

Abstract
In the healing process of myocardial infarction, cardiac fibroblasts are activated to produce collagen, leading to adverse remodeling and heart failure. Our previous study showed that ASPP1 promotes cardiomyocyte apoptosis by enhancing the nuclear trafficking of p53. We thus explored the influence of ASPP1 on myocardial fibrosis and the underlying mechanisms. Here, we observed that ASPP1 was increased after 4 weeks of MI. Both global and myofibroblast knockout of ASPP1 in mice mitigated cardiac dysfunction and fibrosis after MI. Strikingly, ASPP1 produced the opposite influence on p53 level and cell fate in cardiac fibroblasts and cardiomyocytes. Knockdown of ASPP1 increased p53 levels and inhibited the activity of cardiac fibroblasts. ASPP1 accumulated in the cytoplasm of fibroblasts while the level of p53 was reduced following TGF-β1 stimulation; however, inhibition of ASPP1 increased the p53 level and promoted p53 nuclear translocation. Mechanistically, ASPP1 is directly bound to deubiquitinase OTUB1, thereby promoting the ubiquitination and degradation of p53, attenuating myofibroblast activity and cardiac fibrosis, and improving heart function after MI.
Subject terms: Cardiovascular diseases, Post-translational modifications
ASPP1 promotes apoptosis of cardiomyocytes by increasing the trafficking of p53 to the nucleus. Here the authors show that, in fibroblasts, ASPP1 promotes the ubiquitination and degradation of p53, decreasing myofibroblast activity and improving cardiac fibrosis and function after myocardial infarction.
Introduction
Cardiac fibrosis is a typical pathological characteristic observed in a variety of chronic and acute cardiovascular diseases1,2. Cardiac fibrosis is characterized by excessive production and deposition of collagen and other extracellular matrix (ECM) proteins in the heart3,4. This pathological remodeling leads to structural disruption, and myocardial dysfunction, eventually impairing the mechanical, electrical, and vasodilatory function of the heart5–7. Therefore, elucidation of the molecular mechanisms that control cardiac fibrosis is essential to establish effective therapeutic strategies.
The apoptosis stimulating of the p53 protein (ASPP) family is a group of proteins involved in cell apoptosis regulation. Among them, ASPP1 and ASPP2 augment the proapoptotic function of p53 by activating p53-mediated transcription of proapoptotic genes, whereas iASPP has the opposite effect8. In our previous work, we found that ASPP1 exacerbates myocardial dysfunction following ischemia-reperfusion injury by binding to p53 and promoting myocardial apoptosis. However, ASPP1 deficiency exhibits improved cardiac function, decreased infarct size, and reduced cardiomyocyte apoptosis. Mechanistically, ASPP1 facilitates the nuclear translocation of p53 and enhances the transcription of pro-apoptotic genes9. p53 is a typical tumor suppressor which inhibits cancer cell proliferation. Abnormal fibroblast activation is biologically similar to unrestricted cell proliferation in oncogenic malignancies10. Tumor suppressor p53 was also involved in the development of pulmonary and cardiac fibrosis11,12. Interestingly, we preliminarily discovered that in contrast to the promoting effects of ASPP1 on p53 activity in cardiomyocytes, it reduced p53 levels in cardiac fibroblasts and increased fibroblast proliferation and collagen production. This striking observation drove us to investigate the role and molecular mechanisms of ASPP1 in post-infarct cardiac fibrosis.
Deubiquitination is a reversible ubiquitination process that plays a crucial role in regulating protein localization and substrate protein degradation13. The process eliminates ubiquitin molecules from modified substrates. Growing evidence suggests that deubiquitinase Otubain 1 (OTUB1) regulates numerous cancer-associated signaling pathways to promote tumor cell survival, proliferation, invasiveness, and drug resistance14–18. It has been known that OTUB1 can remove ubiquitin or polyubiquitin from p53 and increase the stability of p5319.
In this study, we demonstrated that ASPP1 acted as a positive regulator of cardiac fibrosis by binding to OTUB1 to enhance p53 ubiquitination and degradation. Suppression of ASPP1 in myofibroblasts greatly alleviated cardiac fibrosis. Our study indicates that targeting ASPP1 holds considerable potential in the treatment of cardiac fibrosis.
Results
ASPP1 is upregulated during cardiac fibrosis
To investigate the potential role of ASPP1 in cardiac fibrosis, we first examined its expression change in chronic (4-week) MI model and cultured primary mouse cardiac fibroblasts (PMCFs) treated with TGF-β1. Four weeks after MI, both mRNA and protein levels of ASPP1 were elevated in the infarct zone of mouse hearts. ASPP1 showed a dynamic increase across a period of 4 weeks of MI (Fig. 1a, b). Consistently, ASPP1 was observed to be upregulated in PMCFs treated with TGF-β1 (Fig. 1c, d).
Fig. 1. Upregulation of ASPP1 in fibrotic models.

a, b mRNA and protein of ASPP1 were elevated at 1 week, 2 weeks, and 4 weeks of MI mice. n = 6 independent animals in each group. c mRNA levels of Col1 and ASPP1 in PMCFs treated with TGF-β1 (20 ng/mL), n = 6 independent samples. d Representative immunoblots/densitometric quantitative analysis of Col1 and ASPP1 levels in total lysates of PMCFs treated with TGF-β1 (20 ng/mL). β-actin was used as a loading control, n = 6 independent samples. Data are represented as mean ± SEM. Statistics: one-way ANOVA followed by Tukey post hoc test (a, b). Two-tailed Student’s t-test was used to calculate the presented P values (c, d). Source data are provided as a Source Data file. Ctl control.
Global ASPP1 depletion alleviates cardiac fibrosis after MI
In our previous study, we generated ASPP1 conventional knockout (KO) mice (C57BL/6 background) (7–8 weeks old, 22-25 g) by employing the CRISPR/Cas9 strategy. The ASPP1-KO mice grow normally after birth9. ASPP1-KO mice were used to explore the functional role of ASPP1 in cardiac fibrosis after MI (Fig. 2a). The experimental design and timing of cardiac echocardiography are indicated in Fig. 2b. Successful KO of ASPP1 was confirmed by western blot assay (Fig. 2c).
Fig. 2. Global ASPP1 depletion in mice prevents cardiac fibrosis after MI.

a Strategy for the generation of ASPP1 global KO mice. b Schematic diagram of the experiment. Cardiac function was measured at the second and fourth week after MI surgery, and samples were taken at the fourth week. The diagram was created using Figdraw. c ASPP1 protein level in the heart of ASPP1 global KO mice by western blot analysis, n = 6 independent samples in each group. d Kaplan–Meier analysis of the survival of WT (n = 51) and ASPP1-KO mice (n = 51) after MI for 4 weeks. e Echocardiographic measurement of cardiac function. EF ejection fraction, FS fractional shortening; (WT, n = 13; ASPP1-KO, n = 14). f Representative images of the hearts and quantification of the ratio of heart weight (HW) to body weight (BW) in four groups of mice. (WT, n = 7; ASPP1-KO, n = 8; WT-MI, n = 17; ASPP1-KO, n = 12). Scale bar = 1 mm. Magnification 2×. g Coronal cross-section images of Masson’s trichrome staining of the whole heart and heart border area, and statistical analysis of fibrotic area (infarct size) calculated by Image-Pro Plus. (WT, n = 7; ASPP1-KO, n = 8; WT-MI = 12; ASPP1-KO = 6). Scale bar = 1 mm. Magnification 2× in whole heart view group. Scale bar = 5 mm, Magnification 20× in border area part. h, i Transcriptional level of genes encoding ECM (h) and myofibroblast markers molecules (i) in left ventricular tissue at 4 weeks after MI by qRT-PCR, n = 6 independent samples. Data are represented as mean ± SEM. Statistics: Two-tailed Student’s t-test was used to calculate the presented P values (c). Log-rank (Mantel–Cox) test (d). Two-way ANOVA followed by Tukey post hoc test (e). One-way ANOVA, followed by Tukey post hoc multiple comparisons test (f–i). Source data are provided as a Source Data file.
