

Abstract
Attention deficit hyperactivity disorder (ADHD) is a complex disorder that manifests variability in long-term outcomes and clinical presentations, however, the genetic contributions to such heterogeneity are not well understood. Here we show several genetic links to clinical heterogeneity in ADHD in a case-only study of 14 084 diagnosed individuals. First, we identify one genome-wide significant locus by comparing cases with ADHD and autism spectrum disorder (ASD) to cases with ADHD but not ASD. Second, we show that cases with ASD and ADHD, substance use disorder (SUD) and ADHD, or first diagnosed with ADHD in adulthood have unique polygenic score profiles that distinguish them from complementary case subgroups and controls. Finally, a polygenic score for an ASD diagnosis in ADHD cases predicted cognitive performance in an independent developmental cohort. Our approach uncovered evidence of genetic heterogeneity in ADHD, helping understand its etiology and providing a model for studies of other disorders.
Introduction
Attention deficit hyperactivity disorder (ADHD) is a multifactorial neurodevelopmental disorder with typical symptom onset in childhood, often persisting into adulthood, and affecting many aspects of life through impaired attention, impulsivity and hyperactivity1–3. Clinical presentations are heterogeneous. Diagnosed individuals demonstrate variability in symptom severity, predominance and duration, treatment, age at diagnosis, need for hospitalization, and other long-term outcomes4–6. Some diagnosed individuals experience more negative outcomes like criminality7, premature mortality8, poor educational attainment9, or lower socioeconomic status10. Psychiatric and somatic comorbidities are common but prevalences and (co)occurrences can vary substantially among individuals and across the life-span4,11. Better understanding the etiology of clinical heterogeneity is important for improving long-term outcomes and precision care. Although genetic heterogeneity is a proposed feature of complex disorders12 and important to understand13, insights have proven elusive, especially when considering the extreme polygenic architectures of psychiatric disorders (e.g.,14,15).
Family studies show genetic factors play an important role in ADHD, with estimates of the narrow-sense heritability (h2) around 0.7416. This genetic contribution is complex, including copy number variants (CNVs)17,18, rare protein truncating variants (PTV)19, and polygenes - common variants with small, independent, additive effects on liability20. Estimates of SNP-based heritability (h2SNP) for ADHD imply this polygene contribution is substantial (22%)20. ADHD associated polygenes are pleiotropic, shared broadly across clustered psychiatric, cognitive, and socio-behavioral traits, and concentrated in chromatin with active roles in neural development and functioning20. These insights come predominantly from consortia-driven meta-analyses21,22 that aggregate multiple cohorts sampled via different ascertainment criteria, case-control definitions, and healthcare contexts. Mapping robustly associated individual loci via genome-wide association studies (GWAS) does not appear overly sensitive to such cohort differences, however, inferences from polygenes may be more susceptible23,24. Polygene contributions to clinical heterogeneity in ADHD are plausible and have important implications for study design, across cohort replication, and providing more nuanced pictures of ADHD etiology13.
Recent studies have sought robust evidence of genetic heterogeneity for numerous complex disorders13, but many have been limited by conceptual, methodological, and data-related challenges that are exemplified by, but not limited to, ADHD. When considering ADHD, few cohorts have been ascertained with both the requisite statistical power for genetic analysis and the necessary depth and breadth of adjacent phenotyping for systematic investigations of clinical heterogeneity. As a result, analyses targeting heterogeneity are often conducted post-hoc or secondary to other aims (i.e., mapping disorder associated loci) where they may also be limited to a focus on single traits or selected variants. As examples, the association of previously discovered ADHD risk variants with comorbid substance use disorder (SUD)25, autism spectrum disorder (ASD)26, sex-differences27, or symptom persistence28. More generally, state of the field analytical approaches (e.g.,29) often emphasize variants with an a priori association to disorder onset and may miss or under prioritize contributions from modifier variants30,31 altering clinical trajectories without such prior association (e.g., those disrupting drug metabolizing enzymes). Clinical presentations of complex disorders vary on many dimensions and likely due to diverse sets of genetic factors, so studies that overcome these prior limitations are poised to have wide-reaching impact.
Here, we study 14 084 individuals diagnosed with ADHD from the iPSYCH2012 case-cohort study32 and a wealth of adjacent phenotyping from Danish population-wide health and civil registers33–36. We define a collection of ADHD-adjacent traits, using this term to refer to plausibly relevant, clinical phenotypes of individuals diagnosed with ADHD, that we assess for etiological relevance. We implement a well understood estimator of h2SNP in a unique way to prioritize ADHD-adjacent traits most strongly associated with genetic differences among diagnosed individuals. Then, we conduct GWAS to identify single variants associated with prioritized traits and describe plausible biological mechanisms. We then apply a polygenic profiling approach using multivariate, multinomial logistic regression to simultaneously compare controls and multiple case groups across polygenic scores (PGS) for cognitive, psychiatric, and socio-behavioral traits, accounting for primary disorder (e.g., ADHD) PGS and covariates. We use this to identify evidence of genetic heterogeneity using multiple PGS. Finally, we construct PGS using variant effects from ADHD-adjacent trait GWAS to predict cognitive and behavioral performance in an independent, typically developing cohort. Our study adds robust, new perspectives on existing clinical debates surrounding ADHD etiology and can serve as a model for other complex disorders.
Results
ADHD-adjacent traits associate with genetic differences
We defined 22 ADHD-adjacent traits from register data to broadly describe variability in medication use, psychiatric comorbidity, need of care, and sex recorded at birth for 14 084 individuals diagnosed with ADHD (Methods, Table 1, Supplementary table 1). We estimated the SNP heritability (h2SNP) of each trait within the ADHD case group to prioritize those most relevant for more detailed investigation. (Methods, Figure 1, Supplementary table 2). We observed significant h2SNP (p<0.002, adjusted for 22 tests) for three traits: a first ADHD diagnosis as an adult (≥18 years of age) (h2SNP=0.143, s.e.=0.025, p=1.7×10−10), an ADHD-adjacent diagnosis of ASD (h2SNP=0.091, s.e.=0.024, p=2.7×10−5), and an ADHD-adjacent diagnosis of SUD (h2SNP=0.085, s.e.=0.024, p=1.0×10−4). Seven traits had nominally significant p-values (p<0.05), suggesting with additional samples more traits would be implicated. We used hierarchical clustering to identify patterns in tetrachoric correlations of all pairs of traits observing roughly three groups: male sex-childhood diagnoses, medication traits, and adult first ADHD diagnosis-mood diagnoses (Supplementary table 3, clustered heatmap: Supplementary figure 1). We note prioritized traits are at least partially independent: a first ADHD diagnosis as an adult positively associated with an SUD diagnosis (rTET=0.62, s.e.=0.01, p<1×10−10) and negatively associated with an ASD diagnosis (rTET= −0.43, s.e.=0.017, p<1×10−10). SUD diagnoses were negatively associated with ASD diagnoses (rTET= −0.33, s.e.=0.019, p<1×10−10). Sensitivity analysis demonstrate that our findings were not driven more plausibly by changing clinical guidelines (Supplementary Note, Supplementary figure 2–6 and Supplementary tables 4–6). ADHD-adjacent traits appear robustly associated with genetic differences, especially a first ADHD diagnosis as an adult, an ADHD-adjacent SUD diagnosis, and an ADHD-adjacent ASD diagnosis.
Table 1. 14 084 individuals diagnosed with ADHD vary on a number of clinically relevant adjacent traits.
Dx, diagnosis; Rx, prescription; ICD-10, international classification of disease, 10th revision, unmodified 2019 version; ADHD, attention-deficit hyperactivity disorder. Full description and definition of the traits are presented in Supplementary table 1.
| Category | Trait | Description | Reference Group (0) | N0 | Outcome Group (1) | N1 |
|---|---|---|---|---|---|---|
| Medication | Any ADHD Rx | Recorded prescription for any ADHD medication | never | 1756 | ever | 12328 |
| Stimulant Rx | Recorded prescription for stimulant medication | never | 2167 | ever | 11917 | |
| Atomoxetine Rx | Recorded prescription for atomoxetine | never | 9948 | ever | 4136 | |
| Number of ADHD Rx | Number of recorded prescriptions for any ADHD medication | ≤ 29 | 7052 | > 29 | 7032 | |
| Adjacent diagnosis (Specific) | Any specific dx | Any recorded diagnosis for an iPSYCH ascertained, specific disorder | never | 10478 | ever | 3606 |
| ASD dx | Autism spectrum disorder (ICD-10: F84.0, F84.1, F84.5, F84.8 F84.9) | never | 11800 | ever | 2284 | |
| AFF dx | Affective disorder (ICD-10: F30–39) | never | 12796 | ever | 1288 | |
| ANO dx | Anorexia (ICD-10: F50.0) | never | 14020 | ever | 64 | |
| SCZ/BP dx | Schizophrenia or bipolar disorder (ICD-10: F20, F30–31) | never | 13769 | ever | 315 | |
| Adjacent diagnosis (Broad) | Any broad dx | Any recorded diagnosis for a mental, behavioral, and/or neurodevelopmental disorders according to ICD-10, chapter F | never | 3171 | ever | 10913 |
| Substance use disorder (SUD) dx | Mental and behavioral disorders due to psychoactive substance use (ICD-10: F10-F19) | never | 11457 | ever | 2627 | |
| Psychosis dx | Schizophrenia, schizotypal, delusional, and other non-mood psychotic disorders (ICD-10: F20-F29) | never | 13133 | ever | 951 | |
| Mood dx | Mood/affective disorders (ICD-10: F30-F39) | never | 12250 | ever | 1843 | |
| Anxiety dx | Anxiety, dissociative, stress-related, somatoform and other nonpsychotic mental disorders (ICD-10: F40-F49) | never | 10373 | ever | 3711 | |
| Physio-behavioral dx | Behavioral syndromes from physiological disturbances and physical factors (ICD-10: F50-F59) | never | 13578 | ever | 506 | |
| Personality dx | Disorders of adult personality and behavior (ICD-10: F60-F69) | never | 12391 | ever | 1693 | |
| Intellectual dx | Intellectual disabilities (ICD-10: F70-F79) | never | 12592 | ever | 1492 | |
| Developmental dx | Pervasive and specific developmental disorders (ICD-10: F80-F89) | never | 8713 | ever | 5371 | |
| Childhood dx | Behavioral and emotional disorders with childhood and adolescent onset (ICD-10: F90-F99). Excludes F90.0–9 and F98.8.. | never | 10264 | ever | 3820 | |
| Need of Care | Number of ADHD contacts | Number of recorded hospital contacts with an ADHD diagnosis (ICD-10: F90.0, only) | ≤2 | 9732 | >2 | 4352 |
| First ADHD dx as an adult | Age at first recorded ADHD diagnosis (ICD-10: F90.0, only) | <18 years | 10761 | ≥18 years | 3323 | |
| Sex | Sex | Biological sex recorded at birth | female | 3668 | male | 10416 |
Fig. 1. ADHD-adjacent traits associate with genetic variability among diagnosed individuals.
