

Abstract
Prediction of protein-coding regions and other features of primary DNA sequence have greatly contributed to experimental biology. Significant challenges remain in genome annotation methods, including the identification of small or overlapping genes and the assessment of mRNA splicing or unconventional translation signals in expression. We have employed a combined analysis of compositional biases and conservation together with frame-specific G+C representation to reevaluate and annotate the genome sequences of mouse and rat cytomegaloviruses. Our analysis predicts that there are at least 34 protein-coding regions in these genomes that were not apparent in earlier annotation efforts. These include 17 single-exon genes, three new exons of previously identified genes, a newly identified four-exon gene for a lectin-like protein (in rat cytomegalovirus), and 10 probable frameshift extensions of previously annotated genes. This expanded set of candidate genes provides an additional basis for investigation in cytomegalovirus biology and pathogenesis.
Sequence analysis has been crucial to understanding the biology of cytomegalovirus (CMV) as well as other herpesviruses (13, 30). Human CMV is an important pathogen, causing neurological damage following congenital infection (37) as well as opportunistic infections in immunocompromised individuals. Models of human CMV pathogenesis and immune control have employed related betaherpesviruses that naturally infect guinea pigs (42), rats (8), and mice (21, 25). The initial annotation of a laboratory-propagated human CMV strain, AD169 (10), predicted 194 unique open reading frames (ORFs). Following this report, reevaluation of genome organization has occurred through correction of errors in the AD169 strain sequence (12, 31, 35, 43), recognition of mRNA splicing events (15, 39), and empirical identification of genes that had escaped annotation (3, 24, 26). The human CMV sequence has been updated through analyses of additional strains (9, 16, 17, 33) as well as by comparison to rhesus CMV (20) and chimpanzee CMV (14) genome sequences. Several revisions of the full genome complement of natural CMV have resulted from these studies. The number of genes in human CMV was estimated to range from under 150 to over 200 genes, and the current estimate of 165 genes is considered reasonable (16). Different estimates depend on the information considered, including homology with other genes in available databases, codon bias, preservation of known protein motifs, and the presence of transcription signals (13, 32).
The annotated human CMV (HCMV) genome sequence has formed a basis for comparisons to other betaherpesviruses. Murine CMV (MCMV) (40) and rat CMV (RCMV) (45) retain obvious sequence homologs of about 80 HCMV ORFs, or roughly 50% of the annotated genes in these viruses. Non-CMV betaherpesviruses infecting humans, such as herpesvirus 6 (19) and herpesvirus 7 (36), as well as those infecting lower primates, such as herpesvirus tupaia (2), retain similar core sets of ORFs. Approximately 40 of these 80 betaherpesvirus-specific ORFs are shared with all mammalian and avian herpesviruses (13) and are considered to be herpesvirus common. Despite obvious levels of divergence in the betaherpesviruses, common biological characteristics have emerged from studies of viruses infecting laboratory animals, and these have helped us to define immune control by the host and immune escape by the virus and to accumulate a myriad of additional basic information on replication, pathogenesis, and latency (25, 28-30, 41).
Prediction of the protein-coding potential of genomes is by nature provisional. In particular, herpesviruses and other eukaryotic viruses have been difficult to annotate accurately using conventional criteria, as evidenced by the recognition of additional genes as well as the elimination of ORFs found to be spurious based on additional investigation. For example, evidence suggests that the commonly employed limitations of ORF length (≥100 codons) and maximum ORF overlap (<60%) lead to the exclusion of known CMV gene products, such as the multiply spliced immunomodulatory function, viral interleukin 10 (24, 26), and the 73-amino-acid herpesvirus-conserved smallest capsid protein (18). Similarly, recent efforts to identify structural proteins in MCMV have also resulted in several revisions to genome annotation (23). In addition to the small sizes of ORFs, biologically relevant events that may confound conventional annotation methods include posttranscriptional modification, mRNA splicing, alternate translation initiation sites, and stop codon suppression. Finally, automated annotation procedures may also be confounded due to unrecognized errors in underlying sequencing. Current limitations of analysis might be overcome by new approaches that are less restrictive and provide an extended list of candidate genes for experimental verification.
In the present study, we investigated the protein-coding potential of the MCMV and RCMV genomes, taking into account the conservation of ORFs and genome-specific sequence features. Analogously to the human and chimpanzee CMV genomes (14), MCMV and RCMV retain a remarkable level of evolutionary relatedness and similarity in both functional organization and arrangement of genes (40, 45). Our analysis of genome-specific sequence features will focus on translational “frame analysis” (5), exploiting the differential G+C distribution among codon base positions in genomes of high G+C content (see Materials and Methods and Fig. S1 in the supplemental material). To provide an objective means to evaluate the extent to which G+C content influences the translational frames and to reveal a potential coding region in any sequence, we also defined a new measure of gene compositional bias and a related measure of coding potential. Our approach makes no assumptions about the minimum length of coding sequences, although we focused on ORFs of ≥20 codons, and does not impose restrictions on the degree of overlap between putative protein-coding regions. This procedure represents a marked modification of standard methods and produces a substantial revision of the current annotations for the MCMV and RCMV genomes. Our analysis suggests that CMV genomes likely encode a greater number of overlapping genes than previously thought.
MATERIALS AND METHODS
The genome sequence of the MCMV Smith strain (GenBank accession number U68299) and the genome sequence of the RCMV Maastricht strain (GenBank accession number NC_002512) were compared, and sequence features were analyzed using frame-specific profiles of G+C contents.
Homologies.
Similarity between ORF products of MCMV and RCMV was evaluated by the significant segment pair alignment (SSPA) program, and regions of similarity were identified by the multiple alignment program ITERALIGN (7). The alignment of viral genomes employed ORF products with lengths of ≥20 codons. The predicted products of all ORFs with lengths of ≥20 codons (60 nucleotides [nt]) were queried against a large nonredundant database of protein sequences using the BLASTP program (1).
Frame-specific G+C profiles (S-profiles).
We characterized the G+C contents and distribution of genomic sequences of MCMV and RCMV by three measures of frame-specific G+C content (5). The G+C content of the genome was evaluated within a moving window of fixed length (201 nt or 102 nt) with respect to every third nucleotide of the genome. First, genome positions 1, 4, 7, and so on, up to the end of the genome sequence, were scanned, and then genome positions 2, 5, 8, and so on, were scanned, followed by genome positions 3, 6, 9, and so on (Fig. S1). With this procedure, variations in G+C contents along the genome were represented by three profiles, each representing a frame, referred to as “S-profiles.” The relationships among S-profiles were used to assess the presence of protein-coding genes in genome regions of high G+C content (5), qualitatively by visual examination and quantitatively through the definitions of a bias in a frame-specific G+C distribution (S-bias) and of a related measure of coding potential (see below).
S-bias.
For a potential coding region of G+C content S, we defined a measure of how the G+C contents at codon base positions 1, 2, and 3 (S1, S2, and S3) compared to expectations (S-bias). Expectations Ŝ1(S), Ŝ2(S), and Ŝ3(S) of S1, S2, and S3 for a potential coding sequence of G+C content S were defined by the linear regressions of S1, S2, and S3 over S, measured in a set of 2,813 published herpesvirus genes (Fig. 1B to D). S1, S2, and S3 values were normalized to these expectations by the differences: 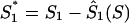 ,
, 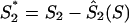 , and
, and 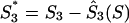 . The obvious relation S1 + S2 + S3 = 3S holds for each gene. Since also Ŝ1(S) + Ŝ2(S) + Ŝ3(S) = 3S, the normalized G+C content values project onto the plane
. The obvious relation S1 + S2 + S3 = 3S holds for each gene. Since also Ŝ1(S) + Ŝ2(S) + Ŝ3(S) = 3S, the normalized G+C content values project onto the plane 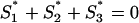 , which can be represented in the two orthogonal dimensions
, which can be represented in the two orthogonal dimensions 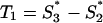 and
and 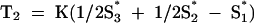 . The scaling factor K was specified so that Var(T1) was equal to Var(T2) for the set of 2,813 published herpesvirus genes (see Results). The S-bias of a putative coding region of G+C contents S, S1, S2, and S3 was defined as the magnitude of the corresponding vector (T1, T2): S-bias(S1, S2, S3 | S) = (T12 + T22)1/2. With this definition, a sequence with a distribution of G+C nucleotides among codon base positions corresponding to expectations will have an S-bias of 0.0, independently of its overall G+C content. As the S-bias increases, the likelihood of an ORF to code for a protein decreases.
. The scaling factor K was specified so that Var(T1) was equal to Var(T2) for the set of 2,813 published herpesvirus genes (see Results). The S-bias of a putative coding region of G+C contents S, S1, S2, and S3 was defined as the magnitude of the corresponding vector (T1, T2): S-bias(S1, S2, S3 | S) = (T12 + T22)1/2. With this definition, a sequence with a distribution of G+C nucleotides among codon base positions corresponding to expectations will have an S-bias of 0.0, independently of its overall G+C content. As the S-bias increases, the likelihood of an ORF to code for a protein decreases.
FIG. 1.

Percent G+C contents in the first, second, and third codon positions of genes across herpesvirus species in relation to genome and gene G+C content. (A) Average G+C contents at the first (red circles), second (green circles), and third (blue circles) codon base positions in coding regions from 28 herpesvirus genomes in relation to the genome G+C content. The point corresponding to all coding regions from MCMV is labeled M, whereas M1 and M2 correspond to genes from the region of high G+C content of MCMV and from the remaining part of the genome, respectively (Fig. 2). Similarly, R, R1, and R2 indicate coding regions from the complete RCMV genome or from corresponding regions of high and low G+C contents (Fig. 2). Linear regressions through these points are shown as solid lines that are color coded to indicate the codon position. A similar set of regression analyses was carried out with 84 prokaryotic genomes for comparison, and results are shown as dashed lines (note that the dashed blue line almost precisely overlaps the solid blue line). (B-D) The G+C contents of 2,813 herpesvirus genes at the first (B), second (C), and third (D) codon base positions in relation to the overall G+C content of each gene. Genes annotated in the MCMV and RCMV genomes are shown in green and red, respectively.
Biases in codon and amino acid usage.
Biases in codon usage were calculated as described in reference 22. Let g(x, y, z) be the frequency of the codon composed of bases (x, y, z) in gene group G (G could be a single gene), normalized for each amino acid codon family such that
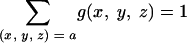 |
where the sum extends over all codons translated to amino acid a. Let f(x, y, z) be the codon frequencies for a gene group F normalized to 1 in each amino acid codon family. The codon usage bias (C-bias) of gene group G relative to F was calculated by the formula (22)
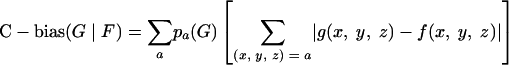 |
where [pa(G)] is the amino acid frequencies of gene group G. For each genome, two reference groups of genes F were chosen to calculate C-biases, one corresponding to all annotated genes inside the regions of high G+C content of the respective genome and a second group including all other annotated genes (see Results for a definition of these regions in the MCMV and RCMV genomes).
