked Immunosorbent Assay
Immunoreactive RvD2 was quantified by ELISA using the supplied manufacturer’s protocol (Cayman Chemical, Ann Arbor, MI).
Flow Cytometry Analysis
Murine aortic tissue was harvested from WT mice after undergoing AAA induction on days 3, 7 and 14, as previously described(34). Tissue was minced and incubated for 15 min at 37°C with collagenase type IA (Sigma Aldrich; St. Louis, MO) in PBS with 0.5% BSA and 2mM EDTA Following red blood cell lysis, the cell suspension was stained with Live/Dead (Aqua; Biolegend, San Diego, CA) diluted in PBS for 30min at 4°C. Cells were then washed in PBS and incubated with FC block (BD Biosciences) for 5 min followed by an extracellular antibody staining cocktail for 30 min. Cells were washed twice and then fixed with fixation buffer (eBioscience; Thermo Fisher Scientific) for 25 min at 4°C. Following fixation, cells were washed in permeabilization buffer (eBioscience) and incubated with intracellular iNOS antibody for 30 min at 4°C. Flow cytometry was performed on a BD FACSymhony™A3 Cell Analyzer (BD Biosciences) and data was analyzed using FlowJo Software. M-MDSCs were identified as CD45+CD11b+CD11c−Ly6C+Ly6G−iNOS+ and G-MDSCs were identified as CD45+CD11b+CD11c−Ly6C−Ly6G+iNOS−, as previously described (35–37) and quantified as a percentage of total CD45+ cells.
In vitro generation of M-MDSCs
M-MDSCs were generated for in vitro experiments as previously described (37, 38). Briefly, cells were isolated from bone marrow of C57Bl/6 mice and cultured with granulocyte-macrophage stimulating factor (100ng or 2×103 units per 3×106 cells, Peprotech, Cranbury, NJ) in RPMI-1640 media containing 10% heat inactivated fetal bovine serum, 1% antibiotic-antimycotic, 1% L-glutamine, and 0.1% β-mercaptoethanol for 3 days. The resulting cell culture, consisting of M- and G-MDSCs, was further separated into Ly6G− and Ly6G+ subpopulations with Anti-Ly6G UltraPure Microbeads (Miltenyi Biotec) via AutoMACS magnetic based cell sorting. The Ly6G− or Ly6G+ population, representing M-MDSCs and G-MDSCs respectively, was further cultured in the presence of LPS (0.1mg/mL; Invitrogen) and IFN-γ (100 U/mL; BioLegend, San Diego, California) for 24hrs and subjected to a Dead Cell Removal kit via magnetic separation (Miltenyi Biotec) and live M- or G-MDSCs were subsequently used for in vitro experiments.
Cell Culture Experiments
C57BL/6 murine M-MDSCs and aortic smooth muscle cells (SMCs) (ATCC, Manassas, VA) were cultured (2×105 cells in 48-well plates) in DMEM F12 with 10% FBS. Cells were transfected for 6 hrs with GPR18 siRNA, or a non-targeting control siRNA as described above, in serum-free media containing a transfection reagent (HiPerFect Transfection Reagent, catalog no. 301704; Qiagen, Valencia, CA). M- or G-MDSC cultures were subjected to a transient elastase treatment (0.4 U) for 5 minutes, washed, and treated with media containing either RvD2 (300nM) or a vehicle control. After 24hrs supernatant was collected and the cytokine expression was analyzed by luminex bead array assay (Millipore Sigma, Burlington, MA). In separate cultures, conditioned media transfer (CMT) was performed using M-MDSCs and SMCs. M-MDSCs were treated with or without RvD2 for 6 hrs, and CMT was performed to SMC cultures treated with/without elastase, and culture supernatants were harvested after 24 hrs for analysis of cytokines (CXCL1 and IL-6) and MMP2 activity (Luminex bead array, Millipore Sigma, St. Louis, MO).
Statistical Analysis
Statistical analysis was performed with GraphPad 10 (GraphPad Software, La Jolla, CA). One-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test was used to compare means between three or more groups. An unpaired Student’s t-test with nonparametric Mann-Whitney or Wilcoxon rank sum test was used for pairwise comparison of groups. Results are displayed as mean ± standard error of mean with p<0.05 considered as statistically significant.
RESULTS
Resolvin D2 attenuates experimental murine AAA formation
Using the topical elastase model, mice were treated with either elastase or heat-inactivated elastase on day 0 and injected with vehicle or RvD2 (300ng/kg bodyweight) daily on post-operative days 1 to 13 and harvested on day 14 (Figure 1A). A significant increase in mean aortic diameter was noted in elastase treated mice compared to heat-inactivated elastase treated control mice (209±4.3% vs 1.2±0.3%; n=10–19/group; p<0.0001; Figure 1B–C). When mice were administered RvD2, elastase-treated mice demonstrated significantly decreased mean aortic dilation compared to mice treated with vehicle alone (141.4±5.0 vs. 209±4.3%; n=19–20/group; p< 0.0001; Figure 1B–C). Additionally, RvD2 treated mice demonstrated significantly increased expression of smooth muscle alpha actin (SM-αA), as well as decreased expression of elastic breaks and immune cell (neutrophils and macrophages) infiltration on day 14 compared to mice treated with vehicle alone (Figure 1D–E). Moreover, RvD2 treatment preserved the contractile phenotype of elastase-treated aortas compared to untreated controls as observed by increase in expression of calponin and myosin expressions (Supplemental Figures S1 and S2). The expression of pro-inflammatory cytokines (IFN-γ, IL-1β, IL-6, TNF-α, IL-17, MCP-1, MIP-2, HMGB1) and matrix metalloproteinase (MMP2) were significantly attenuated by RvD2 treatment (Figure 2). Moreover, the expression of TGF-β2 (38.8±7.4 vs. 5.5±1.6 pg/ml; p<0.001) and IL-10 (235±26.8 vs. 98.5±9.6 pg/ml; p<0.001) were significantly increased in the RvD2-treated mice compared to untreated controls (Figure 2). Analysis of human aortic tissue demonstrated significantly decreased expression of RvD2 and GPR18 expression in human aortic tissue from AAA patients compared to controls (Supplemental Figure S3).