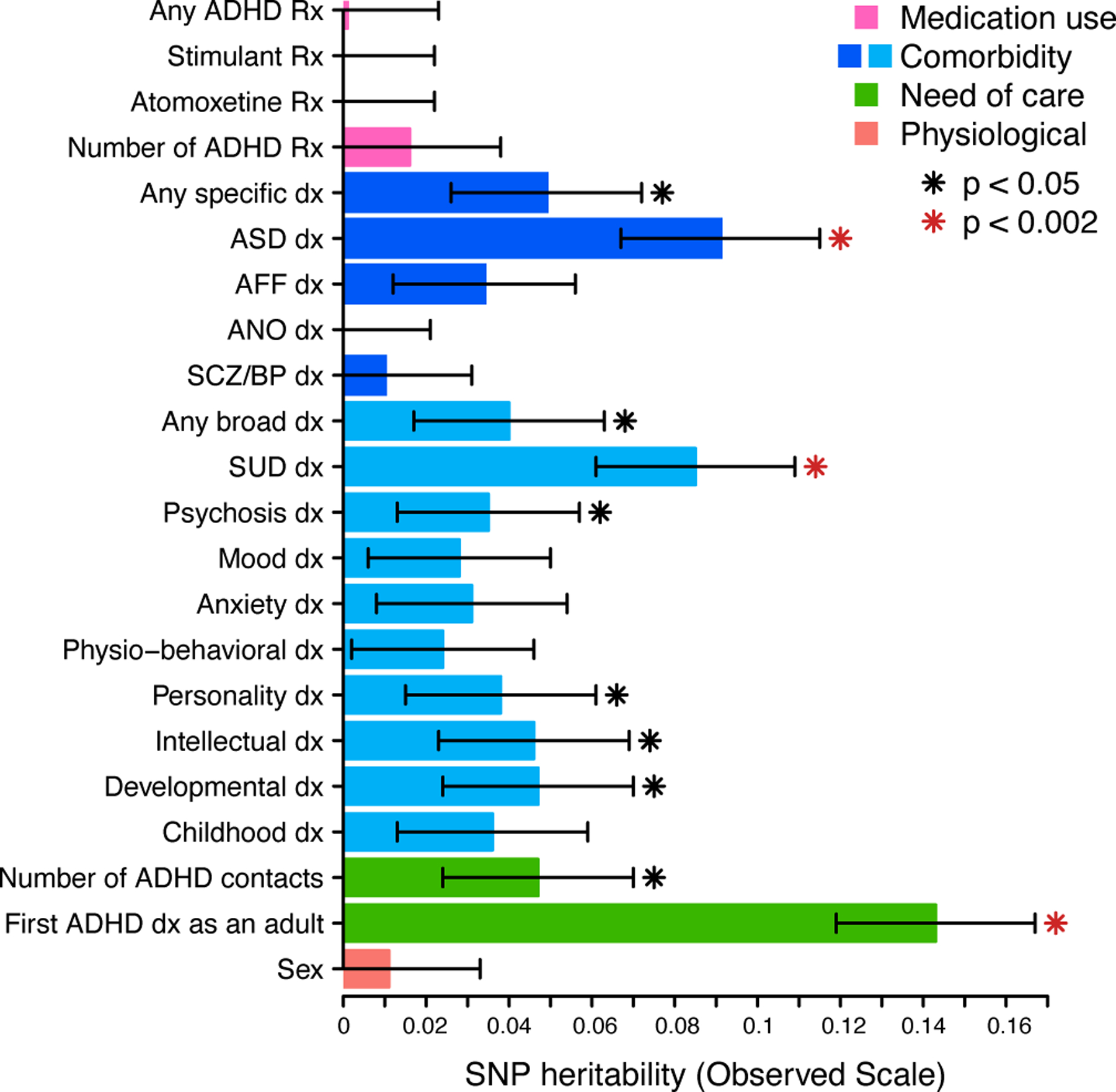
The same 14 084 individuals diagnosed with ADHD were repeatedly partitioned into 22 groups on the basis of ADHD-adjacent traits and the SNP-heritability (bars) of each trait was estimated with GCTA. Full statistical results and sample size for each estimate are available in Supplementary table 2. Significance (red star; p < 0.05/22=0.002) is after Bonferroni correction of one-tailed p-values from z-score tests. Error bars depict standard errors of the estimated observed scale SNP-heritability. Rx, prescription; dx, diagnosis; ADHD, attention-deficit hyperactivity disorder; ASD, autism spectrum disorders; AFF, affective disorders; ANO, anorexia; SCZ/BP, schizophrenia or bipolar disorder; SUD, substance use disorder
rs8178395 is associated with ADHD-adjacent ASD
We performed GWAS to identify single variants associated with the prioritized ADHD-adjacent traits (Methods). No individual SNPs were associated (p>5×10−8) with a first ADHD diagnosis as an adult (Supplementary figure 7) or an ADHD-adjacent SUD diagnosis (Supplementary figure 8). However, one locus on chromosome 17q22 was significantly associated with an ADHD-adjacent ASD diagnosis (hg19: 55,341,733–57,341,733, lead SNP:rs8178395, minor allele (T) frequency (MAF)=0.14, odds ratio (OR)=1.30, s.e. on ln scale =0.05, pGWAS=1.98×10−08; Figure 2a, Supplementary figure 9). This locus was not identified in previous ADHD20 (rs8178395, p=0.44), ASD37 (rs8178395, p=0.26), or across-psychiatric disorders38 (rs8178289, a proxy SNP with r2 LD=0.9, p=0.23) GWAS, although these individuals were included in each meta-analysis. PheWAS for rs8178395 and rs8178289 showed evidence for prior associations with measures of blood cell composition, protein levels, metabolites, cardiovascular complications, and sleep behavior (Supplementary figure 10, Supplementary table 7). rs8178395 appears reliably imputed (Supplementary figure 11–12, Supplementary table 8).
Fig. 2. rs8178395 is specifically associated with an ADHD-adjacent ASD diagnosis.
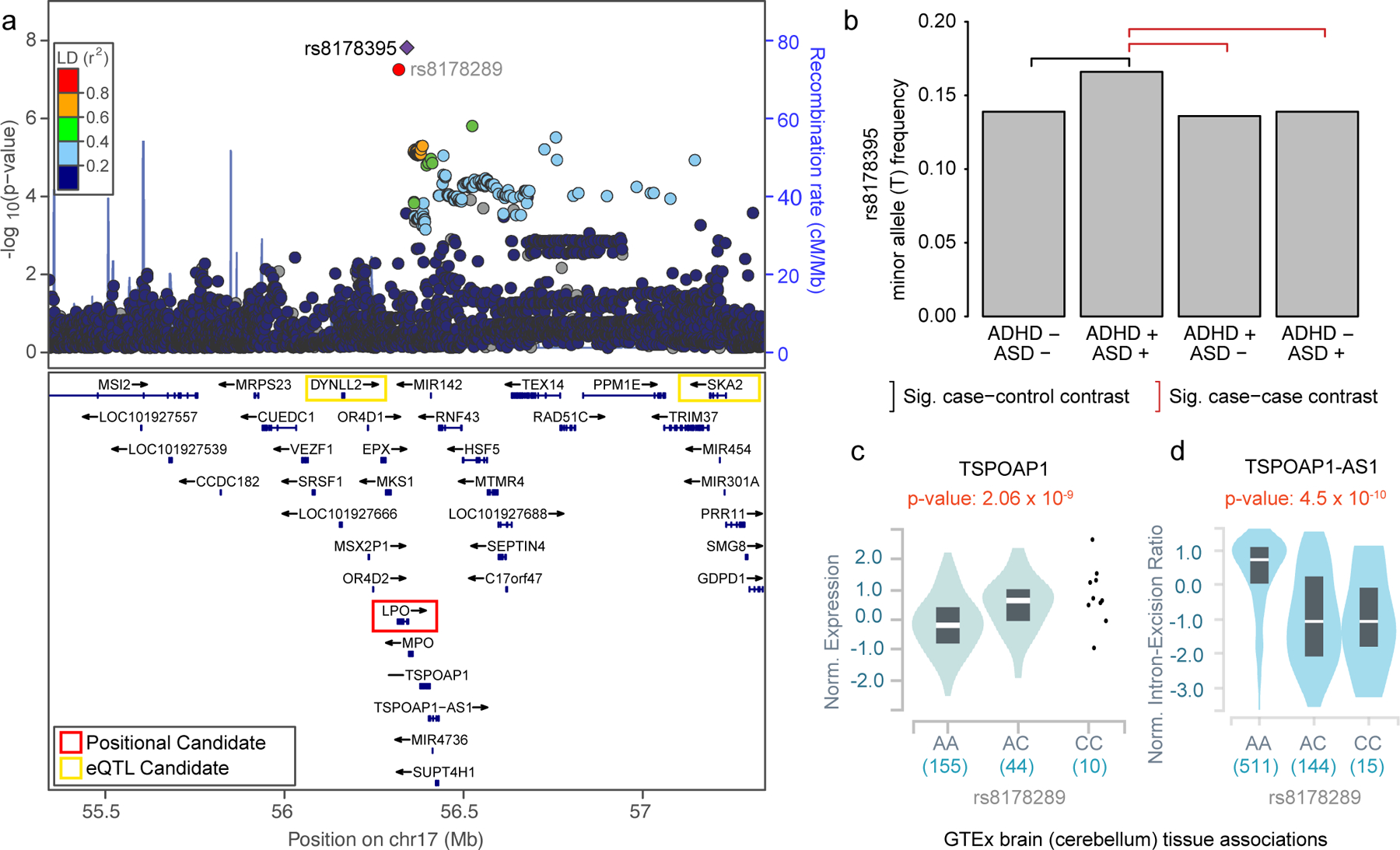
a, Locus zoom of lead SNP (rs8178395) identified as genome-wide significant (two-sided p < 5×10–8) in GWAS using logistic regression comparing individuals diagnosed with both ADHD and ASD (n=2 284) to those only diagnosed with ADHD (n=11 800). LPO, DYNLL2, TSPOAP1, and SKA2 are prioritized as candidate genes according to different criteria. LD, linkage disequilibrium; cM, centimorgans; Mb, megabases; TAS, transcription association study; b, Using multinomial logistic regression, we observed the minor allele (T) of rs8178395 is significantly increased in frequency in the group of individuals diagnosed with both ADHD and ASD (ADHD +, ASD +) relative to those diagnosed with neither ADHD, nor ASD (black bracket; ADHD −, ASD −: two-tailed p=2.9×10−7) and either, exclusively (red brackets; ADHD +, ASD −: two-tailed p=2.4×10−8; ADHD −, ASD +: two-tailed p=1.0×10−6). Significance is based on multinomial logistic regression and Bonferroni correction for the 3 follow up tests. See Supplementary tables 8–9 for additional details. Sig, Significant. c, The proxy SNP for rs8178395 (rs8178289, r2 LD: 0.9) is associated with TSPOAP1 expression in brain (and other) tissue(s). d, rs8178289 is also reported as a member of a haplotype containing a splice QTL for a TSPOAP1 associated antisense RNA, TSPOAP1-AS1, in the same (and other) tissue(s). c,d, eQTL and sQTL results were visualized using the GTEx portal with two-sided, unadjusted p-values from linear regression, center line representing the median, and box spanning 1st to 3rd quartiles.
To determine if this variant is a novel ASD SNP or represents evidence of genetic heterogeneity within ADHD, we employed multinomial logistic regression (MLR) (Methods). This test extends the two-group logistic regression comparing ADHD with or without an adjacent ASD diagnosis to additionally compare individuals with an ASD, but not ADHD diagnosis, and controls with neither diagnosis. The frequency of the minor (T) allele of rs8178395 was significantly increased in the group with ADHD-adjacent ASD relative to all other groups (vs. ADHD only, pMLR=2.4×10−8; vs. ASD only, pMLR=1.0×10−6; vs. controls, pMLR=2.9×10−7; Figure 2b, Supplementary table 9). rs8178395 indexes the first locus reported to have a specific association to individuals diagnosed with both ADHD and ASD. After excluding individuals diagnosed before 2000, 2005, or 2008 this SNP remained genome-wide significant (Supplementary Table 10).
