

Abstract
Breast cancer is the most common malignancy afflicting women from Western cultures. Developments in breast cancer molecular and cellular biology research have brought us closer to understanding the genetic basis of this disease. Recent advances in microarray technology hold the promise of further increasing our understanding of the complexity and heterogeneity of this disease, and providing new avenues for the prognostication and prediction of breast cancer outcomes. These new technologies have some limitations and have yet to be incorporated into clinical use, for both the diagnosis and treatment of women with breast cancer. The most recent application of microarray genomic technologies to studying breast cancer is the focus of this review.
Introduction
Mortality from breast cancer results from the ability of some tumors to metastasize to distant sites. Selecting patients with micrometastases at diagnosis is crucial for clinicians in deciding who should and who should not receive toxic and expensive adjuvant chemotherapy to eradicate these metastatic cells. Although many individual biomarkers were originally attractive, over the years most have failed to become clinically useful. In addition, the management of breast cancer has changed, with most node-negative patients now undergoing systemic adjuvant therapy because we cannot precisely determine an individual's risk of recurrence. A majority of node-negative patients are being unnecessarily overtreated because if they were left systemically untreated, only about 25% of node-negative patients would ever develop recurrence. There is therefore a crucial need to identify patients with a sufficiently low risk of breast cancer recurrence to avoid further treatment. In addition, in patients at risk of recurrence and in need of therapy, optimal therapeutic selection is an increasingly important objective. Recent developments in applying microarray technologies to breast tumor samples suggest that these new techniques might provide for the transition of molecular biological discoveries to clinical application, and will generate clinically useful genomic profiles that more accurately predict long-term outcome for individual breast cancer patients.
Background
Until recently, evaluations of prognostic and predictive factors have considered one factor at a time or have used small panels of markers. However, with the advent of new genomic technologies such as microarrays capable of simultaneously measuring thousands of genes or gene products, we are beginning to construct molecular fingerprints of individual tumors so that accurate prognostic and predictive assessments of each cancer can be made. Clinicians might one day base clinical management on each woman's personal prognosis and predict the best individual therapies from the genetic fingerprint of each individual cancer.
Breast cancer is characterized by a heterogeneous clinical course. A major goal of recent studies is to determine whether RNA microarray expression profiling, or DNA array gene amplification or gene loss patterns, can accurately predict an individual's long-term potential for recurrence from breast cancer, so that appropriate treatment decisions can be made. Microarrays can be used to measure the mRNA expression of thousands of genes at one time or to survey genomic alterations that might distinguish between molecular phenotypes associated with long-term recurrence-free survival or clinical response to treatment. These new technologies have been successfully applied to primary breast cancers and may eventually outperform currently used clinical parameters in predicting disease outcome.
Because the RNA expression microarray technology provides a method for monitoring the RNA expression of many thousands of human genes at one time, there was considerable anticipation that it would quickly and easily revolutionize our approaches to cancer diagnosis, prognosis, and treatment. The reality remains extremely promising but it is also complex. A potential complication in the application of microarray technology to samples of primary human breast tumors is the presence of variable numbers of normal cells, such as stroma, blood vessels, and lymphocytes, in the tumor. Indeed, it has been demonstrated, with the use of gross analysis of human breast cancer specimens compared with breast cancer cell lines, that the tumors expressed sets of genes in common not only with these cell lines but also with cells of hematopoietic lineage and stromal origin [1]. Laser capture microdissection has also been used successfully to isolate pure cell populations from primary breast cancers for array profiling [2]. In their seminal paper, Sgroi and colleagues [2] used laser capture microdissection to isolate morphologically 'normal' breast epithelial cells, invasive breast cancer cells, and metastatic lymph node cancer cells from one patient, and were able to demonstrate the feasibility of using microdissected samples for array profiling, and also for following the potential progression of cancer in this patient. However, with the emerging data supporting important roles for the surrounding stroma in breast cancer progression, and the labor-intensive and technically challenging nature of laser capture technology with subsequent amplification of RNA for quantification, most published investigations so far have evaluated total gene expression to identify prognostic profiles, as will be described in the next section.