Figure 1.
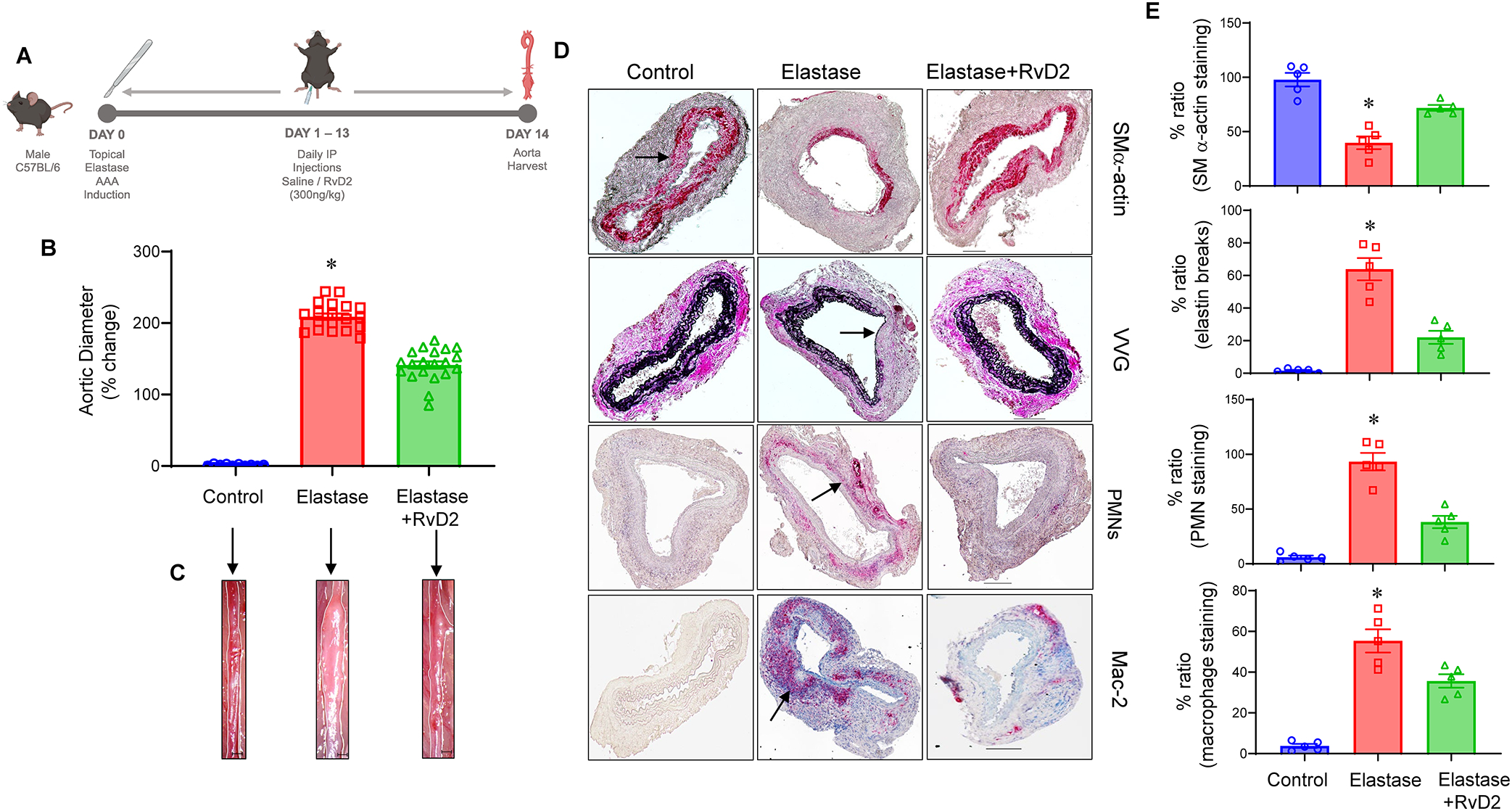
RvD2 administration decreases aortic aneurysm phenotype and preserves morphology to mitigate AAA formation. A, Schematic description of murine topical elastase model with RvD2 treatments. Mice were divided into three groups and treated with either heat-inactivated elastase or elastase on day 0. Mice were then administered either vehicle or RvD2. Aortic diameter was measured on day 14 and tissue harvested for additional analysis. B, RvD2 treated mice demonstrated a significant decrease in aortic diameter compared to vehicle treated mice; *p<0.001 vs. other groups; n=10–20 per group). C, Representative images of aortic phenotype in the respective groups. Scale bar is 100μM. D, Comparative histology performed on day 14 indicates that elastase-treated mice administered with RvD2 have a marked increase in smooth muscle cell α-actin (SM-α actin) expression, decrease in elastic fiber disruption (Verhoeff-Van Gieson staining for elastin) as well as neutrophil (PMN) and macrophage (Mac-2) infiltration, compared to elastase-treated WT mice alone (n=5 per group). Representative histological images from separate animals in the respective groups are depicted. Arrows indicate areas of immunostaining or elastin breaks. E, Quantification of histological staining in respective groups; n=5 per group; *p<0.02 vs. other groups. Representative scale bar is 100μM. Data analyzed by one-way ANOVA and Tukey’s multiple comparisons test.
