

Abstract
The E7 oncoprotein of human papillomavirus type 16 promotes cell proliferation in the presence of antiproliferative signals. Mutagenesis of E7 has revealed that this activity requires three regions, conserved regions 1 and 2 and a C-terminal zinc finger. Binding to the retinoblastoma tumor repressor (Rb) through an LxCxE motif in conserved region 2 is necessary, but not sufficient, for E7 to induce proliferation. We tested the hypothesis that binding to Rb is not sufficient because conserved region 1 and/or the C terminus are required for E7 to functionally inactivate Rb and thus induce proliferation. One mechanism proposed for how E7 inactivates Rb is by blocking Rb-E2F binding. Either conserved region 1 or the C terminus was necessary, in combination with the LxCxE motif, for E7 to block Rb-E2F binding in vitro. While all full-length E7 proteins with mutations outside of the LxCxE motif inhibited Rb-E2F binding, some failed to abrogate cell cycle arrest, demonstrating that blocking Rb-E2F binding is not sufficient for abrogating antiproliferative signals. Another mechanism proposed for how E7 inactivates Rb is by promoting the destabilization of Rb protein. Mutations in conserved region 1 or the LxCxE motif prevented E7 from reducing the half-life of Rb. Though no specific C-terminal residues of E7 were essential for destabilizing Rb, a novel class of mutations that uncouple the destabilization of Rb from the deregulation of keratinocyte proliferation was discovered. Destabilization of Rb correlated with the abrogation of Rb-induced quiescence but was not sufficient for overriding DNA damage-induced cell cycle arrest or for increasing keratinocyte life span. Finally, the same regions of E7 required for destabilizing Rb were required for reducing p107 and p130 levels. Together, these results suggest that inactivation of all three Rb family members is not sufficient to deregulate keratinocyte cell cycle control.
Human papillomavirus (HPV) infects the basal cells of stratified squamous epithelia and replicates in the suprabasal layers. HPV lacks its own replication enzymes and must therefore induce S-phase entry of the normally quiescent suprabasal cells for access to the host replication machinery (71). The ability of HPV to induce S phase in suprabasal layers likely contributes to the pathogenic manifestations of the virus, which range from warts to invasive carcinoma (71). Most, if not all, cervical carcinomas are associated with the so-called high-risk HPVs such as HPV type 16 (HPV16) (71). The E7 oncoprotein of HPV16 is sufficient to induce S-phase entry of suprabasal epithelial cells (5). There are three regions of E7 which are required for abrogating suprabasal quiescence and DNA damage- or transforming growth factor-β-induced cell cycle arrest in epithelial cells (14). These regions also function in rodent cell transformation (3, 19, 55). Two of the regions, termed conserved regions 1 and 2 (CR1 and CR2), share homology with tumor virus oncoproteins such as adenovirus E1A (56). The third region is a C-terminal zinc finger that functions in E7 dimerization (49).
An LxCxE motif within E7 CR2 is required for abrogation of the antiproliferative signals discussed above (14) and for transformation (19, 55). The LxCxE motif is also required for high-affinity binding to the retinoblastoma tumor suppressor (Rb) (3, 52) and to the Rb family members p107 and 130 (18). Rb negatively regulates the transition from G1 to S phase by repressing genes responsive to the E2F family of transcription factors (17). E2F-responsive genes such as those encoding cyclin E, cyclin A, proliferating cell nuclear antigen, and E2F1 itself are limiting for S-phase entry and progression (17). Rb binds E2F and inhibits transcription by blocking the E2F transcriptional activation domain (32) and by recruitment of factors such as histone deacetylase 1 (HDAC1) (7, 45, 46) and human SWI-SNF (16, 70). In cycling cells, Rb is inactivated in mid to late G1 through phosphorylation by several cyclin–cyclin-dependent kinase (CDK) complexes (17). Hyperphosphorylated Rb no longer binds E2F; consequently, repression of E2F-dependent promoters is relieved (17). E7 binds and inactivates the G0/G1-specific, hypophosphorylated form of Rb (24, 38).
There are two mechanisms proposed for the inactivation of Rb by E7 that are not mutually exclusive. One is that E7 physically interferes with Rb-E2F binding (referred to herein as E2F competition). Several pieces of evidence support this mechanism. First, complexes of Rb and E2F are lacking in cells derived from HPV-positive tumors and in cells engineered to express E7 (12, 53). Second, E7 blocks the association of E2F and Rb in vitro (12, 67), though the main E7 and E2F binding sites on Rb differ (43, 67). Third, the LxCxE motif of E7 is important for interference with Rb-E2F binding (12), which correlates with the importance of the LxCxE motif in overriding antiproliferative signals. The role of E7 sequences outside of the LxCxE motif in blocking the association of Rb and E2F is not well defined. Adenovirus CR1 is important for both deregulating cell growth (44) and blocking Rb-E2F binding (21, 37), but a requirement for E7 CR1 in blocking Rb-E2F binding has not been found (36, 67). Sequences within the C terminus of E7 have been reported to interfere with Rb-E2F binding (36, 48, 54), though the specific C-terminal amino acids involved have not been delineated.
The second mechanism proposed for the inactivation of Rb by E7 is the targeted destabilization of Rb (referred to herein as Rb destabilization). Expression of E7 reduces Rb levels in a number of cell types (4, 6, 15, 40), and this reduction occurs posttranslationally (4, 6, 40). Furthermore, both the LxCxE motif and CR1 have been shown to be necessary for efficient reduction of Rb steady-state levels (4, 40, 41), while the role of E7 C-terminal residues has not been explored. The correlation between bypass of cell cycle arrest and destabilization of Rb by E7 may indicate that the reduction in Rb levels contributes to the abrogation of antiproliferative signals. Alternatively, the correlation may indicate that Rb destabilization is a secondary effect of E7 expression; that is, the alteration of cell cycle control by E7 may contribute to a reduction in Rb. Berezutskaya et al. have shown that the reduction in Rb occurs shortly after E7 expression, suggesting that Rb destabilization may be a primary effect of E7 rather than a by-product of E7 expression (4).
The present study was aimed at examining the role of E7 sequences outside of the Rb binding motif in both E2F competition and Rb destabilization. In turn, the role of E2F competition and Rb destabilization in the abrogation of cell cycle arrest was evaluated. A mutation in E7 CR1 that destroyed Rb destabilization and left E2F competition intact demonstrated that E2F competition by E7 is not sufficient to overcome Rb-induced quiescence of SAOS2 cells. However, E7 proteins that could destabilize Rb could overcome Rb-induced quiescence. No specific C-terminal residues of E7 were essential for E2F competition or Rb destabilization. However, several mutations demonstrated that Rb destabilization by E7 was not sufficient to overcome DNA damage-induced cell cycle arrest of keratinocytes or to extend keratinocyte life span. These same mutations provided evidence that inactivation of p107 and p130 by E7 may also be insufficient to overcome keratinocyte cell cycle controls. This represents a novel category of E7 mutations and is the first report to uncouple Rb destabilization and the abrogation of cell cycle arrest.
MATERIALS AND METHODS
Expression constructs.
All constructs were sequenced prior to use. The E7 mutations E46A, H51A, R66A, and R77A were generated using a Clontech Transformer site-directed mutagenesis kit according to the manufacturer's instructions. The template, pLXSN(16E7), was described previously (26). The E7 mutations DST62-64AAA, LRL65-67AAA, CVQ68-70AAA, and THV72-74AAA were generated by using a Morph site-specific plasmid DNA mutagenesis kit (5-Prime 3-Prime, Inc.). pBS(−)16E7 (14) was the template. Oligonucleotides for introducing mutations into E7 were as follows: E46A, 5′ TGGACAAGCAGCCCCGGACAGAGCC 3′; H51A, 5′ CGGACAGAGCCGCTTACAATATTGTAA 3′; R66A, 5′ GACTCTACGCTTGCGTTGTGCGTAC 3′; R77A 5′ CACGTAGACATTGCTACTTTGGAAGAC 3′; DST62-64AAA, 5′GCAAGTGTGCTGCAGCTCTTCGGTTGTGCGTACAAATGACTCACGTA GAC 3′; LRL65-67AAA, 5′ GACTCTACGGCTGCAGCTTGCGTACAAAGTACTCACGTAGAC 3′; CVQ68-70AAA, 5′ CTTCGGTTGGCTGCTGCAAGTACTCACGTAGAC 3′; and THV72-74AAA, 5′ GTACAAAGCGCAGCTGCAGACATTCGAACTTTGGAAG 3′. LRL65-67AAA, CVQ68-70AAA, and THV72-74AAA contain unique restriction sites introduced by silent mutation. H2P was from K. Vousden (19); Δ6–10 and Δ21–24 were from K. Munger (55); Δ65–67, Δ75–77 and Δ79–83 were from L. Banks (47). An HDAC1 cDNA was obtained from S. Schrieber (29). pGST-Rb(379–928) and pET-p27 were from J. M. Roberts. Rb(379–928) is the large pocket domain of Rb and retains Rb functions important for regulating the G1-to-S phase transition (20, 33, 57). pCMV-E2F-1, and pGEX-E2F-2, -3, and -4 were from J. R. Nevins.
