

Abstract
Although tamoxifen has been an effective treatment for breast cancer, several novel anti-oestrogen compounds have been developed with a reduced agonist profile on breast and gynaecological tissues. These include selective oestrogen receptor modulators (SERMs; both 'tamoxifen-like' and 'fixed-ring' SERMs) and selective oestrogen receptor downregulators (SERDs), although none has been proved superior in efficacy to tamoxifen in various advanced breast cancer trials. Thus, many have questioned whether a need for SERMs in breast cancer still exists, although chemoprevention remains a possible niche setting. In contrast, SERDs may have useful efficacy following aromatase inhibitors because of their unique mechanism of action, and clinical trials to determine their optimal use or sequence are ongoing.
Introduction
Oestrogen has important physiological effects on the growth and function of hormone-dependent tissues, including breast epithelium, uterus, vagina and ovaries. In addition, oestrogen preserves bone mineral density and reduces the risk for osteoporosis, protects the cardiovascular system by reducing cholesterol levels, and modulates cognitive function and behaviour. Tamoxifen is a nonsteroidal anti-oestrogen that antagonizes the action of oestrogen and is effective in both the treatment [1,2] and prevention of breast cancer [3]. Although concerns were raised regarding the potential anti-oestrogenic effects on normal tissues, paradoxically tamoxifen acts as an oestrogen on bone, blood lipids and the endometrium [4]. In the adjuvant and prevention settings, this may increase the risk for endometrial cancer in women taking tamoxifen, although the risk has been perceived to be small in relation to the substantial benefit from reduction in breast cancer related events [5]. Likewise, breast epithelial cells and established carcinomas adapt to chronic anti-oestrogen exposure and develop resistance to tamoxifen, which may also result from the drug's partial agonistic activity stimulating tumour regrowth [6].
The term 'selective oestrogen receptor modulator' (SERM) refers to the capacity of separate anti-oestrogens to exert alternative effects on various oestrogen regulated targets. Over the past 10–15 years several strategies were employed to improve or alter the agonist/antagonist profile of tamoxifen. An understanding of structure–function relationships led to chemical modifications of tamoxifen, either by altering the side chains to produce new tamoxifen analogues such as toremifene, idoxifene, droloxifene, lasofoxifene and TAT-59; or by altering the nonsteroidal triphenylethylene ring structure of tamoxifen to produce a nonsteroidal 'fixed ring' structure such as the benzothiophene derivatives raloxifene and arzoxifene, the benzopyran derivative acolbifene, or the indole ERA-923. All of these nonsteroidal anti-oestrogens have been classified as SERMs because they exhibit mixed tissue dependent agonist/antagonist activity.
At the same time the search for a 'pure anti-oestrogen' with no agonist activity and increased antagonist potency compared with tamoxifen led to the discovery of the selective oestrogen receptor downregulators (SERDs; e.g. fulvestrant). Experimental models have shown that the novel steroidal anti-oestrogen fulvestrant, which is devoid of agonist effects, can antagonize tamoxifen-stimulated growth, and as a treatment for hormone sensitive tumours it may delay the emergence of resistance. This led to the hope that these different structural classes of anti-oestrogens (Fig. 1) with an altered agonist/ antagonist profile may overcome this form of resistance and improve further on the efficacy of tamoxifen in treating breast cancer. Central to this approach, however, is an understanding of the molecular biology of the oestrogen receptor (ER) and the differential effects of various SERMs and SERDs in effectively antagonizing the action of ER.
Figure 1.
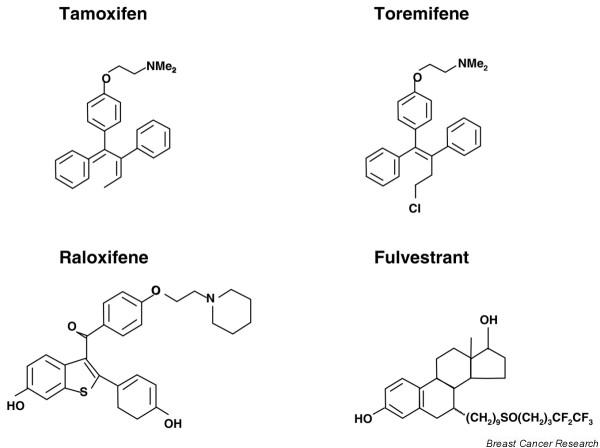
Chemical structures of anti-oestrogen compounds. Shown are the chemical structures of tamoxifen, the 'tamoxifen-like' selective oestrogen receptor modulator (SERM) toremifene, the 'fixed-ring' SERM raloxifene, and the selective oestrogen receptor downregulator (SERD) fulvestrant.
Molecular biology of the eostrogen receptor: differential effects of SERMs and SERDs
Progress in our molecular understanding of ER function has provided insights into the differential effects of various ER ligands, including oestrogen and tamoxifen in different tissues (for review [7]). Oestrogen influences gene expression and cellular phenotype by diffusing into the cell and binding nuclear ER, which in turn activates receptor dimerization; association with various coactivator and corepressor proteins to a greater or lesser extent, respectively; and subsequent DNA binding of liganded ER within promoter regions of DNA upstream of oestrogen regulated target genes. Gene transcription is activated through two separate transactivation domains within ER, termed AF-1 in the amino-terminal A/B region and AF-2 in the carboxyl-terminal E region [8]. At its simplest level tamoxifen functions as a competitive anti-oestrogen to inhibit the action of oestrogen. Tamoxifen-bound ER still dimerizes and binds DNA, but the downstream effects are different as a result of the altered conformational shape of the tamoxifen-ER complex as compared with oestradiol. This results in a change in the receptor bound balance of coactivators and corepressors, such that tamoxifen-liganded ER may block gene transcription through the AF-2 domain while AF-1 mediated gene transcription may still occur [9]. This may explain the partial agonist activity of tamoxifen in addition to its ability to antagonize oestrogen regulated gene transcription (Fig. 2).
Figure 2.
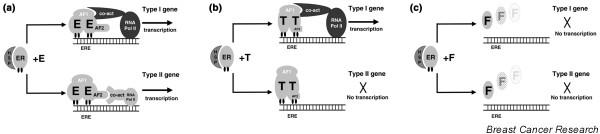
Molecular effects of oestradiol and anti-oestrogen compounds. Shown are the molecular effects of (a) oestradiol, (b) the selective oestrogen receptor modulator (SERM) tamoxifen and (c) the selective oestrogen receptor downregulator (SERD) fulvestrant on oestrogen receptor (ER) dimerization, conformational shape and DNA binding by liganded receptor, AF1/AF2 activation, coactivator recruitment, and subsequent transcriptional activation of type I and type II ER-regulated genes. As shown in panel a, oestradiol binding to ER leads to loss of heat shock proteins (HSPs), dimerization and phosphorylation of receptors, with conformational change leading to coactivator activation at both AF1 and AF2 sites; a full agonist effect is seen. In panel b, SERM (tamoxifen) binding to ER leads to loss of HSPs, dimerization and phosphorylation of receptors, but with different specific conformational change leading to coactivator activation at AF1 only, and not at AF2 sites; therefore, a partial agonist effect is seen. As shown in panel c, SERD (fulvestrant) binding to ER leads to loss of HSPs, but lack of receptor dimerization because of altered conformational change. Thus, receptor degradation is enhanced with no activation at AF1 or AF2 sites; no agonist effect is seen. AF, activating function; E, oestradiol; ERE, oestrogen response element; F, fulvestrant; RNA Pol II, ribonucleic acid polymerase II; T, tamoxifen.
It has become clear that the molecular biology of ER is complex, and that other aspects of its function may mediate the differential ligand effects seen in response to oestrogen or tamoxifen. In addition to classical ER (now called ER-α), a second ER was cloned (ER-β), which shares sequence homology within the DNA binding domain [10] but which differs in that AF-1 activity is considerably less than with ER-α [11]. Equally ER-β lacks much of the carboxyl-terminal F domain of ER-α, which may be an important region in determining an agonist response to tamoxifen [12]. The distribution in normal tissues of ER-β is different from that of ER-α, which implies a distinct physiological role, and some evidence has implicated increased ER-β expression as a mechanism for tamoxifen resistance in breast cancer [13]. It has also been established that, in addition to the classical model of liganded ER that binds DNA at defined oestrogen response elements, other response pathways can become activated by ER. For example AP-1 response elements regulate genes involved in cell proliferation, motility and apoptosis, and liganded ER may indirectly regulate AP-1 gene transcription through direct protein-protein interaction with AP-1 transcription factors (c-fos and c-jun). Tamoxifen was shown to be an agonist on AP-1 regulated genes with either ER-α or ER-β [14], whereas oestrogen liganded with ER-β inhibited AP-1 gene transcription [15]. Enhanced activation of AP-1 by tamoxifen may also be associated with tamoxifen resistance in models of breast cancer [16], and in tumours from breast cancer patients relapsing on tamoxifen [17]. Finally, the relative balance in a given cell type of coactivator and corepressor proteins may also determine the given response of ER to a particular ligand. For example, overexpression of the coactivator SRC-1 has been shown to enhance the agonist response to tamoxifen [18], whereas a reduction in the level of the corepressor N-CoR was associated with development of tamoxifen resistance in breast cancer xenografts [19]. Thus, changes in expression of ER-β relative to ER-α, enhancement of the AP-1 pathway, or a change in the balance of coactivator/corepressor proteins could all account for differential agonist/antagonist responses to anti-oestrogens both in different target tissues and human breast carcinomas.