The amino acid bias (aa-bias) of gene group G relative to gene group F was defined as
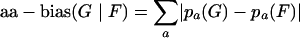 |
where pa(G) and pa(F) are the frequencies of the amino acid a in G and in F, respectively, and the sum extends over all amino acids.
Coding potentials.
Local coding potentials were evaluated from nucleotide composition as follows. For each of the six coding frames (three on the direct strand and three on the complementary strand), an S-bias was evaluated within a window of 102 nt. The probability distribution of S-biases in coding regions was determined from similar windows extracted from all annotated coding regions of 28 herpesvirus genomes. Corresponding probabilities were obtained for random distributions of S1, S2, and S3 given G+C content S. In the case of random distributions, S1, S2, and S3 values have the same expectation (S) and same distribution. The S-bias for random distribution (S-biasrand) was calculated by normalizing S1, S2, and S3 values as follows: 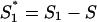 , S2* = S2 − S, and
, S2* = S2 − S, and 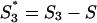 and biases were directly calculated with the equation
and biases were directly calculated with the equation 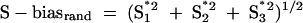 . The conditional probability (coding potential) [P(CODi|F)] of a sequence, F, to be coding in frame i was evaluated as follows:
. The conditional probability (coding potential) [P(CODi|F)] of a sequence, F, to be coding in frame i was evaluated as follows:
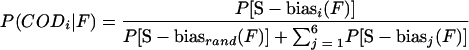 |
where P[(S−biasi(F)] is the frequency of S−biases greater than or equal to S−biasi(F) among all windows extracted from herpesvirus annotated coding regions and P[S−biasrand(F)] is the frequency compared to the random distribution obtained for sequences of the same G+C content.
GeneMark coding potentials (6) were also evaluated based on predictions obtained with the program GeneMarkS (4) as implemented at the website http://opal.biology.gatech.edu/GeneMark/genemarks.cgi.
RESULTS
G+C contents in coding regions of the MCMV and RCMV genomes.
Analysis of genes and genomes of prokaryotic origin has shown that global G+C content correlates distinctively with the G+C content at the first (S1), second (S2), and third (S3) codon positions (5, 34). As expected, similar relationships extended to complete herpesvirus genomes (Fig. 1A) as well as to individual herpesvirus genes (Fig. 1B to D). Variations in global G+C content corresponded to large differences in S3, whereas smaller differences occurred in S1 and, especially, in S2. The relationship S3 ≫ S1 ≫ S2 is typical of the three codon positions of expressed genes in G+C-rich genomes.
The high overall G+C contents of the MCMV and RCMV genomes (58.7% and 61.0%, respectively) and the corresponding high contrasts in G+C usage at different codon positions (Fig. 1A) were expected to provide a means to reliably identify protein-coding regions. However, C and G bases were not evenly distributed across the two genomes (Fig. 2). In both MCMV and RCMV, the G+C contents were greatest (61.7% and 69.2%, respectively) in the large genomic segment containing the betaherpesvirus-conserved protein-coding regions. G+C contents were more varied and generally lower (54.3% and 47.7%, respectively) in the remaining genomic segments. These differences in G+C contents resulted in varied contrasts of G+C usage at each codon position for genes expressed in different genomic regions.
FIG. 2.

Sequence G+C contents in the MCMV and RCMV genomes measured within moving windows with a size of 201 nt. The regions of high G+C contents conserved between the MCMV and RCMV genomes are shaded gray.
Compositional analysis of ORFs of ≥20 codons from the MCMV and RCMV genomes.
We identified a total of 5,541 MCMV and 4,741 RCMV ORFs with lengths of ≥20 codons (defined without regard to AUG codons, from stop codon to stop codon). All ORFs were analyzed in terms of S-bias, C-bias, and aa-bias (see Materials and Methods). The compositional biases of ORFs corresponding to previously annotated coding sequences were determined over the previously reported length (40, 45), and newly annotated ORFs were evaluated over the entire stop codon-to-stop codon distance as well as beginning at AUG codons when these were at least 60 nt upstream of a stop codon.
S-biases.
We devised a scoring system to quantify biases in G+C contents at codon positions across a putative coding region (see Materials and Methods). The expected G+C contents at codon positions 1, 2, and 3 were determined from the regression lines over 2,813 annotated ORFs from 28 herpesvirus genomes (Fig. 1B to D). Normalized G+C values were transformed into the coordinate system T1 and T2 with a scaling factor (K) equal to 1.804 (see Materials and Methods). The distribution of S-biases of all MCMV and RCMV ORFs of ≥20 codons is shown by the black lines in Fig. 3A and B, where we chose the starting position associated with the lowest bias for each newly annotated ORF. The distribution of all such ORFs with G+C contents of >50% (4,348 ORFs from MCMV and 3,095 ORFs from RCMV) is shown by the gray lines. The distribution of biases among previously annotated ORFs is also shown for comparison. A large proportion (46% in MCMV and 51% in RCMV) of all previously nonannotated ORFs had a low S-bias typical of coding regions (lower than the threshold corresponding to 95% of the annotated genes). Virtually all ORFs with high S-biases (>40) had high G+C contents (>50%), as expected from the great asymmetries in frame-specific G+C usage that distinguish G+C-rich coding from noncoding sequences. However, 34% of the ORFs with high G+C contents showed low S-biases. We also evaluated the biases in codon usage and amino acid usage for all ORFs (see Materials and Methods). We computed all biases relative to the average frequencies observed either among previously annotated genes encoded in the region of high G+C content or among all other annotated genes of the respective genome. For each ORF we selected the smaller of the two biases. The distribution of C-biases and aa-biases among ORFs of MCMV and RCMV are shown in Fig. 3C to F. The C-biases and aa-biases of previously annotated ORFs were low compared to those of the ORF sets analyzed here, although, as for S-biases, a large number of these ORFs have C-biases (18% in MCMV and 32% in RCMV) and aa-biases (19% in MCMV and 33% in RCMV) within the 95th percentile range of the corresponding annotated genes.
FIG. 3.

Distribution of compositional biases of ORFs of ≥20 codons from the MCMV and RCMV genomes. The distributions among all ORFs are shown as black lines, and the distributions among ORFs of ≥20 codons and with G+C contents of >50% are shown as gray lines. The distributions among published genes are shaded black. See the text for definitions of S-bias, C-bias, and aa-bias.
We identified 227 ORFs from MCMV and 350 ORFs from RCMV (supplemental Tables S1 and S2) that had not been previously annotated but were characterized by compositional biases typical of annotated genes (within the 95% confidence interval). For each of these ORFs, all starting positions associated with low biases are listed in supplemental Tables S1 and S2. The average length of these ORFs was 108 codons in RCMV and 129 codons in MCMV. Most of the ORFs from MCMV (91.6%) and the majority of those from RCMV (72.3%) were characterized by a high G+C content (>50%), with averages of 59.6% in MCMV and 58.0% in RCMV. By compositional criteria alone, these ORFs were considered potentially expressed.
Conservation between MCMV and RCMV genomes.
We searched for similarities between proteins potentially encoded by ORFs with lengths of ≥20 codons from MCMV and RCMV using the computer protocol SSPA (7). Pairwise comparisons between 5,541 MCMV and 4,734 RCMV ORFs resulted in 73,330 pairs (0.28%) that exhibited statistically significant similarity. As expected, extended similarities (≥50% SSPA similarity) were distributed along the two viral genomes in a collinear fashion. We then applied the ITERALIGN multiple sequence alignment program (7) to identify all ungapped blocks of aligned positions with lengths of >10 codons. To select the most-reliable regions of homology among all blocks, we constructed a pairwise alignment of the MCMV and RCMV genomes, starting from the longest blocks and progressively adding shorter blocks. Blocks that were not collinear with the partial alignment obtained from the longer blocks were excluded. The resulting genome alignment, shown in Fig. 4, involved 107,739 positions, covering about 47% of each genome sequence. Of these, 94,261 (87.5%) coincided with alignments between amino acids of annotated proteins and were plotted in black. A remarkable 13,478 positions (12.5%), plotted in red, corresponded to alignments between ORF pairs involving at least one ORF not appearing in the original genome annotations.
FIG. 4.

Homologous protein-coding positions of the MCMV and RCMV genomes as derived from significant similarities determined by SSPA (7) analysis of all potential protein products of ORFs at least 20 codons long. Red bars highlight segments corresponding to the 13,478 conserved positions that were not previously recognized in published genome annotations. K, thousand. Numbers on the x and y axes indicate positions.
We found 66 ORF pairs corresponding to conserved regions involving nonannotated ORFs (Table 1) , comprising 49 ORFs from MCMV and 56 ORFs from RCMV. ORFs whose alignment regions were not closely interspersed within regions otherwise conserved between annotated genes are shown in Table 1 in boldface. The conservation of 10 MCMV ORFs and 18 RCMV ORFs (Table 1) strongly suggested that they were expressed genes (see alignments in the supplemental material). Among these, two ORFs from MCMV showed similarities to annotated genes of RCMV. One of them (newly annotated as m120.1) was similar to RCMV r119.4 and included a putative start codon (AUG) at position 175665. Based on position and conservation, a second ORF similar to RCMV R31 and annotated as M31b appeared to be a likely 3′ frameshift extension of M31, as recently reported (23). ORF M73.5e2 was not published in the initial report of the MCMV genome sequence but was subsequently described as exon 2 of M73.5 in MCMV TS9 (42a) (GenBank entry L34342). Seven additional ORFs from MCMV (m38.5, m44.1, m44.3, m45.2, m106.1, m106.3, and m123.1) showed similarities to corresponding newly annotated ORFs from RCMV.
TABLE 1.
