

Abstract
Excessive activity of the tyrosinase enzyme during melanogenesis results in hyperpigmentation in the skin. To address this issue, there is a need to develop effective tyrosinase inhibitors as a treatment for hyperpigmentation. In this study, we synthesized some novel 4H-chromene-3-carbonitrile compounds (6a-o) and assessed their inhibitory activities against tyrosinase, comparing them with kojic acid, which is known as a positive control. Compound 6f emerged as the most effective inhibitor, with an IC50 of 35.38 ± 2.12 µM. Kinetic studies of 6f exhibited competitive inhibition, with Ki = 16.15 µM. Molecular docking studies highlighted the importance of π-π stacking and hydrogen bonding interactions within the binding site. Molecular dynamics simulations showed that the R-enantiomer 6f exhibited superior binding stability compared to the S-enantiomer, with a lower standard deviation of RMSD and more persistent interactions with the key active site residues. These findings underscore the potential of the R-enantiomer of compound 6f as a potent tyrosinase inhibitor and provide insights for developing effective treatments for hyperpigmentation and related skin conditions.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13065-024-01305-0.
Keywords: Melanogenesis, 4H-chromene-3-carbonitrile derivatives, Tyrosinase inhibitors, Molecular docking, Molecular dynamics simulation
Introduction
Melanogenesis is a crucial biological process that produces melanin, the natural pigment responsible for protecting organisms from sun-induced skin damage. This process is essential for maintaining skin integrity and preventing damage from ultraviolet radiation [1].
While melanin is crucial for protecting the skin from harmful sun rays, excessive melanin production is a common concern for many individuals, especially in conditions like melasma and lentiginosis [2].
Tyrosinase is a key enzyme in melanin production, catalyzing the hydroxylation of L-tyrosine to form L-dihydroxyphenylalanine (L-Dopa), which is then oxidized to create dopaquinone. This dopaquinone undergoes a series of reactions that lead to melanin formation [3, 4]. Additionally, in the early stages of melanogenesis, the enzyme tyrosinase plays a critical role by catalyzing the oxidation of phenol to o-quinone, a crucial step in the melanin production pathway [5]. Tyrosinase is a copper-containing enzyme with six key histidine residues in its active site, which are essential for its catalytic function [6]. Disruptions in tyrosinase activity can lead to abnormal pigment production, potentially resulting in excessive melanin production and hyperpigmentation. These conditions are associated with various skin disorders, ranging from benign to potentially serious, such as freckles, age spots, pregnancy spots, melasma, acne, and melanoma, one of the most deadly types of cancer [1]. To address these challenges, researchers have been actively exploring the development of tyrosinase inhibitors, which can regulate melanin production and mitigate related skin issues. Most cosmetics and skin-lightening products function by inhibiting tyrosinase, an enzyme crucial for melanogenesis, which is exclusively produced by melanocytes [7].
In this context, synthetic agents such as quinazolinone [8], isopropylquinazolinone [9], indole [10], and α,β-unsaturated carbonyl compounds [11], thiazolopyrimidine [12] and vanillin-benzylidenehydrazine [13] have been developed as potent tyrosinase inhibitors. One of the most well-known examples is kojic acid, an FDA-approved tyrosinase inhibitor, which has been widely reported for its use in reducing melanin production. However, kojic acid’s clinical use is often limited by skin irritation and chemical instability, leading to side effects [14]. To overcome these drawbacks, modifications of kojic acid have been pursued. For instance, kojic acid conjugated with thio-quinazolinones has shown enhanced tyrosinase inhibitory activity and significant potency against the B16F10 melanoma cell line, effectively reducing melanin content (Fig. 1, compound A) [8]. Similarly, kojic acid derivatives linked to an amino-pyridine moiety have emerged as potent tyrosinase inhibitors. The kojic moiety plays a crucial role in these interactions, forming coordination bonds with two Cu2+ ions, hydrogen bonds with His279, and engaging in π-π stacking interactions with His263 (Fig. 1, compound B) [15]. One such compound exhibited high inhibitory activity against tyrosinase with an IC50 of 26.5 µM (compound C) [16].
Fig. 1.

Designed pyran derivatives based on compounds containing pyran and chromene scaffolds
Further structural modifications of the pyran ring embedded in kojic acid derivatives have also been explored, resulting in the development of chromene-based analogs with potent tyrosinase inhibitory effects (Fig. 1, compounds D-G) [17, 18]. These analogs demonstrate that the pyran core structure in the kojic acid derivatives is a crucial feature for tyrosinase inhibition. Recent advancements include novel kojic acid-fused 2-amino-3-cyano-4H-pyrans, which have been evaluated in both R and S isomeric forms. The compound H exhibited mixed-type inhibition with a Ki of 7.57 µM and demonstrated superior interactions compared to the S-enantiomer [17]. The kojopyran moiety was shown to form hydrogen bonds with Gly281 (2.52 Å) and additional critical interactions with Phe264, His244, and metal chelation with Cu, underscoring its potential as a robust tyrosinase inhibitor [19].
Continuing our ongoing investigations into bioactive heterocycles [20–22], with a particular focus on those that inhibit tyrosinase [19, 23, 24], we have designed a new series of 4H-chromene-3-carbonitrile derivatives in this study. The methoxy linker was closed following the methodology of Najafi et al., with various groups substituted at the R1 and R2 moieties. These derivatives were subsequently evaluated for their in-vitro enzyme inhibitory activities. Additionally, we conducted kinetic studies, molecular docking, and molecular dynamics simulations on the most potent compound to further elucidate its mechanism of action.
Results and discussion
Chemistry
The synthesis of the products 6a-o involved multiple stages, as detailed in Scheme 1. Initially, benzyloxy-benzaldehydes 3a-c were synthesized via a nucleophilic reaction. This reaction occurred between 4-hydroxy-benzaldehydes or vanillin (1a-b) and benzyl halide derivatives (2a-h), using potassium carbonate in DMF at ambient temperature. Subsequently, a multicomponent synthesis involving benzyloxy-benzaldehydes 3a-o, dimedone 4, and malononitrile 5 was performed in acetonitrile (5 mL), utilizing triethylamine as a catalyst under reflux conditions. This process successfully produced the desired compounds 6a-o with satisfactory yields (52–69%). The structures of all newly synthesized 4H-chromene-3-carbonitrile derivatives was confirmed using NMR, Mass, and IR spectroscopy.
Scheme 1.

Synthesis of 4H-chromene-3-carbonitrile derivatives 6a-o
The mechanism of 4H-chromene-3-carbonitrile ring formation is shown in Scheme 2. Initially, intermediate VII is generated through the condensation of one equivalent of dimedone with an aldehyde. Subsequently, the malononitrile reacts with intermediate VII and after removing H2O, the 4H-chromene-3-carbonitrile ring is formed.
Scheme 2.

Mechanism of 4H-chromene-3-carbonitrile ring formation
Biological study
In vitro tyrosinase inhibitory activity
The anti-tyrosinase properties of these 4H-chromene-3-carbonitrile derivatives (6a-o) were assessed by comparing their activity against mushroom tyrosinase to that of kojic acid, which served as a positive control (Table 1). The most potent derivatives in this set were 6e and 6f, with IC50 values of 39.09 ± 0.34 µM and 35.38 ± 2.12 µM, respectively.
Table 1.
Inhibitory activities of the new compounds 6a-o against tyrosinasea

To properly evaluate the structure-activity relationship (SAR), the derivatives were divided into two groups based on the R1 substitution. Among compounds 6a-h bearing R1 = H, compound 6h, as an unsubstituted derivative, exhibited weak potency against tyrosinase. To improve potency, different moieties were substituted at the R2 position. Evaluation of derivatives bearing fluorine at the R2 position showed the following order of activity: 6b (R2 = 2-F) > 6a (R2 = 3-F) and compound 6c (R2 = 4-F) exhibited no activity. The lack of activity in 6c indicates that the 4-fluoro substitution might create steric hindrance or disrupt essential interactions within the enzyme’s active site. Next, to improve activity at the para position, derivatives 6d (R2 = 4-Br) and 6g (R2 = 4-Cl) were synthesized, which demonstrated improved potency compared to 6c. The improved activity of 6d and 6g suggests that the optimal size and electronegativity of bromine and chlorine facilitate better interactions with the enzyme and enhance potency. Notably, the presence of a methyl group as an electron-donating group at the para position significantly increased the potency, with an IC50 value of 39.09 ± 0.34 µM. The increased potency of 6e (R2 = 4-CH3) may be attributed to the electron-donating effect of the methyl group, which could enhance the binding affinity by increasing the electron density on the aromatic ring and facilitating stronger interactions with the active site residues. Following this, derivative 6f (R2 = 4-CN) showed the most potent inhibition, displaying an IC50 value of 35.38 ± 2.12 µM. The high potency of 6f can be explained by the presence of the cyano group, which creates a favorable environment for binding to the enzyme’s active site through hydrogen bonding or dipole interactions. Also, it can participate in Cu2+ interaction with the binding site of the enzyme.