Figure 2.

Pro-inflammatory cytokine production is decreased by RvD2 treatment. Aortic tissue from RvD2-treated mice showed a significant attenuation in pro-inflammatory cytokine/chemokine production and MMP2 expression compared to elastase-treated WT mice alone. There was a significant increase in the expression of anti-inflammatory cytokines, TGF-β2 and IL-10, between RvD2-treated mice compared to untreated mice. *p<0.01 vs. other groups; n=6–8 mice per group. Data analyzed by one-way ANOVA and Tukey’s multiple comparisons test.
RvD2-mediated protection is mediated by GPR18-dependent signaling to mitigate AAA formation
In order to assess the molecular target and ligand-receptor interactions of RvD2, the effect of in vivo knockdown of endogenous murine GPR18 was examined. Treatment with GPR18 siRNA obliterated the protective effect of RvD2 as observed by increased aortic diameter compared to mice administered with control siRNA+RvD2 (169.5±8.4% vs. 137.2±5.6%; p=0.004, Figure 3A–B) Expression of smooth muscle-α actin was significantly increased, while elastin fragmentation, as well as neutrophil and macrophage infiltration were significantly decreased in mice administered with control siRNA+RvD2 compared to GPR18-siRNA+RvD2 treated animals (Figure 3C–G). Mice injected with GPR18 siRNA had a significantly reduced GPR18 expression compared to non-targeting controls (Figure 3H). The expression of pro-inflammatory cytokines (IFN-γ, IL-1β, IL-6, TNF-α, IL-17, MCP-1, MIP-2, HMGB1) and matrix metalloproteinase (MMP2) were significantly attenuated, while the expression of anti-inflammatory TGF-β2 (47.4 ±14.9 vs. 10.9±2.0 pg/ml; p<0.01) and IL-10 (223.9±34.7 vs. 29.0±8.1 pg/ml; p=0.007) were significantly increased by RvD2 treatment in control siRNA treated mice but not in GPR18 siRNA-treated mice (Figure 4).
Figure 3.

RvD2 mediated protection is exerted via GPR18 receptors. A, GPR18-siRNA treatment of WT mice demonstrated a significant increase in aortic diameter as compared to control (c)-siRNA treated mice after administration of RvD2 in respective groups (*p=0.004, n=12–13 per group). B, Representative images of aortic phenotype in respective groups. Scale bar is 100μM. C, Expression of smooth muscle-α actin is significantly increased, and elastin fragmentation as well as macrophage and neutrophil infiltration in aortic tissue are significantly decreased in mice treated with c-siRNA+RvD2 compared to mice treated with GPR18 siRNA+RvD2 (n=5 per group). Representative histological images from separate animals in the respective groups are depicted. Arrows indicate areas of immunostaining or elastin breaks. D-G, Quantification of histological staining in respective groups; n=5 per group; *p<0.01 vs. other groups. Representative scale bar is 100μM. H, GPR18 receptor expression was decreased in murine aortic tissue after GPR18 siRNA treatment as compared to control siRNA treatment. *p=0.001; n=4 per group. An unpaired Student’s t-test with nonparametric Mann-Whitney test was used for pairwise comparison of groups.
Figure 4.

Aortic inflammation is mitigated by RvD2/GPR18 signaling. Aortic tissue from elastase-treated WT mice after c-siRNA+RvD2 administration showed a significant attenuation in pro-inflammatory cytokine/chemokine production and MMP2 expression, and a significant upregulation of TGF-β2 and IL-10 expression, compared to GPR18-siRNA+RvD2-treated mice. *p<0.02; n=5 per group. Data analyzed by Mann-Whitney test.
RvD2 treatment induces increase in infiltration and activation of monocytic-MDSCs to mitigate smooth muscle-dependent inflammation and remodeling
Administration of RvD2 in WT mice displayed a significant increase in the M-MDSC population (CD45+CD11b+CD11c−Ly6G−Ly6C+iNOS+) infiltrating in the aortic tissue on day 3 compared to untreated mice (9.5±1.3 vs. 4.1±0.7%; p=0.001; Figure 5). No significant differences were observed in M-MDSC quantification on days 7 and 14 in the RvD2-treated mice compared to untreated controls (Supplemental Figure S4). These results highlight a critical association between RvD2-mediated protection and M-MDSC infiltration during the inflammation-resolution process. To investigate the mechanistic crosstalk between RvD2-activated M-MDSC and aortic parenchyma, we used in vitro studies involving murine M-MDSCs (isolated as shown in Supplemental Figure S5) and SMCs. Transient elastase-treatment with/without RvD2 was performed on cultured M-MDSCs that demonstrated a significant increase in TGF-β2 and IL-10 expression in culture supernatants after 24hrs (Figure 6A–B). Furthermore, the RvD2-dependent increase in TGF-β2 and IL-10 secretion was abolished when cells were pretreated with GPR18-siRNA compared to M-MDSCs treated with RvD2 and GPR18-siRNA (Figure 6A–B). No significant change in expression of TGF-β2 or IL-10 was observed in RvD2-treated cultures of G-MDSCs (Supplemental Figure S6).
