

Abstract
As a viral opportunistic pathogen associated with serious disease among the immunocompromised and congenital defects in newborns, human cytomegalovirus (HCMV) must engage the translational machinery within its host cell to synthesize the viral proteins required for its productive growth. However, unlike many viruses, HCMV does not suppress the translation of host polypeptides. Here, we examine how HCMV regulates the cellular cap recognition complex eIF4F, a critical component of the cellular translation initiation apparatus that recruits the 40S ribosome to the 5′ end of the mRNA. This study establishes that the cap binding protein eIF4E, together with the translational repressor 4E-BP1, are both phosphorylated early in the productive viral growth cycle and that the activity of the cellular eIF4E kinase, mnk, is critical for efficient viral replication. Furthermore, HCMV replication also induces an increase in the overall abundance of eIF4F components and promotes assembly of eIF4F complexes. Notably, increasing the abundance of select eIF4F core components and associated factors alters the ratio of active eIF4F complexes in relation to the 4E-BP1 translational repressor, illustrating a new strategy through which members of the herpesvirus family enhance eIF4F activity during their replicative cycle.
Although innocuous in most healthy individuals, human cytomegalovirus (HCMV) is a widespread opportunistic pathogen responsible for severe disease among the immunocompromised, including bone marrow and solid organ transplant recipients and AIDS patients. In addition, congenital HCMV infection is the leading viral cause of birth defects in newborns (reviewed in references 22 and 24). Finally, it has been suggested, although not proven that HCMV infection might play a role in the pathogenesis of vascular disease, notably atherosclerosis and restenosis (21, 40). A member of the betaherpesvirus subfamily, HCMV establishes a latent infection characterized by a restricted gene expression program in myeloid progenitor cells for the duration of the host's life (6, 12, 32, 33). Periodically, in response to a variety of cues, the virus reemerges from this latent state and replicates productively, expressing many of its >200 genes and ultimately killing the host cell. To achieve this end, HCMV must successfully engage the host cells' translational machinery.
Like all viruses, HCMV is completely dependent upon the translational machinery in the host cell to synthesize the viral proteins necessary for its productive growth. While it does not completely suppress the synthesis of host polypeptides in infected cells (34), it must have a mechanism to ensure that its mRNAs, most of which are capped on their 5′ ends, can compete effectively with host mRNAs for translation initiation factors and ribosomes. Recognition of the 5′ cap structure and recruitment of the 40S ribosome to the mRNA requires the activity of eIF4F, a critical translation initiation factor and a prime target for regulating translation initiation (31).
eIF4F is a complex of cellular polypeptides (2) whose core is composed of a cap binding protein (eIF4E), a large scaffolding subunit (eIF4G), and an RNA helicase (eIF4A). Depending upon their abundance, a family of small eIF4E binding proteins (4E-BPs) can sequester eIF4E in a phosphorylation-dependent manner (3). In their hypophosphorylated form, 4E-BPs inhibit cap-dependent translation. Hyperphosphorylation of the 4E-BPs by the mammalian target of rapamycin (mTOR) kinase results in the release of eIF4E, allowing the cap binding protein to associate with eIF4G and assemble an active eIF4F complex. In addition, the cap binding protein eIF4E is phosphorylated by the eIF4G associated kinase mnk (25, 30, 37). While not essential for translation initiation, eIF4E phosphorylation is thought to play a regulatory role, although the details of how this is achieved and which mRNAs are most susceptible are poorly understood (29). Finally, the cellular poly(A) binding protein (PABP), while itself not a core component of eIF4F, physically interacts with eIF4G in the eIF4F initiation complex, bridging the 5′ and 3′ ends of the mRNA and generating a circular topology which possibly serves as a checkpoint to ensure the mRNA is both capped and polyadenylated prior to translation initiation (7, 10, 38).
Recently, we established that a related human alphaherpesvirus, herpes simplex virus type 1 (HSV-1) can activate the translational machinery in resting, primary human cells through the action of a viral gene product and that this is critical for productive viral growth (36). Despite numerous global similarities between the productive growth cycles of alpha- and betaherpesviruses, considerable differences exist. Notably, in addition to its significantly protracted life cycle compared to HSV-1, host polypeptide synthesis is not suppressed in HCMV-infected cells, whereas it is strongly inhibited in HSV-1-infected cells (34; reviewed in reference 27). Our understanding of how HCMV interacts with the translational machinery of the host and how this influences productive viral growth, however, remains relatively underexplored. Here, we examine how HCMV engages the cellular cap recognition complex, eIF4F, and establish that eIF4E is phosphorylated early in the productive viral growth cycle; moreover, the activity of the cellular eIF4E kinase mnk is critical for viral replication. The translational repressor 4E-BP1 is also phosphorylated in HCMV-infected cells and released from eIF4E. This is followed by an increase in the overall abundance of eIF4E, eIF4G, and PABP, along with enhanced assembly of eIF4F complexes. This is the first demonstration that viral infection can induce the accumulation of eIF4F core and associated components; furthermore, it illustrates how different members of the herpesvirus family use discrete strategies to enhance eIF4F activity during their replicative cycle.
MATERIALS AND METHODS
Cells, viruses, antibodies, and chemicals.