Next, derivatives 6i-o containing methoxy at the R1 position were synthesized. Fluorine derivatives at the R2 position exhibited that compound 6k (R2 = 4-F) recorded better results compared to 6i (R2 = 3-F) and 6j (R2 = 2-F). The better activity of 6k suggests that the 4-fluoro substitution is more favorable when a methoxy group is present at R1, likely due to an optimal electronic and steric fit in the enzyme’s active site. This trend differs from that seen in compounds 6a-c bearing H at R1. It seems that the presence of a methoxy group at the R1 position results in an orientation that made a substitution at the para position more favorable compared to meta and ortho.
However, similar to the previous set, 6n (R2 = 4-Cl) was more potent than 6l (R2 = 4-Br). This suggests that chlorine is more compatible with the enzyme’s active site than bromine when a methoxy group is present at R1, possibly due to its smaller size and favorable electronegativity. However, these derivatives exhibited lower activity compared to their counterparts in the R2 = H set. Similarly, the evaluation of 6m, which bears an electron-donating group, recorded significantly lower activity compared to 6e. In contrast, derivative 6o, with no substitution at the R2 position, exhibited better activity than its 6a counterpart. Overall, it can be suggested that, in the presence of the bulky methoxy group at R1, which increases the electron density on the central benzene ring, the absence of substitution at R2 or the presence of a small group reduces steric hindrance. This reduction in steric hindrance allows for better accommodation within the enzyme’s active site and improves the potency.
Kinetic study
The mechanism and type of inhibition were elucidated through comprehensive kinetic studies. To determine the mode of enzyme inhibition, we performed a Lineweaver-Burk plot analysis using the most effective derivative, compound 6f (Fig. 2). The Lineweaver-Burk plot, which graphs 1/V against 1/[S], was utilized to demonstrate the inhibition of tyrosinase by 6f at various concentrations of both the inhibitor (6f) and the substrate (L-Dopa). In this analysis, Km refers to the Michaelis-Menten constant, while Vmax denotes the maximum reaction velocity.
Fig. 2.

Kinetic study of compound 6f against tyrosinase. (a) The Lineweaver– Burk plot in the absence and presence of different concentrations of compound 6f; (b) The secondary plot of the slope of lines and various concentrations of compound 6f
The data analysis revealed that the Vmax of the enzyme remained unchanged, while the Km values increased with higher concentrations of compound 6f. This pattern indicates that compound 6f acts as a competitive inhibitor. The plot of the slope of the lines versus different concentrations of 6f provided an estimate of the inhibition constant, Ki = 16.15 µM.
Molecular docking study
To elucidate the molecular interactions between compound 6f and the tyrosinase enzyme, docking studies were conducted. Key amino acids in the active site of the tyrosinase enzyme (PDB ID: 2Y9X) are His259, His263, Phe90, His85, Cys83, His61, His94, Phe292, His296, Val283, and Phe264 [25]. Before performing the molecular docking study, the docking protocol was validated to ensure accuracy. Tropolone, the crystallographic ligand, was docked into the active site of tyrosinase, and the Root Mean Square Deviation (RMSD) was determined by comparing the best docking pose with the crystallographic ligand. The RMSD value was found to be less than 2 Å, indicating reliable accuracy of the docking protocol. Tropolone exhibited two interactions with both Cu²⁺ cofactors, as well as a hydrogen bond with His61. Additionally, it showed two π-π stacking interactions with His85 and His263, and two Pi-alkyl interactions with Val283 and Val286 (Fig. 3).
Fig. 3.

3D and 2D interaction of tropolone in complex with tyrosinase
Compound 6f, selected as the most effective inhibitor based on in vitro tyrosinase inhibitory assay, was synthesized as a racemic mixture, and thus, both the R and S isomers were evaluated in the docking studies.
The S-isomer of compound 6f exhibited a predicted free energy of binding of -5.27 kcal/mol when docked into the active site of tyrosinase. This binding energy suggests that the S-isomer may interact strongly with the enzyme. This isomer established a hydrogen bond with Val283 through the oxygen linked to the benzyl group. Furthermore, the phenyl ring connected to the pyran moiety displayed Pi-Pi stacking interactions with Phe264, and the benzyl group exhibited Pi-Pi stacking interactions with the amino acid residue His263.
Regarding the R-isomer, the calculated free energy of binding within the active site of tyrosinase was − 5.44 kcal/mol. The R-isomer forms Pi-Pi stacking interactions with His263 through its benzyl group. These interactions play a vital role in stabilizing the ligand in the active site and positioning it optimally for enzyme inhibition. The docking study highlights the importance of Pi-Pi stacking and hydrogen bonding interactions in the binding of compound 6f to tyrosinase enzyme.
After conducting the molecular docking study, we utilized the bootstrapping method to analyze the confidence interval for each isomer, as the calculated energy values for both the R and S isomers were very close (Figs. 4 and 5). The 95% confidence intervals were calculated and found to be (-5.43, -5.35) for the R isomer and (-5.41, -5.20) for the S isomer. The non-overlapping confidence intervals suggest that the R-isomer, which exhibits a more favorable estimated free energy of binding, represents the best docking outcome.
Fig. 4.

3D and 2D interaction of the S-enantiomer of compound 6f in complex with tyrosinase
Fig. 5.

3D and 2D interaction of the R-enantiomer of compound 6f in complex with tyrosinase
Also, the molecular docking study of all derivatives was also performed and the results are summarized in table S1. Compounds with fluorine substitutions (6a-c) exhibit binding energies ranging from − 4.681 to -4.893 kcal/mol. The electron-withdrawing nature of fluorine reduces the electron density on the aromatic rings, which weakens both π-π stacking and Pi-Alkyl interactions, possibly explaining the reduced anti-tyrosinase activity of these compounds. In contrast, bromine substitutions (6d, 6L) introduce steric hindrance due to bromine’s large size, but they compensate with stabilizing interactions through metal-acceptor coordination with copper, π-π stacking, and hydrogen bonds. These interactions result in moderate binding affinities, with free energies of -5.001 and − 5.136 kcal/mol. Chlorine substitutions (6g, 6n) improve binding affinity due to chlorine’s smaller size, which reduces steric hindrance and enhances stability through Pi-Alkyl and metal-acceptor interactions with copper. Methyl substitutions (6e, 6 m) serve as electron donors, significantly strengthening Pi-Alkyl and Pi-Pi stacking interactions. Compound 6e, with the strongest binding affinity (-7.306 kcal/mol), achieves this through extensive interactions, including multiple hydrogen bonds and π-π stacking. The cyano substitution (6f) shows a strong binding affinity (-5.44 kcal/mol) due to hydrogen bonds and Pi-Alkyl interactions, with cyano’s electron-withdrawing nature stabilizing the interactions within the enzyme’s active site.
Overall, it can be understood that hydrogen bonds are notably observed in compound 6e, which forms bonds with His85 and Asn260, and in compound 6f, where hydrogen bonds form with Arg268 via the cyano group. π-π stacking interactions are evident in compound 6e, where the phenoxy and substituent groups stack with His85 and His263, while compound 6f exhibits stacking interactions with His263 for both the substituted moiety and phenoxy groups. For Pi-Alkyl interactions, compound 6e shows a methyl group on the substituted moiety interacting with Phe264, while both the phenoxy and substituted moiety interact with Val283 and Ala286. In compound 6f, methyl groups on the tetrahydro-5 H-chromen-5-one core interact with His244, while the tetrahydro-5 H-chromen-5-one, phenoxy, and substituted moiety engage in Pi-Alkyl interactions with Val248, His244, Val283, and Ala286.