Figure 5.

RvD2 treatment increases M-MDSCs in aortic tissue during AAA formation. A, Representative flow cytometry gating strategy for M-MDSC identification in aortic tissue. B, The percentage of M-MDSCs was significantly upregulated in elastase-treated mice administered with RvD2 compared to elastase-treated mice alone on day 3. C-E, No significant differences were observed in M-MDSC quantification on days 7 and 14 between these groups. *p=0.001; n=5–15 per group; ns, not significant. Data analyzed by Mann-Whitney test.
Figure 6.

RvD2 upregulates M-MDSC-mediated TGF-β2 and IL-10 secretion to decrease SMC activation. A-B, RvD2 treatment upregulates M-MDSC-mediated TGF-β2 and IL-10 secretion in a GPR18-dependent manner compared to untreated cells. *p<0.01 vs control and elastase; #p<0.01 vs. c-siRNA+elastase+RvD2; n=5/group. C, Schematic of conditioned media transfer (CMT) experiments between M-MDSCs and SMC cultures. D-F, CMT from RvD2-treated M-MDSCs to elastase-treated SMCs significantly mitigated IL-6, CXCL1 and MMP2 expression compared to CMT from untreated MDSCs to elastase-treated SMCs. Inhibition of SMC secreted cytokine and MMP2 expression by RvD2-treated M-MDSCs is dependent on GPR18 receptors. M-MDSCs treated with GPR18-siRNA+RvD2 abolished the decrease in SMC secreted cytokine and MMP2 expression compared to CMT from control (c) siRNA+RvD2. *p<0.001 vs. MDSC+RvD2→SMCelastase and MDSC→SMC; #p<0.001 vs. MDSC+c-siRNA+RvD2→SMCelastase; n=5 per group. SMCelast denotes SMCelastase. Data analyzed by one-way ANOVA and Tukey’s multiple comparisons test.
In a separate group of experiments, conditioned media transfer (CMT) was performed between M-MDSCs and SMCs (Figure 6C). M-MDSCs treated with/without RvD2 underwent CMT to SMCs treated with/without transient elastase followed by analysis of cell culture supernatants for cytokines and MMP2 expression. Elastase-treatment induced significant expression of IL-6, CXCL1, and MMP2 in SMCs which was abolished after CMT from RvD2-treated M-MDSCs in a GPR18-dependent manner (Figure 6D–F). No change in expression of TGF-β2 and IL-10 expression was observed on CMT from elastase-treated SMCs to M-MDSCs, signifying that the crosstalk between M-MDSCs and SMCs is unidirectional (Supplemental Figure S7). Taken together, these results demonstrate the ability of RvD2-mediated immune activation by enhancing the infiltration and activation of GPR18 on M-MDSCs and suggest downstream attenuation of SMC activation that likely leads to mitigation of aortic inflammation and remodeling during AAA formation.
DISCUSSION
The process of inflammation-resolution plays a pivotal role in chronic vascular pathologies in limiting the disease process in the presence of cytotoxic stimuli. In the context of cardiovascular pathologies, dysregulation of endogenous resolution pathways can lead to uncontrolled aortic tissue inflammation and subsequent remodeling (39–41). In this study, we characterized the role of specialized pro-resolving lipid mediator, RvD2, and the downstream signaling executed via the receptor, GPR18, in upregulating resolution pathways via enhancement of myeloid-precursors cell subsets to attenuate AAA formation. Our results demonstrated that RvD2 treatment can effectively decrease aortic inflammation, leukocyte transmigration, and vascular remodeling during AAA formation in a GPR18-dependent manner. Moreover, we observed a significant increase in myeloid-derived suppressor cells due to RvD2 treatment, especially in the early phase of aortic inflammation. Mechanistically, we further defined that RvD2/GPR18 axis treatment can upregulate monocytic(M)-MDSC activation that leads to increase in TGF-β2 and IL-10 secretion. Finally, in vitro experiments suggest that the crosstalk between M-MDSCs and aortic SMCs can be regulated by RvD2/GPR18-dependent signaling that likely attenuates SMC activation and vascular remodeling. Collectively, these results demonstrates the ability of RvD2/GPR18 signaling to result in increased myelopoiesis, enhanced M-MDSC infiltration and activation, thereby promoting the resolution of inflammation during AAA formation.