The development of SERMs, which are structurally different from tamoxifen, has provided further insight into the biology of ER action, and created a new understanding of how modulating the structure–function interaction of ligand with ER could alter the biological effect. A crucial aspect of oestrogen-ER interaction is the complete envelopment of the steroid in a hydrophobic pocket within the ligand binding domain due to the critical positioning of a 'lid' formed by helix 12 of the ER. The position of this helix is also critical for the correct recruitment of coactivators to the AF-2 transactivation site, which allows subsequent initiation of RNA polymerase activity (Fig. 2). Occupation of the ligand-binding domain by tamoxifen, SERMs, or SERDs may result in a qualitatively different conformational shape of the liganded receptor, due to the alkylaminoethoxy side chain of the tamoxifen-like triphenylethylenes (i.e. toremifene), the different structure of the fixed ring benzothiophenes (i.e. raloxifene), or the long side chain of steroidal anti-oestrogens (i.e. fulvestrant; Fig. 1). This imparts a different positioning of the helix 12 'lid', the exact nature of which depends on the conformational shape that each anti-oestrogen imparts to the ligand ER complex [20]. As a result, the relative positioning of helix 12 may influence the likelihood of coactivator/corepressor binding, and as such determine the transcriptional response to liganded ER for a given gene. Likewise, in the endometrium tamoxifen, but not raloxifene, may have oestogenic-like effects due to recruitment or coactivators to a subset of genes, and this aspect may vary in different tissues depending on the background level of expression of coactivators such as SRC-1 [21].
These data provided a foundation for new hypotheses based on the chemical structure and structure–function relationship for each of the different SERMs/SERDs, together with the cell type and promoter specific differences in coregulator recruitment, which together may explain their differential antagonist/agonist profile observed in different tissues. Based on these characteristics, which seem to differentiate these new compounds from tamoxifen, several of the SERMs were developed for breast cancer with the expectation that they would have an improved preclinical and clinical profile (Table 1).
Table 1.
The ideal profile of a novel SERM in comparison with tamoxifen
| Profile | Details |
| Preclinical | Greater binding affinity for ER |
| Ability to antagonize oestrogen dependent growth of breast cancer cells in vitro | |
| Equal or greater inhibition of hormone-dependent xenograft growth in vivo | |
| Activity against tamoxifen dependent (resistant) tumours | |
| Delayed emergence of anti-oestrogen resistance in vivo | |
| Reduced agonist effects in uterotrophic assays | |
| Lack of stimulation of endometrial cancer cells in vitro/in vivo | |
| Lack of DNA adduct formation | |
| Prevention of bone loss in ovariectomized animals | |
| Clinical | Activity in hormone sensitive breast cancer, at least equivalent to tamoxifen |
| Increase in time to disease progression compared with tamoxifen | |
| Activity in tamoxifen resistant breast cancer | |
| Improved side effect profile (i.e. less hot flushes) | |
| No endometrial thickening/hyperplasia/cancer risk | |
| Preservation of bone mineral density | |
| Reduction in serum cholesterol |
ER, oestrogen receptor; SERM, selective oestrogen receptor modulator.
'Tamoxifen-like' triphenylethylene SERMs
For each of the triphenylethylene derivatives preclinical data suggested an improved antagonist/agonist profile compared with tamoxifen. This led to their clinical development in the hope that these may prove safer or more effective anti-oestrogens for the treatment of breast cancer compared with tamoxifen. The preclinical and clinical data were reviewed in detail elsewhere [22], but key aspects of each compound are highlighted below.
Toremifene
Toremifene's only structural difference compared with tamoxifen relates to a single chlorine atom at position 4 (Fig. 1), and as such the pharmacological profiles of these drugs are very similar. Unlike tamoxifen, toremifene was found not to be hepato-carcinogenic in preclinical models, which in part may relate to an inability of toremifene compared with tamoxifen to induce DNA adducts in rat liver [23]. Toremifene had a similar relative binding affinity for ER to tamoxifen and inhibited the growth of ER-positive breast cancer cells in vitro and in vivo [24]. However, toremifene had oestrogenic effects on endometrial cells, similar to tamoxifen [25], although it had a reduced oestrogenic effect on bone [26].
In terms of clinical efficacy, toremifene was no different to tamoxifen as first-line endocrine therapy in five large phase III randomized controlled trials (Table 2) [27-31]. A meta-analysis of these trials [32] showed an overall similar response rate for toremifene compared with tamoxifen (24% versus 25.3%), with no significant difference in time to disease progression or overall survival. Any potential difference in carcinogenicity, which was identified in preclinical studies, was not evaluated in any of these advanced breast cancer studies. Two adjuvant trials were initiated to compare efficacy and in particular long-term tolerability and safety in early breast cancer patients. After a median follow up of 4.4 years in the largest of these studies (1480 postmenopausal node-positive patients) [33] there were no significant differences in relapse-free survival or tolerability, and in particular the number of subsequent second cancers was similar.
Table 2.
Clinical efficacy of toremifene versus tamoxifen
| Toremifene | Tamoxifen | |||||
| Study [ref.] | n | ORR | TTP (months) | n | ORR | TTP (months) |
| Hayes et al. [28] | 221 | 21% | 5.6 | 215 | 19% | 5.8 |
| Pyrhonen et al. [29] | 214 | 31% | 7.3 | 201 | 37% | 10.2 |
| Gershanovich et al. [30] | 157 | 21% | 4.9 | 149 | 21% | 5.0 |
| Nomura et al. [27] | 62 | 24% | 5.1 | 60 | 27% | 5.1 |
| Milla-Santos et al. [31] | 106 | 38% | 11.9 | 111 | 32% | 9.2 |
| Meta-analysisa [32] | 725 | 24.0% | 4.9 | 696 | 25.3% | 5.3 |
Shown is a summary of clinical efficacy data from the randomised phase III trials of toremifene (40–60 mg/day) versus tamoxifen (20–40 mg/day) as first-line endocrine treatment of advanced breast cancer in postmenopausal women (oestrogen receptor status positive or unknown). aThe meta-analysis [32] was published in 1999 and included data from the first four trials [27-30], together with an unpublished small German study, but it did not include the Spanish study [31], which was published in 2001. ORR, objective response rate, including complete response and partial response; TTP, median time to disease progression.
Droloxifene
Droloxifene (or 3-hydroxytamoxifen) had a 10-fold higher relative binding affinity for ER compared with tamoxifen, a shorter half-life, greater growth inhibition of breast cancer cells in vitro, reduced oestrogenicity in the rat uterus, and absence of DNA adduct formation [34]. However, like tamoxifen it also behaved as an oestrogen in bone, preserving bone mineral density [35]. Despite promising phase II data, in which objective responses were seen in both tamoxifen refractory and naïve settings (for review [22]), droloxifene was inferior to tamoxifen in the phase III setting and its development was stopped.
Idoxifene
Idoxifene is metabolically more stable than tamoxifen as a result of a pyrrolidino side chain, with increased binding affinity for ER due to substitution of an iodine atom at the 4 position. Preclinically, idoxifene exhibited reduced stimulation of uterine weight in various uterotrophic assays compared with tamoxifen [36], with a delay in MCF-7 xenograft outgrowth in vivo compared with tamoxifen [37]. Thus, idoxifene was developed in the hope that its reduced agonist profile in breast and gynaecologocal tissues would be an advantage over tamoxifen for breast cancer patients. However, in a randomized phase II study [38], and in two international phase III studies of idoxifene versus tamoxifen as first-line therapy in advanced breast cancer [39,40], no improvements in efficacy or safety profile over tamoxifen were demonstrated, and development of idoxifene was stopped in 2001 (Table 3).
Table 3.
Clinical efficacy of idoxifene versus tamoxifen
| US phase III trial [39] | European phase III trial [40] | |||
| Efficacy measure | Tamoxifen (n = 111) | Idoxifene (n = 108) | Tamoxifen (n = 108) | Idoxifene (n = 112) |
| ORR | 9% | 13% | 19% | 20% |
| CBR | 39% | 34% | 48% | 38% |
| TTP | 166 days | 140 days | 181 days | 127 days |
Shown is a summary of efficacy data from two randomized double-blind phase III trials of idoxifene (40 mg/day) versus tamoxifen (20 mg/day) as first-line therapy in advanced/metasatic breast cancer. CBR, percentage of patients with either objective response or stable disease for 6 months or longer; ORR, percentage of patients with an objective response, including complete response and partial response; TTP, median time to disease progression.
