

Abstract
The European Commission requested EFSA to update the scientific guidance for the preparation of applications for authorisation of novel foods, previously developed following the adoption of Regulation (EU) 2015/2283 on novel foods. This guidance document provides advice on the scientific information needed to be submitted by the applicant towards demonstrating the safety of the novel food. Requirements pertain to the description of the novel food, production process, compositional data, specifications, proposed uses and use levels and anticipated intake of the novel food. Furthermore, information needed in sections on the history of use of the novel food and/or its source, absorption, distribution, metabolism, excretion, toxicological information, nutritional information and allergenicity is also described. The applicant should integrate and interpret the data presented in the different sections to provide their overall considerations on how the information supports the safety of the novel food under the proposed conditions of use. Where potential health hazards have been identified, they are to be discussed in relation to the anticipated intake of the novel food and the proposed target populations. On the basis of the information provided, EFSA will assess the safety of the novel food under the proposed conditions of use.
Keywords: authorisation, EFSA guidance, food innovation, food safety, hazard characterisation, hazard identification, novel foods, risk assessment
BACKGROUND AS PROVIDED BY THE EUROPEAN COMMISSION IN 2015
On 25 November 2015, the European Parliament and the Council adopted the Regulation of the European Parliament and of the Council on novel foods. 1
The Regulation requires that all applications for the authorisation of novel foods shall be submitted to the Commission who may then request a risk assessment from the European Food Safety Authority (EFSA). In assessing the safety of novel foods, EFSA shall, where appropriate, consider the following:
whether the novel food concerned is as safe as food from a comparable food category already existing on the market within the Union;
whether the composition of the novel food and the conditions of its use do not pose a safety risk to human health in the Union;
a novel food, which is intended to replace another food, does not differ from that food in such a way that its normal consumption would be nutritionally disadvantageous for the consumer.
The Regulation also introduces a special procedure for safety assessment for traditional foods from third countries, based on a history of safe food use. In this case, a notification for the placing on the market of a traditional food from a third country is sent to the Commission who forwards it to all the Member States and EFSA. A Member State or EFSA may submit duly reasoned safety objections on the placing on the market of the notified food. In this latter case, the applicant may transform the notification into an application, for which a safety evaluation will be requested from EFSA. In assessing the safety of these types of novel foods, EFSA shall, where appropriate, consider the following:
-
–
whether the history of safe food use in a third country is substantiated by reliable data submitted by the applicant;
-
–
whether the composition of the food and the conditions of its use do not pose a safety risk to human health in the Union;
-
–
where the traditional food from the third country is intended to replace another food, whether it does not differ from that food in such a way that its normal consumption would be nutritionally disadvantageous for the consumer.
The Commission shall adopt implementing rules on administrative and scientific requirements for the preparation and the presentation of the applications for novel foods, as well as for the notifications and applications for traditional foods from third countries for the scientific assessment, respectively, in accordance with Article 13 and Article 20 of the Regulation. These implementing measures need to be complemented with scientific and technical guidance regarding the information that needs to be submitted by the applicants. In this context, the current Commission Recommendation 97/618/EC, 2 which is in place for the additional safety assessment of the novel food applications under the current rules (Regulation (EC) No 258/97 3 ), should serve as the basis for updating the guidance on preparation and presentation of applications for novel foods.
TERMS OF REFERENCE AS PROVIDED BY THE EUROPEAN COMMISSION IN 2015
In accordance with Article 29 of Regulation (EC) No 178/2002, the European Commission asks EFSA to update and develop scientific and technical guidance for the preparation and presentation of applications for authorisation of novel foods, and to develop scientific and technical guidance for notifications and applications for authorisation of traditional foods from third countries.
BACKGROUND AND TERMS OF REFERENCE AS PROVIDED BY THE EUROPEAN COMMISSION IN 2020
The European Commission asked EFSA to update the ‘Guidance on the preparation and submission of an application for authorisation of a novel food in the context of Regulation (EU) 2015/2283 4 ’ (EFSA NDA Panel, 2016) in order to align it to Regulation (EU) 2019/1381 5 on the transparency and sustainability of the EU risk assessment in the food chain (hereinafter ‘Transparency Regulation’), which applies as of 27 March 2021.
The guidance document has been identified to require updating as regards its administrative part. This request does not cover the scientific part of the document that has been left unchanged.
BACKGROUND AS PROVIDED BY THE EUROPEAN COMMISSION IN 2023
Following the adoption of Regulation (EU) 2015/2283 on novel foods, the Commission asked EFSA to update and further develop scientific and technical guidance for the preparation and presentation of applications for authorisation of novel foods (EFSA guidance). EFSA adopted its guidance document on the preparation and submission of an application for authorisation of a novel food in the context of Regulation (EU) 2015/2283 on 21 September 2016.
The EFSA guidance identified the essential elements that need to be part of novel food applications pursuant to Article 10 of Regulation (EU) 2015/2283 to support their safety, and served as the basis for the implementation of Commission Implementing Regulation (EU) 2017/2469. 6 As this Regulation came into effect after the EFSA guidance was developed and implemented, there is a need to ensure full consistency between Regulation (EU) 2015/2283 and Implementing Regulation (EU) 2017/2469 to better assist and support applicants in the preparation of Article 10 novel food applications.
In addition, since the onset of its implementation on 1 January 2018 when Regulation (EU) 2015/2283 came into effect considerable experience has been gained by EFSA in assessing different types of novel foods, including, but not limited to, foods deriving from new sources and foods produced using new production. Therefore, the current EFSA guidance may need to be updated with novel scientific elements and specific requirements for the safety assessment of some novel foods, to take stock of the assessments performed so far by EFSA and the advances in science and technologies since the publication of its current version.
Given the above, there is a need to update the EFSA guidance document on the preparation and submission of an application for authorisation of a novel food in the context of Regulation (EU) 2015/2283.
TERMS OF REFERENCE AS PROVIDED BY THE EUROPEAN COMMISSION IN 2023
In accordance with Article 31 of Regulation (EC) No 178/2002 the European Commission asks the European Food Safety Authority to update the guidance document on the preparation and submission of an application for authorisation of a novel food in the context of Regulation (EU) 2015/2283.
OBJECTIVES
This guidance is intended to explain the type and quality of scientific information EFSA needs to conclude whether or not the novel food is safe under the proposed conditions of use. The scientific requirements for an application for the notification of a traditional food from a third country are dealt with by a separate guidance document by the EFSA NDA Panel (EFSA NDA Panel, 2024a).
The guidance will be kept under review and it will be further updated as appropriate in the light of experience gained from the evaluation of novel food applications or under any legal revision.
SCOPE
The guidance presented in this document is to assist applicants with the scientific requirements in preparing applications for authorisation of a novel food under Article 10 of Regulation (EU) 2015/2283. A separate EFSA guidance document is available to assist applicants in preparing and presenting a notification dossier for a traditional food from a third country under Article 14 of Regulation (EU) 2015/2283 (EFSA NDA Panel, 2024a). The latter document specifically addresses the data required to substantiate the ‘history of safe food use in third country’ of a traditional food, as defined by Article 3 of Regulation (EU) 2015/2283. Under the notification procedure, Regulation (EU) 2015/2283 foresees that a Member State or EFSA may submit to the Commission duly reasoned safety objections to the placing on the market within the Union of the traditional food concerned. In such cases, the present guidance should also serve applicants in preparing an application under Article 16 of Regulation (EU) 2015/2283, where the application concerns data other than those on the ‘history of safe food use in a third country’. An EFSA guidance document on the scientific principles and data requirements for the safety and relative bioavailability assessment of new micronutrient sources is available (EFSA NDA Panel, 2024b).
Procedural aspects linked to the submission of an application for authorisation of a novel food in the context of Regulation (EU) 2015/2283 are not in the scope of this guidance document. Instead, applicants are advised to consult the EFSA Administrative guidance for the preparation of novel food applications in the context of Article 10 of Regulation (EU) 2015/2283 (EFSA, 2024), the EFSA Administrative guidance for the processing of applications for regulated products (EFSA, 2021a), and the EFSA Catalogue of support initiatives during the life cycle of applications for regulated products (EFSA, 2021b).
The present guidance, as well as the guidance on the scientific requirements for a notification and application for authorisation of traditional foods from third countries in the context of Regulation (EU) 2015/2283 and the one on the scientific principles and data requirements for the safety and relative bioavailability assessment of new micronutrient sources, apply as of 1 February 2025.
DEFINITIONS
As per Article 3, paragraph 2 of Regulation (EU) 2015/2283, the following definition applies:
‘Novel food’ means any food that was not used for human consumption to a significant degree within the Union before 15 May 1997 irrespective of the dates of accession of Member States to the Union and that falls under at least one of the following categories:
food with a new or intentionally modified molecular structure, where that structure was not used as, or in, a food within the Union before 15 May 1997;
food consisting of, isolated from or produced from microorganisms, fungi or algae;
food consisting of, isolated from or produced from material of mineral origin;
food consisting of, isolated from or produced from plants or their parts, except when the food has a history of safe food use within the Union and is consisting of, isolated from or produced from a plant or a variety of the same species obtained by:
-
–
traditional propagating practices which have been used for food production within the Union before 15 May 1997; or
-
–
non‐traditional propagating practices which have not been used for food production within the Union before 15 May 1997, where those practices do not give rise to significant changes in the composition or structure of the food affecting its nutritional value, metabolism or level of undesirable substances;
-
v
food consisting of, isolated from or produced from animals or their parts, except for animals obtained by traditional breeding practices which have been used for food production within the Union before 15 May 1997 and the food from those animals has a history of safe food use within the Union;
-
vi
food consisting of, isolated from or produced from cell culture or tissue culture derived from animals, plants, microorganisms, fungi or algae;
-
vii
food resulting from a production process not used for food production within the Union before 15 May 1997, which gives rise to significant changes in the composition or structure of a food, affecting its nutritional value, metabolism or level of undesirable substances;
-
viii
food consisting of engineered nanomaterials as defined in point (f) of Article 3, paragraph 2 of Regulation (EU) 2015/2283;
-
ix
vitamins, minerals and other substances used in accordance with Directive 2002/46/EC, Regulation (EC) No 1925/2006 or Regulation (EU) No 609/2013, where:
-
–
a production process not used for food production within the Union before 15 May 1997 has been applied as referred to in point (a) (vii) of this paragraph; or
-
–
they contain or consist of engineered nanomaterials;
-
x
food used exclusively in food supplements within the Union before 15 May 1997, where it is intended to be used in foods other than food supplements as defined in point (a) of Article 2 of Directive 2002/46/EC.
GENERAL PRINCIPLES
For information on the novel food applications procedure, applicants should consult the EFSA Administrative guidance for the preparation of novel food applications in the context of Article 10 of Regulation 2015/2283 7 (EFSA, 2024). The administrative guidance provides also a full description of the requirements introduced by the Transparency Regulation such as the notification of studies obligations [Article 32b of Regulation (EC) No 178/2022 ’General Food Law‘ 8 ], the possibility to request General Pre‐submission advice (Article 32a of the General Food Law) and the provision of transparency and confidentiality (Articles 38 and 39 of the General Food Law).
Several EFSA scientific guidance documents may also be of relevance for the preparation of novel food applications, especially those of the EFSA Scientific Committee. 9 Some of them are listed throughout the present document. Some EFSA guidance documents may be applicable only in specific cases. Over time, new guidance documents may be developed which may be of relevance for novel food applications. Applicants are therefore advised to consult the EFSA webpage and consider the most up‐to‐date versions of the available and applicable guidance documents.
For novel foods comprising single substances or simple mixtures thereof suggested as new sources of micronutrients (i.e. vitamins and minerals) for addition to foods 10 (including foods for special groups 11 ) and/or to be consumed as food supplements, 12 applicants are also referred to the EFSA guidance on the scientific principles and data requirements for the safety and relative bioavailability of new micronutrients sources (EFSA NDA Panel, 2024b).
The term ‘application’ is defined in Article 2 of Commission Implementing Regulation (EU) 2017/2469 as meaning a stand‐alone dossier containing the information and the scientific data submitted for the authorisation of a novel food pursuant to Article 10(1) of the novel food regulation.
The information required on the identity of the novel food, production process, compositional data, specifications and on proposed uses and use levels and anticipated intake of the novel food constitutes the minimum information and data requirements which must be fulfilled in all applications for marketing authorisation of a novel food. Data and information should also be provided in the other sections concerning the history of use of the novel food and/or of its source, absorption, distribution, metabolism and excretion, toxicological information, nutritional information and allergenicity unless the applicant can provide scientific justification and argumentation as to why new data/information is not needed for one or more of these sections to support the safety of the novel food.
Applications which concern an already authorised novel food may relate to changes in the production process, specifications or the conditions of use, e.g. adding a target population, adding uses (adding new food categories to which a novel food is intended to be added) or use levels. In such cases, not only the changes as such are to be described in detail but also consequences regarding composition (if a production process is changed), exposure (if uses and/or use levels are added), nutritional aspects and the safety of the novel food must be addressed. It is the responsibility of the applicant to provide all of the available (proprietary, confidential or published) scientific data (including both data in favour and not in favour) that are pertinent to the safety of the novel food. As such, an application to demonstrate the safety of the novel food has to be comprehensive and complete.
Data pertinent to the safety of the novel food must be identified and documented to demonstrate that the application covers the complete information package available on the novel food. Information on the search strategy, including the sources used to retrieve pertinent data (databases, other sources), the terms and limits used (e.g. publication dates, publication types, languages, population, default tags) should be provided. Where applicable, the published literature is to be reviewed taking into account systematic review principles (EFSA, 2010). Full study reports should be provided if available.
The applicant should provide their considerations at the end of individual sections on how the information supports the safety of the novel food under the proposed conditions of use. Uncertainties must be addressed, and a critical appraisal of data both in favour and not in favour, of the safety of the novel food is to be provided.
Deviations from the requirements specified in the respective sections of this guidance document must be justified.
Analyses/tests characterising the novel food should be performed in a facility qualified for this purpose. Quality systems in place for control/documentation have to be indicated. Information on the accreditation of involved facilities and certificates of analyses should be provided. Whenever official guidelines (e.g. OECD, EMA and ICH) and quality systems (e.g. GLP, GMP, GCP and applicable ISO systems) were followed, the applicant should indicate compliance.
Referring to Directive 2010/63/EU, 13 Regulation (EU) 2015/2283 emphasises that tests on animals should be replaced, reduced or refined (3 Rs), wherever possible. This goal to reduce animal studies to the minimum needed is also in line with the EU's chemicals strategy for sustainability and EFSA's Strategy 2027 to develop and integrate new scientific developments focusing on NAM 14 ‐based methods and the minimisation of animal testing. When these methodologies are qualified or become validated as alternative approaches, applicants are encouraged to make use of them to provide data on the safety of the novel food. Before performing any study with animals, applicants should, as part of the tiered toxicological approach outlined in this guidance, conduct a comprehensive literature search for relevant published data concerning the novel food or its constituents to allow thorough considerations of the available knowledge regarding the need of performing toxicological studies. If in vivo studies are required to be performed to demonstrate the safety of the novel food, as noted by the Commission Implementing Regulation (EU) 2017/2469, they should be carried out in accordance with the rules set out in Directive 2004/10/EC 15 of the European Parliament and of the Council. If those tests are carried out outside the territory of the Union, they should still follow the OECD Principles of good laboratory practice. 16 In addition, when there is the need to perform in vitro and/or in vivo toxicological studies, they should be conducted in accordance with international guidelines such as OECD or ICH.