Primary normal human diploid fibroblasts (NHDFs; Clonetics, Walkersville, MD), were cultured in Dulbecco's modified Eagle medium (DMEM) containing 5% fetal bovine serum (FBS) and growth arrested by being serum starved in 0.2% serum for 72 h as previously described (36). Under these conditions, >98% of the cells were in the G1 phase of the cell cycle, as revealed by propidium iodide staining followed by flow cytometry (36). The AD169 strain of HCMV was obtained from the American Type Culture Collection and propagated according to standard protocols. Cell-free virus was diluted from concentrated stocks such that a final serum concentration of 0.2% was maintained at all times during infections. Mock lysates were prepared in an identical manner from uninfected cells. Sucrose density gradient-purified HCMV strain AD169 was purchased from ABI, Inc. (Columbia, Maryland) and used where indicated. UV-inactivated virus was prepared as previously described (36). Antibodies and chemicals were as described in reference 36 or were purchased from the following vendors: anti-pp28 (ABI, Inc.), anti-IE1/2 (Mab810; Chemicon, Int.), anti-PABP (ABCAM), anti-pp65 (Virusys), anti-actin (Oncogene, Inc.), and ganciclovir (GCV; Sigma).
Immunoblotting, isoelectric focusing, and analysis of eIF4E binding proteins by batch chromatography on 7-methyl GTP-Sepharose.
These procedures were essentially performed as previously described (36) with the following minor modifications. NLB buffer containing 50 mM HEPES (pH 7.5), 100 mM NaCl, 1.5 mM MgCl2, 2 mM EDTA, 2 mM Na3VO4, 25 mM glycerophosphate, 0.25% NP-40, and complete mini protease inhibitor cocktail (Roche) was used to lyse cells prior to 7-methyl GTP-Sepharose chromatography. To quantify the signal on immunoblots, chemiluminescent images were captured and analyzed with a Bio-Rad Chemidoc XR5 system.
Multicycle growth experiments.
NHDF cells (8 ×105 cells/dish) were seeded, serum starved, and treated with either dimethyl sulfoxide (DMSO) or CGP5730 as previously described (36). At 7 days postinfection with HCMV (multiplicity of infection [MOI] = 0.05), cell-free lysates were prepared by freeze-thawing, and the amount of virus recovered was quantified by plaque assay on NHDF cells.
Analysis of protein synthesis in HCMV-infected cells.
NHDFs were seeded in 35-mm dishes (3 × 105 cells/dish) in DMEM plus 10% FBS. After attaching overnight, the cells were growth arrested by serum starvation for 72 h in DMEM plus 0.2% FBS as described in reference 36. Cells were either mock infected or infected with HCMV (AD169) at an MOI of 5 for 1 h at 37°C in DMEM plus 0.2% FBS. The cells were subsequently refed with DMEM plus 0.2% FBS. One hour prior to the indicated time points, the infected cells were overlaid with DMEM lacking methionine and cysteine but containing 50 to 70 μCi/ml 35S Express (a commercial mixture of 35S-labeled methionine and cysteine from Perkin-Elmer), and the incubation continued for 1 h at 37°C. A master stock of labeling mixture was prepared in advance from which aliquots were removed for each time point and stored frozen at −80°C until needed to ensure that the specific activity of the mixture used for each time point would be identical. Total cellular protein was solubilized in 1× Laemli sample buffer and boiled for 3 min, and a portion was fractionated in a 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Following electrophoresis, the gel was fixed in 25% methanol-10% acetic acid, dried, and exposed to Kodak XAR film.
RESULTS
Enhanced phosphorylation of eIF4E in HCMV-infected cells.
To determine the effect of HCMV infection on eIF4E phosphorylation, lysates from NHDFs growth arrested by serum starvation were fractionated by isoelectric focusing and analyzed by immunoblotting. An increase in the ratio of phosphorylated eIF4E, relative to the faster-migrating unphosphorylated species, was evident as early as 1 h postinfection and was maintained through day 7 (Fig. 1A and data not shown). Although the cellular eIF4E kinase mnk was stimulated by signals transmitted through p38 and extracellular signal-regulated kinases 1 and 2 (ERK1/2) and p38 and ERK1/2 have been reported to be activated in HCMV-infected cells, we felt it was important to evaluate their activation state in our experimental system, as their activation kinetics have not been directly compared in a single study, nor have they been examined with respect to eIF4E phosphorylation (8, 26). The increase in eIF4E phosphorylation correlated with sequential activation of ERK1/2, peaking from 1 to 4 h postinfection, followed by activation of p38 in a tighter peak from 4 to 8 h postinfection (Fig. 1A). Although reduced from their maximum points, activated ERK1/2 and p38 levels greater than those seen with mock-infected cells were detectable in cells as late as 7 days postinfection, while the total abundance of ERK1/2 with p38 remained relatively constant throughout the complete course of the experiment (Fig. 1A). The pattern of cellular and viral polypeptides synthesized during this 7-day period appears in Fig. 1C. Consistent with prior reports (34), the overall pattern of proteins produced was altered following HCMV infection but host protein synthesis was not completely impaired.
FIG. 1.

