

Abstract
Poliovirus provides a well-characterized system for understanding how nonenveloped viruses enter and infect cells. Upon binding its receptor, poliovirus undergoes an irreversible conformational change to the 135S cell entry intermediate. This transition involves shifts of the capsid protein β barrels, accompanied by the externalization of VP4 and the N terminus of VP1. Both polypeptides associate with membranes and are postulated to facilitate entry by forming a translocation pore for the viral RNA. We have calculated cryo-electron microscopic reconstructions of 135S particles that permit accurate placement of the β barrels, loops, and terminal extensions of the capsid proteins. The reconstructions and resulting models indicate that each N terminus of VP1 exits the capsid though an opening in the interface between VP1 and VP3 at the base of the canyon that surrounds the fivefold axis. Comparison with reconstructions of 135S particles in which the first 31 residues of VP1 were proteolytically removed revealed that the externalized N terminus is located near the tips of propeller-like features surrounding the threefold axes rather than at the fivefold axes, as had been proposed in previous models. These observations have forced a reexamination of current models for the role of the 135S particle in transmembrane pore formation and suggest testable alternatives.
Enveloped viruses are able to deliver their nucleocapsids to the cytoplasm of cells by fusing the viral membrane with a host cell membrane at the cell surface or within endocytic vesicles. Lacking a membrane, nonenveloped viruses must provide some other mechanism that allows the viral genome or the nucleocapsid to enter their hosts. In many ways, poliovirus is an excellent model system for studying the cell entry mechanism of simple nonenveloped viruses. A significant human pathogen and the type member of the picornavirus family, it is well characterized biochemically and genetically, and the availability of tissue culture systems that produce high yields of virus makes the virus and several cell entry intermediates amenable to structural studies.
Poliovirus contains a single-stranded plus-sense RNA genome within an icosahedral capsid composed of 60 copies each of four coat proteins, VP1, VP2, VP3, and VP4. The virion surface is dominated by (i) star-shaped mesas at the fivefold axes surrounded by deep canyons and (ii) three-bladed propellers at the threefold axes separated by saddle-like depressions straddling the twofold axes (22). The capsid proteins VP1, VP2, and VP3 share the same eight-stranded β-barrel fold but are distinguished from one another by the shape of their loops and N- and C-terminal extensions. The native form of the capsid is stabilized by an intricate network on its inner (RNA-proximal) surface, formed by VP4 and the intertwined N termini of the other capsid proteins. Much of this network cannot be formed until late in assembly when the precursor, VP0, is cleaved to yield VP4 and VP2 (2).
Poliovirus infection is initiated by binding to a specific receptor, Pvr (poliovirus receptor) or CD155. Pvr is a member of the nectin family of CAM-like cell surface proteins and consists of an N-terminal ectodomain with three immunoglobulin-like domains, a transmembrane domain and either of two splice variant C-terminal cytoplasmic domains. Cryo-electron microscopy (cryo-EM) reconstructions of virus-receptor complexes demonstrate that the receptors bind with their N-terminal ectodomains inserted into the canyons surrounding the fivefold axes (5, 17, 35).
When native virus binds its receptor at physiological temperature, it undergoes an irreversible conformational change, producing the 135S particle. This conformational change, which can also be induced by heating virus in a low-salt buffer (11), results in a decreased affinity for Pvr and externalization of VP4 (which is myristoylated at its N terminus) and the N terminus of VP1 (14). Unlike the native virion, the 135S particle is sensitive to proteases, including the V8 protease of Staphylococcus aureus, which specifically cleaves after a glutamate at position 31 of VP1. Removal of the first 31 amino acids of VP1 prevents the 135S particle from associating with membranes or releases it from preformed 135S-membrane complexes, leaving only the 31-amino-acid peptide attached to the membrane (14). Late in infection, the virus undergoes a second irreversible conformational change resulting in release of the viral RNA, leaving an empty particle which sediments at 80S. The trigger for this final stage of the entry process and where it takes place in the cell are not known.
Electrophysiology experiments have implicated the membrane-associated N terminus of VP1 and VP4 in the formation of channels in lipid bilayers (33). In addition, viruses containing mutations in the N terminus of VP1 are defective in RNA release and viruses with a T-G mutation in residue 28 of VP4 are defective in both channel formation and RNA release (12). Together, these experiments support a model in which these channels facilitate translocation of RNA across cell membranes.
Structures of 135S and 80S particles have been determined by cryo-EM at 22-Å and 23-Å resolution, respectively (3). Computational docking of the β barrels from crystal structures into the 135S density map provided insights into the structural rearrangements that the capsid undergoes during cell entry, which resemble tectonic shifts, i.e., rigid-body movements on a spherical surface. These data were inconsistent with models that envisaged the N terminus of VP1 exiting the virion via the channel at the fivefold axis (15, 18, 25) and, rather, suggested that it exits at the base of the canyon. However, the resolution was insufficient to conclusively identify the point of egress of the N terminus of VP1 or its location in the 135S particle.
To address these questions, we have determined the structures of two forms of the 135S particle to 10-Å resolution by cryo-EM. In one form, the 135S particle was intact; in the other, the first 31 residues of VP1 were proteolytically removed. The improved resolution of the 135S structure has led to a more tightly constrained and confidence-inspiring model that includes loops and N- and C-terminal extensions that could not be placed in the earlier structure (3). Comparison of the cleaved and uncleaved 135S particles shows that the extreme N terminus of VP1 is located at the tip of the propeller-shaped feature on the outer surface, rather than at the fivefold axis, as previously thought. Together with the fitting of pseudoatomic models, these comparisons also permit us to infer the likely path of egress of the N terminus from the interior position that it occupies in the native virion. These results have forced a reexamination of current models for the role of the 135S particle in pore formation and have led to the generation of new testable alternative hypotheses.
MATERIALS AND METHODS
Virus propagation and preparation of 135S particles.
Native poliovirus was propagated in HeLa cell suspension, harvested by centrifugation, released by freeze-thawing the cell pellet, and purified by CsCl density gradient centrifugation as described previously (36). 135S particles were made in vitro from native virus by heating for 3 min at 50°C in a low-salt buffer. (The 135S1 preparation was made by heating in 2 mM CaCl2, 20 mM HEPES, pH 7.4, and the 135S2 sample was prepared in 2 mM CaCl2, 20 mM Tris, pH 7.5.)