Molecular classification of breast cancer
A study of sporadic breast tumor samples by Perou and colleagues [1] was the first to show that breast tumors could be classified into subtypes distinguished by differences in their expression profiles. Using 40 breast tumors, and 20 matched pairs of samples before and after doxorubicin treatment, an 'intrinsic gene set' of 476 genes were selected that were more variably expressed between the 40 sporadic tumors than between the paired samples. This intrinsic gene set was then used to cluster and segregate the tumors into four major subgroups: a 'luminal cell-like' group expressing the estrogen receptor (ER); a 'basal cell-like' group expressing keratins 5 and 17, integrin β4, and laminin, but lacking ER expression; an 'Erb-B2-positive' group; and a 'normal' epithelial group (Fig. 1).
Figure 1.
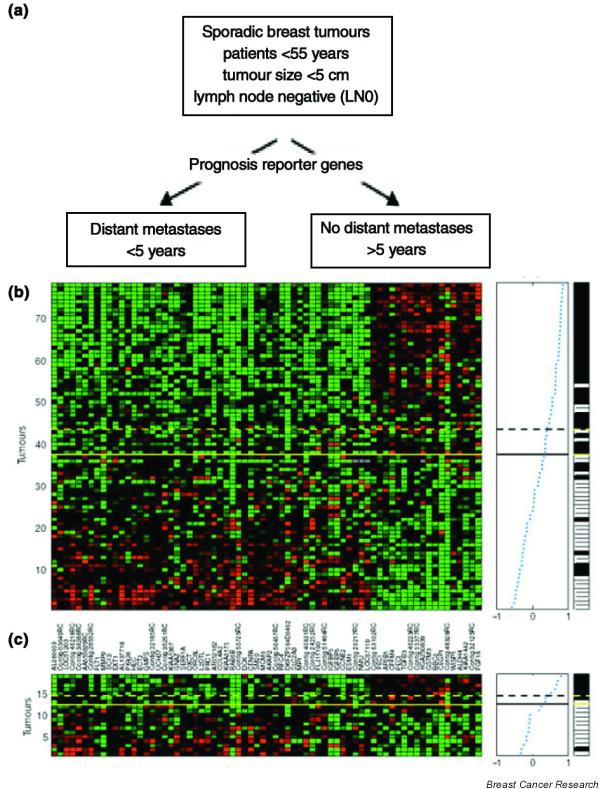
Supervised classification on prognosis signatures (van 't Veer). The 78 tumors are listed vertically, and the 70 'prognostic' genes horizontally. The expression levels are shown in red (expression levels above the mean for the gene) and green (levels below the mean for the gene).
In a subsequent study with 38 additional cancers, the investigators found the same subgroups as before [3], except that the luminal, ER-positive group was further subdivided into subsets with distinctive gene expression profiles. In univariate survival analysis, performed on the 49 patients diagnosed with locally advanced disease but without evidence of distant metastasis, ER positivity was not a significant prognostic factor on its own, but the luminal-type group enjoyed a more favorable survival than the other groups. Conversely, the basal-like group had a significantly poorer prognosis. Although small and exploratory, this study suggests that important differences in outcome can be ascertained from microarray expression profiling.
An interesting study was reported by Gruvberger and colleagues [4], who profiled 58 grossly dissected primary invasive breast tumors and used artificial neural network analysis to predict the ER status of the tumors on the basis of their gene expression patterns. They then determined which specific genes were the most important for ER classification. By comparison with SAGE (serial analysis of gene expression) data from estradiol-stimulated breast cancer cells, they determined that only a few genes of the many genes that were associated with ER expression in tumors were indeed estrogen-responsive in cell culture. This observation lends further support to the hypothesis developed by Perou and colleagues that basic cell lineages, such as the luminal ER-positive cell type, can be partly explained by observed genomic gene expression patterns rather than by downstream effectors of only one pathway, such as the ER.