Activation of mnk by both ERK and p38 is required for eIF4E phosphorylation in HCMV-infected cells. Growth-arrested, serum-starved NHDFs were mock infected (0-h time point) or infected with HCMV AD160 (MOI = 5), and total protein was harvested at the indicated times postinfection. (A) To analyze eIF4E phosphorylation, lysates were fractionated by isoelectric focusing, transferred to a membrane support, and immunoblotted with antisera directed against eIF4E. Chemiluminescent images were captured and analyzed with a Bio-Rad Chemidoc XR5 system to quantify the percentage of phosphorylated eIF4E relative to the total amount of eIF4E present at each time point (% P-4E). To evaluate the abundance and phosphorylation state of ERK1/2 and p38, lysates were fractionated by SDS-PAGE and analyzed by immunoblotting with antisera specific for total ERK1/2, total p38, or their phosphorylated forms [(P) ERK or (P)p38]. The portion of the blot probed with an anti-actin antibody served as a loading control. (B) To assess the impact of inhibiting ERK activation, p38, or mnk on eIF4E phosphorylation in HCMV-infected cells, cultures were treated with either DMSO, PD98059, SB203580, or CGP57380, and eIF4E phosphorylation was analyzed as described above. (C) At the indicated time points, cultures of mock (0-h) or HCMV-infected (MOI = 5) growth-arrested, serum-starved NHDFs were radiolabeled with 35S-labeled methionine and cysteine for 1 h. Total protein was isolated, fractionated by SDS-PAGE, and visualized by autoradiography.
The role of the cellular kinases ERK1/2 and p38 in activating the cellular eIF4E kinase mnk was established through the use of pharmacological inhibitors specific for each kinase. While the mnk inhibitor CGP57380 (11) effectively blocked eIF4E phosphorylation, PD98059, which prevents ERK activation, and the p38 inhibitor SB203580 were unable to completely prevent eIF4E phosphorylation individually. They were, however, effective when combined, demonstrating that ERK1/2 and p38 both participate in activating mnk to phosphorylate eIF4E in HCMV-infected cells (Fig. 1B).
Accumulation of eIF4F core components and associated factors in quiescent cells infected with HCMV.
Significantly, the overall amount of activated ERK1/2 and p38 kinase was reduced by 4 days postinfection. Lower levels of these activated kinases could potentially account for the observed reduction in the ratio of phosphorylated eIF4E to unphosphorylated eIF4E observed from 72 to 120 h postinfection relative to the greater ratios observed at earlier times. Alternatively, a change in the overall abundance of eIF4E in HCMV-infected cells might also contribute to the ratio of phosphorylated to unphosphorylated eIF4E. To determine if this change in the distribution of phosphorylated eIF4E isoforms resulted solely from the observed alterations to ERK and p38 or also involved changes in the total amount of eIF4E present in HCMV-infected cells, the overall steady-state levels of eIF4F core and associated factors were evaluated by immunoblotting. Figure 2 clearly demonstrates that the abundance of eIF4E began increasing somewhere between 24 and 48 h postinfection relative to actin, a control antigen whose abundance is unaffected by HCMV infection. Overall levels of eIF4E increased between four- to fivefold by 96 h postinfection. Moreover, during this same time frame, the levels of the translational repressor 4E-BP1 appeared to decrease by a factor of twofold from 12 to 72 h postinfection before recovering somewhat by 96 to 120 h postinfection. Although small in magnitude, it is likely that this reduction in 4E-BP1 abundance is significant, as it becomes more pronounced at an MOI of 10, where a four- to sixfold reduction in 4E-BP1 levels was observed. In addition, the proteasome inhibitor MG132 prevented this decline of 4E-BP1 abundance (data not shown). Examination of other eIF4F core components revealed that eIF4G levels increased by four- to fivefold at 96 h postinfection, whereas eIF4A abundance only increased by a factor of 2 to 2.5. Finally, levels of PABP, an eIF4G-associated protein that is thought to mediate an interaction between the 5′ and 3′ ends of the mRNA during translation, also increased six- to eightfold. Thus, HCMV induces a substantial increase in eIF4E, eIF4G, and PABP late in the productive growth cycle, increasing the abundance of these components relative to the translational repressor 4E-BP1. In contrast, continued incubation of mock-infected cells in reduced serum resulted in a substantial reduction in the overall abundance of eIF4E, eIF4G, eIF4A, and PABP relative to actin, a control antigen whose abundance remained constant (data not shown). This most likely reflects the continued progression of these growth-arrested, serum-starved cells into a deeper quiescent state.
FIG. 2.

Increased abundance of eIF4G, eIF4E, and PABP in HCMV-infected cells. NHDFs that were growth arrested by serum starvation were mock infected (0-h time point) or infected with HCMV AD169 (MOI = 5), and total protein was harvested at the indicated times postinfection. Lysates were fractionated by SDS-PAGE and analyzed by immunoblotting with the indicated antisera. The illustration at the top of the panel depicts the eIF4F complex and associated factors bound to the mRNA. Upon binding the 7-methyl guanine cap (7m) at the 5′ end of the mRNA, eIF4E (4E) recruits eIF4G and eIF4A to assemble the eIF4F complex. The 40S ribosome is recruited through its association with eIF3. eIF4G also interacts with the poly(A) binding protein (PABP) bound to the 3′ end of the mRNA and the eIF4E kinase mnk-1, which phosphorylates eIF4E (depicted as a “P”).
Phosphorylation of the translational repressor 4E-BP1 in HCMV-infected cells is insensitive to rapamycin.