The locus indexed by rs8178395 contains 38 genes (Figure 2a). rs8178395 falls within an intron of LPO and has been associated with expression of DYNLL2, TSPOAP1, and SKA2 in brain and MPO, RAD51C, RNF43, SEPTIN4, SMG8, SUPT4H1, TEX14, and TRIM37 in other tissues (Methods, Supplementary table 11). Partitioned LD-score regression did not identify significant enrichment in selected adult or fetal brain derived annotations (Methods, Supplementary figures 13–15, Supplementary table 12). We performed a regional transcription association study (TWAS) (Methods) using expression levels imputed via adult39 and fetal brain40 expression quantitative trait loci (eQTLs) (Methods; Supplementary table 13), but did not identify significant candidates after correction (p<0.006); adult-brain SKA2 expression was most significant (p=0.008). The proxy SNP for rs8178395 (rs8178289, r2 LD=0.9) is also associated with TSPOAP1 expression in brain tissue (Figure 2c) and, additionally, is reported by GTEx as a member of a haplotype containing splice QTLs (sQTL) for TSPOAP1-AS1, which encodes an associated antisense RNA active in brain (Figure 2d) and other tissues41. Regulation of TSPOAP1 is a plausible mechanism as TSPOAP1 is a brain expressed gene42 encoding a binding protein coupling voltage gated calcium channels to neurotransmitter vesicles in the presynaptic active zone43, has a role in neurotransmitter release and synaptic transmission44, and exonic CNVs have been associated with ASD in families45.
For comparison we performed five supporting GWAS: case subgroup (e.g. ADHD adjacent ASD) vs. control, Methods, Supplementary Figures 16–18) and cross-trait (e.g ADHD or ASD) vs. control analyses (Methods, Supplementary Figure 19–20). P-values and odds ratios of index SNPs significant in at least one of the nine GWAS (Supplementary Table 14, Supplementary Figures 21–22) and LD-score genetic correlations among the GWAS (Supplementary Figure 23) suggest case-case GWAS emphasize different loci and polygenes.
ADHD-adjacent traits share polygenes with other traits
We estimated genetic correlations (ρG,SNP; Methods) between ADHD-adjacent traits and 46 psychiatric, cognitive, and socio-behavioral traits using GWAS summary statistics (Figure 3, Supplementary table 15). Trends were broadly similar across reference traits for first ADHD diagnosis as an adult and ADHD-adjacent SUD, and generally opposite to those for ADHD-adjacent ASD, consistent with phenotypic correlations (Supplementary figure 1, Supplementary table 3). Genetic correlation trends for case-case GWAS were distinct from case-control, case subgroup-control, and cross-trait vs. control GWAS (Supplementary Figures 24–25; Supplementary Table 16–17). Individually, first diagnosis as an adult was correlated (ρG,SNP, FDR<0.05) with psychiatric traits, alcohol use (i.e., AUDIT, P score), reproductive behaviors, socioeconomic factors, education, and cognitive performance (Figure 3a), and showed a large negative trend with clinically ascertained ADHD (ρG,SNP= −0.5, s.e.=0.27, p=0.052). ADHD-adjacent SUD showed similar estimates of ρG,SNP with psychiatric outcomes, reproductive behaviors, education, and cognitive performance (Figure 3b) and large trends for several SUD related traits (i.e., Alcohol and Opioid dependence). ADHD-adjacent ASD had different ρG,SNP for education and cognitive performance, socioeconomic factors, psychiatric, reproductive traits, sleeping, and smoking traits (Figure 3c). ADHD-adjacent traits share polygenes with psychiatric, cognitive, and sociobehavioral traits that may define genetic heterogeneity in ADHD.
Fig. 3. ADHD-adjacent traits share polygenes with psychiatric, cognitive, and socio-behavioral traits.
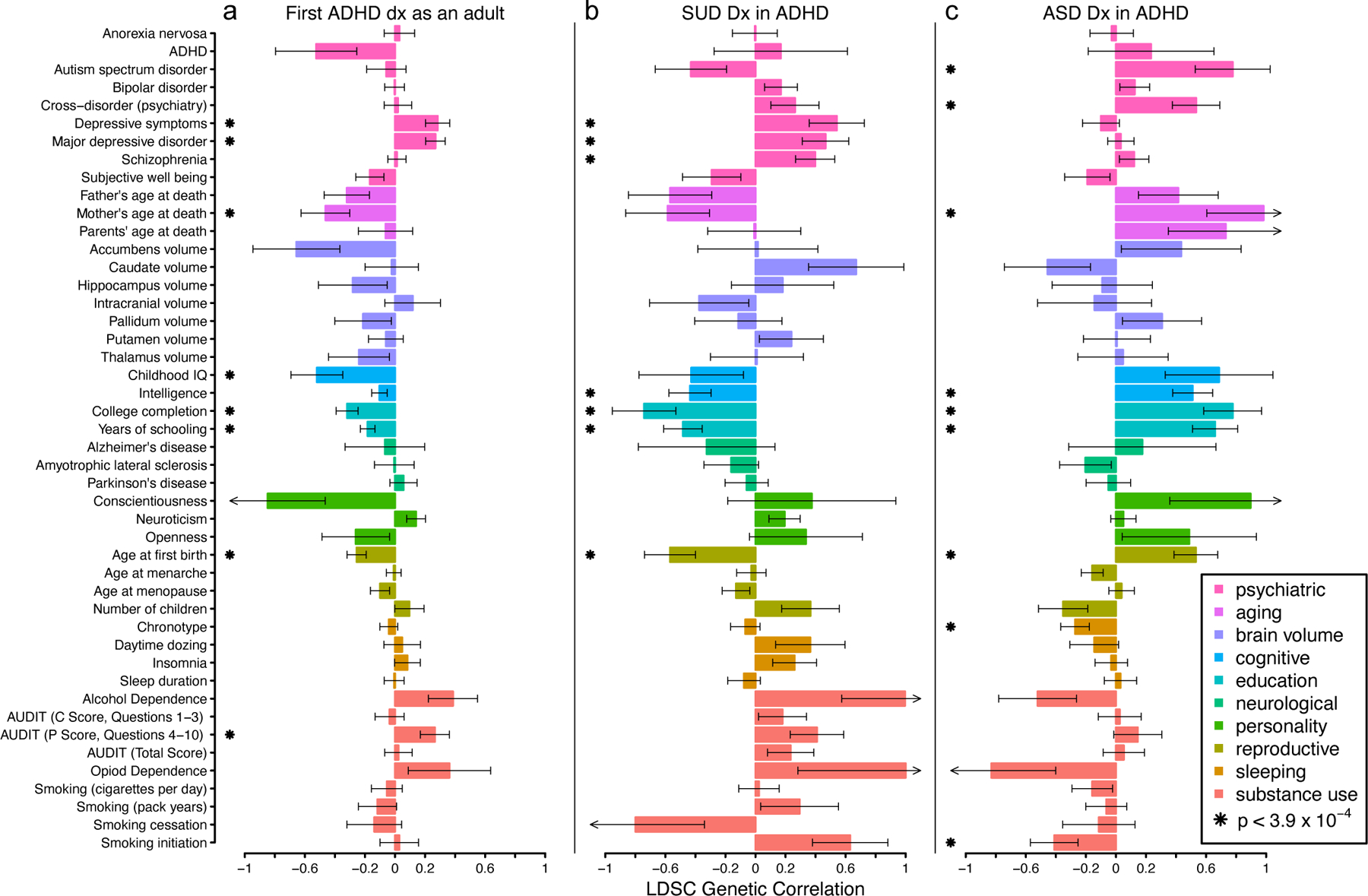
LD score regression was used to estimate the genetic correlations between ADHD-adjacent traits and a reference set of psychiatric, cognitive, and socio-behavioral traits. a, A first diagnosis of ADHD as an adult (n=3 323 cases with adult ADHD dx, n=10 761 cases with childhood ADHD dx), b, an ADHD-adjacent SUD diagnosis (n=2 627 ADHD cases with SUD dx, n=11 457 ADHD cases without SUD dx), and c, an ADHD-adjacent ASD diagnosis (n=2 284 ADHD cases with ASD dx, n=11 457 ADHD cases without ASD dx) show different patterns of genetic correlation (bars) with 46 reference traits.. Error bars denote standard error of the genetic correlation estimates, arrowheads indicate standard errors extend beyond the natural range of the estimand (>1 or < −1), and multiple comparisons were adjusted using FDR < 0.05 (corresponding to two-tailed z-score p-value < 3.9 × 10–4). See Supplementary table 15 for full statistical results and Supplementary table 28 for sample size of 46 reference GWAS. LDSC, LD score regression; FDR, false discovery rate; dx, diagnosis; ADHD, attention-deficit hyperactivity disorder; ASD, autism spectrum disorders; SUD, substance use disorder.
Polygenic profiles are linked to heterogeneity in ADHD
Next, we applied a polygenic profiling approach to test for polygene heterogeneity that can delineate ADHD-adjacent trait subgroups (Methods; Figure 4, Supplementary tables 18–20). Multi-PGS profiles were different among individuals receiving a first diagnosis for ADHD as an adult (1st Dx adult; n=3 323), a first diagnosis for ADHD before adulthood (1st Dx child; n=10 761), and controls (ADHD-; n=21 409). The mean PGS for ADHD, depressive symptoms, major depression, years of schooling, age at first birth, and alcohol use (i.e. AUDIT P score) varied significantly among the groups (p < 0.05/169 =3×10−4; Figure 4a, stars; Supplementary table 18). These three-group-wise differences were driven by specific, highly significant pairwise contrasts (Figure 4a, black and red brackets). The two ADHD case groups generally deviated significantly from controls and in the same direction. Individuals first diagnosed as an adult had significantly less positive PGS (i.e., closer to the population average) for ADHD and more positive PGS (i.e., further from the population average) for major depressive disorder than ADHD cases diagnosed before adulthood. Additional trends suggested more negative cognitive, education, and reproductive PGS for adult diagnosed ADHD cases. Simulations suggest the trends are not broadly consistent with models where adult diagnosis is simply due to ADHD liability associating with onset, a misdiagnosis of MDD, or education as an age dependent, heritable exposure (Supplementary Note, Supplementary Figures 26–30).
Fig. 4. Profiles of polygenic scores for psychiatric, cognitive, and socio-behavioral traits define aspects of heterogeneity in ADHD.