The two 135S preparations were digested with V8 protease to cleave the N-terminal 31 residues of VP1. V8-cleaved 135S1 was produced by incubating 135S particles with Staphylococcus areus-V8 protease (Sigma) at a ratio of 2.3:1 (wt/wt) for 1 hour at room temperature in 2 mM CaCl2, 20 mM HEPES, pH 7.4. After the incubation the reaction was quenched on ice and cleaved virus was purified on a Sephacryl S-100 column. A second V8-cleaved 135S preparation was made by mixing 135S with V8 protease at a ratio of 1.4:1 (wt/wt) for 1 hour at 4°C, after which cleaved particles were purified on an S-300 column (This second preparation was split into two separate samples which differed in ionic strength [see Table 1].) Although these two preparation methods differed slightly, gels showing the extent of cleavage and purification of particles were indistinguishable.
TABLE 1.
Microscopy and image reconstruction data
| Sample | Microscopeb (voltage) | Magnification | No. of micrograph pairs | Pixel size (Å) | Particle imagesc | CTF correctiond | Resolution (Å)e | EMDB IDf |
|---|---|---|---|---|---|---|---|---|
| 135S1 | Tecnai 20 (200 kV) | 50,000 | 22 | 2.7 | 3,641 | CTFMIX | 11 | 3139 |
| 135S2 | CM200 (120 kV) | 38,000 | 3 | 1.8 | 8,224 | Bsoft | 10 | 3142 |
| V8-cleaved 135S1 | Tecnai 20 (200 kV) | 50,000 | 20 | 2.7 | 4,381 | CTFMIX | 11 | 3150 |
| V8-cleaved 135S2a | CM200 (120 kV) | 38,000 | 6 | 1.8 | 6,850 | Bsoft | 9 | 3143 |
| V8-cleaved 135S3a | CM200 (120 kV) | 38,000 | 2 | 1.8 | 3,205 | Bsoft | 10 | 3144 |
Second V8-cleaved preparation: salt was added when adjusting the concentration (final NaCl concentrations of 0.07 M and 0.5 M, for samples 2 and 3, respectively).
FEI, Eindhoven, The Netherlands.
Number of particle images used in three-dimensional reconstruction. Corresponding particle images from each micrograph pair are counted as one.
CTF correction refers to the program package, either CTFMIX (9) or Bsoft (20), used to apply the correction scheme described in Materials and Methods.
Resolution was measured with the Fourier shell correlation criteria (30), 0.3 threshold (27), with images corrected for CTF and decay.
Submission number in the Electron Microscopy Data Bank.
Microscopy and image reconstruction.
The 135S concentrations were adjusted to 0.4 to 0.6 mg/ml, while the V8-cleaved 135S samples were adjusted to 0.7 to 0.9 mg/ml for cryo-EM. Both sets were imaged as described previously (6) (for details, see Table 1). In brief, virus particles were loaded onto glow-discharged holey carbon grids and flash frozen in liquid ethane. The V8-cleaved 135S1 sample required the application of a thin layer of carbon on one side of the holey carbon grid to ensure a good distribution of particles. Images were taken on film using a 200-kV electron microscope equipped with a field emission gun. Focal pairs of micrographs were recorded at ∼10 electrons/Å2 per exposure, with a variety of focal settings from 0.9- to 3-μm underfocus. The close-to-focus image was recorded first.
Micrographs were digitized with a SCAI scanner (Z/I Imaging, Huntsville, Alabama) (pixel sizes defined in Table 1). The images used for orientation determination were corrected for the phase reversal of the contrast transfer function (CTF), while images used in the reconstruction were corrected with a deconvolution of the CTF and decay. Orientations and origins for windowed particles were determined via a modified version of the PFT algorithm (1) altered to use both phase and amplitude information in orientation selection. Three-dimensional reconstructions were computed with a Fourier-Bessel algorithm (10) (Table 1).
Difference map calculation.
All reconstructions used in difference map comparisons were size calibrated to a previously calibrated 135S reconstruction (3) by comparing radial density plots (4). Radial density plots also were used to calibrate densities for difference imaging. Difference density maps were computed between scaled reconstructions using the Bsoft software package (20). Resolution-dependent, bin-scaled (23) difference maps were also calculated and produced identical results.
Model fitting and refinement.
Crystallographic refinement procedures, originally developed for the efficient refinement of highly symmetric atomic models (23), were adapted for an automated rigid-body refinement into the EM density maps, with six adjustable parameters per movable body (i.e., translations and rotations about the x, y, and z axes). Refinement was carried out by the INSOUT program (D. J. Filman, unpublished), a flexible framework for rigid-body fitting that incorporates a user-designed UNIX shell script to select functions from crystallographic refinement programs, computational chemistry software, or locally developed software.
Using the INSOUT algorithm, density corresponding to one protomer (i.e., the VP1, VP2, and VP3 subunits that are cleaved from a single polypeptide during assembly) and its neighboring contacts was boxed out of the EM map and Fourier transformed. Rigid-body transformations of a protein backbone model were carried out for a series of trials. For each trial, the residuals (defined in Table 2) for the Fourier phase error (ΣF|Δφ|) and the sum of Fourier amplitudes squared (ΣF2model) were evaluated and optimized according to the following formula:
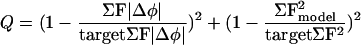 |
(1) |
The ΣF |Δφ| is a measure of the disagreement between the model and the experimental density, while ΣF2model penalizes shifts that result in collisions between the rigid bodies. The denominators in Q contain user-defined target values for each residual. Minimizing Q tends to cause each refinement residual to match its target more closely. Target values were adjusted periodically to produce and maintain acceptable contacts between domains, while improving the agreement between observed and model-based densities. Additional statistics, which are reported by other EM density fitting procedures (29), were calculated after the refinement was finished (Table 2).
TABLE 2.
Results of fitting and refining atomic models of VPs into a cryo-EM map of 135S
| Modela | Phase errorb | ΣF2modelc | % Densityd | % Clashe |
|---|---|---|---|---|
| Native protomer | 34.4 | 8,809 | 5.1 | 0.0 |
| Refinement of native | 25.8 | 8,273 | 4.1 | 0.3 |
| Hybrid | 25.5 | 8,157 | 3.8 | 0.6 |
| Hybrid + tube | 25.4 | 8,134* | 3.8 | 0.2 |
| Hybrid + tube + helix | 24.0 | 8,261* | 3.8 | 0.6 |
| Hybrid + tube + helix + Nterm VP3 | 24.0 | 8,504* | 3.5 | 0.5 |
| Hybrid + tube + helix + Nterm VP3 + VP2 hinge | 23.27 | 8,415 | 4.1 | 0.3 |
The hybrid model consists of the coordinates from the manually docked VP1, combined with VP2 and VP3 coordinates resulting from the refinement of the 160S protomer into the 135S map. Tube refers to shifting the VP3 β tube to better fit the 135S EM density. Helix describes a 10-residue helix docked into the ridge. Nterm VP3 includes residues 14 to 49 of VP3 in the rigid-body refinement of VP1. VP2 hinge indicates that residues 13 to 27 of VP2 are moving as a rigid body with VP3 instead of VP2.