Depending on its abundance relative to the cap binding protein eIF4E, the translational repressor 4E-BP1 can bind to eIF4E and regulate its availability (3). However, only the hypophosphorylated form of 4E-BP1 is able to bind and subsequently sequester eIF4E. To assess the level of phosphorylated 4E-BP1, lysates from HCMV-infected cells were resolved by SDS-PAGE with 17.5% gels and visualized by immunoblotting. Following infection of growth-arrested NHDFs with HCMV, two waves of 4E-BP1 modification were apparent over the first 96 h of infection, as shown by an altered distribution of immunoreactive bands over time (Fig. 3A). The first of these waves occurred between 3 and 4 h postinfection, during which time the hypophosphorylated, faster-migrating forms of 4E-BP1 accumulated. Slower-migrating, hyperphosphorylated forms of 4E-BP1 began to increase at 7 h postinfection and were the predominate form through 96 h. By 72 h, hypophosphorylated forms were again observed, but were by no means as abundant as the hyperphosphorylated forms. Importantly, in all cases, 4E-BP1 phosphorylation in HCMV-infected cells was only partially sensitive to the mTOR inhibitor rapamycin, whereas the induction of 4E-BP1 phosphorylation in response to serum stimulation was completely sensitive to rapamycin (Fig. 3B). Thus, it is possible that the properties of mTOR, the cellular 4E-BP1 kinase, are altered in HCMV-infected cells or that 4E-BP1 phosphorylation is achieved through a different pathway.
FIG. 3.

Phosphorylation of the 4E-BP1 translational repressor in HCMV-infected cells is partially resistant to rapamycin. (A) Quiescent NHDFs were mock infected (0-h time point) or infected with HCMV AD169 (MOI = 5), and total protein was harvested at the indicated times postinfection. Lysates were fractionated by SDS-PAGE and analyzed by immunoblotting with antisera specific for 4E-BP1. (B) Rapamycin, an mTOR inhibitor, was added 1 h prior to infection where indicated. As a control to demonstrate that rapamycin effectively prevents 4E-BP1 phosphorylation, quiescent cells were stimulated with serum in the presence (+) and absence (−) of rapamycin prior to harvesting.
Recruitment of eIF4G and PABP into eIF4F complexes is enhanced in quiescent cells infected with HCMV.
As a single surface on eIF4E is responsible for either engaging 4E-BP1 and establishing a repressive complex or binding to eIF4G and assembling an eIF4F complex (5, 18, 19), the protein composition of eIF4E-containing complexes in HCMV-infected cells was examined by batch chromatography on 7-methyl GTP-Sepharose, followed by immunoblotting. In growth-arrested, serum-starved cells harvested at the 0-h time point, the amount of PABP and eIF4G associated with eIF4E was barely detectable. A clear increase in levels of PABP, together with eIF4G found associated with eIF4E, was observed by 24 h postinfection, while the abundance of 4E-BP1 bound to eIF4E was markedly reduced (Fig. 4A). After the submission of the manuscript, similar findings regarding the increase in eIF4G bound to eIF4E, along with the elevated phosphorylation of 4E-BP1 within the first 24 h of HCMV infection, were published (14). Significantly, eIF4E levels remained constant over this initial 24-h period (Fig. 4A), suggesting that the release of 4E-BP1 from eIF4E was accompanied by the concomitant recruitment of eIF4G and PABP into an eIF4E-containing complex. Furthermore, the increase in abundance of eIF4G and PABP associated with eIFE over the 96-h time course clearly exceeded any increase in eIF4E observed over the same period. Importantly, levels of 4E-BP1 bound to eIF4E rose between 72 and 96 h postinfection (Fig. 4A), reflecting the reappearance of hypophosphorylated forms of 4E-BP1 (Fig. 3A). Nevertheless, the rise in levels of hypophosphorylated 4E-BP1 capable of binding to eIF4E was insufficient to preclude the incorporation of eIF4G into the eIF4F complex. Inclusion of rapamycin in the culture medium did not enhance binding of 4E-BP1 to eIF4E (Fig. 4A), consistent with the partial sensitivity of 4E-BP1 phosphorylation to rapamycin in HCMV-infected cells (Fig. 3B). Strikingly, while the release of 4E-BP1 from eIF4E followed by the recruitment of eIF4G is also observed in quiescent cells infected with the related alphaherpesvirus HSV-1 (36), the overall abundance of PABP retained in an eIF4E-containing complex only increased in HCMV-infected cells (Fig. 4A and B).
FIG. 4.

Assembly of eIF4F complexes and recruitment of PABP in quiescent cells is enhanced following HCMV infection. (A) Serum-starved, growth-arrested, primary human NHDF cells were either mock infected (0-h time point) or infected with wild-type HCMV AD169 in the presence of DMSO or rapamycin. At the indicated times, cell extracts were prepared, and proteins bound to 7-methyl GTP-Sepharose 4B were fractionated by SDS-PAGE, immunoblotted, and visualized with the indicated antibodies. (B) Assembly of eIF4G into an eIF4E-containing complex is not accompanied by PABP recruitment in HSV-1-infected cells. NHDFs growth arrested by serum starvation were either mock infected or infected with HSV-1 (MOI = 5). Cell extracts were prepared at 10 h postinfection, and proteins bound to 7 methyl GTP-Sepharose 4B were fractionated by SDS-PAGE and analyzed by immunoblotting with the indicated antisera. Serum-stimulated cells were included as a control to demonstrate recruitment of both eIF4G and PABP into an eIF4E-containing complex.
Modification of eIF4F core and associated factors in HCMV-infected cells requires viral gene expression and displays a differential sensitivity to GCV.