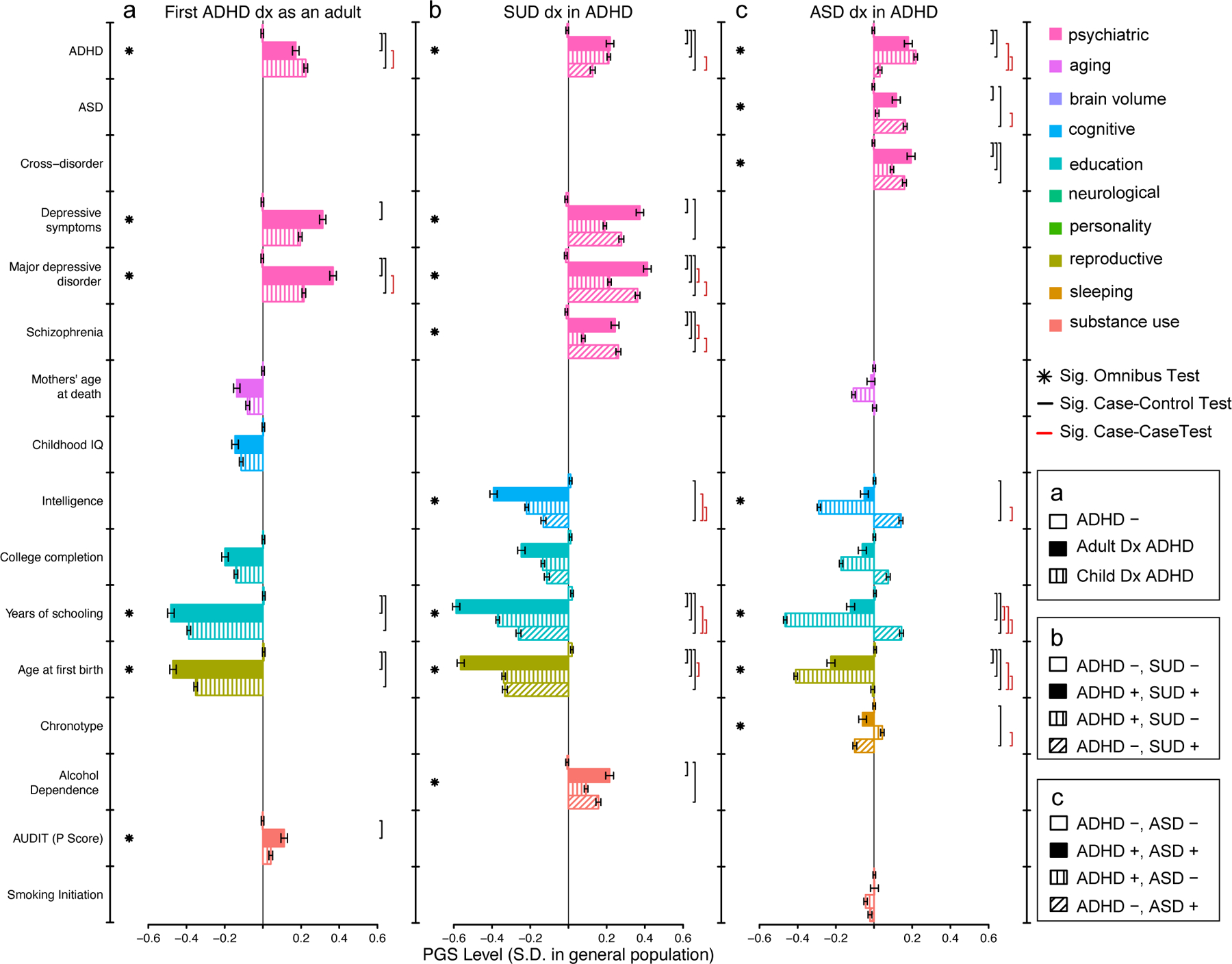
The mean level of polygenic scores (PGS; bars) are displayed for ADHD subgroups, controls, and complementary disorder groups after centering and standardizing based on a population random sample. a. Individuals not diagnosed with ADHD (ADHD −, n=21 409), first diagnosed as an adult (1st Dx Adult, n=3 323), and first diagnosed as a child (1st Dx Child, n=10 761), b. Individuals diagnosed with neither ADHD, nor SUD (ADHD-, SUD-, n=20 509), both ADHD and SUD (ADHD+, SUD+, n=2 627), ADHD but not SUD (ADHD+, SUD-, n=11 457), and SUD but not ADHD (ADHD-, SUD+, n=5 943), and c. Individuals diagnosed with neither ADHD, nor ASD (ADHD-, ASD-, n=21 197), both ADHD and ASD (ADHD+, ASD+, n=2 284), ADHD but not ASD (ADHD+, ASD-, n=11 800), and ASD but not ADHD (ADHD-, ASD+, n=9 804), vary significantly with respect to multivariate profiles of PGS. Stars denote a significant global test (one-tailed p-value) for differences in PGS level among all groups, while brackets denote significant pairwise group contrasts (two-tailed p-value), accounting for all other PGS and 25 ancestry principle components in a joint multinomial logistic regression. Significance determined by Bonferonni correction, p<0.05/169=0.0003. Error bars denote standard error of mean. See Supplementary tables 18–20 for complete statistical results and Supplementary table 28 for details of reference GWAS used for PGS. ADHD, attention deficit hyperactivity disorder; ASD, autism spectrum disorder; SUD, substance use disorder; Dx, diagnosis; Sig., Significant, PGS, polygenic score.
The multi-PGS profiles for individuals with an ADHD-adjacent SUD diagnosis (ADHD+, SUD+; n=2 627), without an ADHD-adjacent SUD diagnosis (ADHD+, SUD-; n=11 457), with an SUD but no ADHD diagnosis (ADHD-, SUD+; n=5 943), and with neither ADHD nor SUD diagnoses (ADHD-, SUD-, controls; n=20 509) were also different and with similar patterns to the previous result (Figure 4). The mean PGS for ADHD, depressive symptoms, major depression, schizophrenia, intelligence, years of schooling, age at first birth, and alcohol dependence varied significantly among the groups (p<4×10−4; Figure 4b, stars; Supplementary table 19). As above, case groups deviated from controls significantly and in the same direction. Notable pairwise differences (Figure 4b, black and red brackets) included individuals with ADHD-adjacent SUD diagnoses showing significantly more positive PGS for major depression and schizophrenia, and significantly more negative PGS for age at first birth, than ADHD without an adjacent SUD diagnosis. Individuals with ADHD-adjacent SUD diagnoses showed, relative to those with SUD diagnoses only (i.e., no ADHD diagnosis), significantly more negative PGS for education and intelligence, but no significant differences for other PGS.
Multi-PGS profiles for individuals with ADHD-adjacent ASD diagnoses (ADHD+, ASD+; n=2 284), with ADHD but no adjacent ASD diagnosis (ADHD+, ASD-; n=11 800), with ASD but not ADHD diagnoses (ADHD-, ASD+; n=9 804), and with neither (ADHD-, ASD- controls; n=21 197) were also different, but trends were broadly different from the previous two analyses. The mean PGS for ADHD, ASD, cross-disorder, intelligence, years of schooling, age at first birth, and chronotype varied significantly among the groups (p<4×10−4; Figure 4c, stars, Supplementary table 20). Individuals with ADHD-adjacent ASD diagnoses had profiles that appeared to reflect both single-diagnosed groups, simultaneously. The ADHD-adjacent ASD group had significantly more positive ADHD and ASD PGS than controls, a trend towards more positive ASD (but not ADHD) PGS than the ADHD only group, and significantly more positive ADHD PGS (but not ASD) PGS than the ASD only group (Figure 4b, black and red brackets). Single diagnosed ASD and ADHD groups had opposite trends (i.e., above vs. below the population average) for intelligence and education PGS, and the ADHD-adjacent ASD group fell in between the two. Simulations suggest the trends are not broadly consistent with misdiagnosis and most consistent with models where diagnosed individuals have diagnoses that relate to an excess of liability for both disorders, simultaneously (Supplementary Note, Supplementary figures 31–39).
Large differences among groups that are not described as statistically significant (e.g., Figure 4b: college completion, ASD PGS for ASD and ADHD vs. ADHD only) are due to collinearity among constituent profile PGS fitted jointly (Methods) and highly significant when fitted separately (Supplementary figure 40–41, Supplementary tables 21–23). Trends depicted as unadjusted levels of PGS in Figure 4 are directionally consistent with trends when depicted as partial PGS effects (i.e., regression coefficients) estimated in the joint models (Supplementary figure 42, Supplementary tables 18–20). Patterns of significant (and not-significant) findings are robust to censoring individuals diagnosed before 2000, 2005, or 2008 (Methods, Supplementary tables 24–26).
ADHD-adjacent trait polygene scores associate with cognition
Finally, we sought external support for our finding that polygenic contributions to clinical heterogeneity in ADHD are shared with psychiatric, cognitive, socio-behavioral traits. We used our three ADHD-adjacent trait GWAS to construct PGS in the Adolescent Brain and Cognitive Development (ABCD) study46, an independent, typically developing cohort. Each PGS was tested for association with each of 51 behavioral and cognitive assessments, adjusting for ADHD PGS and potential confounders (Methods, Figure 5, Supplementary table 27). These analyses confirmed an association between the polygenic basis of an ADHD-adjacent ASD diagnoses and cognitive performance. Specifically, a higher PGS was significantly (p<0.001) associated with increased performance on overall cognitive function that appears driven by assessment in verbal cognition (e.g., crystalized composite, picture vocabulary scores; Figure 5c). No other tests were significant, although a few trends are noted: the signs of the relationships of the PGS and cognitive or psychiatric traits were in line with our previous analyses and multiple individual PGS trends were consistent for specific psychiatric or behavioral symptoms. We find consistent, independent support for a polygenic relationship between an ADHD-adjacent ASD diagnosis and cognitive performance, but small discovery GWAS and indirect, modest replication sample limit power for broader replication.
Fig. 5. Polygenes for ADHD-adjacent ASD are associated with cognitive performance in an independent, typically developing cohort.
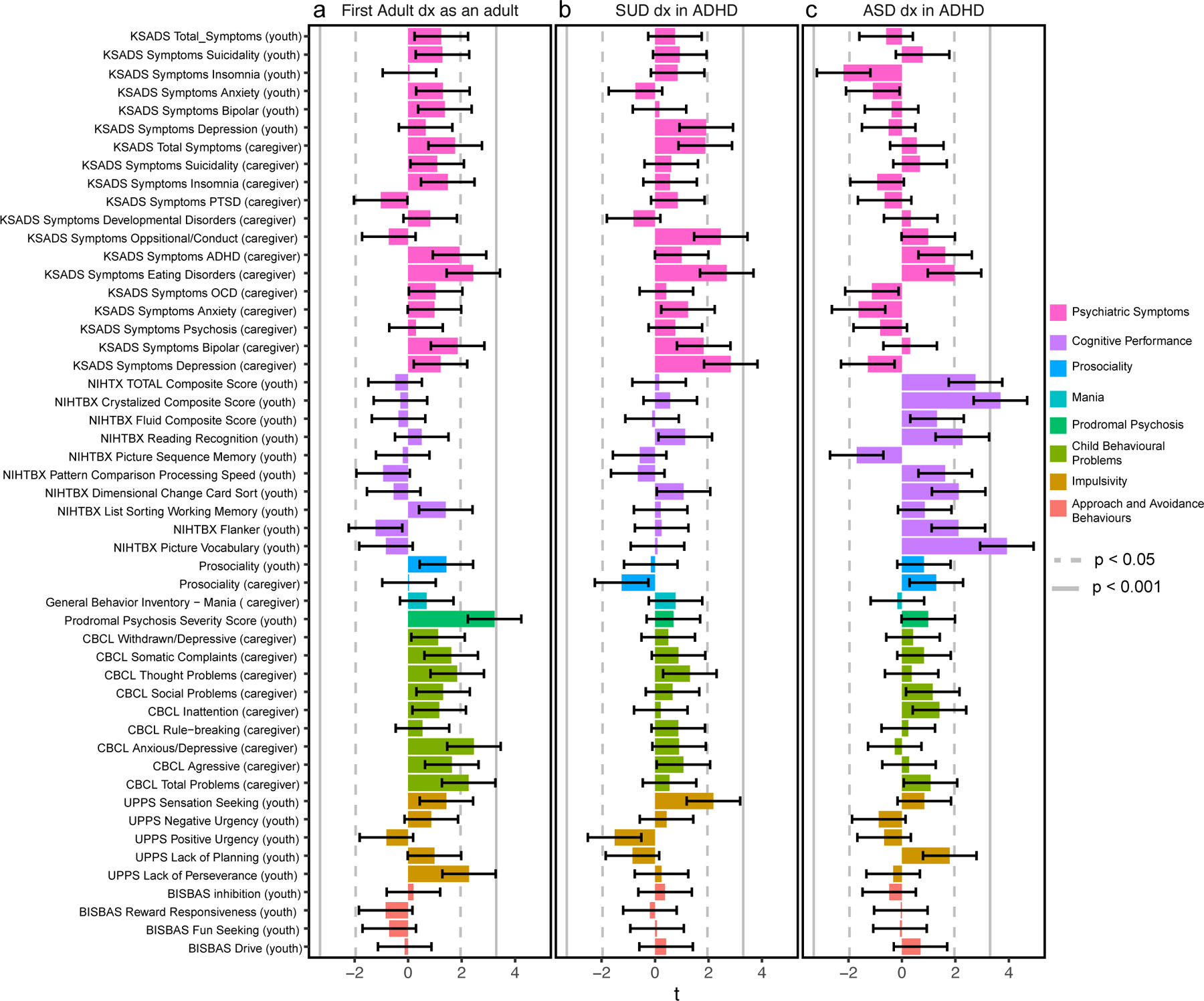
Polygenic scores (PGS) constructed using summary statistics from ADHD-adjacent trait GWAS were tested in generalized linear models (GLM) for association with cognitive, behavioral, and psychiatric traits in n=5 449 nine or ten year-old children from the Adolescent brain cognitive development (ABCD) Study. The t-statistic of each regression (bars) reveals no significant associations for a PGS for adult diagnosed ADHD or b ADHD-adjacent SUD, although consistent trends are prevalent, but c the PGS for an ADHD-adjacent ASD diagnosis associated significantly with cognitive performance, replicating our previous finding that polygenes are shared among these two domains. Error bars depict standard deviation of the t-statistic. Two-tailed p-values were declared significant after Bonferroni correction (p<0.05/51=0.001;solid line) and suggestive with nominal trends(p<0.05; dotted line). Caregiver or youth in parenthesis denotes the informant for the assessment of the child. t, t-statistic. See Supplementary table 27 for complete statistical results and Supplementary table 28 for a description of the questionnaire used for each ABCD assessment.