Other tamoxifen-like derivatives
Other structural analogues of tamoxifen were synthesized, including TAT-59, which has a 10-fold higher affinity for ER than tamoxifen and was more effective at inhibiting human breast cancer xenograft growth in vivo [41]; GW5638, a carboxylic derivative, which demonstrated reduced agonist activity on the uterus in ovariectomized rats [42]; and lasofoxifene, a derivative of tetrahydronapthalene, which maintained bone mineral density in animal models [43]. None of these were developed for use in breast cancer.
'Fixed ring' SERMS
Greater optimism surrounded the profile of second- and third-generation SERMs, in particular because these drugs appeared devoid of any agonist activity in the endometrium while behaving as potent anti-oestrogens in the breast that retained agonist activity in bone. The benzothiophenes raloxifene is the most extensively studied SERM in this class (Fig. 1).
Raloxifene
The binding affinity of raloxifene for ER is similar to that of tamoxifen, and most of the pharmacological data showed similar activity in terms of inhibiting breast cancer cells in vitro and in vivo [44]. In preclinical models the drug maintained bone mineral density but had significantly less oestrogenic activity on endometrial cells than did tamoxifen and could inhibit tamoxifen-stimulated endometrial cancer growth in vivo [45]. Raloxifene was not developed as an anti-oestrogen for breast cancer, and few data exist on the activity of raloxifene in patients with advanced disease (for review [22]). However, during the development of raloxifene for use in osteoporosis it was found to reduce significantly the incidence of breast cancer (in particular ER-positive tumours) in postmenopausal women by 76% (95% confidence interval 56–87%), without any increase in endometrial thickening or risk to the gynaecological tract [46]. This suggested that raloxifene could represent a safer SERM for use in chemoprevention -a theme that has been developed further (see below).
Arzoxifene
Arzoxifene is a benzothiophene analogue; it is a more potent anti-oestrogen, with an improved SERM profile and greater anticancer efficacy as compared with raloxifene [47-49]. Clinical efficacy was reported in a phase II study in hormone-sensitive advanced breast cancer [50]. A second phase II trial compared two doses in 63 tamoxifen-resistant patients, and separately in 49 patients with hormone-sensitive disease [51]. Response rates were low in the tamoxifen-resistant patients (10% for 20 mg, 3% for 50 mg). In contrast, a response rate of 30% was seen with 20 mg arzoxifene in the hormone-sensitive group, with a further 17% having stable disease. The response rate for the 50 mg dose was somewhat lower (8%), and 20 mg dose arzoxifene was taken forward into a large multicentre phase III trial against tamoxifen as first-line therapy.
Acolbifene
EM-800 (SCH-57050) is an orally active prodrug of the active benzopyrene derivative acolbifene (EM-652), a so-called 'pure' nonsteroidal antiestrogen [52]. Preclinically the binding affinity of acolbifene for ER was significantly greater than that of oestradiol, tamoxifen, raloxifene, or fulvestrant, and in vitro acolbifene was more effective than 4-hydroxy-tamoxifen or fulvestrant at inhibiting oestradiol induced breast cancer cell proliferation [53]. In vivo, acolbifene was devoid of any agonist activity in an immature rat uterotrophic assay and on mouse endometrial tissues [54,55]. In an in vivo ZR-75-1 breast cancer xenograft model in ovariectomized mice acolbifene had no agonist effects on tumour growth, and was more effective at inhibiting oestrone-stimulated tumour growth than were five other tested anti-oestrogens (tamoxifen, toremifene, idoxifene, GW-5638 and raloxifene), with complete regressions seen in 65% of acolbifene treated tumours [56]. Likewise EM-800 (the oral precursor of the active metabolite acolbifene) was 30-fold more potent than tamoxifen at inhibiting uterine weight and reducing uterine/ vaginal ER expression [57]. In addition, studies have shown that EM-800 can prevent bone loss in the ovariectomized rat and lower serum cholesterol levels [58].
In terms of clinical development, a phase II study of EM-800 (20 mg or 40 mg) was undertaken in 43 postmenopausal women who had progressed on tamoxifen either in the metastatic or adjuvant setting [59]. There was one complete response and four partial responses (response rate 12%), with a median duration of response of 8 months. An additional seven (16%) patients had stable disease for longer than 6 months. These results in patients with defined tamoxifen-resistant disease are in contrast to those observed with other SERMs described above, for which partial cross-resistance with tamoxifen occurred, and a randomized phase III study in patients who had failed tamoxifen was initiated that will compare the efficacy of EM-800 with the aromatase inhibitor (AI) anastrozole. These data imply that as a 'pure' anti-oestrogen devoid of agonist activity, EM-800 may have an important different mechanism of action to that of other SERMs, and indeed may possess greater similarities to the steroidal anti-oestrogen fulvestrant (see below) than to the other SERMs described above.
ERA-923
The anti-oestrogen zindoxifene (D16726) is a 2-phenylindole structure that was previously shown to have oestrogenic activity in the uterus [60] but was inactive in a breast cancer trial [61]. By making rigid the alkylamino side chain, similar to the structure of raloxifene and EM-800, a new indole SERM called ERA-923 was created that was devoid of uterotrophic activity in immature rats when compared with raloxifene and ZK119010 [62]. ERA-923 had an improved preclinical profile in breast cancer experimental models compared with tamoxifen and raloxifene, and MCF-7 cells that are resistant to tamoxifen retain complete sensitivity to ERA-923 both in vitro and in vivo [63]. Unlike tamoxifen, droloxifene and raloxifene, ERA-923 was not uterotrophic in immature rats or ovariectomized mice. Following initial safety studies in healthy volunteers [64], clinical trials of ERA-923 as second-line therapy were initiated in 100 ER-positive patients with tamoxifen resistant metastatic breast cancer, together with proposals for trials in ER-positive hormone sensitive metastatic breast cancer as first-line therapy.
Role of SERMs in chemoprevention: biomarker studies
Although none of the SERMs outlined above has proved superior in efficacy to tamoxifen in the treatment of established breast cancer, the ability of SERMs to prevent the development of ER-positive breast cancer perhaps remains the greatest opportunity for these drugs to have a major impact on the disease. The evidence that both tamoxifen and raloxifene can prevent the development of breast cancer has provided 'proof of principle' for endocrine intervention as an important manipulation for women at risk for developing breast cancer [3,46,65]. However, important questions remain in the prevention setting, namely the identification of those women who are most likely to benefit from such an intervention, the most appropriate risk parameters to be used and, in particular, the safest and most effective SERM to utilize in this setting. Tamoxifen may reduce breast cancer incidence by 48% in an at-risk population, but it is associated with an increased risk for endometrial cancer and thrombotic events [3]. In contrast, raloxifene yielded an apparent greater risk reduction in breast cancer incidence with a reduced risk for endometrial cancer, albeit in a different population of women who were at risk for osteoporosis [46,65]. The current Study of Tamoxifen And Raloxifene (STAR) chemoprevention trial is comparing the effects of raloxifene with those of tamoxifen with the anticipation that efficacy in risk reduction maybe somewhat similar, but that the toxicity profile in terms of gynaecological problems may be better for raloxifene than for tamoxifen [66].
The development of SERMs as chemoprevention agents with an even better efficacy and improved toxicity profile over tamoxifen or raloxifene remains an important goal. However, conducting large prevention studies in 20,000 women or more over 10–15 years in order to generate results is increasingly expensive and inefficient. An alternative approach to identify novel SERM candidates for chemoprevention is to conduct short-term phase IA/IB preoperative biomarker modulation studies in women with newly diagnosed primary breast cancer. Changes in the proportion of proliferating tumour cells (as indicated by Ki-67) in ER-positive primary breast cancer has been shown to correlate with clinical response following treatment with tamoxifen [67], and more recently a greater reduction in Ki-67 after 2 weeks was observed in patients treated with the AI anastrazole than with tamoxifen [68], which is analogous to the improved outcome seen in the large scale Arimidex, Tamoxifen, Alone or in Combination (ATAC) adjuvant trial [69]. This has also been studied in randomized controlled trials in primary breast cancer with different doses of tamoxifen [70], and with the tamoxifen-like SERM idoxifene [71], raloxifene [72] and more recently arzoxifene [73]. In the placebo-controlled studies of idoxifene and raloxifene, short-term treatment for 2 weeks was associated with mean 35% and 21% reductions in Ki-67, respectively, as compared with a 6–7% mean increase for placebo. In the recent study with arzoxifene, changes in proliferation indices in 58 women were not statistically different from placebo control because of the confounding factor of stopping hormone replacement therapy before study entry, which was not allowed in the other studies. Similar clinical studies may be warranted with the two new SERMs acolbofene and lasofoxifene, given that they appear to be potent anti-oestrogens in the breast, pro-oestrogenic in the bone, and devoid of the unwanted uterotrophoic effects seen with tamoxifen. In addition, experimental studies in carcinogen-induced mammary carcinoma in rats have shown that novel SERMs such as acolbifene [74] and arzoxifene [75] can both effectively prevent mammary cancer development.