The understanding of the process of resolution of inflammation (also known as catabasis) has grown significantly in recent years and increasing numbers of key molecules involved have been identified(42). Recent evidence suggests that resolution is an active process with the critical components involving inactivation and cessation of recruitment of neutrophils as well as promotion of macrophage recruitment, efferocytosis, and clearance of pathogens and cellular debris(43) A special class of lipids called specialized pro-resolving mediators (SPMs) are molecules derived from polyunsaturated fatty acids that are produced during the acute inflammatory phase as well as during efferocytosis, and function to resolve inflammation by promoting the core functions of resolution of inflammation(10, 11). SPMs are derived from ω−3 and ω−6 fatty acids and can be organized into five classes of molecules, lipoxins, E-series and D-series resolvins, protectins and maresins(11). These pro-resolving molecules exert their influence in pico- to low nano- molar concentrations via cell surface G protein-coupled receptors including ALX/FPR2, DRV1/GPR32, DRV2/GPR18, ERV1/ChemR23, GPR37, GPR101, and LGR6 and act on various parenchymal and immune cells including neutrophils, monocytes, macrophages, natural killer cells, dendritic cells, epithelial cells, and endothelial cells(10). One of the key mechanisms of SPM-mediated signaling includes facilitating macrophage phagocytosis of cellular debris and inhibition of pro-inflammatory functions via cytokine and chemokine signaling. In order to promote resolution of inflammation, SPMs have the ability to enhance phenotypic polarization in macrophages(13). However, the mechanism that determines the class switch from pro-inflammatory lipid mediators to resolving lipid mediators is not well defined as the process of aging appears to have an impact on SPM biosynthesis, which inhibits or slows the resolution of inflammation.
Based on the current understanding of the role of inflammation in the underlying pathophysiology of vascular pathologies, a few of the bioactive isoforms of SPMs have been previously investigated. In particular, Resolvin D1 and Maresin-1 appear to have the most prominent and well understood role in controlling vascular diseases via downregulation of pro-inflammatory signaling and upregulation of macrophage-dependent efferocytosis(44–46). It has been demonstrated in human and murine models that SPM expressions in plasma or inflamed tissue is associated with disease incidence indicating a correlative association between lipid mediators and vascular pathologies(47, 48). In the context of AAAs, there is evidence that polyunsaturated fatty acid supplementation in animal models reduces inflammation and degeneration of the matrix. Previous studies from our group and others have shown that RvD1 and RvD2 promote a reparative macrophage phenotype in disease models and reduce matrix metalloproteinases, decreasing the size of aneurysms in mice(13, 45). Additionally, the reparative role of MaR1 has also been shown to significantly attenuate growth of aortic aneurysms by upregulating the process of efferocytosis(34). However, the mechanistic aspects of RvD2-mediated signaling in vascular diseases remains to be deciphered. Recent data suggests that RvD2 may exert bone marrow to exert a pro-resolving function in the liver and endogenously produced within the bone marrow upon inflammation, and can limit atherosclerosis progression via myeloid cell-GPR18 signaling(49, 50). Thus, local production and signaling of SPMs like RvD2 within the bone could be an additional mechanism for the protective actions of RvD2-mediated signaling. Our data in this study also confirms that exogenous RvD2 administration has the ability to enhance bone marrow function by producing monocytic and granulocytic progenitors. The immunosuppression activity of monocytic MDSCs was observed to play a critical role in mediating inhibition of vascular inflammation by acting on SMCs, whereas the granulocytic component of MDSCs failed to do so. These findings are critical to understand the myriad of immunological activities in the biology of myeloid and granulocytic subsets during emergency myelopoiesis that may differ based on disease pathologies, and need to be further investigated.
Monocytic subset of MDSCs are readily recruited to sites of inflammation through canonical trafficking pathways like the CCL2/CCR2 axis (51). These regulatory cells are capable of mediating immune suppression through secretion of anti-inflammatory cytokines, recruitment of regulatory immune cells, exhaustion of pro-inflammatory cells through nutrient sequestering, and facilitating cell polarization to anti-inflammatory phenotypes (20). The functional capabilities of monocytes/macrophages are affected by cleavage of efferocytic receptors, such as TAM (Tyro3, Axl, MerTK), in the inflammatory tissue milieu(52). Therefore, it becomes imperative to discover therapeutic modalities that can enhance endogenous immune regulation. Although previous studies have indicated the role of SPMs like RvD1 in enhancing protective capabilities of macrophages by preserving the efferocytic capabilities, our study pinpoints the previously undescribed role of RvD2-dependent signaling in enhancing endogenous resolution via increase in bone marrow precursors of monocytic compartment that precede the anti-inflammatory macrophage phenotype and readily immunomodulate the aortic tissue microenvironment. This alternate strategy supplements the phenotype switch of mature macrophages for inducing an anti-inflammatory profile, and provides an endogenous resolution to tissue inflammation by providing a monocytic subset that readily provides an immunocompetent profile,
A few limitations of this study should be considered for a translational strategy. The murine elastase AAA model is an excellent experimental tool to delineate the early inflammatory signaling pathways in the aortic wall during AAA, as the important hallmarks of human aneurysm pathology such as macrophage infiltration, matrix degradation, increased MMP activation, elastin fiber degradation, and loss of smooth muscle integrity are recapitulated. However, this experimental model lacks progression of AAA to aortic rupture and chronic vascular inflammation and remodeling as observed clinically (53). Therefore, the findings observed in this study need to be further delineated in chronic aortic aneurysm and rupture models, as we recently described (54, 55). Furthermore, a combined therapeutic strategy using additional bioactive isoforms of SPMs such as MaR1, in combination with RvD2, represents a multi-faceted approach for a combined effective attenuation of aortic inflammation and vascular remodeling to mitigate AAAs (34). Also, the effects of RvD2 may directly alter the differentiation and lineage transformation of vascular SMCs, as previously reported (56, 57). Future studies will be focused on using the mechanistic approach of MaR1-dependent upregulation of efferocytosis in combination with RvD2-mediated decrease in SMC activation, differentiation and lineage transformation during AAA formation.