1. IDENTITY OF THE NOVEL FOOD
Information on the identity of the novel food must be provided considering the requirements outlined in the subsections listed below. There may be cases where two or more subsections could be of relevance to a novel food. In those circumstances, the respective information for all relevant subsections should be provided. The subsections below are to be distinguished from the categories outlined in Article 3 of Regulation (EU) 2015/2283, to which the applicant must assign their novel food upon submission of the application dossier.
The novel food subject to the risk assessment should be the product resulting from the production process, without the addition of non‐novel ingredients/excipients used to formulate the final product intended to be marketed. Although information on the use of such non‐novel compounds is to be provided in the description of the production process, non‐novel compounds should not be considered for the identity of the novel food, the compositional analyses and the proposed specifications, unless they are essential to maintain specific characteristics of the novel food, e.g. stability, physical form. In this context, for example, compounds which serve solely for standardisation of the composition of the novel food should not be considered as part of the novel food.
The name of the novel food in the application submitted has to reflect its characteristic elements, e.g. its source, the main part(s) of organisms used, its form(s) (e.g. dried, frozen, powder), specific elements of the production process. Scientific names according to the most recent taxonomy or scientific nomenclature are to be included; commercial names, including trademarks, are to be avoided.
1.1. Chemical substances, products of mineral origin and polymers
The following information must be provided for novel foods that are single chemical substances, and for each component when the novel food is a simple mixture, i.e. a chemical mixture whose constituents can be fully characterised; in these cases, chemical composition and identity must be reported for each component (including isomers, e.g. stereoisomers, constitutional isomers) as indicated below and in Section 3.2. Such information, in line with Section 3.3, is to be provided for complex mixtures and whole foods (where not all constituents can be fully chemically characterised and/or identified) for the containing chemical substances which are relevant for the identity and/or the safety of the novel food.
Chemical name, when appropriate, according to IUPAC nomenclature rules (IUPAC, 2012, 2015, 2020);
CAS number, European Community (EC) Number – European Chemicals Agency (ECHA) and other relevant identification numbers (e.g. PubChem, E numbers, ChEBI, ChEMBL, Flavis, HMDB/FooDB, Lipidmaps, ChemSpider, IUBMB number), when available;
Synonyms or common names, trade names, abbreviations;
Molecular and structural formulae with stereochemistry;
Molar mass (g/mol) or Molecular mass (Da);
InChI (International Chemical Identifier) and InChIkey (digital representation of the InChI);
Canonical SMILES and isomeric SMILES;
Identity tests of the relevant constituents should be performed with the most relevant analytical techniques (e.g. chromatography, nuclear magnetic resonance, mass spectrometry, FT‐IR, UV, optical rotation in the case of chiral compounds, XRD data and/or melting point for solids and crystals);
Particle size, shape and distribution in the final product; 17
Comparison with chemical standards, certified reference materials, authentic biological specimens, naturally occurring compounds or other relevant materials, when available.
In the case of simple chemical mixtures, if chemical or physical interactions are expected to occur that would alter the properties of the single components or their behaviour in the body e.g. bioavailability, these should be outlined, together with predicted consequences of the interaction.
The relevant requirements detailed above apply also in the case of novel foods that are consisting of, isolated from or produced from material of mineral origin. This pertains to inorganic mineral constituents utilised as inorganic or organic salts or complexes/chelates.
In the case of polymers 18 obtained from natural sources or through chemical or enzymatic synthesis or modification, the identity has to be demonstrated as per the relevant requirements detailed above. Further to those, the following requirements also apply:
Structural formulae of monomers;
Structure of the polymer, degree of polymerisation, number average molecular weight, weight average molecular weight and viscosity average molecular weight;
If the polymer is obtained by chemical synthesis, structural formulae of starting materials and reagents involved in the polymerisation;
If the polymer is obtained by enzyme‐catalysed synthesis, structural formulae of starting materials, enzymes used and information on its source;
In the case of chemical or enzymatic modification of the polymer, the nature and degree of modification of the polymer should be detailed.
Where applicable, the ECHA guidance for identification and naming of substances under REACH 19 and CLP Regulations 20 could be followed. 21
1.2. Foods consisting of, isolated from or produced with microorganisms
The scientific requirements for the taxonomic and hazard identification of microorganisms intentionally used in the food chain (including bacteria, yeasts, filamentous fungi, microalgae/protists and viruses) depend on the particular role of the microorganism and, when applicable, genetic modification and qualified presumption of safety (QPS) status.
In the context of novel foods and for the purpose of this guidance, microorganisms can have different roles:
Novel foods consisting of non‐genetically modified microorganism(s) (non‐GMMs) capable of multiplication are defined as ‘active agent(s)’.
Novel foods made from non‐GMMs in which the inactivated cells, not capable of multiplication, and/or their genetic material may still be detected are defined as ‘biomass(es)’.
Novel foods produced with GMMs or non‐GMMs in which these microorganism(s) are used in the manufacturing of the novel food and defined as ‘production strain(s)’.
Among the four categories of GMMs and their products defined by EFSA for risk assessment purposes (EFSA GMO Panel, 2011; EFSA Scientific Committee, 2022a), only GMM categories 1 and 2 fall under the remit of the Novel Foods Regulation.
The EFSA QPS provides a safety pre‐assessment of microbial strains belonging to QPS taxonomic units (TUs). The lowest TU for which the QPS status is granted is the species level for bacteria, yeasts and microalgae/protists and the family level for viruses. Only unambiguously identified microbial strains belonging to QPS TUs can benefit from the risk assessment approach based on QPS. Safety concerns related to a QPS TU are reflected, when possible, as ‘qualifications’, which should be tested at strain and/or product level. In the case of GMMs for which the species of the recipient strain qualifies for the QPS status, and for which the genetic modification does not give rise to safety concerns, the QPS approach can be extended to the GMM (EFSA BIOHAZ Panel, 2023a).
Overall scientific requirements for the taxonomic and hazard identification of microorganisms as novel foods (active agents and biomasses) or used in the production of novel foods (production strains) are listed below (detailed description in Appendix A), including references to relevant EFSA guidance documents for additional information:
Unambiguous taxonomic identification at species level and certificate of deposition (including accession number) of the microbial strain under assessment in an internationally recognised culture collection having acquired the status of International Depositary Authority following the Budapest Treaty rules (EFSA, 2021c; EFSA FEEDAP Panel, 2018);
Characterisation of genes of potential concern, i.e. acquired antimicrobial resistance (AMR) genes of clinical relevance, toxigenicity and pathogenicity traits (EFSA, 2021c; EFSA BIOHAZ Panel, 2023b; EFSA FEEDAP Panel, 2018);
Assessment of the capacity of the microbial strain to produce antimicrobials of clinical relevance, unless a QPS TU or a TU is known not to produce those antimicrobials (EFSA FEEDAP Panel, 2018);
Purpose, characterisation and structure of the genetic modification(s) for GM production strains (EFSA FEEDAP Panel, 2018; EFSA, 2021c; EFSA GMO Panel, 2024);
Whole genome sequence (WGS) data according to the most up‐to‐date versions of the available and applicable EFSA scientific outputs (e.g. EFSA, 2021c).
Additionally, the presence of viable cells in the novel food has to be analysed for (i) biomasses and non‐QPS TUs as novel foods, (ii) QPS TUs with the qualification ‘for production purposes only’ and (iii) non‐QPS or GM production strains (additional requirements in Section 3.1 of EFSA FEEDAP Panel, 2018).
The presence of DNA from the production strain in the novel food has to be analysed for (i) GM production strains and (ii) non‐GM production strains carrying acquired AMR genes of clinical relevance (additional requirements in Section 3.2 of EFSA FEEDAP Panel, 2018).
1.3. Food consisting of, isolated from or produced from plants, macroscopic fungi and macroalgae, or their parts
The following information must be provided in the case of novel foods consisting of, isolated from or produced from plants, 22 macroscopic fungi and macroalgae (i.e. seaweed) or their parts. In the case of a mixture of source organisms, the information is to be reported for each source and the mass percentages of each source in the mixture must be specified.
Scientific (Latin) name and taxonomy (family, genus, species, and if applicable subspecies, variety with author's name, chemotype, strain) according to the international codes of nomenclature for plants 23 and for macroscopic fungi and macroalgae; 24
Accepted synonyms;
Trivial or common names used to identify the novel food intended to be marketed;
For plants, experimental verification of the identity of the plant (e.g. authentic plant specimen deposit in a recognised herbarium, macroscopic/microscopic verification with comparison to an authentic standard, chemical fingerprint compared to standard, DNA‐based authentication);
For macroscopic fungi and macroalgae, verification of the identity according to internationally recognised databases and methodology and, if available, deposition in an internationally recognised culture collection with access number;
Part(s) used (e.g. flower, seed, root);
Growing region(s) of the source organism (continent, country, region) and, when relevant, season of harvesting;
Growing conditions to produce the source organism (i.e. cultivated or from the wild, conditions of cultivation);
Non‐GMO 25 statement from the applicant accompanied by information on the source material.
1.4. Food consisting of, isolated from or produced from animals or their parts
The following information is to be provided for novel foods isolated from or produced from animals or their parts:
Scientific (Latin) name (family, genus, species, subspecies, breed, if applicable);
Accepted synonyms;
Trivial or common names used to identify the novel food intended to be marketed;
Verification of the identity (e.g. certification, DNA‐based authentication);
Suitability of the animal sources for human consumption according to Commission Regulation (EU) No 2015/1162; 26
Compliance with Regulation (EU) 2017/625 27 on official controls and other official activities and, where applicable, with Regulation (EC) No 853/2004 28 on specific hygiene rules for food of animal origin;
Health status of the source animal, age, access to herd/lot health certification;
Part(s) used (e.g. organ(s) or tissue(s));
Geographical origin (continent, country, region), farming and husbandry conditions;
Origin of the initial livestock (e.g. national repository). In case the source of the novel food is provided by external vendors, supporting documents should be provided;
Non‐GMO statement from the applicant accompanied by information on the source material.
1.5. Foods consisting of, isolated from or produced from cell culture or tissue culture derived from animals, plants, macroscopic fungi or macroalgae
This section concerns cell/tissue cultures derived from multicellular origin (animals, plants including macroscopic fungi and macroalgae). For foods consisting of, isolated from or produced from cell cultures derived from microorganisms (including bacteria, yeasts, filamentous fungi and microalgae/protists), reference is made to the scientific requirements laid down in Section 1.2. The novel foods defined under this category can be the harvested cells, the biomass or the further processed biomass obtained from cell or tissue culture.
1.5.1. Foods consisting of, isolated from or produced from cell culture or tissue culture derived from animals
Identity of the source organism as per the relevant requirements in Section 1.4, including information to attest that the primary cells and tissues used for the preparation of the novel food comply with inspection requirements laid down in that section;
When using established cell lines: genetic and phenotypic identity and stability of cells;
When using primary cells: biopsy location or source material, cell type(s) isolated, genetic and phenotypic identity of cells;
Information to attest the absence of any risks of infectivity from viruses or other zoonotic agents e.g. testing for viruses (species‐specific viruses), testing for prions in the case of limited health information on source animals;
Information on whether the cells or tissues sourced from a non‐GM animal have been genetically modified after biopsy.
1.5.2. Foods consisting of, isolated from or produced from cell culture or tissue culture derived from plants, macroscopic fungi or macroalgae
Identity of the source organism as per the relevant requirements in Section 1.3;
Laboratory or culture collection sourced;
Identity of the cells or cell lines: part(s) of the organism sourced, cell type isolated, genetic and phenotypic identity, genetic and phenotypic stability of the cell lines.
Information on whether the cells or tissues sourced from non‐GM plants, macroscopic fungi or macroalgae, have been genetically modified after collection.
1.6. Foods containing or consisting of engineered nanomaterials
For novel foods containing or consisting of ‘engineered nanomaterials’, 29 the parameters for identification and characterisation to be provided to support the application are outlined in the ‘Guidance on risk assessment of nanomaterials to be applied in the food and feed chain’ (EFSA Scientific Committee, 2021b). For novel foods that may contain small particles including particles within the nanoscale which do not meet the definition of engineered nanomaterials, specific requirements are outlined in Section 3.1.4 of this Guidance.
2. PRODUCTION PROCESS
The process(es) employed to produce the novel food (e.g. chemical synthesis, enzyme catalysis, fermentation or isolation from a natural source) should be comprehensively described. The description of the production process must be detailed enough to ensure understanding of the critical parameters and steps involved, enabling the identification of all potential food safety hazards. This information will form the basis for evaluating the composition, specifications, bioavailability, nutritional value and safety of the novel food.
2.1. General provisions
Information on all input materials used in the manufacturing process of the novel food should be presented, including their functional role, and their regulatory status in the EU (Appendix B). Additionally, information on the specification and quality of the input/raw materials and fermentation aids has to be provided. 30 Moreover, for every material in contact with food during the production process (e.g. plastic containers), a declaration of compliance as laid down by Regulation (EC) No 1935/2004 31 and any other relevant EU provisions should be provided. Considering all steps during the production process, the production yield, i.e. the resulting amount of a novel food from its raw materials, should be calculated, providing also the ‘processing factors, 32 ’ when applicable. Regarding safety, the description must include information on potential by‐products, impurities or contaminants. Formation of processing contaminants should be also considered based on the processes applied and a description of the parameters that may lead to the formation of a given processing contaminant should be included.
The applicant must inform whether a production process is novel, i.e. not used for food production within the EU before 15 May 1997, and characterise the novel aspects of the process.
The implementation of food safety management systems in place to produce the novel food should cover procedures based on the HACCP principles in line with Regulation (EC) No 852/2004 on the hygiene of foodstuffs. 33 Operational limits and key parameters of the production process should be given. Measures implemented for production control and quality and safety assurance should be described (e.g. HACCP, GMP, ISO). These procedures should be detailed, including critical control points, operational prerequisite programmes, monitored parameters, corrective actions, verification procedures, frequency of analysis, analytical methods, etc. A production flow chart should be provided, including quality and safety control checks. Standardisation criteria (e.g. markers for the novel food) should be provided.
If the description on the production process contains information for which a confidentiality request has been submitted, pursuant to Articles 39 to 39e of Regulation (EC) No 178/2002 and EFSA's Practical Arrangements concerning transparency and confidentiality (EFSA, 2021d), a non‐confidential summary of the production process should also be provided, including all steps of the process with a general description of the operational conditions and safety‐related parameters.
2.2. Considerations for specific production process steps
Information must also be provided on the handling of the sources, for example, the propagation, growth and harvesting conditions for plants and fungi (e.g. wild or cultivated, type of cultivation and cultivation practices, composition of fertilisers used, time of harvest in relation to both season and stage of the plant growth); the cultivation conditions for aquacultures (e.g. measures in place to ensure water quality, temperature, length of growth in the water, composition of fertilisers used); the breeding, rearing, feeding and farming conditions along with the description of feed and certificates of feed compliance with EU Regulations for farmed animals or the hunting, catching or collecting and killing of wild living animals; the culture conditions for microorganisms; and cell culture or tissue culture from plants and animals. The parts of the organism used as a raw material must be specified and information on other starting substances or materials should be provided. The description of the cultivation of plants, fungi, macroalgae and microorganisms and the rearing of animals should also include information on the use of pesticides, hormones, veterinary drugs, antimicrobials and antiparasitic agents or feed additives. Biological agents (e.g. parasites, bacteria, endophytes, viruses, prions) that can infect organisms or tissue cultures used to produce the novel food or be hosted by these organisms (animals, plants, fungi, macroalgae and microorganisms) should be considered in the assessment. Information and measures in place to mitigate the respective risks should be provided and the impact of these agents on human health should be discussed.
