

Abstract
The use of human antibodies as biologic therapeutics has revolutionized patient care throughout fields of medicine. As our understanding of the many roles antibodies play within our natural immune responses continues to advance, so will the number of therapeutic indications for which a monoclonal antibody will be developed. The great breadth of function, long half-life, and modular structure allow for nearly limitless therapeutic possibility. Human antibodies can be rationally engineered to enhance their desired immune functions and eliminate those that may result in unwanted effects. Antibody therapeutics now often start with fully human variable regions, either acquired from genetically engineered humanized mice or from the actual human B cell themselves. These variable genes can be further engineered by widely used methods for optimization of their specificity through affinity maturation, random mutagenesis, targeted mutagenesis, and use of in silico approaches. Antibody isotype selection and deliberate mutations are also used to improve efficacy and tolerability by purposeful fine-tuning of their immune effector functions. Finally, improvements directed at binding to the neonatal Fc receptor can endow therapeutic antibodies with unbelievable extensions in their circulating half-life. The future of engineered antibody therapeutics is bright, with the global monoclonal antibody market projected to exhibit compound annual growth, forecasted to reach a revenue of nearly half a trillion dollars in 2030.
Keywords: Allergy, immunology, IgE, monoclonal antibody, therapeutic antibody, antibody engineering
Overview of antibodies as therapeutics
The use of monoclonal antibodies (mAbs) in the biotechnology and pharmaceutical industries have shown logarithmic growth over the last 4 decades (1,2). Since the advent of mAb technology in 1975 (3), the ability to develop antibodies as specific targeting therapeutic molecules has now culminated in antibodies becoming the predominant class of new drugs developed in recent years (1). In the upcoming years, antibody therapies will be a major contributor to overall biopharmaceutical product sales. This growth is due to the nature of antibodies themselves and their long persistence in circulation. They are highly diverse molecules that exhibit great breadth of influence within our immunity. Their structure and functions allow for highly specific targeting and recruitment of a plethora of immune functions which permit rational approaches toward therapeutic engineering of the molecule. After widespread adoption, earlier generations of monoclonal antibodies became associated with being an emergent leading cause of adverse reactions, including anaphylaxis, reported to the FDA (4). Many early generation monoclonal antibody therapeutics were of murine origin, invoking antidrug antibodies (ADAs) against both protein and glycan components, resulting in reduced therapeutic effect and/or adverse reactions ranging from rash to systemic inflammatory responses. Cetuximab, a murine/human chimeric antibody inhibitor of epidermal growth factor receptor, is a great example of how murine derived antibody therapeutics, and the cell-line from which the antibody is expressed, can have tremendous repercussions in the number of adverse reactions which may result. New methods of development, resulting in fully human antibody therapies, are far less immunogenic and result in less antidrug immune responses and adverse events. Consequently, not only can one develop an antibody to bind very specifically to a desired target with high affinity, but one can engineer the antibody to possess or lack a variety of immune functions and induce less unwanted ADAs. Human antibodies now can be rationally engineered as therapeutics with enhanced desired immune functions while eliminating those functions or features that may result in harmful effects.
The structure of an antibody can be broken into two parts, split along its functional regions (Figure 1). The antigen-binding fragment (Fab) contains the variable region of the antibody, which is the product of gene rearrangement and somatic hypermutation, allowing for highly specific antigen binding. The constant or crystallizable fragment (Fc), comes in a variety of types, known as isotypes, and is responsible for binding complement and a diversity of cell receptors (FcRs), resulting in many immune functions (5). There are five human Fc isotypes: IgG, IgM, IgA, IgD, and IgE, as well as subtypes of IgG (IgG1–4) and IgA (IgA1–2). Typically, IgG makes up 70–85% of the total antibody in serum, while IgM and IgA each make up 5–15%, and IgE with IgD <1% (6,7). Each of these Fc isotypes engages a corresponding family of FcRs. Having this modular structure of an Fab and Fc allows antibodies to link bound antigen with both humoral and cellular components of the immune system in a wide array of manners.
Figure 1. Antibody structure, isotype/subtypes, and their major features.

The structure of a human antibody can be broken into two basic parts linked by a hinge, helping separate its functional regions. The antigen-binding fragment (Fab) contains the antibody variable region of the antibody, which is the product of gene rearrangement and somatic hypermutation, allowing for highly specific antigen binding. The constant or crystallizable fragment (Fc), comes in a variety of types, known as isotypes (there are five isotypes: IgM, IgD, IgG, IgA and IgE), and is responsible for binding complement and a diversity of cell receptors (FcRs), resulting in many immune functions. Immune functions are influenced by the structural differences existing between the isotypes and subtypes of antibodies. These differences influence both antigen binding, such as hinge flexibility, and receptor selection, such as glycosylation. The vast array of immune functions, conveyed in an antigen specific fashion, is controlled by these many features of human antibodies.
A great wealth of information about antibodies obtained over time has given investigators the ability to molecularly engineer antibodies with controlled immune functions, elicited through their abilities to interact with various immune cells to induce effector functions (8). One such function is an antibody’s ability to bind complement, initiating its activation, known as complement-dependent cytotoxicity (CDC). Most of the cellular functions of antibodies occur through low affinity interactions with FcRs, requiring interaction with multivalent immune complexes for activation to occur. These antibody mediated immune functions, particularly those driven by IgG type antibodies, directly target pathogens, including antibody-dependent cell-mediated cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP). Antibodies also can result in immune functions via release of cytokines, chemokines, or other cellular immunomodulatory agents (9,10). Finally, antibodies enhance antigen uptake, processing, and presentation by antigen presenting cells (APCs) (11,12), facilitating adaptive responses. Antibodies can modulate adaptive immune responses through B cell activation, antigen presentation, and can even induce tolerance (13). Engineered monoclonal antibodies that enhance or reduce these specific Fc effector functions may be useful for the strategic design of a highly tailored immune response needed for a particular therapeutic.
In the process of developing an antibody therapeutic, candidates undergo many stages of research and development before being considered for clinical studies (14). Typically, this begins with antibody discovery based on antigen binding screens to identify the desired variable sequences. Once specific binding antibody candidates are selected, biological function is assessed and engineered to optimize safety and efficacy. Finally, various characteristics associated with manufacturability are evaluated to ensure necessary stability and shelf-life. Currently all approved antibody therapeutics are of the IgG isotype, though some IgE based therapies have entered stages of clinical development. Many approved antibody therapeutics have engineered Fcs, some possess a combination of mutations to maximize the therapeutic potency and often those to enable improved half-life.
Given the diversity of new engineered antibody therapeutics, reflected in the engineering strategies highlighted in this review, the naming convention has once again changed and future therapeutic antibodies will no longer end in “-mab”. Since the introduction of antibody therapeutics, specific modifiers have been added prior to the “-mab” suffix. A “-u-” for human or “-o-” for mouse was used to indicate the source from which the antibody was originally derived, the modifications “-xi-” for chimeric or “-zu-” for humanized. Moving forward, four new suffixes have been created to replace the use of “-mab” (15). The new suffix “-tug” will be used for monospecific, full-length, and Fc unmodified antibodies. “-bart” will be used for monospecific, full-length antibodies with engineered Fc regions. The suffix “-mig” is now used for bi- or multispecific antibodies. Finally, the suffix “-ment” will now be used for monospecific antibody fragments of any kind.
Antibody immune function diversity
Much of the variety of functions antibodies convey within our immune system stem from the diversity of effector functions necessary for humans to respond optimally to the vast universe of organisms wanting to use us to support their existence. Our innate and adaptive immune responses, orchestrated by innate lymphoid and T cells, can be split into 3 types (Types 1–3) depending on the cells and cytokines involved (16). Type 1 immunity makes use of T-cell cytokines (mainly IFN-γ) and cytotoxic T-cells focusing on protection from intracellular pathogens. Type 3 immunity (IL-17 and/or IL-22), often included in type 1 immunity, involves the cytokine IL17, the recruitment of neutrophils, and induction of antimicrobial responses at the epithelial surface to protect against extracellular bacteria and fungi (17). Finally, type 2 immunity, often opposing the action of type 1 and 3, works through a different array of cytokines (IL-4, IL-5, and IL-13) involving the induction of mast cell, basophil, and eosinophil, and fundamentally helps protect us from large and complicated extracellular pathogens, like helminths (18). Antibody effector functions are tuned across our cellular immunity through their display of Fc receptors.