For generation of His fusion proteins, forward and reverse primers containing BamHI sites were used to PCR the insert for cloning into the BamHI site of pET14B, in frame with an N-terminal tag (Novagen). For in vitro translations, Rb was cut out of pGST with BamHI and ligated into the BamHI site of pBS(+) (Stratagene). HDAC1 was cut out of pBJ5 with BamHI and ligated into pBS(+). For production of GST-E2F1, E2F1 was excised from pCMV with BamHI and ligated into the BamHI site of pGEX-2T (Pharmacia) in frame with an N-terminal glutathione S-transferase (GST) tag. pLXSN was provided by A. D. Miller (50). Cloning of E7 WT, (wild type) H2P, and Δ21–24 into pLXSN has been described previously (14). Forward primers containing an EcoRI site and reverse primers containing a BamHI site were used for PCR amplification of DST62-64AAA, LRL65-67AAA, CVQ68-70AAA, THV72-74AAA, Δ75–77, and Δ79–83. Following restriction digestion, the PCR products were ligated into BamHI/EcoRI-cut pLXSN.
Recombinant protein production.
His- and GST-tagged proteins were bacterially expressed and purified on Ni-nitrilotriacetic acid resin (Qiagen) or glutathione-Sepharose 4B (Amersham-Pharmacia) according to the manufacturer's instructions. Proteins were dialyzed in binding buffer (see “In vitro binding assays”). Protein concentrations were determined by the Bradford assay (Bio-Rad) and by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie blue staining and comparison with bovine serum albumin standards. 35S-labeled in vitro translated (35S-IVT-Rb) Rb was generated by in vitro translation using the TNT coupled reticulocyte lysate system (Promega) according to the manufacturer's instructions.
In vitro binding assays.
To assess the ability of E7 proteins to block Rb-E2F binding, approximately 1 μg of GST or GST-E2F was immobilized on Glutathione-sepharose beads in 50 mM Tris-Cl (pH 7.8)–70 mM NaCl–10% glycerol–0.2% NP-40 plus protease inhibitor cocktail (Roche); 2 μl of 35S-labeled Rb and approximately 4 μg, 200 ng, 10 ng, 300 pg, or 25 pg of His-tagged E7 or control proteins was added, and the reactions were mixed for 90 min at room temperature. The beads were washed several times, and the amount Rb remaining associated was measured via SDS-PAGE and phosphorimaging. Band intensity was quantified with ImageQuant (Molecular Dynamics).
To assess Rb binding by His-tagged E7 or control proteins, approximately 1 μg of His-tagged protein was immobilized on Ni-NTA-agarose resin in His pull-down buffer (50 mM Tris-Cl [pH 7.8], 100 mM NaCl, 10 mM imidazole, 10% glycerol, 1% NP-40, protease inhibitor cocktail); 3 μl of 35S-labeled Rb was added, and the reactions were mixed for 60 min at room temperature. The resin was washed three times in His pull-down buffer and boiled in sample buffer. Rb was resolved by SDS-PAGE. Gels were dried and exposed overnight to Kodak BioMax MR film. The relative amount of Rb bound was estimated by densitometry.
To assess the LxCxE dependence of HDAC1 binding to Rb, GST or GST-Rb was immobilized on glutathione-Sepharose beads in His pull-down buffer minus imidazole. 35S-labeled in vitro-translated HDAC1 and His-p27, His-E7 WT, or His-E7 15–35 were added, and the reactions were mixed for 60 min at room temperature. HDAC1 was resolved by SDS-PAGE. The gel was dried and exposed overnight to Kodak BioMax MR film.
Cells.
Primary human keratinocytes were cultured from neonatal foreskins as previously described (25) and maintained in Keratinocyte-SFM supplemented with bovine pituitary extract and epidermal growth factor (Gibco BRL). Keratinocytes stably expressing E7 proteins were generated by transduction with LXSN retroviruses as previously described (25). Populations were selected in G418 (Sigma) until the mock-transduced cells were dead. SAOS2 human osteosarcoma cells were maintained in Dulbecco modified Eagle medium (Gibco BRL) supplemented with 10% calf serum.
Immunoprecipitation and Western blotting.
For Rb, p107, p130, and actin Western blots, keratinocytes were trypsinized, washed two times in cold phosphate-buffered saline, and lysed in 250 mM NaCl–50 mM Tris-Cl (pH 8)–1% NP-40–0.5 mM sodium orthovanadate–80 μM β-glycerophosphate–50 mM NaF plus protease inhibitor cocktail. Lysate concentration was determined by the Bradford assay (Bio-Rad). For Rb blots, 20 μg of extract was loaded per lane on 6% acrylamide gels. For p107 and p130 blots, 100 μg of extract was loaded per lane on 6% acrylamide gels. Following SDS-PAGE, proteins were transferred to polyvinylidene difluoride membranes (Millipore) and probed with either mouse anti-Rb clone G3-245 (PharMingen), rabbit anti-p107 (C-18; Santa Cruz Biotechnology), or goat anti-p130 (C-20; Santa Cruz). The antibodies used for loading controls were goat anti-actin (I-19; Santa Cruz), rabbit anti-β-tubulin (H-235; Santa Cruz), and mouse antinucleolin clone MS-3 (Santa Cruz). The secondary reagents were goat anti-mouse immunoglobulin G1-horseradish peroxidase (HRP) (Jackson ImmunoResearch Labs, Inc.), donkey anti-goat-HRP (Santa Cruz), and protein A-HRP (Roche). Proteins were visualized by enhanced chemiluminescence (Lumilight, Roche) and exposure to Kodak X-Omat Blue film.
For E7 immunoprecipitation and Western blotting, keratinocyte and SAOS2 extracts were prepared in lysis buffer as described above. Two hundred micrograms of extract was subjected to immunoprecipitation with rabbit anti-16E7 (antibody 16E7NP1 [22]). Immunoconjugates were collected on protein A-agarose (Roche), washed with lysis buffer, and resolved on 15% acrylamide gels. Proteins were transferred to polyvinylidene difluoride and probed with mouse anti-E7 clone 8C9 (Zymed), which recognizes the extreme N terminus of E7. Anti-mouse immunoglobulin G1-HRP was used as the secondary reagent. Proteins were visualized by enhanced chemiluminescence followed by exposure of blots to Kodak X-Omat Blue film.
Pulse-chase analysis.
Keratinocytes were metabolically labeled with Express Label [35S]cysteine-[35S]methionine (DuPont NEN) for 2 h. Cells were chased for various periods of time with Keratinocyte-SFM, washed two times in cold PBS, and lysed on ice for 30 min in lysis buffer (see above). Following a brief sonication, the insoluble material was pelleted and the supernatant was stored at −80°C until completion of the time course. Lysate concentration was determined by the Bradford assay, and labeling efficiency was assessed by trichloroacetic acid precipitation. Lysates were precleared with rabbit anti-L1 (HPV capsid protein) and protein A-agarose. Rb was immunoprecipitated with rabbit anti-Rb (M153; Santa Cruz), and the immunoconjugates were collected on protein A-agarose beads. Beads were washed one time in lysis buffer with 500 mM NaCl, four times in lysis buffer, and one time in 10 mM Tris-Cl–EDTA (pH 7.5). The beads were boiled in sample buffer and loaded onto a 6% acrylamide gel. Following SDS-PAGE, the gels were dried and subjected to phosphorimaging (Molecular Dynamics). Band intensity was measured using ImageQuant.
Flat cell assay.