Newly identified ORF pairs conserved between MCMV and RCMV
| Strand | MCMV ORFa
|
Annotationb
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Start site | End site | Length (nt) | Low bias | Name | Length (nt) | |||||
| C | 20805 | 23147 | 2343 | + | m20 | 2241 | ||||
| C | 23781 | 25136 | 1356 | + | M23 | 1173 | ||||
| C | 29001 | 30701 | 1701 | + | m25.1 | 1281 | ||||
| C | 30248 | 31876 | 1629 | + | m25.2 | 969 | ||||
| D | 38775 | 39065 | 291 | + | M31bc | ∼291 | ||||
| D | 43068 | 45644 | 2577 | + | M34 | 2562 | ||||
| C | 51780 | 52520 | 741 | + | m38.5c | 588 | ||||
| D | 52260 | 52511 | 252 | + | ||||||
| C | 52686 | 53021 | 336 | |||||||
| C | 55155 | 55373 | 219 | |||||||
| C | 57730 | 57906 | 177 | |||||||
| D | 58756 | 60105 | 1350 | + | m44.1 | 1062 | ||||
| D | 59141 | 59425 | 285 | m44.3 | 285 | |||||
| C | 61767 | 63083 | 1317 | + | m45.1 | 1272 | ||||
| D | 62806 | 62886 | 81 | m45.2 | <81 | |||||
| D | 73492 | 73578 | 87 | |||||||
| D | 73541 | 73870 | 330 | m48.1 | 309 | |||||
| D | 73545 | 73628 | 84 | |||||||
| C | 73574 | 73888 | 315 | m48.2 | 294 | |||||
| D | 73632 | 73889 | 258 | |||||||
| D | 82173 | 82853 | 681 | |||||||
| D | 87949 | 88380 | 432 | + | ||||||
| D | 93186 | 94298 | 1113 | |||||||
| D | 93238 | 94260 | 1023 | m59 | 1020 | |||||
| D | 94812 | 95039 | 228 | + | ||||||
| D | 101768 | 102799 | 1032 | + | M71 | 897 | ||||
| D | 101768 | 102799 | 1032 | + | M71 | 897 | ||||
| D | 102558 | 102830 | 273 | |||||||
| D | 105797 | 106069 | 273 | + | M73.5e2c | ≤273 | ||||
| C | 105875 | 106141 | 267 | |||||||
| D | 133519 | 133632 | 114 | |||||||
| C | 135266 | 135766 | 501 | |||||||
| D | 142050 | 143783 | 1734 | + | M98 | 1683 | ||||
| C | 142341 | 142670 | 330 | |||||||
| D | 145587 | 148031 | 2445 | M102 | 2436 | |||||
| C | 146472 | 147881 | 1410 | |||||||
| D | 146726 | 147829 | 1104 | |||||||
| D | 149117 | 149263 | 147 | |||||||
| D | 151159 | 151419 | 261 | |||||||
| C | 153916 | 154473 | 558 | + | m106 | 441 | ||||
| C | 154196 | 154456 | 261 | m106.1 | 153 | |||||
| C | 155781 | 155918 | 138 | m106.3 | 138 | |||||
| D | 156357 | 156458 | 102 | |||||||
| C | 157257 | 157478 | 222 | |||||||
| D | 159576 | 159995 | 420 | + | ||||||
| D | 162826 | 162951 | 126 | |||||||
| C | 167208 | 169154 | 1947 | + | M116 | 1935 | ||||
| D | 167274 | 167450 | 177 | |||||||
| D | 168693 | 168914 | 222 | |||||||
| C | 174640 | 175725 | 1086 | + | m120.1 | 1026 | ||||
| C | 181863 | 182219 | 357 | + | m123.1 | 357 | ||||
| D | 181924 | 182277 | 354 | + | m124 | 345 | ||||
| D | 182007 | 182180 | 174 | |||||||
| C | 182473 | 182733 | 261 | |||||||
| C | 184468 | 184569 | 102 | |||||||
| C | 185598 | 185711 | 114 | |||||||
| D | 185871 | 185984 | 114 | |||||||
| C | 188382 | 188642 | 261 | + | m132e2 | 73 | ||||
| C | 208663 | 208740 | 78 | |||||||
| C | 216869 | 216931 | 63 | |||||||
| C | 217626 | 217706 | 81 | |||||||
| D | 219053 | 219283 | 231 | |||||||
| C | 220323 | 220481 | 159 | |||||||
| D | 226076 | 226204 | 129 | |||||||
| C | 226501 | 226917 | 417 | |||||||
| D | 227183 | 227440 | 258 | + | ||||||
| Strand | RCMV ORFa
|
Annotationb
|
||||||||
| C | 5754 | 7895 | 2142 | + | r4 | ∼1548 | ||||
| C | 7899 | 10040 | 2142 | R23a | ∼108 | |||||
| C | 15207 | 15893 | 687 | |||||||
| C | 15305 | 15961 | 657 | + | r25.3b | ∼197 | ||||
| D | 21662 | 24010 | 2349 | + | R31 | 2319 | ||||
| D | 27839 | 28357 | 519 | |||||||
| C | 35741 | 36997 | 1257 | r38.5 | 582 | |||||
| D | 35837 | 36421 | 585 | + | ||||||
| C | 35741 | 36997 | 1257 | |||||||
| C | 38231 | 38533 | 303 | |||||||
| C | 38231 | 38533 | 303 | |||||||
| C | 40636 | 40896 | 261 | |||||||
| D | 42221 | 42832 | 612 | + | r44.1 | 612 | ||||
| D | 40647 | 42608 | 1962 | r44.3 | 102 | |||||
| C | 44906 | 45646 | 741 | |||||||
| D | 45413 | 45760 | 348 | + | r45.2 | 348 | ||||
| D | 55979 | 56386 | 408 | |||||||
| D | 55979 | 56386 | 408 | |||||||
| D | 56106 | 56732 | 627 | + | ||||||
| C | 56102 | 56731 | 630 | + | r48.2 | 258 | ||||
| D | 56106 | 56732 | 627 | + | ||||||
| D | 64619 | 64765 | 147 | |||||||
| D | 70544 | 75454 | 4911 | r58.1 | >626 | |||||
| D | 76469 | 77419 | 951 | |||||||
| D | 76434 | 77432 | 999 | + | ||||||
| D | 78733 | 78822 | 90 | |||||||
| D | 85539 | 85790 | 252 | |||||||
| D | 85631 | 86485 | 855 | + | R71 | ≤855 | ||||
| D | 88869 | 90035 | 1167 | + | r70.4 | 1020 | ||||
| D | 93695 | 94201 | 507 | + | R73.5e2 | ∼260 | ||||
| C | 94001 | 94225 | 225 | |||||||
| D | 121235 | 122080 | 846 | + | R91 | 720 | ||||
| C | 123046 | 123987 | 942 | |||||||
| D | 129944 | 130225 | 282 | + | R98a | ∼249 | ||||
| C | 129905 | 130570 | 666 | + | ||||||
| D | 135108 | 136100 | 993 | + | R102b | ∼684 | ||||
| C | 135279 | 136370 | 1092 | |||||||
| D | 133472 | 136429 | 2958 | + | R102 | 2958 | ||||
| D | 137109 | 137174 | 66 | |||||||
| D | 136805 | 139774 | 2970 | |||||||
| C | 142139 | 142723 | 585 | + | ||||||
| C | 145051 | 145143 | 93 | r106.1 | 93 | |||||
| C | 148724 | 148822 | 99 | r108.1 | 99 | |||||
| D | 149388 | 149669 | 282 | |||||||
| C | 149837 | 149941 | 105 | |||||||
| D | 150557 | 150619 | 63 | |||||||
| D | 152938 | 153261 | 324 | + | ||||||
| C | 157690 | 158127 | 438 | + | r115.1 | ≤438 | ||||
| D | 157810 | 157869 | 60 | |||||||
| D | 158297 | 158593 | 297 | + | ||||||
| C | 163547 | 164599 | 1053 | + | r119.4 | 1017 | ||||
| C | 174032 | 174328 | 297 | + | r124.2 | 297 | ||||
| D | 173976 | 174329 | 354 | + | r124.1 | 354 | ||||
| D | 173981 | 174262 | 282 | + | ||||||
| C | 174637 | 174867 | 231 | |||||||
| C | 176368 | 176448 | 81 | |||||||
| C | 175409 | 175492 | 84 | |||||||
| D | 179521 | 179628 | 108 | |||||||
| C | 182966 | 183247 | 282 | + | r132e2 | ≤282 | ||||
| C | 199028 | 199108 | 81 | |||||||
| C | 216453 | 216527 | 75 | |||||||
| C | 220451 | 220942 | 492 | + | r158 | 336 | ||||
| D | 217897 | 217962 | 66 | |||||||
| C | 221908 | 221970 | 63 | |||||||
| D | 228283 | 229347 | 1065 | r171 | 864 | |||||
| C | 229243 | 230076 | 834 | |||||||
| D | 228680 | 228760 | 81 | |||||||
Boldface indicates newly annotated ORFs with conserved regions that are distinct from previously annotated ORFs. Newly annotated ORFs with extensive regions of similarity are underlined. Groups of overlapping conserved ORFs are in italics. ORF positions and lengths are from stop codon to stop codon.
Lightface ORF names are from previously annotated genes. Boldface names indicate new or modified ORFs. Lengths of ORFs are as published previously (lightface) or are estimated for newly annotated ORFs (boldfaced) based on the position of the first Met codon and from S-profile information (see Fig. 6 and 7). Asterisks indicate ORFs included in Table 2 or 3.
Experimentally verified (23, 27, 42a).
In RCMV we found 11 newly annotated ORFs (r4, R23a, r25.3b, r48.2, R71, R73.5e2, R98a, R102b, r115.1, r124.1, and r132e2) with similarity to published genes of MCMV. Four of these were interpreted, for their relative positions, as likely to derive from frameshifts (likely resulting from sequencing errors) within the sequence of neighboring annotated genes. In particular, we found a 5′ extension of R23 (R23a), a 3′ extension of r25.3 (r25.3b), a 5′ extension of R98 (R98a), and a 3′ extension of R102 (R102b). The similarity of ORF r4 to MCMV m20 suggested a substantial 5′ extension of this ORF compared to the current annotation (see the supplemental material). Surprisingly, no AUG codon was found in the extended region of r4. Similarly, the strong conservation between r70.1 and MCMV M71 suggested that the coding region of r70.1 may be extended to a region 5′ of the current annotation, which also lacked any AUG codons, and that this gene should be named R71 to emphasize its homology to the gene already published in MCMV. The lack of AUG codons in the extended regions of these ORFs suggests an alternative translation start mechanism, a multiexon gene structure, or the possibility of sequencing errors. We identified a homolog of MCMV M73.5e2 in RCMV that we designated R73.5e2. The short ORF r115.1 showed strong similarity to the C-terminal region of M116 from MCMV, which did not show significant similarity to R116. The newly annotated ORF r132e2 of RCMV was recognized as the second exon of annotated gene r133 for its high similarity to exon m132e2 in MCMV (see the supplemental material). This result suggests that r133 (homologous to m133e1) should be renamed r133e1. Seven newly annotated ORFs from RCMV (r38.5, r44.1, r44.3, r45.2, r106.1, r108.1, and r124.2) showed similarities to corresponding newly annotated ORFs from MCMV. Groups of conserved ORFs overlapping in the respective genomes are italicized in Table 1. Additional sequence features would be needed to distinguish the frame(s) of the coding sequence(s) among these ORFs (see the supplemental material for the cases, e.g., of m48.1 and m48.2).
BLASTP analyses.
We queried the products of all ORFs with lengths of ≥20 codons against the NCBI nonredundant database of 644,068 proteins from coding sequence translations of sequences in GenBank, the Protein Data Bank, Swiss-Prot, and PIR using BLASTP (1). Significant results (E-value, <0.001) from this analysis for ORFs in the MCMV and RCMV genomes not previously annotated are reported in supplemental Tables S3 and S4, respectively. Eleven of these matches (see the footnotes of Tables S3 and S4) involved sequences of low complexity and are likely spurious. Other matches confirmed ORFs M31b, M73.5e2, m120.1, r48.2, R73.5e2, and R102b, newly annotated based on SSPA analysis.