The mortality rate was 49% in WT mice 4-week after MI, which was reduced to 16% in ASPP1-KO mice (Fig. 2d). Echocardiographic examination revealed that there was no difference in contractile function at baseline of WT and ASPP1-KO mice. At 2 weeks and 4 weeks post-MI, deletion of ASPP1 significantly improved cardiac function as indicated by increased left ventricular ejection fraction (EF%) and fractional shortening (FS%) (Fig. 2e). ASPP1 deletion reduced the ratio of heart weight to body weight (HW/ BW) and infarct size 4 weeks after MI (Fig. 2f, g). Of note, ASPP1 KO diminished the expression of ECM proteins Col1, Col3, and Fn1 and myofibroblast markers α-SMA and Postn (Fig. 2h, i). These data indicated that ASPP1 deletion protects against left ventricular adverse remodeling post-MI.
Myofibroblast ASPP1 deletion restricts fibrotic remodeling and preserves the function of the injured hearts
As deletion of ASPP1 is protective on cardiomyocytes during ischemia injury9, the above observed beneficial effects of ASPP1 deletion on chronic MI may be caused by the effects of ASPP1 deficiency in cardiomyocytes. We then generated conditional ASPP1 deletion in myofibroblasts using tamoxifen-inducible Postn promoter-driven MerCreMer transgene (PostnMCM) technology to explore the contribution of ASPP1 to post-infarct cardiac remodeling. PostnMCM mice were crossed with ASPP1fl/fl mice to obtain ASPP1fl/fl PostnMCM mice (Fig. 3a). Tamoxifen (TAM) was administrated to ASPP1fl/flPostnMCM or ASPP1+/+PostnMCM mice for five days after MI surgery to induce the deletion of ASPP1 in myofibroblasts (ASPP1-CKO) and further examinations were performed as indicated (Fig. 3b).
Fig. 3. Myofibroblast ASPP1 deletion prevents cardiac fibrosis in mice.

a Schematic diagram for the construction of myofibroblast ASPP1 KO mice. ASPP1fl/fl mice were crossed with PostnMCM to obtain ASPP1fl/fl PostnMCM mice. b Schematic diagram of the experimental design. ASPP1+/+PostnMCM (PostnMCM) and ASPP1fl/fl PostnMCM (ASPP1-CKO) mice were given TAM continuously for 5 days after MI by intraperitoneal injection. PostnMCM, ASPP1+/+ PostnMCM; ASPP1-CKO, ASPP1fl/fl PostnMCM; TAM, tamoxifen. The diagram was created using Figdraw. c ASPP1 protein level in the heart of ASPP1fl/fl PostnMCM by western blot assay, n = 6 independent samples. d Kaplan–Meier analysis of the survival of ASPP1+/+PostnMCM (PostnMCM) and ASPP1-CKO upon MI 4 weeks. n = 24 in PostnMCM + MI group, n = 37 in ASPP1-CKO + MI group. e Echocardiographic measurement of cardiac function. n = 18 in PostnMCM group, n = 16 in ASPP1-CKO group. f Representative images of the hearts and quantification of the ratio of HW to BW in four groups of mice. n = 8 in PostnMCM group, n = 7 in ASPP1-CKO group, n = 8 in PostnMCM + MI group, n = 7 in ASPP1-CKO + MI group, Scale bar = 1 mm. Magnification 2×. g Coronal cross-section images of Masson’s trichrome staining and statistical analysis of fibrotic area (infarct size) using Image-Pro Plus. n = 7 in the PostnMCM group, n = 7 in the ASPP1-CKO group, n = 7 in the PostnMCM + MI group, n = 10 in ASPP1-CKO + MI group. Scale bar = 1 mm. Magnification 2× in the whole heart view group. Scale bar = 5 mm, Magnification 20× in border area part. h, i. Transcriptional level of genes encoding ECM (h) and myofibroblast markers molecules (i) in left ventricular tissue at 28 days after MI by qRT-PCR, n = 6 independent samples. Data are represented as mean ± SEM. Statistics: A two-tailed Student’s t-test was used to calculate the presented P values (c). Log-rank (Mantel–Cox) test (d). 2-way ANOVA followed by Tukey post hoc test (e). One-way ANOVA, followed by Tukey post hoc multiple comparisons test (f–i). Source data are provided as a Source Data file.
The protein level of ASPP1 was remarkably downregulated in ASPP1-CKO mice (Fig. 3c). Four weeks after MI, ASPP1-CKO mice exhibited a significantly higher survival rate than ASPP1+/+PostnMCM mice (Fig. 3d). Moreover, ASPP1-CKO mice showed improved cardiac function after MI as indicated by increased EF and FS compared with ASPP1+/+PostnMCM mice (Fig. 3e). Myofibroblast KO of ASPP1 significantly reduced HW/BW ratio and infarct size of MI mice (Fig. 3f, g). Additionally, myofibroblast ASPP1 deletion inhibited the expression of ECM proteins Col1, Col3, and Fn1 and fibroblast activation markers α-SMA and Postn, while producing no influence on quiescent marker Tcf21 (Fig. 3h, i).
These findings confirmed that deletion of ASPP1 in myofibroblasts resulted in improved cardiac function and prevented adverse cardiac remodeling post-MI.
ASPP1 promotes cardiac fibroblast activation and collagen production in vitro
To evaluate the role of ASPP1 in fibroblast proliferation, differentiation, and the production of ECM in vitro, we transfected ASPP1 siRNA into cultured PMCFs to knockdown its expression (Fig. 4a). Silencing of ASPP1 decreased Col1 and Fn1 mRNA and protein levels in PMCFs treated with TGF-β1 (Fig. 4b–d). EdU staining indicated a conspicuous cell proliferation inhibition with the silencing of ASPP1 in TGF-β1 treated PMCFs (Fig. 4e). Then, flow cytometry was employed to measure DNA content in each cell cycle phase to evaluate the proliferative state of PMCFs. ASPP1 knockdown led to cell cycle arrest in the G0/G1 phase, and the cells in the S phase were reduced from 29.07% to 15.83% in TGF-β1 treated PMCFs (Fig. S1a). Immunofluorescent staining revealed a significant decrease in the number of α-SMA-positive cells in TGF-β1 treated PMCFs (Fig. 4f).
Fig. 4. ASPP1 facilitates cardiac fibroblast activation and collagen production in vitro.