Flat cell assays were carried out with SAOS2 cells as described by Hinds et al. (34), with some modifications. Cells were counted, plated in 60-mm-diameter dishes, and transfected using Fugene 6 transfection reagent (Roche) with pLXSN alone, 4 μg of pLXSN(Rb), or 4 μg of pLXSN(Rb) plus 4 μg of pLXSN encoding wild-type or mutated E7. One microgram of pCMV(β-gal) was included in all transfections, and the total amount of DNA was equalized with pLXSN. Cells were selected in G418 for 10 to 12 days, and the number of flat cells per 400 to 500 cells total was determined.
Keratinocyte life span extension.
Keratinocytes were mock transduced or transduced with empty LXSN retroviruses or LXSN retroviruses encoding E7 proteins. Cells were selected in G418 until the mock-transduced cells were dead. Cells were then passaged at 1:4, in the presence of G418, until reaching senescence.
DNA damage assays.
Keratinocytes were treated with 0.1 μM adriamycin (ADR; Sigma) or with medium alone for 28 h or irradiated with 16 Gy of ionizing radiation and incubated for 24 h. Cells were pulsed for the last 2 h of incubation with 20 μM bromodeoxyuridine (BrdU; Sigma) and fixed. Following RNase treatment, cells were labeled with anti-BrdU-fluorescein isothiocyanate (Becton Dickinson) to measure DNA synthesis and propidium iodide (Sigma) to determine cell cycle distribution. Two-parameter fluorescence-activated cell sorting (FACS) analysis was carried out with Becton Dickinson FACSCalibur instrumentation and CellQuest software (Becton Dickinson).
RESULTS
Expression of E7 proteins with C-terminal mutations.
The contribution of the E7 C terminus to Rb inactivation has not been extensively studied because mutations in this region, particularly within the zinc-binding motifs C58-X-X-C91 and C91-X-X-C94, often destroy protein stability (55, 65, 66). Amino acid substitution mutations were introduced into the C terminus in an attempt to generate stably expressed, mutated E7 proteins. Several small deletion mutations from Massimi et al. (47) were also included in the study. Figure 1A illustrates the locations of the C-terminal mutations and of representative CR1 and LxCxE mutations. Also shown are E7 truncations generated for use in vitro binding assays. All of the proteins were expressed in bacteria (Fig. 1B). Stable expression of the E7 proteins in human keratinocytes was observed (Fig. 1C), though some C-terminal mutations resulted in reduced expression (Fig. 1C). The CR1 mutation H2P is known to be expressed comparably to E7 WT (2, 14), though expression appears lower here because the E7 antibody used has reduced affinity for H2P (see Materials and Methods).
FIG. 1.

Expression of E7 proteins. (A) Locations of E7 mutations. CR1 and 2 and the C-terminal zinc finger are indicated on E7 WT. Substitution mutations are referred to by the residue changed, the position and the new residue (e.g., His 51 to Ala is H51A). Deletions are indicated by a “Δ” followed by the residues deleted. Truncated E7 proteins are named for the sequences remaining. Substitution mutations in the C terminus and truncations of N- and C-terminal sequences were generated as described in Materials and Methods. The C-terminal deletion mutations were provided by Massimi et al. (47). Representative CR1 and CR2 mutations were included in the study. (B) Recombinant E7 proteins for in vitro studies. Expression of bacterially produced His-tagged E7 proteins was confirmed by SDS-PAGE and Coomassie blue staining. (C) Expression of E7 proteins in keratinocytes. Expression of E7 proteins with small substitution or deletion mutations was confirmed in cells by immunoprecipitation and Western blotting with anti-E7 antibodies. Postimmunoprecipitation lysates were immunoblotted with an actin antibody as a control for lysate input.
Small alterations in either CR1 or the C terminus of E7 do not prevent E7 from blocking Rb-E2F binding.
A number of groups have demonstrated that E7, like adenovirus E1A, interferes with Rb-E2F binding (12, 36, 67). However, the importance of E2F competition in the functional inactivation of Rb by E7 is not clear. E2F competition assays were carried out to clarify the role for specific sequences within CR1 and the C terminus of E7 in blocking Rb-E2F binding. An in vitro assay using recombinant and in vitro-translated proteins was chosen for several reasons. First, input was easily controlled and output was based on the intensity of one band, thus simplifying interpretation. Second, a similar assay was shown to be more sensitive than gel shifts for detecting the ability of E7 to interfere with Rb-E2F binding (48). We generated E7 WT and mutants as His fusion proteins, and tested the ability of E7 and control proteins to bind Rb in vitro (Fig. 2A). All of the recombinant proteins bound efficiently to Rb except for the negative controls p27 and E7 Δ21–24. p27, a CDK inhibitor, was a control for nonspecific binding. Δ21–24 lacks much of the LxCxE motif required for high-affinity binding to Rb.
FIG. 2.

E7 competes with E2F for binding to Rb. (A) Autoradiographs showing binding of recombinant His-tagged proteins to 35S-IVT-Rb. Rb binding was measured by densitometry and expressed relative to the amount bound by E7 WT. (B) E2F competition assay showing requirement for E7 LxCxE motif in blocking Rb-E2F binding. A representative experiment is shown at the top. GST (lane 1) or GST-E2F1 (lanes 2 to 13) was mixed with 35S-IVT-Rb in the absence of competitor (n.c.; lane 2), in the presence of His-p27 (lane 3), or in the presence of decreasing amounts of His-E7 WT (lanes 4 to 8) or His-E7 Δ21–24 (lanes 9 to 13). The amount of Rb bound was measured, and a summary of three experiments was graphed (bottom). Error bars represent standard errors of the means. Lanes 5 and 10 represent reactions where the molar ratio of E2F to E7 WT and Δ21–24 respectively, was 1:1. E2F competition assays with HDAC1 (C) and E7 CR1 mutations (D) were carried out as described above.
E2F competition experiments were carried out with His-tagged E7 or control proteins, GST–E2F-1 and 35S-IVT-Rb. The amount of Rb bound to E2F was measured by phosphorimaging and plotted in bar graphs (Fig. 2B to D). The activity of His-tagged competitors in blocking Rb-E2F binding was determined relative to that of E7 WT as described for Fig. 2. E7 WT blocked Rb-E2F binding even at concentrations 20-fold lower than the E2F input (Fig. 2B to D, lanes 6). In contrast p27 did not interfere with Rb-E2F binding, even at concentrations 20-fold greater than E2F (Fig. 2B-D, lanes 3). Similar results were obtained when 35S-labeled E2F-1 and GST-Rb were used and with 35S-labeled Rb and GST-tagged E2F-2, E2F-3, or E2F-4 (data not shown). As previously shown (48, 67), E7 Δ21–24, which did not bind Rb, did not block Rb-E2F binding at any concentration (Fig. 2B, lanes 9 to 13). Not all proteins that bind Rb through LxCxE-like motifs block the association of E2F. HDAC1 was used as a control that binds Rb but can exist in complex with Rb and E2F (7, 45, 46). HDAC1 contains an IxCxE motif and interacts with Rb in manner that is blocked by LxCxE-containing proteins (references 7 and 46 and data not shown). HDAC1 failed to significantly inhibit Rb-E2F binding (Fig. 2C, lanes 9 to 13), with about 7% of the activity of E7 WT. The activity in blocking Rb-E2F binding was determined by comparing the amount of Rb bound in the presence of HDAC1 to the amount bound in the presence of E7 WT in reactions where the molar ratio of competitor to E2F was 1:1 (Fig. 2C, lanes 5 and 10).
The E7 proteins H2P and Δ6–10, which do not induce epithelial cell proliferation (14), blocked Rb-E2F binding (Fig. 2D, lanes 9 to 13 and 14 to 18) with 100 and 95% of the activity of E7 WT. The E7 proteins E46A, H51A, DST62-64AAA, R66A, LRL65-67AAA, CVQ68-70AAA, THV72-74AAA, R77A, Δ75–77, and Δ79–83 all had wild-type activity in blocking Rb-E2F binding (data not shown).
Both E7 CR1 and the C-terminus function in E2F competition.