Discussion
In this study we used a unique data resource and analysis strategy to identify evidence for genetic heterogeneity underlying clinical heterogeneity in a widely studied complex disorder. We showed multiple, clinically relevant ADHD-adjacent traits were related to genetic variability among diagnosed individuals and that these genetic contributions were complex (i.e., marked by few large effects despite significant h2SNP) but characterizable. Our multivariate analysis of polygenic profiles demonstrated that variability in polygenes associated with psychiatry, cognitive, and socio-behavioral traits underlie differences in clinical presentations of individuals diagnosed with ADHD. Our findings regarding age of first diagnosis and adjacent ASD diagnoses, in particular, make timely contributions to understanding the genetic etiology of ADHD and other results reflect themes relevant for the study of complex disorders more broadly.
Diagnostic practices around ADHD have evolved significantly over recent decades47, coinciding with a dramatic increase in prevalence48. These changes have relevance for our study as guidelines around the adult diagnosis of ADHD and comorbid ASD diagnosis in ADHD are two significant changes with DSM-51,47. In this study, we include ADHD diagnoses made from 1992 through 2012, with a majority (>70%) made after 2007. Even still, our study takes place on the background of evolving clinical guidelines that could introduce diagnosis-year cohort effects. We observed the few, very earliest diagnosed individuals (n=500, <1999) may be subtly different from the rest of the cohort. In this cohort, the genetic relationship to year of diagnosis was smaller and less significant than to having a first diagnosis as an adult and having an ADHD adjacent diagnosis of SUD or ASD. Our primary findings are robust to censoring individuals diagnosed before 2000, before 2005, or before 2008. Future work could more directly test genetic correlates of changing clinical practice and increasing prevalence, which could have a larger impact on meta-cohorts combining diagnoses spanning more years.
Despite evidence of clinical co-occurrence in individuals and families49–51, shared genetic risk factors19,52, and biological overlap49, few GWAS have considered effects related to sharing, discriminating, or comorbidity. We identified a locus that appears novel for both disorders, with effects that appear specific to comorbid ADHD and ASD. This locus was not discovered by previous GWAS that focused on combined analysis of all ADHD and ASD cases53, variants that discriminate between ADHD and ASD53,54, comorbid cases and controls53, or case-control GWAS of each individually20,37, but none tested for association to a comorbid group, in a case-only design. In our bioinformatics characterizations, both eQTL and previous trait associations suggest the locus is functional, has a plausible connection to regulating neurobiology42–44, and is supported in a complementary design using multiplex ASD families45. While this association needs further replication, it suggests studying specific comorbidity patterns or presentations may help identify pleiotropic SNPs and disentangle antagonistic or heterogeneous variant effects.
Until the introduction of DSM-5 in 20131, formal guidelines did not allow ASD and ADHD to be diagnosed comorbid55. The validity of co-occurring syndromes is supported by the recognition of overlap in cognition56, genetics56, symptoms scores57, and neuroanatomic characteristics58. However, the possibility that the core etiologies of both disorders could exist in an individual, simultaneously, is an active area of research and debate55. Our polygenic profile approach suggests individuals with an ADHD-adjacent ASD diagnosis carry, at minimum, the polygenic etiologies of both disorders, a finding that is supported by a parallel, contemporaneous PGS analysis of iPSYCH53. This idea is supported by a recent register study in the Swedish population51 that reported comorbid ADHD and ASD diagnosis were most often made on the same day, by the same clinician, observing concurrent symptoms. This trend stands in contrast to similar studies of bipolar disorder and schizophrenia, that suggest an appreciable level of lifetime comorbidity may relate to evolving symptoms and difficulties in diagnosing first episode psychosis58. Our polygenic profiling approach supports the notion that ASD and ADHD can be experienced simultaneously, consistent with emerging trends in clinical epidemiology and evolving psychiatric nosology.
Adult-onset ADHD, it’s validity, existence, and whether it represents a distinct clinical or biological construct, is a particularly active and contentious research topic4,59. The crux of this debate rests on the timing of symptom onset. As above, it was DSM-51 that introduced criteria for an adult ADHD diagnosis where diagnosis requires retrospective self or informant reported childhood symptoms, but these reports may suffer recall bias, and objective, representative longitudinal studies of premorbid symptoms are sparse. One perspective (e.g.28) is that a first diagnosis as an adult is persistent ADHD, perhaps missed during childhood, and, as symptoms may remit with age, be considered a more severe or biological form of ADHD. Here, we observed that, contrary to this perspective, individuals with a first registered diagnosis as an adult had less ADHD PGS suggesting they may have a more environmental, less reliable, or less severe form of the disorder60. Our polygenic profile approach offers varied support for each of these hypotheses in that adult diagnosed individuals had lower education PGS, higher non-ADHD psychiatric PGS, and similar profiles to individuals with ADHD-adjacent SUD diagnoses. A contemporary study of iPSYCH61 found similar PGS trends for secondary traits, but suggested that differences with regards to ADHD PGS may be driven by a persistent ADHD subgroup with confirmed diagnoses in both childhood and adulthood. Our data and approach implicate genetic heterogeneity in important debates surrounding adult ADHD..
Our study highlights themes that are broadly relevant for the study of complex disorders and associated heterogeneity. First, as demonstrated for MDD23, male pattern baldness62, and in simulations23,62,63, the polygenic background of case groups may become skewed if evolving nosology, case definitions, or censoring are not considered carefully. Here, we expect that evolving guidelines around comorbid ADHD and ASD may change the genetic landscape of typical ADHD and ASD case groups, implicitly, but active ascertainment or censoring mechanisms could have similar consequences. Second, PGS have received a lot of attention as potential clinical instruments for various applications64,65, but the focus is often on primary disorder scores (i.e., here, ADHD) and distinguishing cases from controls. Here, profiles of multiple polygenic scores were powerful in distinguishing among ADHD cases, and the strongest predictors were not primary disorder PGS. The idea that the polygenic background of a patient can alter a clinical trajectory is not new, but as PGS are aimed at more diverse clinical decisions (i.e., beyond case-control discrimination), more comprehensive summaries of polygenic background should be considered. Finally, methods for detecting heterogeneity might benefit from relaxing a priori restrictions to onset associated variants when focusing on polygenes. Our study may inform work on complex disorders beyond ADHD.
Our results must be considered in light of important limitations. We use register diagnoses, which are given with high reliability by trained psychiatrists, but are assigned per hospital contact, and may shift over time as symptoms evolve or clarify66. This could lead to some misdiagnosis. This issue is not unique to register studies but may offer a different perspective from studies of research diagnoses that include retrospective censoring or integration according to a hierarchy. Some individuals may seek treatment exclusively through primary care and those contacts are not available in iPSYCH. This may be especially relevant when considering symptom onset and first diagnosis as earlier contacts could be missed. Registered diagnoses may represent an increase in severity, if not onset, as a hospital referral could represent a change in need of care. iPSYCH is relatively young and may underestimate the prevalence of adult and later onset outcomes and only a combined ADHD subtype (ICD-10: F90.0). Important genetic differences may emerge if inattentive (ICD-10: F98.8) and hyperactive subtypes (ICD-10: F90.1, F90.8, or F90.9) were included. Methodologically, we compared groups of ADHD cases to estimate SNP-heritability and performed GWAS to implicate genetic contributions to clinical heterogeneity. Just as with case-control analysis, these approaches are sensitive to spurious differences due to population stratification or heritable, disorder irrelevant features (e.g., hair color), if used to define patient groups13,29. We mitigate this by adjusting for ancestry related principal components, carefully selecting ADHD adjacent-traits with plausible clinical relevance, and including controls in our polygenic profile approach. Finally, the prevalence and clinical criteria for ADHD are evolving, and this may result in differences in the genetic architecture of cases ascertained in different years meaning our results may generalise less well to cohorts diagnosed in different time-windows.
This work is part of an emerging body of evidence in psychiatric genetics that suggests we are at a point where we can study not just what makes diagnosed individuals different from healthy controls, but also what differentiates diagnosed individuals with respect to outcomes.
Methods
iPSYCH2012 case-cohort study
The Lundbeck Foundation initiative for Integrative Psychiatric Research (iPSYCH)32 is a case-cohort study of individuals born in Denmark between 1981 and 2005 (n=1 472 762). 87 764 individuals were sampled, including a random sample of 30 000 and 57 764 ascertained for diagnoses of attention-deficit hyperactivity disorder (ADHD), autism spectrum disorders (ASD), anorexia (ANO), affective disorder (AFF), bipolar disorder (BP), or schizophrenia (SCZ). DNA was extracted from dried bloodspots in the Danish Neonatal Screening Biobank67. Diagnoses were obtained from the Danish Psychiatric Central Research Register (PCR)35 and for anorexia also from the Danish National Patient Register (DNPR)34 from 1 year birthday or 10 year birthday of study individuals to December 31, 2012. Linkage across registers uses the Danish Civil Registration System36. Diagnoses given by psychiatrists in primary care are not recorded in these registers. After genotype, kinship, and ancestry quality control (see below), this study included 14 084 of 18 726 individuals ascertained for ADHD (ICD-10: F90.0), 12 088 of 16 146 ascertained for ASD (ICD-10: F84.0, F84.1, F84.5, F84.8 or F84.9), and 21 197 of 30 000 controls diagnosed with neither ADHD nor ASD. Furthermore, 8 498 individuals with substance use disorders (SUD) (ICD-10: F1) were selected from among the 30 000 population controls and 57 764 psychiatric cases and finally, 20 509 of 30 000 controls with neither ADHD nor SUD. The use of this data follows standards of the Danish Scientific Ethics Committee, the Danish Health Data Authority, the Danish Data Protection Agency, and the Danish Neonatal Screening Biobank Steering Committee. Data access was via secure portals in accordance with Danish data protection guidelines set by the Danish Data Protection Agency, the Danish Health Data Authority, and Statistics Denmark.