a Efficiency of small interfering RNA (siRNA) of ASPP1 in PMCFs by Western blot, n = 4 independent samples. b Effects of siASPP1 on TGF-β1-induced decreases of Col1, and Fn1 mRNA levels in PMCFs by qRT-PCR analysis, n = 6 independent samples. c, d Effects of ASPP1 knockdown (siASPP1) on TGF-β1-induced decreases of Col1 and Fn1 protein levels in PMCFs by Western blot analysis, n = 5 independent experiments. e Representative photographs of EdU-positive cells by immunofluorescent staining, n = 17 independent fields of view from three independent experiments. Scale bar = 50 μm. Magnification 20×. f Representative images of α-SMA positive cells by immunofluorescent staining and rate of α-SMA positive cells. n = 8 independent fields of view from three independent experiments in the TGF-β1 group, n = 11 independent fields of view from three independent experiments in the TGF-β1 + siASPP1 group. Scale bar = 40 μm. Magnification 40×. g Efficiency of ASPP1 overexpression plasmid in PMCFs by western blot, n = 6 independent samples. h The mRNA levels of Col1 and Fn1 in PMCFs after transfection of ASPP1 overexpression plasmids by qRT-PCR analysis, n = 6 independent samples. i, j The protein levels of Col1 and Fn1 upregulated in the PMCFs after transfection of ASPP1 overexpression plasmids by Western blot analysis, n = 4 independent experiments in Col1 group, n = 5 independent experiments in Fn1. k Representative photographs of EdU-positive cells in the ASPP1 overexpression group. n = 17 independent fields of view from three independent experiments in the NC group, n = 10 independent fields of view from three independent experiments in the oe-ASPP1 group. Scale bar = 50 μm. Magnification 20×. l Immunofluorescent staining of α-SMA and statistical analysis of α-SMA positive cells. n = 8 independent fields of view from three independent experiments. Scale bar = 40 μm. Magnification 40×. Data are represented as mean ± SEM. Statistics: Two-tailed Student’s t-test was used to calculate P values (a–d and f–l). Source data are provided as a Source data file.
We then overexpressed ASPP1 in cultured PMCFs (Fig. 4g). As expected, ASPP1 overexpression increased the mRNA and protein levels of Col1 and Fn1 (Fig. 4h–j). ASPP1 overexpression promoted fibroblast proliferation, as evidenced by more EdU-positive cells. Meanwhile, flow cytometry results showed that 29.6% of cells were in the S phase in the ASPP1 overexpression group and 19.6% of them in the control group (Figs. 4k and S1b). In addition, the rate of α-SMA positive cells was significantly elevated in the ASPP1 overexpression group compared to the control group (Fig. 4l).
ASPP1 modulates p53 signaling in cardiac fibroblast
As p53 is critical in fibroblast activation and cardiac fibrosis11, we speculate that ASPP1 may regulate cardiac fibrosis via its influence on p53. Four weeks after MI, both global and myofibroblast conditional KO of ASPP1 dramatically restored p53 protein level in the infarct zone (Fig. 5a). Knockdown of ASPP1 substantially upregulated p53 level in PMCFs treated with TGF-β1, and overexpression of ASPP1 significantly suppressed p53 expression in PMCFs (Fig. 5b). However, the mRNA level of p53 was not considerably affected in response to up- and down-regulation of ASPP1 (Fig. 5c) and no difference was seen in ASPP1 global KO mice and myofibroblast KO mice (Fig. S2a, b). In addition, myofibroblast KO of ASPP1 in mice with MI or silencing of ASPP1 in PMCFs treated with TGF-β1 significantly promoted the expression of mitotic cyclins Ccna2, Ccnb1, Ccne1, and Cdk1, and inhibited the expression of CDK1 inhibitor Cdkn1a (Figs. 5d and S2c, d). The opposing effects were observed when ASPP1 was overexpressed (Fig. 5e). Notably, the disturbance of ASPP1 had no significant effects on other classic target genes except for p53 involved in the cell cycle (Fig. S2e, f). The immunofluorescent study showed that ASPP1 knockdown led to the upregulation and nuclear accumulation of p53 in TGF-β1 treated PMCFs (Fig. 5f), while overexpression of ASPP1 dramatically reduced the total and nuclear level of p53 (Fig. 5g). Furthermore, we further investigated whether the biological function of ASPP1 depends on p53 in fibrosis. We found that knockdown of p53 significantly prevented the inhibitory effects of ASPP1 suppression on Col1 and Fn1 expression in TGF-β1 treated PMCFs (Fig. 5h). Suppression of p53 further facilitated the cell proliferation by knockdown of ASPP1 and restored the cells proliferation and the expression of mitotic cyclins genes (Fig. S3c, d. In addition, overexpression of p53 canceled the promoting effects of ASPP1 on Col1 and Fn1 expression in PMCFs (Fig. 5i). Furthermore, the overexpression of p53 decreased the rate of fibroblast proliferation and altered the expression of cell cycle-related genes induced by ASPP1. (Fig. S3e, f). Knockdown and overexpression efficiency of p53 were shown in Fig. S1a, b.
Fig. 5. ASPP1 negatively regulates p53 protein level.

a p53 protein levels in ASPP1-KO group (The left) and ASPP1-CKO group (the right) by western blot assay, n = 6 independent samples in ASPP1-KO group in each group, n = 6 independent samples in ASPP1-CKO group in each group. b Effects of ASPP1 knockdown (si-ASPP1) on TGF-β1-induced (The left) or ASPP1 overexpression (The right) negative regulation of p53 protein levels in PMCFs by western blot analysis, n = 6 independent samples. c Effects of ASPP1 knockdown (si-ASPP1) on TGF-β1-induced (The left) or ASPP1 overexpression (the right) on p53 mRNA levels in PMCFs by qRT-PCR, n = 8 independent samples in si-ASPP1 + TGF-β1 group, n = 7 independent samples in ASPP1 overexpression group. d, e qRT-PCR was used to evaluate the expression of genes encoding cell cycle regulators in PMCFs, n = 6 independent samples in each group. f, g Immunofluorescent staining of ASPP1 and p53 in PMCFs. ASPP1 was stained in red, and p53 in green. The nucleus was stained in blue with DAPI, n = 3 independent experiments. Scale bar = 20 μm. Magnification 60×. h The protein levels of Col1 and Fn1 after co-transfection of si-ASPP1 and the siRNA for p53 (si-p53), n = 5 independent samples in each group. i The effect of Col1 and Fn1 after co-transfection of oe-ASPP1 plasmids and p53 overexpression (oe-p53) plasmids, n = 5 independent samples in each group. Data are represented as mean ± SEM. Statistics: A two-tailed Student’s t-test was used to calculate P values (a–e). One-way ANOVA, followed by Tukey post hoc multiple comparisons test (h, i). Source data are provided as a Source Data file.
These findings suggest that ASPP1 exerts fibrogenic function by negatively modulating the protein level of p53.