That small CR1 and C-terminal mutations did not affect the ability of E7 to block Rb-E2F binding suggested that multiple regions of E7 in addition to the LxCxE motif may participate in this activity. This possibility was addressed using truncated E7 proteins generated such that the LxCxE motif was left intact (Fig. 1A). Experiments similar to those described in Fig. 2B to D were carried out with the truncated proteins and are summarized in Table 1. E7 1–40, which retains all of CR1 and CR2 and some sequence C terminal to CR2 (Fig. 1A), blocked Rb-E2F binding at 42% of E7 WT activity (Table 1). Untagged recombinant E7 1–40 was generated to verify that the tag was not contributing to the activity of this protein. His-tagged and untagged E7 1–40 had the same activity in blocking Rb-E2F binding (data not shown). To test whether an even smaller N-terminal portion of E7 would compete with E2F, E7 1–28 was generated. E7 1–28 terminates two residues C terminal to the LxCxE motif (Fig. 1A). Surprisingly, E7 1–28 blocked Rb-E2F binding, with 38% of E7 WT activity (Table 1), which is in contrast to results using comparable fragments of E7 in gel shift assays (36, 67). E7 19–98, which lacks all of CR1 but retains all of the C terminus and most of CR2 (Fig. 1A), was indistinguishable from E7 WT in blocking Rb-E2F (Table 1). Since E7 proteins retaining either the N- or C-terminal sequences blocked Rb-E2F association, E7 proteins lacking both N- and C-terminal sequences were generated to determine the minimal portion of E7 required for competition with E2F. E7 19–57 lacks all of CR1 and terminates immediately N terminal to the zinc finger, and E7 15–35 encompasses mainly CR2 (Fig. 1A). E7 19–57 and E7 15–35 were the least efficient of all of the truncations in blocking Rb-E2F (Table 1); their activities were was 15 and 4%, respectively, when at an approximate 1:1 molar ratio with E2F.
TABLE 1.
Activities of E7 fragments in inhibiting Rb-E2F binding
| Protein | % Inhibitiona |
|---|---|
| E7 1–40 | 42 |
| E7 1–28 | 38 |
| E7 19–98 | 100 |
| E7 19–57 | 15 |
| E7 15–35 | 4 |
Determined by comparing the amount of Rb bound to E2F in the presence of E7 fragments to the amount bound in the presence of E7 WT (taken as 100%). Reactions where the molar ratio of competitor to E2F was 1:1 were used for calculations.
C-terminal mutations do not affect the ability of E7 to destabilize Rb.
CR1 and the LxCxE motif of E7 have been shown to be important for the reduction of cellular Rb protein levels (4, 40, 41). However, the role of the E7 C terminus was not examined in these studies. Western blotting was used to assess Rb steady-state levels in keratinocytes stably expressing E7 proteins with C-terminal mutations. As previously reported, expression of E7 WT reduced Rb steady-state levels (4, 6, 15, 40), while mutations within the LxCxE motif or CR1 resulted in failure to reduce Rb levels (4, 40, 41) (Fig. 3A). Expression of E46A, H51A, DST62-64AAA, R66A, CVQ68-70AAA, THV72-74AAA, R77A, or Δ79–83 reduced Rb levels equivalently (Fig. 3B), despite some differences in expression level of the E7 proteins (Fig. 1C).
FIG. 3.

Rb destabilization by E7. (A and B) Western blots showing Rb (top) and actin (bottom) steady-state levels in E7-expressing keratinocytes. Rb-pp, phosphorylated Rb. (C) Pulse-chase analysis of Rb turnover. Cells were metabolically labeled followed by a 0-, 3-, 6-, or 12-h chase. Rb was immunoprecipitated and visualized by SDS-PAGE and phosphorimaging as described in Materials and Methods. A representative experiment is shown. (D) Rb half-life. Band intensity was used to determine Rb half-life. Three experiments were averaged. Error bars represent standard deviations.
Measurements of Rb half-life were also carried out (Fig. 3C and D). Several E7 C-terminal mutations and representative CR1 and LxCxE mutations were tested. Keratinocytes stably expressing vector alone, E7 WT, or mutated E7 proteins were counted, plated, and metabolically pulse-labeled the following day with [35S]cysteine-[35S]methionine. Following various lengths of chase with unlabeled media, cells were lysed and Rb levels were assessed by immunoprecipitation (Fig. 3C). A summary of three experiments is shown (Fig. 3D). Rb half-life has been reported to be greater than 12 h in other cell types (6, 40). Rb turnover in keratinocytes appeared to be somewhat faster, with a half-life of approximately 11 h. Rb half-life was approximately 10 h in cells expressing E7 with the CR1 mutation H2P or the LxCxE mutation Δ21–24. In keratinocytes expressing E7 WT, E46A, CVQ68-70AAA, or Δ79–83, Rb half-life was consistently reduced to 4 to 5 h. The faster turnover of hypophosphorylated Rb than of the hyperphosphorylated form (Fig. 3C) is consistent with previous reports (4, 6, 40).
The ability to overcome Rb-induced quiescence of SAOS2 cells correlates with Rb destabilization rather than E2F competition.
SAOS2 osteosarcoma cells lack functional Rb, and transfection with either full-length Rb or the large pocket domain of Rb results in the formation of large, flat, quiescent cells. The flat cell assay of Hinds et al. (34) was used to determine whether functional inactivation of Rb correlated with E2F competition or with Rb destabilization. Upon transfection of SAOS2 cells with pLXSN(Rb) and selection in G418, quiescent cells were readily apparent, based on their characteristic morphology (data not shown). Cotransfection experiments indicated that E7 WT, E46A, CVQ68-70AAA, and Δ79–83, all of which reduce Rb half-life, were equally efficient in overcoming flat cell formation by Rb (summarized in Fig. 4). H2P, which did not significantly reduce Rb half-life, failed to efficiently overcome Rb-induced quiescence despite the ability to block Rb-E2F binding. Expression of the E7 proteins was confirmed by immunoprecipitation and Western blotting, and expression levels did not correlate with the ability to overcome Rb-induced quiescence (data not shown).
FIG. 4.

Abrogation of Rb-induced quiescence by E7. Flat cell assays were carried out as described in Materials and Methods. The number of quiescent (flat) cells was determined relative to the number of total cells counted. A summary of three experiments is shown. Lane 1 represents cells transfected with empty vector. Lanes 2 to 7 represent cells transfected with Rb alone (lane 2) or Rb plus various E7 constructs (lanes 3 to 7). The number of flat cells was expressed relative to the number of flat cells following transfection with Rb (lane 2). Error bars represent standard deviations.
Mutations in the C terminus of E7 uncouple Rb destabilization from the extension of keratinocyte life span and from the abrogation of DNA damage-induced cell cycle arrest.
The Rb-p16ink4a pathway is important in senescence of human keratinocytes, and inactivation of this pathway is required for keratinocyte immortalization (42). Expression of E7 alone in primary human cells increases their life span (31, 51) and can lead to immortalization at low frequency (26). It was therefore of interest to determine whether Rb destabilization by E7 was sufficient for extending the life span of keratinocytes. Keratinocyte populations derived from the same foreskin and stably transduced with empty vector or with E7-expressing constructs were passaged until reaching senescence. Cells expressing vector, CVQ68-70AAA, or Δ79–83 ceased proliferating between population doubling levels (PDL) 10 and 14 (Fig. 5). In contrast, populations of cells expressing either E7 WT or E46A have continued to proliferate beyond PDL 26 (Fig. 5). H2P and Δ21–24 were no more efficient than empty vector in extending keratinocyte life span (data not shown).
FIG. 5.

Extension of keratinocyte life span. Keratinocytes expressing empty vector, E7 WT, or mutated E7 (four populations of each) were passaged until reaching senescence (detailed in Materials and Methods). PDL 0 refers to the point at which drug selection was complete. The PDL at which cells ceased to proliferate is marked by the horizontal lines.
Rb has a role in negatively regulating the G1-S transition in response to DNA damage (10, 15, 28, 63). Thus, it was of interest to determine whether the C-terminal mutations would affect the abrogation of DNA damage-induced cell cycle arrest by E7. Keratinocytes were treated with adriamycin (ADR) to induce DNA damage. Cell cycle distribution assessed by FACS analysis of DNA content and BrdU incorporation is shown in Table 2. Cells expressing vector, H2P, Δ21–24, CVQ68-70AAA, or Δ79–83 accumulated in G1 and G2 following ADR treatment; in contrast, ADR-treated cells expressing E7 WT or E46A retained had a significantly smaller G1 population (Table 2). Cells expressing E7 WT or E46A retained 65 to 70% of their S-phase population relative to untreated controls (Fig. 6). As previously shown (14, 64), mutations in CR1 or the LxCxE motif resulted in failure of E7 to induce S phase following DNA damage (Fig. 6). Interestingly, CVQ68-70AAA and Δ79–83 were severely deficient in inducing S phase following ADR treatment (Fig. 6), despite reduction of Rb levels and inactivation of Rb in the flat cell assay. H51A, R66A, DST62-64AAA, THV72-74AAA, and R77A had wild-type activity (data not shown). Results with an alternate form of DNA damage, induced by ionizing radiation, were similar to the ADR data (data not shown).