Such biomarker data strongly support the further clinical development in the chemoprevention setting of these novel SERMs that have antiproliferative effects on breast tissue and reduced agonist effects on the gynaecological tract, but that remain protective of bone mass. Many may feel that the existing experimental and early clinical studies provide sufficient supportive data to merit clinical trials in the chemoprevention setting, albeit that such studies remain large scale, time consuming and expensive. The next step will be to develop risk algorithms to identify those women who have the most to gain from such an intervention, at whom the next generation of chemoprevention trials with a novel SERM that is safer than tamoxifen could be specifically targeted.
SERDs
Mechanism of action
SERDs are distinguishable from tamoxifen and other SERMs, both pharmacologically and in terms of their molecular activity. Although both classes of agent mediate their effects through the ER, they differ significantly in their interaction with ER and the subsequent downstream effects. The steroidal anti-oestrogens bind to the ER but, because of their long bulky side chains at the 7α and 11β positions, receptor dimerization appears to be sterically hindered [76]. There is evidence that ER turnover is increased and nuclear localization is disrupted, with a concomitant reduction in the number of detectable ER molecules in the cell both in vitro and in vivo. This is in marked contrast to the stable or increased levels of ER expression associated with tamoxifen and other related SERMs [77]. Experimental studies suggest that, as a consequence of ER downregulation, ER-mediated transcription is completely attenuated due to inactivation of AF-1 and AF-2, with complete suppression of oestrogen dependent gene expression (Fig. 2c).
The preclinical characteristics of fulvestrant, which define this compound as a SERD devoid of oestrogen-like activity, have been extensively reviewed [78]. These include an affinity for the ER approximately 100 times that of tamoxifen, the specific absence of oestrogen-like activity on the uterus, and the capacity to block completely the stimulatory activities of both oestrogens and anti-oestrogens like tamoxifen with partial agonist activity. The absence of oestrogenic activity has important consequences for the development of resistance, which can limit the effectiveness of long-term tamoxifen therapy. In vitro studies demonstrate that tamoxifen-resistant breast cancer cell lines remain sensitive to growth inhibition by fulvestrant [79], and that in vivo tamoxifen-resistant tumors remain sensitive to fulvestrant [80]. Taken collectively, these data suggest that fulvestrant may be a more effective oestrogen antagonist than tamoxifen that is able to produce a longer response in animal models.
Clinical studies of fulvestrant
The clinical efficacy of fulvestrant has been compared with those of tamoxifen and anastrazole in postmenopausal women with breast cancer. Some of the first clinical data came from a short-term preoperative study conducted in 201 women with operable breast cancer in which the biological effects of fulvestrant were compared with those of tamoxifen [81]. A dose-dependent reduction in the levels of ER and progesterone receptor (PgR) expression was observed across three doses of fulvestrant (50, 125 and 250 mg) administered intramuscularly for 14–21 days before surgery, compared with placebo or tamoxifen. At all three doses fulvestrant reduced proliferation as measured by Ki67 labelling index [82]. These clinical data confirmed that fulvestrant acts as an ER downregulator, with clear anti-oestrogenic and antiproliferative activity. Furthermore, the effect on PgR provided evidence of a more complete blockade of this ER-dependent pathway compared with tamoxifen, which increased PgR levels because of its partial agonist activity.
The efficacy of fulvestrant in tamoxifen-resistant breast cancer was first demonstrated in a small phase II trial conducted in 19 patients with tamoxifen-refractory disease. Thirteen patients (69%) achieved clinical benefit, with a median duration of 25 months, with seven patients demonstrating a partial response and six patients stable disease [83]. These data in tamoxifen-resistant disease are in stark contrast to those with the SERMs outlined above, where cross-resistance with tamoxifen was invariably shown. Two phase III studies then compared the efficacy and tolerability of fulvestrant (250 mg monthly) with anastrozole in post-menopausal women whose disease had progressed on or after prior adjuvant endocrine therapy [84,85]. The median time to disease progression was numerically longer with fulvestrant than with anastrozole for both trials, with a longer duration of response observed in the North American trial [84]. Fulvestrant was also well tolerated and is the first anti-oestrogen reported to be at least as effective as a new generation AI, unlike the trials with the tamoxifen-like or benothiophene SERMs outlined above.
More recently, data from a multinational randomized double-blind study comparing fulvestrant (250 mg monthly, intramuscular) with tamoxifen (20 mg/day, oral) as first-line therapy in metastatic breast cancer were reported [86]. The study randomized a total of 587 postmenopausal women with metastatic breast cancer who were ER and/or PgR positive or in whom receptor status was unknown, and at a median follow up of 14.5 months there was no significant difference between the fulvestrant and tamoxifen groups in terms of time to progression in the whole population (median time to progression: 6.8 months versus 8.3 months, respectively; P = 0.088). However, there was a significant difference in time to treatment failure in favour of tamoxifen (P = 0.026), with the median being 5.9 months for fulvestrant and 7.8 months for tamoxifen. These were unexpected findings that were not obviously explained by imbalance in patient groups, failure to administer intramuscular injections correctly, or undue toxicity. The separation of the Kaplan-Meier curves for TTP occurred almost immediately and was most pronounced at 3 months, suggesting a higher rate of early progression in the fulvestrant group. Pharmacokinetic studies have shown that accumulation of the drug may take 3–6 months to reach steady state plasma levels [87].
New clinical directions for SERDs
The clinical scenario has shifted somewhat with the recent pre-eminence of the AIs as the first-line endocrine therapy of choice both in the metastatic and, increasingly, in the adjuvant setting [69]. As such, there is a need to establish which endocrine agent and sequence is most effective in the post-AI setting. In vitro, long-term oestrogen deprivation (LTED) is a situation analogous to that caused by long-term AI treatment and subsequent AI resistance, and is associated with an adaptive increase in ER expression and intracellular signalling that results in hypersensitivity to low oestradiol levels [88,89]. It is unclear whether tamoxifen or other related SERMs will be effective in this setting given their partial agonist effects, which may be more pronounced in cells that contain these adaptive changes in ER signalling. In contrast, fulvestrant has no agonist activity and has been shown to be more effective than tamoxifen in model systems of both LTED resistance in vitro [88] and resistance to long-term letrozole in vivo [90]. Encouraging clinical data were reported for fulvestrant following progression on AIs in five small phase II studies (Table 4), with clinical benefit seen in 19–52% patients [91-95]. At present two large phase III trials (EFECT and SoFEA) are assessing the true benefit of using a SERD in this setting by comparing the efficacy of fulvestrant with that of the steroidal aromatase inactivator exemestane, which has demonstrated some partial non-cross-resistance with either letrozole or anastrazole; if positive, these studies may help to define the optimal role for fulvestrant in ER-positive postmenopausal metastatic breast cancer [96].
Table 4.
Clinical efficacy of fulvestrant after progression on prior endocrine therapy with AIs
| Study [ref.] | Treatment (setting) | Prior systemic treatments | Number of | Clinical benefita (%) |
| Perey et al. [93] | Fulvestrant (third line) | Include tamoxifen and AIs | 67 | 28 |
| Ingle et al. [92] | Fulvestrant (second and third line) | Prior nonsteroidal AIs, and tamoxifen in 79% of patients | 77 | 29 |
| Petruzelka and Zimovjanova [94] | Fulvestrant (second to fifth line) | Include nonsteroidal AIs, adjuvant tamoxifen, and goserelin formestane | 44 | 52 |
| Franco et al. [91] | Fulvestrant (mean prior endocrine therapies = 3.4) | Include nonsteroidal AIs, tamoxifen, megestrol acetate, exemestane, and chemotherapy | 42 | 19 |
| Steger et al. [95] | Fulvestrant (second to fifth line) | Include nonsteroidal AIs, tamoxifen, exemestane, goserelin, and formestane | 88 | 57 |
Phase II clinical trials with fulvestrant following disease progression on prior endocrine therapy with aromatase inhibitors are summarized. aClinical benefit included patients who had a complete response, partial response, or stable disease for 24 weeks or longer. AI, aromatase inhibitor.
At present, no studies are being conducted to investigate the benefit of fulvestrant in the adjuvant setting. Clinical studies combining fulvestrant with various signal transduction modulators are ongoing, including trastuzumab (Herceptin), EGFR (epidermal growth factor receptor) tyrosine kinase inhibitors, and farnesyltransferase inhibitors. These trials are working on the premise that complete ER blockade combined with effective signal transduction blockade of growth factor pathways may abrogate resistance mechanisms and provide greater control of cancer cells. It also remains to be seen whether two new orally bioavailable pure anti-oestrogens (SR16234 and ZK191703) will have equivalent or superior potency in patients to intramuscularly administered fulvestrant.
