

Abstract
Understanding breast cancer genetic risk relies on identifying causal variants and candidate target genes in risk loci identified by genome-wide association studies (GWAS), which remains challenging. Since most loci fall in active gene regulatory regions, we developed a novel approach facilitated by pinpointing the variants with greater regulatory potential in the disease’s tissue of origin. Through genome-wide differential allelic expression (DAE) analysis, using microarray data from 64 normal breast tissue samples, we mapped the variants associated with DAE (daeQTLs). Then, we intersected these with GWAS data to reveal candidate risk regulatory variants and analysed their cis-acting regulatory potential. Finally, we validated our approach by extensive functional analysis of the 5q14.1 breast cancer risk locus. We observed widespread gene expression regulation by cis-acting variants in breast tissue, with 65% of coding and noncoding expressed genes displaying DAE (daeGenes). We identified over 54 K daeQTLs for 6761 (26%) daeGenes, including 385 daeGenes harbouring variants previously associated with BC risk. We found 1431 daeQTLs mapped to 93 different loci in strong linkage disequilibrium with risk-associated variants (risk-daeQTLs), suggesting a link between risk-causing variants and cis-regulation. There were 122 risk-daeQTL with stronger cis-acting potential in active regulatory regions with protein binding evidence. These variants mapped to 41 risk loci, of which 29 had no previous report of target genes and were candidates for regulating the expression levels of 65 genes. As validation, we identified and functionally characterised five candidate causal variants at the 5q14.1 risk locus targeting the ATG10 and ATP6AP1L genes, likely acting via modulation of alternative transcription and transcription factor binding. Our study demonstrates the power of DAE analysis and daeQTL mapping to identify causal regulatory variants and target genes at breast cancer risk loci, including those with complex regulatory landscapes. It additionally provides a genome-wide resource of variants associated with DAE for future functional studies.
Keywords: Cis-regulation, Polymorphism, Cancer predisposition, Breast cancer
Subject terms: Breast cancer, Cancer genomics, Functional genomics, Genetic markers
Introduction
Genome-wide association studies (GWAS) for breast cancer (BC) have identified hundreds of risk-associated loci and have generated long lists of candidate loci requiring further validation1. Nevertheless, the identification of the causal variants and their target genes, as well as understanding the underlying biological mechanisms, remain challenging. This is because disease risk loci often have many variants in high linkage disequilibrium (LD) with the risk-associated variant, harbour multiple genes and mainly fall in noncoding genome regions2. However, the overrepresentation of potential causal variants at active gene regulatory regions3,4 indicates that variants regulating gene expression levels likely influence BC genetic predisposition, both proximally and over a long range5–11. These variants have commonly been mapped by expression quantitative trait loci (eQTL) analysis, but this approach is impacted by the effects of negative feedback control and environmental factors12. An increasingly popular alternative approach is to detect imbalances in allelic transcript levels—differential allelic expression (DAE). By comparing the relative expression of the two alleles in a heterozygous individual, each allele will serve as an internal standard for the other, thus controlling for trans-regulatory and environmental factors affecting both alleles13,14. Consequently, this directly indicates regulatory variants acting in cis (rSNPs).
Given the importance of cis-regulatory variants for BC susceptibility, a genome-wide map of cis-regulatory variants would be key to interpreting GWAS results and identifying causal variants of risk. Studies in various healthy tissues showed that DAE is a relatively common event13,15–19. Given that gene expression regulation is tissue-specific, performing these studies in the tissue from which the disease arises, namely, normal breast tissue, is essential. Although others have used allelic expression analysis to identify BC risk, this was carried out in tumour tissue or lymphoblastoid cells20,21. This study proposes an integrative approach to identify causal variants of risk that have a cis-regulatory role (Fig. 1): to combine GWAS results with SNPs associated with DAE levels in normal breast tissue. Hence, we first carried out DAE analysis in normal breast tissue samples at a genome-wide level, then mapped the candidate risk regulatory variants for GWAS loci and finally functionally unveiled the mechanisms underlying BC risk at a selected locus.
Fig. 1.

Strategy framework used to identify causal variants and target genes associated with breast cancer risk. aeSNP an SNP that passed quality control and at which allelic expression (AE) was measured, daeSNP an aeSNP showing differential AE (DAE), Genotyped SNP an SNP with genotype information (either genotyped in the study or imputed) and tested for association with AE ratios, daeQTL an SNP associated with AE ratios measured for a daeSNP, risk-daeQTL a daeQTL with a r2 ≥ 0.4 with a GWAS hit variant, candidate risk rSNP a variant with a r2 ≥ 0.95 with the risk-daeQTL.
Methods
SNP and call filtering at the gDNA and cDNA levels
We used an Illumina Infinium Exon510S-Duo arrays dataset of normal breast tissue available from Gene Expression Omnibus (GEO, www.ncbi.nlm.nih.gov/geo/) under accession number GSE3502322. It consists of 66 samples of DNA and cDNA (derived from total RNA) run on Illumina Infinium Exon510S-Duo arrays. These exon-centric microarrays contain probes for 511,354 SNPs, with more than 60% of the markers located within 10 kb of a gene and targeting more than 99.9% of human RefSeq genes. Sample filtering and normalisation were performed as described previously, and 12 samples displaying poor dynamic range of intensities in either channel (IQR of non-normalized log2 X or log2 Y < 1), or showing evidence of clear contamination of RNA with DNA sample were removed from further analysis22. For the remaining 64 samples, we used within-array strip-level quantile normalization to correct for dye-biases between the two channels (in general X = Cy5 = allele A and Y = Cy3 = allele B) from each array to obtain normalized intensities (X* and Y*) for each SNP. The function stripNormalize in the crlmm R package (version 1.10.0) was used to normalize the data. Log-ratios (M = log2 X* − log2 Y*) and average log-intensities [S = 0.5(log2 X* + log2 Y*)] were then calculated for each SNP on each array. We work on the log-ratio scale rather than the β-scale [β = X*/(X* + Y*), as used in Ref.13, as it has been shown to have more desirable properties for statistical testing in other applications of Infinium technology23.
Genome-wide DAE analysis
Allelic expression was measured in the filtered dataset of SNPs and samples in a varying number of heterozygous (AB) individuals for each transcribed SNP (aeSNP). As cDNA was prepared from total RNA, without selection for poly-A mRNAs, AE was measured for variants in fully processed and unspliced primary transcripts. Allelic expression ratios (AE ratios) were defined as the log2 of the ratio between the levels of allele A transcript and the levels of allele B transcript (heterozygote ratio), normalised by the same heterozygote ratio calculated for genomic DNA (gDNA) (Fig. S1), to account for copy number variation and correct for technical biases. Differential allelic expression (DAE) was called at the sample level when AE ratios were greater than 0.58 or less than − 0.58 (corresponding to the log2 of a 1.5-fold difference between alleles).
To define aeSNPs displaying mono-allelic expression (maeSNPs)24 we first identified SNPs presenting extreme allelic expression (AE) ratios, specifically with values greater than 0.58 or lower than − 0.58, without any heterozygous samples showing intermediate values. Next, using a binomial test, we tested the distribution of AE ratios at this subset of aeSNPs, displaying extreme ratios for an equal distribution of heterozygous samples showing positive and negative AE ratios on a variant-by-variant basis. We applied multiple testing correction to p-values, and variants with a false discovery rate (FDR) above 5% were considered to display a random choice of expression between alleles and classified as maeSNPs. Genes with at least one maeSNP were labelled maeGenes.
After filtering out maeSNPs, the remaining aeSNPs were tested for differential allelic expression by applying an Equal or Given Proportions test (prop.test function in R), with the alternative hypothesis that the proportion of heterozygotes with absolute AE ratios ≥ 0.58 is greater than 10% for any given SNP. The resulting p-values were corrected using a false discovery rate of 5% to define daeSNPs (Fig. 1, Suppl Fig. S1) i.e., heterozygous transcribed variants displaying differential allelic expression. Genes with at least one daeSNP were henceforth denominated daeGenes.
Validation of nine daeSNPs was performed by TaqMan® PCR technology, as described previously25, in 25 independent normal breast tissue samples heterozygous for a variable number of individuals per SNP using the following TaqMan® Genotyping Assays predesigned by Applied Biosystems: C___8354687_10; C__29939330_20; C__31232634_10; C___3133316_10; C__11844169_10; C___2627792_10; C___1517694_1_; C____787630_20; C___3108259_10. The prop.test was equally applied to confirm the presence of differential allelic expression.
Genotype imputation
Imputation was run on the Illumina Exon 510 Duo germline genotype data from the 64 samples that passed microarray quality control filters. Before imputation data, quality control was applied to the genotyping data, and SNPs with call rates < 85%, minor allele frequency < 0.01, and Hardy–Weinberg equilibrium with p-value < 1.0E−05 were excluded from the analysis. Imputation was performed using MACH1.026 and the phased haplotypes for HapMap3 release (HapMap3 NCBI Build 36, CEU panel—Utah residents with Northern and Western European ancestry) as a reference panel. We applied the recommended two-step imputation process: model parameters (crossover and error rates) were estimated before imputation using all haplotypes from the study subjects and running 100 iterations of the Hidden Markov Model (HMM) with the command option—greedy and -r 100. Genotype imputation was then carried out using the model parameter estimates from the previous round with command options of -greedy, -mle, and -mldetails specified. Imputation results were assessed by the platform-specific measures of imputation uncertainty for each SNP (rq Score) and filtered for an rq-score ≥ 0.3, as suggested in the author webpage (http://csg.sph.umich.edu/abecasis/mach/tour/) and MAF ≥ 0.01.
This imputation was conducted using HapMap3, which was the most widely used and trusted reference dataset for populations of European ancestry at that time. Since then, more comprehensive reference panels such as the 1000 Genomes Project and the Haplotype Reference Consortium (HRC) have become available, providing improved accuracy. Consequently, for subsequent analyses, including proxy retrieval, we utilized these updated reference panels to enhance the robustness of our findings.