TABLE 2.
Keratinocyte cell cycle of distribution before and after ADR treatmenta
| Cells transfected with: | Distribution (%)
|
|||||
|---|---|---|---|---|---|---|
| G1
|
S
|
G2/M
|
||||
| −ADR | +ADR | −ADR | +ADR | −ADR | +ADR | |
| Vector | 61.4 ± 5.3 | 42.0 ± 6.7 | 17.5 ± 3.3 | 3.5 ± 1.2 | 22.0 ± 3.7 | 51.6 ± 7.9 |
| E7 WT | 58.1 ± 4.1 | 22.9 ± 3.4 | 15.5 ± 2.3 | 10.2 ± 1.4 | 23.5 ± 3.9 | 63.2 ± 7.9 |
| H2P | 64.8 ± 6.0 | 46.0 ± 1.8 | 19.2 ± 5.8 | 2.7 ± 0.1 | 20.9 ± 4.3 | 50.8 ± 1.5 |
| Δ21–24 | 61.2 ± 9.7 | 45.4 ± 3.8 | 18.6 ± 5.5 | 2.2 ± 0.3 | 23.6 ± 8.2 | 50.0 ± 2.4 |
| E46A | 63.7 ± 3.1 | 24.2 ± 5.7 | 15.5 ± 1.1 | 10.6 ± 1.3 | 23.5 ± 1.1 | 68.4 ± 4.6 |
| CVQ68-70AAA | 63.4 ± 3.0 | 42.6 ± 5.3 | 16.6 ± 1.3 | 4.3 ± 0.7 | 19.8 ± 1.3 | 50.7 ± 5.6 |
| Δ79–83 | 64.0 ± 4.6 | 48.3 ± 4.2 | 15.4 ± 3.8 | 3.5 ± 1.1 | 20.0 ± 3.2 | 44.3 ± 5.2 |
Keratinocytes were DNA damaged with ADR and analyzed by two-parameter FACS as described in Materials and Methods. Averages of four experiments ± standard deviation are shown.
FIG. 6.

Induction of S phase following DNA damage. The S-phase population of ADR-treated keratinocytes expressing vector or E7 proteins was normalized to that in untreated controls. Shown is a summary of four experiments (raw data are shown in Table 2). Error bars represent standard deviations.
Mutations in the C terminus of E7 uncouple reduction in p107 and p130 levels from the extension of keratinocyte life span and from the abrogation of DNA damage-induced cell cycle arrest.
p107 and p130 have recently been shown to function in murine cells in both p16-mediated cell cycle arrest (9) and the DNA damage-induced G1 checkpoint (59). E7 was reported to reduce cellular levels of p107 and p130 in the human colon carcinoma cell line RKO (40), though the importance of different E7 regions in this reduction was not addressed. The possibility that E7 CVQ68-70AAA and Δ79–83 do not overcome keratinocyte cell cycle controls because they do not destabilize p107 and/or p130 was addressed. Steady-state levels of p107 and p130 were assessed in keratinocytes stably expressing wild-type or mutated E7. Both p107 and p130 were reduced in E7-expressing keratinocytes, and the integrity of CR1 and the LxCxE motif was necessary for this reduction (Fig. 7A and C). E7 CVQ68-70AAA and Δ79–83 reduced p107 and p130 levels to a similar extent as E7 WT (Fig. 7B and D). All data obtained with the mutated E7 proteins, including those not shown, are summarized in Table 3.
FIG. 7.

Effect of E7 on p107 and p130 steady-state levels. Western blots showing p107 (A and B) and p130 (C and D) steady-state levels in E7-expressing keratinocytes. Blots were stripped and reprobed with antinucleolin (Santa Cruz) or anti-β-tubulin to verify even loading.
TABLE 3.
Summary of results with mutated E7 proteinsa
| Protein | Expression in keratinocytes | E2F competition | Destabilization of:
|
Overrides DNA damage checkpoints | Bypasses Rb-induced quiescenceb | Extends keratinocyte life span | ||
|---|---|---|---|---|---|---|---|---|
| Rb | p107 | p130 | ||||||
| E7 WT | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ |
| H2P | ++ | ++ | − | − | − | − | − | − |
| Δ6–10 | ++ | ++ | −c | NT | NT | −d | NT | NT |
| Δ21–24 | ++ | − | − | − | − | − | NT | − |
| E46A | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ |
| H51A | ++ | ++ | ++ | NT | NT | ++ | NT | NT |
| DST62-64AAA | ++ | ++ | ++ | NT | NT | ++ | NT | NT |
| LRL65-67AAA | + | ++ | NT | NT | NT | NT | NT | NT |
| R66A | ++ | ++ | ++ | NT | NT | ++ | NT | NT |
| CVQ68-70AAA | + | ++ | ++ | ++ | ++ | − | ++ | − |
| THV72-74AA | ++ | ++ | ++ | NT | NT | ++ | NT | NT |
| Δ75–77 | ++ | ++ | ++ | NT | NT | NT | NT | NT |
| R77A | + | ++ | ++ | NT | NT | ++ | NT | NT |
| Δ79–83 | + | ++ | ++ | ++ | ++ | − | ++ | − |
DISCUSSION
There are two mechanisms proposed for how E7 inactivates Rb: E2F competition and Rb destabilization. One objective of this study was to clarify the role of E7 CR1 and C-terminal sequences in each of these mechanisms. E7 CR1 participates in both E2F competition (this study) and Rb destabilization (4, 40, 41; this study). The E7 C terminus is involved in E2F competition (36, 48, 67; this study), though we did not identify any specific C-terminal amino acids essential for this activity. None of the E7 C-terminal mutations had an effect on Rb destabilization (summarized in Table 3), suggesting that the amino acids mutated do not have an essential role in destabilization. Results with a CR1 mutation, H2P, demonstrate that the functional inactivation of Rb correlates with Rb destabilization rather than with E2F competition (Table 3). E7 must bind Rb to destabilize it (4, 40, 41; this study); however, the results with H2P indicate that inactivation of Rb is not mediated solely by E7 binding. The second objective of this study was to determine whether the destabilization of Rb by E7 is sufficient to override keratinocyte cell cycle arrest. E7 proteins with CR1 or LxCxE mutations, such as H2P or Δ21–24, respectively, do not destabilize Rb in keratinocytes (40, 41; this study) and do not abrogate cell cycle arrest (14; this study). We report here that several C-terminal mutations uncouple Rb destabilization from the bypass of cell cycle arrest in keratinocytes. Using the E7 mutations CVQ68-70AAA and Δ79–83, we demonstrate that the destabilization of Rb is not sufficient for bypassing DNA damage-induced cell cycle arrest or for increasing the life span of normal human keratinocytes.
Rb functions in the G1 checkpoint following DNA damage (10, 15, 28, 63); however, it is not clear whether inactivation of Rb alone can result in loss of the checkpoint in human keratinocytes. E7 CVQ68-70AAA and Δ79–83 were severely deficient in inducing S phase in keratinocytes following DNA damage, despite the destabilization of Rb. This suggests that E7 has activities in addition to Rb inactivation that are necessary for complete loss of G1 control. E7 binds to and inactivates the Rb-related proteins p107 and 130 (13, 18). While Harrington et al. showed that Rb, but not p107 and p130, was essential for G1 control in DNA-damaged mouse embryo fibroblasts (28), others have provided evidence that p107 and/or p130 function in murine cell cycle control following DNA damage (59, 63). The mechanism by which E7 inactivates p107 and p130 is not clear and may differ according to cell type (1, 4, 40, 68). In agreement with Jones et al. (40), we find that E7 reduces p107 and p130 levels, and we extended these findings to show that the same regions of E7 are required for reducing Rb, p107, and p130 levels. The ability of E7 CVQ68-70AAA and Δ79–83 to reduce p107 and p130 levels suggests that destabilization of all three pocket proteins is not sufficient for E7 to overcome keratinocyte cell cycle control.