Prognostic implications
Microarrays have also been used to predict lymph node status and very short-term relapse-free survival in two groups (n = 37 and 52, respectively) of heterogeneously treated patients [5]. Although prediction of nodal status is of limited interest clinically, the study uses innovative statistical methods, rigorously generates estimates of future classifier performance and further demonstrates the feasibility of accurate prediction of tumor biology with expression arrays. In a more focused and somewhat more clinically relevant study, van 't Veer and colleagues [6] used RNA expression microarray analyses to identify a 70-gene prognostic gene signature ('classifier') in young, untreated, axillary lymph node-negative patients by using a training set of 44 tumors with a good outcome (disease-free more than 5 years) and 34 with a poor outcome (distant relapse in less than 5 years), and then tested the classifier in a validation set of 19 tumors. The same group [7] has now extended the study to a total of 295 young (less than 53 years of age), stage I to II breast cancer patients with both node-negative and node-positive disease, using the 70-gene classifier [6]. The microarray-based predictions are consistent with, and perhaps better than, estimates that can be obtained with current prognostic indices.
Genetic susceptibility
A few studies have used new genomic approaches for the study of inherited breast cancer (reviewed in [8]). There is accumulating evidence, both epidemiological and histo-logical, that tumors arising as a result of mutations in the two breast cancer susceptibility gene families (BRCA1 and BRCA2) are biologically distinct. For instance, BRCA1 breast cancers are most often ER and progesterone receptor (PR)-negative, but BRCA2 cancers more often tend to be positive for these receptors [9]. In a seminal paper published by Hedenfalk and colleagues [8], seven tumors each from BRCA1 and BRCA2 gene mutation carriers, or sporadic breast cancers, were compared by expression microarray analysis. They found that the gene expression profiles of the three tumor groups differed significantly from each other, underscoring the fundamental differences between BRCA1 and BRCA2 mutation-associated tumors. Of course a potential confounding issue was the differential distribution of ER between the BRCA1 and BRCA2 tumors. However, even after the removal of ER/PR-associated genes from the analysis, the two inherited tumor groups were still discernible. Thus, ER status alone does not fully explain the observed differences in gene expression profiles. Although this study is obviously very small, and other confounding issues such as tumor stage, grade, and treatment were not able to be considered, it does set a foundation for larger validation studies to confirm differential genes that could then provide important clues to the etiology of inheritable breast cancer.
Microarrays are also being studied as a way of predicting response to systemic therapy. The neoadjuvant setting is especially attractive for these studies for several reasons including early assessment of response to therapy, biopsiable access to the primary tumor, and considerably reduced sample sizes compared with those required in the adjuvant setting.
Predictive implications
Methods for assessing response in neoadjuvant trials remain problematic. Clinical response to neoadjuvant chemotherapy is a validated surrogate marker for improved survival [10,11]. Women who achieve pathologic complete response are most likely to have the best clinical outcome, although survival is still improved in those who clinically respond who do not achieve pathologic complete response.
In an early study, Buchholz and colleagues [12] obtained sufficient RNA from core biopsies of five patients to perform serial microarray expression profiles and showed that, despite differences in therapy, patients with good pathological responses to neoadjuvant treatment seemed to have gene profiles that clustered distinctly differently from those of patients who were poor responders to treatment. More recently, Chang and colleagues [13] have shown that gene profiling can be used in accurately predicting response to neoadjuvant docetaxel. The study enrolled 24 subjects, extracted sufficient RNA from all core needle biopsies and constructed a 92-gene predictor of response (Fig. 2). In a complete cross-validation analysis, which gives an unbiased estimate of performance on future samples, the classifier correctly identified 10 of 11 responders and 11 of 13 non-responders for an overall accuracy of 88%. In a small validation set, this 92-gene classifier successfully predicted response in six patients. This compares very favorably with the best existing predictive factors for response to specific therapy, and strongly suggests that after appropriately extensive validation, microarray profiling will be useful for treatment selection. A second neoadjuvant study was recently published with the use of cDNA arrays to develop predictors for paclitaxel, fluorouracil, doxorubicin, and cyclophosphamide, involving 24 samples. A classifier with 74 markers was developed, with 78% accuracy, suggesting that transcriptional profiling has the potential to identify a gene expression pattern in breast cancer that might lead to clinically useful predictors of chemotherapy response [14].