We next investigated the mechanisms underlying the effect of ASPP1 on p53 protein stability. We examined the half-life of p53 by employing cycloheximide. The half-life of p53 is prolonged by suppression of ASPP1, and overexpression of ASPP1 shortens p53’s half-life (Fig. S4a, b). Treatment of PMCFs with proteasome inhibitor MG132 prevented p53 downregulation induced by ASPP1 overexpression (Fig. 6a), indicating that ASPP1 may regulate the degradation of p53 via the ubiquitin-proteasome pathway. We then performed co-immunoprecipitation (co-IP) and mass spectrometry (MS) analysis of TGF-β1-treated PMCFs to screen for proteins that bind to ASPP1 and hold the potential to regulate p53 ubiquitination and degradation (Fig. 6b). A total of 2526 proteins were identified. KEGG analysis showed that a host of proteins were involved in protein folding and degradation, which included the deubiquitinating enzyme OTUB1 that was reported to regulate p53 degradation19. Subsequently, we verified that the antibody for ASPP1 precipitated OTUB1 in TGF-β1 treated PMCFs and vice versa (Fig. 6c). Silencing of OTUB1 suppressed the upregulation of p53 caused by ASPP1 knockdown (Fig. 6d). Knockdown efficiency of OTUB1 was shown in Fig. S5a. In addition, overexpression of OTUB1 completely restored the expression of p53 in ASPP1 treated PMCFs (Fig. 6e). Overexpression efficiency of OTUB1 was shown in Fig. S5b. These data indicate that OTUB1 mediates the regulation of ASPP1 on p53 protein level.
Fig. 6. ASPP1 regulates p53 stability by binding to OTUB1.

a Effect of ASPP1 overexpression on p53 protein in the presence of ubiquitin-proteasome inhibitor MG132 (5 μM) by western blot assay, n = 4 independent samples. b Coomassie Blue staining of proteins immunoprecipitated by ASPP1 antibody, n = 3 independent experiments. Bar plot showing Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of ASPP1-binding genes. c Western blot analysis of OTUB1 and p53 immunoprecipitated by ASPP1 (Upper panel), and ASPP1 immunoprecipitated by OTUB1(Lower panel). The data have been reproduced in 3 independent experiments. d Protein level of p53 after co-transfection of si-ASPP1 and si-OTUB1, n = 4 independent samples. e Protein levels of p53 after co-transfection of oe-ASPP1 plasmid and oe-OTUB1 plasmid, n = 4 independent samples. f Effects of concurrent OTUB1 knockdown on p53 ubiquitination suppressed by siASPP1 in TGF-β1-induced PMCFs by Co-IP, n = 3 independent experiments. g Effect of OTUB1 overexpression on p53 ubiquitination induced by ASPP1 in PMCFs by Co-IP, n = 3 independent experiments. h. Effects of ASPP1 knockdown on the binding of OTUB1 to p53 in TGF-β1-treated PMCFs by Co-IP, n = 3 independent experiments. i Effect of ASPP1 overexpression on p53’s binding to OTUB1, n = 3 independent experiments. Data are represented as mean ± SEM. Statistics: one-way ANOVA, followed by Tukey post hoc multiple comparisons test (a, d, and e). Source data are provided as a Source data file.
Since OTUB1 functions as a deubiquitinase, we examined whether the interaction between ASPP1 and OTUB1 alters the ubiquitination and degradation of p53 in cardiac fibroblasts. A significant reduction in the ubiquitination level of p53 was observed upon suppression of ASPP1, and knockdown of OTUB1 restored the ubiquitination status of p53 (Fig. 6f). Upregulation of ASPP1 substantially increased p53 ubiquitination, whereas OTUB1 overexpression reversed this effect (Fig. 6g). Furthermore, the immunoprecipitation study showed that OTUB1 antibody precipitated more p53 upon ASPP1 knockdown, and less when ASPP1 was overexpressed (Fig. 6h, i). These findings indicate that ASPP1 effectively restricts the interaction between OTUB1 and p53 thus mediating its ubiquitination and degradation.
Discussion
Myocardial fibrosis is a critical pathological process after cardiac MI injury, which determines the clinical outcome of patients. The current study found that ASPP1 was significantly elevated during cardiac fibrosis after MI. ASPP1 deletion in myofibroblasts significantly reduced myocardial fibrosis and alleviated cardiac dysfunction. Mechanistically, ASPP1 promoted the ubiquitinated degradation of p53 by inhibiting the binding of deubiquitinating enzyme OTUB1 to p53. The findings reveal that ASPP1 plays a key role in the development of cardiac fibrosis, and inhibition of ASPP1 holds therapeutic potential in the cardiac remodeling of infarct hearts (Fig. 7).
Fig. 7. The influence of ASPP1 on cardiac fibrosis.
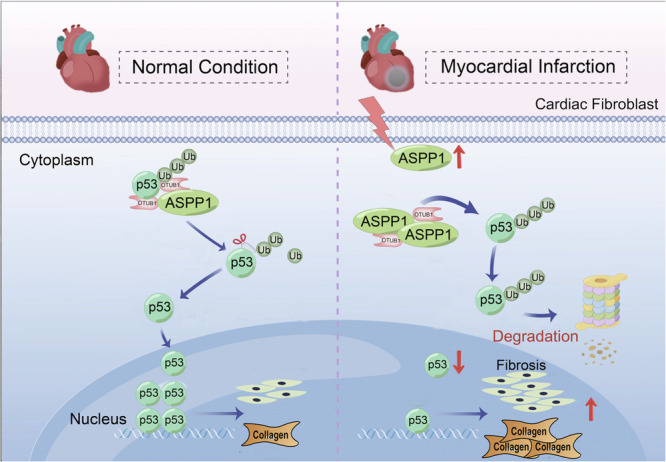
ASPP1 was up-regulated during cardiac fibrosis after MI, which bound more OTUB1 and thus inhibited OTUB1 interaction with p53. The deubiquitination of p53 was therefore decreased resulting in its enhanced ubiquitination and degradation. The degradation of p53 accelerated the cell cycle progression of fibroblasts and promoted cardiac fibrosis. Ub, ubiquitin. The diagram was created using Figdraw.
ASPP1 enhanced apoptosis by promoting BAX expression as well as inhibiting Bcl2 expression, suppressing tumor growth rate20,21. In cardiac ischemia-reperfusion injury, ASPP1 promotes cardiomyocyte apoptosis and severely impairs cardiac function. Different from the pro-apoptotic effects of ASPP1 in cancer cells and cardiomyocytes9,22, we discovered an interesting ability of ASPP1 to promote cardiac fibroblast proliferation and facilitate the development of cardiac fibrosis in the present study. ASPP1 exerts pro-apoptotic effects by trafficking to the nucleus to activate p53 or p53 family members and selectively enhance the transcription of pro-apoptotic genes downstream of p5322,23. In this study, we found that ASPP1 negatively regulated p53 protein levels in cardiac fibroblasts.
Notably, p53 plays a vital role in the development of myocardial fibrosis24. It inhibits the progression of myocardial fibrosis by inhibiting fibroblast proliferation through its cell cycle arrest function during myocardial fibrosis11,12. p53 arrests cell cycle in the G1/G0 phase and prevents it from entering the S phase, inhibiting cell proliferation25,26. Our finding that ASPP1 promotes cardiac fibroblast proliferation and fibrosis can be explained by its negative regulation on p53 in cardiac fibroblasts. p53 primarily acts as a transcription factor in the nucleus, exerting its influence by directly modulating the expression of target genes. In ischemia-reperfusion injury, p53 accumulates and induces cardiomyocyte apoptosis via regulation of Bcl2 and Puma27. On the other hand, PAI-1 induces senescence in alveolar type 2 cells in fibrotic lung disease by elevating p53 expression and subsequently activating the p53–p21–Rb cell cycle inhibitory pathway28. ASPP1 controls cell function by regulating p53. When RAS is activated, ASPP1 accumulates and drives p53 and p53 family members to the nucleus, enhancing p53 transcription and apoptosis function22,23,29. In cardiac ischemia/reperfusion (I/R) injury, the interaction between ASPP1 and p53 leads to their simultaneous translocation into the nucleus through importin-β1, ultimately facilitating cardiomyocyte apoptosis9. In this study, we found that in cardiac fibroblasts ASPP1 inhibits p53 and enhances fibroblast proliferation. The absence of ASPP1 promotes the nuclear localization of p53, and the accumulation of p53 nuclear localization significantly inhibits the progression of fibrosis. The opposing biological effects of ASPP1 in different cell types can be observed due to the opposite regulation of ASPP1 on p53 expression. For example, ASPP1 increases p53 levels in cardiomyocytes to induce apoptosis9.