Genotyping was performed on the Infinium PsychChip v1.0 array with amplified DNA extracted from dried bloodspots. Data quality control is described in detail elsewhere32,68. Briefly, 246 369 of the ~550 000 genotyped SNPs were deemed good quality, phased using SHAPEIT369, and imputed using the 1 000 genomes project phase370 as a reference with Impute271. Imputed additive genotype dosages and best-guess genotypes were checked for imputation quality (INFO>0.2), Hardy–Weinberg equilibrium (HWE; p<1×10−6), association with genotyping wave (p<5×10−8), association with imputation batch (p<5×10−8), differing imputation quality between cases and controls (p<1×10−6), and minor allele frequency (MAF>0.01). 8 019 760 dosages and best-guess genotypes remained. Subjects of homogeneous genetic ancestry were selected after principal components analysis using EIGENSOFT v6.0.172. One from each pair of individuals with closer than third degree kinship as estimated with KING v1.973 was excluded, and no samples had abnormal heterozygosity, high levels of missing genotypes (>1%), nor genotype/recorded sex discordance.
Selecting ADHD-adjacent traits
DNPR34 and PCR35 contain information (e.g., date of admission, ICD diagnostic code, etc.) on all inpatient hospital contacts since 1977 and 1969, respectively, and outpatient and emergency room contacts since 1995. From these registers we defined two sets of ADHD-adjacent traits to capture psychiatric comorbidity. First, we coded the presence of specific psychiatric disorders as defined by the iPSYCH2012 case-cohort ascertainment criteria32, including ASD, AFF, ANO, and combined psychotic disorders (SCZ and BP). As a second set, we summarized psychiatric diagnoses more broadly, recording each ICD-10 Behavioral and Mental Disorders subchapter recorded in either PCR or DNPR until 2016 (F1, F2, F3, F4, F5, F6, F7, F8, F9 excluding ADHD (F90.0–9 and F98.8)). As before, we created variables representing a diagnosis from each subchapter. We also counted the number of hospitalizations with ADHD (ICD-10 F90.0) recorded as the main diagnosis of action in the PCR up until 2016, also dichotomized as a split by the median. A variable splitting cases on the first recorded ADHD diagnosis (ICD-10 F90.0) occurring before or after 18 years of age. Finally, a variable was created recording sex as reported at birth. Note that the version of ICD implemented in Denmark is the research version of ICD-10 (“the green book” from 19933; https://icd.who.int/browse10/2019/en) which codes combined ADHD subtype as F90.0, inattentive as F98.8, and hyperactive subtypes as F90.1, F90.8, or F90.9. These codes are different than in the clinically modified version ICD-10-CM used in the US for coding claims (https://www.icd10data.com/ICD10CM/Codes) which refers to the combined subtype as F90.2 and inattentive as F90.0.
The Danish National Prescription Register (NPR)33 holds information on prescriptions redeemed from pharmacies in Denmark since 1995. The NPR does not cover drugs used during hospital admissions, by certain institutionalized individuals (e.g. psychiatric), or drugs supplied directly by hospitals or treatment centers. We defined three traits from the NPR noting cases with a record of at least two prescriptions after the age of 3 for drugs with the Anatomical Therapeutic Chemical (ATC) codes for stimulants (N06BA01, N06BA12, N06BA02, N06BA04), or atomoxetine (N06BA09), or both (i.e., any ADHD medication). A fourth trait counted the number of prescriptions an individual had filled and dichotomized on the median number (in this cohort) of total prescriptions. Further details are provided in Supplementary table 1.
We used tetrachoric correlations to describe the co-occurrence of ADHD-adjacent traits, estimating them with the R package polycorr v0.874 and adding 0.5 to zero cells75. Hierarchical clustering was performed using the heatmaply v1.376 package.
SNP-based heritability
SNP heritability (h2SNP) was estimated on the observed scale using GREML in GCTA v1.92.1 beta677 with 25 ancestry principal components as fixed effect covariates. The genetic relationship matrix (GRM) was constructed using GCTA and from best-guess genotypes with a MAF greater than 0.01. Significance was adjusted by Bonferroni correction of one-tailed p-values (p<0.05/22=0.002), and nominally significant tests (p<0.05) were deemed suggestive.
Genome-wide association studies
Case-case genome-wide association studies (GWAS) were performed within the ADHD case group (n=14 084). Logistic regression in plink v1.90b3.3478 was used to test the association between imputed additive allele counts and case subgroup membership, with 25 ancestry principal components as covariates. Genomic inflation was estimated as the unconstrained LD-score regression v1.0.179 intercept following: https://github.com/bulik/ldsc/wiki. Individual SNPs with two-tailed p<5×10−8 were declared genome-wide significant. Lead SNPs were defined as the most significant SNP within a 2 megabase (mb) locus. We additionally considered GWAS for: three case subgroups vs. controls (First ADHD diagnosis as an adult (n=3 323) vs. controls (n=21 411), ADHD-adjacent diagnosis of SUD (n=2 627) vs. controls (n=21 411) and ADHD–adjacent diagnosis of ASD (n=2 284) vs. controls (n=21 411)) and two cross-trait meta-analysis vs. controls (ADHD or SUD (n=20 065) vs. controls (n=20 509) and ADHD or ASD (n=23 926) vs. controls (n=21 197).
For genome-wide significant loci, multinomial logistic regression (MLR)80 was performed using the R package nnet v7.381. The MLR tests the logistic regression comparison of allele counts at a lead SNP between ADHD subgroups (i.e., individuals with comorbid ADHD and ASD, n=2 284, and individuals with ADHD but not ASD, n=11 800) to simultaneously include comparisons with individuals diagnosed with relevant complementary disorder (i.e., individuals with ASD but not ADHD, n=9 804) and undiagnosed controls (i.e., individuals with neither ADHD nor ASD, n=21 197).
For genome wide significant loci, candidate genes were selected according to three criteria: 1) positional candidate genes were selected by ANNOVAR82 in FUMA v1.3.5e83 as overlapping a lead SNP’s genomic position, 2) eQTL candidate genes with previous expression association to a lead SNP in one of multiple sources aggregated by FUMA, 3) transcriptional association candidate genes with predicted expression in adult and/or fetal brain. Here a transcription association (TWAS) analysis was used integrating published per-SNP effects on expression for fetal40 and adult84 brain, and FUSION v201885 to generate aggregate predicted expression in each individual. TWAS two-tailed p-values were considered significant after Bonferroni correction (p<0.05/9=0.006). For lead SNPs from genome-wide significant loci, the GWASatlas v2019111586 (https://atlas.ctglab.nl/) was used to pursue a pheWAS across 4 756 studies grouped according to the provided ontology. The proxy SNP for rs8178395 was identified using LDlink v4.0 (https://ldlink.nci.nih.gov/?tab=help#LDproxy)87. eQTL and sQTL analysis were visualized summary data using tools provided by the GTEx Portal on 10 March 2021.
We used stratified LD score regression v1.0.1 (LDSC)88 to partition h2SNP among selected genome annotations, accounting for LD and baseline annotations. Annotations of interest included those defined by eQTLs from fetal brain40, eQTLs from adult39 brain, and regions of open chromatin measured by ATAC sequencing in fetal89 and adult90 brain. A “full baseline model” of 53 functional categories was employed following Finucane et al91. We followed the author protocols for analysis (https://github.com/bulik/ldsc/wiki/Partitioned-Heritability). eQTL and ATAC LD-scores were created for the subset of human SNPs genotyped in HapMap v3 SNPs (HM3) using the LD-score regression software by identifying HM3 SNPs within a 500bp window (±250) around each eQTL SNP or within an ATAC open chromatin window. Category-specific LD-scores were the sum of LD (r2) for each HM3 SNP with SNPs meeting the previous functional criteria. The European subset of the 1000 Genomes Project Phase3 was used as an LD reference. Enrichment was estimated for each annotation as the proportion of heritability explained by each annotation divided by the proportion of SNPs in the genome falling in that category. Enrichment p-value calculated based on the coefficient (τC) of each annotation were declared significant after Bonferroni correction (p<0.05/28=0.0018) or suggestive when p<0.05.
SNP-based genetic correlations
SNP-based genetic correlations (ρG,SNP) were estimated using LD-Score regression comparing summary statistics for the three ADHD adjacent traits GWAS performed for this study and each of 46 traits from 10 psychiatric, cognitive, and socio-behavioral categories.: substance use, neurology, personality, sleeping, cognition, reproduction, education, neuroimaging, psychiatry, and ageing. Prioritized traits had FDR<0.05.
Polygenic scores in iPSYCH
GWAS summary statistics (Supplementary table 28) were downloaded from public repositories, cleaned using cleansumstats v1.6 (https://github.com/BioPsyk/cleansumstats), DENTIST v1.3 (https://github.com/Yves-CHEN/DENTIST), and effect sizes were reweighted using SBayesR92 implemented in GCTB v2.03beta (https://cnsgenomics.com/software/gctb) Reference GWAS were selected to ensure no subject overlap with iPSYCH. SNP inclusion criteria were: MAF>0.05, INFO>0.98, and HWE p-value>1×10−5. Palindromic (A/T, C/G) SNPs, SNPs not uniquely mapped to hg19 positions, and SNPs not having unique IDs in dbSNP v151 were excluded. Details of the SBayesR workflow are shown in Supplementary figure 43.
We used multivariate, multinomial logistic regression (MLR)80 implemented in the R package nnet81 to test for heterogeneity in PGS profiles among ADHD case subgroups, non-diagnosed controls, and individuals diagnosed with ASD or SUD but not ADHD. For each of the three ADHD adjacent traits, we jointly fit primary disorder PGS (i.e., ADHD, ASD), the set of PGS for traits that showed significant ρG,SNP with the ADHD variable in the LDSC analysis described above, and 25 ancestry principal components. MLR models fitting each PGS individually, along with ancestry covariates, are presented in the Supplementary tables 21–23. Three MLR were of primary interest. First, for ADHD-adjacent ASD, we compared individuals diagnosed with neither ADHD nor ASD (n=21 197), both ADHD and ASD (n=2 284), ADHD but not ASD (n=11 800), and ASD but not ADHD (n=9 804) on PGS profiles containing scores for ADHD20, ASD37, Cross-psychiatric disorders93, Mother’s age at death94, intelligence95, college completion96, years of schooling97, age at first birth98, chronotype99, and smoking initiation100. For ADHD-adjacent SUD, we compared individuals with neither ADHD nor SUD (n=20 509), both ADHD and SUD (n=2 627), ADHD but not SUD (n=11 457), and SUD but not ADHD (n=5 943) on PGS profiles containing scores for ADHD20, depressive symptoms101, schizophrenia102, intelligence95, college completion96, years of schooling97, age at first birth98 and alcohol dependence103. For age of first ADHD diagnosis we compared individuals without a diagnosis for ADHD (n=21 409), with a first diagnosis of ADHD as an adult (>18 years of age) n=3 323), and with a first diagnosis of ADHD before age 18 (n=10 761) on PGS profiles containing scores for ADHD20, depressive symptoms101, major depressive disorder104, mother’s age at death94, childhood IQ105, college completion96, years of schooling97, age at first birth98 and alcohol use (AUDIT, P score)106. A likelihood ratio test implemented in R with the anova function was used to compare the goodness of fit of the joint MLR models with and without each individual PGS and one-tailed p-values were used to determine significance of the variability in the mean PGS across groups (pglobal). Two-tailed p-values were used for individual, pairwise contrasts. Significance was declared after Bonferroni correction for 169 p-values (p<0.05/169=3×10−4).