BLASTP analysis identified four additional candidate genes in the MCMV genome, one with similarity to a region of RCMV r5, one overlapping M57 and similar to the single-stranded DNA-binding protein of primate CMVs, one similar to a hypothetical protein of the rhesus macaque CMV, and one similar to RCMV r95.1. In RCMV, 16 ORFs showed interesting BLASTP matches (boldfaced in supplemental Table S4). Three ORFs showed respective similarities to the arabinogalactan protein of maize, to the regulatory protein E2 from human papillomavirus, and to BHLF1 from EBV. ORF r169.1 (overlapping r169) showed extensive similarity to ORF r171, located immediately downstream in the RCMV genome (see the supplemental material). A notable feature in the RCMV genome evidenced by the BLASTP analysis was the existence of multiple similarities between ORFs overlapping in different frames the published genes r121.1, r121.2, and r125. These similarities corresponded to multiple exact repetitions of long DNA elements (supplemental Tables S5 and S6) duplicated in different frames within the same overlapping ORF. The lack of relatedness of these DNA structures to any coding frame suggests that these genome regions may not code for proteins at all.
Three ORFs of RCMV, newly named r153e2, r153e3, and r153e4, showed significant similarity to a lectin-like glycoprotein first identified in the English isolate of RCMV (46), where the protein is encoded by five exons. Similarity analysis of this protein against our collection of peptides suggested that a homologous lectin-like protein is also encoded in the RCMV Maastricht genome, within the region including positions 217034 to 217816. By the alignment of the putative products of these ORFs to the protein identified in the English isolate (Fig. 5) and the identification of putative donor and acceptor sites in the RCMV genome, we suggest that this protein is encoded in RCMV Maastricht but employs four exons and has a total length of 186 aa.
FIG. 5.

Alignment of a lectin-like glycoprotein identified in RCMV English (46) with ORFs from RCMV Maastricht (45). ORFs from RCMV Maastricht are translated and numbered from stop codon to stop codon. Aligned positions are represented in capital letters. Nonaligned positions are represented in lowercase letters. The alignment suggests that in RCMV Maastricht, this protein is encoded by four exons (indicated in red) within ORF C217640.0.217816 (exon 1, C217651-217783), ORF C217366-217680 (exon 2, C217402-217580), ORF C217214-217411 (exon 3, C217221-217327), and ORF C217004-217162 (exon 4, C217004-217142). The N-terminal conservation of a similar highly hydrophobic region within the ORFs including exons 1 and 3 suggests possible alternative splicing.
S-profile analysis.
Figures 6 and 7 display frame-specific profiles of G+C contents (see Materials and Methods) and regions of conservation of the MCMV and RCMV genomes. For frame-specific analysis (5), the frequencies of G+C were calculated with respect to every third position using windows of 102 nt or 201 nt. The G+C contents determined over positions in frame with nt 1, 2, or 3 of the complete genomes are represented by the red, green, and blue curves (see Materials and Methods and the legend of Fig. 6). We refer to the shape and relationship of the three curves at various sequence positions as “S-profiles.” Published genes (40, 45) are represented in Fig. 6 and 7 as colored with reference to the frame of the third base of their codons (see the legend of Fig. 6). The color and orientation of each gene also identify the G+C curves corresponding to the first and second base positions of its codons. For example, the G+C contents of codon base positions 1, 2, and 3 for gene M100, colored in green and encoded on the complementary strand, are shown by the red, blue, and green curves, respectively. For gene M102, colored in green but encoded on the direct strand, positions 1, 2, and 3 instead correspond to the blue, red, and green curves, respectively. Figures 6 and 7 also show all conserved regions identified by SSPA analysis (7), all nonannotated ORFs highlighted by SSPA analysis (from Table 1) or BLASP analysis (from supplemental Tables S3 and S4) (1), and the frames of all regions of high coding potential, determined by S-profile biases and by the GeneMark procedure (6). Finally, Fig. 6 and 7 show all potential coding regions identified by our analysis of S-profiles and conservation among all ORFs with ≥20 codons from the two genomes (see the legend of Fig. 6).
FIG. 6.






Frame-specific G+C profiles (S-profiles [5]) along the MCMV genome (horizontal axis) are represented by red, green, and blue curves within windows with a size of 201 nt (intense red, green, and blue curves, respectively) and with a size of 102 nt (light red, light green, and light blue curves). The overall G+C contents, measured within windows with a size of 201 nt, are represented by the black curve. All positions in the MCMV and RCMV genomes showing significant (P ≤ 0.01) S-profile contrasts that are not consistent with previously annotated genes are identified by shaded areas across the S-profile plot. All ORFs are represented as arrows pointing from the 5′ to the 3′ end and are colored according to the frame of the third position of their codons. ORFs previously annotated in published literature (40) are plotted as “annotated genes,” with filled regions denoting conservation between MCMV and RCMV. “Conserved” indicates other regions conserved between the MCMV and RCMV genomes, evaluated by comparing similarities of all ORFs of ≥20 codons (see Materials and Methods and Results) from the direct strand of the genome (upper line) or from the complementary strand (lower line) and colored according to the frame of the third codon position. All conserved nonannotated ORFs with ungapped blocks of similarity longer than 10 aa that are consistent with the collinear arrangement of the two genomes (shown in Table 1) and ORFs with significant BLASTP (1) hits (from Table S3) are indicated by thin arrows. “Coding potential” indicates genome positions with a coding potential of >0.5, evaluated by S-biases (S) or by the GeneMark procedure (G) (6). The frames of the regions of high coding potential are color coded as for genes, and the genome strand is distinguished by representation on the upper (direct) or lower (complementary) lines. “Newly annotated genes” indicates coding regions newly predicted by our methods. Coding regions predicted with highest confidence are depicted with thick lines and shown in full color.
FIG. 7.






Genome and published annotation of RCMV Maastricht (45). Explanatory material can be found in the legend to Fig. 6.
Previously annotated genes recognized by S-profile analysis.
Within the region of high G+C contents of the MCMV genome and of the RCMV genome, there was a clear correspondence of S-profiles with most previously annotated ORFs (see also supplemental Tables S7 and S8). In this region, S-profile analysis recognized 74 of the 88 annotated ORFs from MCMV and 79 of the 92 annotated ORFs from RCMV. S-profiling was sensitive to G+C content such that in the MCMV regions of low G+C contents, only 50 of the 89 annotated ORFs were identified and in the low-G+C-content region of RCMV, 21 of the 75 annotated ORFs were recognized solely by this method. Considering the G+C compositions of individual ORFs, S-profiles recognized 219 of the 291 annotated ORFs with a G+C content of >50%, compared to 5 of 53 ORFs with a G+C content of <50%. These results indicated that S-profile analysis is a powerful approach to identifying coding sequences with G+C contents as low as 50%. Among the annotated genes of high G+C content (>50%) not identified by S-profiles, some were clearly contradicted by their S-profiles whereas others exhibited high G+C contents (often >60%) in all three codon positions (see below).
To provide an objective means to identify regions in the MCMV and RCMV genomes where S-profiles would predict the presence of protein-coding sequences, we first identified all positions (centered in windows of 102 nt) where frame-specific G+C contents differed by more than 35% (corresponding to a random probability of ≤0.01). We then excluded all regions where these contrasts could be explained by the presence of previously annotated ORFs. The remaining regions of high frame-specific G+C contrasts, shown as shaded blocks in Fig. 6 and 7, suggest the existence of expressed genes.
Newly annotated ORFs.
All ORFs whose expression was consistent with the observed high contrasts in S-profiles (>35%) were shown among the newly annotated ORFs in Fig. 6 and 7 and were listed in Table 2 (MCMV) and Table 3 (RCMV). In these tables we have indicated the genome positions of the annotated coding sequences, their G+C contents, and the newly assigned name for each ORF, and we have also indicated the published genes that overlapped each newly annotated ORF. For each ORF we have also identified the presence of a putative translation initiation codon, low compositional biases, conservation, and overlap to sequence regions of high coding potential measured by S-biases or measured by the GeneMark (6) procedure, as well as weak conservation or overlap only to short regions of high coding potential. The presence of an AUG codon did not apply when ORFs were interpreted as internal or last exons or as 3′ frameshift extensions of a sequence in a different frame. Evidence from S-profiles was distinguished as extending through the full length of the ORF or as partially covering the length of the ORF. We included 33 ORFs in Tables 2 and 3 that were identified by SSPA and/or BLASTP similarity and had been retained after scrutiny through S-profile analysis. ORFs that were most reliably predicted as coding sequences (by the extension and strength of the S-profile signal and/or by strong conservation) are indicated. ORFs supported by conservation (through SSPA and/or BLASTP analysis) and by S-profiles are also indicated.
TABLE 2.