The productive growth cycle of betaherpesviruses such as HCMV is complex, and much more protracted relative to related alphaherpesviruses such as HSV-1 (22, 27). To determine the stage in the HCMV life cycle during which ERK1/2 and p38 are activated, 4E-BP1 is phosphorylated, the abundance of core and associated eIF4F components increase, and the assembly of eIF4F complexes occurs, we monitored the status of these alterations in the presence of GCV, an inhibitor of viral DNA replication that arrests the viral life cycle prior to the late phase of the infectious cycle (reviewed in reference 22). Figure 5A demonstrates that while ERK activation, p38 activation, and 4E-BP1 phosphorylation were unaffected by GCV and likely occurred early in infection, the increase in the abundance of eIF4F core and associated components was partially sensitive to GCV, suggesting that this occurs later in the viral life cycle. Interestingly, modification of eIF4E-containing complexes occurred at both late and early times, as the recruitment of eIF4G and PABP into eIF4F complexes was sensitive to GCV, while the ejection of 4E-BP1 from the complex was resistant to GCV (Fig. 5B). The latter alteration supports our earlier finding that 4E-BP1 phosphorylation is unaffected by GCV (Fig. 5A). pp28, an HCMV-encoded late polypeptide whose accumulation depends upon viral DNA synthesis (1), served as a control for the efficacy of GCV treatment, whereas actin levels were unaffected by the drug and served as a loading control (Fig. 5A).
FIG. 5.

GCV partially prevents changes in the abundance of eIF4F components and the composition of eIF4F complexes, but it has no effect on 4E-BP1 phosphorylation or activation of ERK1/2 and p38. (A) Quiescent NHDFs were mock infected (M) or infected with HCMV AD169 (MOI = 5) in the presence or absence of ganciclovir (150 μM). Total protein was harvested at 72 h postinfection, fractionated by SDS-PAGE, and analyzed by immunoblotting using the indicated antisera. (B) As in panel A, except that detergent extracts were prepared at 72 h postinfection, and proteins bound to 7-methyl GTP-Sepharose 4B were fractionated by SDS-PAGE, immunoblotted, and visualized with the indicated antibodies. (C) NHDFs were either mock infected (M), infected with gradient purified, UV-inactivated HCMV AD169 (MOI = 5) (UV), or infected with wild-type untreated virus (WT). Total protein was isolated at 12 h postinfection, fractionated by isoelectric focusing (for eIF4E) or SDS-PAGE, and analyzed by immunoblotting with the indicated antisera. At 2 h postinfection, cells were fixed and stained with a monoclonal antibody against the pp65 tegument protein to demonstrate that the UV-treated virus was still capable of entering cells.
To evaluate how early in the HCMV life cycle eIF4E and 4E-BP1 were phosphorylated, infections were performed using gradient-purified, UV-inactivated virus. In contrast to our findings with nonirradiated virus, we were unable to detect eIF4E phosphorylation in cells infected with UV-inactivated virus, and 4E-BP1 phosphorylation was substantially impaired (Fig. 5C). While we also did not observe IE1 and IE2 expression following infection of cells with UV-inactivated virus, we routinely detected the tegument protein pp65 in nuclei. These controls established that the UV treatment effectively inhibited viral gene expression and preserved the ability of the virus to enter and deliver its tegument cargo into cells.
Efficient replication of HCMV in quiescent cells requires the activity of the cellular eIF4E kinase mnk.
To evaluate the impact of eIF4E phosphorylation on the productive growth cycle of HCMV, quiescent NHDFs were infected in the presence and absence of CGP57380, an inhibitor of the cellular eIF4E kinase mnk (11). After 7 days, cell-free lysates were prepared, and the amount of virus produced was quantified by plaque assay. Figure 6A establishes that CGP57380 reduced HCMV replication by >200 fold. The accumulation of pp28, a late viral protein, was also exquisitely reduced by CGP57380 or by a combination of SB203580 plus PD98059 (Fig. 6B). Individually, the p38 inhibitor SB203580 had little effect on overall pp28 levels, while preventing ERK activation with PD98059 achieved a modest reduction in pp28 abundance. The fluctuations of pp28 levels in cultures treated with either PD98059 or SB203580 paralleled the changes in eIF4E phosphorylation presented in Fig. 1, while the more drastic reduction in pp28 levels resulting from SB203580 plus PD98059 or CGP57380 treatment were consistent with our earlier demonstration that both ERK1/2 and p38 activation led to eIF4E phosphorylation in HCMV-infected cells. Together, this establishes that the activity of the eIF4E kinase mnk is required for efficient HCMV replication in quiescent NHDFs and the accumulation of wild-type levels of late proteins exemplified by pp28.
FIG. 6.