The phase error is defined as Σ|Freference∥φreference − φmodel|, where Freference and φreference come from the Fourier transform of the reconstruction and φmodel comes from the transform of the model-based electron density. It is a measure of agreement between the model and reference densities.
ΣF2model is the sum of the squares of terms in the Fourier transform of the model-based electron density. It is a measure of intra- and intermolecular contacts. It is expected to increase as more residues are added to a given model (*). Decreases in ΣF2model correspond to reduction in overlap of model parts.
% Density refers to the percentage of atoms in the model that lie in “negative”density.
The percentage of atoms in the model whose neighbors are less than 3.6 Å away. Neighbors exclude atoms from the same symmetry copy of the same protein chain (i.e., distances that are unaffected by rigid-body movement of the chain) but include all other contacts in the 60-fold symmetry-expanded model of the virus.
Backbone models of native poliovirus capsid proteins, including N, Cα, and C atoms, were used in the 135S refinement. The refinement was initiated with three movable rigid bodies: a model consisting of VP1, VP2, and VP3 β barrels (residues 71 to 302, 28 to 264, and 50 to 323, respectively) truncated to remove potentially flexible loops and N- and C-terminal extensions. In addition, the VP3 β tube, composed of five intertwining N-terminal residues (1 to 12), was docked in the density visually but was not refined. To improve the visual fit to the density, the program O (24) was used to adjust VP1 manually and to incorporate additional residues. Additional residues included stepwise in the refinement included the N terminus of VP3 (residues 14 to 49), the N-terminal hairpin of VP2 (residues 13 to 27), and a 10-residue helix (a portion of the N terminus of VP1). To test the robustness of the refinement procedure, the native (160S) protomer was refined independently into each of the 135S maps, while incorporating the VP1 transformation and hinge points discovered during the stepwise refinement.
Protein structure accession numbers.
The 135S model (PDB ID, 1XYR) and 135S reconstructions (Table 1) were deposited in the Protein Data Bank and Electron Microscopy Data Bank, respectively.
RESULTS
Cryo-EM reconstructions of 135S and V8-cleaved 135S.
The previously reported low-resolution reconstruction of the 135S particle demonstrated that the 135S particle is ∼4% larger than the native 160S virion (3), and the current reconstructions were size scaled to that standard. The general features of the new 135S map at 10-Å resolution are consistent with those of the previous reconstruction at 22 Å. The capsid forms a closed shell surrounding the viral RNA. The RNA shell also expands to meet the swollen capsid, making contacts at the base of the fivefold axis and at the quasithreefold axis. As noted above, the 135S particle has a star-shaped mesa at the fivefold axis surrounded by a canyon (Fig. 1B, 2, and 3A). The reconstruction shows that the 135S capsid thins at the canyon as well as at the saddle-like depression at the twofold axis. The apparent radius of the mesa is noticeably larger than that of the 160S virion, and there is a linear ridge, extending from each point of the mesa to the tip of the nearest propeller, that is not present in the native virus (Fig. 2). The shape of this density feature is suggestive of an α helix.
FIG. 1.

V8 protease cleaves the exposed N terminus of VP1 at the propeller tip of 135S. (A) Coomassie blue-stained sodium dodecyl sulfate-polyacrylamide gel whose lanes represent different stages in the preparation of V8-cleaved 135S. (Lane 1) The 135S particle before digestion showing the migration of the capsid proteins VP1, VP2, and VP3. (Lane 2) Blank. (Lane 3) The 135S particle after digestion with the V8 protease. Cleaved VP1 comigrates with VP2 (arrow) (14). (Lane 4) An aliquot from the void volume from a Sephacryl S-100 column demonstrating the homogeneity of the purified V8-135S particles. (Lane 5) An aliquot from the inclusion volume of the S-100 column (50 ml after the void volume peak) containing the V8 protease. (B to D) Surface renderings of poliovirus cell entry intermediate 135S and difference images (scale bar, 100 Å). Difference electron density maps between reconstructions were contoured at 2.5 σ, while both 135S and V8-cleaved 135S reconstructions were contoured at 0.75 σ (σ is the root-mean-square deviation of the electron density values). Figures were produced with DINO (http://www.dino3d.org). (B) A stereo representation of the 135S1 cryo-EM reconstruction (grey). Differences between the 135S2 and 135S1 reconstructions (green) were insignificant. The ridge between the mesa and propeller tip is labeled (arrowhead). (C) A reconstruction of the V8-proteolyzed 135S (grey) particle. A difference electron density map calculated between two V8-cleaved 135S reconstructions showed no visible differences on the outer surface. (D) Significant difference density features were seen when each V8-cleaved 135S map (grey) was subtracted from either unmodified 135S particle reconstruction. The largest differences (green) were located at the propeller tip.
FIG. 2.

Close-up view of the surface of the 135S reconstruction (left) and of native poliovirus (right) in the vicinity of the fivefold axis. Electron density for the native virus was calculated to 11-Å resolution from the atomic coordinates. In each panel, one propeller tip (red dot) and a nearby point of the fivefold mesa (blue dot) are indicated. In the 135S reconstruction a prominent linear ridge connects the mesa with the propeller and rises to a substantial elevation above the floor of the canyon. No such feature is observed in the native virus.
FIG. 3.