Several hypotheses could explain why E7 CVQ68-70AAA and Δ79–83 do not overcome antiproliferative signals in keratinocytes. Perhaps CVQ68-70AAA and/or Δ79–83 do not inactivate p21, a CDK inhibitor important for cell cycle arrest following DNA damage (62). E7 was shown to inactivate p21 (23, 39) through direct binding of the E7 C terminus to a region in the p21 C terminus (23). The possibility that E7 CVQ68-70AAA and Δ79–83 do not inactivate p21 is currently being addressed. However, preliminary results showed that both CVQ68-70AAA and Δ79–83 bound p21 as well as E7 WT (A. Helt, unpublished results). Alternatively, the failure of E7 CVQ68-70AAA or Δ79–83 to override cell cycle control in keratinocytes may be related to other factors. Δ79–83 does not transform rodent cells (47), and the region encompassed by this mutation has been implicated in binding to TATA-binding protein (47), Mi-2β (8), M2 pyruvate kinase (73), and acid α-glucosidase (72). The interaction of E7 with one or several of these factors, or as yet unidentified factors, may be important for deregulating keratinocyte cell cycle control.
Although results with CR1 mutations suggest that E2F competition is not sufficient for efficiently inactivating Rb or for overcoming cell cycle arrest in keratinocytes, there is evidence that competition may be sufficient for induction of some E2F-dependent genes. For instance, E7 H2P was reported to activate cyclin E and, to a lesser extent, cyclin A expression in rodent cell lines (60, 69). E7 is rapidly turned over (58, 61), and perhaps there is not enough E7 to completely inactivate the more stable Rb protein by a stoichiometric mechanism like E2F competition (40, 48). It is interesting that relatively small LxCxE-containing proteins like E7 and adenovirus E1A interfere with Rb-E2F binding, while larger cellular proteins containing LxCxE or LxCxE-related motifs, like HDAC1, do not block the interaction. The reason for this is unclear, though it is possible that CR1 and/or the C terminus of E7, and CR1 of E1A, make contact at or near the E2F binding site on Rb. Interestingly, phosphorylation of Rb by cyclin E-CDK2 was reported to alter the structure of Rb such that E2F no longer binds (27). Perhaps E7 binding induces a similar structural shift in Rb that disrupts E2F binding.
We suggest in Fig. 8 a model where E2F competition acts in concert with Rb destabilization. Perhaps Rb is stabilized by E2F, and E7 may first block Rb-E2F binding to target Rb for proteolysis. There is precedence for this in the observation that E2F is stabilized by binding to Rb (11, 30, 35). At this stage, the binding of other factors, such as HDAC, to Rb may be blocked as well (7, 45, 46). Following isolation of Rb, CR1 may recruit factors necessary to target Rb for destruction, though there are few data on CR1 binding partners. We suggest that p107 and p130 may be inactivated by a similar mechanism. Finally, E7 must activate (or inactivate) an additional pathway(s) to deregulate the G1/S transition.
FIG. 8.
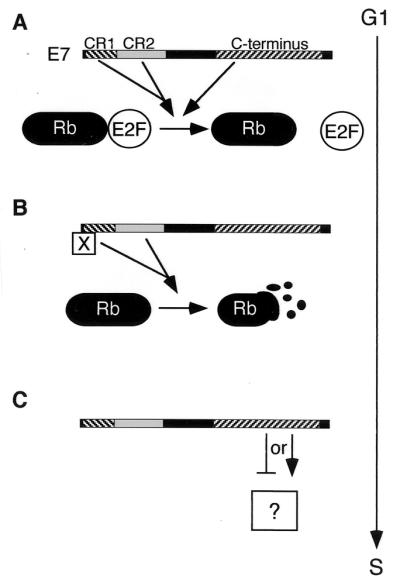
Model for induction of S phase by E7. (A) CR1, the LxCxE motif in CR2, and the C terminus of E7 block the interaction of Rb (and, possibly, p107 and p130) and E2F. (B) CR1 and LxCxE motif of E7 then target Rb, p107, and p130 for degradation. Possibly CR1 recruits factors (represented by “X”) necessary for targeting the pocket proteins. (C) The C terminus of E7 is involved in an additional activity (represented by the question mark) required for abrogating keratinocyte G1/S control.
ACKNOWLEDGMENTS
We thank Maxine Linial and Christopher Meiering for critical review of the manuscript and members of the Galloway lab, past and present, for helpful discussions. We are grateful for plasmids from Lawrence Banks, Karl Munger, Karen Vousden, James M. Roberts, Joseph R. Nevins, and Stuart L. Schreiber.
This work was supported by grant CA 64795 from the National Cancer Institute to D.A.G. A.-M.H. was supported by a fellowship from the UW/STD Predoctoral and Postdoctoral Training Program (NIAID grant AI0714).
REFERENCES
- 1.Arroyo M, Bagchi S, Raychaudhuri P. Association of the human papillomavirus type 16 E7 protein with the S-phase-specific E2F-cyclin A complex. Mol Cell Biol. 1993;13:6537–6546. doi: 10.1128/mcb.13.10.6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banks L, Edmonds C, Vousden K H. Ability of the HPV16 E7 protein to bind RB and induce DNA synthesis is not sufficient for efficient transforming activity in NIH3T3 cells. Oncogene. 1990;5:1383–1389. [PubMed] [Google Scholar]
- 3.Barbosa M S, Edmonds C, Fisher C, Schiller J T, Lowy D R, Vousden K H. The region of the HPV E7 oncoprotein homologous to adenovirus E1a and SV40 large T antigen contains separate domains for Rb binding and casein kinase II phosphorylation. EMBO J. 1990;9:153–160. doi: 10.1002/j.1460-2075.1990.tb08091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berezutskaya E, Yu B, Morozov A, Raychaudhuri P, Bagchi S. Differential regulation of the pocket domains of the retinoblastoma family proteins by the HPV16 E7 oncoprotein. Cell Growth Differ. 1997;8:1277–1286. [PubMed] [Google Scholar]
- 5.Blanton R A, Coltrera M D, Gown A M, Halbert C L, McDougall J K. Expression of the HPV16 E7 gene generates proliferation in stratified squamous cell cultures which is independent of endogenous p53 levels. Cell Growth Differ. 1992;3:791–802. [PubMed] [Google Scholar]
- 6.Boyer S N, Wazer D E, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56:4620–4624. [PubMed] [Google Scholar]
- 7.Brehm A, Miska E A, McCance D J, Reid J L, Bannister A J, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- 8.Brehm A, Nielsen S J, Miska E A, McCance D J, Reid J L, Bannister A J, Kouzarides T. The E7 oncoprotein associates with Mi2 and histone deacetylase activity to promote cell growth. EMBO J. 1999;18:2449–2458. doi: 10.1093/emboj/18.9.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bruce J L, Hurford R K, Classon M, Koh J, Dyson N. Requirements for cell cycle arrest by p16INK4a. Mol Cell. 2000;6:737–742. doi: 10.1016/s1097-2765(00)00072-1. [DOI] [PubMed] [Google Scholar]
- 10.Brugarolas J, Bronson R T, Jacks T. p21 is a critical CDK2 regulator essential for proliferation control in Rb-deficient cells. J Cell Biol. 1998;141:503–514. doi: 10.1083/jcb.141.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campanero M R, Flemington E K. Regulation of E2F through ubiquitin-proteasome-dependent degradation: stabilization by the pRB tumor suppressor protein. Proc Natl Acad Sci USA. 1997;94:2221–2226. doi: 10.1073/pnas.94.6.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chellappan S, Kraus V B, Kroger B, Munger K, Howley P M, Phelps W C, Nevins J R. Adenovirus E1A, simian virus 40 tumor antigen, and human papillomavirus E7 protein share the capacity to disrupt the interaction between transcription factor E2F and the retinoblastoma gene product. Proc Natl Acad Sci USA. 1992;89:4549–4553. doi: 10.1073/pnas.89.10.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies R, Hicks R, Crook T, Morris J, Vousden K. Human papillomavirus type 16 E7 associates with a histone H1 kinase and with p107 through sequences necessary for transformation. J Virol. 1993;67:2521–2528. doi: 10.1128/jvi.67.5.2521-2528.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Demers G W, Espling E, Harry J B, Etscheid B G, Galloway D A. Abrogation of growth arrest signals by human papillomavirus type 16 E7 is mediated by sequences required for transformation. J Virol. 1996;70:6862–6869. doi: 10.1128/jvi.70.10.6862-6869.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demers G W, Foster S A, Halbert C L, Galloway D A. Growth arrest by induction of p53 in DNA damaged keratinocytes is bypassed by human papillomavirus 16 E7. Proc Natl Acad Sci USA. 1994;91:4382–4386. doi: 10.1073/pnas.91.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dunaief J L, Strober B E, Guha S, Khavari P A, Alin K, Luban J, Begemann M, Crabtree G R, Goff S P. The retinoblastoma protein and BRG1 form a complex and cooperate to induce cell cycle arrest. Cell. 1994;79:119–130. doi: 10.1016/0092-8674(94)90405-7. [DOI] [PubMed] [Google Scholar]