Molecular dynamic simulation
To further investigate the stability and interaction of the R and S isomers of compound 6f with tyrosinase enzyme, we accomplished molecular dynamics (MD) simulations. These simulations were performed over 100 nanoseconds to evaluate the dynamic behavior and binding stability of the isomers within the enzyme’s active site. The standard deviation of the root mean square deviation (RMSD) values, which indicate fluctuations in the enzyme’s three-dimensional structure, was 0.19 for the R isomer and 0.31 for the S isomer (Fig. 6). Additionally, the average RMSD values for the R and S isomers were 0.77 and 1.46, respectively. The lower standard deviation observed for the R isomer suggests a more stable enzyme conformation during the simulation period compared to that of the S isomer.
Fig. 6.
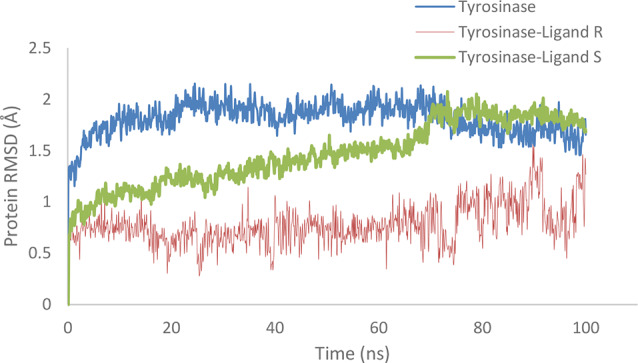
RMSD of tyrosinase (in blue), the complex of tyrosinase with the R-enantiomer 6f (in red), complex of tyrosinase with S- S-enantiomer 6f (in green) over a molecular dynamics simulation time of 100 ns
Figure 7 illustrates that the root mean square fluctuation (RMSF) values for key residues in the active site including His259, His263, Phe90, His85, Cys83, His94, Phe292, His296, Val283, and Phe264 were lower in the presence of the R isomer compared to the S isomer. This observation suggests that the R isomer enhances stability and decreases flexibility in these residues, indicating a more effective binding interaction.
Fig. 7.

RMSF of tyrosinase (in blue), complex of tyrosinase with the R-enantiomer 6f (in orange), complex of tyrosinase with S- enantiomer 6f (in gray) for a 100 ns molecular dynamics simulation time
The R isomer demonstrated sustained interactions with the active site, which are crucial for ensuring binding stability. Notably, the cyano (CN) group of the benzyl ring of the R isomer engaged in chelation with the copper ion in the active site for 56% of the simulation period (Fig. 8b). This interaction also affected the coordination of the copper ions with adjacent amino acids, including His259, His263, His296, and Glu256. Among these, the interaction between the benzyl moiety and His263 was particularly significant, persisting for 92% of the simulation time. This interaction seems crucial for determining both the orientation and effectiveness of the R enantiomer within the binding site.
Fig. 8.

(a) A timeline representation of the interactions and contacts; (b) Protein residue interactions with R- enantiomer atom
As shown in Fig. 8a, the R enantiomer demonstrated a notably greater number of interactions with the enzyme’s active site throughout the simulation period. These interactions play a crucial role in enhancing the stability of the enzyme-ligand complex, as supported by the RMSD results. In the case of the S enantiomer, the phenyl attached to the pyran moiety of the S isomer interacted with His85 for 23% of the simulation time which indicates that the S enantiomer does not interact as effectively with other key residues compared to the R enantiomer (Fig. 9).
Fig. 9.

(a) A timeline representation of the interactions and contacts; (b) Protein residue interactions with S- enantiomer atom
The MD simulations indicate that the R isomer of compound 6f exhibits better binding stability and interaction with the tyrosinase enzyme when compared to the S isomer. The R isomer’s enhanced stability and effective interactions highlight its potential as a more potent inhibitor of tyrosinase.
Drug‑likeness prediction
The concept of drug-likeness was introduced by Lipinski, known as the Rule of Five. This rule outlines specific physicochemical parameters that are typically associated with orally active drugs. The criteria include: a molecular weight (MW) of no more than 500 daltons, a maximum of 10 hydrogen bond acceptors (HBA), a maximum of 5 hydrogen bond donors (HBD), a lipophilicity index (logP) not exceeding 5, up to 10 rotatable bonds (RBC), and a polar surface area (PSA) of 140 Ų or less [26]. To assess these parameters, two online tools, pkCSM and SwissADME, were utilized [27, 28]. Compounds 6e and 6f were identified as the most promising antityrosinase agents and were selected for further evaluation of their physicochemical properties. The data presented in Table 2 demonstrate that two lead compounds successfully met the criteria outlined in Lipinski’s rule of five.
Table 2.
Physicochemical properties of the selected compounds (6e, 6f)
| Compound | TPSA | RBC | Log P | HBD | HBA | MW |
|---|---|---|---|---|---|---|
| 6e | 85.34 | 4 | 3.64 | 1 | 4 | 414.50 |
| 6f | 109.13 | 4 | 3.45 | 1 | 5 | 425.48 |
Abbreviations: MW, molecular weight; HBA, number of H-bond acceptors; HBD, number of H-bond donors; Log P, the octanol-water partition coefficient; RBC, rotatable bond count; TPSA, topological polar surface area
Prediction of ADMET properties
The ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties of compounds 6e and 6f were predicted using pkCSM, SwissADME, and ProTox online servers [27–29]. Compound 6e demonstrated a blood-brain barrier (BBB) permeability (Log BB) of -0.227, excellent intestinal absorption at 93.38%, and a steady-state volume of distribution (VDss) of 0.286, indicating moderate distribution characteristics. It is a substrate and inhibitor for CYP3A4 as well as an inhibitor of CYP2C9 and CYP2C19 (Table 3). This suggests that drug interactions should be carefully considered, particularly with drugs that have narrow therapeutic indices that are metabolized by these enzymes. Furthermore, 6e is neither a substrate nor an inhibitor for CYP2D6, reducing the risk of interactions with other drugs in this metabolic pathway. Notably, 6e is not a substrate for renal OCT2, which means that compound 6e is unlikely to be involved in transporter-mediated renal drug-drug interactions and may have a favorable safety profile concerning nephrotoxicity. In addition, compound 6e does not pose risks of cytotoxicity, skin sensitization, or hERG1 inhibition. Compound 6f showed a Log BB of -0.133, intestinal absorption of 100%, and a VDss of 0.140 log L/kg, indicating moderate distribution. Similarly, it shares metabolic, excretion, and toxicity profiles similar to those of 6e. These ADMET profiles indicate that both compounds have promising pharmacokinetic properties, supporting their potential as safe and effective therapeutic agents [30, 31].
Table 3.
ADMETa prediction of the selected compounds 6e and 6f
| Compound | Absorption | Distribution | Metabolism | Excretion | Toxicity | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BBB permeability (Log BB) |
Intestinal absorption (%Absorbed) |
VDss (log L/Kg) |
CYP3A4 substrate |
CYP3A4 Inhibitor |
CYP2C9 inhibitor |
CYP2C19 inhibitor |
CYP2D6 Substrate |
CYP2D6 inhibitor |
Renal OCT2 substrate | cytotoxicity | Skin Sensitization | hERG1 inhibitor | |
| 6e | -0.227 | 93.388 | 0.286 | Yes | Yes | Yes | Yes | No | No | No | No | No | No |
| 6f | -0.133 | 100 | 0.140 | Yes | Yes | Yes | Yes | No | No | No | No | No | No |
aHIA (Human Intestinal Absorption): > 80% is high and < 30% is poor; VDss (steady-state volume of distribution): log L/Kg: > 0.45 is high and < − 0.15 is low. A compound is considered to have low skin permeability if it has a logKp > − 2.5. PkCSM and SwissADME online servers calculated all data. bThe software could not predict parameters for these compounds (−)
Pan-assay Interference compounds (PAINS) filters
Pan-Assay Interference Compounds (PAINS) represent a significant challenge in medicinal chemistry and high-throughput screening (HTS) due to their tendency to produce false positive results in biological assays. These compounds often bind nonspecifically to multiple targets, leading to misleading outcomes that can divert efforts away from more promising drug candidates. Identifying and excluding PAINS early in the drug discovery process is crucial to avoid wasting time and resources on compounds that are unlikely to lead to successful therapeutics [31–33]. In this study, compounds 6e and 6f were subjected to PAINS screening to evaluate their potential for producing false-positive results. Both compounds 6e and 6f showed no PAINS alerts in the screenings. This outcome suggests that these compounds do not possess the promiscuous binding characteristics typically associated with PAINS; therefore, they are unlikely to interfere with the biological assay results through known nonspecific interactions. The absence of PAINS alerts for compounds 6e and 6f is a positive indication of their specificity and potential as viable drug candidates. By passing these initial PAINS filters, the compounds are less likely to be false positives in the early stages of drug discovery, thereby justifying further investigation into their efficacy and safety profiles. This finding supports the continued development of 6e and 6f as potential inhibitors of tyrosinase, with a reduced risk of misleading assay results.