Post‐harvest handling, e.g. transport, drying techniques and storage conditions (duration, light, moisture and temperature) of unprocessed foods and the raw materials for further processing should be described.
When food enzymes are used as processing aids for the production of the novel food, the presence or absence of the enzymes in the novel food has to be demonstrated experimentally in at least three representative batches of the novel food that have been independently produced (preferably with independent batches of raw materials). If the enzyme is present in the novel food, the enzymatic activity and potential residual activity should be reported in at least three representative batches of the novel food that have been independently produced (EFSA CEP Panel, 2021). If the enzyme has been inactivated or removed, the processes and operational conditions in place for the inactivation/removal are to be provided. Removal or inactivation of the enzyme should be demonstrated in case of safety concerns. The safety of the food enzyme(s) used in the manufacture of the novel food is subject to the provisions of Regulation (EC) No 1332/2008, 34 and therefore, it is outside the scope of this guidance, which concerns the assessment of the safety of the novel food according to the provisions of Regulation (EU) 2015/2283. Therefore, the applicant is requested to provide information about the status of the enzyme(s) according to Regulation (EC) No 1332/2008. Food enzymes used in the production of novel food should preferably have been already assessed with a positive outcome by the EFSA Panel on Food Contact Materials, Enzymes and Processing Aids (EFSA CEP Panel, 2021). In case the food enzymes have not been assessed or the risk assessment is still in progress, additional data could be requested to establish the safety of the novel food (EFSA CEP Panel, 2021). For enzymes of microbial origin, the requested data will be in line with the scientific criteria outlined in relevant EFSA guidance documents (EFSA, 2021c; EFSA CEP Panel, 2021; EFSA FEEDAP Panel, 2018). The assessment of the novel food will be without prejudice to the safety assessment of the food enzyme per se.
With regard to the use of food additives in the production of a novel food, it should be noted that such additives must be authorised and listed with conditions of use in the EU's positive list based on Regulation (EC) No 1333/2008. 35 Any unauthorised additives cannot be used.
2.3. Considerations for specific novel food categories
For novel foods obtained via chemical synthesis, the reaction sequence, side reactions and purification steps are to be described. Information on reaction conditions (e.g. reagents, temperature, duration of the reaction and catalysts), chemical or physical purification methods (e.g. solvent extraction and crystallisation) are to be reported. Directive 2009/32/EC 36 on extraction solvents used in the production of foodstuffs and food ingredients should be considered.
Regarding production processes employing microorganisms, the techniques used to remove/inactivate microbial cells during downstream processing should be described in detail, with full provision of operational conditions (e.g. time, temperature, kinetics, etc.). In the case of a novel food consisting of viable cells, information on the techniques/methods and operational conditions used to ensure microbial viability must also be reported. The applicant should investigate, and report whether the specific production conditions of the novel food (e.g. due to processing aids or component of the media) may trigger the formation of toxic compounds by microorganisms.
For novel foods consisting of, isolated from or produced from plants, macroscopic fungi, macroalgae or animals, a detailed description of the process(es) by which the raw material is converted into an ingredient or a food product, must be provided. Examples may include e.g. heat treatment, extraction, distillation, fractionation, purification, concentration, fermentation or other procedure(s). Information on substances used in the manufacturing process, e.g. identity and purity of the extraction solvents, the ratio of extraction solvent to the material, reagents, additives, residues remaining in the final product and any special precautions (e.g. protection from light and controlled temperature) should be provided.
For foods consisting of, isolated from or produced from cell culture or tissue culture derived from animals, plants, macroscopic fungi or macroalgae, information is to be provided on the type of cells used as source (e.g. primary cells or established cell lines). In case primary cells are used, information on the source, purification steps, cell isolation, cell selection, cell subculture, absence of pathogens and microbial contaminants is to be provided. If cells from established cell lines are used, information must be provided on the source, the cell line preparation, the cell banking process, as well as the passage number of aliquot of cells used. Description of any changes made to the cells used (e.g. selection, differentiation, immortalisation, adaptations, reprogramming), and the link of such changes with the production of substances of possible concern must be included. All processes applied for the treatment, extraction, screening and selection of cell lines or tissues must be provided in detail including all chemicals and biological materials used, and including the impurities that may result from their use. The genetic stability of the cells throughout the production process should be investigated by comparison of the starting material (i.e. initially selected cells from biopsy/cell line) and the cells at different steps of the production process (e.g. propagation step). Also changes of the morphology, markers of differentiation and other phenotypic features of the cells at the start and at the end of the production process should be investigated and described. Information on the compliance with good cell culture practices 37 should be provided, as well as on the compliance with applicable relevant standards, such as those outlined in the EMA Guidance document on the derivation and characterisation of cell substrates used for production of biotechnological/biological products. 38 The safety of growth factors of microbial origin (e.g. recombinant proteins, vitamins, amino acids) used in the production of, e.g. novel foods consisting of, isolated from or produced from cell culture or tissue culture will be assessed to establish the safety of the novel food, taking into consideration the scientific requirements for the taxonomic and hazard identification of microorganisms intentionally used in the food chain, as listed in Section 1.2 and Appendix A according to relevant EFSA guidance documents (EFSA, 2021c; EFSA FEEDAP Panel, 2018).
2.4. Additional considerations
In case the novel food dossier contains analytical data on novel food batches manufactured by different producers (e.g. the application is submitted by a consortium of producers) or by processes involving steps that can be different (e.g. drying the raw material using various methods), such differences shall be described, equivalency substantiated and consistency in production methods among different producers/processes demonstrated. Food safety management systems (e.g. HACCP plan) should be provided from all producers/processes covering the entire production process. The variability of the supplying starting materials is to be investigated and be covered by the analytical data provided. Any changes to the production process during the risk assessment must be notified to EFSA by the applicant.
3. COMPOSITIONAL DATA
Compositional data serve as a tool to characterise the novel food and its constituents, encompassing both qualitative and quantitative information on the chemical, physicochemical, microbiological and nutritional attributes of the novel food. They should facilitate an in‐depth exploration of the compositional characteristics of the novel food, linked to its source and employed production process. Variability of compositional data between different batches should be analysed and discussed, towards investigating the ability of the food business operator to produce the novel food in a consistent and reproducible manner, while being the basis for hazard identification and establishment of the specification parameters. Section 3.1 outlines the general data requirements applicable to all novel foods, while Sections 3.2 and 3.3 set specific requirements, depending on whether the novel food is a single substance or a simple mixture, a complex mixture or a whole food. 39
3.1. General requirements
3.1.1. Analytical methods
Validated methods, preferably nationally or internationally recognised (e.g. Association of Official Analytical Chemists, European Pharmacopoeia, International Organization for Standardization, European Committee for Standardization) should be used for the analyses. The respective methods of analysis should be described alongside their references. The limits of detection (LOD) and quantification (LOQ) should be mentioned. Certificates of analyses and information on the matrix accreditation 40 and the scope of accreditation of the laboratories should be provided. If in‐house methods are employed, the analytical protocols implemented should be fully described, and the results of the respective method validation procedures should be provided. If an analytical method is used for a food matrix beyond the scope 41 of accreditation/standardisation, it should be treated as in‐house method (the same applies in cases that standard methods are modified). If the analyses are not performed in accredited laboratories, a justification should be provided. A table with all the analytical methods employed and the corresponding analytes should be provided. The table should include the name of the method, the reference, the main analytical technique(s) employed, as well as the respective LOD and/or LOQ.
3.1.2. Addressing compositional variability
Compositional data and their variability should support the setting of specifications of the novel food 42 (Section 4). The analytical information should be provided on at least five representative batches of the novel food that have been independently produced (preferably with independent batches of raw materials), 43 unless a different number of batches is explicitly requested in this guidance. The analyses should preferably be performed on the same group of batches, to obtain a comprehensive picture of their composition. It is expected that the analysed batches are produced either at an industrial production scale or at one representative of it. Representativeness shall be justified. The examined batches should be sampled in a manner adequate to address potential compositional variations (e.g. seasonal) of the raw materials. Additional batches of the novel food may also be needed to explore the variability of potentially hazardous substances present in the novel food or its source. When several production processes are proposed, such data should be provided for each process. Moreover, compositional data should also cover the whole variability spectrum of the production process parameters (e.g. highest and lowest amount of solvents used, range of temperatures applied). The compositional variability should be discussed, highlighting the reasons for the variation in results. If the application pertains to various forms of the novel food (e.g. dried, frozen, powder), all analyses must be conducted on at least five representative batches of each form, produced independently. Any deviations from this requirement must be justified. Analytical data from publications can also be used if the publications provide sufficient information on the laboratory where analyses have been carried out, the methods utilised and if the studies were performed with representative samples of the novel food. Available published data can also contribute to providing information on the variability of the composition of the novel food.
3.1.3. Sampling practices
Principles of representative sampling should be applied (e.g. sample size, containers, conditions), and the rationale on why the employed sampling plan is considered representative should be provided. Information on any relevant existing legally defined or standard sampling protocols should be considered and provided. On each certificate of analysis, the name as well as the dates of production and analysis of the batch must be stated.
3.1.4. Compositional analytes
Information on the identity and the quantity of impurities or by‐products, residues and chemical and microbiological contaminants should be provided (e.g. heavy metals, mycotoxins, PCBs/dioxins, pesticides, microbial hygiene indicators and pathogens). The potential target analytes should be selected considering the sources and the production process, regulatory levels as well as the information available in the scientific literature. For example, for substances obtained by chemical synthesis, residual starting materials and by‐products anticipated from side reactions should be analysed; for substances produced by microbial fermentation, the presence of metabolites of safety concern should be investigated; for substances isolated by extraction, data on residual solvents should be provided.
The protein content of the novel food should be quantified using the 6.25 nitrogen‐to‐protein conversion factor. In case the protein content of the novel food is substantial, 44 it should also be calculated as the sum of the anhydrous amino acids, to account for the presence of non‐protein nitrogen and the complete quantitative amino acid profile should be provided. When novel foods consist of or are enriched in specific proteins or peptides, characterisation of the individual protein/peptide profile (e.g. sequence, degree of hydrolysis) is additionally requested. Moreover, considering the allergenicity‐related analytical requirements described in Section 10, further analyses for the characterisation of the protein profile may be necessary.
When a novel food application concerns a material that meets the definition of engineered nanomaterial as set out in the Novel Food Regulation (EU) 2015/2283, the chemical identification and physicochemical characterisation need to be provided as per the Guidance on risk assessment of nanomaterials to be applied in the food and feed chain (EFSA Scientific Committee, 2021b).
Novel foods which do not meet the definition of engineered nanomaterial may contain small particles including particles within the nanoscale, formed naturally or as by‐products in the production process or during handling and processing of the foods (EFSA Scientific Committee, 2021a). For such novel foods, the applicants need to demonstrate that a fraction of small particles (as defined in the Guidance) is either not present after food consumption 45 or covered by the conventional risk assessment using the appraisal routes given in the EFSA Guidance on technical requirements for regulated food and feed product applications to establish the presence of small particles including nanoparticles (EFSA Scientific Committee, 2021a). The applicants should select, according to the available information on the substance under assessment, the best appraisal route or combination of appraisal routes, to demonstrate whether the novel food requires a nano‐specific toxicological risk assessment (Sections 2 and 3 of EFSA Scientific Committee, 2021a).
Considering their nature and in order to avoid unnecessary testing, some categories of novel foods do not require a priori nano‐specific risk assessment, e.g. (i) microorganisms (e.g. bacteria, yeasts, fungi, microalgae), (ii) unmodified proteins (including enzymes) and amino acids, (iii) whole foods (e.g. seeds, fruits, insects). Therefore, if the manufacturing process does not include any step that may lead to the presence of small particles, and if it can be demonstrated that a novel food falls under one of the above or similar categories, the novel food may qualify for exemption from the characterisation and/or demonstration of the absence of small particles (as defined in EFSA Scientific Committee (2021a)).
3.2. Single substances and simple mixtures
Simple mixtures are mixtures whose components can be fully chemically characterised. For simple mixtures of defined substances, information on the identities and the relative ratios of all components should be provided. This should allow the elaboration of a mass balance. For single substances and substances in simple mixtures, the identity‐relevant analytical data outlined in Section 1.1 should be provided.
For single substances and simple mixtures produced with GMMs, applicants are referred to the requirements for GMMs Category 1 (EFSA GMO Panel, 2011).
3.3. Complex mixtures and whole foods
Complex mixtures (e.g. extracts, protein hydrolysates, active agents, biomasses) and whole foods (e.g. milk, meat, fruits, seeds, insects) are defined as those novel foods where not all constituents can be fully characterised and/or identified.
A qualitative and quantitative characterisation of the main constituents is to be performed, at least via sum parameters. For whole foods, this should include proximate analyses (i.e. ash, moisture, protein, fat, carbohydrates). On the basis of these data, a mass balance should be calculated. The amount of unidentified components should be indicated and should be as low as possible.
For the classes of naturally or chemically derived components which characterise the novel food (e.g. amino acids, peptides, phospholipids, carotenoids, phenolics, sterols), comprehensive qualitative and quantitative data should be provided. Additionally, when it is anticipated that the production process may result in the emergence of new proteins, a thorough characterisation of the protein profile is required (Section 3.1.4).
Qualitative and quantitative data on nutritionally relevant inherent constituents such as micronutrients, antinutrients 46 and dietary fibre 47 should be provided.
Information on the occurrence and occurrence levels of inherent substances of possible concern to human health (e.g. toxic, allergenic) should be provided. The impact of processing on the compositional profile of the novel food (e.g. occurrence of heat‐induced processing contaminants) should also be considered.
In addition to the batch‐to‐batch analysis, a comprehensive literature search should be performed according to the methodology developed by EFSA (2010) to retrieve published compositional data (chemical, physicochemical and microbiological) for the source and the part(s) used in/as novel food, as well as for compositional aspects linked to the production process. Information on the keywords and applied inclusion/exclusion criteria for the literature search should be provided. Considering the retrieved information, the applicant should provide a rationale on the compositional analysis strategy followed.
Any substances of concern (e.g. toxins, heavy metals) potentially present in the starting materials, should be analysed in the novel food. Particular attention should be given to the possible presence of genotoxic and/or carcinogenic substances.
For plants, levels at which the constituents are present in the respective part of the botanical or botanical preparation should be given where available. It is recommended that chemical fingerprinting of the botanical material is undertaken for this purpose.
The following non‐exhaustive list of tools can help identifying the possible substances of concern in a botanical material:
The EFSA Compendium of Botanicals, 48 , 49 which provides information on naturally occurring substances that may be of concern for human health (EFSA, 2012),
The EFSA Chemical Hazard Database (OpenFoodTox). 50
For complex mixtures produced with GMMs, applicants are referred to the requirements for GMMs Category 2 (i.e. complex products in which both GMMs and newly introduced genes are no longer present) (EFSA GMO Panel, 2011).
The EFSA Scientific Committee has identified potential hazards related to the use of farmed insects as food (EFSA Scientific Committee, 2015). These should be considered in applications for novel foods which consist of, are isolated from or are produced from farmed insects, considering the species and substrates to be used, as well as methods for farming and processing.
For active agents and biomasses, the respective concentration of viable cells and non‐viable cells in the novel food should be reported.