It has been demonstrated that the same molecule or pathway may exert different or opposing biological functions in different cell or tissue types. It has been reported that ubiquitin-specific peptidase 7 (USP7) yields multiple functions through interacting with p53 in cellular processes. USP7 promotes colorectal cancer cell apoptosis by directly stabilizing p5330,31. However, USP7 induces fibroblast activation and myocardial fibrosis by binding to and stabilizing MDM2, leading to p53 degradation12. We previously reported that p53 exacerbates ischemia-reperfusion injury by upregulating Puma, Bax, and Noxa to promote myocardial cell apoptosis32. Interestingly, in the current study, p53 mainly upregulates Cdkn1a and inhibits cell cycle progression, eventually leading to cell cycle arrest rather than apoptosis, thereby preventing the abnormal proliferation of cardiac fibroblasts and reducing the burden on the heart11,12. Our findings provide a new insight that ASPP1 is a novel regulator in cardiac fibrosis, and, more importantly, the same genetic manipulation strategy for silencing ASPP1, which inhibits p53 in cardiomyocytes while promoting p53 in fibroblasts, can produce cardiac protective effects as reflected by the reduced cardiac fibrosis and increased cardiomyocyte survival through differential regulatory mechanisms of p53 expression. Different cells or tissues exhibit different biological functions and responses to the stimulation/stress, which primarily depend on different cell-specific components and signaling pathways. The different response of cells/tissue to one stimulation confers significant potential and implications in clinical research and treatment.
The ubiquitination modification of p53 is a crucial post-translational modification that alters its protein level. Deubiquitination modifications serve as a reversible mechanism of ubiquitination, which governs protein levels continuously and maintains protein homeostasis in vivo. A previous study has demonstrated that the deubiquitinating protease USP9X participates in interaction with MDM2, a classical E3 ubiquitin ligase of p53 to induce the accumulation of MDM2, and consequently p53 ubiquitination and degradation33. OTUB1 functions as a deubiquitinating enzyme through its interaction with p53. OTUB1 directly suppresses p53 ubiquitination in the cytoplasm, stabilizing and activating p53, and induces p53-dependent apoptosis and drastic cell growth inhibition19. In this study, we found that an abundance of ASPP1 restricted the interaction between p53 and OTUB1, ultimately resulting in the ubiquitination and degradation of p53.
Following myocardial infarction, cardiac fibroblasts undergo a phenotypic switch from a quiescent to an activated state. These activated CFs or myofibroblasts play an important role in healing phases and collagen deposition responsible for cardiac fibrosis development34,35. The abundant presence of myofibroblasts results in fibrosis due to ECM accumulation, which greatly increases morbidity and mortality. Here, we found that myofibroblast KO of ASPP1 after the onset of myocardial infarction, had a strong inhibitory effect on the progression of myocardial fibrosis in mice. Inhibition of ASPP1 expression in myofibroblasts leads to a significant improvement in cardiac function and a reduction in infarct size following myocardial infarction. In the current study, the PostnMCM strategy was employed to delete ASPP1 resulting in significant alleviation of myocardial fibrosis. However, Postn is also expressed in other cell types such as vascular smooth muscle cells, endothelial cells, etc. Therefore, it cannot be ruled out the potential contribution of these cells to cardiac protection in response to ASPP1 deficiency. Hence, ASPP1 represents a potential therapeutic target of cardiac fibrosis post-MI for clinical implementation.
Our findings revealed that inhibiting ASPP1 in both cardiomyocytes and cardiac myofibroblasts produced beneficial effects on cardiac function in mice. Thus, suppressing ASPP1 could be an excellent strategy for preventing myocardial infarction or reducing the injury caused by ischemia-reperfusion. Currently, there is no small molecular inhibitors for ASPP1 available except of its siRNA or shRNA. Inhibition of ASPP1 may be accomplished by utilizing Proteolysis Targeting Chimeras (PROTAC) technology to specifically decrease the expression of ASPP1 in fibroblasts and cardiomyocytes.
The application of p53 stabilizers, such as the nutlin MDM2 inhibitor, prevents the degradation of p53 and results in an increase of p53 protein in both cardiomyocytes and myofibroblasts. It can be speculated that cardiac fibrosis is suppressed due to the inhibition of fibroblast activity by p53, while cardiomyocyte apoptosis may be promoted due to the pro-apoptotic function of p53. However, inhibition of ASPP1 may suppress fibrosis by increasing p53 in myofibroblasts and alleviate heart injury by decreasing p53 and cardiomyocyte apoptosis. Thereby, inhibition of ASPP1 produces beneficial effects from both fibroblasts and cardiomyocytes and is likely a better strategy than only inhibition of p53 degradation by nutlin an MDM2 inhibitor. However, this needs to be demonstrated with experimental data.
Despite of the interesting findings, there remain some limitations of the study. Firstly, we confirmed that ASPP1 negatively regulated p53 stability via interaction with OTUB1, while we cannot dismiss the involvement of other proteins in this process. Secondly, we verified that inhibition of ASPP1 exerted anti-fibrotic effects. However, we did not find an ASPP1-specific inhibitor that inhibits cardiac fibrosis. Finally, the specific mechanisms of how ASPP1 is elevated in cardiac fibrosis and how it promotes fibroblast to myofibroblast transformation remain to be investigated in the future.
In summary, our study indicates that ASPP1 plays a crucial role in the development of pathological myocardial fibrosis. Inhibition of ASPP1 mitigates cardiac dysfunction in myocardial infarction and prevents cardiac fibrosis. The interaction between ASPP1 and OTUB1 facilitates the ubiquitination and subsequent degradation of p53, thereby contributing to the activation of cardiac fibroblasts and the development of myocardial fibrosis. These findings offer unique insights into the molecular mechanism of pathological myocardial fibrosis and present new therapeutic strategies for myocardial fibrosis.
Methods
Animals
All mice were maintained in a temperature-controlled facility with 12 h light/dark cycle at 23 ± 3 °C and 30–70% humidity. All animal experiments were approved by the Ethic Committees of the College of Pharmacy, Harbin Medical University (IRB3005821), and in accordance with the Guide for the Care and Use of Laboratory Animals in Harbin Medical University.
Generation of ASPP1 KO mice
ASPP1 KO mice were generated by Cyagen (Guangzhou, China) using CRISPR/Cas9 strategy9. Briefly, gRNA1, gRNA2, and Cas9 expression plasmids were designed to delete exon 2 of ASPP1. Cas9 mRNA and gRNAs (guide RNAs) were generated by in vitro transcription. Two gRNAs (gRNA1 and gRNA2) were used to identify the target sequence that contains ASPP1 in the present study (Supplementary Table 1). Cas9 mRNA and gRNAs were microinjected into fertilized eggs. All mice were compared with non-transgenic or wild-type gender-matched littermates. The primers used for verifying the flox gene in mice are: forward 5’-TGTGGTTCCCCTGTCAAACTC-3’ and reverse 5’-CGTCCAGAAGAACTGAGCTAAC-3’.