Conclusion
In conclusion, we successfully synthesized a novel series of 4H-chromene-3-carbonitrile derivatives (6a-o) and evaluated their inhibitory activity against tyrosinase compared to the standard kojic acid. Among them, compound 2-amino-4-(4-((4-cyanobenzyl)oxy)phenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6f), exhibited the most potent inhibition of tyrosinase with an IC50 value of 35.38 ± 2.12 µM, indicating a competitive inhibition mechanism. Kinetic studies further clarified the inhibition mechanism, confirming that compound 6f functions as a competitive inhibitor, as demonstrated by the Lineweaver-Burk plot. Additionally, docking studies revealed significant π-π stacking and hydrogen bonding interactions, particularly with His263, Val283, and Phe264, which are essential for the ligand’s inhibitory potency.
The consistent interactions between the R-isomer and key residues, including His263 and the copper ions, underscore its promise as a more effective tyrosinase inhibitor. MD simulations provided further evidence of the R-isomer’s superior stability within the enzyme’s active site. The R-isomer exhibited a more stable conformation, as evidenced by a lower standard deviation in RMSD values compared to the S-isomer. The consistent interactions between the R-isomer and key residues, including His263 and the copper ions. Overall, our findings highlight the potential of the synthesized 4H-chromene-3-carbonitrile derivatives as powerful tyrosinase inhibitors. These results offer valuable insights for the design and development of novel tyrosinase inhibitors aimed at treating melanogenesis-related disorders. Continued research and optimization of these compounds may lead to the development of effective therapeutic agents for managing hyperpigmentation and related skin conditions.
Materials and methods
Experimental
All chemicals used in the experiments were sourced from Merck and Aldrich. Melting points were measured using a Kofler hot stage apparatus. Nuclear Magnetic Resonance (NMR) spectra, including both 1H NMR and 13C NMR, were acquired with Bruker 400 and 300 MHz NMR spectrometers. Infrared (IR) spectra were recorded using a Bruker ALPHA FT-IR spectrometer on KBr disks. Mass spectrometry (MS) data were collected with an Agilent Technologies (HP) mass spectrometer, operating at an ionization potential of 70 eV. 1H, 13C NMR were performed by the central lab of Mashhad University of Medical Sciences, Mashhad, Iran, Mass spectra were recorded by the central lab of Tehran University, Tehran, Iran, and IR spectra were recorded by the central lab of Hamadan University of Medical Sciences, Hamadan, Iran.
Chemistry
General procedure for the synthesis of (benzyloxy)benzaldehydes (3a-o)
In a 25 mL round-bottom flask, 1 mmol of hydroxyl benzaldehyde (1a and 1b) and 3 mmol of benzyl halides (2a-h) were dissolved in 3 mL of DMF. To this solution, 1 mmol of potassium carbonate was added, and the mixture was stirred at room temperature for 24 h. The progress of the reaction was monitored using TLC (n-hexane/EtOAc, 4:1). Once completed, the reaction mixture was poured into an ice-water mixture. The resulting precipitates were collected, dried, and utilized in subsequent reactions without additional purification [34].
General procedure for the synthesis of 4H-chromene-3-carbonitrile derivatives (6a-o )
To synthesize the 4H-chromene-3-carbonitrile derivatives (6a-o), we began by combining 0.5 mmol of the benzyloxy-benzaldehyde derivatives (3a-o) with 0.5 mmol of dimedone in a 50 ml round-bottom flask. Next, we added 0.6 mmol of malononitrile, two drops of triethylamine, and 4 ml of acetonitrile to the reaction mixture. The mixture was stirred at room temperature for 24 h to ensure the complete reaction. The progress of the reaction was monitored by TLC (n-hexane/EtOAc, 3:2). Once the reaction was finished, the mixture was allowed to cool to room temperature, and the resulting precipitate was filtered out.
2-Amino-4-(4-((3-fluorobenzyl)oxy)phenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6a)
White solid; Yield: 61%, Rf =0.45 (n-hexane: EtOAc (60: 40)); mp: 210–212 °C. 1H NMR (301 MHz, DMSO-d6) δ 7.46 (td, J = 8.0, 6.0 Hz, 1H), 7.34–7.25 (m, 2 H), 7.22–7.13 (m, 1H), 7.11–7.05 (m, 2 H), 6.99 (s, 2 H), 6.97–6.90 (m, 2 H), 5.10 (s, 2 H), 4.15 (s, 1H), 2.53 (s, 2 H), 2.31–2.07 (m, 2 H), 1.05 (s, 3 H), 0.97 (s, 3 H). 13C NMR (76 MHz, DMSO-d6) δ 196.19, 164.27, 162.68, 161.05, 158.89, 157.34, 140.65, 140.55, 137.75, 131.00, 130.89, 128.75, 124.02, 123.98, 120.30, 115.17, 114.97, 114.90, 114.84, 114.56, 113.38, 68.80, 58.91, 50.44, 35.21, 32.27, 28.85, 27.25. Ms m/z (%): 109.1(100), 83.1(15), 66.1(6), 161.1(3), 217.2(12), 243.2(19), 281.1(5.5), 309.2(23), 352.2(10), 419.3(8), 418.3(13.5), 133.1(2) .IR (KBr, cm− 1) v: 3357.4(N-H), 3185.91(C-H aromatic), 2190.61(C ≡ N), 1654.03 (C = O), 1604.96 (C = C aromatic), 1507.98 (C = C aromatic).
2-Amino-4-(4-((2-fluorobenzyl)oxy)phenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6b).
White solid; Yield: 60%, Rf =0.42 (n-hexane: EtOAc (60: 40)); mp: 214–217 °C. 1H NMR (301 MHz, DMSO-d6) δ 7.57 (td, J = 7.5, 1.8 Hz, 1H), 7.49–7.39 (m, 1H), 7.32–7.21 (m, 2 H), 7.12–7.05 (m, 2 H), 7.00 (s, 2 H), 6.96 (d, J = 8.6 Hz, 2 H), 5.11 (s, 2 H), 4.15 (s, 1H), 2.53 (s, 2 H), 2.30–2.08 (m, 2 H), 1.05 (s, 3 H), 0.97 (s, 3 H). 13C NMR (76 MHz, DMSO- d6) δ 196.18, 162.67, 162.49, 159.23, 158.90, 157.39, 137.80, 131.23, 131.18, 130.91, 130.80, 128.77, 125.03, 124.99, 124.45, 124.25, 120.29, 115.99, 115.72, 114.85, 113.40, 63.95, 63.90, 58.93, 50.46, 35.23, 32.26, 28.85, 27.26. Ms m/z (%): 109.1(100), 83.1(10), 66.1(5), 161.1(3), 217.2(13), 243.2(6.5), 281.1(5), 309.2(25), 352.2(5.5), 419.3(4), 418.3(13.5), 133.1(2) IR (KBr, cm− 1) v: 3357.94(N-H), 3179.32(C-H aromatic), 2187.61(C ≡ N), 1652.44 (C = O), 1604.88 (C = C aromatic), 1508.59 (C = C aromatic).
2-Amino-4-(4-((4-fluorobenzyl)oxy)phenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6c).
White solid; Yield: 62%, Rf =0.42 (n-hexane: EtOAc (60: 40)); mp: 200–203 °C. 1H NMR (301 MHz, DMSO-d6) δ 7.56–7.45 (m, 2 H), 7.28–7.19 (m, 2 H), 7.10–7.05 (m, 2 H), 6.99 (s, 2 H), 6.97–6.92 (m, 2 H), 5.05 (s, 2 H), 4.14 (s, 1H), 2.53 (s, 2 H), 2.29–2.08 (m, 2 H), 1.05 (s, 3 H), 0.97 (s, 3 H). 13C NMR (75 MHz, DMSO-d6) δ 196.18, 163.83, 162.66, 160.61, 158.89, 157.46, 137.64, 133.86, 133.82, 130.50, 130.39, 128.72, 120.30, 115.86, 115.58, 114.94, 113.40, 68.92, 58.93, 50.45, 35.22, 32.26, 28.86, 27.25. Ms m/z (%): 109.2(100), 83.2(7), 66.2(1.5), 161.2(2), 217.2(9), 243.2(4), 281.3(4), 309.3(23), 352.3(8.5), 419.3(5.5), 418.3(21), 133.2(1.5) IR (KBr, cm− 1) v: 3362.21(N-H), 3179.42(C-H aromatic), 2185.48(C ≡ N), 1653.54(C = O), 1604.07 (C = C aromatic), 1512.66 (C = C aromatic).