When it could be relevant to further substantiate the safety (e.g. for novel foods obtained using a novel production process), it is recommended that a comparative compositional analysis of a novel food to its potential conventional comparators is conducted (e.g. UV‐treatment in EFSA NDA Panel, 2023a). While a comparative approach can be useful in some cases, it may not always be adequate for addressing specific risks associated with the novel food itself.
3.4. Stability testing
3.4.1. Stability of the novel food
The stability of the novel food has to be evaluated to ensure both the compositional integrity and the safety of the novel food. Hazards that might arise during storage and transport must be identified and the nature of degradation products should be characterised.
Stability tests should consider compositional qualifiers, as well as constituents and parameters of the novel food which may be susceptible to changes during storage and which may affect its safety and/or its identity or serve as indicators for alterations that could have an impact on the safety and/or the integrity of the novel food. The rationale for the parameters selected to be monitored during the stability testing, as well as for those parameters disregarded as not relevant, should be provided.
Depending on the nature, production process and composition of the novel food, the testing is to address the chemical, physicochemical and microbiological stability of the novel food under the intended conditions of storage, taking into account the effect of packaging and the storage environmental parameters (temperature, light exposure, oxygen, moisture, relative humidity). Information on the intended storage conditions, including the proposed shelf‐life, of the novel food must be provided as well as on the conditions under which the stability testing was performed. The stability testing has to be provided on at least five representative batches of the novel food that have been independently produced (preferably with independent batches of raw materials). When the application pertains to various forms of the novel food (e.g. dried, frozen, powder), such data should be provided for each form. Any deviations from this requirement must be justified. Testing of a lower number of batches should be justified by scientific arguments. The novel food batches selected to be monitored at the beginning of the stability testing have to be those monitored for the whole duration of the stability testing. The stability testing results can be taken into consideration when establishing the limits of relevant specification parameters. On the other hand, compliance of the novel food with the specification parameters throughout the proposed shelf‐life should be demonstrated.
The monitoring period of the stability test has to cover at least the end of the proposed shelf‐life. Intermediate intervals of testing must be considered, depending on the nature of the novel food, its composition, as well as the intended shelf‐life. Although it is advisable to submit stability testing studies under intended conditions of storage, accelerated conditions may be used as an alternative. Such approaches, usually conducted at higher temperatures, could be applicable only in cases where chemical parameters are monitored. In cases where results from accelerated conditions are extrapolated to predict results under the intended storage conditions, scientific evidence must be provided to justify the validity of this extrapolation. Information on ingredients added to the novel food to improve its stability has to be provided.
3.4.2. Impact of processing on the novel food in the proposed‐for‐use matrices
If the novel food is used as an ingredient added to other foods the manufacture of which requires further processing (e.g. heating), the impact on the novel food of this processing is to be investigated. Also alterations in the processed foods due to the presence of the novel food should be investigated in foods or in relevant model systems (mimicking the food matrix and the respective processing conditions), taking also into consideration at least the extremes of the possible processing conditions (e.g. highest temperature to which the novel food will be exposed when used as a food ingredient, lowest and highest pH) as resulting from the proposed uses (Section 6.2). More specifically, it should be investigated what happens to relevant components of the novel food, when it is used as a food ingredient. Interactions with other constituents in the processed foods and the formation of processing contaminants should be investigated. The use of proper controls (e.g. the product manufactured with the same process/recipe without containing the novel food as ingredient) is necessary.
Moreover, when the novel food is subject to further processing that differs from the conventionally applied processing methods, any hazards potentially arising are to be identified and characterised.
4. SPECIFICATIONS
Specifications comprise chemical, physicochemical, nutritional and microbiological parameters that characterise and substantiate the identity and safety of the novel food, including the respective numerical ranges or limits.
Specifications serve as a tool for risk managers, i.e. the European Commission and Member States, who decide which of the proposed specification parameters and respective limits will be considered for inclusion and updating of the Union list of novel foods 51 in accordance with Article 9 of Regulation (EU) 2015/2283, when a novel food is granted marketing authorisation. Given that risk managers may consider not only compositional aspects, applicants should propose also a brief but comprehensive description of the novel food, incorporating identity parameters such as the name of the source or relevant parts thereof, and the microbial strain used as novel food or in the production of novel foods. It is also advisable to provide key descriptors related to the production process.
Applicants must provide a comprehensive set of compositional specification parameters in a tabulated format. Depending on the identity and composition of the novel food, the table should include the following:
proximate analytes (protein, lipids, carbohydrates, ash and moisture),
the major groups of constituents within the food,
more characteristic components (e.g. carotenoids, polyphenols, terpenes, alkenyl benzenes, lignin, saponins, chitin, micronutrients, number of viable/non‐viable microorganisms),
parameters relevant for the safety of the novel food at the proposed uses and use levels (e.g. toxins, alkaloids, phytic acid and other antinutrients, heavy metals, pathogens, impurities or degradation products from the production process),
parameters related to the quality and/or stability that may have an impact on the safety of the novel food (e.g. markers of lipid oxidation, microbial hygiene indicators or water activity).
The rationale for each proposed specification parameter and respective limits has to be provided.
The table must include minimum and/or maximum specification limits for each selected parameter. The specifications, including their limits, should be supported by the available information on the chemical, physicochemical and microbiological composition of the novel food including the results from the available batch‐to‐batch analysis and the stability testing. They should be verifiable by means of the analytical techniques as indicated in Section 3. Information on the employed analytical techniques and their sensitivity (LOD/LOQ) should be provided.
In general, the proposed maximum specification limits for undesirable substances should be as low as possible. Existing health‐based guidance values (HBGV) for substances of potential toxicological concern, but also dietary reference values (DRV) including tolerable upper intake levels (UL) for micronutrients and exposure estimates to such compounds, should be considered when proposing the maximum specification limits. Minimum specification limits for nutrients may be necessary to ensure that a minimum level is present in the novel food, especially when a novel food represents a potential alternative or is intended to replace an existing food on the market, which provides a relevant contribution to the intake of certain nutrients. If EU regulatory limits are applicable for the novel food, then they do not necessarily have to be listed in the specifications.
For novel foods consisting of engineered nanomaterials, the specific provisions on specifications given in the Guidance on risk assessment of nanomaterials to be applied in the food and feed chain have to be considered (Table 1 and Section 5.1.3 of EFSA Scientific Committee, 2021b). For novel foods which are conventional materials containing a fraction of small particles, the considerations on specifications given in the respective guidance document should be considered and addressed (in particular Section 3.4.3, EFSA Scientific Committee, 2021a).
5. HISTORY OF USE OF THE NOVEL FOOD AND/OR OF ITS SOURCE
5.1. History of use of the source
Data on the composition, production and experience from the use of products from the source (other than the novel food itself) may provide relevant aspects for further consideration, for example, regarding critical substances contained in the source, potential hazards or precautions of handling the source of the novel food (e.g. the food consumed only after being cooked). With respect to foods derived from plants, relevant information may be found in EFSA's Compendium on Botanicals (EFSA, 2012).
5.2. History of use of the novel food
Data may be available on the use of the novel food as food in countries outside the EU and on non‐food uses. Such data may provide information which could be relevant for assessing the safety of the novel food. Such information could include a description of the extent of use as a food and/or for non‐food purposes (e.g. quantity of consumption, information on the serving size(s), average, high and if available maximum intake levels per person should be provided), the population group for which the food has been a part of their diet, its role in the diet (e.g. main dish, consumed as a snack, pattern of consumption), the handling and preparation of the food (e.g. cooking methods, storage conditions) and any other relevant information. A comprehensive literature review of human studies reporting on relevant safety outcomes should be performed. Information on the search strategy, including the sources used to retrieve pertinent data (databases, other sources), the terms and limits used (e.g. publication dates, publication types, languages, population, default tags) should be provided. Where applicable, the published literature should be reviewed by taking into account systematic review principles (EFSA, 2010).
The applicant should not only consider and limit the literature search to the novel food itself but should also consider searching for studies with specific and safety‐relevant components of the novel food and for studies with similar foods from the same or other closely related sources (e.g. other varieties or subspecies or related species of the same genus or family) or on foods with similar chemical composition.
6. PROPOSED USES AND USE LEVELS AND ANTICIPATED INTAKE OF THE NOVEL FOOD
Estimates of novel food intake by the EU population are necessary to evaluate its dietary and nutritional significance and to conduct risk characterisation. These estimates are based on the proposed uses and use levels of the novel food and data on actual food consumption.
This section should provide information on the target population, proposed uses and use levels and precautions and restrictions of use, with cross‐referencing to relevant safety data.
If potential health hazards have been identified based on the composition, toxicological or other data, they should be discussed and adequately addressed in the proposed conditions of use to ensure that the consumption of the novel food is safe for the target population. When setting up proposed uses and use levels, applicants should consider appropriate margins of exposure (‘uncertainty factors’) as suggested by the Scientific Committee (2012a).
For novel foods proposed as new sources of micronutrients (i.e. vitamins and minerals) for addition to foods 52 (including foods for special groups 53 ) and/or to be consumed as food supplements, 54 applicants are referred to the EFSA guidance on the scientific principles and data requirements for the safety and relative bioavailability assessment of new micronutrient sources (EFSA NDA Panel, 2024b). The guidance outlines specific considerations regarding the target population and the calculation of anticipated intakes.
6.1. Target population
The applicant should unambiguously specify the intended target population (e.g. general population, adolescents, adults).
Where it cannot be excluded that a novel food intended for a particular group of the population would also be consumed by other groups of the population (e.g. a novel food added as an ingredient to foods or be consumed as a whole food), the safety data provided shall also cover those groups in accordance with Article 5(6) of Commission Implementing Regulation (EU) 2017/2469.
In certain cases, the consumption of the novel food can be restricted to a particular group of the general population. In such cases, the applicant must specify the intended target population (e.g. adults; individuals above 10 years of age; or other age population groups as defined in the EFSA Guidance on the Comprehensive European Food Consumption Database, 2011). Examples include:
when the novel food is intended to be used in food supplements;
when the novel food is intended to be used in foods for special groups as defined in Regulation (EU) No 609/2013 (e.g. if the novel food is intended to be used in ‘Total diet replacements for weight control’ in accordance with Regulation (EU) 609/2013′, the target population is adults as defined by the Commission Delegated Regulation (EU) 2017/1798; if the novel food is intended to be used in ‘Infant formula and follow‐on formula’ and/or in ‘processed cereal‐based food and baby food’ in accordance with Regulation (EU) 609/2013′, the target population is infants and young children as defined in such Regulation).
6.2. Proposed uses and use levels
The applicant has to specify the intended uses of the novel food (e.g. as a whole food, ingredient, food supplement).
If the novel food is intended to be added as an ingredient to foods, the applicant should provide the following information in a tabulated format:
The food categories in which the novel food is proposed to be added. Food categories can be specified according to the EFSA Food Additive Intake Model (FAIM) tool 55 or the Dietary Exposure (DietEx) tool. 56 All intended uses should be expressed with the use of a unique classification system (i.e. either FAIM tool categories or DietEx tool categories). Codes and names of the proposed food categories should be provided and presented as per Tables 1 and 2;
When selecting the FAIM tool categories, applicants are advised to refer to the instructions available on the website 57 particularly in relation to the unspecified food categories displayed in the FAIM tool;
When using the DietEx tool, the applicant is advised to use broad food categories instead of overly specific ones (e.g. yoghurts in general rather than certain types of yoghurts; biscuits in general rather than certain types of biscuits);
The proposed maximum use levels (i.e. maximum concentrations) of the novel food in each food category as consumed (e.g. expressed as mg/kg or mg/100 g or mg/100 mL);
If the novel food is proposed in different forms (e.g. dried, frozen, powder), the food categories and maximum use levels should be proposed for each form of the novel food as requested in points above. It should be specified whether the different forms of the novel food are meant to be utilised singularly and/or in combination in a specific food category.
TABLE 1.
Proposed uses and use levels according to the FAIM tool.
| FAIM tool code | FAIM tool category | Maximum level |
|---|---|---|
| 01.7.2 | Ripened cheese | 100 mg/kg |
TABLE 2.
Proposed uses and use levels according to the DietEx tool.
| FoodEx code | FoodEx category | Maximum level |
|---|---|---|
| A00EY | Cereal bars | 10 mg/100 g |
Tables 1 and 2 display examples of how the applicant should present the information on the proposed uses and use levels as described in the points above.
When the novel food is intended to be used as a whole food, the applicant has to indicate food(s) already consumed in the EU (using either a category in the FAIM or the DietEx tool) which can reasonably reflect the anticipated consumption pattern of the novel food. Applicants should provide their considerations and explanations as to why it is reasonable to expect that the novel food corresponds to specific food(s) consumed in the EU.
If the novel food is intended to be used in food supplements, ‘Total diet replacements for weight control’, ‘Meal replacements for weight control’ and ‘Food for special medical purposes’, the applicant should not select these food categories in the FAIM tool or the DietEx tool. The following instructions should be followed when these food categories are intended to be used.
If the novel food is intended to be used in food supplements, the applicant is requested to indicate the proposed maximum daily intake of the novel food for the groups of the target population (e.g. 50 mg/day for adolescents [from 10 to 17 years of age]; 100 mg/day for adults [18 years of age and above]).
If the novel food is intended to be used in ‘Total diet replacements for weight control’ in accordance with Regulation (EU) No 609/2013, the applicant should indicate the maximum daily intake of the novel food in mg/day.
If the novel food is intended to be used in ‘meal replacement for weight control,’ 58 the applicant should specify the amount of the novel food to be used in a single meal replacement.
If the novel food is intended to be used in ‘Food for special medical purposes’, its conditions of use should be in accordance with Regulation (EU) No 609/2013.
6.3. Anticipated intake of the novel food
On the basis of the information provided in Sections 6.1 and 6.2, the applicant should estimate the chronic daily intake of the novel food. This estimate should present both the amount of novel food consumed per kilogram of body weight and the total absolute amount of novel food consumed per day. If estimates are not provided on total absolute intake, the applicant should use the mean default body weights as reported in the EFSA guidance on selected default values to be used by the EFSA Scientific Committee, Scientific Panels and Units in the absence of actual measured data (EFSA Scientific Committee, 2012a). The applicant should provide estimates of the mean and high (95th percentile) anticipated daily intakes of the novel food for each target population group, including specific population groups such as pregnant and lactating women if available.
The FAIM tool and the DietEx tool are available to applicants to perform the chronic intake estimate of the novel food when added to foods. When estimating the intake, the applicant should consider all food categories to which the novel food is intended to be added for a conservative scenario. Both FAIM and DietEx tools use individual consumption data from the EFSA Comprehensive Food Consumption Database to generate estimates (mean and 95th percentile) for population groups (infants, young and other children, adolescents, adults) throughout several EU countries. It is noted that the DietEx tool provides more refined food categories as compared to the FAIM tool which uses broader food categories. Thus, DietEx allows a more refined selection of food categories and intake estimates of the novel food.
If the available toxicological data, human data, data on chemical composition or literature review raise concerns regarding an acute effect, the applicant should also consider acute intake estimates of the novel food.
When the intended uses are expressed as maximum daily intakes of the novel food (e.g. for food supplements (Section 6.2), the applicant should also provide the maximum daily intake expressed on a per kilogram body weight basis for each population group of the target population. For this, the applicant should use the mean default body weights as reported in the EFSA Guidance on selected default values to be used by the EFSA Scientific Committee, Scientific Panels and Units in the absence of actual measured data (EFSA Scientific Committee, 2012a) for each population group of the target population (e.g. safety of paramylon EFSA NDA Panel, 2023b).