The receptors bound by IgG antibodies differ from one another in their function, cellular distribution, glycosylation, and strength of binding to the IgG Fc subtypes (19). They are roughly characterized into three groups known as FcγRI, FcγRII, and FcγRIII (Figure 2). FcγRI, FcγRIII and FcγRIIa receptors are capable of activating functions within the cell though immunoreceptor tyrosine-based activation motifs (ITAMs), while only the FcγRIIb receptor transmits an inhibitory signal through an immunoreceptor tyrosine-based inhibitory motif (ITIM) (20). Receptor aggregation by an immune complex induces phosphorylation of the ITAM tyrosine residues, ultimately transmitting a signal to induce cellular activation through the PI3K pathway. Co-ligation between the FcγRIIb inhibitory and a heterologous activating receptor by immune complexes, by contrast, promotes the recruitment of inositol phosphatases, ultimately leading to abrogation of the activation signal. FcγRI is the only high affinity receptor for monomeric IgG, partly due to its interaction with Fc glycans. To trigger signaling with the low-affinity FcγRs requires cross-linking with IgG immune complexes. Just as Fab development is important in the design of an antibody therapeutic, rational selection of an IgG subtype, along with fine-tuning of the Fc domain to adjust FcR interactions, is needed to achieve optimal efficacy. In general, due to their weak FcγR-binding profile, IgG2 and IgG4 are considered the least inflammatory (6).
Figure 2. Fc receptor profiles for effector cell induction and inhibition.

Receptors bound by IgG antibodies differ from one another in their function, cellular distribution, glycosylation, and affinity to which IgG Fc subtypes they bind. Originally named for its role in IgG transport from the mother to the fetus or neonate, FcRn binds to, transports, and recycles IgG, playing a central role in cellular trafficking and serum half-life of IgG antibodies. FcRn is expressed primarily on vascular endothelial cells but also on epithelial cells, macrophages, and other cell types. Fc receptors present on immune effector cells varies greatly by cell type, but are roughly categorized into three groups: FcγRI, FcγRII, and FcγRIII. FcγRI, FcγRIII and FcγRIIa receptors are capable of activating functions within the cell though ITAM cytoplasmic domains – highlighted in red. Only the FcγRIIb receptor can modulate effects of the other Fc receptors by transmitting an inhibitory signal through their ITIM cytoplasmic domain – highlighted in green. Most of the cellular functions arise through low affinity interactions between Fc and FcRs, requiring multivalent immune complexes for signal transduction to occur, allowing for an additional layer of control. The summation of signal generated by the diversity of interactions ultimately dictate the activation state of the effector cell.
If the effector functions of naturally occurring antibodies are misdirected, the result is autoimmunity or allergy. Type 1 and 3 immunity play a pathogenic role in several human diseases, most notably autoimmunity. When type 2 immune functions are inappropriately unleashed, allergic diseases result. Activation and regulation are critical for directing the proper function of our immunity while not allowing for allergy and autoimmunity. Engineered antibodies that inappropriately cause autoimmunity or induce allergic diseases have not been reported. However, some patients who are experiencing active autoimmunity and are treated with antibodies specific to individual inflammatory cytokines have been reported to demonstrate a detrimental change in their inflammatory pattern or an evolution to a new, different inflammatory pathology, such as the occasional development of psoriasis upon initiation of inflammatory bowel disease treatment with TNF-alpha antagonists (21,22).
Within type 2 immune responses, classical mast cell and basophil activation occurs through the high-affinity IgE receptor, FcεRI. Type 2 effector cells can also express activating receptors FcγRIII (CD16) (23) and can transiently express FcγRI (CD64). These receptors allow activation of effector cells by immune complexes. Since these cells have the capacity to trigger strong proinflammatory responses their activation needs to be controlled. Most importantly, expression of FcγRIIb (CD32B), the ITIM-associated receptor, is capable of suppressing mast cell activation (24). FcγRIIb crosslinking has been shown to reduce IgE-mediated mast cell activation. FcγRIIb seems to display different binding affinity and selectivity toward the various IgG subtypes, but how this translates to effector cell inhibition remains under active investigation. Several experiments have been performed demonstrating that co-aggregation of FcεRI and FcγRIIb inhibits IgE-dependent activation and mediator release from mast cells and basophils (25). Finally, Siglecs (sialic acid-binding immunoglobulin-like lectins) are also expressed on human mast cells and basophils and can mediate cellular inhibitory activity upon engagement with a specific sialylated glycan ligand (26,27). In fact, strategies to artificially ligate Siglec-8 with an antibody inhibit mast cell activation and prevented anaphylaxis in an animal model (28,29). All of these receptor mediated features must be considered carefully when engineering antibodies for therapeutic use.
Generation/engineering of the variable region
The earliest monoclonal antibody drugs, those targeting CD3, highlighted the potential of mAbs as drugs, but also showed the risks of severe immune responses toward non-human antibodies (30,31). The most obvious approach to avoid these concerns is to use completely human variable regions, which can then be molecularly grafted onto appropriate human constant regions. Methods for attaining human antibodies either by human hybridoma technology or single B cell sequencing are readily available (32,33). These approaches have become standard for antibody therapeutics developed to infectious diseases, however, antibodies targeting human proteins are typically produced in experimental animals and must be humanized to avoid the presence of non-human antigens/allergens such as galactose-alpha-1,3-galactose and reduce the likelihood of developing ADAs (34,35). This typically involves grafting of the complementary determining regions of both heavy and light chains onto a human variable domain with subsequent mutagenesis to regain antigen binding either by rational design or phage display (36). Many antibody therapeutics have been developed in mice and were humanized, a couple of examples are omalizumab and mepolizumab. Two alternative approaches to develop human antibodies as drugs include the use of humanized mice and diverse, in vitro, human antibody libraries. Humanized mice of various types in which mouse antibody gene loci have been replaced with human loci have been used for several decades and have the advantages that antibodies from these animals undergo affinity maturation and self-tolerance (2,37,38). Many new antibody therapeutics have been developed making use of humanized mice, two examples are dupilumab and tezepelumab. Antibody display methods that use naïve or designed libraries allow antibody identification by phage or yeast display and subsequent affinity improvement by directed evolution (39–41). Finally, single chain variable (scFv) regions either as linked variable heavy and light chains, camelid derived single domain variable regions (sdFv), or human derived sdFv’s from phage display (42–44) offer significant advantages when bispecific or poly-specific antibodies are needed.
Antibody improvement, by diversity-based approaches, knowledge-based approaches, or most recently by harnessing artificial intelligence (45,46) is necessary both to optimize affinity, and to improve “developability” as future therapeutics. Typical considerations are stability, solubility, aggregation, off-target binding, and chemical modification (47). The variable region is typically the only non-human portion of an engineered antibody with the remainder being modular constant regions, thus optimization of the variable region can occur independently of other design considerations. A critical consequence of the modularity of antibodies is that unique valencies can be designed. Bispecific antibodies, with different Fab arms connected to similar Fc’s can be engineered to heterodimerize (approaches reviewed in 48). Beyond these bispecific designs, it is possible to append either scFv’s or sdFv’s to CH1 and/or CL domains (49,50). A few examples of FDA-approved bispecific antibody therapeutics are blinatumomab, faricimab, and teclistamab.
Engineering efforts have recently been extended to IgE antibodies (51,52) making it now possible to begin drug development targeting IgE mediated diseases using the variable gene sequences of an allergen specific human IgE. By harnessing the specific allergen targeting that naturally occurs in allergic diseases, antibody engineering techniques can be applied to create a unique therapeutic molecule. Expressing the IgE variable sequences as an IgG isotype antibody, one can take a pathogenic antibody capable of inducing mediator release from mast cells and basophils and create a specific targeting therapeutic, able to block “like” IgE molecules from binding allergen.
There remains much debate as to the origins and relationships exhibited by IgE and populations of IgG which bind similar epitopes (53,54). The root of this debate focuses on how IgE antibodies arise, either through direct class switching from naïve IgM, or indirect class switching via populations of allergen selected IgG. Evidence has been accumulating that suggest the later; populations of allergen specific IgG undergo class switch recombination to become the pathogenic IgE molecule (55–57). However, it is important to note that IgE cannot class switch back, due to the nature of class switch recombination and the resulting excision of portions of the antibody heavy chain locus from the chromosome. If IgG antibodies are responsible for the tolerogenic effects seen with allergen immunotherapy, how then do populations of IgG, which are potentially the predecessors of the pathogenic IgE, expand to be capable of overwhelming and abating the allergic response? If only a select population of allergen specific IgG give rise to IgE, studies of the IgE antibody directly are needed to remove possible doubt that a given IgG is relevant in induction of allergic disease or conversely tolerance.