Pseudoatomic modeling of the 135S particle. (A) The structure of the 160S particle serves as a reference for prominent structural features. (Left panel) A radially depth-cued rendering of the atomic model (22) of the 160S particle. (Right panel) An expanded representation of a single protomer showing ribbon diagrams of VP1 (blue), VP2 (yellow), VP3 (red), and VP4 (green) overlaid on an icosahedral framework. The fivefold, threefold, and twofold axes are indicated by numbers. Cyan lines point to prominent surface features in the two panels. (B to E) Pseudoatomic models of VP1 (cyan), VP2 (yellow), and VP3 (red) resulting from rigid-body docking and refinement of native virus coordinates into the 135S density maps (black mesh). Residues outside the β-barrel cores that were not modeled previously (3) were included in the refinement (dark blue). Green diamonds signify low-density regions within the core of protein subunits. (B) A model-density overlay in the vicinity of a fivefold axis (pentagon). The RNA and protein shells make contacts (arrows) at the fivefold axis near the VP3 β tube (red star) and at the base of the canyon. The truncated N terminus of VP1 (residue 71) is indicated (*). (C) A model-density overlay in the vicinity of a threefold axis (inverted triangle). Loops at the narrow ends of the VP3 β barrel and the β barrels from symmetry-related VP2s and VP3s alternate around the threefold axis. The newly modeled VP2 β hairpin (dark blue) is seen to contact VP3. (D) Model-density overlay in the vicinity of the propeller tip (arrow) showing VP2, the GH loop and C terminus of VP1 (cyan), and the newly modeled VP2 β hairpin (dark blue) from panel C fitting into a crevice in the EM envelope. (E) A slice through the model-density overlay of a protomer. A helix occupying the external linear ridge, possibly from the residue range 41 to 53 of VP1, is shown in magenta. The newly modeled VP3 N terminus (residues 14 to 49, dark blue) can be seen within the density envelope on the underside of the capsid in panels B and E.
Difference maps between 135S and V8-cleaved 135S reconstructions.
We sought to localize the N-terminal extension of VP1 in the 135S intermediate by comparing it with a modified 135S particle in which the first 31 residues of VP1 were removed by the V8 protease (14). The proteolyzed 135S sample was separated from the enzyme and from released fragments by gel filtration (Fig. 1A) and then imaged by cryo-EM. The resulting three-dimensional reconstruction is shown in Fig. 1C.
The reconstructions to be compared had the same resolution and were statistically equivalent in terms of the numbers of contributing particles. Nevertheless, to assess the scale of residual noise fluctuations—which are pertinent to the interpretation of subtle features in cryo-EM difference maps—several control reconstructions were calculated. Two 135S samples were generated and processed separately. The difference map between these density maps showed no significant features (Fig. 1B). Similarly, three different V8-cleaved 135S reconstructions were computed from independent sample preparations, and again no significant differences were observed in pairwise difference maps (Fig. 1C). These experiments attest to the robustness of the sample preparation, imaging, and reconstruction processes.
Difference maps comparing all possible combinations of 135S and V8-cleaved 135S reconstructions were calculated. They consistently showed peaks at the tips of the threefold propeller and in the saddle-shaped depression at the twofold axes. The relative intensities of these two difference features in the various 135S/V8-cleaved 135S comparisons did vary somewhat, possibly due to slight differences in corrections for the CTF and scaling. The positive difference density on the outer surface of the particle in the vicinity of the twofold axis (not visible in Fig. 1D because its density level is below the chosen contour) is paired with an obvious negative difference feature on the inner surface. Furthermore, the thickness of the shell in this region is nearly identical in the 135S and V8-135S reconstructions (data not shown). Thus, both the difference maps and direct comparison of the two reconstructions are consistent with a small inward shift of VP2 in this region, upon V8 cleavage, rather than the excision of density by proteolysis. The connection between proteolysis and this small shift is not clear. In contrast, the obvious positive difference feature at the tip of the propeller in the 135S-135S(V8) difference map (Fig. 1D) is not paired with a negative density feature (data not shown). It coincides with a prominent bump in the reconstructions of the 135S particle that is absent in reconstructions of the V8-cleaved particle (Fig. 1), leading us to attribute this feature to a portion of the first 31 amino acids of VP1. The difference feature is large enough to accommodate only a few amino acids, indicating that most of the 31 residues are disordered.
Docking and refinement of atomic coordinates.
Atomic coordinates corresponding to main-chain models of the native poliovirus capsid proteins VP1, VP2, and VP3 were manually docked and rigid-body refined into the 10-Å EM density maps. The 135S model evolved through a succession of stages to achieve the lowest possible refinement residuals, which coincided with independent statistics (Table 2) that were not explicitly minimized in the refinement. The best statistical values also corresponded to the most satisfactory fits by visual inspection. The refined model consisted of β barrels corresponding to VP1, VP2, and VP3; several loops; and terminal extensions of the capsid proteins. In the final stages of refinement, a 10-residue helix was added to fill the linear ridge.
β barrels.
Overall, the model fits the density for the 135S reconstructions well. The VP1 and VP3 β barrels match the contours of the map as rigid bodies (Fig. 3B and C). Because the distance between β sheets is greater than 10 Å at the wide end of the β sandwiches of VP1, VP2, and VP3, each of the subunits is seen to have a clear low-density core. These features impose additional constraints that improved the accuracy and reliability of fitting the atomic model into the EM maps (Fig. 3).
VP3 plug.
In all 135S-related reconstructions, there are two distinct low-density regions, one larger than the other, at different radii along the fivefold axis. Between these two regions, there is a cylindrical piece of density that accommodates a β tube formed by intertwining of five copies of the extreme N terminus of VP3 (Fig. 3B and E). This feature is also present in the 160S virion (3, 22). To ensure that the chemically reasonable hydrogen-bonding pattern of the β tube would not be disrupted, the tube was positioned manually along the fivefold axis.
Additional residues.
Because of the greater information content of the new reconstructions, residues from most of the loops and terminal extensions of the native virus could be included in the model, in addition to the β barrels. The current 135S model accounts for 93% of the atomic coordinates of the major capsid proteins compared with 73% previously (3). The additional residues include an N-terminal β hairpin of VP2 (residues 13 to 27), the N terminus of VP3 (residues 14 to 49), the GH loop and C terminus of VP1 (residues 210 to 229 and 273 to 302, respectively), and a helical peptide (modeled as a portion of the N terminus of VP1, residues 42 to 52).
VP2 N-terminal hairpin.
In the native virus, the β hairpin at the N terminus of VP2 hydrogen bonds with a symmetry-related copy of VP3 from a neighboring pentamer (five copies of the protomer arranged about a fivefold axis, where a protomer is defined as the VP1, VP2, and VP3 subunits cleaved from a single polypeptide chain during assembly), adding two additional strands to the four-stranded β sheet containing the C, H, E, and F strands of VP3. In our earliest fitting into the 135S map, the β hairpin of VP2 was omitted. After the hairpin was added back to the model as part of VP2, the fit to the density was good, but not optimal. Separating the core of VP2 from the hairpin at residue 28, and allowing the hairpin to move as a rigid body with VP3, improved the statistics and produced a better visual fit to the density (Table 2; Fig. 3C and D). These modeling results suggest that the native-like hydrogen-bonding pattern of the extended β sheet is maintained during the 135S transition.