When a novel food is reasonably expected to be used as an alternative to another food already consumed in EU (e.g. when the novel food is a whole food – Section 6.2), the applicant should use the consumption data of food(s) already consumed in EU to estimate the anticipated intake of the novel food (e.g. safety of edible kernels of Jatropha curcas L. which would reasonably be consumed as an alternative to peanuts (EFSA NDA Panel, 2022b)).
Based on the intended uses of the novel food (e.g. whole food, ingredient, food supplement), the applicant should provide the combined intake scenarios resulting from the different uses of the novel food, considering the highest 95th percentile of anticipated intakes of the novel food for each group of the target population (as indicated in Section 6.1) (e.g. safety of paramylon, EFSA NDA Panel, 2023b; safety of edible kernels of Jatropha curcas L., EFSA NDA Panel, 2022b; safety of frozen and freeze‐dried formulations of the lesser mealworm (Alphitobius diaperinus larva), EFSA NDA Panel, 2022a).
6.4. Combined intake considering other sources of the novel food or its main constituents
The applicant should consider a combined intake of the novel food or its main constituent(s) from other sources. The applicant should take into account other potential sources of intake of the novel food which may derive from other uses (e.g. as a food additive) or from natural occurrence in foods (i.e. from the background diet) (e.g. opinion on the safety of synthetic lycopene, EFSA NDA Panel, 2008). Furthermore, when relevant, the applicant should consider the combined exposure of constituents from the novel food with other potential sources of that constituent (e.g. from the background diet) (e.g. safety of vitamin D2 mushroom powder, EFSA NDA Panel, 2022c).
The combined intake should be estimated, taking into account:
high daily intakes (95th percentile) of the novel food/its constituent from the proposed uses and maximum use levels (as estimated in Section 6.3);
mean and high daily intakes from natural sources (i.e. from the background diet) derived from literature;
daily intake from other uses (e.g. food additive) derived from literature.
There might be also cases where a novel food is added to foods that may partly replace foods that significantly contribute to the intake of specific compounds (e.g. vitamins, minerals) in the diet. In these cases, applicants should consider the potential double accounting of these compounds from the novel food and the background diet.
When relevant, the applicant should provide considerations on the exposure from fortified foods and/or food supplements and/or authorised novel foods already on the market.
With regard to nutrients and antinutrients potentially present in the novel food, please refer to Section 9.
Data on exposure from other potential non‐dietary sources (e.g. from consumer products such as cosmetics, from pharmaceuticals) should be provided, where relevant.
6.5. Estimate of exposure to substances of safety concern
Exposure estimates should be provided for substances of safety concern identified in the compositional analysis (e.g. secondary plant metabolites, residues, contaminants or degradation products, Section 3). These substances may be present in the novel food due to its source or the manufacturing process, as well as due to its use and storage.
The same approach as that used for the anticipated intake of the novel food should be followed to estimate the exposure to substances of safety concern from the novel food for the proposed target population. To anticipate the exposure to these substances from the novel food, the applicant should consider the maximum amount of these substances expected to occur in the novel food (e.g. maximum limit set in the specifications or, in the case that specifications are not established, the maximum level reported among the batch‐to‐batch analytical data) and the highest estimated daily intake (i.e. 95th percentile) of the novel food for the proposed target population. The applicant should also consider the exposure to those substances from the background diet. The exposure to substances of safety concern from the novel food (plus from the background diet when relevant) should be compared with HBGVs (e.g. ADI or TDI), when available. When relevant, applicants should consider the potential double accounting of substances of safety concern from the novel food and the background diet, especially in those cases in which the exposure to substances of safety concern from the diet already exceeds the HBGV.
6.6. Precautions and restrictions of use
When proposing precautions (including directions for its preparation and/or use) and restrictions of use, all available information on safety should be taken into consideration.
The applicant should specify the population groups (including population groups with certain physiological conditions) which should avoid consumption of the novel food and include the rationale.
7. ABSORPTION, DISTRIBUTION, METABOLISM AND EXCRETION
7.1. General considerations
Data on absorption, distribution, metabolism and excretion (ADME) in humans and animals are relevant for both nutritional and toxicological assessment of a novel food. ADME studies inform about the extent of absorption of the novel food or its components from the gastrointestinal (GI) tract, the nature and extent of metabolism and elimination and the potential for bioaccumulation. Differences in ADME between animals and humans may affect the adequacy and interpretation of experimental animal studies.
Prior to planning and conducting ADME studies, applicants should consider the chemical, physicochemical and microbiological characteristics of the novel food, including its nutritionally and toxicologically relevant components. Additionally, a comprehensive literature review of existing ADME data on the novel food or its relevant components should be conducted, and the collected evidence should be critically appraised.
Information on ADME or specific ADME studies is necessary for novel foods composed of new single substances. Consideration of potential matrix effects in foods in which the novel food will be added is required, especially in cases where it is foreseen that the food matrix may influence the novel food's ADME behaviour, thus possibly enhancing or diminishing its bioavailability. In cases where the novel food is a mixture, the ADME of toxicologically relevant constituents should be investigated. Similarly, if in the case of simple chemical mixtures, chemical or physical interactions are expected to occur that would alter the properties of the single components or their behaviour in the body, the impact on bioavailability should be assessed.
In some cases, ADME studies may not be needed, for example, when the novel food is composed of substances described to be commonly found in the body or in the diet. However, in the case of nutritionally relevant constituents, ADME assessments should be conducted in accordance with the information provided in Section 9. The (relative) bioavailability of a nutrient needs to be specifically assessed and quantified when the novel food is also a new nutrient source (Section 7.3) (EFSA NDA Panel, 2024b).
Where there is a potential concern about the protein in the novel food, appropriate protein digestibility studies should be performed as part of the weight of evidence approach for the assessment of the nutritional, toxicological and allergenic properties (e.g. EFSA GMO Panel, 2017, 2021, 2022).
Where the novel food consists of polymers > 1000 Da and where evidence is provided that they are not degraded in the GI tract to fragments < 1000 Da, ADME studies may not be needed.
With respect to novel foods containing or consisting of ‘engineered nanomaterials’ applicants have to consider the specific requirements and follow the approach as set out in the EFSA Scientific Committee guidance on risk assessment of nanomaterials to be applied in the food and feed chain (in particular, sections on in vitro degradation tests and ADME studies) (EFSA Scientific Committee, 2021b: Sections 7.2 and 7.6 therein). For conventional materials containing small particles including particles at the nanoscale formed naturally or as by‐products in the production process or during the handling and processing of the foods, the ADME studies have to comply with the requirements as set out in Section 4 of the EFSA Guidance on technical requirements for regulated food and feed product applications to establish the presence of small particles including nanoparticles (EFSA Scientific Committee, 2021a).
7.2. Tiered approach to conducting ADME studies
ADME studies are to be conducted following a tiered testing approach. 59 Guidance on how to conduct in vivo ADME studies in animals can be found in OECD TG 417 (OECD, 2010). A description of the data requirements for the different tiers for ADME and the triggers prompting the need to move to a higher tier of data requirements are described below and illustrated in Figure 1.
FIGURE 1.
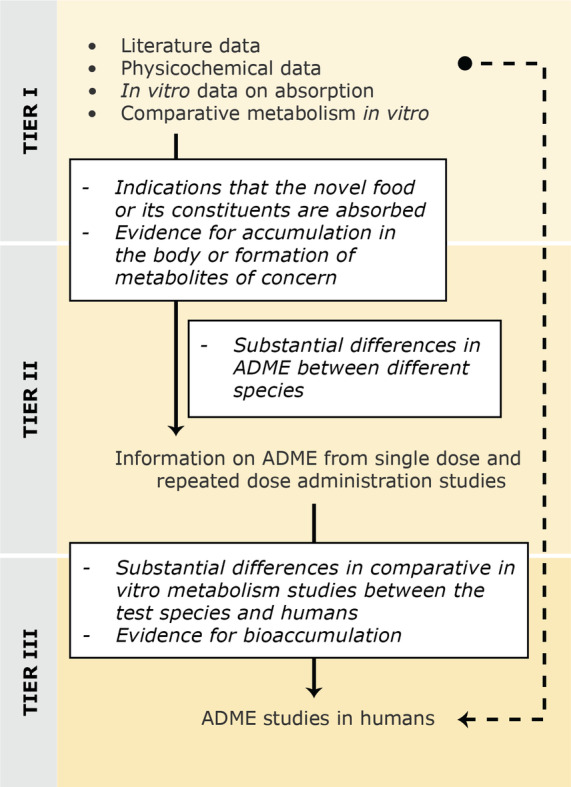
Overview of the tiered approach for assessing the ADME characteristics of novel foods and/or their components. The boxes in white represent triggers that will call for higher‐tier studies as indicated by the arrows. Solid lines refer to the standardised way of conducting a tiered‐based approach by moving from lower to higher tier assessments; dashed lines refer to a deviated tiered‐based approach where the existing data require Tier III studies.
The need to conduct ADME studies may be waived provided that a scientific rationale is given.
7.2.1. Tier I ADME testing
Assessing all relevant data from the published literature documenting in vitro and in vivo studies on the ADME of the novel food or its components and potential metabolites belongs to Tier 1. Chemical and physicochemical data may predict the dissociation characteristics of a novel food under GI conditions, which may have an impact on intestinal absorption.
Progress has been made in recent years with the development of human‐relevant in vitro models to quantify transport across the intestinal membrane and assess metabolism (EFSA PPR Panel, 2021; ICH, 2024; OECD, 2021). Existing models include cell‐based systems of various levels of complexity (e.g. MDCK, Caco‐2, human small intestinal and liver organotypic 3D culture models). Such in vitro models could complement or, when shown to have a similar level of predictivity, replace in vivo models to assess absorption and metabolism, noting the interrelationship between the tiers.
Tier I includes also the assessment of the comparative metabolism of the novel food. This is to establish, where applicable, that the pattern of metabolites formed in human in vitro test systems (as a surrogate of the in vivo situation) is comparable to that in the animal species tested (EFSA PPR Panel, 2021).
Gut microbiota and their associated enzymes can potentially have an impact on biotransformation, activation or detoxification of the chemicals of the novel food. Therefore, in case there is evidence that the novel food per se or its derived components are not absorbed in the small intestine, in vitro (e.g. M‐ARCOL, SHIME, Triple co‐culture) studies mimicking the human gut and its microbiota dynamics should be conducted by the applicant to identify or quantify relevant novel food‐derived metabolites which could potentially be of safety concern.
If Tier I data do not provide sufficient information for the ADME assessment, or if there are triggers prompting higher tier data requirements (Sections 7.2.2 and 7.2.3), Tier II or Tier III studies are required.
7.2.2. Tier II ADME testing
The triggers leading to Tier II testing in animals include one or more of the following:
indications that the novel food or its constituents are absorbed or systemically available;
evidence for the accumulation in the body or formation of metabolites of concern.
In Tier II, ADME information from both single‐dose administration and repeated dose studies (e.g. satellite groups from a sub‐chronic toxicity study) in vivo is needed. Tier II ADME information would also be required where e.g. a prolonged half‐life or enzyme induction is observed or expected. For the repeated dose ADME study, it is advised to use appropriate samples generated in the subchronic toxicity study (OECD TG 408) (OECD, 2018a) (Section 9.4.1.2). Guidance for the Tier II ADME assessment is also provided in the OECD TG 417 (OECD, 2010).
7.2.3. Tier III ADME testing
The purpose of Tier III is to generate ADME information on the novel food in humans when one or more of the triggers described below is activated.
The triggers leading to Tier III testing (ADME studies in humans) include one or more of the following:
substantial differences in ADME between different species
substantial differences in comparative in vitro metabolism studies between the test species and humans (where no suitable animal models exist)
evidence for bioaccumulation of the novel food, its components or metabolites thereof in the test species.
7.3. Specific considerations for novel foods that are new nutrient sources
For novel foods that are also new nutrient sources, the bioavailability of the nutrient from the new source needs to be demonstrated. Conclusions on bioavailability should consider the information provided in Sections 7.1 and 7.2 and the following:
For new sources of micronutrients, i.e. vitamins (including metabolites and new vitamers) and minerals, the relative bioavailability of the micronutrient from the new source needs to be quantified following the principles outlined in the EFSA Guidance on the scientific principles and data requirements for the safety and relative bioavailability assessment of new micronutrient sources (EFSA NDA Panel, 2024b).
For nutrients that are not micronutrients, bioavailability needs to be assessed and demonstrated but not quantified (EFSA NDA Panel, 2024b).
8. TOXICOLOGICAL INFORMATION
8.1. General considerations
The purpose of conducting toxicological studies on a novel food is to identify and characterise its potential hazards and to support establishing safe intake levels for humans.
Before designing and conducting toxicological studies, applicants have to consider the compositional data of the novel food (Section 3). Moreover, a comprehensive literature review on the toxicological properties of the novel food and/or its relevant components should be performed. All relevant available knowledge on the novel food should be thoroughly considered to determine the need for toxicity studies, and if so, the corresponding toxicological testing strategy, a thorough description of which, is to be provided.
Important elements to be considered for the toxicological testing strategy include
the source, production process, identity and composition of the novel food,
available ADME information,
available toxicological information (in silico, in vitro, in vivo studies) on the novel food, its constituents (including nutrients, Section 9.1) or its metabolites,
available human studies and case reports,
available relevant information and safety assessments from non‐food uses (e.g. chemicals, pharmaceuticals, cosmetics).
When, despite a comprehensive characterisation and literature review, potential data gaps for the hazard identification or hazard characterisation of a novel food remain, appropriate toxicological studies should be conducted aiming to fill the gaps. Such studies should be carried out with a representative test material, i.e. the test material should be derived from the production process (Section 2), be in accordance with the compositional data (Section 3) and meet the proposed specifications (Section 4). In some cases (e.g. some whole foods or extracts), toxicity tests using the novel food as intended to be placed on the market may lack the necessary sensitivity to identify potential toxicological properties. In such cases, a concentrate or (an) appropriate fraction(s) of the novel food may be used to increase the sensitivity of a toxicological study. In all cases where the test material is not the novel food, the applicant has to provide the rationale for its use, detailed information on the preparation of the test material and a comprehensive compositional analysis.
Toxicological studies performed with the novel food should be conducted in accordance with international guidelines (e.g. OECD) and according to the OECD principles of GLP (Organisation for Economic Co‐operation and Development principles of Good Laboratory Practices (OECD, 1997); Commission Implementing Regulation (EU) 2017/2469.
For the safety assessment of nanomaterials, additional requirements need to be considered as detailed in the Guidance on risk assessment of nanomaterials to be applied in the food and feed chain: human and animal health (EFSA Scientific Committee, 2021b). If a material is not a nanomaterial, but contains a fraction of small particles including nanoparticles, the provisions in Section 4 of the applicable guidance apply (EFSA Scientific Committee, 2021a); this section focuses on the information to be provided on safety studies originally designed for addressing conventional materials in order to ensure that the possible adverse effects linked to the fraction of small particles can be identified.
Toxicological data on structurally related substances and their metabolic profiles may be considered for applying a read‐across approach. The applicability of read‐across to novel food may be limited to defined organic chemicals or simple mixtures.
The threshold of toxicological concern (TTC) approach may be helpful when assessing the risk of substances at low exposure levels (such as impurities, metabolites and degradation products present in (or derived from) the novel food) for which toxicity data are not available. Applicants are advised to consult the EFSA Guidance on the use of the TTC approach in food safety assessment (EFSA Scientific Committee, 2019b).