Engineering of the Fc region
Natural differences in the attributes of IgG Fc subtypes associated with receptor binding must first be considered when engineering a therapeutic antibody (6,58). IgG1 is normally the most abundant subtype and is the most frequently used for engineering purposes. IgG2 is thought to be prominent in responses against bacterial capsular polysaccharide antigens. IgG3 antibodies are particularly effective in the induction of effector functions and are called upon to defend against many viral and bacterial infections. This is partly due to their unusually large and elongated hinge region, which conveys greater molecular flexibility and additional glycosylation, which contribute to its remarkable FcR engagement. IgG4 antibodies are often formed following repeated and/or long-term exposure to antigen. A classic example of such an antigen exposure is the response seen after allergy immunotherapy (59). IgG2 or IgG4 subtypes or engineered IgG1 antibodies with mutations that preclude FcR or C1q binding to avoid cytotoxicity are often used for therapeutic antibody development. C1q primarily binds to IgG1 and IgG3, but not IgG2 or IgG4. IgG3 antibodies are generally not considered for use in therapeutic antibody development given their potent pro-inflammatory properties.
Engineering the Fc region of a therapeutic monoclonal antibody allows the generation of molecules that are better suited to the pharmacologic activity required of them. The Fc is critical to the functioning of an antibody and has been the focus of many engineering efforts (Figure 3). Alterations in the Fc that impact FcR engagement have been shown to drastically affect the efficacy of antibody therapeutics in three main ways: increased and decreased Fc functions, and prolonged half-life (60). One of the earliest Fc engineered antibody contained a specific LALA-PG mutation (L234A/L235A/P329G) (61,62). This mutation resulted in elimination of complement binding and fixation as well as silenced ADCC. Another early mutation, K322A, which removes binding to C1q and prevents activation of complement, has been used to reduce complement-dependent lysis resulting in less pain as an adverse side effect (63,64). Cocrystal structures of human IgG1 Fc with the low affinity FcγRs have allowed high resolution mapping of the binding interfaces which in turn assist with the selective mutation of residues involved in their interaction.
Figure 3. Fc engineering features for enhancement of therapeutic effects.
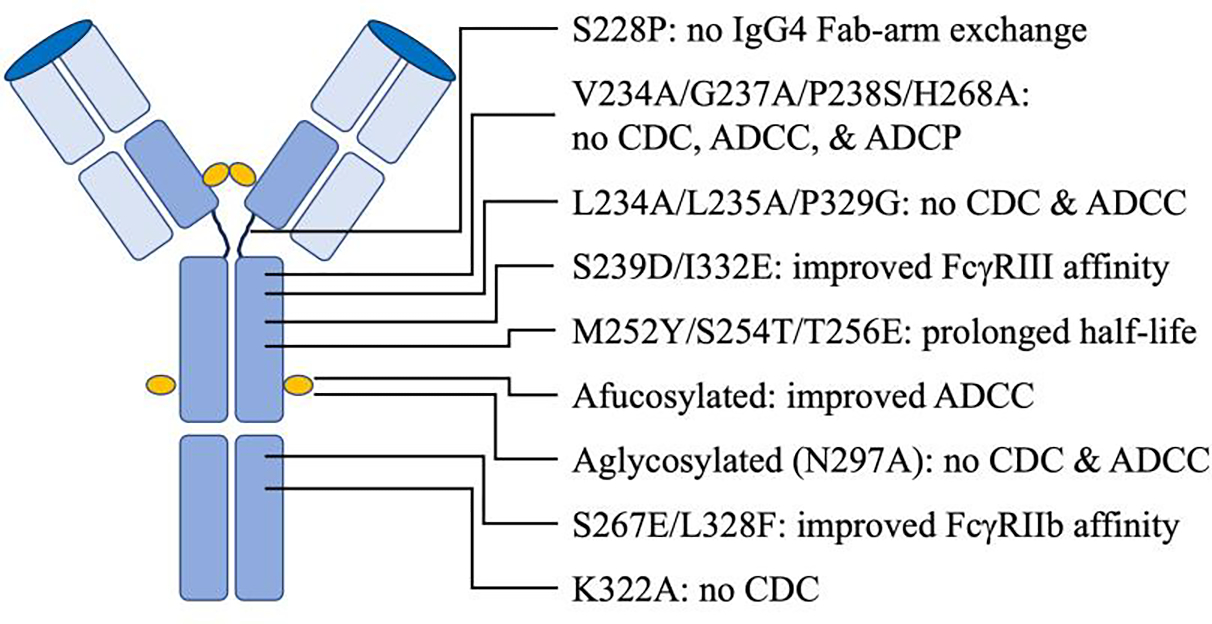
Engineering the Fc region of a therapeutic monoclonal antibody allows for generation of unique drugs rationally designed for a strategic pharmacological purpose. Alterations of Fc affinity, such as the engineered YTE mutation, for FcRn can have dramatic effects on serum half-life by greatly facilitating antibody recycling. Simple changes can be performed which result in greater stability and prevention of arm-swapping often seen as a problem for IgG4 subtype antibodies frequently used for their natural tendency to be minimally inflammatory. Modification of Fc residues can result in dramatic increases or decreases in various effector cell functions and their tendency to fix complement. Glycan alteration can also be engineered to improve pharmacokinetics, efficacy, and safety of therapeutic antibodies. A wide array of Fc engineering features must be considered carefully when designing antibodies for therapeutic use.
Numerous engineering efforts have worked toward both reducing and/or enhancing effector functions to allow for the ultimate tailoring of an antibody’s functional profile. A variant was reported that contained the IgG2 to IgG4 cross-subtype mutations V309L/A330S/P331S combined with the non-germline mutations V234A/G237A/P238S/H268A, which resulted in reduction of CDC, ADCC, and ADCP to undetectable levels (65). Alternatively, engineering mutations which enhance binding of Fc receptors can significantly enhance cellular cytotoxicity (66). Pharmaceutical researchers identified the mutations S239D/A330L/I332E (termed 3M) (67), which resulted in enhanced ADCC. Furthermore, additional mutations of the Fc residue G236A resulted in enhanced ADCC and ADCP (68). An example of an Fc engineered antibody with reduced FcR binding properties is teplizumab, an anti-CD3 therapeutic used to delay onset of type 1 diabetes.
IgG4 has two isomers differing in the disulfide bonding of hinge cysteines which coexist, resulting in half-molecule exchange often referred to as Fab-arm exchange (69). This results in functionally monovalent but bispecific antibodies which may not be desired when developing a therapeutic. Mutation to prevent Fab-arm swapping of IgG4 have been reported. One mutation, S228P, prevents IgG4 Fab-arm exchange (70). Three additional mutations, Y219C, G220C, and S228P, in the Fc has been shown to have an additive benefit resulting in an increase in stability while preventing Fab-arm exchange (71). A few examples of modified IgG4 being used for their subdued Fc-mediated effector functions include the therapeutic PD-1 binding antibodies cemiplimab, nivolumab, and pembrolizumab.
Antibodies are glycoproteins, and several studies have shown that mAbs which have only a difference within their glycosylation pattern assume different binding affinities for FcγR, thus glycosylation is an important factor in the design of antibody-based therapeutics (72). The binding affinity of IgG1 to FcγRs is highly dependent on the N-linked glycan at asparagine 297 (N297) in its CH2 domain (73). Antibody glycoengineering, has been employed to achieve complete effector silencing of blocking antibodies. Aglycosylation of IgG1 has been used to completely remove the unwanted Fc-mediated functions (74). However, aglycosylated antibodies are often unstable and aggregation prone, which enhance the development of anti-drug antibodies (75). The Fc core fucosylation naturally decreases ADCC functions of IgG antibodies, producing antibodies with knockout of the core fucose, afucosylation, results in increased affinity only to FcγRIII receptors, a result in increased phagocytosis by macrophages and increased ADCC activity. FcγRIIIa binding affinities for sialylated Fc parts are reduced, thus hypersialysation was shown to reduce binding properties and consequently ADCC and CDC activity (76). An example of an afucosylated therapeutic antibody is benralizumab.