Ancestry analysis
Genetic similarity, as a proxy for global ancestry, was determined by Principal Component Analysis on imputed genotypes from 64 samples, as implemented in PLINK (v.2.00a6LM) using 1000 Genomes Project data (release 20181203) as reference. Data wrangling was performed using bcftools (v.1.10.2, htslib v1.10.2–3).
daeQTL mapping
daeSNPs are not necessarily the regulatory variants (rSNPs) responsible for the observed allelic imbalance. Therefore, mapping of candidate rSNPs associated with the DAE observed—henceforth designated as daeQTLs (differential allelic expression quantitative trait loci) (Fig. S1, Fig. 1) was performed to identify neighboring variants that are statistically associated with the AE ratios observed at the daeSNPs. This approach took into consideration the pattern of AE ratio distribution displayed by each daeSNP. This pattern is highly dependent on the LD between the daeSNP and the rSNP acting upon the gene27.
To test the association between candidate SNP zygosity and the allelic expression of a daeSNP, henceforth designated daeQTL analysis (differential allelic expression quantitative trait loci analysis), we considered the pattern of the allelic expression (AE) ratio distribution displayed at each daeSNP, as this is dependent on the linkage disequilibrium between the daeSNP and the rSNP. When a single rSNP is in strong LD (r2 ~ 1) with the daeSNP, the normalised AE ratios for all heterozygotes will be unidirectional, with all samples preferentially expressing the same allele (i.e., all samples exhibiting either positive AE ratios or negative AE ratios). In this case, a one-sample Wilcox test was used to compare the mean normalised AE ratios for samples heterozygous for the candidate rSNP to 0.
When the rSNP is not in r2 ~ 1 with the daeSNP, the distribution of the AE ratios will depend on the rSNP-daeSNP haplotypes present in the analysed samples. In this scenario we applied, we applied a two-sample Wilcox test for the null hypothesis that the absolute AE ratios at the samples heterozygous for the candidate rSNP are higher than the absolute AE ratios at the samples homozygous for the tested rSNP. These tests were performed for rSNP-daeSNP pairs located within 500 kb of each other. p-values were adjusted with the Benjamini‒Hochberg method28, using all daeSNP/tested SNP pairs, with the distance between them as a covariate (package ihw, R)29 and reported as significant when the false discovery rate was below 5%.
Breast cancer GWAS data retrieval
One thousand and eleven GWAS-significant risk-associated SNPs for BC published until April 2018 were retrieved from the NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog)30 using the gwasrapidd R package31. We applied a suggestive p-value threshold (p ≤ 1.0E−05) to capture a broader set of potentially significant variants, including those identified in meta-analyses and replication studies, to ensure the inclusion of biologically relevant findings. This approach aimed to provide a comprehensive and robust analysis. Additionally, we included only studies involving European populations and the following reported traits: “Breast cancer”, “Breast cancer (early onset)”, “Breast cancer (estrogen-receptor negative)”, “Breast cancer (male)”, “Breast cancer in BRCA1 mutation carriers”, “Breast cancer in BRCA2 mutation carriers”, “Breast cancer male”, and “Breast cancer and/or colorectal cancer”. The complete list of SNPs is presented in Table S1.
Proxy SNP retrieval
Variants in LD with index SNPs were retrieved from Ensembl32 using the function get_ld_variants_by_window() from the ensemblr R package (https://github.com/ramiromagno/ensemblr) using the 1000 GENOMES project data (phase_3) for the CEU population and a genomic window size of 500 kb (250 kb upstream and downstream of the queried variant). The r2 cut-off used varied between 0.2 and 0.95 depending on the analysis and is indicated in each analysis description.
Annotation of variants
Variants were annotated according to hg38/GRCh38 with biomaRt v 2.40.5. aeSNP consequence types were categorised as follows: UTR if classified as 3_prime_UTR_variant or 5_prime_UTR_variant; coding if classified as coding_sequence_variant, incomplete_terminal_codon_variant, missense_variant, stop_retained_variant, synonymous_variant, stop_lost, start_lost, stop_gained, splice_region_variant, splice_acceptor_variant or splice_donor_variant; intronic if classified as intron_variant; and noncoding_transcript_variant if classified as noncoding_transcript_variant, noncoding_transcript_exon_variant or mature_miRNA_variant. We classified aeSNPs further according to gene biotype as follows: pseudogene if located in IG_C_pseudogene, processed_pseudogene, transcribed_unprocessed_pseudogene, transcribed_unitary_pseudogene, translated_unprocessed_pseudogene, unprocessed_pseudogene, unitary_pseudogene, transcribed_processed_pseudogene, polymorphic_pseudogene or rRNA_pseudogene; protein-coding gene if located in protein_coding, IG_V_gene, TR_C_gene, TR_J_gene, TR_V_gene or TEC; and noncoding_rna if located in lncRNA, miRNA, misc_RNA, snRNA, snoRNA, scaRNA or ribozyme.
To test whether classes of consequence type and gene biotype were overrepresented (i.e., enriched) in the list of daeSNPs, we applied two-tailed Fisher’s exact tests. Information from imprinted genes was retrieved from a comprehensive study of genomic imprinting in the breast33 and from the geneimprint database (http://www.geneimprint.com) searching for Imprinted Genes: by Species: Human.
Retrieval of previously suggested BC target genes
Genes previously suggested as targets of cis-acting regulatory variation in post-GWAS studies for BC, with extensive fine-scale mapping and in silico prediction or functional analysis, and those classified as Inquisit 1 by Fachal et al.4 are indicated in Table S2.
GTEx eQTL and gene expression data retrieval
The Genotype-Tissue Expression (GTEx) project identified expression quantitative trait loci (eQTL) using normal mammary tissue samples34. eGenes (genes with at least one SNP in cis significantly associated, at a false discovery rate (FDR) of ≤ 0.05, with expression differences of that gene) and significant variant-gene associations based on permutations were downloaded from GTEx Analysis V8 (dbGaP Accession phs000424.v8.p2, available on 18/07/2019).
All SNP-gene associations tested for breast mammary tissue, including nonsignificant and gene expression levels (TPM), were downloaded from GTEx Analysis V7 (available on 2016-01-15).
Comparison of daeGenes, eGenes and gwasGenes
First, the list of publicly available eGenes was compared with the daeGenes identified in our study, restricting this comparison to genes analysed in both datasets. Then, we investigated the percentage of gwasGenes, defined as genes containing variants in moderate to strong LD (r2 ≥ 0.4) with GWAS index SNPs, displaying evidence of cis-regulation by either DAE or eQTL analysis.
Functional characterisation of candidate risk SNPs
daeQTLs in moderate to strong linkage disequilibrium (LD) (r2 ≥ 0.4) with GWAS index SNPs were defined as risk-daeQTLs. These variants, along with their proxies (r2 ≥ 0.95), were considered candidate risk rSNPs in the subsequent analysis. Candidate risk rSNPs were examined for regulatory potential by assessing the overlap of the variant’s location with epigenetic marks derived from the ENCODE35 and NIH Roadmap Epigenomics project data36 using the R package haploR (https://github.com/cran/haploR). Candidate causal variants (variants overlapping with DNase I hypersensitivity sites and H3K4me1 or H3K4me3 or histone modifications in normal breast or breast tumour cell lines) at the 5q14.1–14.2 locus were further analysed regarding their genomic context and transcription factor (TF) binding using the UCSC Genome Browser37,38, HaploReg v4.139 and RegulomeDB v1.140 tools. Emphasis was given to overlapping with transcription factor (TF) binding identified in breast myoepithelial cells (BR. MYO, E027), human mammary epithelial cells (HMECs, E119), variant human mammary epithelial cells (vHMECs, E028) and two BC cell lines (MCF-7 and T47D). Allele-specific epigenetic modifications (H3k4me3 and DNase I), RNA polymerase II (POL2) and transcription factors (TF) binding with alignment data available in HMEC, MCF-7 and MCF-10A breast cancer cell lines from ENCODE were retrieved and visualised using the Integrative Genomics Viewer (IGV Version 2.3.71) tool41, to analyse protein‒DNA interactions and allelic preferential binding. Differential allelic binding was analysed in heterozygous candidate risk rSNPs located within TF binding peaks in experiments with a read coverage at the SNP site ≥ 20. We applied a two-tailed binomial test with the null hypothesis assuming no bias (balanced binding of the protein to the two alleles of the variant). The p-value was corrected for multiple testing using the R package qvalue42. When multiple tracks for the same SNP, trait and cell line existed, only the p-value for the experiment with higher total read counts was reported in the main manuscript.
Analysis related to alternative transcription at the 5q14.1–14.2 locus was carried out in three ways. First, sQTLseekeR (v1.4)43 was used to test the association of genetic variants with alternative isoform expression in both normal breast and tumour tissue using total read counts derived from RNA-seq data from the TCGA (TCGA-BRCA, hg19) and GTEx (phs000424.v6.p1, hg38) projects. Only ATG10 displayed sufficient alternative transcription dispersion to allow sQTL analysis. Additionally, all SNPs within 5 kb upstream or downstream of ATG10 were included in the analysis, not only the candidate risk rSNPs, to increase the stringency of the association exercise. P-values for all SNPs tested for ATG10 sQTL analysis were controlled for multiple testing using a 5% FDR. Correlation analyses between -log10 (FDR q-value) and LD (r2) with rs7707921 were performed using Pearson’s test. Then, the overlapping of variant location with RNA processing-associated proteins was assessed using CLIP data retrieved from POSTAR2 (http://lulab.life.tsinghua.edu.cn/postar/)44 and from RBP-Var (http://www.rbp-var.biols.ac.cn/)45, which additionally informed on riboSNitch potential46. Finally, allele-specific RBP binding predictions were performed with RBPmap47 using the analysed variant flanking sequence (30 nucleotides on each side, with the variant at index 31) using all available human RBP motifs.
Haplotype analysis
Haplotypes in the 5q14.1–14.2 region were analysed on Haploview 4.2 using the imputed genotypes from the 64 normal breast tissue samples48. For candidate risk SNPs whose genotype was not possible to determine (because it was neither genotyped nor imputed), a proxy SNP in strong LD (r2 ≥ 0.95) was used instead. Haplotype blocks were generated using the default algorithm.
TCGA-BRCA gene expression analysis
Processed gene expression and isoform expression from RNA-Seq data for 113 normal solid tissues and 1102 primary solid tumours from the TCGA-BRCA project, together with corresponding clinical data, were retrieved from the Genomic Data Commons archive using the R package TCGAbiolinks49 accessed in October 2018. Isoform expression was annotated according to the genome assembly hg19, and total gene expression was annotated according to hg38. We applied two-sample Wilcoxon tests to compare the mean expression of ATG10 isoforms between normal-solid tissues (normal-matched) and breast tumours, correcting for multiple testing with the Benjamini and Hochberg (BH) procedure. We applied Pearson’s test to correlate gene expression among ATG10, RPS23, and ATP6AP1L. Spearman’s test was applied to correlate ATG10, RPS23, and ATP6AP1L with MYC and MAX gene expression.