2-Amino-4-(4-((4-bromobenzyl)oxy)phenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6d).
White solid; Yield: 69%, Rf =0.42 (n-hexane: EtOAc (60: 40)); mp: 208–209 °C. 1H NMR (301 MHz, DMSO-d6) δ 7.61 (d, J = 8.4 Hz, 1H), 7.42 (d, J = 8.1 Hz, 2 H), 7.08 (d, J = 8.4 Hz, 2 H), 6.99 (s, 2 H), 6.93 (d, J = 8.4 Hz, 2 H), 5.06 (s, 2 H), 4.15 (s, 1H), 2.52 (s, 2 H), 2.30–2.08 (m, 2 H), 1.05 (s, 3 H), 0.97 (s, 3 H). 13C NMR (76 MHz, DMSO-d6) δ 196.26, 162.72, 158.90, 157.35, 137.71, 137.10, 131.83, 130.27, 128.73, 121.37, 120.30, 114.98, 113.38, 68.85, 58.95, 50.44, 35.20, 32.26, 28.84, 27.25. Ms m/z (%): 109.1(100), 90.2(16.5), 66.2(5.5), 217.2(7), 243.2(7), 281.1(3), 309.2(16.5), 414.2(5.5), 480.2(5.5), 478.2(5.5), 479.3(2), 481.2(1.5) IR (KBr, cm− 1) v: 3327.45 (N-H), 3216.72 (C-H aromatic), 2194.68(C ≡ N), 1669 (C = O), 1599.43 (C = C aromatic), 1508.99 (C = C aromatic).
2-Amino-7,7-dimethyl-4-(4-((4-methylbenzyl)oxy)phenyl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6e).
White solid; Yield: 68%, Rf =0.42 (n-hexane: EtOAc (60: 40)); mp: 222–223 °C. 1H NMR (301 MHz, DMSO-d6) δ 7.34 (d, J = 7.8 Hz, 2 H), 7.21 (d, J = 7.7 Hz, 2 H), 7.07 (d, J = 8.6 Hz, 2 H), 6.99 (s, 2 H), 6.96–6.90 (m, 2 H), 5.02 (s, 2 H), 4.14 (s, 1H), 2.52 (s, 2 H), 2.30–2.08 (m, 2 H), 1.05 (s, 3 H), 0.97 (s, 3 H). 13C NMR (76 MHz, DMSO-d6) δ 196.18, 162.65, 158.89, 157.58, 137.52, 137.48, 134.58, 129.45, 128.69, 128.27, 120.30, 114.94, 113.43, 69.54, 58.97, 50.46, 35.21, 32.26, 28.85, 28.38, 27.27, 21.25. Ms m/z (%): 105.3(100), 77.3(7), 55.3(2), 161.2(2.5), 217.3(4.5), 243.3(2.5), 309.3(7), 348.3(4.5), 414.4(13.5) IR (KBr, cm− 1) v: 3323.18(N-H), 3211.77(C-H aromatic), 2190.45(C ≡ N), 1659.26 (C = O), 1602.92 (C = C aromatic), 1507.66 (C = C aromatic).
2-Amino-4-(4-((4-cyanobenzyl)oxy)phenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6f).
White solid; Yield: 67%, Rf =0.41 (n-hexane: EtOAc (60: 40)); mp: 208–209 °C. 1H NMR (301 MHz, DMSO-d6) δ 7.93–7.83 (m, 2 H), 7.65 (d, J = 8.1 Hz, 2 H), 7.13–7.06 (m, 2 H), 7.00 (s, 2 H), 6.98–6.86 (m, 2 H), 5.19 (s, 2 H), 4.15 (s, 1H), 2.53 (s, 2 H), 2.29 − 2.08 (m, 2 H), 1.05 (s, 3 H), 0.97 (s, 3 H). 13C NMR (76 MHz, DMSO- d6) δ 196.18, 162.70, 158.88, 157.17, 143.51, 137.91, 132.91, 128.78, 128.54, 119.26, 114.99, 113.35, 110.90, 68.69, 35.20, 32.27, 28.85, 27.24. Ms m/z (%): 116.2(100), 89.2(16.5), 55.2(3.5), 159.2(11.5), 201.2(18.5), 243.3(56), 359.3(34), 285.1(3.5), 309.3(3.5), 359.3(35), 425.3(2), 262.1(2), 140.2(2) IR (KBr, cm− 1) v: 3330.14(N-H), 2186.44(C ≡ N), 1672.72(C = O), 1603.14(C = C aromatic), 1509.36(C = C aromatic).
2-Amino-4-(4-((4-chlorobenzyl)oxy)phenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6g).
White solid; Yield: 64%, Rf =0.45 (n-hexane: EtOAc (60: 40)); mp: 207–208 °C. 1H NMR (301 MHz, DMSO-d6) δ 7.48 (s, 4H), 7.11–7.05 (m, 2 H), 6.99 (s, 2 H), 6.96–6.91 (m, 2 H), 5.07 (s, 2 H), 4.15 (s, 1H), 2.52 (s, 2 H), 2.30–2.08 (m, 2 H), 1.05 (s, 3 H), 0.97 (s, 3 H). 13C NMR (76 MHz, DMSO-d6) δ 196.18, 162.67, 158.89, 157.37, 137.71, 136.68, 132.85, 129.97, 128.91, 128.74, 120.31, 114.96, 113.40, 68.80, 58.94, 50.45, 35.22, 32.25, 28.86, 27.25. Ms m/z (%): 125.1(100), 89.2(8), 66.2(4), 161.1(2), 217.2(7), 243.2(4), 281.1(2), 309.2(15), 368.2(5), 434.3(7.5) IR (KBr, cm− 1) v: 3323.22(N-H), 2194.57(C ≡ N), 1655.30(C = O), 1605.95 (C = C aromatic), 1507.92 (C = C aromatic).
2-Amino-4-(4-(benzyloxy)phenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6 h).
White solid; Yield: 69%, Rf =0.57 (n-hexane: EtOAc (60: 40)); mp: 221–222 °C. 1H NMR (301 MHz, DMSO-d6) δ 7.47–7.40 (m, 5 H), 7.22–7.16 (m, 2 H), 6.96–6.90 (m, 2 H), 5.05 (s, 2 H), 4.63 (d, J = 20.9 Hz, 2 H), 4.40 (s, 1H), 2.48 (s, 2 H), 2.26 (d, J = 2.8 Hz, 2 H), 1.14 (s, 3 H), 1.07 (s, 3 H). 13C NMR (76 MHz, DMSO-d6) δ 196.09, 161.36, 157.96, 157.45, 137.09, 135.77, 128.69, 128.61, 127.98, 127.64, 127.58, 118.84, 114.88, 114.24, 77.26, 70.04, 63.72, 50.71, 40.69, 34.77, 32.23, 28.89, 27.73. Ms m/z (%): 91.2(100), 65.2(33.5), 112.2(20), 140.2(26), 217.2(4), 169.1(3.5), 243.2(7), 260.1(20), 309.2(8.5), 334.3(8), 400.3(6.5) .IR (KBr, cm− 1) v: 3323.18(N-H), 3210.66(C-H aromatic), 2187.16(C ≡ N), 1659.30 (C = O), 1603.04(C = C aromatic), 1508.73 (C = C aromatic).
2-Amino-4-(4-((3-fluorobenzyl)oxy)-3-methoxyphenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6i).