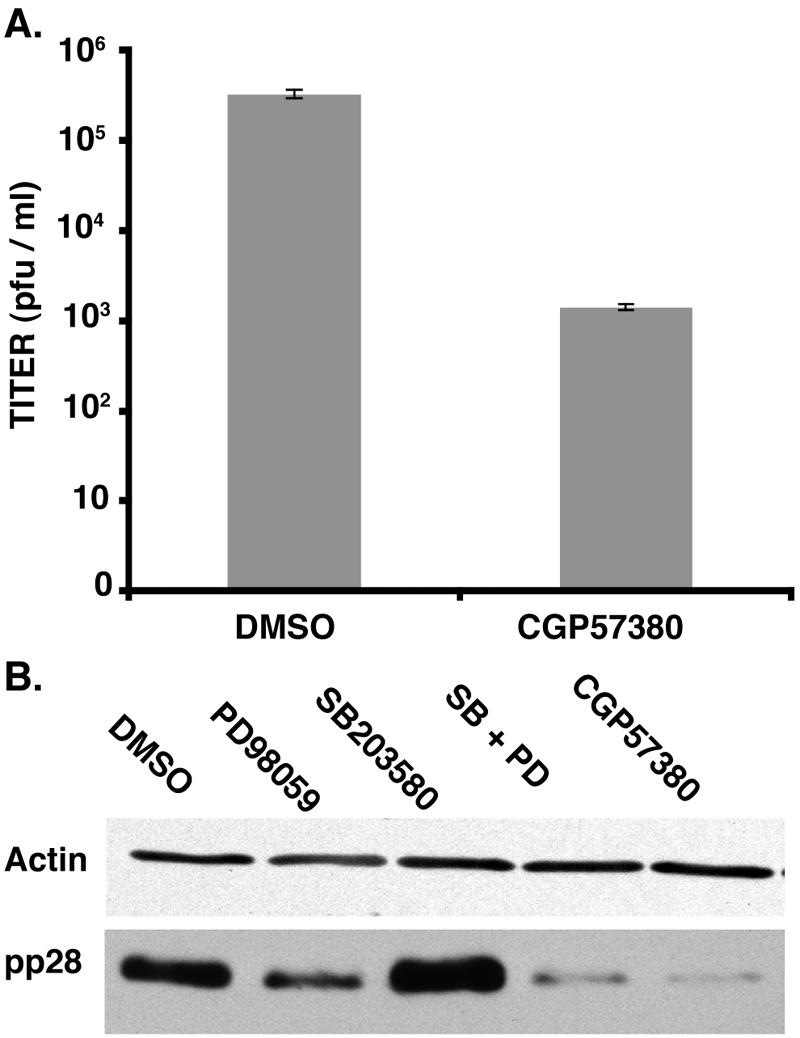
Inhibiting eIF4E phosphorylation significantly reduces HCMV replication and the accumulation of late viral proteins in quiescent cells infected at low input doses of virus. (A) Primary human fibroblasts (NHDF cells) were growth arrested by serum starvation and infected (MOI = 0.05) in the presence or absence of the mnk inhibitor CGP57380. After 7 days, cell-free lysates were prepared by freeze-thawing, and the titer in NHDF cells was determined. (B) Cells were infected as described in the legend to panel A. After 7 days, total protein was isolated, fractionated by SDS-PAGE, and analyzed by immunoblotting using antisera directed against pp28 or actin.
DISCUSSION
To ensure that their mRNAs can successfully engage the cellular translational machinery, many viruses impair the synthesis of host proteins by inactivating the cellular translation factor eIF4F (31). HCMV is particularly interesting in this regard, as it does not significantly inhibit host protein synthesis (34), and as we establish in this study, it activates (as opposed to inhibits) eIF4F. In addition to promoting the assembly of eIF4F complexes, the overall abundance of core eIF4F components and associated factors relative to the translational repressor 4E-BP1 increases in HCMV-infected cells. While other viruses decrease the abundance of cellular translation factors (4, 15, 39), this represents the first report of a virus that induces the accumulation of eIF4F components, perhaps improving the ability of HCMV mRNAs to compete with cellular mRNAs for translation initiation factors. Indeed, increasing the availability of a rate-limiting factor can have a larger impact on the efficiency with which less effectively translated mRNAs are translated, as opposed to mRNAs that are translated at normal levels (9, 13, 17, 20, 23). Finally, 4E-BP1 and eIF4E are phosphorylated in HCMV-infected cells. Significantly, the activity of the cellular eIF4E kinase mnk is critical for efficient viral replication in quiescent cells.
Like the related alphaherpesvirus HSV-1 (36), HCMV stimulates mnk-1, resulting in eIF4E phosphorylation; moreover, that mnk activation is required for efficient replication of both alpha- and betaherpesviruses in resting cells illustrates its importance, together with the phosphorylation of eIF4E, the only mnk substrate identified to date in herpesvirus biology. Indeed, both viruses establish life-long latent infections in different cell types within their human host, characterized by a restricted viral gene expression program (22, 27). Periodically, in response to various stimuli, herpesviruses initiate an alternate gene expression program that results in lytic viral replication, the release of large amounts of progeny virus, and the destruction of the host cell. Perhaps their ability to phosphorylate eIF4E and promote the assembly of eIF4F complexes plays a crucial role in the ability of these viruses to engage the translational machinery of the host and reprogram it with viral mRNAs upon commencing their productive growth cycle.
Although we have identified common themes by which alpha- and betaherpesviruses manipulate eIF4F core and associated factors, HSV-1 and HCMV clearly use different strategies to achieve these goals. In quiescent primary human cells, HCMV stimulates both ERK1/2 and p38 to activate mnk, whereas HSV-1 relies exclusively on p38 (36). While the ratio of eIF4F components to the 4E-BP1 translational repressor is increased in cells infected with either HSV-1 or HCMV, this is achieved solely through proteasome-mediated degradation of 4E-BP1 in HSV-1-infected cells (36), while the abundance of eIF4F core and associated components increases markedly in HCMV-infected cells. Likewise, eIF4G and PABP are recruited into eIF4F complexes in HCMV-infected cells, whereas only eIF4G is recruited into eIF4F complexes in cells infected with HSV-1. It is likely that these variations reflect fundamental differences in the life cycles of these two related viruses or the distinct differentiated cell types that they normally colonize. For example, the finding that eIF4G, but not PABP, is recruited into eIF4F complexes in HSV-1-infected cells might be related to the inhibition of host protein synthesis characteristic of HSV-1 lytic replication. The relative dependence of HSV-1 mRNAs on PABP remains to be explored. However, PABP is recruited into eIF4F complexes in HCMV-infected cells, and host protein synthesis proceeds relatively unimpaired. Finally, it is intriguing that both viruses promote 4E-BP1 phosphorylation, albeit by apparently different mechanisms. In HSV-1 infected cells, rapamycin prevents 4E-BP1 phosphorylation, implicating the involvement of the mTOR-raptor complex (36). However, rapamycin does not inhibit HSV-1 replication in quiescent fibroblasts, as the overall level of 4E-BP1 in these cells is not sufficient to prevent eIF4E from engaging eIF4G and assembling eIF4F complexes. 4E-BP1 phosphorylation is resistant to rapamycin in HCMV-infected cells; not surprisingly, the drug does not significantly impair HCMV replication (14; C. Perez and I. Mohr, unpublished observations). This could reflect the involvement of an alternative cellular mTOR complex, perhaps involving rictor (28) or an HCMV-encoded component that is insensitive to rapamycin.