Results
Cis-regulatory variation is common in normal breast tissue
Genome-wide allelic expression (AE) analysis was performed using microarray data from 64 normal breast tissue samples. Of these, 61 samples were of European ancestry (EUR), two samples were of American ancestry (AMR) and one sample was of African ancestry (AFR) (Fig. S2). Normalised allelic expression ratios were calculated for SNPs in coding and noncoding regions upon filtering for the cDNA signals’ expression level and allelic discrimination potential. Overall, we identified 91,467 autosomal allelic-expressed SNPs (aeSNPs) located in 21,527 annotated Ensembl genes (median of three aeSNPs per gene) (Fig. S3). Unsurprisingly, the number of aeSNPs analysed per gene correlated with the annotated gene length (rho = 0.60, p-value < 2.2e−16, Fig. S4).
We found that almost one-third of the aeSNPs (26,266 out of 91,467) displayed biallelic differential expression (daeSNPs, q-value ≤ 0.05) (Table 1, Table S3), while 84 SNPs displayed monoallelic expression (maeSNPs). TaqMan PCR validated seven out of nine daeSNPs (Fig. S5) that showed significant DAE and concordant preferential expression (Fisher’s exact test p-value > 0.05).
Table 1.
Summary of the genome-wide breast tissue allelic expression analysis results.
| Set of SNPs | n | Ensembl gene IDs |
|---|---|---|
| All aeSNPs | 91,467 | 21,527 |
| maeSNPs | 84 | 44 |
| daeSNPs | 26,266 | 13,689 |
The daeSNPs are distributed across the genome, with low interchromosomal variability (ranging from 26 to 35%, Fig. S6). They overlapped 13,688 (65%) annotated genes (daeGenes), of which 3666 (17%) harboured three or more daeSNPs (Fig. 2a, Table 1, Table S3). When considering daeSNPs mapping exclusively to one gene, we identified 8193 daeGenes (out of 12,944) that showed evidence of being under the control of allele-specific cis-acting factors, either genetic or epigenetic. In terms of consistency of DAE detection across the length of these genes, we found that in the majority of daeGenes, the frequency of daeSNPs was higher than 40% (7476 in 13,688), with 3894 daeGenes presenting imbalances in all the analysed aeSNPs (Fig. 2b). The aeSNPs showed a large distribution of mean |AE ratios|, with daeSNPs centred at 0.60 (corresponding to a difference between alleles of 1.5) and non-daeSNPs centred at 0.26 (corresponding to a difference of 1.2). Twelve per cent of daeSNPs showed average absolute AE ratios between 1 and 5, corresponding to average allelic fold changes ranging from 2 to 34 (Fig. 2c, Table S3). The amplitude of the imbalances measured at aeSNPs correlated negatively with the average expression level of both alleles (rho = − 0.4, p-value < 2.2e−16) (Fig. 2d) but not with the standard deviation across individuals (Fig. S7). The aeSNPs are located mainly in intronic regions and noncoding transcript genes, but non-daeSNPs and daeSNPs showed differences in class distribution for consequence type, with daeSNPs enriched at unannotated regions and depleted at intronic, non-coding regions and UTRs (p-value < 0.05, Fig. 2e). Although most of the aeSNPs analysed were in protein-coding genes, daeSNPs were relatively more common in noncoding genes and pseudogenes when compared to non-daeSNPs and depleted in protein-coding genes (p-value < 0.05, Fig. 2f).
Fig. 2.

Characterisation of aeSNPs. (a) Histogram of the rank number of daeSNPs identified per gene across 17,135 annotated genes. (b) Histogram of the rank proportion of daeSNPs per aeSNPs identified per gene. (c) Box plot with the distribution of the mean of the absolute values of AE ratios across heterozygous individuals measured at non-daeSNPs and daeSNPs. (d) Distribution of the mean absolute values of AE ratios at aeSNPs according to the average intensity of both alleles at aeSNPs in the microarray Spearman’s results of a Spearman’s correlation test are shown. (e,f) Relative frequency of aeSNPs and daeSNPs according to consequence type and gene biotype, respectively.
Monoallelic expression in breast tissue
Regarding monoallelic expression, maeSNPs were annotated to 44 Ensembl genes (Table 1, Table S4, Fig. S8), the majority of which were previously reported as imprinted in breast tissue (e.g., IGF2 or ZDBF2) or in other tissues (e.g., KCNQ1, KCNQ1OT1, RTL1, NAA60, ZIM2, and L3MBTL1), validating our AE analysis. Interestingly, we detected maeSNPs in a region containing the lncRNA MEG9 and a cluster of miRNA genes that had only previously been reported as imprinted in nonhuman species50–52. Additionally, we found unreported monoallelic expression at an intergenic region (22q11.23), suggesting the existence of unannotated transcripts in this region. Notably, we observed two groups of heterozygotes preferentially expressing opposite alleles of rs17122278, an intronic variant of ARCN1, suggesting the latter as a candidate novel monoallelically expressed protein-coding gene in breast tissue.
Mapping of daeQTLs in normal breast tissue
Evidence of DAE supports that a gene’s expression is controlled by cis-regulatory variation, which can be mapped using AE ratios as a quantitative trait—in what we termed DAE quantitative trait loci (daeQTL) analysis. Here, we found a minority of daeSNPs (6928 out of 26,266) for which all the heterozygotes preferentially expressed the same allele. This pattern indicates moderate to strong linkage disequilibrium between the daeSNP and the rSNPs acting on it27. Hence, our mapping approach considered the allelic expression (AE) ratio distribution pattern displayed at each daeSNP, and one-sample or two-sample Wilcox tests were applied accordingly. We identified 54,357 daeQTLs (5% FDR) for 6761 (26%) daeGenes (Table S5), primarily located within 20 kb from the corresponding daeSNP but as far as the 500 kb window used for the analysis (Fig. 3a). daeQTLs for MROH8 and ZNF132, two coding genes located on chromosomes 19 and 20, respectively, were among the most significant ones found (Fig. 3b,c), but we identified other highly significant daeQTLs (adjusted p-values smaller than 5.0E−04) for 2507 genes.
Fig. 3.

Mapping of variants associated with differential allelic expression. (a) Empirical cumulative distribution for the distance between the daeSNP and corresponding mapped daeQTL. (b,c) daeQTL mapping result for the most significant daeQTL identified for MROH8 using a one-sample Wilcox test and for ZNF132 using a two-sample Wilcox test. The AE ratios calculated at the daeSNPs are represented on the y-axis in the two panels and stratified according to genotype at the candidate SNP (black dots represent heterozygous individuals, and red dots represent homozygous individuals).
Identification of target genes within BC risk loci
To pinpoint the most likely candidate target genes within BC risk loci, a main post-GWAS challenge, we identified the genes within previously reported GWAS loci (gwasGenes) displaying the most robust evidence of being under the control of cis-regulatory variation, provided either by DAE (daeGenes) or eQTL (eGenes) analysis. We found that most gwasGenes (783 out of 948) showed such evidence, with 69% of these with evidence via DAE analysis (358 genes identified solely by DAE and 300 by both analyses) (Table S6). Compared to all genes studied, gwasGenes presented a significant enrichment of Fisher’s significance of DAE (Fisher’s exact test = 2.48e−05). Finally, we successfully mapped daeQTLs for 385 gwasGenes (Fig. 4, Table S6).
Fig. 4.

Summary of colocalisation analyses for daeGenes and risk-daeQTLs. (a) UpSet plot for 15,706 genes tested for DAE and eQTL (GTEx breast mammary tissue). daeGenes genes identified as having differential allelic expression in normal breast tissue; eGenes genes reported as being eQTL genes in GTEx mammary tissue data (q-value ≤ 0.05); gwasGenes genes where GWAS index SNPs or proxies (r2 ≥ 0.4) are located, daeGenes with daeQTL mapping daeGenes for which daeQTLs were identified. (b) UpSet plot for 424 variants located in DHSs, according to the presence of protein binding and location in active promoters and/or enhancers in breast cell lines.
Next, we verified our ability to identify 178 previously proposed breast cancer target genes (Table S2). We found that 44% of these genes were exclusively daeGenes (e.g., ELL, TOX3, RNF115), 23% were both daeGenes and eGenes (e.g., CASP8, POU5F1B, STXBP4) and 14% were exclusively eGenes (e.g., RMND1, HELQ, PRKRIP1). However, we did not find evidence supporting other genes, such as CITED4, IGFBP5 and MYC. (Table S2). As total levels of gene expression may confound the ability to identify daeGenes and eGenes, it is noteworthy that eGenes showed higher median levels overall than daeGenes, and only 4.7% of exclusive daeGenes showed low median levels (< 0.1 TPM) (Fig. S9).
Identification of causal variants within BC risk loci
Another post-GWAS challenge we addressed was the identification of the causal variants within risk loci. We first identified 1416 daeQTLs in moderate to strong LD (r2 ≥ 0.4) with GWAS index SNPs (Fig. 1) (GWAS p-value < 1.0E−05), henceforth referred to as risk-daeQTLs. These were distributed across 93 loci in 19 chromosomes, primarily in introns, followed by intergenic regions (Table S7, Fig. S10). Then, we assessed these risk-daeQTLs plus their proxies (r2 ≥ 0.95) for their cis-acting regulatory potential. We started by identifying 425 variants located in DNase I hypersensitivity sites (DHS), of which the majority (69%) mapped to regions with histone marks associated with active regulatory elements (Fig. 4b, Fig. S11). More specifically, 149 risk-daeQTLs localised in both active promoter (H3K4me3 and H3K9ac) and regions with active enhancer-associated (H3K4me1 and H3K27ac) histone marks, 76 localised exclusively in regions with active enhancer-associated marks and another 67 exclusively localised in regions with active promoter-associated marks. Of these, 122 risk-daeQTLs also showed protein binding evidence, thus representing strong candidate causal variants within 41 of the initial 93 BC risk loci (Table S8).