White solid; Yield: 58%, Rf =0.52 (n-hexane: EtOAc (60: 40)); mp:174–175 °C. 1H NMR (301 MHz, DMSO-d6) δ 7.49–7.42 (m, 1H), 7.32–7.25 (m, 2 H), 7.18 (td, J = 7.3, 1.7 Hz, 1H), 6.99 (s, 2 H), 6.96 (d, J = 8.3 Hz, 1H), 6.75 (d, J = 2.1 Hz, 1H), 6.66 (dd, J = 8.2, 2.1 Hz, 1H), 5.08 (s, 2 H), 4.15 (s, 1H), 3.76 (s, 3 H), 2.54 (s, 2 H), 2.31–2.11 (m, 2 H), 1.06 (s, 3 H), 1.00 (s, 3 H). 13C NMR (76 MHz, DMSO-d6) δ 196.22, 162.86, 158.85, 149.25, 146.82, 140.75, 138.44, 130.98, 130.87, 124.05, 120.30, 119.53, 115.17, 114.87, 114.58, 113.99, 113.20, 111.69, 69.60, 58.87, 55.99, 50.44, 35.51, 32.24, 28.96, 27.11. Ms m/z (%): 109.2(100), 83.2(22.5), 55.2(4.5), 161.2(8.5), 189.2(8.5), 231.2(14.5), 273.3(76.5), 311.3(13.3), 339.3(34.5), 382.3(46.5), 448.3(15), 449.3(4.5) IR (KBr, cm− 1) v: 314.49(N-H), 3190.11(C-H aromatic), 2191.59(C ≡ N), 1655.30(C = O), 1607.68(C = C aromatic), 1514.57 (C = C aromatic).
2-Amino-4-(4-((2-fluorobenzyl)oxy)-3-methoxyphenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6j).
White solid; Yield: 56%, Rf =0.52 (n-hexane: EtOAc (60: 40)); mp: 200–201 °C. 1H NMR (301 MHz, DMSO-d6) δ 7.56 (td, J = 7.5, 1.7 Hz, 1H), 7.44 (dtd, J = 7.4, 6.4, 1.9 Hz, 1H), 7.27 (tdd, J = 7.5, 5.6, 1.3 Hz, 2 H), 7.03 (s, 1H), 7.00 (d, J = 2.6 Hz, 2 H), 6.74 (d, J = 2.1 Hz, 1H), 6.68 (dd, J = 8.2, 2.1 Hz, 1H), 5.08 (s, 2 H), 4.16 (s, 1H), 3.73 (s, 3 H), 2.55 (s, 2 H), 2.32–2.10 (m, 2 H), 1.06 (s, 3 H), 1.00 (s, 3 H). 13C NMR (76 MHz, DMSO-d6) δ 196.21, 162.85, 162.49, 159.23, 158.86, 149.21, 146.88, 138.48, 131.35, 131.30, 130.91, 130.80, 125.02, 124.98, 124.54, 124.34, 120.29, 119.56, 115.97, 115.69, 113.81, 113.22, 111.71, 64.60, 63.84, 58.88, 55.93, 50.46, 35.52, 32.23, 28.97, 27.11. Ms m/z (%): 109.2(100), 83.2(18), 55.2(3.5), 140.2(3), 161.2(5), 189.2(5), 217.2(8), 245.3(4), 273.3(31), 339.3(12), 308.2(4) 382.3(31), 448.3(4), 449.3(2) IR (KBr, cm− 1) v: 3317.34(N-H), 3208.57(C-H aromatic), 2191.17(C ≡ N), 1653.42(C = O), 1606.69(C = C aromatic), 1514.78 (C = C aromatic).
2-Amino-4-(4-((4-fluorobenzyl)oxy)-3-methoxyphenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6k).
White solid; Yield: 58%, Rf =0.52 (n-hexane: EtOAc (60: 40)); mp: 184–186 °C. 1H NMR (301 MHz, DMSO-d6) δ 7.54–7.46 (m, 2 H), 7.29–7.20 (m, 2 H), 6.99 (s, 2 H), 6.96 (s, 1H), 6.74 (d, J = 2.0 Hz, 1H), 6.66 (dd, J = 8.2, 2.1 Hz, 1H), 5.03 (s, 2 H), 4.15 (s, 1H), 3.74 (s, 3 H), 2.55 (s, 2 H), 2.32–2.11 (m, 2 H), 1.06 (s, 3 H), 1.00 (s, 3 H). 13C NMR (76 MHz, DMSO- d6) δ 196.22, 163.85, 162.85, 160.62, 158.86, 149.24, 146.95, 138.30, 133.95, 133.91, 131.81, 130.54, 130.43, 120.29, 119.52, 115.83, 115.55, 113.93, 113.23, 111.68, 69.70, 58.92, 55.96, 50.45, 35.51, 32.23, 28.96, 27.10. Ms m/z (%): 109.2(100), 83.2(12.5), 55.2(3.5), 140.2(2), 161.1(3), 189.1(1.5), 217.2(11), 245.2(1.5), 273.1(3.5), 311.2(6), 339.2(16), 382.3(10), 448.3(6) IR (KBr, cm− 1) v: 3324.67(N-H), 3210.39(C-H aromatic), 2194.46(C ≡ N), 1649.40(C = O), 1603.53(C = C aromatic), 1507.18(C = C aromatic).
2-Amino-4-(4-((4-bromobenzyl)oxy)-3-methoxyphenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6L).
White solid; Yield: 63%, Rf =0.52 (n-hexane: EtOAc (60: 40)); mp: 197–198 °C. 1H NMR (301 MHz, DMSO-d6) δ 7.61 (d, J = 8.3 Hz, 1H), 7.41 (d, J = 8.4 Hz, 1H), 6.99 (s, 2 H), 6.95 (d, J = 8.3 Hz, 1H), 6.74 (d, J = 2.1 Hz, 1H), 6.65 (dd, J = 8.2, 2.1 Hz, 1H), 5.03 (s, 2 H), 4.15 (s, 1H), 3.74 (s, 3 H), 2.54 (s, 2 H), 2.31–2.10 (m, 2 H), 1.06 (s, 3 H), 1.00 (s, 3 H). 13C NMR (76 MHz, DMSO-d6) δ 196.23, 162.87, 158.86, 149.23, 146.84, 138.38, 137.20, 131.81, 130.32, 121.38, 120.30, 119.51, 113.97, 113.21, 111.68, 69.61, 58.89, 55.97, 50.44, 35.50, 32.23, 28.96, 27.11. Ms m/z (%): 169.2(100), 90.2(15.5), 55.2(4.5), 146.2(2), 115.2(2), 191.2(3), 231.2(6.5), 273.3(44.5), 442.2(24.5) IR (KBr, cm− 1) v: 3316.38(N-H), 2202.25(C ≡ N), 1654.32(C = O), 1514.89(C = C aromatic).
2-Amino-4-(3-methoxy-4-((4-methylbenzyl)oxy)phenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6 m).
White solid; Yield: 62%, Rf =0.52 (n-hexane: EtOAc (60: 40)); mp: 180–182 °C. 1H NMR (301 MHz, DMSO-d6) δ 7.36–7.31 (m, 2 H), 7.21 (d, J = 7.8 Hz, 2 H), 6.99 (s, 2 H), 6.96 (d, J = 8.3 Hz, 1H), 6.73 (d, J = 2.1 Hz, 1H), 6.65 (dd, J = 8.3, 2.1 Hz, 1H), 4.99 (s, 2 H), 4.15 (s, 1H), 3.73 (s, 3 H), 2.52 (s, 2 H), 2.31–2.10 (m, 2 H), 1.06 (s, 3 H), 1.00 (s, 3 H). 13C NMR (76 MHz, DMSO-d6) δ 196.20, 162.83, 158.86, 149.21, 147.10, 138.08, 137.52, 134.65, 129.42, 128.38, 120.30, 119.52, 113.79, 113.25, 111.67, 70.27, 58.93, 55.96, 50.46, 35.49, 32.23, 28.96, 27.13, 21.25. Ms m/z (%): 105.2(100), 77.2(5), 55.2(2.5), 273.1(5), 378.3(14) IR (KBr, cm− 1) v: 3323.93(N-H), 2194.48(C ≡ N), 1652.29(C = O), 1602.64(C = C aromatic), 1503.82 (C = C aromatic).
2-Amino-4-(4-((4-chlorobenzyl)oxy)-3-methoxyphenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6n).