Characteristics of newly annotated ORFs in MCMV
| Coding regiona | % G+C content | Name | Presence of Met | Low bias | Conservation | Sd | Ge | Evidencef | Published overlapping gene |
|---|---|---|---|---|---|---|---|---|---|
| 4205-4378 | 48.9 | m4.1 | + | + | F | m5 | |||
| C4216-4374 | 49.1 | m4.2 | + | F | m5 | ||||
| C6600-6731 | 53.0 | m7.1 | P | m7 | |||||
| C6735-6815 | 49.4 | m7.2 | + | + | P | m7 | |||
| 6942-7115 | 54.6 | m7.3 | + | F | m7 | ||||
| C7023-7202 | 55.0 | m7.4 | + | + | + | + | P | m7 | |
| 7956-8219 | 60.6 | m8.1 | + | + | F | m8 | |||
| C8477-8608 | 54.5 | m8.2 | + | + | P | m8 | |||
| 9075-9191 | 49.6 | m9.1 | + | + | + | F | m9 | ||
| 11131-11304 | 49.4 | m11.1 | + | F | m11 | ||||
| 12042-12239 | 41.9 | m12.1 | + | F | m12 | ||||
| 12505-12738 | 53.4 | m12.2 | + | + | + | P | m13 | ||
| 14143-14259 | 51.3 | m15.1 | + | P | m15 | ||||
| 14657-15673 | 44.1 | m16 | + | (+) | (+) | P | m16 | ||
| 15265-15432 | 50.0 | m16.1 | + | F | m16 | ||||
| 15481-15609 | 51.9 | m16.2 | + | + | F | m16 | |||
| 17053-17478 | 62.2 | m18.1 | + | + | + | P | m18 | ||
| C17897-18412 | 66.9 | m18.2 | + | + | P | m18 | |||
| 17963-18361 | 67.7 | m18.3 | + | + | P | m18 | |||
| 18937-19086 | 62.7 | m18.4 | + | + | + | F | m18 | ||
| C19124-19279 | 64.1 | m18.5 | + | + | P | m18 | |||
| 19727-20041 | 65.1 | m18.6 | + | + | P | m18 | |||
| 20591-20890 | 65.7 | m19.1 | + | + | + | P | m19 | ||
| C20582-20958c | 65.8 | m20b | NAb | + | + | + | F | m19, m20 | |
| 23620-23973 | 61.6 | m22.1 | + | + | + | P | m22, M23 | ||
| 26835-27278 | 64.2 | m25.01 | + | + | P | M25 | |||
| C27248-27454 | 60.4 | m25.02 | + | + | + | F | M25 | ||
| 29443-29481 | 79.5 | m25.15 | + | P | m25.1 | ||||
| C31263-31346 | 69.0 | m25.3 | + | + | F | ||||
| 31794-32099 | 63.1 | m26.1 | + | + | + | + | P | M26 | |
| C31910-32065 | 67.9 | m26.2 | + | + | + | F | M26 | ||
| <35647-35784 | 56.5 | m28.1 | + | + | F | M28 | |||
| 35934-36029 | 67.7 | m28.2 | + | + | F | m29 | |||
| C36826-37011 | 58.1 | m29.2 | + | + | F | m30 | |||
| C37105-37644 | 65.4 | m30.1 | + | + | + | + | P | m30 | |
| C37251-37379 | 67.4 | m30.2 | + | + | + | F | m30 | ||
| 38775-39065c | 60.8 | M31b* | NA | + | + | + | + | F | M31 |
| 40027-40527 | 60.3 | m32.1 | + | + | P | M32 | |||
| 41486-41605 | 68.3 | m32.2 | + | + | + | P | |||
| C43067-43297 | 67.1 | m33.1 | + | + | + | P | M34 | ||
| C44822-45280 | 66.2 | m34.1 | (+) | + | P | M34 | |||
| C51780-52367 | 58.5 | m38.5* | + | + | + | + | P | M38 | |
| 51879-52049 | 59.1 | m38.6 | + | (+)c | + | + | F | M38 | |
| C54019-54189 | 63.2 | m41.1 | + | (+) | (+) | m41 | |||
| 54470-54685 | 64.4 | m42.1 | + | + | + | F | m42 | ||
| C54867-54989 | 51.2 | m42.2 | + | + | + | F | |||
| 55382-55630 | 64.3 | m43.1 | + | + | P | M43 | |||
| C57059-57166 | 63.0 | m43.2 | + | F | M43 | ||||
| C57467-57748 | 49.6 | m43.3 | + | + | P | ||||
| 59044-60105 | 61.6 | m44.1* | + | + | + | (+) | M45 | ||
| 59141-59425 | 60.7 | m44.3* | + | ||||||
| 62806-62886 | 49.4 | m45.2* | + | m45.1 | |||||
| 63077-63199 | 67.5 | m46.1 | (+) | + | + | P | M46 | ||
| 74589-74756 | 70.8 | m49.1 | + | (+) | + | + | P | M49 | |
| 76198-76269 | 59.7 | m50.1 | (+) | + | F | M50 | |||
| C85515-85664 | 59.3 | m55.1 | + | + | F | M55 | |||
| C91750-92352 | 64.7 | m58.1 | + | + | (+) | + | + | P | M58 |
| 101387-101473 | 60.9 | m70.1 | + | (+) | + | P | M71 | ||
| C105629-105691 | 46.0 | m74.1 | + | + | P | m74 | |||
| >105797-106069d | 56.0 | M73.5e2* | NA | + | + | + | + | F | m74 |
| C105907-106092 | 54.8 | m74.2 | + | + | P | ||||
| 117995-118600 | 64.9 | m83.1 | + | + | + | P | M83 | ||
| 118607-118819 | 61.5 | m83.2 | (+) | + | + | P | M83 | ||
| 120497-120742 | 71.5 | m84.1 | + | (+) | + | + | P | M84 | |
| C140098-140514 | 61.9 | m96.1 | + | + | (+) | (+) | P | M97 | |
| 148138-148275 | 42.0 | m102.1 | P | M103 | |||||
| 148330-148437 | 56.5 | m103.1 | + | (+) | + | P | M103 | ||
| C152720-153478 | 58.8 | m105.1 | + | (+) | (+) | (+) | P | M105 | |
| C154196-154363 | 68.5 | m106.1* | + | + | (+) | m106 | |||
| 154213-154365 | 69.9 | m106.2 | + | + | P | m106 | |||
| C155781-155918 | 35.5 | m106.3* | + | ||||||
| C159096-159176 | 51.9 | m106.4 | (+) | + | F | ||||
| 160169-160219 | 39.2 | m106.5 | + | F | |||||
| 162129-162293 | 63.0 | m107.1 | + | + | P | m107, m108 | |||
| C162095-162478 | 62.2 | m107.2 | + | + | P | m107, m108 | |||
| C162605-162862 | 69.0 | m107.3 | + | + | + | + | P | m107, m108 | |
| C163222-163527 | 66.7 | m112.1 | + | + | (+) | + | + | P | M112e1 |
| C163771-163911 | 65.2 | m112.2 | (+) | + | + | P | M112e1 | ||
| 167209-167970 | 57.1 | m116.1 | + | + | (+) | + | + | P | M116 |
| C168283-168438 | 51.3 | m116.2 | (+) | + | + | P | M116 | ||
| C168442-168543 | 51.0 | m116.3 | + | + | P | M116 | |||
| 169930-170400 | 62.6 | m117.2 | + | + | P | m117, m117.1 | |||
| 173438-173542 | 57.1 | m119.31 | + | (+) | + | + | P | m119.3 | |
| C173525-173608 | 46.4 | m119.32 | + | + | P | m119.3 | |||
| C174640-175665 | 52.3 | m120.1* | + | + | + | (+) | |||
| 175550-175612 | 49.2 | m120.2 | + | + | P | ||||
| C175875-176105 | 59.3 | m121.1 | + | + | P | M121 | |||
| C176053-177123 | 59.2 | m121.2 | + | + | (+) | + | P | M121 | |
| C176109-176384 | 60.1 | m121.3 | + | P | M121 | ||||
| 176251-176475 | 65.3 | m121.4 | + | + | + | P | M121 | ||
| 176479-176640 | 56.2 | m121.5 | + | P | M121 | ||||
| 178320-178439 | 60.0 | m122.1 | + | + | (+) | + | P | M122e5 | |
| 178653-179081 | 55.9 | m122.2 | + | (+) | + | + | P | M122e5 | |
| C179095-179217 | 52.0 | m122.3 | + | + | F | M122e5 | |||
| 179843-179896 | 63.0 | m122.4 | + | P | M122e4 | ||||
| C179996-180232 | 54.0 | m122.5 | + | + | + | P | M122e4 | ||
| 180108-180377 | 55.9 | m122.6 | + | + | F | M122e4 | |||
| C181863-182219 | 48.7 | m123.1* | + | + | m124-m124.1 | ||||
| 185677-185715 | 56.4 | m127.1 | + | + | + | P | |||
| 190220-190444 | 58.7 | m135.1 | + | + | + | P | m134-m136 | ||
| 190728-190958 | 50.2 | m135.2 | + | (+) | + | P | m136 | ||
| 192243-192377 | 63.0 | m137.1 | + | + | P | m138 | |||
| 192531-192770 | 62.1 | m138.1 | + | (+) | + | + | P | m138 | |
| C200667-201368 | 68.6 | m142.1 | + | + | + | + | P | m142, m143 | |
| C200966-201385> | 74.5 | m143b | NA | + | + | F | m143 | ||
| 204291-204500 | 54.8 | m145.1 | + | + | + | P | m145 | ||
| C204393-204515 | 53.7 | m145.2 | + | + | P | m145 | |||
| 204765-204863 | 60.6 | m145.3 | (+) | + | F | m145 | |||
| 207855-208160 | 62.1 | m150.1 | + | + | + | + | P | m149, m150 | |
| 208508-208660 | 53.6 | m150.2 | + | + | + | P | m150 | ||
| 209049-209306 | 56.2 | m151.1 | + | + | + | + | P | m151 | |
| 212933-213043 | 56.8 | m153.1 | + | F | m154 | ||||
| 212964-213428 | 57.6 | m154.1 | + | + | P | m154 | |||
| C213414-213617 | 57.4 | m154.2 | (+) | + | + | F | m154 | ||
| 216145-216369 | 48.9 | m157.1 | + | (+) | + | + | P | m157 | |
| C217486-217686 | 49.3 | m158.1 | + | (+) | + | + | P | m158 | |
| 217730-217801 | 50.0 | m158.2 | + | P | m158 | ||||
| 218648-218734 | 47.1 | m159.1 | + | (+) | + | P | m159 | ||
| 222109-222177 | 66.7 | m163.1 | + | + | + | + | F | m163 | |
| 223391-223609 | 66.7 | m165.1 | (+) | + | + | P | m165 | ||
| 224046-224123 | 69.2 | m165.2 | + | + | P | m165 | |||
| 226603-226764 | 68.5 | m167.1 | + | + | P | m167 | |||
| C227760-227789 | 66.7 | m167.2 | + | F | |||||
| 228747-228872 | 57.1 | m169.1 | + | + | F | ||||
| 229438-229662 | 70.7 | m170.1 | + | + | P | m170 | |||
| 230127-230261 | 59.3 | m171 | + | + | P |
“C” refers to complementary strand. ORFs identified by SSPA similarity are marked with an asterisk (see Table 1). *, from Table 1; <, uncertainty in boundary of the coding region.
NA, not applicable.
(+), weak levels.
Coding potential based on S-profiles.
Coding potential based on GeneMark protocol.
S-profile evidence over partial (P) or complete (F) sequence.
TABLE 3.