Generation of myofibroblast ASPP1 KO mice
ASPP1flox/flox (ASPP1fl/fl) mice were generated using the CRISPR/ Cas9 system by Cyagen Biosciences Inc (Guangzhou, China). The second exon was flanked by loxP sites, and thus, two single guide RNAs (sgRNA1 and sgRNA2) targeting ASPP1 exons 2 were designed. The donor vector containing exon 2 flanked by two loxP sites and the two homology arms were used as a template. The primers used for verifying flox gene in mice are: forward 5’-TGGTGTAGGTGTGTATGTCCTGG−3’ and reverse 5’-TTGTAGGTTGTTGAAATGGCTC−3’. PostnMerCreMer+/− mice (Jackson Laboratory, USA) were bred with ASPP1fl/fl mice to generate myofibroblast ASPP1 KO mice. TAM (75 mg/kg) was administered by intraperitoneal (i.p.) injection every 24 h for a total of five consecutive days to induce the deletion of ASPP1. As a protective measure, the injection site was sterilized with 70% ethanol prior to injection. ASPP1+/+PostnMerCreMer littermates were used as controls. The PostnMerCreMer+/−; ASPP1fl/fl progeny were viable, fertile, and reproduced at expected Mendelian ratios, and showed no overt pathological phenotypes.
Mouse model of myocardial infarction
Mice were anesthetized with Avertin (100 mg/kg, Sigma-Aldrich, USA) via intraperitoneal injection and ventilated using a rodent ventilator with a tidal volume of 200 μl and a frequency of 110 breaths per minute (R415; RWD life science, China). The skin surface of the left chest was disinfected and a thoracotomy through the 3,4 intercostal area was performed to expose the heart. The left anterior descending coronary artery was occluded by tying a ligation with a 7–0 silk suture 1–2 mm from the lower edge of the left atrium. After the operation, the general condition of the mice was monitored daily, and the mice were sacrificed at 4 weeks after MI. The experimenters were blind to treatment/genotype grouping information during the experiment and quantification. Group sizes were determined according to our previous experience with the establishment of a mouse model of myocardial ischemia36.
Echocardiography
To determine the cardiac function of mice, M-mode echocardiography of the heart was acquired by Vevo2100 Imaging System (VisualSonics, Toronto, Canada) equipped with a 10-MHz phased-array transducer. Briefly, after removing the hair from the chest of mice using NairTM depilatory cream (Church & Dwight Co., Inc., Princeton, NJ, USA), mice were smeared with medical ultrasonic couplant on the chest (Tianjin Yajie Medical Material Co., Ltd., Tianjin, China). Two-dimensional targeted M-mode traces were recorded from the parasternal short-axis view at the level of the mid-papillary muscles and from the parasternal long-axis view at the level of immediately under of the papillary muscle. A minimum of six consecutive cardiac cycles were obtained, and the parameters including left ventricular EF, and FS were calculated from M-mode recording. The data are presented as the average of measurements of three consecutive beats.
Masson’s trichrome staining
The hearts of mice were fixed in 4% paraformaldehyde for 24 h, embedded in paraffin, and cut longitudinally into 5 μm thick cross-sectional slices. The slices were stained with Masson’s trichrome staining kit. The extent of fibrotic area was measured by Image-Pro Plus 6.0.
Isolation and culture of cardiac fibroblasts
PMCFs were isolated from 1- to 3-day-old C57BL/6 mice via the following procedures. After disinfecting the mice with 75% alcohol, the hearts were rapidly excised with high-temperature roasted scissors and tweezers and washed with pre-cooling PBS. Then the hearts were digested by pancreatin and placed on a rocking bed at 4 °C for 8 h to 12 h. After digestion with Type II Collagenase, the obtained cells were centrifuged at 1000 g for 5 min and resuspended by high glucose DMEM complete medium containing 10% fetal bovine serum and 1% penicillin/streptomycin. After 2 h incubation (5% CO2, 95% humidified air, 37 °C), adherent cells were called fibroblasts, which needed to be incubated for another 48 h under the same condition.
Transfection procedure and TGF-β1 administration
ASPP1, p53, and OTUB1 plasmids were constructed by Genechem Co., Ltd. (Shanghai, China). Transfection of plasmids was carried out by mixing with LipofectamineTM 2000 reagent (Invitrogen, America). siRNAs of ASPP1 (5′-GCUGCUGUGGGUCCUUAUATT-3′, and 5′-UAUAAGGACCCACAGCAGCTT-3′), p53 (5′-CAUUUUCAGGCUUAUGGAATT-3′, and 5′-UUGAGUUCUGCCUUAUAC CTT-3′), OTUB1 (5′-AGGUGGAACCCAUGUGCAATT-3′, and 5′- UUGCACAUGGGUUCCACCUTT-3′), and negative control (NC) for siRNA (5′-UUCUCCGAACGUGUCACGUTT-3′, and 5′-ACGUGACACGUUCG GAGAATT-3′) were synthesized by General Biosystems Co., Ltd. (Anhui, China). X-treme GENE siRNA Transfection Reagent (Roche, Basel, Switzerland) was used to transfect siRNA. Twenty-four hours after transfection, cells were treated with TGF-β1 (20 ng/mL) for another 48 h37.
Western blot
Total protein samples were extracted from cardiac fibroblasts and infarcted zones of mouse hearts using RIPA buffer (Beyotime, China) adding 1% protease inhibitor (Roche, Switzerland). The concentration of the protein sample was determined by a BCA kit (Beyotime, Shanghai, China). After denaturing at 100 °C for 7 min, 80 μg protein samples were subjected to 8–10% SDS-PAGE and transferred onto nitrocellulose membranes. After 20 min of blocking in 5% milk, the nitrocellulose membranes were incubated with primary antibodies overnight at 4 °C. After washing with PBST (0.05% Tween in phosphate-buffered saline), the membranes were incubated with the secondary anti-rabbit or anti-mouse (1:10,000) polyclonal antibody at room temperature for 50 min without light. The membranes were scanned and analyzed by the Odyssey infrared scanning system (LI-COR, American). The primary antibodies used were anti-ASPP1 (1:1000, A4355, Sigma, America), anti-p53 (1:1000, #2524, Cell Signaling Technology, America), anti-OTUB1 (1:1000, A11656, ABclonal, China), anti-Fn1 (1:1000, 5613-1-AP, Proteintech, China), anti-CollagenI (1:750, WL0088, Wanleibio, China), anti-Ubiquitin (1:500, Sc-166553, Stanta Cruz Biotechnology, America), and anti-β-actin (1:5000, 66009-1-Ig, Proteintech, China).
qRT-PCR assay
RNA samples were extracted using TRIzol reagent according to standard protocol (Invitrogen, Carlsbad, CA, USA). For each sample, 800 ng total RNA was reversely transcribed into cDNA using TransScript All-inOne First-Strand cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China), and then amplified with SYBR Green method using LightCycler® 96 Instrument (Roche, Mannheim, Germany). The relative mRNA expression was determined using the 2−ΔΔct method. β-actin was used as an internal control. The primer pairs were synthesized by Invitrogen and listed in Supplementary table 2.