While mnk activity appears critical for efficient replication of alpha- and betaherpesviruses in resting cells, it is paradoxical that it may in fact be dispensable for proper development. Mice lacking functional mnk-1 and -2 genes develop normally, as do Drosophila melanogaster flies containing mutant eIF4E alleles that cannot be phosphorylated (16, 35). The flies, while normal, are significantly smaller than animals with WT eIF4E alleles, although no such defect has been identified in the mnk knockout mice, cells from which do not contain detectable levels of phosphorylated eIF4E. These studies clearly support the notion that eIF4E phosphorylation is not essential for translation. However, they do not to address the potential importance of eIF4E phosphorylation in controlling translation in response to various stimuli. Such a regulatory role is consistent with our findings on the importance of mnk activity for herpesvirus replication in quiescent cells.
Acknowledgments
We thank Eain Murphy, Tom Jones, and Hua Zhu for many valuable discussions on HCMV methodology; Hermann Gram for generously providing us with CGP5730; and Matthew Mulvey together with Angus Wilson for their critical comments on the manuscript.
This work was supported by a grant from the NIH to I.M. J.N. was supported in part by an NIH training grant, and C.P. was supported in part by an ASM Watkins Fellowship.
REFERENCES
- 1.Depto, A. S., and R. M. Stenberg. 1992. Functional analysis of the true late human cytomegalovirus pp28 upstream promoter: cis-acting elements and viral trans-acting proteins necessary for promoter activation. J. Virol. 66:3241-3246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gingras, A.-C., B. Raught, and N. Sonenberg. 1999. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 68:913-963. [DOI] [PubMed] [Google Scholar]
- 3.Gingras, A.-C., B. Raught, and N. Sonenberg. 2001. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 15:807-826. [DOI] [PubMed] [Google Scholar]
- 4.Gradi, A., Y. V. Svitkin, H. Imataka, and N. Sonenberg. 1998. Proteolysis of human eukaryotic translation initiation factor eIF4GII, but not eIF4GI, coincides with the shutoff of host protein synthesis after poliovirus infection. Proc. Natl. Acad. Sci. USA 95:11089-11094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haghighat, A., S. Mader, A. Pause, and N. Sonenberg. 1995. Repression of cap-dependent translation by 4E-binding protein 1: competition with p220 for binding to eukaryotic initiation factor-4E. EMBO J. 14:5701-5709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hahn, G., R. Jores, and E. S. Mocarski. 1998. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. USA 95:3937-3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Imataja, H., A. Gradi, and N. Sonenberg. 1998. A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. EMBO J. 17:7480-7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson, R. A., S-M. Huong, and E-S. Huang. 2000. Activation of the mitogen activated protein kinase p38 by human cytomegalovirus infection through two distinct pathways: a novel mechanism for the activation of p38. J. Virol. 74:1158-1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kabat, D., and M. R. Chappell. 1977. Competition between globin messenger ribonucleic acids for a discriminating initiation factor. J. Biol. Chem. 252:2684-2690. [PubMed] [Google Scholar]
- 10.Kahvejian, A., G. Roy, and N. Sonenberg. 2001. The mRNA closed-loop model: the function of PABP and PABP-interacting proteins in mRNA translation. Cold Spring Harbor Symp. Quant. Biol. 66:293-300. [DOI] [PubMed] [Google Scholar]
- 11.Knauf, U., C. Tschopp, and H. Gram. 2001. Negative regulation of protein translation by mitogen-activated protein kinase-interacting kinases 1 and 2. Mol. Cell. Biol. 21:5500-5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kondo, K., J. Xu, and E. S. Mocarski. 1996. Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals. Proc. Natl. Acad. Sci. USA 93:11137-11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koromilas, A. E., A. Lazaris-Karatzas, and N. Sonenberg. 1992. mRNAs containing extensive secondary structure in their 5′ non-coding region translate efficiently in cells overexpressing initiation factor eIF-4E. EMBO J. 11:4153-4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kudchodkar, S. B., Y. Yu, T. G. Maguire, and J. C. Alwine. 2004. Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinase. J. Virol. 78:11030-11039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuyumcu-Martinez, N. M., M. E. Van Eden, P. Younan, and R. E. Lloyd. 2004. Cleavage of poly(A)-binding protein by poliovirus 3C protease inhibits host cell translation: a novel mechanism for host translation shutoff. Mol. Cell. Biol. 24:1779-11790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lachance, P. E., M. Miron, B. Raught, N. Sonenberg, and P. Lasko. 2002. Phosphorylation of eukaryotic translation initiation factor 4E is critical for growth. Mol. Cell. Biol. 22:1656-1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lodish, H. F. 1974. Model for the regulation of mRNA translation applied to haemoglobin synthesis. Nature 251:385-388. [DOI] [PubMed] [Google Scholar]