Fc variants with selectively enhanced FcγRIIb binding also have been described. A strategy to inhibit FcεRI-mediated reactions has been to take advantage of the inhibitory FcγRIIb (77). Co-aggregation of FcεRI and FcγRIIb is a viable strategy to limit allergic responses (25). The dual mutation S267E/L328F was shown to increase affinity between IgG1 and FcγRIIb by 430-fold. However, these engineered antibodies were shown to be rapidly cleared from circulation by liver sinusoidal endothelial cells (LSECs), which express high levels of FcγRIIb. As a result, antibodies formatted with the S267E/L328F mutations exhibit a serum half-life of only 3 to 4 days in humans. Enhanced FcγRIII binding has also been shown with mutation of S298A/E333A/K334A or S239D/I332E targeted IgG1 Fc mutations (65,78). Enhanced FcγRIII and reduced FcγRIIb binding was engineered into the HER2 targeting therapeutic antibody margetuximab.
The neonatal Fc receptor (FcRn) is a major histocompatibility complex class I-type molecule that binds to, transports, and recycles IgG and plays a central role in the cellular trafficking and serum half-life of IgGs. FcRn is expressed primarily on vascular endothelial cells but also on epithelial cells, macrophages, and other cell types (79). Originally named for its role in IgG transport from the mother to the fetus or neonate, FcRn is responsible for the long half-life of IgG in circulation. FcRn acts as a transport receptor, IgG is internalized by pinocytosis. IgG in early acidic endosomes remains bound to FcRn and is recycled back to the plasma membrane where it is released at the physiological pH of the extracellular space. The process protects IgG from lysosomal degradation. FcRn also transports IgG across epithelial barriers, allowing for IgG delivery on mucosal surfaces and is the main factor that maintains IgG antibodies time in circulation. Antibody interaction with FcRn occurs at the interface between the CH2 and CH3 domains of Fc. Screening of a phage display Fc variant library identified mutations (M252Y/S254T/T256E, termed YTE) with enhanced binding to human FcRn (80). The YTE variant had 10-fold increased binding affinity resulting in extension of half-life in some cases to 100 days. Several groups have investigated Fc variants with enhanced pharmacokinetics, with mutants M428L/N434S (81), and H433K/N434F (82) shown to increase IgG1 half-life in humans by more than two-fold. Fc modification to enhance FcRn binding was recently used to create the long-acting antibody combination tixagevimab/cilgavimab, used for pre-exposure prophylaxis to protect against CoVID-19 infection.
Lastly, antibody Fc conjugates are antibody therapeutics designed to deliver a compound specifically to a target – often called antibody drug conjugates (ADCs) as their payloads are often drugs (83). This type of antibody engineering is most studied in oncology, where cytotoxic compounds are covalently conjugated to an antibody to allow for highly specific delivery of a payload to the malignant cell. The payload can be conjugated to the antibody using either a cleavable, where the payload ultimately separates from the mAb, or non-cleavable linker. ADCs are a rapidly growing group of antibody therapeutics. In fact, the FDA recently approved several ADCs for use in the treatment of breast cancer, trastuzumab emtansine, trastuzumab deruxtecan, and sacituzumab govitecan.
Novel antibody engineering strategies to combat allergy
Several antibody engineering strategies are under active investigation that make use of Siglec receptors and their ability to inhibit activation of mast cells. Siglec ligand conjugated anti-receptor antibodies have been reported that can suppress B cell activation through anti-IgD surface receptor antibodies. Further, anti-IgE conjugated with a mast cell Siglec ligand can even suppress IgE mediated mast cell degranulation and anaphylaxis in an animal model (84). Siglec ligand conjugated antibody approaches might be enhanced by using the allergen specific human IgE variable sequences to direct the antibody conjugated therapeutic mAb to the specific allergen immune complex.
A paradigm under examination in the field of allergy is the notion that antibodies of different isotypes, specific to the same epitope, arising from the same repertoire of B cells, can have effects that interact, regulate, block, or interfere with one another. In particular, there is increasing attention being paid to the presence of IgG4 subtype antibodies specific to key allergens as being a biomarker for tolerance to those allergens, such as peanut (85), egg (86,87) and cow’s milk (88). With no direct mechanistic evidence that the effect is driven by blocking, the clinical utility of an IgE/IgG4 ratio remains under investigation for the assessment of whether a sensitized patient will demonstrate allergic symptoms upon completion of immunotherapy (87).
The corollary of this paradigm is that therapeutic antibodies which interfere with specific pathological IgE mediated responses could potentially be developed. Attempts at antibody-based treatments have mostly been nonspecific up to this point, removing IgE from the circulation in wholesale fashion. Studies up to now have been essentially agnostic to the component based molecular binding interactions by which the disease causing IgE attach to their key allergens, which may have limited their utility. When broken down to epitope level specificity, the utility of IgE and IgG4 based responses to predict unresponsiveness to a challenge appears to go up (89). It may therefore be that only certain key subsets of allergen specific antibodies determine the worst pathogenicity (90,91). As the specificity of each pathogenic IgE’s binding to a key allergenic epitope becomes clearer over time, this knowledge may allow for the rational engineering of other isotypes of highly specific “like” targeting antibodies which could block, interfere, or incapacitate the IgE-mediated, allergen specific response.
Rational engineering of specific allergen blocking therapeutics
Sometimes referred to as passive immunotherapy, engineered IgG mAbs directed against IgE epitopes of major allergens are delivered by injection. These antibodies are thought to be analogous to those IgE-blocking antibodies hypothesized to be induced by allergen immunotherapy. Support for passive immunotherapy first came from two recombinant anti-Fel d 1 IgG4 antibodies, produced in mice engineered to be humanized for their immunoglobulin genes, which conferred protection against nasal challenge with whole cat allergen extract (92). Similarly, a cocktail of three mAbs directed against the major birch allergen Bet v 1 was effective in inhibiting the clinical response to birch pollen nasal challenge (93). To date, given the constraints on the number of antibodies which can feasibly make up a therapeutic cocktail developed for this purpose, allergic diseases driven by a single major allergen have been investigated.
Along similar lines, monoclonal IgE that recapitulate patients’ exact allergen specific IgE binding have been created from patients with a wide variety of IgE mediated allergies and even following helminth infections (51,94–98). The IgE mAbs then can be used for their variable regions, one that engages the target allergen exactly where an IgE binds, as it was taken from an IgE. Antibody engineering based upon these allergenic binding sequences presents the possibility of antibodies with the exact same molecular specificity that could interfere with or inhibit IgE. To summarize many of the antibody engineering approaches highlighted in this article we can specifically use this as a case-study to exemplify the develop of a novel human antibody therapeutic. As such, in theory, therapeutic IgG molecules, created from the IgE variable sequence (often referred to as cross-isotype or switched-variant antibodies) could be engineered to have several desirable therapeutic effects (Figure 4): 1. therapeutic IgG molecules could be engineered to have higher affinity for FcRn, resulting in prolonged serum half-life and allergen blocking effects; 2. allergen protein would be bound by the therapeutic IgG, bispecific IgG, or cocktail of mAbs preventing IgE binding and reducing the activation signal caused by the allergen cross-linking FcεRI; 3. IgG or bispecific IgG bound to allergen could engage/cross-link FcγRIIb or co-aggregate with FcεRI sending an inhibitory signal; 4. IgG bound allergen could be taken up by APCs via FcγRIII-dependent manner and presented to create/reprogram a tolerogenic immune response toward the allergen.
Figure 4. Rational engineering of theoretical allergen blocking therapeutics.

The IgE molecule bound to its high affinity receptor provides specific control for the release of the mediators responsible for the symptoms of allergic diseases. Blocking antibody therapeutics, created from the variable sequences of allergen specific IgE, in theory can be engineered for the purpose of passive immunotherapy, to mitigate against anaphylaxis, or augmenting immunotherapy, to foster immune reprogramming – targeting immune mechanisms that could in theory provide advantages for treating allergy. Such antibodies could have engineered features geared toward enhancement of such overall therapeutic goals: 1. improved affinity for FcRn to increase serum half-life, 2. variable regions taken from an allergen specific IgE binds an IgE epitope to block IgE binding, 3. improved affinity for FcγRIIb to send inhibitory signaling via allergen specific cross-linking or co-aggregation of FcεRI and FcγRIIb, 4. improve antigen uptake/presentation by APCs through FcγRIII, to facilitate reprogramming of the allergen specific adaptive immune response. Use of IgG4 subtype and additional alterations of the Fc could also improve tolerability and reduce potential side-effects caused by complement and unwanted pro-inflammatory effector functions. Additional engineered mAbs, antibody cocktails, or bispecific antibodies may be needed to block sufficient allergen epitopes to prevent allergic symptoms.