In summary, these findings suggest that the pro-resolving lipid mediator RvD2 plays an important role in enhancing the endogenous inflammation-resolution pathways by enhancing the monocytic compartment of macrophage precursors that mitigate SMC-regulated vascular remodeling and AAA formation. Administration of RvD2-mediated protection is attributable to GPR18-dependent signaling via increase in infiltration and activation of M-MDSCs to mitigate downstream tissue inflammation and remodeling of the aortic tissue. The enhancement of resolution via therapeutic strategies represents an important clinical translational approach as anti-inflammatory treatments can reduce host defense mechanisms leading to enhanced susceptibility of infections that are especially critical in the elderly population. Our results suggest a previously undescribed mechanism to boost endogenous resolution pathways by using bioactive SPMs like RvD2, that can be harnessed to alter the inflammation-resolution dysregulation, for management of chronic inflammatory vascular diseases.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Aravinthan Adithan and Jeff Valisno for help with histology. This study was supported by NIH R01HL153341 and R01HL138931 (GRU and AKS). Schematic figures were made using Biorender.
Abbreviations:
- AAA
abdominal aortic aneurysm
- RvD2
resolvin D2
- GPR18
formyl peptide receptor
- MDSCs
myeloid-derived suppressor cells
- α-SMA
alpha-smooth muscle cell actin
- HMGB1
high mobility group box 1
Footnotes
DISCLOSURES
The authors have no conflicts of interest related to this article.
DATA AVAILABILITY STATEMENT
All relevant data within the manuscript and in Supporting information are available upon request from corresponding authors.
REFERENCES
- 1.Ailawadi G, Eliason JL, and Upchurch GR Jr. (2003) Current concepts in the pathogenesis of abdominal aortic aneurysm. J Vasc Surg 38, 584–588 [DOI] [PubMed] [Google Scholar]
- 2.Bengtsson H, Sonesson B, and Bergqvist D (1996) Incidence and prevalence of abdominal aortic aneurysms, estimated by necropsy studies and population screening by ultrasound. Ann N Y Acad Sci 800, 1–24 [DOI] [PubMed] [Google Scholar]
- 3.Dimick JB, Stanley JC, Axelrod DA, Kazmers A, Henke PK, Jacobs LA, Wakefield TW, Greenfield LJ, and Upchurch GR Jr. (2002) Variation in death rate after abdominal aortic aneurysmectomy in the United States: impact of hospital volume, gender, and age. Ann Surg 235, 579–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnston KW, Rutherford RB, Tilson MD, Shah DM, Hollier L, and Stanley JC (1991) Suggested standards for reporting on arterial aneurysms. Subcommittee on Reporting Standards for Arterial Aneurysms, Ad Hoc Committee on Reporting Standards, Society for Vascular Surgery and North American Chapter, International Society for Cardiovascular Surgery. J Vasc Surg 13, 452–458 [DOI] [PubMed] [Google Scholar]
- 5.Anidjar S, Dobrin PB, Eichorst M, Graham GP, and Chejfec G (1992) Correlation of inflammatory infiltrate with the enlargement of experimental aortic aneurysms. J Vasc Surg 16, 139–147 [DOI] [PubMed] [Google Scholar]
- 6.Aziz F, and Kuivaniemi H (2007) Role of matrix metalloproteinase inhibitors in preventing abdominal aortic aneurysm. Ann Vasc Surg 21, 392–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freestone T, Turner RJ, Coady A, Higman DJ, Greenhalgh RM, and Powell JT (1995) Inflammation and matrix metalloproteinases in the enlarging abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol 15, 1145–1151 [DOI] [PubMed] [Google Scholar]
- 8.Crowther M, Goodall S, Jones JL, Bell PR, and Thompson MM (2000) Increased matrix metalloproteinase 2 expression in vascular smooth muscle cells cultured from abdominal aortic aneurysms. J Vasc Surg 32, 575–583 [DOI] [PubMed] [Google Scholar]
- 9.Xiong W, Knispel R, Mactaggart J, and Baxter BT (2006) Effects of tissue inhibitor of metalloproteinase 2 deficiency on aneurysm formation. J Vasc Surg 44, 1061–1066 [DOI] [PubMed] [Google Scholar]
- 10.Serhan CN (2014) Pro-resolving lipid mediators are leads for resolution physiology. Nature 510, 92–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Basil MC, and Levy BD (2016) Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol 16, 51–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiang N, de la Rosa X, Libreros S, and Serhan CN (2017) Novel Resolvin D2 Receptor Axis in Infectious Inflammation. J Immunol 198, 842–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pope NH, Salmon M, Davis JP, Chatterjee A, Su G, Conte MS, Ailawadi G, and Upchurch GR Jr. (2016) D-series resolvins inhibit murine abdominal aortic aneurysm formation and increase M2 macrophage polarization. FASEB J 30, 4192–4201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang J, Hodges A, Chen SH, and Pan PY (2021) Myeloid-derived suppressor cells as cellular immunotherapy in transplantation and autoimmune diseases. Cell Immunol 362, 104300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scalea JR, Lee YS, Davila E, and Bromberg JS (2018) Myeloid-Derived Suppressor Cells and Their Potential Application in Transplantation. Transplantation 102, 359–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang W, Li J, Qi G, Tu G, Yang C, and Xu M (2018) Myeloid-derived suppressor cells in transplantation: the dawn of cell therapy. J Transl Med 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yaseen MM, Abuharfeil NM, Darmani H, and Daoud A (2020) Mechanisms of immune suppression by myeloid-derived suppressor cells: the role of interleukin-10 as a key immunoregulatory cytokine. Open Biol 10, 200111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sinha P, Clements VK, Bunt SK, Albelda SM, and Ostrand-Rosenberg S (2007) Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol 179, 977–983 [DOI] [PubMed] [Google Scholar]