N terminus of VP3.
The higher-resolution 135S maps enabled the placement of a nearly complete N terminus of VP3. Initially, residues 14 to 49 were omitted from the model because their conformation in the native virus depends on their contacts with VP1. After the locations of VP1 and the VP3 β tube (residues 1 to 12) were optimized, we found that the VP3 N terminus (residues 14 to 49) could be modeled to adopt a native-like association with the VP1 β barrel, to occupy a vacant density feature on the inner surface of the capsid. Grouping residues 14 to 49 of VP3 together with VP1 in a single rigid body improved or maintained all refinement residuals (Table 2). This step optimized the fit of the model to the EM envelope on the underside of the capsid (Fig. 3B and E). After refinement, residue 14 of VP3 was located only 7.2 Å away from residue 12 of VP3, and residue 49 was 6.3 Å from residue 50. These distances correspond approximately to the length of two peptide bonds.
Residues corresponding to the propeller tip.
The latest model also included the C-terminal 30 residues and the GH loop (residues 210 to 230) of VP1, which lie on either side of the EF loops of VP2 (residues 132 to 177). Both the C terminus and the GH loop were treated as rigid bodies with the rest of VP1 in the refinement. The C terminus continues to lie across the surface of the capsid, contacting VP2 and VP3 at the “propeller tip” as in the native virus. The GH loop flanks the two large EF loops of VP2. The volume corresponding to density at the propeller tip is large enough to accommodate the GH loop of VP1 and EF loops of VP2, as well as residues corresponding to some part of the N terminus of VP1 (Fig. 3D and 4). Although the two large EF loops of VP2 extend beyond the density at the propeller tip, the observation that the low-density features in the capsid coincide with the hydrophobic center of the VP2 β barrel confirms correct placement of the β barrel (Fig. 3D). Efforts to refine the EF loops of VP2 treating them as independent rigid bodies failed to produce models that fit the density. Although a better fit might be obtained by relaxing rigid-body constraints, the resolution is not yet sufficient to warrant more detailed modeling.
FIG. 4.

Localization and characterization of the N terminus of VP1. (A) Two views of 135S model coordinates, VP1 (cyan), VP2 (yellow), and VP3 (red), are overlaid on the cryo-EM 135S reconstruction (grey semitransparent). Trajectory for the N-terminal 70 residues of VP1 (tan) begins at residue 71 (orange sphere), emerges from a gap between fivefold symmetry-related copies of VP1, forms a helix (magenta) through the ridge, and connects to the V8-cleavage site (green sphere). The helix originates at the points of the star-shaped mesa. For clarity, only one protomer is shown in each panel. (B) Sequence alignment of the N-terminal extension of VP1 from different picornaviruses (coxsackievirus B3, echovirus 1, poliovirus 1 Mahoney strain, poliovirus 1 Sabin strain, rhinovirus 14, rhinovirus 2, foot-and-mouth-disease virus 10, and mengovirus, top to bottom, respectively) reveals a highly conserved stretch of residues (41 to 53, mostly dark highlighting) that is predicted by the Chou-Fasman algorithm to include an alpha helix. The algorithm also identified the extreme N-terminal 20 residues as a helix. This amphipathic helix was previously identified, and it was proposed to form a pore in the host cell membrane (14). A third helical stretch, located near residue 70, is observed in the native virus making contacts with the VP1 β barrel. All predicted helices are depicted as grey cylinders and labeled with their respective residue numbers. The V8 cleavage site is labeled (*). (C) A helical wheel representation (generated using EMBOSS Pepwheel, www.hgmp.mrc.ac.uk/Software/EMBOSS/Apps/pepwheel.html) of VP1 residues 41 to 52 reveals an amphipathic arrangement of side chains. (D) Schematic of the externalization of the VP1 N terminus showing the capsid (blue), VP3 β tube (red), RNA (purple), and the N terminus of VP1 (residues 71 to 54 [cyan], 53 to 41 [magenta helix], V8 cleavage site [green sphere], and 31 to 1 [cyan helix]). The N-terminal 20 residues that are thought to form an amphipathic helix (14) could be either disordered or mobile in the absence of lipid (cyan helix).
Linear ridge.
Although the current model fits the basic contour of the 135S map, the truncated VP1, VP2, VP3, and VP3 β tube models do not account for the density corresponding to the linear ridge now seen on the outer surface (Fig. 4). This ridge, ∼15 Å in length, commences at the point of the fivefold mesa and stretches across the canyon to the propeller tip where the extreme N-terminal residues of VP1 are exposed (Fig. 2 and 4). We have modeled this ridge as a 10-residue helix. In the rigid-body fitting, VP1 was truncated at residue 71, an approximate dividing point between the core of VP1 and its N-terminal extension. Residue 71 is located directly below one end of the linear ridge. No other residues missing from the atomic model, except for the N terminus of VP1, are close enough to the ridge to explain it.
Robustness of refinement.
To test the robustness of the refinement, the procedure was carried out from scratch using the native (160S) coordinates while imposing the hinge points identified by the piecewise refinement. The resulting VP1 coordinates were then substituted for the docked VP1 from the original refinement, and further refined. This procedure was done in parallel for V8-cleaved and uncleaved 135S reconstructions. Atomic coordinates refined into the two uncleaved 135S reconstructions differed by a root-mean-square deviation (RMSD) of 0.6 Å, while those refined into the V8-cleaved 135S reconstructions differed by 0.9 Å. The results of this test were compared with the original piecewise refinement, yielding an RMSD range of 0.7 to 1.4 Å. Models refined into the 135S reconstructions differed from the native virus coordinates by RMSD values of 5.0 to 5.1Å, which are considerably greater than the differences among 135S models.
Native-to-135S transition.
Overall, the 160S-to-135S transition is characterized by an expansion of the capsid shell. This expansion can be described as both an “iris-like” twisting of the VP1 β barrels and an “umbrella-like” opening of the mesa. These movements result in both a broadening of the mesa and an opening at the interface between VP1 and VP3 at the base of the canyon (Fig. 5B; also see movie S1 in the supplemental material). Conformational changes at the fivefold axis coincide with changes at the twofold axis observed by a thinning of the capsid (Fig. 5A and B; also see movie S1 in the supplemental material) and a corresponding gap between pentamers in the pseudoatomic model. Model building into the 135S density maps revealed that native-like contacts were preserved (i) between the N-terminal hairpin of VP2 and the core of VP3 from a symmetry-related protomer (Fig. 3C) and (ii) between the N terminus of VP3 and the VP1 β barrel (Fig. 3E).