Among these 41 risk loci, we detected 47 novel candidate target genes in 29 loci with no previous report of target genes, such as SMC2 in 9q31.1, MLLT10 in 10p12.32 and MAN2C1 and PTPN9 in 15q24.2. We confirmed previously reported target genes in nine loci and identified eight novel genes, including NASP and IPP in 1p34.1 and ATP6AP1L in 5q14.1. Finally, we identified strong candidate causal variants at three loci but could not discern the target gene due to a lack of genomic annotation (Table 2, Table S9).
Table 2.
Loci with candidate risk rSNPs and novel suggested target genes.
| Chr_band | GWAS variant | GWAS nearest gene | Candidate causal variants | daeQTL gene | Regulatory feature |
|---|---|---|---|---|---|
| A. Loci with novel suggested target genes | |||||
| 1p36.23 | rs225132 | ERRFI1-DT | rs12757968 | ERRFI1-DT | Active enhancer |
| 1q22 | rs10796944, rs7524950 | ASH1L, PKLR | rs1046188 | FDPS, RUSC1-AS1 | Active promoter and active enhancer |
| 1q22 | rs348196, rs10796944, rs12091730, rs7524950 | DAP3, ASH1L, MSTO1, DAP3P1, PKLR | rs2048431 | FDPS, RUSC1-AS1 | Active promoter and active enhancer |
| 1q22 | rs348196 | DAP3 | rs3841838 | ARHGEF2, RIT1 | Active promoter and active enhancer |
| 1p23.2 | rs67073037 | WDR43 | rs4407214 | WDR43 | Active promoter and active enhancer |
| 4p12 | rs199501877 | NIPAL1, TXK | rs98270 | NIPAL1 | Active promoter and active enhancer |
| 5q11.1 | rs145106188 | EMB | rs4865698 | EMB | Active enhancer |
| 5q11.1 | rs145106188 | EMB | rs4865699 | EMB | Active enhancer |
| 5q11.1 | rs145106188 | EMB | rs28528780 | EMB | Active promoter and active enhancer |
| 5q31.1 | rs6860806 | SLC22A4 | rs1979981 | SLC22A4, MIR3936HG, SLC22A5 | Active promoter and active enhancer |
| 5q31.1 | rs6860806 | SLC22A4 | rs162887 | SLC22A4, MIR3936HG, SLC22A5 | Active promoter and active enhancer |
| 5q31.1 | rs6860806 | SLC22A4 | rs460089 | SLC22A4, MIR3936HG, SLC22A5 | Active promoter and active enhancer |
| 5q31.1 | rs6860806 | SLC22A4 | rs460271 | SLC22A4, MIR3936HG, SLC22A5 | Active promoter and active enhancer |
| 5q31.1 | rs6860806 | SLC22A4 | rs367805 | SLC22A4, MIR3936HG, SLC22A5 | Active enhancer |
| 5q31.1 | rs6860806 | SLC22A4 | rs2631369 | SLC22A4, MIR3936HG, SLC22A5 | Active promoter and active enhancer |
| 5q31.1 | rs6860806 | SLC22A4 | rs2631368 | SLC22A4, MIR3936HG, SLC22A5 | Active promoter and active enhancer |
| 5q31.1 | rs736801 | ENSG00000283782 | rs2070721 | ENSG00000283782 | Active promoter and active enhancer |
| 5q31.1 | rs736801 | ENSG00000283782 | rs2548998 | ENSG00000283782 | Active promoter and active enhancer |
| 6p22.2 | rs17598658, rs13195401 | H2BC6, BTN2A1 | rs9467701 | BTN3A2 | Active promoter and active enhancer |
| 6p22.2 | rs17598658, rs13195401 | H2BC6, BTN2A1 | rs6923139 | BTN3A2 | Active promoter and active enhancer |
| 6p22.2 | rs17598658, rs13195401 | H2BC6, BTN2A1 | rs6903015 | BTN3A2 | Active promoter and active enhancer |
| 6p22.2 | rs17598658, rs13195401 | H2BC6, BTN2A1 | rs68112369 | BTN3A2 | Active promoter |
| 6p22.2 | rs17598658, rs13195401 | H2BC6, BTN2A1 | rs66827971 | BTN3A2 | Active promoter |
| 6p22.2 | rs13195401 | BTN2A1 | rs9379873 | BTN3A2 | Active promoter and active enhancer |
| 6p22.2 | rs71557345, rs13195401 | ENSG00000285571, BTN2A1 | rs36162392 | BTN3A2 | Active promoter and active enhancer |
| 6p22.1 | rs3094146, rs1611579 | ZNRD1ASP, ENSG00000285799 | rs707910 | HLA-A | Active promoter and active enhancer |
| 6p22.1 | rs3094146, rs1611579 | ZNRD1ASP, ENSG00000285799 | rs415137 | HLA-A | Active promoter and active enhancer |
| 6p22.1 | rs3094146, rs1611579 | ZNRD1ASP, ENSG00000285799 | rs438610 | HLA-A | Active promoter and active enhancer |
| 6p22.1 | rs3094054, rs3094146, rs3132615 | UBQLN1P1, ZNRD1ASP, ENSG00000288805 | rs2188100 | HCG17, HLA-L | Active promoter and active enhancer |
| 6p22.1 | rs3129984, rs3132610, rs9262142, rs3132615 | ENSG00000288805, HCG20, ABCF1, PPP1R18 | rs9262142 | C6orf136 | Active promoter and active enhancer |
| 7q21.3 | rs847577 | LMTK2 | rs1874343 | TECPR1 | Active promoter and active enhancer |
| 9q31.1 | rs4742903, rs718857 | SMC2 | rs3818625 | SMC2 | Active promoter |
| 9q31.1 | rs4742903, rs718857 | SMC2 | rs3818626 | SMC2 | Active promoter |
| 9q31.1 | rs4742903, rs718857 | SMC2 | rs10820599 | SMC2 | Active promoter |
| 9q31.1 | rs4742903, rs718857 | SMC2 | rs10820600 | SMC2 | Active promoter |
| 9q31.1 | rs4742903, rs718857 | SMC2 | rs4742903 | SMC2 | Active promoter |
| 10p12.31 | rs7072776, rs10828247, rs11012730, rs10828249, rs7098100 | MLLT10, SKIDA1 | rs10828247 | MLLT10 | Active promoter |
| 10p12.1 | rs7918232 | ANKRD26 | rs7907988 | YME1L1 | Active promoter and active enhancer |
| 10q21.2 | rs10822013 | ENSG00000285837, ZNF365 | rs10822013 | ENSG00000285837 | Active enhancer |
| 11p11.2 | rs11039183 | MADD | rs7947450 | SPI1 | Active promoter |
| 11q13.1 | rs617791 | DRAP1, TSGA10IP | rs14157 | SART1 | Active promoter |
| 11q13.1 | rs617791 | DRAP1, TSGA10IP | rs12794370 | SART1 | Active promoter and active enhancer |
| 11q13.2 | rs1783730 | TMEM151A | rs33981819 | ENSG00000254458 | Active promoter and active enhancer |
| 11q13.2 | rs11344495, rs55908905 | CTSF, SPTBN2 | rs11110 | DPP3-DT, B4GAT1-DT | Active promoter and active enhancer |
| 11q13.2 | rs11344495, rs55908905 | CTSF, SPTBN2 | rs55853079 | DPP3-DT, B4GAT1-DT | Active promoter and active enhancer |
| 11q22.3 | rs11374964 | POGLUT3 | rs228589 | NPAT | Active promoter |
| 11q22.3 | rs11374964 | POGLUT3 | rs189037 | NPAT | Active promoter |
| 11q23.1 | rs505372 | SIK2 | rs541198 | NA | Active promoter and active enhancer |
| 11q23.1 | rs505372 | SIK2 | rs7107213 | CRYAB | Active promoter and active enhancer |
| 14q13.2 | rs58327846 | PRORP | rs1056879 | PRORP | Active promoter and active enhancer |
| 14q13.2 | rs58327846 | PRORP | rs2236167 | PRORP | Active promoter and active enhancer |
| 14q32.33 | rs60226654 | COA8 | rs3759586 | KLC1, XRCC3 | Active enhancer |
| 15q21.1 | rs1876206 | FBN1 | rs8029993 | FBN1 | Active enhancer |
| 15q24.2 | rs60381548, rs8027365 | SIN3A, PTPN9 | rs11637068 | MAN2C1, PTPN9 | Active promoter and active enhancer |
| 15q24.2 | rs60381548, rs8027365 | SIN3A, PTPN9 | rs75219778 | MAN2C1, PTPN9 | Active promoter |
| 15q24.2 | rs60381548, rs8027365 | SIN3A, PTPN9 | rs62027209 | MAN2C1, PTPN9 | Active promoter and active enhancer |
| 15q26.1 | rs77554484 | PRC1, ENSG00000284946 | rs867468 | PRC1, PRC1-AS1, ENSG00000284946 | Active promoter and active enhancer |
| 15q26.1 | rs77554484 | PRC1, ENSG00000284946 | rs2001216 | PRC1, PRC1-AS1, ENSG00000284946 | Active promoter and active enhancer |
| 15q26.1 | rs77554484 | PRC1, ENSG00000284946 | rs12905855 | PRC1, PRC1-AS1, ENSG00000284946 | Active promoter and active enhancer |
| 16q12.2 | rs17817449, rs62033406, rs7193144, rs62048402 | FTO | rs9940128 | FTO | Active enhancer |
| 16q12.2 | rs17817449, rs62033406, rs7193144, rs62048402 | FTO | rs11642015 | FTO | Active enhancer |
| 16q12.2 | rs17817449, rs62033406, rs7193144, rs62048402 | FTO | rs17817497 | FTO | Active enhancer |
| 16q12.2 | rs17817449, rs62033406, rs7193144, rs62048402 | FTO | rs8050136 | FTO | Active enhancer |
| 16q13 | rs2303282, rs2432539 | BBS2, AMFR | rs2440467 | AMFR | Active enhancer |