When microorganisms used as novel foods (active agents and biomasses) or in the production of novel foods (production strains) meet the criteria for the QPS approach to safety assessment (namely, (i) unambiguous taxonomic identification as belonging to a species included in the QPS list, (ii) any QPS qualification is met and (iii) for productions strains, no concerns are raised by genetic modification(s)), toxicological studies with the novel food will only be required in relation to possible safety concerns identified elsewhere in the assessment process, e.g. production process.
8.2. Tiered approach to conducting toxicological studies
Appropriate toxicity studies should be conducted following tiered testing approaches both for genotoxicity and repeated dose toxicity. When the data requirements of Tier I, the lowest tier, provide sufficient information for the toxicity assessment (including from published literature and reports), and in the absence of triggers prompting the need to move to a higher tier of data requirements, no further studies are required. A description of the data requirements for the different tiers for the testing of genotoxicity and repeated dose testing and the triggers prompting the need to move to a higher tier of data requirements are described below.
While the approach for the genotoxicity testing generally follows a tiered approach as described below in Section 8.3, the testing strategy for the repeated dose toxicity testing requires more flexibility in order to address various different situations regarding data available in the scientific literature, on the production process, the compositional characterisation, intended uses and use levels and resulting intake estimates, available toxicological and human studies and existing HBGV relevant for the novel food. For instance findings from a Tier I subchronic toxicity study may trigger the need for performing Tier II reproductive and developmental toxicity studies. In cases where the data in the literature raise concerns regarding reproductive and developmental toxicity, a Tier III extended one‐generation reproductive toxicity study (EOGRTS), which covers also subchronic toxicity, may be more appropriate. This would be more efficient regarding time and the number of animals needed, as compared to performing a Tier I subchronic toxicity followed by a Tier II reproduction and developmental study. There may also be situations where the available data from Tier I or Tier II concern potential chronic toxicity or carcinogenicity. Such cases may require follow‐up investigations such as mechanistic studies and/or in exceptional cases by a Tier III chronic toxicity or carcinogenicity study.
The tiered approach to genotoxicity and repeated dose toxicity testing, their interrelationships and their connection to ADME information are illustrated in Figure 2 and are further explained below.
FIGURE 2.
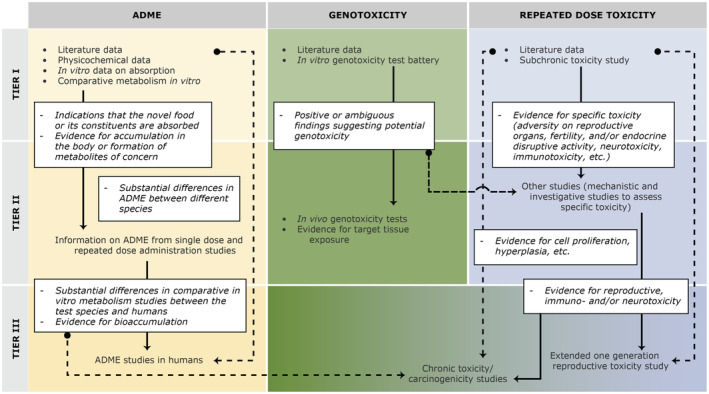
Tiered approach for ADME & toxicological assessment of novel foods. The boxes in white represent triggers that will call for higher‐tier studies as indicated by the arrows. Solid lines refer to the standardised way of conducting a tiered‐based approach by moving from lower to higher tier assessments; dashed lines refer to a deviated tiered‐based approach where the existing data require Tier III studies, from ADME to toxicity or from genotoxicity to repeated dose toxicity.
8.3. Genotoxicity
The assessment of the genotoxic potential is a basic component of chemical risk assessment in food and feed safety (EFSA Scientific Committee, 2011b). Accordingly, genotoxicity testing of novel foods or their constituents should aim at identifying substances which could cause genetic damage in humans.
The Scientific Committee recommended a tiered approach for the generation and evaluation of data on the genotoxic potential (EFSA Scientific Committee, 2011b).
Specific approaches should be followed based on the characteristics and composition of the novel food. If the novel food is a mixture, the suggested approach is described in the EFSA statement on Genotoxicity assessment of chemical mixtures (EFSA Scientific Committee, 2019a) where different scenarios are reported based on the chemical characterisation of the mixture. Similarly, for whole foods, it may be necessary to focus on specific constituents of the novel food.
If the novel food is a nanomaterial or contains a fraction of small particles, the genotoxicity assessment should follow the respective EFSA guidance documents (EFSA Scientific Committee, 2021a, 2021b).
In case microorganisms are used as novel foods, either as active agents or biomasses (Section 1.2), the applicant may be requested to perform genotoxicity testing depending on the taxonomic and hazard identification of the microorganisms. The recommended approach to evaluate genotoxicity is to test both the supernatant (from at least three independently produced batches of the novel food pooled together) and the cell lysate (from at least one batch of the novel food). Proof of efficient lysis of the cells/spores must be provided.
EFSA notes that there is currently no generally agreed‐upon genotoxicity testing strategy for novel foods produced with GMMs or derived from cell culture. Therefore, case‐by‐case evaluations are necessary.
8.3.1. Tier I genotoxicity testing
A basic battery of in vitro tests, comprising both a bacterial reverse mutation assay (OECD TG 471), (OECD, 2020) and an in vitro micronucleus assay (OECD TG 487) (OECD, 2023a), should be performed as a first step, before conducting in vivo studies (EFSA Scientific Committee, 2011b). Special considerations are needed for Tier 1 genotoxicity testing when the novel food is e.g. a nanomaterial or proteins/peptides, for which for example mammalian gene mutation assays (EFSA Scientific Committee, 2021b) and/or the bacterial reverse ‘treat and plate’ methodology (EFSA CEP Panel, 2021) might be more appropriate. In case a of positive outcome of the in vitro micronucleus test, the applicant will be requested to further investigate whether the novel food induces aneugenicity by performing a kinetochore staining or fluorescence in situ hybridisation (FISH). Therefore, the applicant is advised to store relevant samples for further analysis testing.
8.3.2. Tier II genotoxicity testing
In case of positive or ambiguous results for genotoxicity from the Tier I in vitro test battery, the follow‐up approaches, as well as recommendations on test types, interpretations of results, evidence of target tissue exposure and other issues in testing in vivo the genotoxicity of substances present in food, are described in detail in the Opinions of the Scientific Committee (EFSA Scientific Committee, 2011b, 2017a, 2017b, 2021b).
8.3.3. Tier III genotoxicity testing
If the novel food results in positive in vivo genotoxicity tests, no further testing is necessary, and the substance should be considered as genotoxic in vivo.
8.4. Repeated‐dose toxicological studies
8.4.1. Tier I repeated‐dose toxicological studies
8.4.1.1. Subacute toxicity
Subacute studies (e.g. 14‐day) may be conducted providing the basis for the selection of appropriate doses to be used in the subchronic setting. In this case, the full technical report of the dose range‐finding study should be submitted.
8.4.1.2. Subchronic toxicity
A subchronic (90‐day) toxicity study is often needed as part of Tier I. Such a study is usually required when the novel food concerns a substance or contains components of unknown toxicity, when there are no HBGVs for the components of a novel food, or when an uncharacterised fraction of the novel food may cause safety concerns despite thorough compositional analyses. If a subchronic study is not conducted, a well‐reasoned justification should be provided.
When conducting such a study, the main objective is to identify any adverse effects following prolonged exposure to the novel food via an appropriate oral route. Ideally, the study should allow the determination of a reference point (RP), namely either BMDL (the lower bound of the benchmark dose – EFSA Scientific Committee, 2022a) or a NOAEL (no‐observed‐adverse‐effect‐level), that is subsequently used for the risk characterisation of the novel food. The RP is generally used in the risk assessment process to verify whether a sufficient margin with the estimated novel food intake (or its relevant components) exists. With this aim, an uncertainty factor (UF) of 200 (100 as the overall default factor for inter‐species and intra‐human variability and 2 to extrapolate from subchronic to chronic duration studies) is usually applied to the RP identified in the 90‐day study (EFSA Scientific Committee, 2012a). On a case‐by‐case basis and upon scientific justification, a different UF may be considered. The results obtained in the subchronic toxicity study can also provide indications of the need for additional studies on specific effects (Section 8.2).
The study should normally cover a period of at least 90 days according to the OECD TG 408 (OECD, 2018a) and a satellite recovery group can be included for follow‐up observations.
Deviations to the relevant TG may be accepted provided that they are scientifically justified (e.g. modified groups and doses for BMD modelling, administration of test the item using pre‐weaning animals for novel food intended for infants). With reference to the test material to be used, the considerations reported in Section 8.1 apply. A rationale for the administration route selected (e.g. by gavage, via the diet) in consideration of the conditions of use of the novel food should be provided.
In addition to the full study report, the applicant should provide a summary table reporting all the statistically significant and/or dose‐dependent results (males and females separately, at all dose levels, with mean and standard deviation for each parameter). Relevant historical control data for the species/strain used should be also included.
In consideration of physicochemical properties or results from ADME (e.g. suggestive of possible accumulation), satellite groups for possible toxicokinetic determinations should be considered. If there are concerns about effects on specific systems such as endocrine, reproductive or cardiovascular systems (e.g. based on literature or in vitro data), optional endpoints covering these effects should be considered and included in the experimental design (e.g. for specific hormonal measurements). It is recommended to store biological samples (e.g. serum samples) collected at the end of the treatment period for possible additional determinations.
Other measurements considered optional by OECD TG 408 (OECD, 2018a), such as water consumption (also when the novel food is orally administered) and urine analyses, might also be helpful for the interpretation of findings in the 90‐day study.
For whole foods or novel foods, mainly composed of macronutrients, specific considerations may be required with regard to dose selection and the avoidance of possible nutritional imbalances due to a high incorporation level of the test item into the animals' feed (also Section 8.1). For further guidance on the conduction of subchronic oral toxicity studies with whole foods, the applicant is advised to consult the relevant guidance from the Scientific Committee (EFSA Scientific Committee, 2011a).
Safety studies originally designed for addressing conventional materials are considered, in general, to sufficiently cover the hazard assessment of small particles, including nanoparticles, provided that specific precautions and adaptations are implemented according to the relevant guidance document (EFSA Scientific Committee, 2021a, Section 4).
When indications of reproductive and/or endocrine effects are identified (from the literature, in vitro, in vivo and/or human studies), the applicant is advised to include additional endpoints in the 90‐day subchronic toxicity study (Section 8.4.2.1).
8.4.2. Tier II repeated‐dose toxicological studies
8.4.2.1. Reproductive, endocrine and developmental toxicity
Decisions on whether tests for reproductive and developmental toxicity are necessary should take into account the kinetic and toxicity data, including read‐across data from structurally related compounds and available data from the literature.
Any indications of effects on reproductive organs or parameters, as observed in vitro and/or in vivo, may trigger the need for testing for reproductive and developmental toxicity. Potential additional tests include, but are not limited to, studies covered by OECD TG 414 (OECD, 2018b), 416 (OECD, 2001), 421 (OECD, 2016), 422 (OECD, 2015), 426 (OECD, 2007a), 440 (OECD, 2007b), 441 (OECD, 2009), 455 (OECD, 2021b), 456 (OECD, 2023b) and 493 (OECD, 2024). Reproductive and developmental toxicity testing may not be required if scientifically justified on a case‐by‐case basis.
When indications of reproductive and/or endocrine effects are identified (from the literature, in vitro, in vivo and/or human studies), the applicant is advised to include at least the following endpoints in the 90‐day subchronic toxicity study: oestrous cycling for at least 15 days prior to necropsy, sperm quality analyses, histopathological analyses of reproductive organs and hormonal measurements at necropsy (including gonadotropins, sex and thyroid hormones). The applicant is advised to store samples to allow for such analyses should endocrine or reproductive effects be identified in the subchronic toxicity study itself.
8.4.2.2. Other tier II studies
The need for other studies, e.g. studies on neurotoxicity, cardiovascular effects, immunotoxicity, hypersensitivity and food intolerance, mechanism (mode of action), may be triggered by findings reported in the literature or in Tier I or II.
8.4.3. Tier III repeated‐dose toxicological studies
Tier III studies comprise toxicological studies of high complexity regarding the duration and the required number of animals.
8.4.3.1. Extended one‐generation reproductive toxicity study
The extended one‐generation reproductive toxicity study (EOGRTS), as conducted according to OECD TG 443 (OECD, 2018c), is designed to evaluate reproductive and developmental effects that may occur as a result of pre‐ and postnatal chemical exposure, as well as an evaluation of systemic toxicity in pregnant and lactating females and young and adult offspring. The study covers reproductive/developmental toxicity, developmental neurotoxicity and developmental immunotoxicity. Indications of such toxic effects (from the literature, in vitro, in vivo and/or human studies) may trigger the request for an EOGRTS without the need for tier II studies.
8.4.3.2. Chronic toxicity and carcinogenicity
Chronic toxicity or carcinogenicity studies are normally not required. However, in exceptional cases, such studies may be needed (e.g. accumulation of the substance, or hyperplasia observed in subchronic toxicity studies) and should follow the respective OECD Test Guidelines (OECD TG 451 (OECD, 2018d), 452 (OECD, 2018e) or 453 (OECD, 2018f)).
8.5. Human data
Human intervention studies, if available, should be provided, regardless of the primary objective of the study, as long as safety aspects were also investigated. For example, efficacy studies with the novel food, even though not directly pertinent for safety, may provide information relevant for the safety assessment, such as physical examination (including blood pressure, heart rate, body weight and height), blood chemistry, haematology, urine analysis, organ function tests and/or monitoring of adverse reactions/tolerance. Relevant data may also be derived from the use of the novel food in observational studies.
In some cases, human studies may be required to establish the safety of the novel food, for example, to investigate further potentially adverse effects observed in toxicological studies (e.g. conjugated linoleic acid (CLA)‐rich oil (EFSA NDA Panel, 2010a, 2010b) or for effects that cannot be investigated in animals (e.g. psychological outcomes, mental health). In those cases, human studies are required to demonstrate that the proposed use of the novel food does not raise safety concerns. When performing such safety studies, applicants need to consider carefully elements such as study design, sample size, study population (representative of the target population of the novel food), study duration, safety endpoints and dose range(s) of the novel food.
For the risk assessment of novel foods added to foods intended for infants below 16 weeks of age, the respective EFSA Guidance needs to be followed (EFSA Scientific Committee, 2017a, 2017b).
The data from intervention studies and observational studies in humans should be organised and considered according to a hierarchy of study designs, and reflecting the relative strength of evidence which may be obtained from different types of studies (EFSA Scientific Committee, 2017a, 2017b).
For novel foods that are also new sources of micronutrients, human intervention studies may be required to assess both the safety and the relative bioavailability of the micronutrient from the new source (EFSA NDA Panel, 2024b).
9. NUTRITIONAL INFORMATION
It must be investigated whether the novel food could be nutritionally disadvantageous for consumers under the proposed conditions of use. The general framework for the assessment of the nutritional impact of the novel food is outlined in Figure 3. The information provided should demonstrate that, at the anticipated levels of intake, the introduction of the novel food in the diet is not expected to contribute to an excess intake of nutrients (Section 9.1) or to adversely affect the nutritional status 60 of consumers by increasing the risk of inadequate nutrient intakes (Section 9.2).