Unlike the common bivalent monospecific IgG1 isotype, antibody engineering strategies and technologies have been developed to allow for antibodies to have an array of valencies and specificities (99,100). Since cross-linking of the FcεRI is often achieved by several IgE antibodies binding nonoverlapping epitopes on the same allergen protein molecule, one might foresee a benefit of multivalent, multispecific IgG blocking therapeutics to mitigate against anaphylaxis from accidental allergen exposure. In its simplest form, this would be a bispecific antibody, this type of molecule could be devised to be biepitopic, that is binding two different epitopes on the same allergen protein, or bispecific, binding two different allergen proteins (48,101). One must first ponder the possibilities involved in the design of such antibodies, considering how they would impact allergen complex formation and crosslinking of FcγRs and possibly FcεRI. Lastly, one could conceivably conjugate Siglec ligands to the therapeutic IgG molecules to further down-regulate allergic effector cells in the presence of allergen protein.
In conclusion, when considering the future possibilities presented by antibody engineering in allergy, it is worth entertaining the idea that as crucial, allergen-specific pathogenic IgE antibody sequences can now be identified from within the repertoire of all antibodies, those sequences in turn might serve as specific therapies to block, down-regulate, and potentially even reprogram the pathogenic response to become more tolerant of these innocuous allergen proteins. With many new advances in antibody engineering, the growth of highly targeted disease specific therapeutics is sure to continue. Adding to this the recent development of methods to now capture naturally occurring allergen specific IgE antibody variable sequences from allergic humans will allow this therapeutic boom to expand deeper into allergic diseases.
ACKNOWLEDGEMENTS
This work was supported by research grants from NIH/National Institute of Allergy and Infectious Disease R01AI155668, R01AI130459, R21AI123307 (to SAS), and Vanderbilt University CTSA UL1TR000445 and the AAAAI Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
ABBREVIATIONS
- Ab
Antibody
- ADA
Anti-drug antibodies
- ADCC
Antibody-dependent cell-mediated cytotoxicity
- ADCP
Antibody-dependent cellular phagocytosis
- ADC
Antibody drug conjugate
- APC
Antigen presenting cell
- CDC
Complement-dependent cytotoxicity
- Fab
Antigen-binding fragment
- Fc
Constant or crystallizable fragment
- FcR
Fc receptor
- ITAM
Immunoreceptor tyrosine-based activation motifs
- ITIM
Immunoreceptor tyrosine-based inhibitory motif
- LSEC
Liver sinusoidal endothelial cells
- mAb
Monoclonal Antibody
- Siglecs
Sialic acid-binding immunoglobulin-like lectins
- scFv
Single-chain variable fragment
- sdFv
Single domain variable regions
Footnotes
Author conflicts of interest:
SAS receives royalties for intellectual property licenses with InBio Inc. and consulting fees from IgGenix Inc. SAS is an inventor on a patent entitled “Generation of human allergen- and helminth-specific IgE monoclonal antibodies for diagnostic and therapeutic use” (U.S. patent no. 11709167), with royalties paid and on a pending patent entitled “Generation of Human Peanut Allergen-Specific IgE Monoclonal Antibodies for Diagnostic and Therapeutic Use” (PCT/US2022/019503), with royalties paid. BWS is a co-founder and principle at Turkey Creek Biotechnology (TCB). TCB was not involved in this work.
REFERENCES
- 1.Wang Z, Wang G, Lu H, Li H, Tang M, Tong A. Development of therapeutic antibodies for the treatment of diseases. Mol Biomed. 2022. Nov 22;3(1):35. doi: 10.1186/s43556-022-00100-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu RM, Hwang YC, Liu IJ, Lee CC, Tsai HZ, Li HJ, et al. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci. 2020. Jan 2;27(1):1. doi: 10.1186/s12929-019-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. 1975. J Immunol. 2005. Mar 1;174(5):2453–5. [PubMed] [Google Scholar]
- 4.Yu RJ, Krantz MS, Phillips EJ, Stone CA Jr. Emerging Causes of Drug-Induced Anaphylaxis: A Review of Anaphylaxis-Associated Reports in the FDA Adverse Event Reporting System (FAERS). J Allergy Clin Immunol Pract. 2021. Feb;9(2):819–829.e2. doi: 10.1016/j.jaip.2020.09.021. Epub 2020 Sep 28.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu LL, Suscovich TJ, Fortune SM, Alter G. Beyond binding: antibody effector functions in infectious diseases. Nat Rev Immunol. 2018. Jan;18(1):46–61. doi: 10.1038/nri.2017.106. Epub 2017 Oct 24.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol. 2014. Oct 20;5:520. doi: 10.3389/fimmu.2014.00520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stoop JW, Zegers BJ, Sander PC, Ballieux RE. Serum immunoglobulin levels in healthy children and adults. Clin Exp Immunol. 1969. Jan;4(1):101–12. [PMC free article] [PubMed] [Google Scholar]
- 8.Delidakis G, Kim JE, George K, Georgiou G. Improving Antibody Therapeutics by Manipulating the Fc Domain: Immunological and Structural Considerations. Annu Rev Biomed Eng. 2022. Jun 6;24:249–274. doi: 10.1146/annurev-bioeng-082721-024500. Epub 2022 Apr 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bournazos S, Ravetch JV. Fcγ receptor pathways during active and passive immunization. Immunol Rev. 2015. Nov;268(1):88–103. doi: 10.1111/imr.12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bournazos S, Gupta A, Ravetch JV. The role of IgG Fc receptors in antibody-dependent enhancement. Nat Rev Immunol. 2020. Oct;20(10):633–643. doi: 10.1038/s41577-020-00410-0. Epub 2020 Aug 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herrada AA, Contreras FJ, Tobar JA, Pacheco R, Kalergis AM. Immune complex-induced enhancement of bacterial antigen presentation requires Fcgamma receptor III expression on dendritic cells. Proc Natl Acad Sci U S A. 2007. Aug 14;104(33):13402–7. doi: 10.1073/pnas.0700999104. Epub 2007 Aug 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Junker F, Gordon J, Qureshi O. Fc Gamma Receptors and Their Role in Antigen Uptake, Presentation, and T Cell Activation. Front Immunol. 2020. Jul 3;11:1393. doi: 10.3389/fimmu.2020.01393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amigorena S, Salamero J, Davoust J, Fridman WH, Bonnerot C. Tyrosine-containing motif that transduces cell activation signals also determines internalization and antigen presentation via type III receptors for IgG. Nature. 1992. Jul 23;358(6384):337–41. doi: 10.1038/358337a0. [DOI] [PubMed] [Google Scholar]
- 14.Wang B, Gallolu Kankanamalage S, Dong J, Liu Y. Optimization of therapeutic antibodies. Antib Ther. 2021. Feb 18;4(1):45–54. doi: 10.1093/abt/tbab003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Balocco R, De Sousa Guimaraes Koch S, Thorpe R, Weisser K, Malan S. New INN nomenclature for monoclonal antibodies. Lancet. 2022. Jan 1;399(10319):24. doi: 10.1016/S0140-6736(21)02732-X. [DOI] [PubMed] [Google Scholar]
- 16.Annunziato F, Romagnani C, Romagnani S. The 3 major types of innate and adaptive cell-mediated effector immunity. J Allergy Clin Immunol. 2015. Mar;135(3):626–35. doi: 10.1016/j.jaci.2014.11.001. Epub 2014 Dec 18. [DOI] [PubMed] [Google Scholar]