FIG. 5.

Comparison of 135S with native virus. The transition from native poliovirus (A) to the 135S intermediate (B) is characterized by an expansion of the virus capsid causing gaps in the model between symmetry-related copies of the protomer. The most pronounced openings occur between VP1 and VP3 at the base of the canyon (*) and at the twofold axis. VP1, VP2, and VP3 are cyan, yellow, and red, respectively. In the 135S model, the helix proposed to occupy the external linear ridge is magenta; in native virus, VP4 is green. The arrow indicates the six-stranded β sheet formed by the hairpin of VP2 and the four-stranded β sheet from VP3 of a different, symmetry-related pentamer. (C) Electron density calculated for Pvr (yellow) overlaid with density calculated for the pseudoatomic 135S model (grey) does not clash with the ridge corresponding to the externalized N terminus of VP1 (magenta) or with the V8 cleavage site (green).
DISCUSSION
A number of lines of evidence support the view that the 135S poliovirus particle is an infectious intermediate in the cell entry pathway. In the early stages of infection, 135S accumulates and is the major cell-associated form of the virus (16). Small molecules that bind to the capsid and inhibit 135S formation also inhibit infection (26). Isolated 135S alone can cause a receptor-independent infection in otherwise poliovirus-resistant cells such as CHO or murine L cells (11). To date, it has not been possible to solve the structure of the 135S particle by X-ray crystallography. In this study, we have extended the resolution of cryo-EM analysis to 10 Å, at which point finer details of the molecular envelope become distinct, including low-density regions that correspond to the hydrophobic cores of the capsid protein β barrels.
Receptor-mediated conversion of virions to 135S particles involves expansion of the capsid and externalization of internal components, specifically VP4 and the N terminus of VP1, which insert into membranes to form channels (12, 14, 32, 33). It still remains to be discovered how these components break free of the stabilizing network on the RNA-proximal side of the capsid and reach the surface of the virus to insert into membranes. In a widely accepted model, Rossmann and others have proposed that receptor binding induces changes that widen the channel at the fivefold axes and displace the VP3 plug, allowing VP4, the N terminus of VP1, and the viral RNA to exit the capsid at the particle fivefold axes (15, 18, 25). However, both our earlier and the present reconstructions of the 135S particle demonstrate that the VP3 plugs remain in place on the fivefold axes and that there is insufficient room for five copies of the N terminus of VP1 to exit at the fivefold axes. We and others have proposed an alternative model in which VP4 and the N terminus of VP1 exit via pores at the base of the canyon and the RNA subsequently exits via the channel at the fivefold axis (3, 13, 19, 34).
Several lines of evidence support this second model: Mutations in the interfaces between capsid protein subunits, particularly between VP1 and VP3, are important for entry (8) and viral stability (13). The expansion observed in the 135S transition may be similar to that seen in plant viruses such as tomato bushy stunt virus (28) and cowpea chlorotic mottle virus (31). In tomato bushy stunt virus, the N termini of A subunits, which are structurally analogous to VP1, are externalized through openings at a corresponding interface at the quasithreefold axes.
The current 135S reconstruction supports the second model and provides a more detailed picture of the externalized N terminus of VP1. There is direct evidence for the positions of both ends of the N-terminal polypeptide. Identification of the propeller tip as the site where the N terminus of VP1 emerges from the capsid shell was based on difference mapping with 135S particles trimmed of the N-terminal 31 residues of VP1 (Fig. 1). Residue 71, which corresponds to the point where the N-terminal extension joins the VP1 β barrel, is located on the inner surface of the capsid at the bottom of the canyon, near the quasithreefold axis, where there is a thinning of the density in the reconstruction and a noticeable gap between subunits in the model. The connection between residues 31 and 71 appears to be well ordered and includes a linear ridge connecting the propeller tip with the fivefold mesa (Fig. 4), which passes above residue 71 in the pseudoatomic model. The linear ridge is the most striking difference between the outer surface of the native virus and the 135S particle (Fig. 2), and therefore it is likely to correspond to a portion of the virus that is externalized during the 135S transition. As all long stretches of amino acids have been accounted for in the model except for the N terminus of VP1, it represents the most likely candidate to fill the ridge.
The linear ridge could be modeled as an alpha helix. In fact the inclusion of a 10-residue helical peptide as an additional rigid body late in refinement improved the fitting statistics of the model, while not increasing the number of unfavorable nonbonded contacts (Table 2). In an effort to define regions of the N-terminal extension that might be helical and occupy the linear ridge, residues 1 to 75 were characterized using a panel of secondary structure prediction tools (Fig. 4B). All of them identified residues 1 to 23 as having strong helical potential. Most of the algorithms also identified residues in the ranges 41 to 53 and 63 to 69 as possible helices. (The decision to model the ridge as 10 residues, based on the appearance of the density, was made prior to and independently of the secondary structure predictions.) Residues 63 to 69 form a helix in the native virus, but its connection to the VP1 β barrel is much too short to allow it to reach the linear ridge (22). Residues 1 to 23 can also be excluded because their removal by V8 cleavage leaves the linear ridge intact (Fig. 1C) and because V8-cleaved 135S cannot be precipitated with antibodies against residues 7 to 24 (14). This suggests that the ridge is most likely to be occupied by some or all of residues 41 to 53 of VP1. Both the polypeptide segments 1 to 23 and 41 to 53 are predicted to include amphipathic helices, an attribute that is conserved among picornaviruses with similar cell entry mechanisms (14) (Fig. 4C). The sequence of residues 41 to 53 is much better conserved than that of the N-terminal helix (Fig. 4B), but that conservation could be related to their role in the native virion.
Once the N terminus of VP1 has been externalized, it is unclear from the biochemical evidence whether the receptor (Pvr) plays a continuing role in helping to anchor 135S particles to the host cell. Even a low-affinity binding to the receptor would be amplified by multiple binding and would enhance the efficiency of membrane association. Human rhinovirus 3, a related virus, continues to bind its receptor, ICAM-1, after the transitions to 135S and 80S empty particles (34). In poliovirus, the 135S transition reduces the affinity for Pvr (11) but may not eliminate it. To address this possibility, we looked at the surface of the 135S reconstruction to see if the altered structure, including the 10-residue helix, would preclude continued binding of Pvr. Residues on the native poliovirus surface located within the footprint of Pvr (as identified previously [5]) were superimposed on corresponding α carbons in the 135S model using LSQKAB (7). When the resulting least squares transformation was applied to the Pvr coordinates from the pseudoatomic model (5), there were no obvious clashes with the 135S surface (Fig. 5C). Slight changes in the footprint on the virus may account for the reduced affinity of the 135S particle for Pvr. However, the lack of gross steric clashes, particularly with the linear ridge and V8 cleavage site, allows for the possibility that the receptor remains attached to the virus while the N terminus of VP1 inserts into the membrane (Fig. 6A).