| 17q21.31 | rs2732699 | ARL17B | rs76594404 | CRHR1, KANSL1, LINC02210, LINC02210-CRHR1, MAPT, MAPT-AS1, ARL17B | Active promoter and active enhancer |
| 17q21.31 | rs2732699 | ARL17B | rs80233201 | CRHR1, KANSL1, LINC02210, LINC02210-CRHR1, MAPT, MAPT-AS1, ARL17B | Active promoter and active enhancer |
| 17q21.31 | rs2732699 | ARL17B | rs62056778 | CRHR1, KANSL1, LINC02210, LINC02210-CRHR1, MAPT, MAPT-AS1, ARL17B | Active promoter and active enhancer |
| 17q21.31 | rs2732699 | ARL17B | rs11575895 | CRHR1, KANSL1, LINC02210, LINC02210-CRHR1, MAPT, MAPT-AS1, ARL17B | Active promoter |
| 17q21.31 | rs2732699 | ARL17B | rs62056779 | CRHR1, KANSL1, LINC02210, LINC02210-CRHR1, MAPT, MAPT-AS1, ARL17B | Active promoter |
| 17q21.31 | rs2732699 | ARL17B | rs74548327 | CRHR1, KANSL1, LINC02210, LINC02210-CRHR1, MAPT, MAPT-AS1, ARL17B | Active promoter |
| 17q21.31 | rs2732699 | ARL17B | rs111972148 | CRHR1, KANSL1, LINC02210, LINC02210-CRHR1, MAPT, MAPT-AS1, ARL17B | Active promoter |
| 17q21.31 | rs2732699 | ARL17B | rs242561 | CRHR1, KANSL1, LINC02210, LINC02210-CRHR1, MAPT, MAPT-AS1, ARL17B | Active promoter |
| 17q21.31 | rs2732699 | ARL17B | rs2316951 | CRHR1, KANSL1, LINC02210, LINC02210-CRHR1, MAPT, MAPT-AS1, ARL17B | Active enhancer |
| 17q21.31 | rs2732699 | ARL17B | rs11079733 | CRHR1, KANSL1, LINC02210, LINC02210-CRHR1, MAPT, MAPT-AS1, ARL17B | Active promoter |
| 17q21.31 | rs2732699 | ARL17B | rs2696633 | CRHR1, KANSL1, LINC02210, LINC02210-CRHR1, MAPT, MAPT-AS1, ARL17B | Active promoter |
| 17q21.31 | rs2732699 | ARL17B | rs143625699 | ARL17B, KANSL1 | Active promoter |
| 17q21.31 | rs2732699 | ARL17B | rs113417378 | CRHR1, KANSL1, LINC02210, LINC02210-CRHR1, MAPT, MAPT-AS1, ARL17B | Active promoter |
| 17q21.31 | rs2732699 | ARL17B | rs143191191 | CRHR1, KANSL1, LINC02210, LINC02210-CRHR1, MAPT, MAPT-AS1, ARL17B | Active enhancer |
| 22q13.31 | rs28512361 | ENSG00000235091 | rs134847 | ATXN10 | Active promoter |
| 22q13.31 | rs28512361 | ENSG00000235091 | rs2071872 | ATXN10 | Active promoter and active enhancer |
| B. Loci with previously suggested target genes | |||||
| 1p34.1 | rs12077974 | MAST2 | rs6697821 | NASP#, IPP# | Active promoter and active enhancer |
| 1p34.1 | rs12077974, rs1707302 | MAST2, PIK3R3 | rs1707303 | NASP#, IPP#, PIK3R3 | Active promoter and active enhancer |
| 1p34.1 | rs12077974 | MAST2 | rs1707302 | NASP#, IPP#, PIK3R3 | Active enhancer |
| 2q33.1 | rs10931936, rs1035142, rs1830298, rs700635, rs3769821 | CASP8 | rs3769823 | CASP8 | Active promoter and active enhancer |
| 3q12.1 | rs9837602, rs9833888, rs9289981 | CMSS1, FILIP1L | rs793463 | CMSS1#, FILIP1L | Active enhancer |
| 3q12.1 | rs9837602, rs9833888 | CMSS1, FILIP1L | rs28714363 | CMSS1#, FILIP1L | Active enhancer |
| 5q14.1 | rs7707921, rs111549985, rs2407064, rs146817970, rs2407156 | ATG10, ATP6AP1L | rs111549985 | ATG10, ATP6AP1L | Active promoter and active enhancer |
| 5q14.1 | rs7707921, rs2407064, rs146817970, rs2407156 | ATG10 | rs226198 | ATG10, ATP6AP1L | Active promoter and active enhancer |
| 5q14.1 | rs7707921, rs2407064, rs146817970, rs2407156 | ATG10 | rs6880209 | ATG10, ATP6AP1L | Active promoter and active enhancer |
| 5q14.1 | rs7707921, rs2407064, rs146817970, rs2407156 | ATG10 | rs11325430 | ATG10, ATP6AP1L | Active promoter and active enhancer |
| 5q14.1 | rs7707921, rs2407064, rs146817970 | ATG10 | rs17247678 | ATG10, ATP6AP1L | Active enhancer |
| 7q21.2 | rs6964587, rs10644111, rs35417517 | AKAP9, LRRD1, CYP51A1-AS1 | rs1011372 | AKAP9 | Active promoter and active enhancer |
| 7q21.2 | rs35522438, rs35417517, rs10644111, rs6964587 | AKAP9, LRRD1, CYP51A1-AS1, KRIT1 | rs5885795 | AKAP9 | Active promoter and active enhancer |
| 7q21.2 | rs35522438, rs35417517, rs10644111, rs6964587 | AKAP9, LRRD1, CYP51A1-AS1, KRIT1 | rs4727266 | AKAP9 | Active promoter and active enhancer |
| 7q21.2 | rs35522438, rs35417517, rs10644111, rs6964587 | AKAP9, LRRD1, CYP51A1-AS1, KRIT1 | rs4727267 | AKAP9 | Active promoter and active enhancer |
| 7q21.2 | rs35522438, rs35417517, rs10644111, rs6964587 | AKAP9, LRRD1, CYP51A1-AS1, KRIT1 | rs6465339 | AKAP9 | Active promoter and active enhancer |
| 7q21.2 | rs35522438, rs35417517, rs10644111, rs6964587 | AKAP9, LRRD1, CYP51A1-AS1, KRIT1 | rs4279 | AKAP9 | Active promoter and active enhancer |
| 7q21.2 | rs35522438, rs35417517, rs10644111, rs6964587 | AKAP9, LRRD1, CYP51A1-AS1, KRIT1 | rs12704637 | AKAP9 | Active promoter and active enhancer |
| 11p15.5 | rs6597981 | PIDD1 | rs7942564 | GATD1 | Active promoter and active enhancer |
| 11p15.5 | rs6597981 | PIDD1 | rs7948070 | GATD1 | Active promoter and active enhancer |
| 11q13.1 | rs3903072 | SNX32, OVOL1 | rs4621 | CFL1, SNX32# | Active promoter and active enhancer |
| 11q13.1 | rs3903072 | SNX32, OVOL1 | rs7125986 | CFL1, SNX32# | Active promoter |
| 11q13.1 | rs3903072 | SNX32, OVOL1 | rs7947929 | CFL1, SNX32# | Active promoter |
| 11q13.1 | rs3903072 | SNX32, OVOL1 | rs7947741 | CFL1, SNX32# | Active promoter |
| 11q13.1 | rs3903072 | SNX32, OVOL1 | rs13817 | CFL1, SNX32# | Active promoter and active enhancer |
| 19p13.11 | rs8170 | BABAM1 | rs3745187 | ABHD8 | Active promoter |
| 19p13.11 | rs2965183, rs2304098 | GATAD2A, YJEFN3 | rs3934667 | MAU2 | Active promoter |
| 19p13.11 | rs2965183, rs2304098 | GATAD2A, YJEFN3 | rs2916068 | MAU2 | Active promoter |
| 19p13.11 | rs2965183, rs2304098 | GATAD2A, YJEFN3 | rs80007081 | YJEFN3#, CILP2#, ENSG00000258674# | Active promoter and active enhancer |
| 19p13.11 | rs2965183, rs2304098 | GATAD2A, YJEFN3 | rs17684164 | YJEFN3#, CILP2#, ENSG00000258674# | Active promoter and active enhancer |
| 19p13.11 | rs2965183, rs2304098 | GATAD2A, YJEFN3 | rs77254326 | YJEFN3#, CILP2#, ENSG00000258674# | Active promoter and active enhancer |
| C. Loci for which no target gene could be pointed | |||||
| 6p22.1 | rs9257408 | KRT18P1 | rs209174 | NA | Active promoter and active enhancer |
| 6p22.1 | rs9257408 | KRT18P1 | rs209173 | NA | Active promoter and active enhancer |
| 6p22.1 | rs9257408 | KRT18P1 | rs3135315 | NA | Active promoter and active enhancer |
| 6p22.1 | rs9257408 | KRT18P1 | rs184093 | NA | Active promoter and active enhancer |
| 6p22.1 | rs9257408 | KRT18P1 | rs209138 | NA | Active promoter |
| 6p22.1 | rs9257408 | KRT18P1 | rs3131102 | NA | Active promoter |
| 17q23.1 | rs61495451 | VMP1 | rs3803863 | chr17:59,852,174 | Active promoter |
| 17q23.1 | rs61495451 | VMP1 | rs2333562 | chr17:59,852,174 | Active promoter and active enhancer |
| 17q23.1 | rs61495451 | VMP1 | rs138148328 | chr17:59,852,174 | Active enhancer |
| 19p13.11 | rs4808801, rs172032, rs7258465 | ELL, SSBP4 | rs28375303 | chr19:18,416,447 | Active promoter |
| 19p13.11 | rs4808801, rs172032, rs7258465 | ELL, SSBP4 | rs271621 | chr19:18,416,447 | Active promoter and active enhancer |
*Reported as rs116095464 in the original GWAS.
#Linkage disequilibrium (LD) values r2 between the daeQTL and the GWAS risk variant in the European population.
§Gene not expressed in breast mammary tissue or without expression information in GTEx.
#Novel suggested target gene in loci with previously suggested target genes.
§Gene not expressed in breast mammary tissue or without expression information in GTEx.
Notably, 2222 daeQTLs were also in lower LD with GWAS hits (0.2 ≤ r2 < 0.4), representing a valuable dataset warranting further exploration (Table S10).