Conclusion
The search for a better version of tamoxifen for the treatment and prevention of breast cancer has yielded many compounds of potential interest, but none has replaced tamoxifen in the clinical arena, despite all the effort involved; as such, many may feel that SERMs and SERDs have lost their way. The reality in the treatment of breast cancer is that they have been surpassed by the third generation AIs, which have shown better tolerability than tamoxifen, with substantial gains in efficacy both in the advanced and adjuvant settings. However, this change in the treatment sequence has created new challenges for development of novel endocrine therapies. It is possible that SERMs that retain a small partial agonist activity may or may not be effective in tumours that become resistant/hypersensitive to low oestradiol levels induced by LTED. In contrast, this may be an ideal opportunity for the SERD fulvestrant to demonstrate its unique endocrine activity because of its ability to down-regulate the hypersensitive and activated ER present in LTED-resistant tumour cells; ongoing clinical trials in advanced disease will determine whether this preclinical promise holds forth. As for SERMs, although their clinical development may have fallen on stony ground to date, if nothing else they have given us a new opportunity to improve our understanding of the complex molecular biology of ER signalling in breast and other tissues. Their clinical resurgence may still occur in the long-term chemoprevention setting, where they could deliver an improved safety profile compared with tamoxifen, combined with effective risk reduction. SERMs may still have an impact to make, and so their development is not yet over.
Abbreviations
AI = aromatase inhibitor; ER = oestrogen receptor; LTED = long-term oestrogen deprivation; PgR = progesterone receptor; SERD = selective oestrogen receptor downregulator; SERM = selective oestrogen receptor modulator; TTP = time to progression.
Note
This article is part of a review series on Endocrinology and hormone therapy in breast cancer, edited by James N Ingle and V Craig Jordan. Other articles in the series can be found online at http://breast-cancer-research.com/articles/review-series.asp?series=bcr_endocrinology.
Competing interests
SRDJ has received research support in the past from AstraZeneca and from SmithKline Beecham.
References
- Cole MP, Jones CT, Todd ID. A new anti-oestrogenic agent in late breast cancer. An early clinical appraisal of ICI46474. Br J Cancer. 1971;25:270–275. doi: 10.1038/bjc.1971.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Early Breast Cancer Trialists Group Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists' Collaborative Group. Lancet. 1998;351:1451–1467. doi: 10.1016/S0140-6736(97)11423-4. [DOI] [PubMed] [Google Scholar]
- Fisher B, Costantino JP, Wickerham DL, Redmond CK, Kavanah M, Cronin WM, Vogel V, Robidoux A, Dimitrov N, Atkins J, et al. Tamoxifen for prevention of breast cancer: report of the National Surgical Adjuvant Breast and Bowel Project P-1 Study. J Natl Cancer Inst. 1998;90:1371–1388. doi: 10.1093/jnci/90.18.1371. [DOI] [PubMed] [Google Scholar]
- Jordan VC. The development of tamoxifen for breast cancer therapy. In: Jordan VC, editor. Long-term Tamoxifen Treatment for Breast Cancer. Madison: University of Wisconsin Press; 1994. pp. 3–26. [Google Scholar]
- Fisher B, Costantino JP, Redmond CK, Fisher ER, Wickerham DL, Cronin WM. Endometrial cancer in tamoxifen-treated breast cancer patients: findings from the National Surgical Adjuvant Breast and Bowel Project (NSABP) B-14. J Natl Cancer Inst. 1994;86:527–537. doi: 10.1093/jnci/86.7.527. [DOI] [PubMed] [Google Scholar]
- Johnston SR. Acquired tamoxifen resistance in human breast cancer: potential mechanisms and clinical implications. Anti-cancer Drugs. 1997;8:911–930. doi: 10.1097/00001813-199711000-00002. [DOI] [PubMed] [Google Scholar]
- Johnston SR, Dowsett M. Aromatase inhibitors for breast cancer: lessons from the laboratory. Nat Rev Cancer. 2003;3:821–831. doi: 10.1038/nrc1211. [DOI] [PubMed] [Google Scholar]
- Kumar V, Green S, Stack G, Berry M, Jin JR, Chambon P. Functional domains of the human estrogen receptor. Cell. 1987;51:941–951. doi: 10.1016/0092-8674(87)90581-2. [DOI] [PubMed] [Google Scholar]
- Tzukerman MT, Esty A, Santiso-Mere D, Danielian P, Parker MG, Stein RB, Pike JW, McDonnell DP. Human estrogen receptor transactivational capacity is determined by both cellular and promoter context and mediated by two functionally distinct intramolecular regions. Mol Endocrinol. 1994;8:21–30. doi: 10.1210/me.8.1.21. [DOI] [PubMed] [Google Scholar]
- Kuiper GGJM, Enmark E, Pelto-Huikko M, Nilsson S, Gustafsson J-A. Cloning of a novel estrogen receptor expressed in rat prostate and ovary. Proc Natl Acad Sci USA. 1996;93:5925–5930. doi: 10.1073/pnas.93.12.5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley SM, Parker MG. A comparison of transcriptional activation by ER alpha and ER beta. J Steroid Biochem Mol Biol. 1999;69:165–175. doi: 10.1016/S0960-0760(99)00055-2. [DOI] [PubMed] [Google Scholar]
- Montano MM, Muller V, Trobaugh A, Katzenellenbogen BS. The carboxy-terminal F domain of the human estrogen receptor: role in the transcriptional activity of the receptor and the effectiveness of antiestrogens as estrogen antagonists. Mol Endocrinol. 1995;9:814–825. doi: 10.1210/me.9.7.814. [DOI] [PubMed] [Google Scholar]
- Speirs V, Parkes AT, Kerin MJ, Walton DS, Carleton PJ, Fox JN, Atkin SL. Coexpression of estrogen receptor alpha and beta: poor prognostic factors in human breast cancer? Cancer Res. 1999;59:525–528. [PubMed] [Google Scholar]
- Webb P, Lopez GN, Uht RM, Kushner PJ. Tamoxifen activation of the estrogen receptor/AP-1 pathway: potential origin for the cell-specific estrogen-like effects of antiestrogens. Mol Endocrinol. 1995;9:443–456. doi: 10.1210/me.9.4.443. [DOI] [PubMed] [Google Scholar]
- Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS. Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. 1997;277:1508–1510. doi: 10.1126/science.277.5331.1508. [DOI] [PubMed] [Google Scholar]
- Schiff R, Reddy P, Coronado E, Osborne CK. Development of tamoxifen-stimulated growth in-vivo is associated with changes in AP-1 activity. Breast Cancer Res Treat. 1997;46:A347. [Google Scholar]
- Johnston SR, Lu B, Scott GK, Kushner PJ, Smith IE, Dowsett M, Benz CC. Increased activator protein-1 DNA binding and c-Jun NH2-terminal kinase activity in human breast tumors with acquired tamoxifen resistance. Clin Cancer Res. 1999;5:251–256. [PubMed] [Google Scholar]
- Smith CL, Nawaz Z, O'Malley BW. Coactivator and corepressor regulation of the agonist/antagonist activity of the mixed antiestrogen, 4-hydroxytamoxifen. Mol Endocrinol. 1997;11:657–666. doi: 10.1210/me.11.6.657. [DOI] [PubMed] [Google Scholar]
- Lavinsky RM, Jepsen K, Heinzel T, Torchia J, Mullen TM, Schiff R, Del-Rio AL, Ricote M, Ngo S, Gemsch J, et al. Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proc Natl Acad Sci USA. 1998;95:2920–2925. doi: 10.1073/pnas.95.6.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzozowski AM, Pike ACW, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson J-A, Carlquist M. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465–2468. doi: 10.1126/science.1068537. [DOI] [PubMed] [Google Scholar]
- Johnston SR. Endocrine manipulation in advanced breast cancer: recent advances with SERM therapies. Clin Cancer Res. 2001. pp. 4376s–4387s. discussion: 4411s-4412s. [PubMed]