FIG. 6.

Working models for poliovirus entry. A cross section of a portion of the capsid is shown in dark blue, VP4 is green, and the N terminus of VP1 is cyan and magenta. The V8 cleavage site is shown as a green circle. (A) Native poliovirus binds its receptor, Pvr (ectodomains 1 to 3, tan; transmembrane domain, black helix), and at physiological temperature undergoes an irreversible change to the 135S particle. The path of egress of the N terminus of VP1, suggested by the present 135S reconstruction, would not preclude the continued binding of Pvr. At this stage, the VP3 β tube (red) blocks an otherwise open channel along the fivefold axis. (B to D) Alternative models for the direct anchoring of the virus to the membrane via the N terminus of VP1 and formation of a transmembrane pore for RNA translocation. To accommodate the passage of RNA (purple), the VP3 β tube has shifted, and the channel has expanded, becoming continuous with a pore through the membrane. The V8 cleavage site remains accessible. (B) Amphipathic helices at the N terminus of VP1 (cyan) may form a five-helix bundle close to the fivefold axis, which would require the magenta helix to dissociate from the body of the virus. Alternatively, VP4 may play a more central role in pore formation (C and D). In that case, VP1 may participate directly in forming the pore (D) or serve as a nonspecific membrane anchor (C).
Finally, the current data on the 135S particle provide new insights into our working model for poliovirus entry (Fig. 6). In previous models (21) the N terminus of VP1 was located at or near the fivefold axes of the altered particle and five copies of the amphipathic helix were envisaged to insert into the host cell membrane to form a five-helix channel (5, 15, 21, 25). In that scenario, VP4 was assigned a passive role, perhaps in assisting the insertion of the VP1 amphipathic helix. The membrane-embedded pore abutted a channel in the capsid connecting the inside of the virus with the cytoplasm. Expansion of both of these channels together with the displacement of the VP3 plug would allow RNA to be translocated across the bilayer.
The observations reported here—that the N-terminal 31 residues of VP1 are docked at the tip of the propeller, far from the fivefold axes, and are linked to the 135S particle by a second amphipathic helix—suggest alterations to the prior model. The most conservative modification retains the idea that the N-terminal helix inserts directly in the membrane to make a channel at the fivefold axis. This would require that the amphipathic helix in the linear ridge be dissociated from its location in the membrane-free 135S particle and become associated with the mesa (Fig. 6B).
Recent data show that mutations in residue 28 of VP4 significantly alter conductance properties of channels and the ability of the virus to translocate its RNA (12). This finding, together with the lack of sequence conservation in the extreme N terminus of VP1 (Fig. 4A) (it is the least conserved region in the capsid protein among poliovirus serotypes while VP4 is well conserved), raises concerns about previous proposals and suggests two alternative models in which VP4 plays a more central role in forming the RNA translocation pore while VP1 either acts as a membrane anchor (Fig. 6C) or acts in concert with VP4 in forming the pore (Fig. 6D). In either case, Glu-31 of VP1 is likely to remain in the periphery of the pore complex and accessible to enzymes since V8 protease can cleave VP1-anchored 135S particles off the membrane (14).
The higher-resolution structure of the 135S particle, in combination with recent biochemical evidence, suggests testable alternative hypotheses for the mechanism of RNA translocation. This will provide a framework for experimental design and improve our understanding of the cell entry mechanisms of nonenveloped viruses.
Supplementary Material
Acknowledgments
We thank Simon Tsang and Lily Lee for help with sample preparation and James Conway, Bernard Heymann, and Marie Chow for helpful discussions. We thank Thomas Walz for access to instruments in the HMS molecular electron microscopy laboratory, established by funds from the Giovanni Armenise-Harvard Foundation and maintained by NIH grant P01-GM62580 (to David DeRosier), and we thank Yifan Cheng for instruction and assistance in their use.
This work was supported by NIH grants AI20566 (to J.M.H.) and an NSF predoctoral fellowship (to D. Bubeck).
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1.Baker, T. S., and R. H. Cheng. 1996. A model-based approach for determining orientations of biological macromolecules imaged by cryoelectron microscopy. J. Struct. Biol. 116:120-130. [DOI] [PubMed] [Google Scholar]
- 2.Basavappa, R., R. Syed, O. Flore, J. P. Icenogle, D. J. Filman, and J. M. Hogle. 1994. Role and mechanism of the maturation cleavage of VP0 in poliovirus assembly: structure of the empty capsid assembly intermediate at 2.9Å resolution. Protein Sci. 3:1651-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belnap, D. M., D. J. Filman, B. L. Trus, N. Cheng, F. P. Booy, J. F. Conway, S. Curry, C. N. Hiremath, S. K. Tsang, A. C. Steven, and J. M. Hogle. 2000. Molecular tectonic model of virus structural transitions: the putative cell entry states of poliovirus. J. Virol. 74:1342-1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Belnap, D. M., W. D. Grochulski, N. H. Olson, and T. S. Baker. 1993. Use of radial density plots to calibrate image magnification for frozen-hydrated specimens. Ultramicroscopy 48:347-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Belnap, D. M., B. M. McDermott, Jr., D. J. Filman, N. Cheng, B. L. Trus, H. J. Zuccola, V. R. Racaniello, J. M. Hogle, and A. C. Steven. 2000. Three-dimensional structure of poliovirus receptor bound to poliovirus. Proc. Natl. Acad. Sci. USA 97:73-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng, N., J. F. Conway, N. R. Watts, J. F. Hainfeld, V. Joshi, R. D. Powell, S. J. Stahl, P. E. Wingfield, and A. C. Steven. 1999. Tetrairidium, a four-atom cluster, is readily visible as a density label in three-dimensional cryo-EM maps of proteins at 10-25Å resolution. J. Struct. Biol. 127:169-176. [DOI] [PubMed] [Google Scholar]