Mapping of cis-regulatory risk variants at the 5q14.1–14.2 locus
To further show the potential use of our integrated approach, we focused our follow-up studies on the BC risk locus 5q14.1–14.2, where some of the most significant risk-daeQTLs and candidate causal variants were identified. In this locus, rs7707921 was previously associated with BC risk in two meta-analyses (OR for alternative A allele = 1.07, 95% CI [1.05–1.1], p = 5E−11)9,53. The region containing this intronic variant of ATG10, its proxy variants (r2 ≥ 0.4) and other risk-associated variants reported in this locus spans three genes (ATG10, RPS23, and ATP6AP1L), hindering the identification of the causal variant(s) and their target gene(s) in this locus.
First, all three genes showed DAE, supporting their regulation by cis-regulatory variants: 10 daeSNPs out of 37 aeSNPs at ATG10, one daeSNP out of two aeSNPs at RPS23 and three daeSNPs out of five aeSNPs at ATP6AP1L (Fig. S12). The highest mean |AE ratios| detected at daeSNPs in these genes was 1.27 (2.4-fold) at RPS23, followed by 0.92 (1.9-fold) at ATP6AP1L (Fig. 5—panel 2, Fig. S11). By daeQTL mapping analysis, we identified daeQTLs for all three genes: 56 for ATG10 (spreading along the ATG10-ATP6AP1L region), 4 for RPS23 (limited to RPS23-ATP6AP1L) and 26 for ATP6AP1L (spreading along the ATG10-ATP6AP1L region) (Fig. 5—panels 3 to 5). Additionally, we classified as risk-daeQTLs the 38 ATG10 daeQTLs and 24 ATP6AP1L daeQTLs (22 of which are common to the two genes) in moderate to strong LD (r2 ≥ 0.4) with the risk-associated variants. Furthermore, both ATG10 and ATP6AP1L daeQTL analysis results strongly correlated with the corresponding LD with the GWAS lead-SNP rs7707921 (Fig. S13), further supporting the role of variants regulating the expression of these two genes in the risk for breast cancer.
Fig. 5.

Evidence of DAE and daeQTL analysis at the 5q14.1 BC risk locus. The top track shows the mean values of the absolute AE ratios measured at aeSNPs across the region, with the non-daeSNPs shown in grey, the daeSNPs in black and the daeSNPs with mapped daeQTLs in red. The subsequent tracks show the daeQTL mapping corrected p-values for ATG10, RPS23 and ATP6AP1L.
Cis-regulatory risk variants act via two different mechanisms on genes in the 5q14.1–14.2 locus
The overlap analysis of the risk-daeQTLs with epigenetic marks in breast cell lines identified seven candidate causal variants for ATG10 and ATP6AP1L (Tables S8, S11). We investigated these variants further for allelic differences in transcription factor binding and association with histone modifications and DHSs. One of these SNPs, rs111549985, overlies the active promoter of ATG10 (Fig. S14), and its minor G-allele is preferentially associated with the H3K4Me3 modification in HMECs (2.7-fold, p = 3.7e−03) and shows robust preferential binding by POL2 in MCF7 cells (ninefold, p = 4.0E−04). However, DHS was more significantly associated with the major/reference C allele in T47D cells (0.5-fold, p = 4.6e−05) (Fig. 6a, Table S12). Another two candidate causal variants, rs226198 (intronic to RPS23) and rs688025ʹ UTR (located at RPS23 5ʹ UTR), overlay the shared promoter of RPS23 and ATP6AP1L and a predicted enhancer interacting with the ATG10 promoter (Fig. S15). The minor C-allele of rs226198 showed preferential binding by MYC and MAX transcription factors, which are known to cooperate in cancer54 (12.6-fold and 7.9-fold difference, respectively, p < 2.2e−16) and preferential H3K4me3 marking (2.7-fold, p = 1.4e−02) in MCF-7 cells (Fig. 6b, Table S12). It would be interesting to elucidate whether rs226198 impacts the binding of both factors and H3K4me3 deposition or whether this epigenetic mark is a consequence of altered transcription, as previously suggested55,56. The minor T-allele of rs6880209 also showed preferential binding by MYC (4.8-fold, p < 2.2e−16) and MAX (2.4-fold, p = 2.7e−03), with smaller fold-change differences than rs226198, and additional preferential binding by POL2 (2.6-fold, p = 1.27e−06) in MCF7 cells. However, similar to rs111549985, DHS preferentially occurred in the major/reference C-allele in T47D cells (5.3-fold, p = 9.1e−04) (Fig. 6c, Table S12). Interestingly, the expression of MAX correlated with ATG10, RPS23 and ATP6AP1L, and the expression of MYC correlated with the expression of ATG10 (Fig. S16). Furthermore, the expression levels of ATG10 and ATP6AP1L were positively correlated in breast tissue from healthy women (top 2.5% quantile of 500,000 pairwise tests) and in normal-matched tissue from patients with BC (Fig. S17). The observation that ATG10 and ATP6AP1L are in different topologically associating domains (TADs) and that the candidate causal variants rs226198 and rs6880209 fall on the boundary between them (Fig. S18) suggests that a shared pattern of chromatin condensation does not drive the correlated gene expression but instead by a shared cis-regulatory sequence.
Fig. 6.

Variants at the 5q14.1 risk locus associated with differential transcription factor binding. Allele-specific analysis of the effect of three candidate risk rSNPs—(a) rs111549985, (b) rs226198 and (c) rs6880209—on RNA polymerase II (POL2) and transcription factor (TF) binding, DNase I targeting (DHS) and H3K4me3 modification in different heterozygous cell lines. An asterisk indicates statistically significant imbalances (two-sided binomial test, p-value ≤ 0.05). HMEC human mammary epithelial cells, MCF7 human breast (adenocarcinoma) cell line, T47D human breast tumour cell line, MCF10A human breast epithelial cell line.
Since genetic variants affecting mRNA decay or alternative splicing57 can cause allelic expression imbalances, we aimed to explore further the alternative transcription’s role in gene expression regulation and driving risk at the 5q14.1 locus. To accomplish this, we performed an sQTL analysis for ATG10 that was not restricted to the candidate risk rSNPs but included all SNPs located within 5 kb upstream and downstream of ATG10 to increase the stringency of the exercise.
We identified six sQTLs (FDR ≤ 5%) in the tumour data, whose minor alleles were associated with changes in the expression of two protein-coding isoforms: decreased expression of ENST00000458350 (one extra exon) and increased expression of ENS3ʹUTR0282185 (longer 3ʹ UTR) (Fig. 7, Fig. S19a, Table S13). Interestingly, ENST00000282185 is expressed at significantly lower levels in tumours than in normal-matched tissue, in line with the reported oncogenic effect of UTR length58, although with a small effect size (fold-change = 1.20) (Fig. S20). The strong correlation between sQTL q-values and LD with the lead GWAS SNP rs7707921 (r = 0.94, p-value = 3.15E−12, Fig. S18b) supports the contribution of alternative transcription of ATG10 to BC risk. Although no sQTL was detected for ATG10 in normal breast data (Table S13), sQTL nominal p-values and LD with rs7707921 were still correlated in normal matched breast samples (r = 0.59, p-value = 0.002) (Fig. S21). ATP6AP1L did not display sufficient alternative transcription dispersion to allow the sQTL analysis. Subsequent functional analysis of ATG10’s sQTLs, and their proxy SNPs (LD r2 ≥ 0.95), revealed the prediction of rs111549985 (5ʹ UTR) and rs6884232 (3ʹ UTR) to cause a riboSNitch (a functional RNA structure disrupted by an SNP46). Although RBP binding data for breast tissue do not exist, these variants have been reported to disrupt the binding of Xrn2 (involved in termination by RNA polymerase II) and of Igf2bp1 (a translation regulator) in K562 cells (Tables S14, S15), which would require confirmation in breast cells.
Fig. 7.
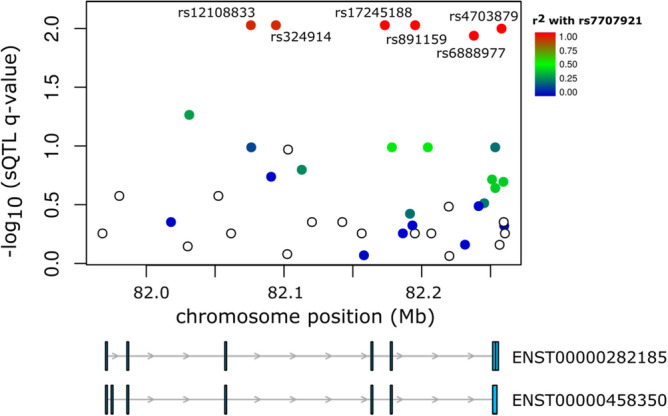
Variants at the 5q14.1 risk locus associated with alternative transcription. Six sQTLs in high LD with rs7707921 were identified for ATG10. The − log10(q-value) for the sQTL analysis (y-axis) is shown for the 5q14.1–14.2 region (hg38). Colour intensity represents the LD (r2) between the analysed variants and the GWAS lead SNP rs7707921. Below are two ATG10 transcripts whose expression ratios are associated with the sQTLs.
Risk model for 5q14.1 links higher expression of ATG10 and ATP6AP1L with protection against BC
Haplotype analysis of the samples included herein revealed two common haplotypes: one harbouring the major alleles of all proposed risk-rSNPs and the GWAS lead SNP rs7707921 (frequency of 71.1%) and another with the corresponding minor alleles (frequency of 21.9%) (Fig. S22). The proposed risk-rSNPs are among the most significant eQTLs for the two genes: rs111549985 for ATG10 and rs6880209 for ATP6AP1L (Fig. S23)59. Therefore, the most common haplotype is associated with an increased risk for BC and lower expression of ATG10 and ATP6AP1L (Fig. S22).
Our proposed model for risk at 5q14.1 (Fig. 8) establishes that the minor alleles of rs111549985, rs226198, and rs6880209 confer protection against BC by (1) increasing the binding of POL2 II to the promoter of ATG10 (driven by rs111549985), (2) the binding of POL2 to the shared promoter of RPS23/ATP6AP1L (driven by rs6880209), and (3) the binding of cMYC and MAX to a regulatory region (possible enhancer) (driven by rs226198), increasing the expression of ATG10 and ATP6AP1L. These results reveal a complex regulatory landscape at the 5q14.1–14.2 locus, with multiple independent causal variants.
Fig. 8.