The adolescent brain cognitive development (ABCD) study
The Adolescent Brain and Cognitive Development (ABCD) study48,103 (http://abcdstudy.org) is an observational study following 11 875 children from across the US, starting at age nine or ten with limited exclusion criteria to create a socio-economically and demographically representative cohort. ABCD administers biennial assessments of physical health, mental health, neuro cognition, family, cultural and environmental variables, SUD, genetic and other biomarkers, and multi-modal neuroimaging. Genotyping used the Smokescreen array, imputations used the Michigan Imputation Server, and quality control is described in Loughnan et al.107. We used measures of 51 assessments (Supplementary table 29) covering categorical and dimensional psychopathology, mania symptoms, prodromal psychosis, impulsivity, and behavioral activation and inhibition collected at enrollment46. To protect against confounding by population structure, we focus on the homogenous, European ancestry sub-cohort of 5 455 children selected following genetic ancestry estimation using fastStructure108.
Polygenic scores in ABCD
PGS in ABCD were calculated using SBayesR92 to reweight summary statistics from each of the three ADHD adjacent trait GWAS described above and following the workflow shown in Supplementary figure 43. Generalized linear models (GLMs) were used to test the association of each of the three PGS across each of 51 assessments, separately. GLMs included fixed effects of ADHD case-control PGS, age, sex at birth, data collection site, and 10 ancestry principal components. To control for family effects (twins and siblings within the sample), we iteratively fit 100 models for each test taking a random selection on singletons (down-sampling to one individual from each family, n=4 622) and report the median across iterations. The distribution of each assessment (response variable) was analyzed to ensure normality or to handle zero inflation and right skewness. Non-zero inflated distributions were rank normalized (as was performed for PGS) and the GLM was fitted using the family=gaussian option. For the right-skewed, zero-inflated distributions, data were shifted to ensure non-negativity and GLM were fitted with a gamma distribution and a log link function. Summary measures making up the Kiddie Schedule Disorders and Schizophrenia (K-SADS), except for the two K-SADS Total Symptoms measures, were binarized using a median split and fit using logistic regression, due to convergence issues using a gamma distribution. Bonferroni adjustment of two-tailed p-values was used to declare significance (p<0.05/51=0.001).
Supplementary Material
Acknowledgments
Data used in the preparation of this article were obtained from the Adolescent Brain Cognitive Development (ABCD) Study (https://abcdstudy.org), held in the NIMH Data Archive (NDA). This is a multisite, longitudinal study designed to recruit more than 10,000 children aged 9-10 and follow them over 10 years into early adulthood. The ABCD Study is supported by the National Institutes of Health and additional federal partners under award numbers U01DA041022, U01DA041028, U01DA041048, U01DA041089, U01DA041106, U01DA041117, U01DA041120, U01DA041134, U01DA041148, U01DA041156, U01DA041174, U24DA041123, and U24DA041147. A full list of supporters is available at https://abcdstudy.org/nih-collaborators. A listing of participating sites and a complete listing of the study investigators can be found at https://abcdstudy.org/principal-investigators.html. ABCD consortium investigators designed and implemented the study and/or provided data but did not necessarily participate in analysis or writing of this report. This manuscript reflects the views of the authors and may not reflect the opinions or views of the NIH or ABCD consortium investigators. The ABCD data repository grows and changes over time. The iPSYCH Initiative is funded by the Lundbeck Foundation (grant numbers R102-A9118 and R155-2014-1724), the Mental Health Services Capital Region of Denmark, University of Copenhagen, Aarhus University and the university hospital in Aarhus. Genotyping of iPSYCH samples was supported by grants from the Lundbeck Foundation, the Stanley Foundation, the Simons Foundation (SFARI 311789), and NIMH (5U01MH094432-02). The IPSYCH Initiative utilize the Danish National Biobank resource that is supported by the Novo Nordisk Foundation. IPSYCH data was stored and analyzed at the Computerome HPC facility (http://www.computerome.dtu.dk/) and authors are grateful for continuous support from the HPC team led by A. Syed of DTU Bioinformatics, Technical University of Denmark. This work acknowledges funding from Lundbeckfonden under the Fellowship R335-2019-2318 (A.J.S), the National Institute for Aging of the National Institutes of Health under awards U19AG023122, U24AG051129S1, UH2AG064706, and UH2AG064706S1 (A.J.S), the Research Fund of the Mental Health Services - Capital Region of Denmark R4A92 (S.L.), The Lundbeck Foundation R208-2015-3951 (S.L) and Fonden for Faglig Udvikling af Speciallægepraksis 38850/16 (S.L), the European Commission Horizon 2020, grant no 667302 (S.D), Helsefonden grant no 19-8-0260(S.D) and the European Union’s Horizon 2020 research and innovation programme under grant agreement No 847879 (S.D).
Footnotes
Code Availability
Code for the multinomial regression tests and supplementary simulations are available at https://github.com/AndrewSchork/. Other software used for analysis are publicly available as described in the methods and reporting summary. Wrappers and pipelines used to link tools with individual level data are available on request and interested parties should contact A.J.S.
Competing Interests
The Authors declare no competing interests.
Data Availability
The consent structure of iPSYCH and Danish law prevent individual genotype and phenotype data from being shared publicly. Reasonable requests to access individual level data to verify the findings in this paper can be accommodated with permission from the Danish Scientific Ethics Committee, the Danish Health Data Authority, the Danish Data Protection Agency, and the Danish Neonatal Screening Biobank Steering Committee and interested parties can contract T.M.W and expect a response within one week. Approvals for access can take several months and must be governed by strict data use agreements. ABCD study data can be accessed by request from the NIMH Data Archive (NDA; https://nda.nih.gov/abcd). GWAS summary statistics used for this work were downloaded from and available in public repositories as described in Supplementary table 28. Leave-one-study-out meta-analysis summary statistics for psychiatric disorders are available on request from the psychiatric genetic consortium (PGC) disorder working group chairs (https://pgc.unc.edu/for-researchers/data-access-committee/data-access-information/). The eQTL and sQTL visualizations and data used for the analyses described in this manuscript were obtained from the GTEx Portal (https://gtexportal.org/) on 10 March 2021 and 23 October 2023.
References
- 1.American Psychiatric Association. DSM-5 Task Force. Diagnostic and Statistical Manual of Mental Disorders: DSM-5. (CBS Publishers & Distributors, Pvt. Limited, 2017). [Google Scholar]
- 2.World Health Organization. The ICD-10 Classification of Mental and Behavioural Disorders: Clinical Descriptions and Diagnostic Guidelines. (World Health Organization, 1992). [Google Scholar]
- 3.World Health Organization. The ICD-10 Classification of Mental and Behavioural Disorders: Diagnostic criteria for research. 1. ed. (WHO, 1993). [Google Scholar]
- 4.Franke B et al. Live fast, die young? A review on the developmental trajectories of ADHD across the lifespan. Eur. Neuropsychopharmacol 28, 1059–1088 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luo Y, Weibman D, Halperin JM & Li X A Review of Heterogeneity in Attention Deficit/Hyperactivity Disorder (ADHD). Front. Hum. Neurosci 13, 42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thapar A, Cooper M & Rutter M Neurodevelopmental disorders. Lancet Psychiatry 4, 339–346 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Dalsgaard S, Mortensen PB, Frydenberg M & Thomsen PH Long-term criminal outcome of children with attention deficit hyperactivity disorder. Crim. Behav. Ment. Health 23, 86–98 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Dalsgaard S, Østergaard SD, Leckman JF, Mortensen PB & Pedersen MG Mortality in children, adolescents, and adults with attention deficit hyperactivity disorder: a nationwide cohort study. Lancet 385, 2190–2196 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Dalsgaard S et al. Association of Mental Disorder in Childhood and Adolescence With Subsequent Educational Achievement. JAMA Psychiatry 77, 797–805 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Daley D, Jacobsen RH, Lange A-M, Sørensen A & Walldorf J Costing Adult Attention Deficit Hyperactivity Disorder: Impact on the Individual and Society. (OUP Oxford, 2015). [Google Scholar]
- 11.Plana-Ripoll O et al. Exploring Comorbidity Within Mental Disorders Among a Danish National Population. JAMA Psychiatry 76, 259–270 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McClellan J & King M-C Genetic heterogeneity in human disease. Cell 141, 210–217 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Dahl A & Zaitlen N Genetic Influences on Disease Subtypes. Annu. Rev. Genomics Hum. Genet 21, 413–435 (2020). [DOI] [PubMed] [Google Scholar]
- 14.Charney AW et al. Evidence for genetic heterogeneity between clinical subtypes of bipolar disorder. Transl. Psychiatry 7, e993 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bipolar Disorder and Schizophrenia Working Group of the Psychiatric Genomics Consortium. Genomic Dissection of Bipolar Disorder and Schizophrenia, Including 28 Subphenotypes. Cell 173, 1705–1715.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faraone SV & Larsson H Genetics of attention deficit hyperactivity disorder. Mol. Psychiatry 24, 562–575 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams NM et al. Rare chromosomal deletions and duplications in attention-deficit hyperactivity disorder: a genome-wide analysis. Lancet 376, 1401–1408 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Olsen L et al. Prevalence of rearrangements in the 22q11.2 region and population-based risk of neuropsychiatric and developmental disorders in a Danish population: a case-cohort study. Lancet Psychiatry 5, 573–580 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Satterstrom FK et al. Autism spectrum disorder and attention deficit hyperactivity disorder have a similar burden of rare protein-truncating variants. Nat. Neurosci 22, 1961–1965 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Demontis D et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet 51, 63–75 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sullivan PF et al. Psychiatric Genomics: An Update and an Agenda. Am. J. Psychiatry 175, 15–27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sullivan PF & Geschwind DH Defining the Genetic, Genomic, Cellular, and Diagnostic Architectures of Psychiatric Disorders. Cell 177, 162–183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cai N et al. Minimal phenotyping yields genome-wide association signals of low specificity for major depression. Nat. Genet 52, 437–447 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wray NR, Lee SH & Kendler KS Impact of diagnostic misclassification on estimation of genetic correlations using genome-wide genotypes. Eur. J. Hum. Genet 20, 668–674 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wimberley T et al. Genetic liability to ADHD and substance use disorders in individuals with ADHD. Addiction 115, 1368–1377 (2020). [DOI] [PubMed] [Google Scholar]