- 17.Mills KHG. IL-17 and IL-17-producing cells in protection versus pathology. Nat Rev Immunol. 2023. Jan;23(1):38–54. doi: 10.1038/s41577-022-00746-9. Epub 2022 Jul 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vacca F, Le Gros G. Tissue-specific immunity in helminth infections. Mucosal Immunol. 2022. Jun;15(6):1212–1223. doi: 10.1038/s41385-022-00531-w. Epub 2022 Jun 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nimmerjahn F, Ravetch JV. Fcgamma receptors as regulators of immune responses. Nat Rev Immunol. 2008. Jan;8(1):34–47. doi: 10.1038/nri2206. [DOI] [PubMed] [Google Scholar]
- 20.Getahun A, Cambier JC. Of ITIMs, ITAMs, and ITAMis: revisiting immunoglobulin Fc receptor signaling. Immunol Rev. 2015. Nov;268(1):66–73. doi: 10.1111/imr.12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nehring P, Przybyłkowski A. Is Psoriasis Treatment a Risk Factor for Inflammatory Bowel Disease? Pharmaceut Med. 2020. Aug;34(4):257–262. doi: 10.1007/s40290-020-00340-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iborra M, Beltrán B, Bastida G, Aguas M, Nos P. Infliximab and adalimumab-induced psoriasis in Crohn’s disease: a paradoxical side effect. J Crohns Colitis. 2011. Apr;5(2):157–61. doi: 10.1016/j.crohns.2010.11.001. Epub 2010 Dec 7. [DOI] [PubMed] [Google Scholar]
- 23.Meknache N, Jönsson F, Laurent J, Guinnepain MT, Daëron M. Human basophils express the glycosylphosphatidylinositol-anchored low-affinity IgG receptor FcgammaRIIIB (CD16B). J Immunol. 2009. Feb 15;182(4):2542–50. doi: 10.4049/jimmunol.0801665. [DOI] [PubMed] [Google Scholar]
- 24.Kanagaratham C, El Ansari YS, Lewis OL, Oettgen HC. IgE and IgG Antibodies as Regulators of Mast Cell and Basophil Functions in Food Allergy. Front Immunol. 2020. Dec 11;11:603050. doi: 10.3389/fimmu.2020.603050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ekoff M, Möller C, Xiang Z, Nilsson G. Coaggregation of FcepsilonRI with FcgammaRIIB Inhibits Degranulation but Not Induction of Bcl-2 Family Members A1 and Bim in Mast Cells. Allergy Asthma Clin Immunol. 2006. Sep 15;2(3):87–97. doi: 10.1186/1710-1492-2-3-87. Epub 2006 Sep 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Youngblood BA, Leung J, Falahati R, Williams J, Schanin J, Brock EC, et al. Discovery, Function, and Therapeutic Targeting of Siglec-8. Cells. 2020. Dec 24;10(1):19. doi: 10.3390/cells10010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smiljkovic D, Herrmann H, Sadovnik I, Gamperl S, Berger D, Stefanzl G, et al. Expression and regulation of Siglec-6 (CD327) on human mast cells and basophils. J Allergy Clin Immunol. 2023. Jan;151(1):202–211. doi: 10.1016/j.jaci.2022.07.018. Epub 2022 Aug 8. [DOI] [PubMed] [Google Scholar]
- 28.Youngblood BA, Brock EC, Leung J, Falahati R, Bryce PJ, Bright J, et al. AK002, a Humanized Sialic Acid-Binding Immunoglobulin-Like Lectin-8 Antibody that Induces Antibody-Dependent Cell-Mediated Cytotoxicity against Human Eosinophils and Inhibits Mast Cell-Mediated Anaphylaxis in Mice. Int Arch Allergy Immunol. 2019;180(2):91–102. doi: 10.1159/000501637. Epub 2019 Aug 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schanin J, Gebremeskel S, Korver W, Falahati R, Butuci M, Haw TJ, et al. A monoclonal antibody to Siglec-8 suppresses non-allergic airway inflammation and inhibits IgE-independent mast cell activation. Mucosal Immunol. 2021. Mar;14(2):366–376. doi: 10.1038/s41385-020-00336-9. Epub 2020 Aug 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kimball JA, Norman DJ, Shield CF, Schroeder TJ, Lisi P, Garovoy M, et al. The OKT3 Antibody Response Study: a multicentre study of human anti-mouse antibody (HAMA) production following OKT3 use in solid organ transplantation. Transpl Immunol. 1995. Sep;3(3):212–21. doi: 10.1016/0966-3274(95)80027-1. [DOI] [PubMed] [Google Scholar]
- 31.Chatenoud L CD3-specific antibody-induced active tolerance: from bench to bedside. Nat Rev Immunol. 2003. Feb;3(2):123–32. doi: 10.1038/nri1000. [DOI] [PubMed] [Google Scholar]
- 32.Pedrioli A, Oxenius A. Single B cell technologies for monoclonal antibody discovery. Trends Immunol. 2021. Dec;42(12):1143–1158. doi: 10.1016/j.it.2021.10.008. Epub 2021 Nov 4. [DOI] [PubMed] [Google Scholar]
- 33.Smith SA, Crowe JE Jr. Use of Human Hybridoma Technology To Isolate Human Monoclonal Antibodies. Microbiol Spectr. 2015. Feb;3(1):AID-0027–2014. doi: 10.1128/microbiolspec.AID-0027-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung CH, Mirakhur B, Chan E, Le QT, Berlin J, Morse M, et al. Cetuximab-induced anaphylaxis and IgE specific for galactose-alpha-1,3-galactose. N Engl J Med. 2008. Mar 13;358(11):1109–17. doi: 10.1056/NEJMoa074943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs. 2010. May-Jun;2(3):256–65. doi: 10.4161/mabs.2.3.11641. Epub 2010 May 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lo BK. Antibody humanization by CDR grafting. Methods Mol Biol. 2004;248:135–59. doi: 10.1385/1-59259-666-5:135. [DOI] [PubMed] [Google Scholar]
- 37.Lee EC, Liang Q, Ali H, Bayliss L, Beasley A, Bloomfield-Gerdes T, et al. Complete humanization of the mouse immunoglobulin loci enables efficient therapeutic antibody discovery. Nat Biotechnol. 2014. Apr;32(4):356–63. doi: 10.1038/nbt.2825. Epub 2014 Mar 16. [DOI] [PubMed] [Google Scholar]
- 38.Lonberg N Human antibodies from transgenic animals. Nat Biotechnol. 2005. Sep;23(9):1117–25. doi: 10.1038/nbt1135. [DOI] [PubMed] [Google Scholar]
- 39.Valldorf B, Hinz SC, Russo G, Pekar L, Mohr L, Klemm J, et al. Antibody display technologies: selecting the cream of the crop. Biol Chem. 2021. Mar 23;403(5–6):455–477. doi: 10.1515/hsz-2020-0377. [DOI] [PubMed] [Google Scholar]
- 40.Beerli RR, Rader C. Mining human antibody repertoires. MAbs. 2010. Jul-Aug;2(4):365–78. doi: 10.4161/mabs.12187. Epub 2010 Jul 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sheehan J, Marasco WA. Phage and Yeast Display. Microbiol Spectr. 2015. Feb;3(1):AID-0028–2014. doi: 10.1128/microbiolspec.AID-0028-2014. [DOI] [PubMed] [Google Scholar]
- 42.Bird RE, Hardman KD, Jacobson JW, Johnson S, Kaufman BM, Lee SM, et al. Single-chain antigen-binding proteins. Science. 1988. Oct 21;242(4877):423–6. doi: 10.1126/science.3140379. Erratum in: Science 1989 Apr 28;244(4903):409. [DOI] [PubMed] [Google Scholar]
- 43.Muyldermans S Nanobodies: natural single-domain antibodies. Annu Rev Biochem. 2013;82:775–97. doi: 10.1146/annurev-biochem-063011-092449. Epub 2013 Mar 13. [DOI] [PubMed] [Google Scholar]
- 44.Lee CM, Iorno N, Sierro F, Christ D. Selection of human antibody fragments by phage display. Nat Protoc. 2007;2(11):3001–8. doi: 10.1038/nprot.2007.448. [DOI] [PubMed] [Google Scholar]
- 45.Thornton JM, Laskowski RA, Borkakoti N. AlphaFold heralds a data-driven revolution in biology and medicine. Nat Med. 2021. Oct;27(10):1666–1669. doi: 10.1038/s41591-021-01533-0. [DOI] [PubMed] [Google Scholar]
- 46.Xu Z, Davila A, Wilamowski J, Teraguchi S, Standley DM. Improved Antibody-Specific Epitope Prediction Using AlphaFold and AbAdapt. Chembiochem. 2022. Sep 16;23(18):e202200303. doi: 10.1002/cbic.202200303. Epub 2022 Aug 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bailly M, Mieczkowski C, Juan V, Metwally E, Tomazela D, Baker J, et al. Predicting Antibody Developability Profiles Through Early Stage Discovery Screening. MAbs. 2020. Jan-Dec;12(1):1743053. doi: 10.1080/19420862.2020.1743053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs. 2017. Feb/Mar;9(2):182–212. doi: 10.1080/19420862.2016.1268307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wu L, Seung E, Xu L, Rao E, Lord DM, Wei RR, et al. Trispecific antibodies enhance the therapeutic efficacy of tumor-directed T cells through T cell receptor co-stimulation. Nat Cancer. 2020. Jan;1(1):86–98. doi: 10.1038/s43018-019-0004-z. Epub 2019 Nov 18. [DOI] [PubMed] [Google Scholar]