FIGURE 3.
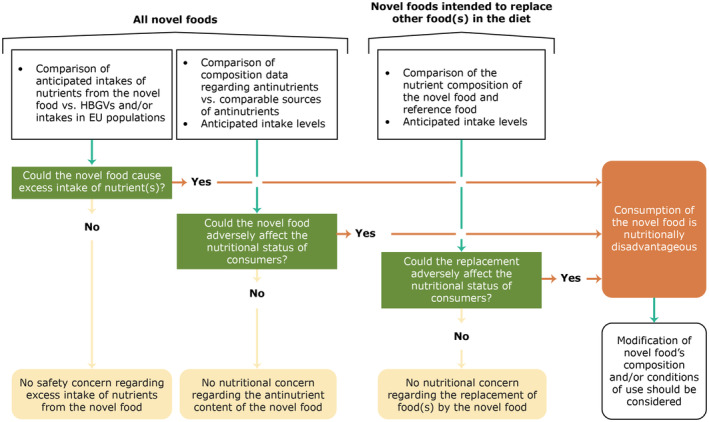
Flowchart for investigating a potential detrimental nutritional impact of the novel food under the proposed conditions of use. The assessment is restricted to nutrient and non‐nutrient components (e.g. dietary fibre) with an established nutritional role in the diet, i.e. for which dietary reference values (DRVs) have been established by EFSA. Safety concerns regarding excess intake of nutrients are assessed by comparing the anticipated intake of nutrients from the novel food to the applicable health‐based guidance values (HBGVs), typically tolerable upper intake levels (ULs). Regarding the assessment of the impact of the novel food on the nutritional status of consumers (due to its antinutrient content or the replacement of other food(s)), particular attention should be paid to the essential nutrients for which intakes are below recommended levels in European populations (EFSA NDA Panel, 2022d).
Specific requirements regarding nutritional information also apply in case the novel food is a new source of micronutrients (Section 9.3) or is a novel protein source (Section 9.4).
9.1. Excess intake of nutrients
A novel food is considered nutritionally disadvantageous if its consumption, under the proposed conditions of use, could lead to an excess intake of nutrients for one or more population groups, i.e. exceeding the ULs. 61 When no UL is available for a nutrient, other HBGVs (e.g. ADI for copper (EFSA Scientific Committee, 2023)) should be considered. The anticipated intake of relevant nutrients through the novel food consumption should be estimated for the target population group(s) (Section 6.1), considering the maximum specification limits 62 and the maximum proposed use levels (Section 6.2). The applicant should consider the combined nutrient intakes from the novel food and other sources of that nutrient (Section 6.4) (e.g. nutrient from the background diet, as in the safety assessment of vitamin D2 mushroom powder, EFSA NDA Panel, 2022c). The resulting nutrient intake estimates are to be compared to the applicable HBGV. The combined intake of the nutrient from the novel food and the background diet should be considered where relevant (e.g. narrow margin between the background intake of the nutrient and its HBGV). 63 In case no HBGV is available, the extent to which the novel food may increase the intake of the nutrient as compared to intakes from the background diet should be discussed and justified.
9.2. Inadequate intakes of essential nutrients
A novel food is considered nutritionally disadvantageous if its consumption, under the proposed conditions of use, has the potential to adversely affect the nutritional status of consumers by increasing the risk of inadequate nutrient intakes. In that context, particular attention is to be paid to essential nutrients for which intakes are below recommended levels in European populations (EFSA NDA Panel, 2022d).
The evaluation of the potential of the novel food to lead to inadequate intakes of essential nutrients should be based on compositional data (Section 3) and comparisons with other foods as described below.
9.2.1. Antinutrient content
Antinutrients are compounds that can interfere with the absorption of essential nutrients. Novel foods can contain antinutrients 64 such as tannins, lectins, trypsin inhibitors, amylase inhibitors, phytic acid and phytates, oxalates or saponins, among others. Considering the source of the novel food, information on the antinutrient content in the novel food as consumed must be provided and compared to the antinutrient content of comparable foods (e.g. partially hydrolysed protein from spent barley and rice (EFSA NDA Panel, 2023c)).
9.2.2. Replacement of food(s) in the diet
When a novel food is intended to replace a conventional food (e.g. novel food UV‐treated milk vs. conventional milk (EFSA NDA Panel, 2016), the applicant has to demonstrate that the nutritional composition of the novel food does not differ from that of the conventional food in a way that would be nutritionally disadvantageous for consumers under the proposed conditions of use. This includes the application of a novel production process that can affect the nutrient composition of a food. In such cases, the nutrient composition of the conventional counterpart should also be provided, focusing on the nutrient(s) for which it represents a significant source in diets. Indicatively, foods which contain 15% per 100 g or 100 mL (or 7.5% per 100 mL for beverages) of the reference intakes for vitamins and minerals are considered significant sources. 65
9.3. Specific considerations for novel foods proposed as new sources of micronutrients
For novel foods comprising single substances or simple mixtures thereof suggested as new sources of micronutrients (i.e. vitamins and minerals) for addition to foods 66 (including foods for special groups 67 ) and/or to be consumed as food supplements, 68 applicants are referred to the EFSA guidance on the scientific principles and data requirements for the safety and relative bioavailability assessment of new micronutrient sources on new sources of micronutrients (EFSA NDA Panel, 2024b). The guidance outlines the principles for assessing and quantifying the micronutrient bioavailability from the new source compared to a reference source, as well as the potential impact of the new source on nutrient adequacy and/or excess.
9.4. Specific considerations regarding novel protein sources
The protein quality of the novel food must be investigated if the highest mean consumption of the novel food under the proposed conditions of use could substantially contribute to the average requirement (AR) for protein for one or more population groups (EFSA NDA Panel, 2012). A substantial contribution is defined as at least 15% of the population‐specific AR for protein (Appendix C). Additionally, protein quality must be assessed when the novel food is intended to serve as a source of protein in ‘single meal replacement products for weight control’ or foods for special medical purposes. 69 Specific regulatory requirements 70 apply to evaluating protein sources used in manufacturing total diet replacement for weight control products and infant and follow‐on formula.
Protein quality is to be assessed using the digestible indispensable amino acid score (DIAAS) method, following FAO recommendations (FAO, 2013, FAO & IAEA, 2024). This involves investigating the true ileal digestibility of each indispensable amino acid (IAA) and combining the respective digestibility coefficients with IAA composition data, in order to derive the digestible IAA content of the novel food. A comparison with an age‐specific IAA reference scoring pattern should be made (FAO, 2013) to derive the final DIAAS value. The protein content should be calculated as described in Section 3.1.4.
The information on protein quality will inform of the nutritional characteristics of the protein in the novel food. Additional aspects such as the expected contribution of the novel food to total protein dietary intake must be taken into account to determine if consumption of the novel food could be nutritionally disadvantageous. This is important, especially in cases where the novel food is intended to be the sole dietary protein source (e.g. FSGs).
The test material for the digestibility study must be the novel food itself. Information on the validation of the method should be provided. Methods to measure the true ileal digestibility of amino acids in vivo have been established in animals and humans (FAO & IAEA, 2024). In vitro models have also been developed but yet not validated (FAO & IAEA, 2024). If an in vitro method is employed, the suitability of the method in consideration will be examined during the risk assessment. The suitability of the method will be assessed based on the following minimum requirements:
standardised test conditions that reflect the environment of the upper GI tract (e.g. relevant enzymes and activity, pH, time, temperature);
inclusion of digestion of a blank sample (i.e. protein‐free comparator) and a reference protein (e.g. casein, whey protein);
suitable methods to differentiate the absorbable and non‐absorbable fraction of the digesta.
The certificates of analysis for the test materials (batches) used in protein digestibility studies should be provided.
The protein quality of a food depends on several factors such as the total protein content, the content of IAAs, the presence of antinutrients and processing. Depending on the variability of these parameters in the novel food, measurements of protein digestibility on several batches may be necessary for a more precise characterisation of its protein quality. The above factors must be considered on a case‐by‐case basis, and the number of batches tested should be justified.
9.5. Additional information
In specific cases, data from investigations in in vitro, in silico and/or in animal models and/or human studies may be needed to address the interaction of the novel food with the diet and nutrients. The necessity for such studies may arise from information on the source, the composition and the production of the novel food, from documented experience on the use, preparation and/or handling of the novel food (e.g. foods consumed in non‐EU countries) and outcomes of ADME, pharmacological, mechanistic, feeding, toxicological or human studies.
10. ALLERGENICITY
Data requirements in this section aim to collect the available information related to the allergenicity of the novel food and to provide guidance on generating data sets on the potential allergenic properties. Such evidence could support regulatory decision‐making by risk managers, including possible labelling requirements. Data requirements may vary across novel foods, depending on the nature of the novel foods, the production process, which includes their source, and their allergen labelling status as per Regulation (EU) No 1169/2011. 71 The general framework for investigating the allergenic potential of the novel food is outlined in Figure 4.
FIGURE 4.
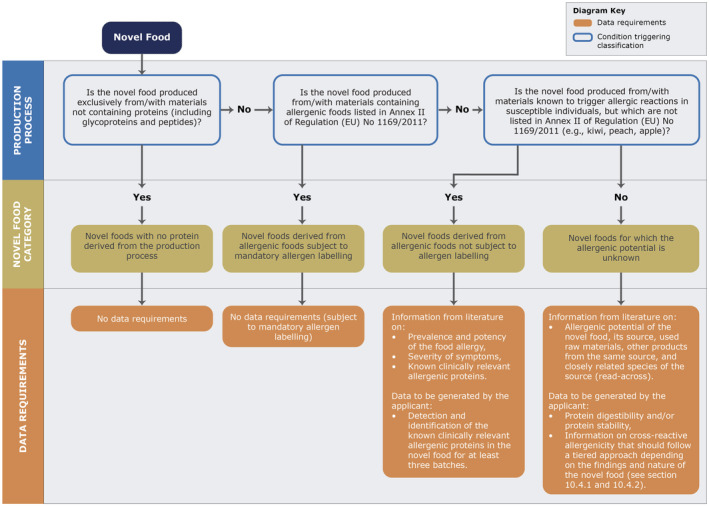
Flowchart for investigating the allergenic potential of the novel food.
10.1. Novel foods with no protein derived from the production process
Food allergens are mostly proteins (including glycoproteins and peptides). Hence, there are no allergenicity‐specific data requirements for novel foods which are exclusively produced from/with raw materials, which do not contain or use proteins (including glycoproteins, enzymes and others) or peptides, e.g. novel foods produced by chemical synthesis from high purity chemicals not containing proteins or novel foods of mineral origin.
10.2. Novel foods derived from allergenic foods subject to mandatory allergen labelling
Novel foods which either consist of, or are isolated from, or are produced from allergenic foods and/or with processing aids containing allergenic foods listed in Annex II of Regulation (EU) No 1169/2011 are by default assumed to retain the allergenic potential of the source and are subject to mandatory allergen labelling under that Regulation.
However, if such novel foods also contain proteins from sources that are not subject to mandatory labelling, the potential allergenic properties of these proteins should be investigated. Depending on the knowledge about these proteins and their source(s), the requirements in Sections 10.3 and 10.4 also apply to such novel foods.
10.3. Novel foods derived from allergenic foods not subject to mandatory allergen labelling
This section addresses novel foods derived from sources (or products thereof) known to trigger allergic reactions in susceptible individuals, but which are not listed in Annex II of Regulation (EU) No 1169/2011 (e.g. kiwi, peach, apple). The default assumption is that the allergenic potential of these novel foods is at least that of the source. The information outlined in points 1–4 should be provided if available in the literature, whereas the evidence requested in the last point is to be generated by the applicant:
Prevalence of the food allergy related to the novel food source;
Type and severity of symptoms triggered by the allergenic food (i.e. the source);
Potency of the allergenic food (i.e. the source). For example, from the clinical history, the food portion that caused a reaction may be used to calculate the amount of protein able to trigger a reaction (i.e. minimal eliciting doses of total protein in the food triggering allergic reactions in susceptible individuals);
Known clinically relevant allergenic proteins of the source;
Detection and identification of the known clinically relevant allergenic proteins in the novel food in at least three batches using appropriate immunological or proteomic approaches, methods of analysis, the LOD of the methods and the complete protocol for protein extraction.
10.4. Novel foods for which the allergenic potential is unknown
The information required here mainly addresses the allergenic potential of novel foods due to potential cross‐reactivity and not the potential for de novo sensitisation, for which there are no validated predictive methods currently available. The information that should be provided for novel foods not falling under categories pertinent to Sections 10.2 and 10.3 is as follows.
A comprehensive literature search and review that cover the novel food, its source, used raw materials, other products from the same source and closely related species of the source. It should include all types of studies such as in silico, in vitro, in vivo and human studies including case reports and all types of evidence which could be an indication for the allergenic potential of the novel food, such as in vitro and in vivo reactivity, cross‐reactivity, elicitation dose, sensitisation and clinical effects, also considering different routes of exposure (e.g. oral intake, skin contact, inhalation);
Protein digestibility and/or protein stability performed in the context of other sections (Sections 7.1 and 9.4; EFSA GMO Panel, 2021, 2022) should be also considered in the allergenicity assessment;
Information on cross‐reactive allergenicity that should follow a tiered approach. Testing for cross‐allergenicity should be considered only if cross‐reactivity of the novel food is demonstrated for known allergens subject to mandatory labelling and/or known to trigger severe allergic reactions (e.g. anaphylaxis) in sensitive individuals.
The type and extent of the required analyses depend on the nature of the product, i.e. novel foods with single protein/simple protein mixtures vs. whole foods or novel foods containing complex protein mixtures.
10.4.1. Single proteins and simple protein mixtures
This section concerns novel foods containing either a single protein or a mixture of a few identified proteins. A tiered approach should be followed by default to investigate potential cross‐allergenicity, as outlined in the subsections below and schematically presented in Figure 5.
FIGURE 5.

Tiered approach to investigate potential cross‐allergenicity of single proteins and simple protein mixtures.
10.4.1.1. Tier I allergenicity testing of single proteins and simple protein mixtures
Information on the allergenicity of the source of the novel food should be based on a comprehensive literature search and review, as described above, and on the phylogenetic relationships with sources containing known food allergens.
10.4.1.2. Tier II allergenicity testing of single proteins and simple protein mixtures
To predict cross‐reactivity, various algorithms and allergen‐sequence databases are available. Currently, there is no consensus on the most appropriate approach to use for risk assessment (EFSA GMO Panel, 2022). By default, amino acid sequence (AAS) homology comparison should be performed using publicly available search engines such as the FASTA local alignment algorithm or the Basic Local Alignment Search Algorithm (BLAST), and a default threshold of 35% identity over at least 80 amino acids was established by FAO/WHO in 2001. Such an approach was adopted by Codex Alimentarius (FAO/WHO, 2009) and, subsequently, by EFSA (EFSA GMO Panel, 2010, 2011). However, this approach in isolation is considered highly conservative and criticised for triggering a high number of false positives. Therefore, complementary approaches such as 3D protein structure similarity searches, machine learning strategies based on IgE epitope matching could be also used (EFSA GMO Panel, 2022).