- 19.Veglia F, Perego M, and Gabrilovich D (2018) Myeloid-derived suppressor cells coming of age. Nat Immunol 19, 108–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gabrilovich DI, and Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol 9, 162–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P, and Segal DM (2002) Myeloid Suppressor Lines Inhibit T Cell Responses by an NO-Dependent Mechanism. The Journal of Immunology 168, 689. [DOI] [PubMed] [Google Scholar]
- 22.Bronte V, Serafini P, Mazzoni A, Segal DM, and Zanovello P (2003) L-arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol 24, 302–306 [DOI] [PubMed] [Google Scholar]
- 23.Gabrilovich DI, and Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9, 162–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nagaraj S, Schrum AG, Cho HI, Celis E, and Gabrilovich DI (2010) Mechanism of T cell tolerance induced by myeloid-derived suppressor cells. J Immunol 184, 3106–3116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srivastava MK, Sinha P, Clements VK, Rodriguez P, and Ostrand-Rosenberg S (2010) Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res 70, 68–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nagaraj S, Youn J-I, and Gabrilovich DI (2013) Reciprocal Relationship between Myeloid-Derived Suppressor Cells and T Cells. The Journal of Immunology 191, 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schrijver IT, Théroude C, and Roger T (2019) Myeloid-Derived Suppressor Cells in Sepsis. Front Immunol 10, 327–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dysthe M, and Parihar R (2020) Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Adv Exp Med Biol 1224, 117–140 [DOI] [PubMed] [Google Scholar]
- 29.Melero-Jerez C, Ortega MC, Moliné-Velázquez V, and Clemente D (2016) Myeloid derived suppressor cells in inflammatory conditions of the central nervous system. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 1862, 368–380 [DOI] [PubMed] [Google Scholar]
- 30.Yi H, Guo C, Yu X, Zuo D, and Wang XY (2012) Mouse CD11b+Gr-1+ myeloid cells can promote Th17 cell differentiation and experimental autoimmune encephalomyelitis. J Immunol 189, 4295–4304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ostrand-Rosenberg S, Sinha P, Beury DW, and Clements VK (2012) Cross-talk between myeloid-derived suppressor cells (MDSC), macrophages, and dendritic cells enhances tumor-induced immune suppression. Semin Cancer Biol 22, 275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Filiberto AC, Spinosa MD, Elder CT, Su G, Leroy V, Ladd Z, Lu G, Mehaffey JH, Salmon MD, Hawkins RB, Ravichandran KS, Isakson BE, Upchurch GR Jr., and Sharma AK (2022) Endothelial pannexin-1 channels modulate macrophage and smooth muscle cell activation in abdominal aortic aneurysm formation. Nat Commun 13, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Filiberto AC, Ladd Z, Leroy V, Su G, Elder CT, Pruitt EY, Hensley SE, Lu G, Hartman JB, Zarrinpar A, Sharma AK, and Upchurch GR Jr. (2022) Resolution of inflammation via RvD1/FPR2 signaling mitigates Nox2 activation and ferroptosis of macrophages in experimental abdominal aortic aneurysms. FASEB J 36, e22579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Elder CT, Filiberto AC, Su G, Ladd Z, Leroy V, Pruitt EY, Lu G, Jiang Z, Sharma AK, and Upchurch GR Jr. (2021) Maresin 1 activates LGR6 signaling to inhibit smooth muscle cell activation and attenuate murine abdominal aortic aneurysm formation. FASEB J 35, e21780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bronte V, Brandau S, Chen S-H, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, Rodriguez PC, Sica A, Umansky V, Vonderheide RH, and Gabrilovich DI (2016) Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nature Communications 7, 12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eckert I, Ribechini E, and Lutz MB (2021) In Vitro Generation of Murine Myeloid-Derived Suppressor Cells, Analysis of Markers, Developmental Commitment, and Function. In Myeloid-Derived Suppressor Cells (Brandau S, and Dorhoi A, eds) pp. 99–114, Springer; US, New York, NY: [DOI] [PubMed] [Google Scholar]