Flow cytometry
The transfected cardiac fibroblasts were digested with trypsin to form a single-cell suspension. After centrifugation to precipitate the cells, the supernatant was discarded. The precipitated cells were resuspended by adding 1.0 mL of pre-cooled PBS. The cells were centrifuged again and the supernatant was discarded. The cells were properly dispersed by gently flicking the bottom of the centrifuge tube. Then, 0.5 mL of propidium iodide staining solution was added, the cell precipitate was gently mixed and resuspended, and incubated at 37 °C away from light for 30 min. Sieving was performed to remove oversized cell clusters. Flow-through assay was carried out with an excitation wavelength of 488 nm, and light scattering was detected at the same time. Examples of gating strategy are provided in Supplementary Fig. 6.
Co-Immunoprecipitation assay
To determine the interaction between proteins, the Pierce Co-Immunoprecipitation Kit was used. Briefly, cells were lysed by lysis buffer. The protein complex was captured by anti-ASPP1 (HPA006394, Sigma-Aldrich, America) antibody, anti-p53 (10442-1-AP, Proteintech, China) antibody, anti-OTUB1 (NBP-149934, Novus, America) antibody, anti-Ubiquitin (Sc-166553, Santa Cruz Biotechnology, America) and anti-rabbit IgG antibody. IgG was used as a negative control. Protein A/G PLUS-Agarose was used to precipitate the protein complex combined with the antibody.
Half-life and ubiquitylation assays
To determine the mode of p53 degradation, the inhibitor of protein synthesis in eukaryotes, Cycloheximide (20 μg/mL) (HY-12320, MedChemExpress, America) and proteasomes inhibitor, MG-132 (20 μM) (HY-13259, MedChemExpress, America) were used.
Immunostaining assay
Cells and tissues with different treatments were fixed with 4% paraformaldehyde for 15 min and permeabilized in 0.4% Triton X-100 for 15 min, then blocked by normal goat serum for 1 h. The cells and tissues were incubated with primary antibody overnight and then incubated with secondary antibody for 1 h in dark conditions. DAPI was used to stain cell nuclei. Images of fluorescence were captured using a fluorescence microscope (Zeiss, Jena, Germany). The antibodies used for the immunostaining assay were anti-α-SMA (1:500; Abcam, Cambridge, USA) followed by DyLight 488 (1:500, 35552, Thermo Fisher, America), anti-ASPP1 (1:100, HPA006394, Sigma-Aldrich, America) followed by DyLight 488 (1:500, 35552, Thermo Fisher, America) in cells, and followed by DyLight 647 (1:500, A32795, Thermo Fisher, America) in tissue, anti-p53 (1:100, AF1355, R&D, America) followed by DyLight 594 (1:500, A23430, AmyJet, China) followed by DyLight 488 (1:1000, 35502, Thermo Fisher, America).
Mass spectrometry
Mass spectrometry was performed at Novogene Co. Ltd. In detail, bands of interest were cut off and washed with ddH2O2 for 10 min. The cut gel was destained by 50 mM TEAB and 50% acetonitrile (ACN) in 50 mM triethylammonium bicarbonate (TEAB) and dehydrated upon washing with 100% ACN. Proteins were digested by lug trypsin overnight at 37 °C. After centrifugation in low speed, the supernatant was collected and centrifuged then lyophilized. The powder was dissolved and mixed in 0.1% of formic acid. After eluting in the C18 desalting column, the eluents were collected and lyophilized for further MS analysis.
UHPLC-MS/MS analyses were performed using the EASY-nLCTM 1200 UHPLC system (Thermo Fisher, Germany) coupled with a Q ExactiveTM HF-X mass spectrometer (Thermo Fisher, Germany) for analysis of peptides. Mobile phase A is 0.1% formic acid in water, mobile phase B is 0.1% formic acid in 80% acetonitrile.
Liquid chromatography (LC) flow rate was set at 0.6 mL/min with a gradient program and full scan ranges set as needed as follows: 0–2 min = 6–10% B; 2–18 min = 10–30% B; 18–20 min = 30–50% B; 20–21 min = 50–100% B; 21–30 min = 00% B. Full scan ranges from m/z 350 to 1500 with resolution of 60,000 (at m/z 200). The top 40 precursors of the highest abundance in the full scan were selected and fragmented by higher energy collisional dissociation (HCD) and analyzed in MS/MS. The mass tolerance for precursor ion was 10 ppm and the mass tolerance for production was 0.02 Da. Carbamidomethyl was specified as fixed modifications, Oxidation of methionine (M) was specified as dynamic modification, and loss of methionine at the N-terminal. A maximum of two missed cleavage sites were allowed. Peptide Spectrum Matches (PSMs) with a credibility of more than 99% were identified. The identified protein contains at least 1 unique peptide. The identified PSMs and protein were retained and performed with FDR no more than 1.0%.
Statistical analysis
Data are expressed as mean ± SEM in at least three independent experiments for each group. For normally distributed data, a two-tailed Student’s t-test was used to compare two groups; one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc multi-comparison test was used to compare differences among multiple groups; statistical analyses comparing two genotypes (WT and ASPP1-KO, ASPP1+/+PostnMCM and ASPP1-CKO or ASPP1+/+PostnMCM + MI and ASPP1-CKO + MI), two manipulations (Sham and MI) were done using a two-way analysis of variance (ANOVA) followed by Tukey’s post-hoc multiple comparison test. For non-normally distributed or small sample size (n < 6) data, the Mann–Whitney test (two-tailed) was used for two groups, and the Kruskal–Wallis, followed by the false discovery rate (FDR) method of Benjamini and Hochberg test was used for multiple groups. A value of P < 0.05 was considered statistically significant. No experiment-wide/across-test multiple test correction was applied and only within-test corrections were made. The representative image was selected from one of the repeated experiments that best matched the mean value.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Supplementary information
Source data
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82430017, 82070344, 82270245 to Z.P., 82330011 to B.Y., 82070283 to Y.L., and 81930009 to Z.Z., 82170393 to Y.Z.), Key Research and Development Plan Project of Heilongjiang Province (2023ZX06C13 to D.Z.), Heilongjiang Touyan Innovation Team Program and CAMS Innovation Fund for Medical Sciences (CIFMS, 2019-I2M-5-078), HMU Marshal Initiative Funding (HMUMIF-21017 to Z.P.), and Natural Science Foundation of Heilongjiang (LH2023H047).
Author contributions
Z.W.P., Y.J.L. B.F.Y., and Y.Z. designed the research. S.X.L., M.Y., and Y.F.Z. supervised all aspects of the research. H.Y.Z., C.S.S, L.M.Z, T.T., Y.P., J.L.L., C.H.L., and L.N.X performed cellular experiments S.X.L., Y.F.Z, Y.Y., X.F.Z., X.D.L., and Y.G. conducted animal experiments. H.K.Y. revised the manuscript. Z.R.Z. finalized the manuscript. S.X.L., D.L.Z., Y.J.L., and Z.W.P. wrote and finalized the manuscript. All persons have made contributions to this work.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD055651. All data generated or analyzed in this study are available within the article and its supplementary information files. Source data are provided with this paper.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Shangxuan Li, Meng Yang, Yinfeng Zhao.