Characteristics of newly annotated ORFs in RCMV
| Coding region | % G+C | Name | Presence of Met | Low bias | Conserved | S-bias | G | Evidence | Published overlapping gene(s) |
|---|---|---|---|---|---|---|---|---|---|
| 14-1030 | 69.4 | r1.1 | + | P | r1 | ||||
| C2-610 | 73.7 | r1.2 | + | + | P | r1 | |||
| 2597-2791 | 74.9 | r2.05 | + | + | + | + | F | r2 | |
| C3228-4145 | 75.6 | r2.2 | + | + | + | + | P | r2, r2.1 | |
| 3637-3837 | 72.6 | r2.3 | + | + | P | r2, 2.1 | |||
| 5049-5156 | 71.3 | r2.4 | + | + | + | + | P | r2.1 | |
| C5754-7301 | 74.7 | r4* | + | + | + | + | P | r4, r4.1, r5, r5.1 | |
| C8594-8776 | 78.1 | r6.1 | + | + | F | r6 | |||
| C<9753-9860 | 75.9 | R23a* | + | + | + | + | F | r5.1, r6, r23, r23.1 | |
| C11891-12664 | 73.6 | r24.1 | + | + | + | + | P | R25 | |
| C13723-13881 | 71.1 | r24.2 | + | F | |||||
| C13969-14046 | 61.5 | r24.3 | + | F | r25.1 | ||||
| C15305-15961 | 69.4 | r25.3b* | NA | + | + | + | + | F | r25.2, r25.3 |
| C<19321-19491 | 73.3 | R27a | + | (+) | + | + | F | R27, r27.1 | |
| C21727-22476 | 70.8 | r31.1 | + | + | + | P | R31 | ||
| C28905-29168 | 67.4 | r34.1 | + | + | + | P | R34 | ||
| C29490-29753 | 75.8 | r34.2 | + | + | + | F | R34 | ||
| 34340-34414 | 72.0 | r37.1 | + | F | R37 | ||||
| 34613-35245 | 72.2 | r37.2 | + | + | + | P | R37, R38 | ||
| C35741-36322 | 69.2 | r38.5* | + | + | + | + | P | R38 | |
| C37859-38023 | 69.7 | r41.1 | + | + | (+) | + | + | F | r41 |
| 42221-42832 | 66.8 | r44.1* | + | + | R44, R45 | ||||
| 42507-42608 | 61.8 | r44.3* | + | + | |||||
| C42344-42424 | 67.9 | r44.2 | + | + | + | P | |||
| 45413-45760 | 76.1 | r45.2* | + | + | (+) | R45 | |||
| C56102-56359 | 68.6 | r48.2* | + | + | + | + | + | F | R49 |
| 67187-67228 | 54.8 | r55.1 | + | + | P | R55 | |||
| 67232-67276 | 55.6 | r55.2 | + | P | R55 | ||||
| 67913-68161 | 71.1 | r55.3 | + | + | + | + | P | R55, R56 | |
| 68883-69371 | 74.6 | r56.1 | + | (+) | + | + | P | R56 | |
| C70732-71136 | 79.5 | r56.2 | (+) | + | + | P | R57 | ||
| <74828-75454 | 72.7 | r58.1*§ | + | (+) | + | r58 | |||
| C79221-79499 | 69.9 | r58.2 | + | + | + | P | |||
| 79389-79490 | 76.5 | r58.3 | + | + | P | ||||
| 85631-86485 | 72.5 | R71* | + | + | + | + | F | r70.1 | |
| C89948-90370 | 62.9 | r70.41 | + | + | + | P | r70.4, r70.5 | ||
| 93941-94201 | 60.2 | R73.5e2*§ | + | + | + | + | + | F | |
| C99062-99271 | 71.4 | r77.1 | + | + | + | P | R78 | ||
| C104114-104320 | 50.7 | r80.1 | + | + | P | ||||
| C104157-104363 | 49.8 | r80.2 | + | P | |||||
| 109407-109607 | 70.6 | r84.1 | + | + | + | P | R84 | ||
| C120680-122236 | 73.7 | r90a | (+) | + | + | P | r90, R91, R92 | ||
| 125254-125511 | 67.1 | r94.1 | + | (+) | + | P | R94, R89e1 | ||
| C127377-127640 | 75.0 | r95.2 | + | + | (+) | + | + | P | R95, r95.1, R97 |
| 129977-130225 | 69.1 | R98a* | + | + | + | + | + | F | R98 |
| 135108-136100 | 74.0 | R102b* | NA | + | + | + | + | F | R102, R103 |
| C145051-145143 | 33.3 | r106.1* | + | ||||||
| C148724-148822 | 32.3 | r108.1* | + | ||||||
| 149409-149669 | 34.1 | r109.1 | + | + | (+) | P | r109 | ||
| 151708-151881 | 62.6 | r110.4 | + | + | F | ||||
| C154891-155019 | 70.5 | r113.1 | + | (+) | + | + | P | R113 | |
| 155325-155513 | 76.2 | r113.2 | + | + | P | R113 | |||
| 155517-155795 | 67.0 | r113.3 | + | + | P | R113 | |||
| C157690-158127 | 50.0 | r115.1* | + | + | (+) | P | R115, R116 | ||
| C162330-162656 | 45.3 | r119.25 | + | + | P | r119.2, r119.3 | |||
| C163299-163541 | 47.3 | r119.35 | + | + | P | ||||
| 167192-167371 | 52.8 | r121.05 | + | F | R121 | ||||
| C171187-171216 | 46.7 | r122.1 | + | P | R122e5 | ||||
| C171220-171300 | 50.6 | r122.2 | + | P | R122e5 | ||||
| C171344-171529 | 46.2 | r122.3 | + | P | R122e5 | ||||
| 171899-172225 | 45.3 | r123.1 | + | P | r123e4 | ||||
| 172253-172426 | 52.3 | r123.2 | + | + | + | F | r123e4 | ||
| 173140-173202 | 31.7 | r123.3 | P | r134e4 | |||||
| 173976-174329 | 54.5 | r124.1* | + | + | (+) | P | |||
| C174032-174328 | 55.6 | r124.2* | + | + | |||||
| C174463-174612 | 48.7 | r124.3 | P | ||||||
| 179064-179381 | 53.1 | r127.1 | + | + | + | P | r127 | ||
| C180014-180376 | 51.2 | r128.1 | + | (+) | + | P | r128 | ||
| C181724-181843 | 35.0 | r128.2 | P | ||||||
| 182081-182224 | 51.4 | r128.3 | + | P | r131 | ||||
| C182966-183247 | 50.0 | r132e2* | NA | + | + | ||||
| 185337-185423 | 57.5 | r136.1 | (+) | + | P | r136 | |||
| 187159-187227 | 62.3 | r138.1 | + | P | r138 | ||||
| C188705-188806 | 55.9 | r139.1 | + | + | F | r139 | |||
| C189002-189061 | 63.3 | r139.2 | + | F | r139 | ||||
| 189132-189209 | 53.8 | r139.3 | + | F | r139 | ||||
| C190223-190285 | 57.1 | r139.4 | + | P | r139 | ||||
| 193785-193892 | 50.0 | r141.1 | + | + | P | r142 | |||
| C194033-194350 | 58.2 | r142.1 | + | + | + | P | r142 | ||
| 194172-194369 | 57.1 | r142.2 | + | + | (+) | + | + | F | r142 |
| C194534-194620 | 65.5 | r142.3 | + | + | + | P | r142 | ||
| 194586-194729 | 59.0 | r142.4 | + | + | F | r142 | |||
| 195451-195531 | 59.3 | r142.5 | + | + | P | r142, r143 | |||
| 196564-196689 | 52.4 | r143.1 | + | P | r143 | ||||
| 196693-196776 | 59.5 | r143.2 | + | F | r143 | ||||
| 200183-200296 | 46.5 | r146.1 | (+) | P | |||||
| C200204-200425 | 44.6 | r146.2 | + | + | P | ||||
| 200300-200425 | 44.4 | r146.3 | (+) | P | |||||
| 201099-201314 | 44.0 | r147.1 | + | P | r147 | ||||
| 204792-205004 | 47.4 | r151.05 | + | P | r151 | ||||
| C208116-208301 | 39.2 | r151.4 | + | P | r151.3 | ||||
| C217004-217142b | 41.7 | r153e4§ | NA | + | |||||
| C217221-217327b | 39.3 | r153e3§ | NA | + | P | ||||
| C217402-217580b | 36.9 | r153e2§ | NA | + | |||||
| C217651-217783b | 44.4 | r153e1 | + | + | + | P | |||
| 221116-221319 | 52.9 | r160.1 | + | P | r160 | ||||
| C221139-221210 | 51.4 | r160.2 | + | + | P | r160 | |||
| 222602-222811 | 55.2 | r162.1 | + | + | P | r162 | |||
| 225438-225629 | 59.4 | r166.1 | + | + | + | + | P | r166 | |
| 226322-226519 | 63.6 | r166.2 | + | + | P | r166, r167 | |||
| C226475-226777 | 63.7 | r166.3 | (+) | + | + | P | r167 | ||
| C226578-226901 | 65.4 | r166.4 | + | P | r167 | ||||
| C226906-227073 | 64.3 | r166.5 | + | + | P | r167 | |||
| 227459-227746 | 53.1 | r168.1 | + | + | + | P | r168, r169 | ||
| 227742-228377c | 59.0 | r169.1§ | + | + | + | (+) | P | r169, r170 |
A total of 126 ORFs from the MCMV genome and 105 ORFs from the RCMV genome were newly annotated as a result of this analysis. In 101 of these ORFs, we could identify a putative start codon; 87 ORFs showed conservation through SSPA or BLASP analyses, and 77 ORFs were characterized by low compositional biases. We observed substantial concordance between regions of high coding potential identified by S-profiles or by the GeneMark procedure. In either viral genome, 175 ORFs included regions of high coding potential based on S-biases and a majority (140 ORFs) were also supported by the coding potential evaluated by GeneMark. A total of 61 ORFs coincided with the underlying contrasts in S-profiles, whereas 152 ORFs were characterized by partial S-profiles.
S-profiles of ORFs identified by SSPA similarity.
All annotated ORFs identified by SSPA similarity retained in Tables 2 and 3 are marked. Among the potential coding regions identified by SSPA similarity analysis, 2 newly annotated ORFs from MCMV (M31b and M73.5e2) and 10 newly annotated ORFs from RCMV (R23a, r25.3b, r38.5, r48.2, R73.5e2, R98a, R102b, r115.1, r153e3, and r153e1) were also confirmed by strong contrasts in S-profiles. S-profiles also distinguished the most likely reading frame within some of the groups of overlapping MCMV and RCMV ORFs with similarity to one another (Table 1). Within two of these groups, S-profiles favored as coding regions RCMV ORF r48.2, homologous to published ORF m48.2, and ORF r124.1, homologous to published ORF m124. In a third group of ORFs overlapping M116 and R116, S-profiles identified in MCMV an ORF borne on the direct strand (m116.1), whereas RCMV conservation and S-profiles favored an ORF on the complementary strand (r115.1). Seven ORFs from MCMV (m44.1, m44.3, m45.2, m106.1, m106.3, m120.1, and m123.1) and seven ORFs from RCMV (r44.1, r44.3, r45.2, r106.1, r108.1, r124.2, and r133e2), identified by extended SSPA similarity, were not recognized by S-profiles. Of these, ORF m120.1 from MCMV and ORF r132e2 from RCMV showed particularly strong conservation. All 14 ORFs are listed in Tables 2 and 3 as potential protein-coding sequences.
S-profiles of ORFs identified by BLASTP similarity.
All annotated ORFs identified by BLASTP analysis and retained in Tables 2 and 3 are indicated. Among four ORFs in MCMV and eight ORFs in RCMV that were identified as candidate genes by BLASTP analysis (Tables S3 and S4 in the supplemental material), three ORFs from RCMV, corresponding to two exons (r153e1 and r153e3) of the lectin-like gene and to a paralog (r169.1) of published ORF r171, were also supported by S-profiles. Exons 2 and 4 of r153 could not be confirmed by their S-profiles due to low G+C contents. Another ORF from RCMV (r58.1) was identified by BLASTP for its similarity to the regulatory protein E2 of human papillomavirus. Although S-profiles did not support the expression of this ORF over its entire length, the expression of the C-terminal portion (corresponding to the conserved region) was supported by extended GeneMark coding potentials and by a weak S-profile signal. Other BLASTP-identified coding regions were not supported by S-profiles, which strongly supported the authenticity of previously annotated ORFs in the same regions. These findings suggest that a reevaluation of other published proteins matching these ORFs (mostly hypothetical proteins from various herpesviruses [see supplemental Tables S3 and S4]) would be valuable.
Nonconserved ORFs identified by S-profile analysis.