White solid; Yield: 60%, Rf =0.55 (n-hexane: EtOAc (60: 40)); mp: 194–195 °C. 1H NMR (301 MHz, DMSO-d6) δ 7.47 (s, 4H), 6.99 (s, 2 H), 6.95 (d, J = 8.2 Hz, 1H), 6.74 (d, J = 2.0 Hz, 1H), 6.65 (dd, J = 8.2, 2.1 Hz, 1H), 5.05 (s, 2 H), 4.15 (s, 1H), 3.74 (s, 3 H), 2.54 (s, 2 H), 2.31–2.10 (m, 2 H), 1.06 (s, 3 H), 1.00 (s, 3 H). 13C NMR (76 MHz, DMSO-d6) δ 196.21, 162.86, 158.86, 149.25, 146.86, 136.79, 132.83, 130.02, 128.89, 119.51, 113.99, 113.21, 111.70, 69.59, 58.90, 55.98, 50.44, 35.50, 32.23, 28.96, 27.12. Ms m/z (%): 125.2(100), 83.2(10), 55.2(3.5), 161.2(2), 189.2(2), 231.2(3), 273.3(16.5), 324.2(2), 398.3(12). IR (KBr, cm− 1) v: 3317.91(N-H), 3189.90(C-H aromatic), 2203.94(C ≡ N), 1654.44(C = O), 1607.68(C = C 1514.70 (C = C aromatic).
2-Amino-4-(4-(benzyloxy)-3-methoxyphenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (6o).
White solid; Yield: 64%, Rf =0.50 (n-hexane: EtOAc (60: 40)); mp: 224–226 °C. 1H NMR (301 MHz, DMSO-d6) δ 7.48–7.34 (m, 5 H), 6.99 (d, J = 1.9 Hz, 2 H), 6.96 (s, 1H), 6.74 (d, J = 2.1 Hz, 1H), 6.66 (dd, J = 8.2, 2.1 Hz, 1H), 5.05 (s, 2 H), 4.16 (s, 1H), 3.74 (s, 3 H), 2.54 (s, 2 H), 2.32–2.10 (m, 2 H), 1.06 (s, 3 H), 1.00 (s, 3 H). 13C NMR (76 MHz, DMSO-d6) δ 196.22, 162.85, 158.86, 149.22, 147.08, 138.18, 137.69, 128.88, 128.29, 120.31, 119.53, 113.80, 113.24, 111.66, 70.40, 58.93, 55.96, 50.46, 35.51, 32.24, 28.97, 27.12. Ms m/z (%): 91.2(100), 65.2(6), 161.2(5.5), 133.2(2.5), 189.2(2.5), 217.3(19.5), 245.2(2.5), 273.3(23), 311.3(11.5), 339.3(29), 364.3(24), 430.3(15) IR (KBr, cm− 1) v: 3314.58(N-H), 3192.12(C-H aromatic), 2191.41(C ≡ N), 1653.93(C = O), 1608.17(C = C aromatic), 1514.90 (C = C aromatic).
In vitro tyrosinase inhibition assay
We modified a previously described method [19] to conduct the mushroom tyrosinase (EC 1.14.18.1) assay using L-Dopa as the substrate. To measure the enzyme’s diphenolase activity, we employed a spectrophotometric approach to monitor the production of dopachrome at 490 nm. All compounds being evaluated, including kojic acid, were dissolved in dimethyl sulfoxide (DMSO-d6) and then diluted to the desired concentrations. In a 96-well microplate, 10 µl (µl) of each sample was combined with 160 µl of 50 millimolar (mM) phosphate buffer at a pH of 6.8. Next, 10 µl of tyrosinase (at a concentration of 600 U mL− 1) was added [35]. After a 20-minute pre-incubation period at 28 °C, 20 µl of L-Dopa solution was introduced, achieving a final concentration of 0.7 mM. After an additional 10-minute incubation period, the absorbance was recorded. The inhibitory activity of the compounds was expressed as IC50 values, which represent the concentration required to inhibit 50% of the maximum response.
The percentage inhibition was calculated using the following equation:
Inhibition (%) = 100 (Abscontrol − Abscompound )/Abscontrol.
Determination of the inhibition type
To conduct the kinetic analysis, we focused on compound 6f, which was identified as the most effective derivative. The inhibitor concentrations ranged from 5 to 50 µM, while the substrate (L-Dopa) concentrations ranged from 0.1 mM to 1.5 mM in all kinetic experiments. We followed the pre-incubation and measurement durations outlined in the mushroom tyrosinase inhibition assay protocol [4]. The maximum initial velocity was determined from the initial linear segment of absorbance within a 10-minute window after adding L-Dopa, with measurements taken at 1-minute intervals. The Michaelis constant (Km) and the maximum velocity (Vmax) of tyrosinase activity were calculated using the Lineweaver–Burk plot method, which involved varying concentrations of L-Dopa as the substrate. To determine the type of enzyme inhibition, we utilized Lineweaver–Burk plots that displayed the reciprocal of velocities (1/V) against the reciprocal of substrate concentrations (1/[S] mM− 1) [36].
Drug‑likeness, ADMET properties and PAINS filters: in silico prediction
The parameters for the selected compounds (6e and 6f), including the number of hydrogen bond acceptors (HBA) and donors (HBD), logP values, topological polar surface area (tPSA), and the count of rotatable bonds (RBC), were determined utilizing the online tools SwissADME and pkCSM [27, 28, 37]. Additionally, the ADMET properties of these compounds were evaluated through the pkCSM software and ProTox online servers. Also, The Pan-Assay Interference Compounds (PAINS) screening was conducted using the SwissADME tool and further validated with the “False Positive Remover” software (http://www.cbligand.org/PAINS/) [38].
Docking study
The crystallographic structure of tyrosinase enzyme (PDB ID: 2Y9X) with a resolution of 2.78 Å was obtained from the RCSB Protein Data Bank. The 3D structure of compound 6f was optimized using density functional theory (DFT) with the BP86/DEF2-TZVP basis set in the ORCA quantum chemistry package [39]. Rigid-ligand molecular docking studies of the R and S enantiomers of 6f were performed using AutoDock Tools 1.5.4 (ADT) [40].
For the preparation of the enzyme, water molecules and other non-essential components were removed from the enzyme’s structure. Hydrogen atoms were added, and non-polar hydrogens were merged with their corresponding carbon atoms. Kollman partial atomic charges were assigned using ADT. An AutoGrid calculation generated a grid map, with the grid box centered at coordinates x = -30.194, y = -4.738, and z = -93.823. The cubic box size was 60 × 60 × 60, featuring a grid spacing of 0.375 Å. Docking simulations were conducted utilizing the Lamarckian genetic algorithm, which began with a population size of 150, a maximum number of evaluations of 25,000,000, a maximum number of generations of 27,000, and 100 independent runs.
MD simulations
Molecular dynamics simulations were conducted using the Desmond v5.3 module within the Maestro interface from the Schrödinger 2018-4 suite [41]. The optimal pose for the molecular dynamics simulation of compound 6f was determined through the IFD method. The system was prepared by solvating the protein-ligand complex with explicit water molecules, and counter-ions, along with NaCl were added to achieve cellular ionic concentrations.
The molecular dynamics protocol included minimization, pre-production, and production molecular dynamics steps. The simulations were performed in the NPT ensemble, which maintains a constant number of particles (N), pressure (P), and temperature (T) throughout the simulation. The system underwent a 100 ns molecular dynamics simulation, during which the dynamic behavior and structural changes were analyzed using the root mean square deviation (RMSD). Additionally, protein-ligand interactions were investigated using the energy-minimized structure from the equilibrated trajectory [10, 12].
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
The authors of this article would like to thank the Vice-Chancellor of Research and Technology of Hamedan University of Medical Sciences and the Vice-Chancellor for Research of Shiraz University of Medical Sciences for the financial support of this research.
Author contributions
AB and MA synthesized all compounds. MA wrote the draft of the manuscript. AI performed molecular dynamics and anti-tyrosinase study and edited the manuscript. ZN was the scientific advisor of this research. GC designed, analyzed, and edited the manuscript.
Funding
Funds for this study were provided by the Research and Technology Vice-chancellor of Hamadan University of Medical Sciences (Grant Number: IR.SUMS.REC.1403.205) related to Azimi’s thesis and the Vice-Chancellor for Research of Shiraz University of Medical Sciences (Grant Number: 31285).
Data availability
No datasets were generated or analysed during the current study.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Gholamabbas Chehardoli, Email: cheh1002@gmail.com.
Aida Iraji, Email: iraji@sums.ac.ir.