- 18.Mader, S., H. Lee, A. Pause, and N. Sonenberg. 1995. The translation initiation factor eIF-4E binds to a common motif shared by the translation factor eIF-4G and the translational repressors 4E-binding proteins. Mol. Cell. Biol. 15:4990-4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcotrigiano, J., A. C. Gingras, N. Sonenberg, and S. K. Burley. 1999. Cap-dependent translation initiation in eukaryotes is regulated by a molecular mimic of eIF4G. Mol. Cell 3:707-716. [DOI] [PubMed] [Google Scholar]
- 20.McKeehan, W. L. 1974. Regulation of hemoglobin synthesis. Effect of concentration of messenger ribonucleic acid, ribosome subunits, initiation factors, and salts on ratio of alpha and beta chains synthesized in vitro. J. Biol. Chem. 249:6517-6526. [PubMed] [Google Scholar]
- 21.Melnick, J. C., E. Adam, and M. E. DeBakey. 1995. Cytomegalovirus and atherosclerosis. Bioessays 17:899-903. [DOI] [PubMed] [Google Scholar]
- 22.Mocarski, E. S., Jr., and C. T. Courcelle. 2001. Cytomegaloviruses and their replication, p. 2629-2673. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus, Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 23.Nudel, U., B. Lebleu, and M. Revel. 1973. Discrimination between messenger ribonucleic acids by a mammalian translation initiation factor. Proc. Natl. Acad. Sci. USA 70:2139-2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pass, R. F. 2001. Cytomegalovirus, p. 2675-2706. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus, Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 25.Pyronnet, S., H. Imataka, A.-C. Gingras, R. Fukunaga, T. Hunter, and N. Sonenberg. 1999. Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J. 18:270-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodems, S. M., and D. H. Spector. 1998. Extracellular signal-regulated kinase activity is sustained early during human cytomegalovirus infection. J. Virol. 72:9173-9180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roizman, B., and D. Knipe. 2001. Herpes simplex viruses and their replication, p. 2239-2459. In D. M. Knipe, P. M. Howley, D. E. Griffin, R. A. Lamb, M. A. Martin, B. Roizman, and S. E. Straus, Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 28.Sarbassov, D. D., S. M. Ali, D. H. Kim, D. A. Guertin, R. R. Latek, H. Erdjument-Bromage, P. Tempst, and D. M. Sabatini. 2004. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14:1296-1302. [DOI] [PubMed] [Google Scholar]
- 29.Scheper, G. C., and C. G. Proud. 2002. Does phosphorylation of the cap-binding protein eIF4E play a role in translation initiation? Eur. J. Biochem. 269:5350-5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheper, G. C., N. A. Morrice, M. Kleijn, and C. G. Proud. 2001. The mitogen-activated protein kinase signal-integrating kinase Mnk2 is a eukaryotic initiation factor 4E kinase with high levels of basal activity in mammalian cells. Mol. Cell. Biol. 21:743-754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider, R. J., and I. Mohr. 2003. Translation initiation and viral tricks. Trends Biochem. Sci. 28:130-136. [DOI] [PubMed] [Google Scholar]
- 32.Sinclair, J., and P. Sissons. 1996. Latent and persistent infections of monocytes and macrophages. Intervirology 39:293-301. [DOI] [PubMed] [Google Scholar]
- 33.Soderberg-Naucler, C., and J. Y. Nelson. 1999. Human cytomegalovirus latency and reactivation—a delicate balance between the virus and its host's immune system. Intervirology 42:314-321. [DOI] [PubMed] [Google Scholar]
- 34.Stinski, M. F. 1977. Synthesis of proteins and glycoproteins in cells infected with human cytomegalovirus. J. Virol. 23:751-767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ueda, T., R. Watanabe-Fukunaga, H. Fukuyama, S. Nagata, and R. Fukunaga. 2004. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol. Cell. Biol. 24:6539-6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walsh, D., and I. Mohr. 2004. Phosphorylation of eIF4E by mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev. 18:660-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waskiewicz, A. J., J. C. Johnson, B. Penn, M. Mahalingam, S. R. Kimball, and J. A. Cooper. 1999. Phosphorylation of the cap-binding protein eukaryotic translation factor 4E by protein kinase mnk1 in vivo. Mol. Cell. Biol. 19:1871-1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wells, S. E., P. E. Hillner, R. D. Vale, and A. B. Sachs. 1998. Circularization of mRNA by eukaryotic translation initiation factors. Mol. Cell 2:135-140. [DOI] [PubMed] [Google Scholar]
- 39.Zamora, M., W. E. Marissen, and R. E. Lloyd. 2002. Multiple eIF4GI-specific protease activities present in uninfected and poliovirus-infected cells. J. Virol. 76:165-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou, Y. F., M. B. Leon, M. W. Waclawiw, J. J. Popma, Z. X. Yu, T. Finkel, and S. E. Epstein. 1996. Association between prior cytomegalovirus infection and the risk of restenosis after coronary atherectomy. N. Engl. J. Med. 335:624-630. [DOI] [PubMed] [Google Scholar]