- 37.Leroy V, Manual Kollareth DJ, Tu Z, Valisno JAC, Woolet-Stockton M, Saha B, Emtiazjoo AM, Rackauskas M, Moldawer LL, Efron PA, Cai G, Atkinson C, Upchurch GR, and Sharma AK (2024) MerTK-dependent efferocytosis by monocytic-MDSCs mediates resolution of post-lung transplant injury. bioRxiv [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eckert I, Ribechini E, and Lutz MB (2021) In Vitro Generation of Murine Myeloid-Derived Suppressor Cells, Analysis of Markers, Developmental Commitment, and Function. Methods Mol Biol 2236, 99–114 [DOI] [PubMed] [Google Scholar]
- 39.Maskrey BH, Megson IL, Whitfield PD, and Rossi AG (2011) Mechanisms of resolution of inflammation: a focus on cardiovascular disease. Arterioscler Thromb Vasc Biol 31, 1001–1006 [DOI] [PubMed] [Google Scholar]
- 40.Fredman G (2019) Can Inflammation-Resolution Provide Clues to Treat Patients According to Their Plaque Phenotype? Front Pharmacol 10, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Halade GV, and Lee DH (2022) Inflammation and resolution signaling in cardiac repair and heart failure. EBioMedicine 79, 103992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Widgerow AD (2012) Cellular resolution of inflammation--catabasis. Wound Repair Regen 20, 2–7 [DOI] [PubMed] [Google Scholar]
- 43.Panigrahy D, Gilligan MM, Serhan CN, and Kashfi K (2021) Resolution of inflammation: An organizing principle in biology and medicine. Pharmacol Ther 227, 107879. [DOI] [PubMed] [Google Scholar]
- 44.Olivares-Silva F, De Gregorio N, Espitia-Corredor J, Espinoza C, Vivar R, Silva D, Osorio JM, Lavandero S, Peiro C, Sanchez-Ferrer C, and Diaz-Araya G (2021) Resolvin-D1 attenuation of angiotensin II-induced cardiac inflammation in mice is associated with prevention of cardiac remodeling and hypertension. Biochim Biophys Acta Mol Basis Dis 1867, 166241. [DOI] [PubMed] [Google Scholar]
- 45.Spinosa M, Su G, Salmon MD, Lu G, Cullen JM, Fashandi AZ, Hawkins RB, Montgomery W, Meher AK, Conte MS, Sharma AK, Ailawadi G, and Upchurch GR Jr. (2018) Resolvin D1 decreases abdominal aortic aneurysm formation by inhibiting NETosis in a mouse model. J Vasc Surg 68, 93S–103S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Viola JR, Lemnitzer P, Jansen Y, Csaba G, Winter C, Neideck C, Silvestre-Roig C, Dittmar G, Doring Y, Drechsler M, Weber C, Zimmer R, Cenac N, and Soehnlein O (2016) Resolving Lipid Mediators Maresin 1 and Resolvin D2 Prevent Atheroprogression in Mice. Circ Res 119, 1030–1038 [DOI] [PubMed] [Google Scholar]
- 47.Salazar J, Pirela D, Nava M, Castro A, Angarita L, Parra H, Duran-Aguero S, Rojas-Gomez DM, Galban N, Anez R, Chacin M, Diaz A, Villasmil N, De Sanctis JB, and Bermudez V (2022) Specialized Proresolving Lipid Mediators: A Potential Therapeutic Target for Atherosclerosis. Int J Mol Sci 23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim AS, and Conte MS (2020) Specialized pro-resolving lipid mediators in cardiovascular disease, diagnosis, and therapy. Adv Drug Deliv Rev 159, 170–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fitzgerald H, Bonin JL, Sadhu S, Lipscomb M, Biswas N, Decker C, Nabage M, Bossardi R, Marinello M, Mena AH, Gilliard K, Spite M, Adam A, MacNamara KC, and Fredman G (2023) The Resolvin D2-GPR18 Axis Enhances Bone Marrow Function and Limits Hepatic Fibrosis in Aging. bioRxiv [Google Scholar]
- 50.Lipscomb M, Walis S, Marinello M, Mena HA, MacNamara KC, Spite M, and Fredman G (2024) Resolvin D2 limits atherosclerosis progression via myeloid cell-GPR18. FASEB J 38, e23555. [DOI] [PubMed] [Google Scholar]
- 51.Serbina NV, and Pamer EG (2006) Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol 7, 311–317 [DOI] [PubMed] [Google Scholar]
- 52.Vago JP, Amaral FA, and van de Loo FAJ (2021) Resolving inflammation by TAM receptor activation. Pharmacol Ther 227, 107893. [DOI] [PubMed] [Google Scholar]
- 53.Bhamidipati CM, Mehta GS, Lu G, Moehle CW, Barbery C, DiMusto PD, Laser A, Kron IL, Upchurch GR Jr., and Ailawadi G (2012) Development of a novel murine model of aortic aneurysms using peri-adventitial elastase. Surgery 152, 238–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fashandi AZ, Hawkins RB, Salmon MD, Spinosa MD, Montgomery WG, Cullen JM, Lu G, Su G, Ailawadi G, and Upchurch GR Jr. (2018) A novel reproducible model of aortic aneurysm rupture. Surgery 163, 397–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu G, Su G, Davis JP, Schaheen B, Downs E, Roy RJ, Ailawadi G, and Upchurch GR Jr. (2017) A novel chronic advanced stage abdominal aortic aneurysm murine model. J Vasc Surg 66, 232–242 e234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miyahara T, Runge S, Chatterjee A, Chen M, Mottola G, Fitzgerald JM, Serhan CN, and Conte MS (2013) D-series resolvin attenuates vascular smooth muscle cell activation and neointimal hyperplasia following vascular injury. FASEB J 27, 2220–2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao M, Zheng Z, Yin Z, Zhang J, Qin J, Wan J, and Wang M (2023) Resolvin D2 and its receptor GPR18 in cardiovascular and metabolic diseases: A promising biomarker and therapeutic target. Pharmacol Res 195, 106832. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data within the manuscript and in Supporting information are available upon request from corresponding authors.