References
- 1.Li J, et al. Recent advances in the design and discovery of synthetic tyrosinase inhibitors. Eur J Med Chem. 2021;224:113744. [DOI] [PubMed] [Google Scholar]
- 2.Choi YJ, et al. Synthesis and biological evaluation of kojic acid derivatives as tyrosinase inhibitors. Bull Korean Chem Soc. 2014;35(12):3647–50. [Google Scholar]
- 3.Alizadeh N, et al. Evaluating the effects of disubstituted 3-hydroxy-1H-pyrrol-2 (5H)-one analog as novel tyrosinase inhibitors. Bioorg Chem. 2022;126:105876. [DOI] [PubMed] [Google Scholar]
- 4.Hosseinpoor H, et al. Anti-melanogenesis and anti-tyrosinase properties of aryl-substituted acetamides of phenoxy methyl triazole conjugated with thiosemicarbazide: design, synthesis and biological evaluations. Bioorg Chem. 2021;114:104979. [DOI] [PubMed] [Google Scholar]
- 5.Solano F. On the metal cofactor in the tyrosinase family. Int J Mol Sci. 2018;19(2):633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Obaid RJ, et al. Natural and synthetic flavonoid derivatives as new potential tyrosinase inhibitors: a systematic review. RSC Adv. 2021;11(36):22159–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pillaiyar T, Manickam M, Namasivayam V. Skin whitening agents: Medicinal chemistry perspective of tyrosinase inhibitors. J Enzyme Inhib Med Chem. 2017;32(1):403–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sepehri N, et al. The natural-based optimization of kojic acid conjugated to different thio-quinazolinones as potential anti-melanogenesis agents with tyrosinase inhibitory activity. Bioorg Med Chem. 2021;36:116044. [DOI] [PubMed] [Google Scholar]
- 9.Hashemi A, et al. Synthesis and tyrosinase inhibitory activities of novel isopropylquinazolinones. BMC Chem. 2023;17(1):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iraji A, et al. Design, synthesis, spectroscopic characterization, in vitro tyrosinase inhibition, antioxidant evaluation, in silico and kinetic studies of substituted indole-carbohydrazides. Bioorg Chem. 2022;129:106140. [DOI] [PubMed] [Google Scholar]
- 11.Ullah S, et al. Tyrosinase inhibitors: a patent review (2011–2015). Expert Opin Ther Pat. 2016;26(3):347–62. [DOI] [PubMed] [Google Scholar]
- 12.Ghasemi N, et al. Thiazolopyrimidine derivatives as novel class of small molecule tyrosinase inhibitor. BMC Chem. 2023;17(1):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Iraji A, et al. Synthesis, biological evaluation and molecular docking analysis of vaniline–benzylidenehydrazine hybrids as potent tyrosinase inhibitors. BMC Chem. 2020;14:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saeedi M, Eslamifar M, Khezri K. Kojic acid applications in cosmetic and pharmaceutical preparations. Biomed Pharmacother. 2019;110:582–93. [DOI] [PubMed] [Google Scholar]
- 15.Rezapour Niri D et al. Design, synthesis, in vitro, and in silico evaluations of kojic acid derivatives linked to amino pyridine moiety as potent tyrosinase inhibitors. Heliyon. 2023;9(11). [DOI] [PMC free article] [PubMed]
- 16.Kang SS, et al. Synthesis of tyrosinase inhibitory (4-oxo-4H-pyran-2-yl) acrylic acid ester derivatives. Bioorg Med Chem Lett. 2009;19(1):188–91. [DOI] [PubMed] [Google Scholar]
- 17.Debbabi M, et al. Design and synthesis of novel potent anticoagulant and anti-tyrosinase pyranopyrimidines and pyranotriazolopyrimidines: insights from molecular docking and SAR analysis. Bioorg Chem. 2019;82:129–38. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, et al. Inhibition of tyrosinase activity by polyphenol compounds from Flemingia philippinensis roots. Bioorg Med Chem. 2014;22(3):1115–20. [DOI] [PubMed] [Google Scholar]
- 19.Najafi Z, et al. Design, synthesis, and molecular dynamics simulation studies of some novel kojic acid fused 2-amino-3-cyano-4H-pyran derivatives as tyrosinase inhibitors. BMC Chem. 2024;18(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Babaee S, et al. Design, synthesis, and Molecular Docking of some Novel Tacrine based cyclopentapyranopyridine-and tetrahydropyranoquinoline‐Kojic acid derivatives as anti‐acetylcholinesterase agents. Chem Biodivers. 2021;18(6):e2000924. [DOI] [PubMed] [Google Scholar]
- 21.Sadafi Kohnehshahri M et al. Novel tacrine-based acetylcholinesterase inhibitors as potential agents for the treatment of Alzheimer’s disease: quinolotacrine hybrids. Mol Divers. 2022;26:1–15. [DOI] [PubMed]
- 22.Ebadi A, et al. Novel Xanthene-1, 8‐dione derivatives containing the Benzylic Ether tail as potent cytotoxic agents: design, synthesis, in Vitro, and in Silico studies. J Chem. 2024;2024(1):6612503. [Google Scholar]
- 23.Najafi Z, et al. Synthesis and molecular modeling of new 2-benzylidenethiobarbituric acid derivatives as potent tyrosinase inhibitors agents. J Chin Chem Soc. 2022;69(4):692–702. [Google Scholar]
- 24.Najafi Z, et al. Design, synthesis, in vitro, and in silico studies of novel benzylidene 6-methoxy-1-tetralone linked to benzyloxy and benzyl-1, 2, 3-triazole rings as potential tyrosinase inhibitors. J Mol Struct. 2023;1271:134018. [Google Scholar]
- 25.Ismaya WT, et al. Crystal structure of Agaricus Bisporus mushroom tyrosinase: identity of the tetramer subunits and interaction with tropolone. Biochem. 2011;50(24):5477–86. [DOI] [PubMed] [Google Scholar]
- 26.Lipinski CA, et al. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 1997;23(1–3):3–25. [DOI] [PubMed] [Google Scholar]
- 27.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7(1):42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pires DE, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58(9):4066–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banerjee P et al. ProTox 3.0: a webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2024;52:gkae303. [DOI] [PMC free article] [PubMed]
- 30.Sidhom PA, et al. Mechanistic insight of synthesized 1,4-Dihydropyridines as an Antidiabetic Sword against reactive oxygen species. J Med Chem. 2023;66(1):991–1010. [DOI] [PubMed] [Google Scholar]
- 31.Sidhom PA, et al. Revisiting ageless antiques; synthesis, biological evaluation, docking simulation and mechanistic insights of 1, 4-Dihydropyridines as anticancer agents. Bioorg Chem. 2021;114:105054. [DOI] [PubMed] [Google Scholar]
- 32.Jasial S, Hu Y, Bajorath Jr. How frequently are pan-assay interference compounds active? Large-scale analysis of screening data reveals diverse activity profiles, low global hit frequency, and many consistently inactive compounds. J Med Chem. 2017;60(9):3879–86. [DOI] [PubMed] [Google Scholar]
- 33.Sidhom PA, et al. Mechanistic insight of synthesized 1, 4-dihydropyridines as an antidiabetic sword against reactive oxygen species. J Med Chem. 2022;66(1):991–1010. [DOI] [PubMed] [Google Scholar]
- 34.Ebadi A, et al. Design, synthesis, molecular modeling and DNA-binding studies of new barbituric acid derivatives. J Iran Chem Soc. 2022;19(9):3887–98. [Google Scholar]
- 35.Iraji A, et al. Synthesis, biological evaluation and molecular docking analysis of vaniline-benzylidenehydrazine hybrids as potent tyrosinase inhibitors. BMC Chem. 2020;14(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hosseinpoor H, et al. A series of Benzylidenes Linked to Hydrazine-1-carbothioamide as tyrosinase inhibitors: synthesis, Biological evaluation and structure-activity relationship. Chem Biodivers. 2020;17(8):e2000285. [DOI] [PubMed] [Google Scholar]
- 37.Hussein HY, et al. Novel pyrazoline-thiazole hybrids containing azo group as antibacterial agents: design, synthesis, in vitro bioactivity, in silico molecular docking, ADME profile and DFT studies. Res Chem Intermed. 2024;50(9):4551–78. [Google Scholar]
- 38.Bogoyavlenskiy A, et al. Naturally occurring isorhamnetin glycosides as potential agents against Influenza viruses: antiviral and molecular Docking studies. ACS Omega. 2023;8(50):48499–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neese F. The ORCA program system. Wiley Interdiscip Rev Comput Mol Sci. 2012;2(1):73–8. [Google Scholar]
- 40.Morris GM, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chinnasamy S, et al. Molecular docking and molecular dynamics simulation studies to identify potent AURKA inhibitors: assessing the performance of density functional theory, MM-GBSA and mass action kinetics calculations. J Biomol Struct Dyn. 2020;38(14):4325–35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No datasets were generated or analysed during the current study.