Eight ORFs from MCMV (m20b, m116.1, m122.5, m122.6, m143b, m154.1, m154.2, and m163.1) and three ORFs from RCMV (r2.2, R27a, and r41.1), although not or poorly conserved, corresponded to strong contrasts in S-profiles and to extended regions of high coding potential. Among these, ORF m20b has been experimentally verified as a frameshift 3′ extension of m20 (23). We also interpreted ORF m143b as a frameshift 3′ extension (or possibly a second exon) of m143, consistent with the lack of an AUG codon. ORF R27a was interpreted as a frameshift 5′ extension of R27 and terminated at the corresponding approximate position (see the supplemental material). ORF r41.1 was similar to m41.1 mostly in a region coincident with a corresponding region of conservation with the overlapping published genes r41 and m41. However, strong contrasts in S-profiles and the presence of a conserved initiation codon (AUG) strongly suggest that this ORF (and its MCMV homolog, m41.1) is expressed.
S-profiles and overlapping ORFs.
S-profiles yielded useful verification of the position of most previously annotated ORFs of high (>50%) G+C content (see above). However, among these ORFs we identified 99 sequences that were only partially matched by S-profiles. The S-profile evidence for these sequences was classified as “partial” in the “evidence” column of Tables S7 and S8, where it was also diagrammatically represented (e.g., for ORF m25.2 “++−” indicates that over approximately the first two thirds of the annotated sequence, S-profiles conform to the expression of this ORF but not over the last third). Many partial S-profile inconsistencies observed in previously annotated genes coincided with the overlap of newly annotated sequences. In MCMV, 35 previously annotated genes overlapped 58 newly identified ORFs (Table S7), and in RCMV, 24 previously annotated genes overlapped 35 newly identified ORFs (Table S8). Irregular S-profiles were observed in these regions of overlap. In 36 of these situations, the identification of a new ORF fully explained the irregularity (supplemental Tables S7 and S8). Irregular regions could be partly explained in 18 other cases.
Alternative start of translation of previously annotated ORFs.
The use of an alternative translation start site was suspected when consistent S-profiles failed to coincide with the most 5′-end-proximal AUG in annotated genes. Alternative initiation sites have already been characterized for some genes, such as MCMV m131, a short first exon of the mck gene, where the fourth AUG codon in the full-length ORF is where translation starts (27).
Using S-profile analysis, we identified 25 ORF candidates in the MCMV genome that may employ alternative translation start sites located upstream of the previously annotated site (Table S7). A different initiation codon downstream of the previous annotation was predicted in 19 cases (m9, M25, m25.1, M31, M34, M43, M51, M53, M55, M69, M71, M72, M73, M77, M102, m119.1, m131, and m139) (Table S7). An upstream start site was suggested by S-profiles for ORF m16, although no AUG codon was found in this region.
In RCMV we found 11 ORF candidates for alternative start sites (Table S8), 9 of these apparently starting downstream and 2 (r4 and r70.1) apparently starting upstream of previously suggested start sites. Six of the nine ORFs for which S-profiles suggested a downstream start of translation (R31, r41, r74, R91, R115, and r171.1) also encoded a putative initiation codon (AUG) in corresponding positions. For three ORFs (R77, R122e5, and r166) in which an alternative start site could not be predicted, overlap to other coding sequences was found to explain the observed S-profiles. In the case of R122e5, S-profiles were also consistent with an alternative exon structure. In the cases of r4 and r70.1, S-profiles strongly confirmed evidence from sequence conservation that the coding regions of these genes should be extended 5′ of the original annotation (see also the section on SSPA similarity analysis and the supplemental material).
Other contributing evidence from S-profiles.
S-profile inconsistencies were found in published ORFs of high G+C content from MCMV (M24, m25.2, M46, m48.2, M50, M69, M71, M87, M93, M112e1, m129, m131, m144, m159, m163, m165, and m170) and from RCMV (R43, r70.4, R77, r133, and r171) that could not be explained by overlapping sequences or alternative translation initiation. Most striking examples of these arrangements were found in genes M69, M87, and R77. Although the nature of these anomalous regions was unclear, in specific cases these might correspond to proteins of peculiar amino acid compositions or to the presence of introns.
Annotated genes not evidenced by S-profiles.
Fourteen previously annotated genes from MCMV and 42 from RCMV could not be confirmed by S-profiles due to low G+C content (indicated in Tables S7 and S8 as not applicable). Virtually all of these genes belonged to the regions of low G+C content of the corresponding genomes. The only exception was gene m74, which had uncharacteristically low G+C content despite its location in the high-G+C region of MCMV. Among annotated genes of high G+C content, 39 genes from MCMV and 28 genes from RCMV did not show the expected S-profile contrasts even though their G+C contents were often >60%. In the case of m19, m48.1, m108, M116, m119.5, and m134 from MCMV and r2.1, r4.1, r25.2, r95.1, and r167 from RCMV (indicated as “contradicted” in Tables S7 and S8), S-profiles largely contradicted their expression, providing evidence for expression of overlapped ORFs in different frames. Fifty-six other annotated genes (classified by “no evidence” in Tables S7 and S8) had high G+C contents in all three codon positions. The atypical S-profiles underlying these genes may be a consequence of corresponding gene products of atypical amino acid composition. It must be noted that most of these genes were not conserved between MCMV and RCMV and that their expression and functionality have not been characterized as yet in any direct investigation.
DISCUSSION
The sequencing of genomes and the use of different annotation methods have brought considerable revisions to the definition of the gene complement of cytomegalovirus. In particular, the human CMV genome sequence has been updated with analyses of additional strains (9, 17, 33) and comparisons of cytomegaloviruses from related species (14, 32, 33). We have compared the gene contents of the closely related MCMV Smith strain and RCMV Maastricht strain genomes, emphasizing sequence conservation and frame-specific G+C content (5). As a consequence of this analysis, we propose substantial revision to the annotation of these genomes. In MCMV, 14 newly identified ORFs were convincingly characterized as protein coding and 113 other ORFs showed evidence of a protein-coding capacity. Our analyses suggest the reevaluation of the translation start site of at least 18 previously annotated protein-coding sequences and provide S-profile evidence that contradicts the expression of six previously annotated genes. In RCMV we found strong evidence of at least 20 newly identified protein-coding sequences and the potential for 85 other coding sequences. We propose alternative translation start sites for at least eight previously annotated genes and provide evidence that suggests that five others are not expressed. At least two (up to four) examples in MCMV genes and at least five (up to six) examples in RCMV genes were found where frameshifts extend previously annotated genes. Among these newly annotated genes we have included in the MCMV genome a protein identified (23, 42a) as an alternative spliced form of M73 (M73.5e2) and we have identified its homolog in RCMV, designated R73.5e2. In RCMV we have also identified a second exon to ORF r133 (r132e2) and a homolog to a lectin-like protein (46) encoded by four exons (r158e1 to e4).
We have avoided conventional criteria of minimum ORF length (≥100 codons) and maximum ORF overlap (<60%) to prevent the a priori exclusion of a class of genes that has previously been found only by direct experimental investigation. We also have not required an AUG codon to be present in potential coding regions. This allowed us to uncover small and overlapping ORFs, mRNA splicing, use of alternative translation initiation sequences in the coding complement of herpesvirus genomes, and several frameshifts within coding sequences. In fact, two of the frameshift extensions identified by our analysis in the MCMV genome, m20b and M31b, have been experimentally verified as correct 3′-terminal sequences of genes m20 and M31 (23). Alternative translation initiation signals are apparently used in a bona fide complete gene (R71, renamed r70.1) and in ORF r4, whose conservation and S-profile signals clearly extend 5′ of their first AUG codon. Other examples of herpesvirus coding regions not initiated by an AUG codon have been reported (46). A coding sequence lacking an initiation codon may also relate to a potential multiexonic structure of the corresponding genes or appear as a consequence of sequencing errors.
Our analyses revealed intriguing anomalies and potential in the G+C distribution (S-profiles) within annotated genes. Within annotated genes showing N-terminal anomalies in S-profiles, AUG codons were often located near the ends of these regions. This suggested the possibility of an alternative start of translation. An interesting example is M25 from MCMV, encoding tegument protein pM25. This protein presents extensive low-complexity regions 5′ of several possible alternative starts of translation identified by our analysis. Intriguingly, in viral preparations, pM25 is found in forms of different molecular masses, identified as a true late 130-kDa peptide (included in the tegument) and two early 105-kDa and 95-kDa peptides (47) and later also as a 200-kDa, 52-kDa, or 48-kDa peptide (23). Peptides translated from the AUG codons corresponding to the region of conservation between MCMV and RCMV and to consistent S-profiles have predicted molecular masses of 57.2 kDa and 45.2 kDa. The sizes of these peptides are consistent with the smaller peptides isolated from viral preparations before replication (the annotated gene has a predicted molecular mass of 103 kDa). We suggest that some peptides from M25 may result from alternative transcription and translation start sites rather than from posttranslational proteolysis.
Interesting anomalies in G+C distribution were evident also in gene M55 (glycoprotein B) from MCMV. This gene shows striking differences in S-profiles between its 5′-terminal, central, and 3′-terminal parts (Fig. 6). The functional form of this protein is generated by cleavage in the central part of the protein (38, 44). The 3′ part of the gene, corresponding to the region of highest S-profile contrasts, is preceded by an AUG codon and two putative TATA box sequences (see the supplemental material). This suggests that the C-terminal part of glycoprotein B may also be alternatively translated from a shortened transcript.
Many anomalies in S-profiles involving the central or C-terminal parts of annotated sequences cannot be explained by alternative start codons. In many cases these coincide with parts of the protein that are not conserved and often include low-complexity sequences, as, for example, in the pairs of homologs M34/R34, M56/R56, M69/R69, M83/R83, and M105/R105. The hydrophilic amino acid composition and lack of sequence conservation of these regions suggests that they may function as flexible linkers between separate functional domains of a protein or that they may correspond to loops or, for terminal elements, to nonfunctional tails. In the case of MCMV m45.1, the entire sequence has an anomalous composition (see the supplemental material). It is possible that m45.1 evolved from a seemingly nonfunctional N-terminal sequence of M45, still present in the homologous sequence R45 from RCMV.
The herpesvirus capsid limits the size of the genome that can be packaged. From this perspective, it seems unlikely that nonfunctional regions of DNA can be retained in a genome where genes tend to be densely packed. While noncoding regions may be involved as control elements in transcription or DNA replication, we speculate that the presence of regions of weak selection in herpesvirus proteins may allow these viruses to encode overlapped genes to a greater extent than presently described. Frame analysis of G+C content suggests that the MCMV and RCMV genomes contain ORFs of high coding potential that overlap.
Annotation is a process of prediction and confirmation by methods that provide a working set of data for additional empirical experimental studies. We believe that there is a need to relax the criteria used in conventional annotation methods in the study of eukaryotic viruses, where overlapping genes and posttranscriptional regulation, such as mRNA splicing and the use of nonconventional translation signals, are relevant biological processes. The increased ease of current experimental techniques in verifying the expression of coding sequences makes striving for coverage, perhaps with a reduction in specificity, a reasonable approach to gene prediction. Our application of different methods of sequence analysis identified a plethora of candidate genes that are excluded by more conventional criteria of annotation, providing a more comprehensive picture of the coding potential of these genomes for experimental verification.