- 7.Collaborative Computational Project. 1994. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D 50:760-763. [DOI] [PubMed] [Google Scholar]
- 8.Colston, E., and V. R. Racaniello. 1994. Soluble receptor-resistant poliovirus mutants identify surface and internal capsid residues that control interaction with the cell receptor. EMBO J. 13:5855-5862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conway, J. F., and A. C. Steven. 1999. Methods for reconstructing density maps of “single” particles from cryoelectron micrographs to subnanometer resolution. J. Struct. Biol. 128:106-118. [DOI] [PubMed] [Google Scholar]
- 10.Crowther, R. A., L. A. Amos, J. T. Finch, D. J. De Rosier, and A. Klug. 1970. Three dimensional reconstructions of spherical viruses by fourier synthesis from electron micrographs. Nature 226:421-425. [DOI] [PubMed] [Google Scholar]
- 11.Curry, S., M. Chow, and J. M. Hogle. 1996. The poliovirus 135S particle is infectious. J. Virol. 70:7125-7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Danthi, P., M. Tosteson, Q. H. Li, and M. Chow. 2003. Genome delivery and ion channel properties are altered in VP4 mutants of poliovirus. J. Virol. 77:5266-5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Filman, D. J., R. Syed, M. Chow, A. J. Macadam, P. D. Minor, and J. M. Hogle. 1989. Structural factors that control conformational transitions and serotype specificity in type 3 poliovirus. EMBO J. 8:1567-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fricks, C. E., and J. M. Hogle. 1990. Cell-induced conformational change of poliovirus: externalization of the amino terminus of VP1 is responsible for liposome binding. J. Virol. 64:1934-1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Giranda, V. L., B. A. Heinz, M. A. Oliveira, I. Minor, K. H. Kim, P. R. Kolatkar, M. G. Rossmann, and R. R. Rueckert. 1992. Acid-induced structural changes in human rhinovirus 14: possible role in uncoating. Proc. Natl. Acad. Sci. USA 89:10213-10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gromeier, M., and K. Wetz. 1990. Kinetics of poliovirus uncoating in HeLa cells in a nonacidic environment. J. Virol. 64:3590-3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He, Y., V. D. Bowman, S. Mueller, C. M. Bator, J. Bella, X. Peng, T. S. Baker, E. Wimmer, R. J. Kuhn, and M. G. Rossmann. 2000. Interaction of the poliovirus receptor with poliovirus. Proc. Natl. Acad. Sci. USA 97:79-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hewat, E. A., and D. Blaas. 2004. Cryoelectron microscopy analysis of the structural changes associated with human rhinovirus type 14 uncoating. J. Virol. 78:2935-2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hewat, E. A., E. Neumann, and D. Blaas. 2002. The concerted conformational changes during human rhinovirus 2 uncoating. Mol. Cell 10:317-326. [DOI] [PubMed] [Google Scholar]
- 20.Heymann, J. B. 2001. Bsoft: image and molecular processing in electron microscopy. J. Struct. Biol. 133:156-169. [DOI] [PubMed] [Google Scholar]
- 21.Hogle, J. M. 2002. Poliovirus cell entry: common structural themes in viral cell entry pathways. Annu. Rev. Microbiol. 56:677-702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hogle, J. M., M. Chow, and D. J. Filman. 1985. Three-dimensional structure of poliovirus at 2.9 A resolution. Science 229:1358-1365. [DOI] [PubMed] [Google Scholar]
- 23.Jacobson, D. H., J. M. Hogle, and D. J. Filman. 1996. A pseudo-cell based approach to efficient crystallographic refinement of viruses. Acta Crystallogr. D 52:693-711. [DOI] [PubMed] [Google Scholar]
- 24.Jones, T. A. 1978. A graphics model building and refinement system for macromolecules. J. Appl. Crystallogr. 11:268-272. [Google Scholar]
- 25.Kolatkar, P. R., J. Bella, N. H. Olson, C. M. Bator, T. S. Baker, and M. G. Rossmann. 1999. Structural studies of two rhinovirus serotypes complexed with fragments of their cellular receptor. EMBO J. 18:6249-6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McSharry, J. J., L. A. Caliguiri, and H. J. Eggers. 1979. Inhibition of uncoating of poliovirus by arildone, a new antiviral drug. Virology 97:307-315. [DOI] [PubMed] [Google Scholar]
- 27.Miyazawa, A., Y. Fujiyoshi, M. Stowell, and N. Unwin. 1999. Nicotinic acetylcholine receptor at 4.6 Å resolution: transverse tunnels in the channel wall. J. Mol. Biol. 288:765-786. [DOI] [PubMed] [Google Scholar]
- 28.Robinson, I. K., and S. C. Harrison. 1982. Structure of the expanded state of tomato bushy stunt virus. Nature 297:563-568. [Google Scholar]
- 29.Rossmann, M. G. 2000. Fitting atomic models into electron-microscopy maps. Acta Crystallogr. D 56:1341-1349. [DOI] [PubMed] [Google Scholar]
- 30.Saxton, W. O., and W. Baumeister. 1982. The correlation averaging of regularly arranged bacterial cell envelope protein. J. Microsc. 127:127-138. [DOI] [PubMed] [Google Scholar]
- 31.Speir, J. A., S. Munshi, G. Wang, T. S. Baker, and J. E. Johnson. 1995. Structures of the native and swollen forms of cowpea chlorotic mottle virus determined by X-ray crystallography and cryo-electron microscopy. Structure 3:63-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tosteson, M., H. Wang, A. Naumov, and M. Chow. 2004. Poliovirus binding to its receptor in lipid bilayers results in particle-specific, temperature-sensitive channels. J. Gen. Virol. 86:1581-1589. [DOI] [PubMed] [Google Scholar]
- 33.Tosteson, M. T., and M. Chow. 1997. Characterization of the ion channels formed by poliovirus in planar lipid membranes. J. Virol. 71:507-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xing, L., J. M. Casasnovas, and R. H. Cheng. 2003. Structural analysis of human rhinovirus complexed with ICAM-1 reveals the dynamics of receptor-mediated virus uncoating. J. Virol. 77:6101-6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xing, L., K. Tjarnlund, B. Lindqvist, G. G. Kaplan, D. Feigelstock, R. H. Cheng, and J. M. Casasnovas. 2000. Distinct cellular receptor interactions in poliovirus and rhinoviruses. EMBO J. 19:1207-1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeates, T. O., D. H. Jacobson, A. Martin, C. Wychowski, M. Girard, D. J. Filman, and J. M. Hogle. 1991. Three-dimensional structure of a mouse-adapted type 2/type 1 poliovirus chimera. EMBO J. 10:2331-2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.