Contributor Information
Yang Zhang, Email: zhangyang0421@hrbmu.edu.cn.
Baofeng Yang, Email: yangbf@ems.hrbmu.edu.cn.
Zhiren Zhang, Email: zhirenz@hrbmu.edu.cn.
Zhenwei Pan, Email: panzw@ems.hrbmu.edu.cn.
Yanjie Lu, Email: yjlu@hrbmu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-52739-y.
References
- 1.Hinderer, S. & Schenke-Layland, K. Cardiac fibrosis—a short review of causes and therapeutic strategies. Adv. Drug Deliv. Rev.146, 77–82 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Park, S., Nguyen, N. B., Pezhouman, A. & Ardehali, R. Cardiac fibrosis: potential therapeutic targets. Transl. Res.209, 121–137 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Travers, J. G., Kamal, F. A., Robbins, J., Yutzey, K. E. & Blaxall, B. C. Cardiac fibrosis: the fibroblast awakens. Circ. Res.118, 1021–1040 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.From the American Association of Neurological Surgeons ASoNC. et al. Multisociety consensus quality improvement revised consensus statement for endovascular therapy of acute ischemic stroke. Int J. Stroke13, 612–632 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Burstein, B. & Nattel, S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J. Am. Coll. Cardiol.51, 802–809 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Rockey, D. C., Bell, P. D. & Hill, J. A. Fibrosis-a common pathway to organ injury and failure. N. Engl. J. Med372, 1138–1149 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Sun, J. et al. Human relaxin-2 fusion protein treatment prevents and reverses isoproterenol-induced hypertrophy and fibrosis in mouse heart. J. Am. Heart Assoc.8, e013465 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trigiante, G. & Lu, X. ASPP [corrected] and cancer. Nat. Rev. Cancer6, 217–226 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Yang, Y. et al. Interdependent nuclear co-trafficking of ASPP1 and p53 aggravates cardiac ischemia/reperfusion injury. Circ. Res.132, 208–222 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalluri, R. & Zeisberg, M. Fibroblasts in cancer. Nat. Rev. Cancer6, 392–401 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Liu, X. et al. p53 Regulates the extent of fibroblast proliferation and fibrosis in left ventricle pressure overload. Circ. Res.133, 271–287 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burke, R. M. et al. Small proline-rich protein 2B drives stress-dependent p53 degradation and fibroblast proliferation in heart failure. Proc. Natl. Acad. Sci. USA115, E3436–E3445 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burger, A. M. & Seth, A. K. The ubiquitin-mediated protein degradation pathway in cancer: therapeutic implications. Eur. J. Cancer40, 2217–2229 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Yu, Y. et al. SDF-1/CXCR7 axis enhances ovarian cancer cell invasion by MMP-9 expression through p38 MAPK pathway. DNA Cell Biol.33, 543–549 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Stanisic, V., Malovannaya, A., Qin, J., Lonard, D. M. & O’Malley, B. W. OTU domain-containing ubiquitin aldehyde-binding protein 1 (OTUB1) deubiquitinates estrogen receptor (ER) alpha and affects ERalpha transcriptional activity. J. Biol. Chem.284, 16135–16145 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edelmann, M. J., Kramer, H. B., Altun, M. & Kessler, B. M. Post-translational modification of the deubiquitinating enzyme otubain 1 modulates active RhoA levels and susceptibility to Yersinia invasion. FEBS J.277, 2515–2530 (2010). [DOI] [PubMed] [Google Scholar]
- 17.Wang, Y. et al. OTUB1-catalyzed deubiquitination of FOXM1 facilitates tumor progression and predicts a poor prognosis in ovarian cancer. Oncotarget7, 36681–36697 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao, L. et al. OTUB1 protein suppresses mTOR complex 1 (mTORC1) activity by deubiquitinating the mTORC1 inhibitor DEPTOR. J. Biol. Chem.293, 4883–4892 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun, X. X., Challagundla, K. B. & Dai, M. S. Positive regulation of p53 stability and activity by the deubiquitinating enzyme Otubain 1. EMBO J.31, 576–592 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samuels-Lev, Y. et al. ASPP proteins specifically stimulate the apoptotic function of p53. Mol. Cell8, 781–794 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Patel, S. et al. Molecular interactions of ASPP1 and ASPP2 with the p53 protein family and the apoptotic promoters PUMA and Bax. Nucleic Acids Res.36, 5139–5151 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang, Y. et al. ASPP1 and ASPP2 bind active RAS, potentiate RAS signalling and enhance p53 activity in cancer cells. Cell Death Differ.20, 525–534 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aylon, Y. et al. The Lats2 tumor suppressor augments p53-mediated apoptosis by promoting the nuclear proapoptotic function of ASPP1. Genes Dev.24, 2420–2429 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao, L. et al. TNAP inhibition attenuates cardiac fibrosis induced by myocardial infarction through deactivating TGF-beta1/Smads and activating P53 signaling pathways. Cell Death Dis.11, 44 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schade, A. E., Fischer, M. & DeCaprio, J. A. RB, p130 and p107 differentially repress G1/S and G2/M genes after p53 activation. Nucleic Acids Res.47, 11197–11208 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Men, H. et al. The regulatory roles of p53 in cardiovascular health and disease. Cell Mol. Life Sci.78, 2001–2018 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mandl, A., Huong Pham, L., Toth, K., Zambetti, G. & Erhardt, P. Puma deletion delays cardiac dysfunction in murine heart failure models through attenuation of apoptosis. Circulation124, 31–39 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Jiang, C. et al. Serpine 1 induces alveolar type II cell senescence through activating p53–p21–Rb pathway in fibrotic lung disease. Aging Cell16, 1114–1124 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson, A. M. et al. ASPP1/2 regulate p53-dependent death of retinal ganglion cells through PUMA and Fas/CD95 activation in vivo. J. Neurosci.33, 2205–2216 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang, Z. et al. STAT3 repressed USP7 expression is crucial for colon cancer development. FEBS Lett.586, 3013–3017 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Liu, J. et al. The phosphorylation-deubiquitination positive feedback loop of the CHK2-USP7 axis stabilizes p53 under oxidative stress. Cell Rep.43, 114366 (2024). [DOI] [PubMed] [Google Scholar]
- 32.Jiang, Y. et al. Cytoplasmic sequestration of p53 by lncRNA-CIRPILalleviates myocardial ischemia/reperfusion injury. Commun. Biol.5, 716 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vucic, D., Dixit, V. M. & Wertz, I. E. Ubiquitylation in apoptosis: a post-translational modification at the edge of life and death. Nat. Rev. Mol. Cell Biol.12, 439–452 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Frangogiannis, N. G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol.11, 255–265 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeda, N. et al. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J. Clin. Invest.120, 254–265 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhuang, Y. et al. MetBil as a novel molecular regulator in ischemia-induced cardiac fibrosis via METTL3-mediated m6A modification. FASEB J.37, e22797 (2023). [DOI] [PubMed] [Google Scholar]
- 37.Sun, F. et al. LncRNA PCFL promotes cardiac fibrosis via miR-378/GRB2 pathway following myocardial infarction. J. Mol. Cell Cardiol.133, 188–198 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD055651. All data generated or analyzed in this study are available within the article and its supplementary information files. Source data are provided with this paper.