- 17.Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998;12:2245–2262. doi: 10.1101/gad.12.15.2245. [DOI] [PubMed] [Google Scholar]
- 18.Dyson N, Guida P, Munger K, Harlow E. Homologous sequences in adenovirus E1A and human papillomavirus E7 proteins mediate interaction with the same set of cellular proteins. J Virol. 1992;66:6893–6902. doi: 10.1128/jvi.66.12.6893-6902.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edmonds C, Vousden K H. A point mutational analysis of human papillomavirus type 16 E7 protein. J Virol. 1989;63:2650–2656. doi: 10.1128/jvi.63.6.2650-2656.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edwards G M, Huber H E, DeFeo-Jones D, Vuocolo G, Goodhart P J, Maigetter R Z, Sanyal G, Oliff A, Heimbrook D C. Purification and characterization of a functionally homogeneous 60-kDa species of the retinoblastoma gene product J. Biol Chem. 1992;267:7971–7974. . (Erratum, 267:13780.) [PubMed] [Google Scholar]
- 21.Fattaey A R, Harlow E, Helin K. Independent regions of adenovirus E1A are required for binding to and dissociation of E2F-protein complexes. Mol Cell Biol. 1993;13:7267–7727. doi: 10.1128/mcb.13.12.7267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Firzlaff J M, Galloway D A, Eisenman R N, Luscher B. The E7 protein of human papillomavirus type 16 is phosphorylated by casein kinase II. New Biol. 1989;1:44–53. [PubMed] [Google Scholar]
- 23.Funk J O, Waga S, Harry J B, Espling E, Stillman B, Galloway D A. Inhibition of CDK activity and PCNA-dependent DNA replication by p21 is blocked by interaction with the HPV-16 E7 oncoprotein. Genes Dev. 1997;11:2090–2100. doi: 10.1101/gad.11.16.2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gage J R, Meyers C, Wettstein F O. The E7 proteins of the nononcogenic human papillomavirus type 6b (HPV-6b) and of the oncogenic HPV-16 differ in retinoblastoma protein binding and other properties. J Virol. 1990;64:723–730. doi: 10.1128/jvi.64.2.723-730.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halbert C L, Demers G W, Galloway D A. The E6 and E7 genes of human papillomavirus type 6 have weak immortalizing activity in human epithelial cells. J Virol. 1992;66:2125–2134. doi: 10.1128/jvi.66.4.2125-2134.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Halbert C L, Demers G W, Galloway D A. The E7 gene of human papillomavirus type 16 is sufficient for immortalization of human epithelial cells. J Virol. 1991;65:473–478. doi: 10.1128/jvi.65.1.473-478.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harbour J W, Luo R X, Dei Santi A, Postigo A A, Dean D C. Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell. 1999;98:859–869. doi: 10.1016/s0092-8674(00)81519-6. [DOI] [PubMed] [Google Scholar]
- 28.Harrington E A, Bruce J L, Harlow E, Dyson N. pRB plays an essential role in cell cycle arrest induced by DNA damage. Proc Natl Acad Sci USA. 1998;95:11945–11950. doi: 10.1073/pnas.95.20.11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hassig C A, Tong J K, Fleischer T C, Owa T, Grable P G, Ayer D E, Schreiber S L. A role for histone deacetylase activity in HDAC1-mediated transcriptional repression. Proc Natl Acad Sci USA. 1998;95:3519–3524. doi: 10.1073/pnas.95.7.3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hateboer G, Kerkhoven R M, Shvarts A, Bernards R, Beijersbergen R L. Degradation of E2F by the ubiquitin-proteasome pathway: regulation by retinoblastoma family proteins and adenovirus transforming proteins. Genes Dev. 1996;10:2960–2970. doi: 10.1101/gad.10.23.2960. [DOI] [PubMed] [Google Scholar]
- 31.Hawley-Nelson P, Vousden K H, Hubbert N L, Lowy D R, Schiller J T. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J. 1989;8:3905–3910. doi: 10.1002/j.1460-2075.1989.tb08570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Helin K, Harlow E, Fattaey A. Inhibition of E2F-1 transactivation by direct binding of the retinoblastoma protein. Mol Cell Biol. 1993;13:6501–6508. doi: 10.1128/mcb.13.10.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hiebert S W. Regions of the retinoblastoma gene product required for its interaction with the E2F transcription factor are necessary for E2 promoter repression and pRb-mediated growth suppression. Mol Cell Biol. 1993;13:3384–3391. doi: 10.1128/mcb.13.6.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hinds P W, Mittnacht S, Dulic V, Arnold A, Reed S I, Weinberg R A. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992;70:993–1006. doi: 10.1016/0092-8674(92)90249-c. [DOI] [PubMed] [Google Scholar]
- 35.Hofmann F, Martelli F, Livingston D M, Wang Z. The retinoblastoma gene product protects E2F-1 from degradation by the ubiquitin-proteasome pathway. Genes Dev. 1996;10:2949–2959. doi: 10.1101/gad.10.23.2949. [DOI] [PubMed] [Google Scholar]
- 36.Huang P S, Patrick D R, Edwards G, Goodhart P J, Huber H E, Miles L, Garsky V M, Oliff A, Heimbrook D C. Protein domains governing interactions between E2F, the retinoblastoma gene product, and human papillomavirus type 16 E7 protein. Mol Cell Biol. 1993;13:953–960. doi: 10.1128/mcb.13.2.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ikeda M A, Nevins J R. Identification of distinct roles for separate E1A domains in disruption of E2F complexes. Mol Cell Biol. 1993;13:7029–7035. doi: 10.1128/mcb.13.11.7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imai Y, Matsushima Y, Sugimura T, Terada M. Purification and characterization of human papillomavirus type 16 E7 protein with preferential binding capacity to the underphosphorylated form of retinoblastoma gene product. J Virol. 1991;65:4966–4972. doi: 10.1128/jvi.65.9.4966-4972.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones D L, Alani R M, Munger K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21Cip1-mediated inhibition of cdk2. Genes Dev. 1997;11:2101–2111. doi: 10.1101/gad.11.16.2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jones D L, Munger K. Analysis of the p53-mediated G1 growth arrest pathway in cells expressing the human papillomavirus type 16 E7 oncoprotein. J Virol. 1997;71:2905–2912. doi: 10.1128/jvi.71.4.2905-2912.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones D L, Thompson D A, Munger K. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology. 1997;239:97–107. doi: 10.1006/viro.1997.8851. [DOI] [PubMed] [Google Scholar]
- 42.Kiyono T, Foster S A, Koop J I, McDougall J K, Galloway D A, Klingelhutz A J. Both Rb/pl6INK4a inactivation and telomerase activity are required to immortalize human epithelial cells. Nature. 1998;396:84–88. doi: 10.1038/23962. [DOI] [PubMed] [Google Scholar]
- 43.Lee J O, Russo A A, Pavletich N P. Structure of the retinoblastoma tumour-suppressor pocket domain bound to a peptide from HPV E7. Nature. 1998;391:859–865. doi: 10.1038/36038. [DOI] [PubMed] [Google Scholar]
- 44.Lillie J W, Loewenstein P M, Green M R, Green M. Functional domains of adenovirus type 5 Ela proteins. Cell. 1987;50:1091–1100. doi: 10.1016/0092-8674(87)90175-9. [DOI] [PubMed] [Google Scholar]