Complex risk regulatory landscape of the 5q14.1 locus. (a) Levels of expression of ATG10 and ATP6AP1L genes differ between the haplotypes containing either the minor alleles of rs111549985, rs226198 and rs6880209 (above) or the major ones (below). Coloured arrows indicate the direction of transcription of the individual genes, the saturation of the corresponding colours indicates the strength of protein binding, the number of green circles indicates the level of H3K4me3 and the coloured curvy lines indicate the relative levels of transcript produced. (b) Schematic representation of the proposed model for the positive correlation between ATG10 and ATP6AP1L via a shared regulatory region.
Discussion
Here, we present the first genome-wide map of differentially expressed allelic genes (daeGenes) and their genetic determinants (daeQTLs) in normal breast tissue. We found widespread differential allelic expression (DAE) across the genome and identified daeQTLs for 26% of daeGenes. By intersecting this map with GWAS data, we identified risk-daeQTLs and target genes for 93 BC risk loci. Next, we retrieved epigenetic regulatory annotations on all candidate risk-rSNPs (risk-daeQTLs and their proxies in high LD) to prioritise variants with regulatory potential for further functional analysis. We identified 406 variants with strong regulatory potential annotated to 55 different chromosomal bands and candidates for regulating the expression levels of 96 genes. Our results represent a practical and valuable resource for prioritising loci for follow-up GWASs. As a proof of concept, we functionally characterised the 5q14.1–14.2 BC risk locus in depth and proposed four causal regulatory variants targeting the genes ATG10 and ATP6AP1L acting via multiple allele-specific mechanisms. Our results suggest a complex regulatory landscape underlying BC aetiology.
We show that cis-acting variants regulate the expression of 65% of genes in normal breast tissue, with some genes displaying extreme allelic differences of up to 32-fold. Notably, we identified a novel gene with monoallelic expression, ARCN1, which warrants further inspection to confirm its imprinting status. An enrichment of daeSNPs at intergenic and intronic regions, as well as noncoding transcripts, noncoding genes, and pseudogenes, concurs with previous reports of predominant allelic imbalances of expression at gene-depleted regions and genes under fewer evolutionary constraints60,61.
To overcome the lack of phasing information, we applied two different tests in the daeQTL mapping, according to the AE ratio distribution, which led to the identification of 54,357 variants associated with AE ratios for 6761 genes, both coding and noncoding for proteins. The stringent statistical correction and the use of distance as a covariate in the second mapping approach increased its confidence level but limited the statistical power to identify regulatory variants in lower LD with the daeSNP or located more distally.
We found evidence of expression regulation by cis-acting variants for most reported GWAS loci and believe that alternative mechanisms are at play in the remainder. We identified risk-daeQTLs at 93 different loci, including 72 loci with novel candidate risk target genes (including NEK10 at 3p24.1 and ZBED6 and ZC3H11A at 1q32.1). Moreover, the initial daeQTL map in normal breast tissue can be further mined whenever new risk variants are identified through GWAS. These results offer a resource platform for functional studies of causal variants and target genes and can help uncover the role of cis-regulatory variation in BC risk.
Finally, we conducted an in silico functional analysis of the 5q14.1–14.2 BC risk locus and identified three strong candidate causal variants: rs111549985, rs226198, and rs6880209. We predict that these variants functionally impact TF binding, chromatin state, and gene expression levels of ATG10 and ATP6AP1L. A similar involvement of diverse regulatory mechanisms has been suggested previously for other BC risk loci4,62,63. Both ATG10 (involved in autophagy) and the ATP6AP1L pseudogene have been suggested to have roles in cancer64–67. A variant at ATG10 (rs7313473) was previously associated with BC risk by regulating promoter activity, and ATG10 was suggested to act as a tumour suppressor gene in breast tissue68. For ATP6AP1L, another variant (rs10514231) was reported to lead to ATP6AP1L downregulation by decreasing the binding affinity of TCF7L2 in an intronic regulatory region67. Although we did not find supporting evidence for the same variants, our results show an indirect association between the lower expression of ATG10 and ATP6AP1L and BC risk, suggesting that the downregulation of these two genes may contribute to tumorigenesis.
The advantages of our analysis compared to previous reports of AE in normal breast and tumour tissue16,18,19,69 include using the most significant number of normal breast tissue samples, the genome-wide approach, and the mapping of candidate regulatory variants. We found a similar frequency of daeSNPs to previous reports in other tissues/cell lines but a higher frequency of daeGenes13,17,18,70. This higher frequency of daeGenes could be due to our ability to identify genes regulated by common cis-acting variants with weak to large effect sizes19, a consequence of the imposed conditions to call DAE (allelic change difference of 1.5-fold and the minimum number of heterozygotes). Additionally, we did not integrate the AE ratios of multiple daeSNPs in the same gene due to the absence of phase data and to maximise the information withdrawn from daeSNPs that might be located in different LD blocks. The complex regulatory landscape we identified at the 5q14.1 locus, with multiple cis-acting variants located in the same haplotypes and AE likely resulting from the sum of the effects of each variant, supports this analysis approach. Furthermore, as we propose, a global measure of the AE imbalance at each gene would impair the mapping of daeQTLs at individual daeSNPs and restrict the analysis to genes with multiple daeSNPs. Finally, besides the more commonly studied protein-coding genes, we analysed noncoding genes and pseudogenes, such as ATP6AP1L.
Our results confirm the advantage of using DAE analysis to detect the effect of rSNPs compared to eQTL analysis, as shown by the higher number of daeGenes than eGenes among gwasGenes71–73. As a minority of gwasGenes were exclusively eGenes, we believe that DAE and eQTL analyses are complementary and should be used in parallel when possible.
Our use of microarray data could be seen as a limitation compared to RNA-seq data, which have more extensive transcriptome coverage and high quantification accuracy for more extreme allelic imbalances. However, microarrays are a widely used and precise technology for measuring AE13,16,22, as we confirmed with our validated monoallelic expression of known imprinted genes and with independent PCR analysis. The only publicly available RNA-seq dataset with normal breast tissue is from the GTEx project. However, our approach presents several advantages: (1) we processed and hybridised the DNA and RNA samples in parallel to minimise technical issues, (2) we used total RNA, which includes coding/noncoding genes and spliced/unspliced transcripts, and (3) we showed that the range of gene expression levels of daeGenes was comparable to that of the eGenes from the GTEx dataset. The following steps will be to carry out matched RNA-seq and DNA-seq to combine all the advantages mentioned above and expand the discovery of daeGenes and rSNPs.
While our approach of defining risk-daeQTLs based on LD with GWAS index SNPs provides valuable insights and a potential link, it does not establish causality. Future work should address this limitation by performing co-localization analysis for all identified risk-daeQTL loci.
Our study predominantly analysed samples from individuals of European ancestry, and we combined these results with GWAS findings that included European populations for consistency. However, leveraging a multi-ancestry study design in future research is essential. Different ancestral populations possess distinct genetic backgrounds, unique sets of genetic variants and varying frequencies of shared variants. Incorporating data from multiple ancestries can broaden our understanding of the genetic and regulatory mechanisms underlying breast cancer risk19,74.
Here, we provide a genome-wide list of variants with strong regulating potential for normal breast tissue, a valuable resource for researchers prioritising GWAS results for functional characterisation and those interested in other BC-related traits. The extensive characterisation of the regulatory landscape at the 5q14.1 BC risk locus identified candidate causal variants and revealed the multiple mechanisms involved. Further studies of this locus will elucidate the mechanisms involved and the relative contributions of each variant and target gene to the genetic risk. Overall, our results reinforce the importance of cis-regulatory variation as a major player in BC susceptibility and the power of identifying these variants in the disease’s tissue of origin—normal breast tissue. They also show that multiple causal variants may co-occur and act via independent cis-regulatory mechanisms at BC risk loci, supporting a broader approach to functional studies.
Supplementary Information
Acknowledgements
The authors would also like to thank Dr Nuno Barbosa-Morais at IMM for the excellent scientific discussions and the support given by the Unidade de Apoio à Investigação (UAIC) at Universidade do Algarve (UAlg), particularly Mr. Vitor Morais, and the Informatics Services of UAlg.
Author contributions
BAJP and ATM conceived the work, and RR, BAJP and ATM designed the work. BPA, CLR, NR, MOR, and ATM carried out laboratory experiments and acquired data. JMX, RM, BPA, AJF, RR, SS, MD, AMM and ATM analysed and interpreted data. JMX and ATM wrote the main manuscript text, and all authors reviewed the manuscript.
Funding
This work was supported by national Portuguese funding through FCT—Fundação para a Ciência e a Tecnologia and CRESC ALGARVE 2020, institutional support CBMR-UID/BIM/04773/2013, RISE—LA/P/0053/2020, UIDB/4255/2020—CINTESIS, POCI “01-0145-” EDER-022184 “GenomePT”, the contract DL 57/2016/CP1361/CT0042 (J.M.X.) and individual postdoctoral fellowship SFRH/BPD/99502/2014 (J.M.X.). The research leading to these results has received funding from the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme FP7/2007-2013/under REA Grant Agreement No. 303745 (A.T.M.), a Maratona da Saúde Award (A.T.M.) and a BCRF project Grant.
Data availability
The datasets supporting the conclusions of this article are available in the following repositories: GWAS Catalog [https://www.ebi.ac.uk/gwas], Ensembl database (annotated to GRCh38.p13) [https://www.ensembl.org], GTEx Project (v7 and v8) [https://www.gtexportal.org], microarray data from GEO [https://www.ncbi.nlm.nih.gov/geo/] under accession number GSE35023.
Code availability
We used R to carry out the DAE and daeQTL mapping analyses, the code of which is available on GitHub (https://github.com/maialab/DaeBreastMicroarrays).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Joana M. Xavier, Email: jgxavier@ualg.pt
Ana-Teresa Maia, Email: atmaia@ualg.pt.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-72163-y.