Supplementary Material
Acknowledgments
This work was supported by PHS grant 2 RO1 GM010452 to S.K., PHS grants RO1A120211 and RO1AI30363 to E.S.M., and an Alfred Benzon Foundation fellowship to T.N.K.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schaffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bahr, U., and G. Darai. 2001. Analysis and characterization of the complete genome of tupaia (tree shrew) herpesvirus. J. Virol. 75:4854-4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baldick, C. J., Jr., and T. Shenk. 1996. Proteins associated with purified human cytomegalovirus particles. J. Virol. 70:6097-6105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Besemer, J., A. Lomsadze, and M. Borodovsky. 2001. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29:2607-2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bibb, M. J., P. R. Findlay, and M. W. Johnson. 1984. The relationship between base composition and codon usage in bacterial genes and its use for the simple and reliable identification of protein-coding sequences. Gene 30:157-166. [DOI] [PubMed] [Google Scholar]
- 6.Borodovsky, M., and J. McIninch. 1993. GeneMark: parallel gene recognition for both DNA strands. Comput. Chem. 17:123-133. [Google Scholar]
- 7.Brocchieri, L., and S. Karlin. 1998. A symmetric-iterated multiple alignment of protein sequences. J. Mol. Biol. 276:249-264. [DOI] [PubMed] [Google Scholar]
- 8.Bruggeman, C. A., H. Meijer, F. Bosman, and C. P. van Boven. 1985. Biology of rat cytomegalovirus infection. Intervirology 24:1-9. [DOI] [PubMed] [Google Scholar]
- 9.Cha, T. A., E. Tom, G. W. Kemble, G. M. Duke, E. S. Mocarski, and R. R. Spaete. 1996. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 70:78-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chee, M. S., A. T. Bankier, S. Beck, R. Bohni, C. M. Brown, R. Cerny, T. Horsnell, C. A. Hutchison III, T. Kouzarides, J. A. Martignetti, et al. 1990. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol. 154:125-169. [DOI] [PubMed] [Google Scholar]
- 11.Reference deleted.
- 12.Dargan, D. J., F. E. Jamieson, J. MacLean, A. Dolan, C. Addison, and D. J. McGeoch. 1997. The published DNA sequence of human cytomegalovirus strain AD169 lacks 929 base pairs affecting genes UL42 and UL43. J. Virol. 71:9833-9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davison, A. J. 2002. Evolution of the herpesviruses. Vet. Microbiol. 86:69-88. [DOI] [PubMed] [Google Scholar]
- 14.Davison, A. J., A. Dolan, P. Akter, C. Addison, D. J. Dargan, D. J. Alcendor, D. J. McGeoch, and G. S. Hayward. 2003. The human cytomegalovirus genome revisited: comparison with the chimpanzee cytomegalovirus genome. J. Gen. Virol. 84:17-28. [DOI] [PubMed] [Google Scholar]
- 15.Davis-Poynter, N. J., D. M. Lynch, H. Vally, G. R. Shellam, W. D. Rawlinson, B. G. Barrell, and H. E. Farrell. 1997. Identification and characterization of a G protein-coupled receptor homolog encoded by murine cytomegalovirus. J. Virol. 71:1521-1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dolan, A., C. Cunningham, R. D. Hector, A. F. Hassan-Walker, L. Lee, C. Addison, D. J. Dargan, D. J. McGeoch, D. Gatherer, V. C. Emery, P. D. Griffiths, C. Sinzger, B. P. McSharry, G. W. Wilkinson, and A. J. Davison. 2004. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 85:1301-1312. [DOI] [PubMed] [Google Scholar]
- 17.Dunn, W., C. Chou, H. Li, R. Hai, D. Patterson, V. Stolc, H. Zhu, and F. Liu. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. USA 100:14223-14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibson, W., K. S. Clopper, W. J. Britt, and M. K. Baxter. 1996. Human cytomegalovirus (HCMV) smallest capsid protein identified as product of short open reading frame located between HCMV UL48 and UL49. J. Virol. 70:5680-5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gompels, U. A., J. Nicholas, G. Lawrence, M. Jones, B. J. Thomson, M. E. Martin, S. Efstathiou, M. Craxton, and H. A. Macaulay. 1995. The DNA sequence of human herpesvirus-6: structure, coding content, and genome evolution. Virology 209:29-51. [DOI] [PubMed] [Google Scholar]
- 20.Hansen, S. G., L. I. Strelow, D. C. Franchi, D. G. Anders, and S. W. Wong. 2003. Complete sequence and genomic analysis of rhesus cytomegalovirus. J. Virol. 77:6620-6636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hudson, J. B. 1979. The murine cytomegalovirus as a model for the study of viral pathogenesis and persistent infections. Arch. Virol. 62:1-29. [DOI] [PubMed] [Google Scholar]
- 22.Karlin, S., and J. Mrazek. 2000. Predicted highly expressed genes of diverse prokaryotic genomes. J. Bacteriol. 182:5238-5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kattenhorn, L. M., R. Mills, M. Wagner, A. Lomsadze, V. Makeev, M. Borodovsky, H. L. Ploegh, and B. M. Kessler. 2004. Identification of proteins associated with murine cytomegalovirus virions. J. Virol. 78:11187-11197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kotenko, S. V., S. Saccani, L. S. Izotova, O. V. Mirochnitchenko, and S. Pestka. 2000. Human cytomegalovirus harbors its own unique IL-10 homolog (cmvIL-10). Proc. Natl. Acad. Sci. USA 97:1695-1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krmpotic, A., I. Bubic, B. Polic, P. Lucin, and S. Jonjic. 2003. Pathogenesis of murine cytomegalovirus infection. Microbes Infect. 5:1263-1277. [DOI] [PubMed] [Google Scholar]
- 26.Lockridge, K. M., S. S. Zhou, R. H. Kravitz, J. L. Johnson, E. T. Sawai, E. L. Blewett, and P. A. Barry. 2000. Primate cytomegaloviruses encode and express an IL-10-like protein. Virology 268:272-280. [DOI] [PubMed] [Google Scholar]
- 27.MacDonald, M. R., M. W. Burney, S. B. Resnick, and H. W. I. Virgin. 1999. Spliced mRNA encoding the murine cytomegalovirus chemokine homolog predicts a beta chemokine of novel structure. J. Virol. 73:3682-3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27a.McCormick, A. L., A. Skaletskaya, P. A. Barry, E. S. Mocarski, and V. S. Goldmacher. 2003. Differential function and expression of the viral inhibitor of caspase 8-induced apoptosis (vICA) and the viral mitochondrial-localized inhibitor of apoptosis (vMIA) cell death suppressors conserved in primate and rodent cytomegaloviruses. Virology 316:221-233. [DOI] [PubMed] [Google Scholar]
- 28.Mocarski, E. S. 2004. Immune escape and exploitation strategies of cytomegaloviruses: impact on and imitation of the major histocompatibility system. Cell. Microbiol. 6:707-717. [DOI] [PubMed] [Google Scholar]
- 29.Mocarski, E. S. 2002. Immunomodulation by cytomegaloviruses: manipulative strategies beyond evasion. Trends Microbiol. 10:332-339. [DOI] [PubMed] [Google Scholar]
- 30.Mocarski, E. S., Jr., and C. T. Courcelle. 2001. Cytomegaloviruses and their replication, p. 2629-2673. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 31.Mocarski, E. S., M. N. Prichard, C. S. Tan, and J. M. Brown. 1997. Reassessing the organization of the UL42-UL43 region of the human cytomegalovirus strain AD169 genome. Virology 239:169-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy, E., I. Rigoutsos, T. Shibuya, and T. E. Shenk. 2003. Reevaluation of human cytomegalovirus coding potential. Proc. Natl. Acad. Sci. USA 100:13585-13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murphy, E., D. Yu, J. Grimwood, J. Schmutz, M. Dickson, M. A. Jarvis, G. Hahn, J. A. Nelson, R. M. Myers, and T. E. Shenk. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. USA 100:14976-14981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muto, A., and S. Osawa. 1987. The guanine and cytosine content of genomic DNA and bacterial evolution. Proc. Natl. Acad. Sci. USA 84:166-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neote, K., D. DiGregorio, J. Y. Mak, R. Horuk, and T. J. Schall. 1993. Molecular cloning, functional expression, and signaling characteristics of a C-C chemokine receptor. Cell 72:415-425. [DOI] [PubMed] [Google Scholar]
- 36.Nicholas, J. 1996. Determination and analysis of the complete nucleotide sequence of human herpesvirus 7. J. Virol. 70:5975-5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pass, R. F. 2001. Cytomegalovirus, p. 2675-2705. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 38.Rapp, M., M. Messerle, B. Buhler, M. Tannheimer, G. M. Keil, and U. H. Koszinowski. 1992. Identification of the murine cytomegalovirus glycoprotein B gene and its expression by recombinant vaccinia virus. J. Virol. 66:4399-4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rawlinson, W. D., and B. G. Barrell. 1993. Spliced transcripts of human cytomegalovirus. J. Virol. 67:5502-5513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rawlinson, W. D., H. E. Farrell, and B. G. Barrell. 1996. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 70:8833-8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reddehase, M. J. 2002. Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2:831-844. [DOI] [PubMed] [Google Scholar]
- 42.Schleiss, M. R. 2002. Animal models of congenital cytomegalovirus infection: an overview of progress in the characterization of guinea pig cytomegalovirus (GPCMV). J. Clin. Virol. 25(Suppl. 2):S37-S49. [DOI] [PubMed] [Google Scholar]
- 42a.Scalzo, A. A., P. B. Dallas, C. A. Forbes, A. S. J. Mikosza, P. Fleming, L. J. Lathbury, P. A. Lyons, S. Laferté, M. M. Craggs, and L. C. Loh. 2004. The murine cytomegalovirus M73.5 gene, a member of a 3′ co-terminal alternatively spliced gene family, encodes the gp24 virion glycoprotein. Virology 329:234-250. [DOI] [PubMed] [Google Scholar]
- 43.Smith, J. A., and G. S. Pari. 1995. Human cytomegalovirus UL102 gene. J. Virol. 69:1734-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Spaete, R. R., R. M. Thayer, W. S. Probert, F. R. Masiarz, S. H. Chamberlain, L. Rasmussen, T. C. Merigan, and C. Pachl. 1988. Human cytomegalovirus strain Towne glycoprotein B is processed by proteolytic cleavage. Virology 167:207-225. [DOI] [PubMed] [Google Scholar]
- 45.Vink, C., E. Beuken, and C. A. Bruggeman. 2000. Complete DNA sequence of the rat cytomegalovirus genome. J. Virol. 74:7656-7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Voigt, S., G. R. Sandford, L. Ding, and W. H. Burns. 2001. Identification and characterization of a spliced C-type lectin-like gene encoded by rat cytomegalovirus. J. Virol. 75:603-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wu, C. A., M. E. Carlson, S. C. Henry, and J. D. Shanley. 1999. The murine cytomegalovirus M25 open reading frame encodes a component of the tegument. Virology 262:265-276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.