- Hard GC, Iatropoulos MJ, Jordan K, Radi L, Kaltenberg OP, Imondi AR, Williams GM. Major difference in the hepatocarcinogenicity and DNA adduct forming ability between toremifene and tamoxifen in female Crl:CD(BR) rats. Cancer Res. 1993;53:4534–4541. [PubMed] [Google Scholar]
- Robinson SP, Jordan VC. Antiestrogenic action of toremifene on hormone-dependent, -independent, and heterogeneous breast tumor growth in the athymic mouse. Cancer Res. 1989;49:1758–1762. [PubMed] [Google Scholar]
- O'Regan RM, Cisneros A, England GM, MacGregor JI, Muenzner HD, Assikis VJ, Bilimoria MM, Piette M, Dragan YP, Pitot HC, et al. Effects of the antiestrogens tamoxifen, toremifene, and ICI 182,780 on endometrial cancer growth. J Natl Cancer Inst. 1998;90:1552–1558. doi: 10.1093/jnci/90.20.1552. [DOI] [PubMed] [Google Scholar]
- Marttunen MB, Hietanen P, Tiitinen A, Ylikorkala O. Comparison of effects of tamoxifen and toremifene on bone biochemistry and bone mineral density in postmenopausal breast cancer patients. J Clin Endocrinol Metab. 1998;83:1158–1162. doi: 10.1210/jc.83.4.1158. [DOI] [PubMed] [Google Scholar]
- Nomura Y, Tominaga T, Abe O, Izuo M, Ogawa N. Clinical evaluation of NK 622 (toremifene citrate) in advanced or recurrent breast cancer: a comparative study by a double blind method with tamoxifen [in Japanese] Gan To Kagaku Ryoho. 1993;20:247–258. [PubMed] [Google Scholar]
- Hayes DF, Van Zyl JA, Hacking A, Goedhals L, Bezwoda WR, Mailliard JA, Jones SE, Vogel CL, Berris RF, Shemano I, et al. Randomized comparison of tamoxifen and two separate doses of toremifene in postmenopausal patients with metastatic breast cancer. J Clin Oncol. 1995;13:2556–2566. doi: 10.1200/JCO.1995.13.10.2556. [DOI] [PubMed] [Google Scholar]
- Pyrhonen S, Valavaara R, Modig H, Pawlicki M, Pienkowski T, Gundersen S, Bauer J, Westman G, Lundgren S, Blanco G, et al. Comparison of toremifene and tamoxifen in post-menopausal patients with advanced breast cancer: a randomized double-blind, the 'nordic' phase III study. Br J Cancer. 1997;76:270–277. doi: 10.1038/bjc.1997.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershanovich M, Garin A, Baltina D, Kurvet A, Kangas L, Ellmen J. A phase III comparison of two toremifene doses to tamoxifen in postmenopausal women with advanced breast cancer. Eastern European Study Group. Breast Cancer Res Treat. 1997;45:251–262. doi: 10.1023/A:1005891506092. [DOI] [PubMed] [Google Scholar]
- Milla-Santos A, Milla L, Rallo L, Solano V. Phase III randomized trial of toremifene vs tamoxifen in hormonodependant advanced breast cancer. Breast Cancer Res Treat. 2001;65:119–124. doi: 10.1023/A:1006440802709. [DOI] [PubMed] [Google Scholar]
- Pyrhonen S, Ellmen J, Vuorinen J, Gershanovich M, Tominaga T, Kaufmann M, Hayes DF. Meta-analysis of trials comparing toremifene with tamoxifen and factors predicting outcome of antiestrogen therapy in postmenopausal women with breast cancer. Breast Cancer Res Treat. 1999;56:133–143. doi: 10.1023/a:1006250213357. [DOI] [PubMed] [Google Scholar]
- Holli K. Tamoxifen versus toremifene in the adjuvant treatment of breast cancer. Eur J Cancer. 2002;(Suppl 6):S37–S38. doi: 10.1016/S0959-8049(02)00279-4. [DOI] [PubMed] [Google Scholar]
- Hasmann M, Rattel B, Loser R. Preclinical data for droloxifene. Cancer Lett. 1994;84:101–116. doi: 10.1016/0304-3835(94)90364-6. [DOI] [PubMed] [Google Scholar]
- Ke HZ, Simmons HA, Pirie CM, Crawford DT, Thompson DD. Droloxifene, a new estrogen antagonist/agonist, prevents bone loss in ovariectomized rats. Endocrinology. 1995;136:2435–2441. doi: 10.1210/en.136.6.2435. [DOI] [PubMed] [Google Scholar]
- McCague R, Leclerq G, Legros N, Goodman J, Blackburn GM, Jarman M, Foster AB. Derivatives of tamoxifen; dependence of antiestrogenicity on the 4-substituent. J Med Chem. 1989;32:2527–2533. doi: 10.1021/jm00132a006. [DOI] [PubMed] [Google Scholar]
- Johnston SR, Riddler S, Haynes BP, A'Hern R, Smith IE, Jarman M, Dowsett M. The novel anti-oestrogen idoxifene inhibits the growth of human MCF-7 breast cancer xenografts and reduces the frequency of acquired anti-oestrogen resistance. Br J Cancer. 1997;75:804–809. doi: 10.1038/bjc.1997.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SR, Gumbrell LA, Evans TR, Coleman RE, Smith IE, Twelves CJ, Soukop M, Rea DW, Earl HM, Howell A, et al. A Cancer Research (UK) randomized phase II study of idoxifene in patients with locally advanced/metastatic breast cancer resistant to tamoxifen. Cancer Chemother Pharmacol. 2004;53:341–348. doi: 10.1007/s00280-003-0733-6. [DOI] [PubMed] [Google Scholar]
- Arpino G, Nair Krishnan M, Doval Dinesh C, Bardou VJ, Clark GM, Elledge RM. Idoxifene versus tamoxifen: a randomized comparison in postmenopausal patients with metastatic breast cancer. Ann Oncol. 2003;14:233–241. doi: 10.1093/annonc/mdg097. [DOI] [PubMed] [Google Scholar]
- Johnston SRD, Gorbunova V, Lichinister M, Manikas G, Koralewski P, Pluznaska A, Garin A, Harvey E. A multicentre double-blind randomised phase III trial of idoxifene versus tamoxifen as first-line endocrine therapy for metastatic breast cancer [abstract] Proc Am Soc Clin Oncol. 2001;20:29a. (A113) [Google Scholar]
- Toko T, Sugimoto Y, Matsuo K, Yamasaki R, Takeda S, Wierzba K, Asao T, Yamada Y. TAT-59, a new triphenylethylene derivative with antitumor activity against hormone-dependent tumors. Eur J Cancer. 1990;26:397–404. doi: 10.1016/0277-5379(90)90241-k. [DOI] [PubMed] [Google Scholar]
- Willson TM, Norris JD, Wagner BL, Asplin I, Baer P, Brown HR, Jones SA, Henke B, Sauls H, Wolfe S, et al. Dissection of the molecular mechanism of action of GW5638, a novel estrogen receptor ligand, provides insights into the role of estrogen receptor in bone. Endocrinology. 1997;138:3901–3911. doi: 10.1210/en.138.9.3901. [DOI] [PubMed] [Google Scholar]
- Ke HZ, Foley GL, Simmons HA, Shen V, Thompson DD. Long-term treatment of lasofoxifene preserves bone mass and bone strength and does not adversely affect the uterus in ovariectomized rats. Endocrinology. 2004;145:1996–2005. doi: 10.1210/en.2003-1481. [DOI] [PubMed] [Google Scholar]
- Gottardis MM, Jordan VC. The antitumor action of keoxifene (raloxifene) and tamoxifen in the N-nitromethylurea-induce rat mammary carcinoma model. Cancer Res. 1987;47:4020–4024. [PubMed] [Google Scholar]
- Delmas PD, Bjarnason NH, Mitlak BH, Ravoux AC, Shah AS, Huster WJ, Draper M, Christiansen C. Effects of raloxifene on bone mineral density, serum cholesterol concentrations, and uterine endometrium in postmenopausal women. N Engl J Med. 1997;337:1641–1647. doi: 10.1056/NEJM199712043372301. [DOI] [PubMed] [Google Scholar]
- Cummings SR, Eckert S, Krueger KA, Grady D, Powles TJ, Cauley JA, Norton L, Nickelsen T, Bjarnason NH, Morrow M, et al. The effect of raloxifene on risk of breast cancer in postmenopausal women: results from the MORE randomized trial. Multiple Outcomes of Raloxifene Evaluation. JAMA. 1999;281:2189–2197. doi: 10.1001/jama.281.23.2189. [DOI] [PubMed] [Google Scholar]
- Sato M, Turner CH, Wang T, Adrian MD, Rowley E, Bryant HU. LY353381.HCl: a novel raloxifene analog with improved SERM potency and efficacy in vivo. J Pharmacol Exp Ther. 1998;287:1–7. [PubMed] [Google Scholar]
- Detre S, Riddler S, Salter J, A'Hern R, Dowsett M, Johnston SR. Comparison of the selective estrogen receptor modulator arzoxifene ( LY353381) with tamoxifen on tumor growth and biomarker expression in an MCF-7 human breast cancer xenograft model. Cancer Res. 2003;63:6516–6522. [PubMed] [Google Scholar]
- Rowley E, Adrian MD, Bryant H, et al. The new SERM LY353381.HCL has advantages over estrogen, tamoxifen, and raloxifene in reproductive and non-reproductive tissues of aged ovariectomised rats. Proc Am Soc Bone Min Res. 1997. p. A490.