- 45.Luo R X, Postigo A A, Dean D C. Rb interacts with histone deacetylase to repress transcription. Cell. 1998;92:463–473. doi: 10.1016/s0092-8674(00)80940-x. [DOI] [PubMed] [Google Scholar]
- 46.Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P, Lorain S, Le Villain J P F, Troalen, Trouche D, Harel-Bellan A. Retinoblastoma protein represses transcription by recruiting a histone deacetylase. Nature. 1998;391:601–605. doi: 10.1038/35410. [DOI] [PubMed] [Google Scholar]
- 47.Massimi P, Pim D, Banks L. Human papillomavirus type 16 E7 binds to the conserved carboxy-terminal region of the TATA box binding protein and this contributes to E7 transforming activity. J Gen Virol. 1997;78:2607–2613. doi: 10.1099/0022-1317-78-10-2607. [DOI] [PubMed] [Google Scholar]
- 48.Mavromatis K O, Jones D L, Mukherjee R, Yee C, Grace M, Munger K. The carboxyl-terminal zinc-binding domain of the human papillomavirus E7 protein can be functionally replaced by the homologous sequences of the E6 protein. Virus Res. 1997;52:109–118. doi: 10.1016/s0168-1702(97)00090-7. [DOI] [PubMed] [Google Scholar]
- 49.McIntyre M C, Frattini M G, Grossman S R, Laimins L A. Human papillomavirus type 18 E7 protein requires intact Cys-X-X-Cys motifs for zinc binding, dimerization, and transformation but not for Rb binding. J Virol. 1993;67:3142–3150. doi: 10.1128/jvi.67.6.3142-3150.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller A D, Rosman G J. Improved retroviral vectors for gene transfer and expression. BioTechniques. 1989;7:980–982. , 984–986, 989–990. [PMC free article] [PubMed] [Google Scholar]
- 51.Munger K, Phelps W C, Bubb V, Howley P M, Schlegel R. The E6 and E7 genes of the human papillomavirus type 16 together are necessary and sufficient for transformation of primary human keratinocytes. J Virol. 1989;63:4417–4421. doi: 10.1128/jvi.63.10.4417-4421.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Munger K, Werness B A, Dyson N, Phelps W C, Harlow E, Howley P M. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. EMBO J. 1989;8:4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pagano M, Durst M, Joswig S, Draetta G, Jansen-Durr P. Binding of the human E2F transcription factor to the retinoblastoma protein but not to cyclin A is abolished in HPV-16-immortalized cells. Oncogene. 1992;7:1681–1686. [PubMed] [Google Scholar]
- 54.Patrick D R, Oliff A, Heimbrook D C. Identification of a novel retinoblastoma gene product binding site on human papillomavirus type 16 E7 protein. J Biol Chem. 1994;269:6842–6850. [PubMed] [Google Scholar]
- 55.Phelps W C, Munger K, Yee C L, Barnes J A, Howley P M. Structure-function analysis of the human papillomavirus type 16 E7 oncoprotein. J Virol. 1992;66:2418–2427. doi: 10.1128/jvi.66.4.2418-2427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Phelps W C, Yee C L, Munger K, Howley P M. The human papillomavirus type 16 E7 gene encodes transactivation and transformation functions similar to those of adenovirus E1A. Cell. 1988;53:539–547. doi: 10.1016/0092-8674(88)90570-3. [DOI] [PubMed] [Google Scholar]
- 57.Qian Y, Luckey C, Horton L, Esser M, Templeton D J. Biological function of the retinoblastoma protein requires distinct domains for hyperphosphorylation and transcription factor binding. Mol Cell Biol. 1992;12:5363–5372. doi: 10.1128/mcb.12.12.5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reinstein E, Scheffner M, Oren M, Ciechanover A, Schwartz A. Degradation of the E7 human papillomavirus oncoprotein by the ubiquitin-proteasome system: targeting via ubiquitination of the N-terminal residue. Oncogene. 2000;19:5944–5950. doi: 10.1038/sj.onc.1203989. [DOI] [PubMed] [Google Scholar]
- 59.Sage J, Mulligan G J, Attardi L D, Miller A, Chen S, Williams B, Theodorou E, Jacks T. Targeted disruption of the three Rb-related genes leads to loss of G1 control and immortalization. Genes Dev. 2000;14:3037–3050. doi: 10.1101/gad.843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schulze A, Mannhardt B, Zerfass-Thome K, Zwerschke W, Jansen-Durr P. Anchorage-independent transcription of the cyclin A gene induced by the E7 oncoprotein of human papillomavirus type 16. J Virol. 1998;72:2323–2334. doi: 10.1128/jvi.72.3.2323-2334.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Selvey L A, Dunn L A, Tindle R W, Park D S, Frazer I H. Human papillomavirus (HPV) type 18 E7 protein is a short-lived steroid-inducible phosphoprotein in HPV-transformed cell lines. J Gen Virol. 1994;75:1647–1653. doi: 10.1099/0022-1317-75-7-1647. [DOI] [PubMed] [Google Scholar]
- 62.Sherr C J, Roberts J M. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 63.Slebos R J, Lee M H, Plunkett B S, Kessis T D, Williams B O, Jacks T, Hedrick L, Kastan M B, Cho K R. p53-dependent G1 arrest involves pRB-related proteins and is disrupted by the human papillomavirus 16 E7 oncoprotein. Proc Natl Acad Sci USA. 1994;91:5320–5324. doi: 10.1073/pnas.91.12.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Song S, Gulliver G A, Lambert P F. Human papillomavirus type 16 E6 and E7 oncogenes abrogate radiation-induced DNA damage responses in vivo through p53-dependent and p53-independent pathways. Proc Natl Acad Sci USA. 1998;95:2290–2295. doi: 10.1073/pnas.95.5.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Storey A, Almond N, Osborn K, Crawford L. Mutations of the human papillomavirus type 16 E7 gene that affect transformation, transactivation and phosphorylation by the E7 protein. J Gen Virol. 1990;71:965–970. doi: 10.1099/0022-1317-71-4-965. [DOI] [PubMed] [Google Scholar]
- 66.Watanabe S, Kanda T, Sato H, Furuno A, Yoshiike K. Mutational analysis of human papillomavirus type 16 E7 functions. J Virol. 1990;64:207–214. doi: 10.1128/jvi.64.1.207-214.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu E W, Clemens K E, Heck D V, Munger K. The human papillomavirus E7 oncoprotein and the cellular transcription factor E2F bind to separate sites on the retinoblastoma tumor suppressor protein. J Virol. 1993;67:2402–2407. doi: 10.1128/jvi.67.4.2402-2407.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zerfass K, Levy L M, Cremonesi C, Ciccolini F, Jansen-Durr P, Crawford L, Ralston R, Tommasino M. Cell cycle-dependent disruption of E2F-p107 complexes by human papillomavirus type 16 E7. J Gen Virol. 1995;76:1815–1820. doi: 10.1099/0022-1317-76-7-1815. [DOI] [PubMed] [Google Scholar]
- 69.Zerfass K, Schulze A, Spitkovsky D, Friedman V, Henglein B, Jansen-Durr P. Sequential activation of cyclin E and cyclin A gene expression by human papillomavirus type 16 E7 through sequences necessary for transformation. J Virol. 1995;69:6389–6399. doi: 10.1128/jvi.69.10.6389-6399.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang H S, Gavin M, Dahiya A, Postigo A A, Ma D, Luo R X, Harbour J W, Dean D C. Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell. 2000;101:79–89. doi: 10.1016/S0092-8674(00)80625-X. [DOI] [PubMed] [Google Scholar]
- 71.zur Hausen H. Papillomavirus infections—a major cause of human cancers. Biochim Biophys Acta. 1996;1288:F55–F78. doi: 10.1016/0304-419x(96)00020-0. [DOI] [PubMed] [Google Scholar]
- 72.Zwerschke W, Mannhardt B, Massimi P, Nauenburg S, Pim D, Nickel W, Banks L, Reuser A J, Jansen-Durr P. Allosteric activation of acid alpha-glucosidase by the human papillomavirus E7 protein. J Biol Chem. 2000;275:9534–9541. doi: 10.1074/jbc.275.13.9534. [DOI] [PubMed] [Google Scholar]
- 73.Zwerschke W, Mazurek S, Massimi P, Banks L, Eigenbrodt E, Jansen-Durr P. Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16 E7 oncoprotein. Proc Natl Acad Sci USA. 1999;96:1291–1296. doi: 10.1073/pnas.96.4.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]