References
- 1.Wendt, C. & Margolin, S. Identifying breast cancer susceptibility genes—A review of the genetic background in familial breast cancer. Acta Oncol.58, 1–12 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Gallagher, M. D. & Chen-Plotkin, A. S. The post-GWAS era: From association to function. Am. J. Hum. Genet.102, 717–730 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fachal, L. et al. Fine-mapping of 150 breast cancer risk regions identifies 191 likely target genes. Nat. Genet.52, 56–73 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyer, K. B. et al. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol.6, e108 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Udler, M. S. et al. Fine scale mapping of the breast cancer 16q12 locus. Hum. Mol. Genet.19, 2507–2515 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meyer, K. B. et al. A functional variant at a prostate cancer predisposition locus at 8q24 is associated with PVT1 expression. PLoS Genet.7, e1002165 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Darabi, H. et al. Polymorphisms in a putative enhancer at the 10q21.2 breast cancer risk locus regulate NRBF2 expression. Am. J. Hum. Genet.97, 22–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michailidou, K. et al. Association analysis identifies 65 new breast cancer risk loci. Nature551, 92–94 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghoussaini, M. et al. Evidence that the 5p12 variant rs10941679 confers susceptibility to estrogen-receptor-positive breast cancer through FGF10 and MRPS30 regulation. Am. J. Hum. Genet.99, 903–911 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dunning, A. M. et al. Breast cancer risk variants at 6q25 display different phenotype associations and regulate ESR1, RMND1 and CCDC170. Nat. Genet.48, 374–386 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pastinen, T., Ge, B. & Hudson, T. J. Influence of human genome polymorphism on gene expression. Hum. Mol. Genet.15, R9–R16 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Ge, B. et al. Global patterns of cis variation in human cells revealed by high-density allelic expression analysis. Nat. Genet.41, 1216–1222 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Forton, J. T. et al. Localization of a long-range cis-regulatory element of IL13 by allelic transcript ratio mapping. Genome Res.17, 82–87 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bjornsson, H. T. et al. SNP-specific array-based allele-specific expression analysis. Genome Res.18, 771–779 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao, C. et al. Identifying breast cancer risk loci by global differential allele-specific expression (DASE) analysis in mammary epithelial transcriptome. BMC Genom.13, 570 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Romanel, A., Lago, S., Prandi, D., Sboner, A. & Demichelis, F. ASEQ: Fast allele-specific studies from next-generation sequencing data. BMC Med. Genom.8, 9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Przytycki, P. F. & Singh, M. Differential allele-specific expression uncovers breast cancer genes dysregulated by cis noncoding mutations. Cell Syst.10, 193–203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aguet, F. et al. Genetic effects on gene expression across human tissues. Nature550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamdi, Y. et al. Association of breast cancer risk with genetic variants showing differential allelic expression: Identification of a novel breast cancer susceptibility locus at 4q21. Oncotarget5, 80140–80163 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang, Y. et al. Integrative genomic analysis predicts causative cis-regulatory mechanisms of the breast cancer-associated genetic variant rs4415084. Cancer Res.78, 1579–1591 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu, R. et al. Allele-specific expression analysis methods for high-density SNP microarray data. Bioinformatics28, 1102–1108 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Du, P. et al. Comparison of Beta-value and M-value methods for quantifying methylation levels by microarray analysis. BMC Bioinform.11, 587 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gimelbrant, A., Hutchinson, J. N., Thompson, B. R. & Chess, A. Widespread monoallelic expression on human autosomes. Science318, 1136–1140 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Maia, A.-T. et al. Extent of differential allelic expression of candidate breast cancer genes is similar in blood and breast. Breast Cancer Res.11, R88 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li, Y., Willer, C. J., Ding, J., Scheet, P. & Abecasis, G. R. MaCH: Using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol.34, 816–834 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao, R. & Scott, L. J. Detection of cis-acting regulatory SNPs using allelic expression data. Genet. Epidemiol.35, 515–525 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hochberg, Y. & Benjamini, Y. More powerful procedures for multiple significance testing. Stat. Med.9, 811–818 (1990). [DOI] [PubMed] [Google Scholar]
- 29.Ignatiadis, N., Klaus, B., Zaugg, J. & Huber, W. Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat. Methods13, 577–580 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacArthur, J. et al. The new NHGRI-EBI catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res.45, D896–D901 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Magno, R. & Maia, A.-T. gwasrapidd: An R package to query, download and wrangle GWAS catalog data. Bioinformatics36, 649 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yates, A. et al. The ensembl REST API: Ensembl data for any language. Bioinformatics31, 143–145 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goovaerts, T. et al. A comprehensive overview of genomic imprinting in breast and its deregulation in cancer. Nat. Commun.9, 4120 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Consortium TGte et al. The genotype-tissue expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science348, 648–60 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dunham, I. et al. An integrated encyclopedia of DNA elements in the human genome. Nature489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kundaje, A. et al. Integrative analysis of 111 reference human epigenomes. Nature518, 317–330 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kent, W. J. et al. The human genome browser at UCSC. Genome Res.12, 996–1006 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonzalez, J. N. et al. The UCSC genome browser database: 2021 update. Nucleic Acids Res.49, D1046–D1057 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ward, L. D. & Kellis, M. HaploReg: A resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res.40, D930–D934 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyle, A. P. et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res.22, 1790–1797 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thorvaldsdóttir, H., Robinson, J. T. & Mesirov, J. P. Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform.14, 178–192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Storey, J., Bass, A., Dabney, A. & Robinson, D. qvalue: Q-Value Estimation for False Discovery Rate Control. http://github.com/jdstorey/qvalue (2021).
- 43.Monlong, J., Calvo, M., Ferreira, P. G. & Guigó, R. Identification of genetic variants associated with alternative splicing using sQTLseekeR. Nat. Commun.5, 4698 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu, Y. et al. POSTAR2: Deciphering the post-transcriptional regulatory logics. Nucleic Acids Res.47, D203–D211 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mao, F. et al. RBP-Var: A database of functional variants involved in regulation mediated by RNA-binding proteins. Nucleic Acids Res.44, D154–D163 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Corley, M., Solem, A., Qu, K., Chang, H. Y. & Laederach, A. Detecting riboSNitches with RNA folding algorithms: A genome-wide benchmark. Nucleic Acids Res.43, 1859–1868 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paz, I., Kosti, I., Ares, M., Cline, M. & Mandel-Gutfreund, Y. RBPmap: A web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res.42, W361–W367 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barrett, J. C., Fry, B., Maller, J. & Daly, M. J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics21, 263–265 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Colaprico, A. et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res.44, e71 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seitz, H. et al. Imprinted microRNA genes transcribed antisense to a reciprocally imprinted retrotransposon-like gene. Nat. Genet.34, 261–262 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Tierling, S. et al. High-resolution map and imprinting analysis of the Gtl2–Dnchc1 domain on mouse chromosome 12. Genomics87, 225–235 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Hagan, J. P., O’Neill, B. L., Stewart, C. L., Kozlov, S. V. & Croce, C. M. At least ten genes define the imprinted Dlk1-Dio3 cluster on mouse chromosome 12qF1. PLoS ONE4, e4352 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Michailidou, K. et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet.47, 373–380 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dang, C. V. MYC on the path to cancer. Cell149, 22–35 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Floc’hlay, S. et al. Cis-acting variation is common across regulatory layers but is often buffered during embryonic development. Genome Res.31, 120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Howe, F. S., Fischl, H., Murray, S. C. & Mellor, J. Is H3K4me3 instructive for transcription activation? Bioessays39, 1–12 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Robles-Espinoza, C. D., Mohammadi, P., Bonilla, X. & Gutierrez-Arcelus, M. Allele-specific expression: Applications in cancer and technical considerations. Curr. Opin. Genet. Dev.66, 10–19 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mayr, C. & Bartel, D. P. Widespread shortening of 3′ UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell138, 673–684 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lonsdale, J. et al. The genotype-tissue expression (GTEx) project. Nat. Genet.45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Campbell, C. D., Kirby, A., Nemesh, J., Daly, M. J. & Hirschhorn, J. N. A survey of allelic imbalance in F1 mice. Genome Res.18, 555–563 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tung, J., Fédrigo, O., Haygood, R., Mukherjee, S. & Wray, G. A. Genomic features that predict allelic imbalance in humans suggest patterns of constraint on gene expression variation. Mol. Biol. Evol.26, 2047–2059 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cox, D. G. et al. Common variants of the BRCA1 wild-type allele modify the risk of breast cancer in BRCA1 mutation carriers. Hum. Mol. Genet.20, 4732–4747 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Maia, A.-T. et al. Effects of BRCA2 cis-regulation in normal breast and cancer risk amongst BRCA2 mutation carriers. Breast Cancer Res.14, R63 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jo, Y. K. et al. Increased expression of ATG10 in colorectal cancer is associated with lymphovascular invasion and lymph node metastasis. PLoS ONE7, e52705 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang, Y., Huang, J.-W., Castella, M., Huntsman, D. G. & Taniguchi, T. p53 is positively regulated by miR-542-3p. Cancer Res.74, 3218–3227 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jo, Y. K. et al. Polypyrimidine tract-binding protein 1-mediated down-regulation of ATG10 facilitates metastasis of colorectal cancer cells. Cancer Lett.385, 21–27 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Ma, S., Ren, N. & Huang, Q. rs10514231 leads to breast cancer predisposition by altering ATP6AP1L gene expression. Cancers13, 3752 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo, X. et al. A comprehensive cis-eQTL analysis revealed target genes in breast cancer susceptibility loci identified in genome-wide association studies. Am. J. Hum. Genet.102, 890–903 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang, K. et al. Digital RNA allelotyping reveals tissue-specific and allele-specific gene expression in human. Nat. Methods6, 613–618 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ma, X. et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature555, 371–376 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Adoue, V. et al. Allelic expression mapping across cellular lineages to establish impact of non-coding SNPs. Mol. Syst. Biol.10, 754 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Almlöf, J. C. et al. Powerful identification of cis-regulatory SNPs in human primary monocytes using allele-specific gene expression. PLos ONE7, e52260 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pastinen, T. & Hudson, T. J. Cis-acting regulatory variation in the human genome. Science306, 647–650 (2004). [DOI] [PubMed] [Google Scholar]
- 74.Martin, A. R. et al. Human demographic history impacts genetic risk prediction across diverse populations. Am. J. Hum. Genet.100, 635–649 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets supporting the conclusions of this article are available in the following repositories: GWAS Catalog [https://www.ebi.ac.uk/gwas], Ensembl database (annotated to GRCh38.p13) [https://www.ensembl.org], GTEx Project (v7 and v8) [https://www.gtexportal.org], microarray data from GEO [https://www.ncbi.nlm.nih.gov/geo/] under accession number GSE35023.
We used R to carry out the DAE and daeQTL mapping analyses, the code of which is available on GitHub (https://github.com/maialab/DaeBreastMicroarrays).