- Baselga J, Llombart-Cussac A, Bellet M, Guillem-Porta V, Enas N, Krejcy K, Carrasco E, Kayitalire L, Kuta M, Lluch A, et al. Randomized, double-blind, multicenter trial comparing two doses of arzoxifene ( LY353381) in hormone-sensitive advanced or metastatic breast cancer patients. Ann Oncol. 2003;14:1383–1390. doi: 10.1093/annonc/mdg368. [DOI] [PubMed] [Google Scholar]
- Buzdar A, O'Shaughnessy JA, Booser DJ, Pippen JE, Jr, Jones SE, Munster PN, Peterson P, Melemed AS, Winer E, Hudis C. Phase II, randomized, double-blind study of two dose levels of arzoxifene in patients with locally advanced or metastatic breast cancer. J Clin Oncol. 2003;21:1007–1014. doi: 10.1200/JCO.2003.06.108. [DOI] [PubMed] [Google Scholar]
- Labrie F, Labrie C, Belanger A, Simard J, Gauthier S, Luu-The V, Merand Y, Giguere V, Candas B, Luo S, et al. EM-652 (SCH 57068), a third generation SERM acting as pure antiestrogen in the mammary gland and endometrium. J Steroid Biochem Mol Biol. 1999;69:51–84. doi: 10.1016/S0960-0760(99)00065-5. [DOI] [PubMed] [Google Scholar]
- Simard J, Labrie C, Belanger A, Gauthier S, Singh SM, Merand Y, Labrie F. Characterization of the effects of the novel non-steroidal antiestrogen EM-800 on basal and estrogen-induced proliferation of T-47D, ZR-75-1 and MCF-7 human breast cancer cells in vitro. Int J Cancer. 1997;73:104–112. doi: 10.1002/(SICI)1097-0215(19970926)73:1<104::AID-IJC16>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Johnston SRD, Detre S, Riddler S, Dowsett M. SCH 57068 is a selective estrogen receptor modulator (SERM) without uterotrophic effects compared with either tamoxifen or raloxifene. Breast Cancer Res Treat. 2000;Dec 2000:A163. [Google Scholar]
- Gutman M, Couillard S, Roy J, Labrie F, Candas B, Labrie C. Comparison of the effects of EM-652 ( SCH57068), tamoxifen, toremifene, droloxifene, idoxifene, GW-5638 and raloxifene on the growth of human ZR-75-1 breast tumors in nude mice. Int J Cancer. 2002;99:273–278. doi: 10.1002/ijc.10302. [DOI] [PubMed] [Google Scholar]
- Roy J, Couillard S, Gutman M, Labrie F. A novel pure SERM achieves complete regression of the majority of human breast cancer tumors in nude mice. Breast Cancer Res Treat. 2003;81:223–229. doi: 10.1023/A:1026118602273. [DOI] [PubMed] [Google Scholar]
- Luo S, Martel C, Sourla A, Gauthier S, Merand Y, Belanger A, Labrie C, Labrie F. Comparative effects of 28-day treatment with the new anti-estrogen EM-800 and tamoxifen on estrogen-sensitive parameters in intact mice. Int J Cancer. 1997;73:381–391. doi: 10.1002/(SICI)1097-0215(19971104)73:3<381::AID-IJC13>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Martel C, Picard S, Richard V, Belanger A, Labrie C, Labrie F. Prevention of bone loss by EM-800 and raloxifene in the ovariectomized rat. J Steroid Biochem Mol Biol. 2000;74:45–56. doi: 10.1016/S0960-0760(00)00087-X. [DOI] [PubMed] [Google Scholar]
- Labrie F, Champagne P, Labrie C, Roy J, Laverdiere J, Provencher L, Potvin M, Drolet Y, Pollak M, Panasci L, et al. Activity and safety of the antiestrogen EM-800, the orally active precursor of acolbifene, in tamoxifen-resistant breast cancer. J Clin Oncol. 2004;22:864–871. doi: 10.1200/JCO.2004.05.122. [DOI] [PubMed] [Google Scholar]
- von Angerer E, Prekajac J, Strohmeier J. 2-Phenylindoles. Relationship between structure, estrogen receptor affinity, and mammary tumor inhibiting activity in the rat. J Med Chem. 1984;27:1439–1447. doi: 10.1021/jm00377a011. [DOI] [PubMed] [Google Scholar]
- Stein RC, Dowsett M, Cunningham DC, Davenport J, Ford HT, Gazet JC, von Angerer E, Coombes RC. Phase I/II study of the anti-oestrogen zindoxifene ( D16726) in the treatment of advanced breast cancer. A Cancer Research Campaign Phase I/II Clinical Trials Committee study. Br J Cancer. 1990;61:451–453. doi: 10.1038/bjc.1990.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CP, Collini MD, Tran BD, Harris HA, Kharode YP, Marzolf JT, Moran RA, Henderson RA, Bender RH, Unwalla RJ, et al. Design, synthesis, and preclinical characterization of novel, highly selective indole estrogens. J Med Chem. 2001;44:1654–1657. doi: 10.1021/jm010086m. [DOI] [PubMed] [Google Scholar]
- Greenberger LM, Annable T, Collins KI, Komm BS, Lyttle CR, Miller CP, Satyaswaroop PG, Zhang Y, Frost P. A new anti-estrogen, 2-(4-hydroxy-phenyl)-3-methyl-1-[4-(2-piperidin-1-yl-ethoxy)-benzyl]-1H-in dol-5-ol hydrochloride (ERA-923), inhibits the growth of tamoxifen-sensitive and -resistant tumors and is devoid of uterotropic effects in mice and rats. Clin Cancer Res. 2001;7:3166–3177. [PubMed] [Google Scholar]
- Cotreau MM, Stonis L, Dykstra KH, Gandhi T, Gutierrez M, Xu J, Park Y, Burghart PH, Schwertschlag US. Multiple-dose, safety, pharmacokinetics, and pharmacodynamics of a new selective estrogen receptor modulator, ERA-923, in healthy post-menopausal women. J Clin Pharmacol. 2002;42:157–165. doi: 10.1177/00912700222011193. [DOI] [PubMed] [Google Scholar]
- Cauley JA, Norton L, Lippman ME, Eckert S, Krueger KA, Purdie DW, Farrerons J, Karasik A, Mellstrom D, Ng KW, et al. Continued breast cancer risk reduction in postmenopausal women treated with raloxifene: 4-year results from the MORE trial. Multiple outcomes of raloxifene evaluation. Breast Cancer Res Treat. 2001;65:125–134. doi: 10.1023/A:1006478317173. [DOI] [PubMed] [Google Scholar]
- Vogel VG, Costantino JP, Wickerham DL, Cronin WM. National surgical adjuvant breast and bowel project update: prevention trials and endocrine therapy of ductal carcinoma in situ. Clin Cancer Res. 2003;9:495S–501S. [PubMed] [Google Scholar]
- Chang J, Powles TJ, Allred DC, Ashley SE, Makris A, Gregory RK, Osborne CK, Dowsett M. Prediction of clinical outcome from primary tamoxifen by expression of biologic markers in breast cancer patients. Clin Cancer Res. 2000;6:616–621. [PubMed] [Google Scholar]
- Dowsett M, Smith IE, Ebbs SR, Dixon JM, Skene A, Griffith C, Boeddinghaus I, Salter J, Detre S, Hills M, et al. Short-term changes in Ki-67 during neoadjuvant treatment of primary breast cancer with anastrozole or tamoxifen alone or combined correlate with recurrence-free survival. Clin Cancer Res. 2005;11:951s–958s. [PubMed] [Google Scholar]
- Howell A, Cuzick J, Baum M, Buzdar A, Dowsett M, Forbes JF, Hoctin-Boes G, Houghton J, Locker GY, Tobias JS. Results of the ATAC (Arimidex, Tamoxifen, Alone or in Combination) trial after completion of 5 years' adjuvant treatment for breast cancer. Lancet. 2005;365:60–62. doi: 10.1016/S0140-6736(04)17666-6. [DOI] [PubMed] [Google Scholar]
- Decensi A, Robertson C, Viale G, Pigatto F, Johansson H, Kisanga ER, Veronesi P, Torrisi R, Cazzaniga M, Mora S, et al. A randomized trial of low-dose tamoxifen on breast cancer proliferation and blood estrogenic biomarkers. J Natl Cancer Inst. 2003;95:779–790. doi: 10.1093/jnci/95.11.779. [DOI] [PubMed] [Google Scholar]
- Dowsett M, Dixon JM, Horgan K, Salter J, Hills M, Harvey E. Antiproliferative effects of idoxifene in a placebo-controlled trial in primary human breast cancer. Clin Cancer Res. 2000;6:2260–2267. [PubMed] [Google Scholar]