- 26.Jansen AG et al. Psychiatric Polygenic Risk Scores as Predictor for Attention Deficit/Hyperactivity Disorder and Autism Spectrum Disorder in a Clinical Child and Adolescent Sample. Behav. Genet 50, 203–212 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martin J et al. A Genetic Investigation of Sex Bias in the Prevalence of Attention-Deficit/Hyperactivity Disorder. Biol. Psychiatry 83, 1044–1053 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rovira P et al. Shared genetic background between children and adults with attention deficit/hyperactivity disorder. Neuropsychopharmacology 45, 1617–1626 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liley J, Todd JA & Wallace C A method for identifying genetic heterogeneity within phenotypically defined disease subgroups. Nat. Genet 49, 310–316 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nadeau JH Modifier genes in mice and humans. Nat. Rev. Genet 2, 165–174 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Fanous AH & Kendler KS Genetic heterogeneity, modifier genes, and quantitative phenotypes in psychiatric illness: searching for a framework. Mol. Psychiatry 10, 6–13 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Pedersen CB et al. The iPSYCH2012 case-cohort sample: new directions for unravelling genetic and environmental architectures of severe mental disorders. Mol. Psychiatry 23, 6–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kildemoes HW, Sørensen HT & Hallas J The Danish National Prescription Registry. Scand. J. Public Health 39, 38–41 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Lynge E, Sandegaard JL & Rebolj M The Danish National Patient Register. Scand. J. Public Health 39, 30–33 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Mors O, Perto GP & Mortensen PB The Danish Psychiatric Central Research Register. Scand. J. Public Health 39, 54–57 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Pedersen CB The Danish Civil Registration System. Scand. J. Public Health 39, 22–25 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Grove J et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet 51, 431–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cross-Disorder Group of the Psychiatric Genomics Consortium. Genomic Relationships, Novel Loci, and Pleiotropic Mechanisms across Eight Psychiatric Disorders. Cell 179, 1469–1482.e11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang D et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walker RL et al. Genetic Control of Expression and Splicing in Developing Human Brain Informs Disease Mechanisms. Cell 179, 750–771.e22 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat. Genet 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mittelstaedt T & Schoch S Structure and evolution of RIM-BP genes: identification of a novel family member. Gene 403, 70–79 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Hibino H et al. RIM binding proteins (RBPs) couple Rab3-interacting molecules (RIMs) to voltage-gated Ca(2+) channels. Neuron 34, 411–423 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Acuna C, Liu X, Gonzalez A & Südhof TC RIM-BPs Mediate Tight Coupling of Action Potentials to Ca(2+)-Triggered Neurotransmitter Release. Neuron 87, 1234–1247 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Bucan M et al. Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 5, e1000536 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barch DM et al. Demographic, physical and mental health assessments in the adolescent brain and cognitive development study: Rationale and description. Dev. Cogn. Neurosci 32, 55–66 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Epstein JN & Loren REA Changes in the Definition of ADHD in DSM-5: Subtle but Important. Neuropsychiatry 3, 455–458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu G, Strathearn L, Liu B, Yang B & Bao W Twenty-Year Trends in Diagnosed Attention-Deficit/Hyperactivity Disorder Among US Children and Adolescents, 1997–2016. JAMA Netw Open 1, e181471 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Martin J et al. Biological overlap of attention-deficit/hyperactivity disorder and autism spectrum disorder: evidence from copy number variants. J. Am. Acad. Child Adolesc. Psychiatry 53, 761–70.e26 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.LaBianca S et al. Brief Report: Clusters and Trajectories Across the Autism and/or ADHD Spectrum. J. Autism Dev. Disord 48, 3629–3636 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Ghirardi L et al. The familial co-aggregation of ASD and ADHD: a register-based cohort study. Mol. Psychiatry 23, 257–262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rommelse NNJ, Franke B, Geurts HM, Hartman CA & Buitelaar JK Shared heritability of attention-deficit/hyperactivity disorder and autism spectrum disorder. Eur. Child Adolesc. Psychiatry 19, 281–295 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mattheisen M et al. Identification of shared and differentiating genetic architecture for autism spectrum disorder, attention-deficit hyperactivity disorder and case subgroups. Nat. Genet 54, 1470–1478 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peyrot WJ & Price AL Identifying loci with different allele frequencies among cases of eight psychiatric disorders using CC-GWAS. Nat. Genet 53, 445–454 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Young S et al. Guidance for identification and treatment of individuals with attention deficit/hyperactivity disorder and autism spectrum disorder based upon expert consensus. BMC Med. 18, 146 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pinto R, Rijsdijk F, Ronald A, Asherson P & Kuntsi J The Genetic Overlap of Attention-Deficit/Hyperactivity Disorder and Autistic-like Traits: an Investigation of Individual Symptom Scales and Cognitive markers. J. Abnorm. Child Psychol 44, 335–345 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Panagiotidi M, Overton PG & Stafford T Co-Occurrence of ASD and ADHD Traits in an Adult Population. J. Atten. Disord 23, 1407–1415 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Aoki Y et al. Association of White Matter Structure With Autism Spectrum Disorder and Attention-Deficit/Hyperactivity Disorder. JAMA Psychiatry 74, 1120–1128 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Asherson P & Agnew-Blais J Annual Research Review: Does late-onset attention-deficit/hyperactivity disorder exist? J. Child Psychol. Psychiatry 60, 333–352 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Zaitlen N et al. Informed conditioning on clinical covariates increases power in case-control association studies. PLoS Genet. 8, e1003032 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rajagopal VM et al. Differences in the genetic architecture of common and rare variants in childhood, persistent and late-diagnosed attention-deficit hyperactivity disorder. Nat. Genet 54, 1117–1124 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yap CX et al. Misestimation of heritability and prediction accuracy of male-pattern baldness. Nat. Commun 9, 2537 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Rheenen W, Peyrot WJ, Schork AJ, Lee SH & Wray NR Genetic correlations of polygenic disease traits: from theory to practice. Nat. Rev. Genet 20, 567–581 (2019). [DOI] [PubMed] [Google Scholar]
- 64.Wray NR et al. From Basic Science to Clinical Application of Polygenic Risk Scores: A Primer. JAMA Psychiatry 78, 101–109 (2021). [DOI] [PubMed] [Google Scholar]
- 65.Lewis CM & Vassos E Polygenic risk scores: from research tools to clinical instruments. Genome Med. 12, 44 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Laursen TM, Agerbo E & Pedersen CB Bipolar disorder, schizoaffective disorder, and schizophrenia overlap: a new comorbidity index. J. Clin. Psychiatry 70, 1432–1438 (2009). [DOI] [PubMed] [Google Scholar]
- 67.Nørgaard-Pedersen B & Hougaard DM Storage policies and use of the Danish Newborn Screening Biobank. J. Inherit. Metab. Dis 30, 530–536 (2007). [DOI] [PubMed] [Google Scholar]
- 68.Schork AJ et al. A genome-wide association study of shared risk across psychiatric disorders implicates gene regulation during fetal neurodevelopment. Nat. Neurosci 22, 353–361 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.O’Connell J et al. Haplotype estimation for biobank-scale data sets. Nat. Genet 48, 817–820 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Howie BN, Donnelly P & Marchini J A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 5, e1000529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Patterson N, Price AL & Reich D Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Manichaikul A et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fox J Polycor: polychoric and polyserial correlations. R package version 0.7–8. [Google Scholar]
- 75.Savalei V What to Do About Zero Frequency Cells When Estimating Polychoric Correlations. Struct. Equ. Modeling 18, 253–273 (2011). [Google Scholar]
- 76.Galili T, O’Callaghan A, Sidi J & Sievert C heatmaply: an R package for creating interactive cluster heatmaps for online publishing. Bioinformatics 34, 1600–1602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang J, Lee SH, Goddard ME & Visscher PM GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet 88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Purcell S et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bulik-Sullivan BK et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Morris AP et al. A powerful approach to sub-phenotype analysis in population-based genetic association studies. Genet. Epidemiol 34, 335–343 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ripley B nnet: Feed-Forward Neural Networks and Multinomial Log-Linear Models v. 7.3–16. (2021).
- 82.Wang K, Li M & Hakonarson H ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Watanabe K, Taskesen E, van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nat. Commun 8, 1826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gusev A et al. Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat. Genet 50, 538–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gusev A et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet 48, 245–252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Watanabe K et al. A global overview of pleiotropy and genetic architecture in complex traits. Nat. Genet 51, 1339–1348 (2019). [DOI] [PubMed] [Google Scholar]
- 87.Machiela MJ & Chanock SJ LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.de la Torre-Ubieta L et al. The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 172, 289–304.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bryois J et al. Evaluation of chromatin accessibility in prefrontal cortex of individuals with schizophrenia. Nat. Commun 9, 3121 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Finucane HK et al. Heritability enrichment of specifically expressed genes identifies disease-relevant tissues and cell types. Nat. Genet 50, 621–629 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lloyd-Jones LR et al. Improved polygenic prediction by Bayesian multiple regression on summary statistics. Nat. Commun 10, 5086 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pilling LC et al. Human longevity is influenced by many genetic variants: evidence from 75,000 UK Biobank participants. Aging 8, 547–560 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Savage JE et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet 50, 912–919 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Davies G et al. Genome-wide association study of cognitive functions and educational attainment in UK Biobank (N=112 151). Mol. Psychiatry 21, 758–767 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Okbay A et al. Polygenic prediction of educational attainment within and between families from genome-wide association analyses in 3 million individuals. Nat. Genet 54, 437–449 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mills MC et al. Identification of 371 genetic variants for age at first sex and birth linked to externalising behaviour. Nat Hum Behav 5, 1717–1730 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jansen PR et al. Genome-wide analysis of insomnia in 1,331,010 individuals identifies new risk loci and functional pathways. Nat. Genet 51, 394–403 (2019). [DOI] [PubMed] [Google Scholar]
- 100.Erzurumluoglu AM et al. Meta-analysis of up to 622,409 individuals identifies 40 novel smoking behaviour associated genetic loci. Mol. Psychiatry 25, 2392–2409 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nagel M et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat. Genet 50, 920–927 (2018). [DOI] [PubMed] [Google Scholar]
- 102.Trubetskoy V et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature 604, 502–508 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Walters RK et al. Transancestral GWAS of alcohol dependence reveals common genetic underpinnings with psychiatric disorders. Nat. Neurosci 21, 1656–1669 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Howard DM et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci 22, 343–352 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Benyamin B et al. Childhood intelligence is heritable, highly polygenic and associated with FNBP1L. Mol. Psychiatry 19, 253–258 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sanchez-Roige S et al. Genome-Wide Association Study Meta-Analysis of the Alcohol Use Disorders Identification Test (AUDIT) in Two Population-Based Cohorts. Am. J. Psychiatry 176, 107–118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Loughnan RJ et al. Intelligence Polygenic Score Is More Predictive of Crystallized Measures: Evidence From the Adolescent Brain Cognitive Development (ABCD) Study. Psychol. Sci 34, 714–725 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Raj A, Stephens M & Pritchard JK fastSTRUCTURE: variational inference of population structure in large SNP data sets. Genetics 197, 573–589 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The consent structure of iPSYCH and Danish law prevent individual genotype and phenotype data from being shared publicly. Reasonable requests to access individual level data to verify the findings in this paper can be accommodated with permission from the Danish Scientific Ethics Committee, the Danish Health Data Authority, the Danish Data Protection Agency, and the Danish Neonatal Screening Biobank Steering Committee and interested parties can contract T.M.W and expect a response within one week. Approvals for access can take several months and must be governed by strict data use agreements. ABCD study data can be accessed by request from the NIMH Data Archive (NDA; https://nda.nih.gov/abcd). GWAS summary statistics used for this work were downloaded from and available in public repositories as described in Supplementary table 28. Leave-one-study-out meta-analysis summary statistics for psychiatric disorders are available on request from the psychiatric genetic consortium (PGC) disorder working group chairs (https://pgc.unc.edu/for-researchers/data-access-committee/data-access-information/). The eQTL and sQTL visualizations and data used for the analyses described in this manuscript were obtained from the GTEx Portal (https://gtexportal.org/) on 10 March 2021 and 23 October 2023.