- 50.Misson Mindrebo L, Liu H, Ozorowski G, Tran Q, Woehl J, Khalek I, et al. Fully synthetic platform to rapidly generate tetravalent bispecific nanobody-based immunoglobulins. Proc Natl Acad Sci U S A. 2023. Jun 13;120(24):e2216612120. doi: 10.1073/pnas.2216612120. Epub 2023 Jun 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wurth MA, Hadadianpour A, Horvath DJ, Daniel J, Bogdan O, Goleniewska K, et al. Human IgE mAbs define variability in commercial Aspergillus extract allergen composition. JCI Insight. 2018. Oct 18;3(20):e123387. doi: 10.1172/jci.insight.123387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Croote D, Darmanis S, Nadeau KC, Quake SR. High-affinity allergen-specific human antibodies cloned from single IgE B cell transcriptomes. Science. 2018. Dec 14;362(6420):1306–1309. doi: 10.1126/science.aau2599. [DOI] [PubMed] [Google Scholar]
- 53.Akdis M, Akdis CA. IgE class switching and cellular memory. Nat Immunol. 2012. Mar 19;13(4):312–4. doi: 10.1038/ni.2266. [DOI] [PubMed] [Google Scholar]
- 54.Gould HJ, Wu YB. IgE repertoire and immunological memory: compartmental regulation and antibody function. Int Immunol. 2018. Aug 30;30(9):403–412. doi: 10.1093/intimm/dxy048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Looney TJ, Lee JY, Roskin KM, Hoh RA, King J, Glanville J, et al. Human B-cell isotype switching origins of IgE. J Allergy Clin Immunol. 2016. Feb;137(2):579–586.e7. doi: 10.1016/j.jaci.2015.07.014. Epub 2015 Aug 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoh RA, Joshi SA, Lee JY, Martin BA, Varma S, Kwok S, et al. Origins and clonal convergence of gastrointestinal IgE+ B cells in human peanut allergy. Sci Immunol. 2020. Mar 6;5(45):eaay4209. doi: 10.1126/sciimmunol.aay4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He JS, Subramaniam S, Narang V, Srinivasan K, Saunders SP, Carbajo D, et al. IgG1 memory B cells keep the memory of IgE responses. Nat Commun. 2017. Sep 21;8(1):641. doi: 10.1038/s41467-017-00723-0. Erratum in: Nat Commun. 2018 Mar 1;9(1):968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Taeye SW, Rispens T, Vidarsson G. The Ligands for Human IgG and Their Effector Functions. Antibodies (Basel). 2019. Apr 25;8(2):30. doi: 10.3390/antib8020030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shamji MH, Ljørring C, Francis JN, Calderon MA, Larché M, Kimber I, et al. Functional rather than immunoreactive levels of IgG4 correlate closely with clinical response to grass pollen immunotherapy. Allergy. 2012. Feb;67(2):217–26. doi: 10.1111/j.1398-9995.2011.02745.x. Epub 2011 Nov 14. [DOI] [PubMed] [Google Scholar]
- 60.Kamat V, Boutot C, Rafique A, Granados C, Wang J, Badithe A, et al. High affinity human Fc specific monoclonal antibodies for capture kinetic analyses of antibody-antigen interactions. Anal Biochem. 2022. Mar 1;640:114455. doi: 10.1016/j.ab.2021.114455. Epub 2021 Nov 14. [DOI] [PubMed] [Google Scholar]
- 61.Chappel MS, Isenman DE, Everett M, Xu YY, Dorrington KJ, Klein MH. Identification of the Fc gamma receptor class I binding site in human IgG through the use of recombinant IgG1/IgG2 hybrid and point-mutated antibodies. Proc Natl Acad Sci U S A. 1991. Oct 15;88(20):9036–40. doi: 10.1073/pnas.88.20.9036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes JM, Zhang Y, et al. Effector-attenuating Substitutions That Maintain Antibody Stability and Reduce Toxicity in Mice. J Biol Chem. 2017. Mar 3;292(9):3900–3908. doi: 10.1074/jbc.M116.767749. Epub 2017 Jan 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Isaacs JD, Greenwood J, Waldmann H. Therapy with monoclonal antibodies. II. The contribution of Fc gamma receptor binding and the influence of C(H)1 and C(H)3 domains on in vivo effector function. J Immunol. 1998. Oct 15;161(8):3862–9. [PubMed] [Google Scholar]
- 64.Sorkin LS, Otto M, Baldwin WM 3rd, Vail E, Gillies SD, Handgretinger R, et al. Anti-GD(2) with an FC point mutation reduces complement fixation and decreases antibody-induced allodynia. Pain. 2010. Apr;149(1):135–142. doi: 10.1016/j.pain.2010.01.024. Epub 2010 Feb 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vafa O, Gilliland GL, Brezski RJ, Strake B, Wilkinson T, Lacy ER, et al. An engineered Fc variant of an IgG eliminates all immune effector functions via structural perturbations. Methods. 2014. Jan 1;65(1):114–26. doi: 10.1016/j.ymeth.2013.06.035. Epub 2013 Jul 17. [DOI] [PubMed] [Google Scholar]
- 66.Mimoto F, Igawa T, Kuramochi T, Katada H, Kadono S, Kamikawa T, et al. Novel asymmetrically engineered antibody Fc variant with superior FcγR binding affinity and specificity compared with afucosylated Fc variant. MAbs. 2013. Mar-Apr;5(2):229–36. doi: 10.4161/mabs.23452. Epub 2013 Feb 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oganesyan V, Damschroder MM, Leach W, Wu H, Dall’Acqua WF. Structural characterization of a mutated, ADCC-enhanced human Fc fragment. Mol Immunol. 2008. Apr;45(7):1872–82. doi: 10.1016/j.molimm.2007.10.042. [DOI] [PubMed] [Google Scholar]
- 68.Richards JO, Karki S, Lazar GA, Chen H, Dang W, Desjarlais JR. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol Cancer Ther. 2008. Aug;7(8):2517–27. doi: 10.1158/1535-7163.MCT-08-0201. [DOI] [PubMed] [Google Scholar]
- 69.van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martínez-Martínez P, Vermeulen E, et al. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007. Sep 14;317(5844):1554–7. doi: 10.1126/science.1144603. [DOI] [PubMed] [Google Scholar]
- 70.Silva JP, Vetterlein O, Jose J, Peters S, Kirby H. The S228P mutation prevents in vivo and in vitro IgG4 Fab-arm exchange as demonstrated using a combination of novel quantitative immunoassays and physiological matrix preparation. J Biol Chem. 2015. Feb 27;290(9):5462–9. doi: 10.1074/jbc.M114.600973. Epub 2015 Jan 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Handlogten MW, Peng L, Christian EA, Xu W, Lin S, Venkat R, et al. Prevention of Fab-arm exchange and antibody reduction via stabilization of the IgG4 hinge region. MAbs. 2020. Jan-Dec;12(1):1779974. doi: 10.1080/19420862.2020.1779974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lu J, Chu J, Zou Z, Hamacher NB, Rixon MW, Sun PD. Structure of FcγRI in complex with Fc reveals the importance of glycan recognition for high-affinity IgG binding. Proc Natl Acad Sci U S A. 2015. Jan 20;112(3):833–8. doi: 10.1073/pnas.1418812112. Epub 2015 Jan 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Arnold JN, Wormald MR, Sim RB, Rudd PM, Dwek RA. The impact of glycosylation on the biological function and structure of human immunoglobulins. Annu Rev Immunol. 2007;25:21–50. doi: 10.1146/annurev.immunol.25.022106.141702. [DOI] [PubMed] [Google Scholar]
- 74.Tao MH, Morrison SL. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J Immunol. 1989. Oct 15;143(8):2595–601. [PubMed] [Google Scholar]