Figure 2.
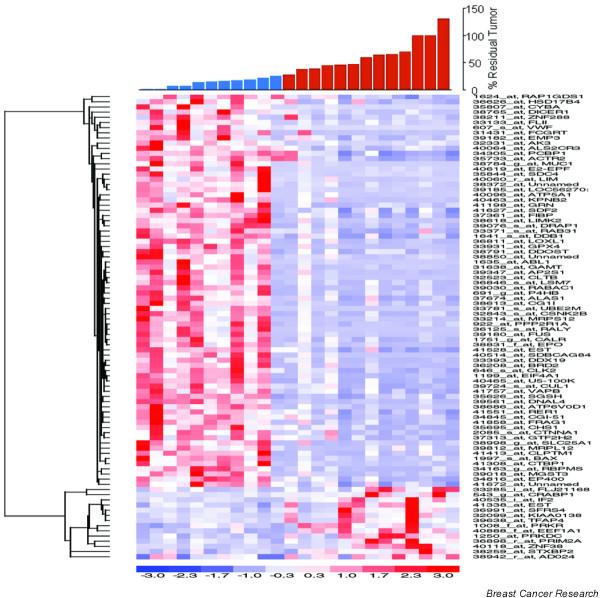
Hierarchical clustering of genes correlated with response to docetaxel. Sensitive tumors (S) are defined as those with 25% residual disease or less (shown as blue bars), and resistant tumors (R) are defined as those with greater than 25% residual disease (shown as red bars). The expression levels are shown in red (expression levels above the mean for the gene) and blue (levels below the mean for the gene).
However, we acknowledge that studies to construct and validate array-based prognostic and predictive 'markers' are complex. These studies must address all of the concerns associated with ordinary, single-gene markers as well as several considerations unique to array studies. Recommendations for the development of array-based prognostic classifiers have recently been enunciated by Simon and colleagues [15]. Among the most important points, they recommend that studies should include the following: first, adequately large sample sizes in both training and validation sets; second, a complete iteration of the entire classifier construction process in estimating cross-validated prediction rates; third, head-to-head comparison of alternative classifiers on the same data set; and fourth, inclusion of the full diversity of cases in any validation sets. In addition, gene expression patterns can be confounded by several other factors including ovarian ablation in premenopausal ER-positive patients, and different mechanisms of action of combination chemotherapies. Groups of patients with different characteristics such as menopausal and ER status or HER-2 over-expression might be necessary for the definitive determination of classifying patterns in these subsets of patients.
Conclusion
The goal of comprehensive, genome-wide approaches is to identify clinically useful genetic profiles that will accurately predict the outcome of therapy and the prognosis of patients with breast cancer. Despite improvements in technology, complex mechanisms driving the evolution of breast cancer continue to present challenges for the use of genomic approaches in the better understanding of breast and other cancers. This, combined with our use of different markers, methods, tumors (for example differing ER and HER2), and measurements of clinical outcomes, impedes the development of a consensus about predictive and prognostic markers for breast cancer.
As this field matures, genomic studies examining identical breast tumor sets with multiple complementary technologies (for example loss of heterozygosity, comparative genetic hybridization, and gene expression array analyses) will prove essential in unraveling the genetic heterogeneity characteristic of this disease. A combined genomic approach is necessary to define the underlying heterogeneous complexity that is characteristic of breast cancer. These data should lead to the identification and characterization of breast cancer subtypes, the definition of the malignant potential of a given lesion, and the prediction of its sensitivity to specific therapies. These multidisciplinary approaches should contribute to a better biological understanding of, and therefore improved clinical management of, breast cancer.
Abbreviations
ER = estrogen receptor; PR = progesterone receptor.
Competing interests
The author(s) declare that they have no competing interests.
Acknowledgments
Acknowledgements
This work was supported by funding provided by NIH grants P50 CA58183, P01 CA 30195, the Breast Cancer Research Foundation, the Emma Jacobs Clinical Breast Cancer Fund, and the US Army Medical Research and Materiel Command DAMD17-01-0132.