- Dowsett M, Bundred NJ, Decensi A, Sainsbury RC, Lu Y, Hills MJ, Cohen FJ, Veronesi P, O'Brien ME, Scott T, et al. Effect of ralox-ifene on breast cancer cell Ki67 and apoptosis: a double-blind, placebo-controlled, randomized clinical trial in postmenopausal patients. Cancer Epidemiol Biomarkers Prev. 2001;10:961–966. [PubMed] [Google Scholar]
- Fabian CJ, Kimler BF, Anderson J, Tawfik OW, Mayo MS, Burak WE, Jr, O'Shaughnessy JA, Albain KS, Hyams DM, Budd GT, et al. Breast cancer chemoprevention phase I evaluation of bio-marker modulation by arzoxifene, a third generation selective estrogen receptor modulator. Clin Cancer Res. 2004;10:5403–5417. doi: 10.1158/1078-0432.CCR-04-0171. [DOI] [PubMed] [Google Scholar]
- Luo S, Labrie C, Belanger A, Candas B, Labrie F. Prevention of development of dimethylbenz(a)anthracene (DMBA)-induced mammary tumors in the rat by the new nonsteroidal antiestrogen EM-800 ( SCH57050) Breast Cancer Res Treat. 1998;49:1–11. doi: 10.1023/A:1005928814521. [DOI] [PubMed] [Google Scholar]
- Suh N, Glasebrook AL, Palkowitz AD, Bryant HU, Burris LL, Starling JJ, Pearce HL, Williams C, Peer C, Wang Y, et al. Arzoxifene, a new selective estrogen receptor modulator for chemoprevention of experimental breast cancer. Cancer Res. 2001;61:8412–8415. [PubMed] [Google Scholar]
- Parker MG. Action of 'pure' antiestrogens in inhibiting estrogen receptor action. Breast Cancer Res Treat. 1993;26:131–137. doi: 10.1007/BF00689686. [DOI] [PubMed] [Google Scholar]
- Pink JJ, Jordan VC. Models of estrogen receptor regulation by estrogens and antiestrogens in breast cancer cell lines. Cancer Res. 1996;56:2321–2330. [PubMed] [Google Scholar]
- Howell A, Osborne CK, Morris C, Wakeling AE. ICI 182,780 (Faslodex): development of a novel, 'pure' antiestrogen. Cancer. 2000;89:817–825. doi: 10.1002/1097-0142(20000815)89:4<817::AID-CNCR14>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Hu XF, Veroni M, De Luise M, Wakeling A, Sutherland R, Watts CK, Zalcberg JR. Circumvention of tamoxifen resistance by the pure anti-estrogen ICI 182,780. Int J Cancer. 1993;55:873–876. doi: 10.1002/ijc.2910550529. [DOI] [PubMed] [Google Scholar]
- Osborne CK, Coronado-Heinsohn EB, Hilsenbeck SG, McCue BL, Wakeling AE, McClelland RA, Manning DL, Nicholson RI. Comparison of the effects of a pure steroidal antiestrogen with those of tamoxifen in a model of human breast cancer. J Natl Cancer Inst. 1995;87:746–750. doi: 10.1093/jnci/87.10.746. [DOI] [PubMed] [Google Scholar]
- Robertson JF, Nicholson RI, Bundred NJ, Anderson E, Rayter Z, Dowsett M, Fox JN, Gee JM, Webster A, Wakeling AE, et al. Comparison of the short-term biological effects of 7alpha-[9-(4,4,5,5,5-pentafluoropentylsulfinyl)-nonyl]estra-1,3,5, (10)-triene-3,17beta-diol (Faslodex) versus tamoxifen in post-menopausal women with primary breast cancer. Cancer Res. 2001;61:6739–6746. [PubMed] [Google Scholar]
- Howell A, DeFriend DJ, Robertson JF, Blamey RW, Anderson L, Anderson E, Sutcliffe FA, Walton P. Pharmacokinetics, pharmacological and anti-tumour effects of the specific anti-oestrogen ICI 182780 in women with advanced breast cancer. Br J Cancer. 1996;74:300–308. doi: 10.1038/bjc.1996.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell A, DeFriend D, Robertson J, Blamey R, Walton P. Response to a specific antioestrogen (ICI 182780) in tamoxifen-resistant breast cancer. Lancet. 1995;345:29–30. doi: 10.1016/S0140-6736(95)91156-1. [DOI] [PubMed] [Google Scholar]
- Osborne CK, Pippen J, Jones SE, Parker LM, Ellis M, Come S, Gertler SZ, May JT, Burton G, Dimery I, et al. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: results of a North American trial. J Clin Oncol. 2002;20:3386–3395. doi: 10.1200/JCO.2002.10.058. [DOI] [PubMed] [Google Scholar]
- Howell A, Robertson JF, Quaresma Albano J, Aschermannova A, Mauriac L, Kleeberg UR, Vergote I, Erikstein B, Webster A, Morris C. Fulvestrant, formerly ICI 182,780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J Clin Oncol. 2002;20:3396–3403. doi: 10.1200/JCO.2002.10.057. [DOI] [PubMed] [Google Scholar]
- Howell A, Robertson JF, Abram P, Lichinitser MR, Elledge R, Bajetta E, Watanabe T, Morris C, Webster A, Dimery I, et al. Comparison of fulvestrant versus tamoxifen for the treatment of advanced breast cancer in postmenopausal women previously untreated with endocrine therapy: a multinational, double-blind, randomized trial. J Clin Oncol. 2004;22:1605–1613. doi: 10.1200/JCO.2004.02.112. [DOI] [PubMed] [Google Scholar]
- Erikstein B, Robertson JFR, Osborne CK, Pippen J, Harrison M. ICI 182,780 ('Faslodex') 250 mg monthly intramuscular (IM) injection shows consistent PK profile when given as either 1 × 5ml or 2 × 2.5 ml injections in postmenopausal women with advanced breast cancer (ABC) Proc Am Soc Clin Oncol. 2001;20:A2025. [Google Scholar]
- Martin LA, Farmer I, Johnston SR, Ali S, Marshall C, Dowsett M. Enhanced estrogen receptor (ER) alpha, ERBB2, and MAPK signal transduction pathways operate during the adaptation of MCF-7 cells to long term estrogen deprivation. J Biol Chem. 2003;278:30458–30468. doi: 10.1074/jbc.M305226200. [DOI] [PubMed] [Google Scholar]
- Jeng MH, Shupnik MA, Bender TP, Westin EH, Bandyopadhyay D, Kumar R, Masamura S, Santen RJ. Estrogen receptor expression and function in long-term estrogen-deprived human breast cancer cells. Endocrinology. 1998;139:4164–4174. doi: 10.1210/en.139.10.4164. [DOI] [PubMed] [Google Scholar]
- Long BJ, Jelovac D, Thiantanawat A, Brodie AM. The effect of second-line antiestrogen therapy on breast tumor growth after first-line treatment with the aromatase inhibitor letrozole: long-term studies using the intratumoral aromatase postmenopausal breast cancer model. Clin Cancer Res. 2002;8:2378–2388. [PubMed] [Google Scholar]
- Franco S, Perez A, Tan-Chiu E, Frankel C, Vogel CL. Fulvestrant demonstrates clinical benefit in heavily pre-treated postmenopausal women with advanced breast cancer. Breast Cancer Res Treat. 2003;82:S105. [Google Scholar]
- Ingle JN, Rowland KM, Suman VJ, Mirchandani D, Bernath AM, Camoriano JK, Perez EA. Evaluation of fulvestrant in women with advanced breast cancer and progression on prior aromatase inhibitor therapy: a phase II trial of the North Central Cancer Treatment Group. Breast Cancer Res Treat. 2004;88:S38. doi: 10.1200/JCO.2005.04.1053. [DOI] [PubMed] [Google Scholar]
- Perey L, Paridaens R, Nole F, Pagani O, Bonnefoi H, Aebi S, Diet-rich D, Goldhirsch A, Thurlimann B. Fulvestrant as hormonal treatment in postmenopausal women with advanced breast cancer progressing after treatment with tamoxifen and aromatase inhibitors: update of a phase II SAKK trial. Breast Cancer Res Treat. 2004;18:S236. [Google Scholar]
- Petruzelka L, Zimovjanova M. Fulvestrant in postmenopausal women with metastatic breast cancer progressing on prior endocrine therapy: results from an expanded access programme. Eur J Cancer. 2004;132:A264. [Google Scholar]
- Steger GG, Bartsch R, Wenzel C, Pluschnig U, Locker G, Mader RM, Zielinksi CC. Fulvestrant beyond the second hormonal treatment line in metastatic breast cancer. Proc Am Soc Clin Oncol. 2003;22:A20. [Google Scholar]
- Johnston S. Fulvestrant and the sequential endocrine cascade for advanced breast cancer. Br J Cancer. 2004;(Suppl 1):S15–S18. doi: 10.1038/sj.bjc.6601632. [DOI] [PMC free article] [PubMed] [Google Scholar]