- 75.Li M, Zhao R, Chen J, Tian W, Xia C, Liu X, et al. Next generation of anti-PD-L1 Atezolizumab with enhanced anti-tumor efficacy in vivo. Sci Rep. 2022. Mar 28;12(1):5255. doi: 10.1038/s41598-022-09081-4. Erratum for: Sci Rep. 2021 Mar 11;11(1):5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Quast I, Keller CW, Maurer MA, Giddens JP, Tackenberg B, Wang LX, et al. Sialylation of IgG Fc domain impairs complement-dependent cytotoxicity. J Clin Invest. 2015. Nov 2;125(11):4160–70. doi: 10.1172/JCI82695. Epub 2015 Oct 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chu SY, Vostiar I, Karki S, Moore GL, Lazar GA, Pong E, et al. Inhibition of B cell receptor-mediated activation of primary human B cells by coengagement of CD19 and FcgammaRIIb with Fc-engineered antibodies. Mol Immunol. 2008. Sep;45(15):3926–33. doi: 10.1016/j.molimm.2008.06.027. Epub 2008 Aug 8. [DOI] [PubMed] [Google Scholar]
- 78.Lazar GA, Dang W, Karki S, Vafa O, Peng JS, Hyun L, et al. Engineered antibody Fc variants with enhanced effector function. Proc Natl Acad Sci U S A. 2006. Mar 14;103(11):4005–10. doi: 10.1073/pnas.0508123103. Epub 2006 Mar 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007. Sep;7(9):715–25. doi: 10.1038/nri2155. Epub 2007 Aug 17. [DOI] [PubMed] [Google Scholar]
- 80.Dall’Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem. 2006. Aug 18;281(33):23514–24. doi: 10.1074/jbc.M604292200. Epub 2006 Jun 21. [DOI] [PubMed] [Google Scholar]
- 81.Zalevsky J, Chamberlain AK, Horton HM, Karki S, Leung IW, Sproule TJ, et al. Enhanced antibody half-life improves in vivo activity. Nat Biotechnol. 2010. Feb;28(2):157–9. doi: 10.1038/nbt.1601. Epub 2010 Jan 17.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vaccaro C, Zhou J, Ober RJ, Ward ES. Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat Biotechnol. 2005. Oct;23(10):1283–8. doi: 10.1038/nbt1143. Epub 2005 Sep 25.. [DOI] [PubMed] [Google Scholar]
- 83.Khongorzul P, Ling CJ, Khan FU, Ihsan AU, Zhang J. Antibody-Drug Conjugates: A Comprehensive Review. Mol Cancer Res. 2020. Jan;18(1):3–19. doi: 10.1158/1541-7786.MCR-19-0582. Epub 2019 Oct 28.. [DOI] [PubMed] [Google Scholar]
- 84.Islam M, Arlian BM, Pfrengle F, Duan S, Smith SA, Paulson JC. Suppressing Immune Responses Using Siglec Ligand-Decorated Anti-receptor Antibodies. J Am Chem Soc. 2022. Jun 1;144(21):9302–9311. doi: 10.1021/jacs.2c00922. Epub 2022 May 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kanchan K, Grinek S, Bahnson HT, Ruczinski I, Shankar G, Larson D, et al. HLA alleles and sustained peanut consumption promote IgG4 responses in subjects protected from peanut allergy. J Clin Invest. 2022. Jan 4;132(1):e152070. doi: 10.1172/JCI152070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Okamoto S, Taniuchi S, Sudo K, Hatano Y, Nakano K, Shimo T, et al. Predictive value of IgE/IgG4 antibody ratio in children with egg allergy. Allergy Asthma Clin Immunol. 2012. Jun 7;8(1):9. doi: 10.1186/1710-1492-8-9. Erratum in: Allergy Asthma Clin Immunol. 2013 Sep 09;9(1):34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wright BL, Kulis M, Orgel KA, Burks AW, Dawson P, Henning AK, et al. ; Consortium of Food Allergy Research. Component-resolved analysis of IgA, IgE, and IgG4 during egg OIT identifies markers associated with sustained unresponsiveness. Allergy. 2016. Nov;71(11):1552–1560. doi: 10.1111/all.12895. Epub 2016 Jun 13.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Caubet JC, Lin J, Ahrens B, Gimenez G, Bardina L, Niggemann B, et al. Natural tolerance development in cow’s milk allergic children: IgE and IgG4 epitope binding. Allergy. 2017. Nov;72(11):1677–1685. doi: 10.1111/all.13167. Epub 2017 Jun 9.. [DOI] [PubMed] [Google Scholar]
- 89.Suárez-Fariñas M, Suprun M, Chang HL, Gimenez G, Grishina G, Getts R, et al. Predicting development of sustained unresponsiveness to milk oral immunotherapy using epitope-specific antibody binding profiles. J Allergy Clin Immunol. 2019. Mar;143(3):1038–1046. doi: 10.1016/j.jaci.2018.10.028. Epub 2018 Dec 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.van Erp FC, Knol EF, Pontoppidan B, Meijer Y, van der Ent CK, Knulst AC. The IgE and basophil responses to Ara h 2 and Ara h 6 are good predictors of peanut allergy in children. J Allergy Clin Immunol. 2017. Jan;139(1):358–360.e8. doi: 10.1016/j.jaci.2016.06.041. Epub 2016 Aug 8. [DOI] [PubMed] [Google Scholar]
- 91.Kukkonen AK, Pelkonen AS, Mäkinen-Kiljunen S, Voutilainen H, Mäkelä MJ. Ara h 2 and Ara 6 are the best predictors of severe peanut allergy: a double-blind placebo-controlled study. Allergy. 2015. Oct;70(10):1239–45. doi: 10.1111/all.12671. Epub 2015 Jul 1.. [DOI] [PubMed] [Google Scholar]
- 92.Orengo JM, Radin AR, Kamat V, Badithe A, Ben LH, Bennett BL, et al. Treating cat allergy with monoclonal IgG antibodies that bind allergen and prevent IgE engagement. Nat Commun. 2018. Apr 12;9(1):1421. doi: 10.1038/s41467-018-03636-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gevaert P, De Craemer J, De Ruyck N, Rottey S, de Hoon J, Hellings PW, et al. Novel antibody cocktail targeting Bet v 1 rapidly and sustainably treats birch allergy symptoms in a phase 1 study. J Allergy Clin Immunol. 2022. Jan;149(1):189–199. doi: 10.1016/j.jaci.2021.05.039. Epub 2021 Jun 11.. [DOI] [PubMed] [Google Scholar]
- 94.Pena-Castellanos G, Smith BRE, Pomés A, Smith SA, Stigler MA, Widauer HL, et al. Biological activity of human IgE monoclonal antibodies targeting Der p 2, Fel d 1, Ara h 2 in basophil mediator release assays. Front Immunol. 2023. May 9;14:1155613. doi: 10.3389/fimmu.2023.1155613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Smith SA, Chruszcz M, Chapman MD, Pomés A. Human Monoclonal IgE Antibodies-a Major Milestone in Allergy. Curr Allergy Asthma Rep. 2023. Jan;23(1):53–65. doi: 10.1007/s11882-022-01055-w. Epub 2022 Dec 2.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Suber J, Zhang Y, Ye P, Guo R, Burks AW, Kulis MD, et al. Novel peanut-specific human IgE monoclonal antibodies enable screens for inhibitors of the effector phase in food allergy. Front Immunol. 2022. Sep 29;13:974374. doi: 10.3389/fimmu.2022.974374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Khatri K, Richardson CM, Glesner J, Kapingidza AB, Mueller GA, Zhang J, et al. Human IgE monoclonal antibody recognition of mite allergen Der p 2 defines structural basis of an epitope for IgE cross-linking and anaphylaxis in vivo. PNAS Nexus. 2022. Jun 2;1(3):pgac054. doi: 10.1093/pnasnexus/pgac054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hadadianpour A, Daniel J, Zhang J, Spiller BW, Makaraviciute A, DeWitt ÅM, et al. Human IgE mAbs identify major antigens of parasitic worm infection. J Allergy Clin Immunol. 2022. Dec;150(6):1525–1533. doi: 10.1016/j.jaci.2022.05.022. Epub 2022 Jun 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chiu ML, Gilliland GL. Engineering antibody therapeutics. Curr Opin Struct Biol. 2016. Jun;38:163–73. doi: 10.1016/j.sbi.2016.07.012. Epub 2016 Aug 12.. [DOI] [PubMed] [Google Scholar]
- 100.Klein C, Schaefer W, Regula JT, Dumontet C, Brinkmann U, Bacac M, et al. Engineering therapeutic bispecific antibodies using CrossMab technology. Methods. 2019. Feb 1;154:21–31. doi: 10.1016/j.ymeth.2018.11.008. Epub 2018 Nov 16.. [DOI] [PubMed] [Google Scholar]
- 101.Coloma MJ, Morrison SL. Design and production of novel tetravalent bispecific antibodies. Nat Biotechnol. 1997. Feb;15(2):159–63. doi: 10.1038/nbt0297-159. [DOI] [PubMed] [Google Scholar]