See related Commentary by Ahmed and Brenton: http://breast-cancer-research.com/content/7/3/96
References
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Sgroi DC, Teng S, Robinson G, LeVangie R, Hudson JR, Jr, Elkahloun AG. In vivo gene expression profile analysis of human breast cancer progression. Cancer Res. 1999;59:5656–5661. [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruvberger S, Ringner M, Chen Y, Panavally S, Saal LH, Borg A, Ferno M, Peterson C, Meltzer PS. Estrogen receptor status in breast cancer is associated with remarkably distinct gene expression patterns. Cancer Res. 2001;61:5979–5984. [PubMed] [Google Scholar]
- Huang E, Cheng SH, Dressman H, Pittman J, Tsou MH, Horng CF, Bild A, Iversen ES, Liao M, Chen CM, et al. Gene expression predictors of breast cancer outcomes. Lancet. 2003;361:1590–1596. doi: 10.1016/S0140-6736(03)13308-9. [DOI] [PubMed] [Google Scholar]
- van 't Veer LJ, Dai H, van De Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van Der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van 't Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- Hedenfalk I, Duggan D, Chen Y, Radmacher M, Bittner M, Simon R, Meltzer P, Gusterson B, Esteller M, Kallioniemi OP, et al. Gene-expression profiles in hereditary breast cancer. N Engl J Med. 2001;344:539–548. doi: 10.1056/NEJM200102223440801. [DOI] [PubMed] [Google Scholar]
- Verhoog LG, Brekelmans CTM, Seynaeve C, Dahmen G, van Geel AN, Bartels CC, Tilanus-Linthorst MM, Wagner A, Devilee P, Halley DJ, et al. Survival in hereditary breast cancer associated with germline mutations of BRCA2. J Clin Oncol. 1999;17:3396–3402. doi: 10.1200/JCO.1999.17.11.3396. [DOI] [PubMed] [Google Scholar]
- Fisher B, Bryant J, Wolmark N, Mamounas E, Brown A, Fisher ER, Wickerham DL, Begovic M, DeCillis A, Robidoux A, et al. Effect of preoperative chemotherapy on the outcome of women with operable breast cancer. J Clin Oncol. 1998;16:2672–2685. doi: 10.1200/JCO.1998.16.8.2672. [DOI] [PubMed] [Google Scholar]
- Chang J, Powles TJ, Allred DC, Ashley SE, Clark GM, Makris A, Assersohn L, Gregory RK, Osborne CK, Dowsett M. Biologic markers as predictors of clinical outcome from systemic therapy for primary operable breast cancer. J Clin Oncol. 1999;17:3058–3063. doi: 10.1200/JCO.1999.17.10.3058. [DOI] [PubMed] [Google Scholar]
- Buchholz TA, Stivers DN, Stec J, Ayers M, Clark E, Bolt A, Sahin AA, Symmans WF, Hess KR, Kuerer HM, et al. Global gene expression changes during neoadjuvant chemotherapy for human breast cancer. Cancer J. 2002;8:461–468. doi: 10.1097/00130404-200211000-00010. [DOI] [PubMed] [Google Scholar]
- Chang J, Wooten EC, Tsimelzon A, Hilsenbeck SG, Gutierrez MC, Elledge R, Mohsin S, Osborne CK, Chamness GC, Allred DC, et al. Gene expression profiles for docetaxel chemosensitivity. Proc Am Soc Clin Oncol. 2002;43 Abstract 1700. [Google Scholar]
- Ayers M, Symmans WF, Stec J, Damokosh AI, Clark E, Hess K, Lecocke M, Metivier J, Booser D, Ibrahim N, et al. Gene expression profiles predict complete pathologic response to neoadjuvant paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide chemotherapy in breast cancer. J Clin Oncol. 2004;22:2284–2293. doi: 10.1200/JCO.2004.05.166. [DOI] [PubMed] [Google Scholar]
- Simon R, Radmacher MD, Dobbin K, McShane LM. Pitfalls in the use of DNA microarray data for diagnostic and prognostic classification. J Natl Cancer Inst. 2003;95:14–18. doi: 10.1093/jnci/95.1.14. [DOI] [PubMed] [Google Scholar]