10.4.1.3. Tier III allergenicity testing of single proteins and simple protein mixtures
If the results of bioinformatic analyses suggest potential cross‐allergenicity with a known food allergen that triggers severe IgE‐mediated allergic reactions in sensitive individuals, a follow‐up analysis of specific IgE binding using human serum should be performed, on at least three batches of the novel food. Serum should be sourced from allergic individuals who have a confirmed food allergy to the known allergen with which the novel food may cross‐react. Confirmation should include relevant history, symptoms and time of onset consistent with an IgE‐mediated food allergy to a relevant food, evidence of sensitisation to that food and diagnosis confirmed via oral food challenge. Immunoassay methods such as ELISA or electrophoresis combined with immunoblotting with serum IgE sera are considered adequate to assess cross‐reactivity with known allergens. If obtaining the necessary serum proves challenging, the applicant should thoroughly document their efforts.
10.4.1.4. Tier IV allergenicity testing of single proteins and simple protein mixtures
Positive results from Tier III necessitate further investigation into the potential for cross‐allergenicity in humans, i.e. whether individuals allergic to the known food allergen, which shares high AAS homology with the novel food protein(s), could also exhibit allergic reactions to the novel food. In such cases, skin‐prick tests and an oral food challenge (preferably double‐blind placebo‐controlled) are required in subjects with confirmed food allergy to the known allergen.
10.4.2. Complex protein mixtures and whole foods
The allergenicity assessment strategy for complex protein mixtures and whole foods should follow a tiered approach, similar to what is described in Section 10.4.1.
In a first step, a phylogenetic analysis of the novel food source should be performed to gain initial information on the potential for cross‐reactive allergenicity. On a case‐by‐case basis and depending on the phylogenetic differences identified, cross‐reactivity analysis may not be needed. However, if a phylogenetic relationship with known food allergens triggering severe IgE‐mediated allergic reactions in sensitive individuals is identified, a sequence analysis of the most representative proteins of the novel food should be conducted. Depending on the protein similarities identified with known allergens and their associated clinical relevance, the investigation should proceed as outlined in Tier III and Tier IV of Section 10.4.1.
11. CONCLUDING REMARKS
The applicant should integrate and interpret the data presented in the previous sections and provide overall considerations on how the provided body of evidence supports the safety of the novel food under the proposed conditions of use.
Where potential health hazards have been identified, they should be discussed in relation to the anticipated intakes of the novel food and the proposed target populations.
In particular, the applicant should address:
the relevance of toxicologically and nutritionally relevant components (e.g. impurities, by‐products, residues, chemical or microbiological contaminants and nutrients) in relation to their estimated intakes, possible background exposure and their health‐based guidance values;
the results of toxicity studies;
any adverse effects identified through the human data;
sources of uncertainties.
The main findings of each toxicity study (both unpublished and published) should be highlighted, together with the method for the identification of the reference point (e.g. BMDL or NOAEL), and any other relevant information. Where necessary, the conclusions should include an interpretation of the importance of the findings in terms of possible mechanisms underlying any effects observed, a discussion of whether these are relevant to humans and, if so, the possible importance of the extrapolation of such findings to humans. Where available, human intervention studies assessing safety endpoints should also be discussed in this context. For novel foods that are also proposed as nutrient sources, conclusions on both safety and (relative) bioavailability should be discussed.
STEPS TAKEN BY EFSA
On 16 June 2023 EFSA received a mandate from the European Commission with the request to update the guidance document on the preparation and submission of an application for authorisation of a novel food in the context of Regulation (EU) 2015/2283. [Ref. Ares (2023) 4194279].
During its meeting on 31 January 2024, the NDA Panel endorsed the draft guidance document for public consultation.
The public consultation was open from 15 February 2024 to 14 April 2024.
The draft guidance document has been amended in view of the comments received during the public consultation. All comments have been addressed by the NDA Panel and are available alongside the respective replies in Annex A of this guidance document.
During its meeting on 27 June 2024, the NDA Panel adopted the guidance on the scientific requirements for an application for authorisation of a novel food in the context of Regulation (EU) 2015/2283.
GLOSSARY
- Acceptable daily intake (ADI)
An estimate of the amount of a substance in food or drinking water that can be consumed daily over a lifetime without presenting an appreciable risk to health. It is usually expressed as milligrams of the substance per kilogram of body weight per day and applies to chemical substances such as food additives, pesticide residues and veterinary drugs (EFSA Scientific Committee, 2021c).
- Adverse effect
Change in the morphology, physiology, growth, development, reproduction or lifespan of an organism, system or (sub)population that results in an impairment of functional capacity to compensate for additional stress or an increase in susceptibility to other influences (FAO/WHO, 2009; EFSA Scientific Committee, 2017c).
- Antimicrobial
An active substance of synthetic or natural origin which destroys microorganisms, suppressing their growth or their ability to reproduce in animals or humans, excluding antivirals and antiparasitic agents. For the purpose of the assessment of antimicrobial susceptibility and production in this guidance, the antimicrobial substances considered are those of clinical relevance (EUCAST, 2024).
- Antinutrients
Antinutrients are naturally occurring or synthetic compounds that can interfere with the absorption of essential nutrients.
- Average requirement (AR)
The level of intake of a defined group of individuals estimated to satisfy the physiological requirement or metabolic demand, as defined by the specified criterion for adequacy for that nutrient, in half of the healthy individuals in a life stage or sex group, on the assumption that the supply of other nutrients and energy is adequate (EFSA NDA Panel, 2022e).
- Benchmark dose (BMD)
A dose level, estimated from the fitted dose–response curve, associated with a specified change in response relative to the control group (background response), the benchmark response (BMR) (EFSA Scientific Committee, 2022b).
- Bioavailability
Nutrient fraction which is absorbed and becomes available to normal metabolic and physiological processes (EFSA NDA Panel, 2022e).
- Dietary reference values (DRV)
A set of nutrient reference values that includes the average requirement, the population reference intake, the adequate intake and the reference intake range for macronutrients (EFSA NDA Panel, 2017).
- Engineered nanomaterial
Any intentionally produced material that has one or more dimensions of the order of 100 nm or less, or that is composed of discrete functional parts, either internally or at the surface, many of which have one or more dimensions of the order of 100 nm or less, including structures, agglomerates or aggregates, which may have a size above the order of 100 nm but retain properties that are characteristic of the nanoscale (Novel Food Regulation (EU) No 2015/2283, point (f) of Article 3(2)). Properties that are characteristic of the nanoscale include: (i) those related to the large specific surface area of the materials considered; and/or (ii) specific physico‐chemical properties that are different from those of the non‐nanoform of the same material (EFSA Scientific Committee, 2021a, 2021b).
- Health‐base guidance value (HBGV)
Umbrella term for values that are established as the result of the risk assessment of chemical substances and provides guidance on the safe consumption of substances, taking into account current safety data, uncertainties in these data, and the likely duration of consumption. Depending on their nature and applications, an HBGV for oral exposure may be termed tolerable upper intake level (UL) (nutrients), acceptable daily intake (ADI) (food additives, pesticides), tolerable daily intake (TDI) (contaminants) or acute reference dose (ARfD) (EFSA Scientific Committee, 2021c).
- Margin of exposure (MoE):
The MOE is the ratio of the no‐observed‐adverse‐effect level (NOAEL) or benchmark dose lower confidence limit (BMDL) for the critical effect to the theoretical, predicted, or estimated exposure dose or concentration to a substance for a given population (EFSA Scientific Committee, 2022b).
- No‐observed‐adverse‐effect‐level (NOAEL)
Highest dose of a substance, found by experiment or observation, that does not cause a statistically significant or biologically relevant adverse effect.
- Read‐across
Approach used in chemical risk assessment that allows for the screening, classification, prioritisation and hazard assessment of chemicals based on the toxicological data for similar chemicals, and is the most common alternative to animal testing. In read‐across, known information from one or more source (data‐rich) substances is used to predict the same property for a (data‐poor) target substance.
- Taxonomic units (TUs)
The lowest taxonomic level for which the QPS status is granted ‐ the species level for bacteria, yeasts and protists/algae, and the family level for viruses.
- Threshold of Toxicological Concern (TTC)
The TTC approach is a screening and prioritisation tool for the risk assessment of chemicals when hazard data are incomplete and human exposure can be estimated (EFSA Scientific Committee, 2019a, 2019b). For substances with exposures below their corresponding TTC values, the probability that they would cause adverse health effects is low. If the estimated exposure to a substance is higher than the relevant TTC value, a non‐TTC approach is required to reach a conclusion on potential adverse health effects.
- Tolerable upper intake levels (UL)
The maximum level of total chronic daily intake of a nutrient (from all sources) which is not expected to pose a risk of adverse health effects to humans. (EFSA NDA Panel, 2022e).
- Uncertainty
General term referring to all types of limitations in available knowledge that affect the range and probability of possible answers to an assessment question. Available knowledge refers here to the knowledge (evidence, data, etc.) available to assessors at the time the assessment is conducted and within the time and resources agreed for the assessment (EFSA, 2019).
- Weight of evidence
A process in which evidence is integrated to determine the relative support for possible answers to a question. The weight of evidence assessment comprises three basic main steps: (1) assembling the evidence into lines of evidence of similar type, (2) weighing the evidence, (3) integrating the evidence (EFSA Scientific Committee, 2017a, 2017b).
ABBREVIATIONS
- AAS
amino acid sequence
- ADI
acceptable daily intake
- ADME
absorption, distribution, metabolism, excretion
- AMR
antimicrobial resistance
- ARfD
acute reference dose
- AR
average requirement
- BIOHAZ
Panel on Biological Hazards
- BLAST
basic local alignment search tool
- BMD
benchmark dose
- BMDL
lower confidence limit for a benchmark dose
- BMR
benchmark response
- bw
body weight
- CAS
Chemical Abstracts Service
- CEP
Panel on Food Contact Materials, Enzymes and Processing Aids
- ChEBI
Chemical Entities of Biological Interest database
- ChEMBL
database of bioactive molecules with drug‐like properties. It is maintained by the European Bioinformatics Institute (EBI) of the European Molecular Biology Laboratory (EMBL)
- CLA
conjugated linoleic acid
- CLP
classification
labelling and packaging
- COL
Catalogue of Life
- DIAAS
digestible indispensable amino acid score
- DietEx
Dietary Exposure tool
- DNA
deoxyribonucleic acid
- DRV
dietary reference value
- ECHA
European Chemicals Agency
- EEA
European Economic Area
- ELISA
enzyme‐linked immunosorbent assay
- EMA
European Medicines Agency
- EOL
Encyclopaedia of Life
- EOGTRS
extended one‐generation reproductive toxicity study
- EUCAST
European Committee on Antimicrobial Susceptibility Testing
- FAIM
Food Additives Intake Model
- FAO
Food and Agriculture Organisation of the United Nations
- FASTA
sequence similarity search with a protein query
- FEEDAP
Panel on Additives and Products or Substances used in Animal Feed
- FISH
fluorescence in situ hybridisation
- FooDB
database of the macro and micronutrients of a wide range of non‐processed foods
- FoodEx
EFSA Food Classification System
- FSG
goods for specific groups
- FSMP
foods for special medical purposes
- FT‐IR
Fourier transform infrared spectroscopy
- GCP
good clinical practice
- GBIF
Global Biodiversity Information Facility
- GI
gastrointestinal
- GLP
good laboratory practice
- GM
genetically modified
- GMM
genetically modified microorganism
- GMO
genetically modified organism
- GMP
good manufacturing practice
- GRIN
Germplasm Resources Information Network
- HACCP
hazard analysis critical control point
- HBGV
health‐based guidance values
- HMDB
Human Metabolome Database
- IAA
indispensable amino acid
- IAEA
International Atomic Energy Agency
- ICH
International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use
- IgE
immunoglobulin E
- InChI
International Chemical Identifier
- IPNI
International Plant Names Index
- ITIS
Integrated Taxonomic Information System
- IVIVE
in vitro to in vivo extrapolation
- ISO
International Organization for Standardization
- IUBMB
International Union of Biochemistry and Molecular Biology
- IUPAC
International Union of Applied and Pure Chemistry
- LOD
limit of detection
- LOQ
limit of quantification
- M‐ARCOL
mucosal artificial colon
- MDCK
Madin‐Darby canine kidney
- NAM
new approach methodology
- NDA
EFSA Panel on nutrition novel foods and food allergens
- NMR
nuclear magnetic resonance
- NOAEL
no observed adverse effect level
- OECD
Organisation for Economic Co‐operation and Development
- PBTK
Physiologically based toxicokinetic
- PCBs
polychlorinated biphenyls
- PPR
Panel on Plant Protection Products and their Residues
- QPS
qualified presumption of safety
- REACH
Registration, Evaluation, Authorisation and Restriction of Chemicals
- RP
reference point
- SCF
Scientific Committee on Food
- SHIME
Simulator of the Human Intestinal Microbial Ecosystem
- SMILES
simplified molecular input line entry system
- TDI
tolerable daily intake
- TG
Test Guideline
- TR
Transparency Regulation
- TTC
threshold of toxicological concern
- TU
taxonomic unit
- UF
uncertainty factor
- UL
tolerable upper intake level
- UV
ultraviolet spectroscopy
- USDA‐ARS
United States Department of Agriculture‐Agricultural Research Service
- UV–VIS
ultraviolet–visible spectroscopy
- XRD
X‐ray diffraction
- WGS
whole genome sequence
- WHO
World Health Organization
CONFLICT OF INTEREST
If you wish to access the declaration of interests of any expert contributing to an EFSA scientific assessment, please contact interestmanagement@efsa.europa.eu.
REQUESTOR
European Commission
QUESTION NUMBER
EFSA‐Q‐2023‐00442
COPYRIGHT FOR NON‐EFSA CONTENT
EFSA may include images or other content for which it does not hold copyright. In such cases, EFSA indicates the copyright holder and users should seek permission to reproduce the content from the original source.
PANEL MEMBERS
Panel members: Dominique Turck, Torsten Bohn, Jacqueline Castenmiller, Stefaan de Henauw, Karen‐Ildico Hirsch‐Ernst, Helle Katrine Knutsen, Alexandre Maciuk, Inge Mangelsdorf, Harry J McArdle, Androniki Naska, Kristina Pentieva, Alfonso Siani, Frank Thies, Sophia Tsabouri and Marco Vinceti.
Supporting information
Outcome of Public Consultation
ACKNOWLEDGEMENTS
The Panel wishes to thank the following for the support provided to this scientific output: hearing experts Ivan Dimitrov, Clare Mills, Daniel Tomé, member of the EFSA Working Group on Novel Foods Rosangela Marchelli, members of the EFSA Working Group on Traditional Foods, and the EFSA colleagues Ana L. Afonso, Elisa Beneventi, Gabriele Bondi, Paolo Colombo, Sara de Berardis, Agnès de Sesmaisons Lecarré, Alessandro Delfino, Silvia Federici, Esther Garcia Ruiz, José Ángel Gómez Ruiz, Dafni Kagli, Maura Magani, Catalina Manieu, Carla Martino, Federico Morreale, Gianluca Rossi, Pablo Rodriguez Fernandez, Francesco Suriano, Alexandra Tard, Panagiota Zakidou.
APPENDIX A.
A.1.
TABLE A.1.
Requirements for the taxonomic and hazard identification of microorganisms as novel foods (active agents and biomasses) or used in the production of novel foods (production strains).
| Requirements | Microorganisms as novel foods (active agents and biomasses) | Microorganisms used in the production of novel foods (production strains) |
|---|---|---|
|
Unambiguous taxonomic identification at species level (EFSA, 2021c; EFSA FEEDAP Panel, 2018) Certificate of deposition (including accession number) of the microbial strain under assessment in an internationally recognised culture collection having acquired the status of International Depositary Authority following the Budapest Treaty rules (EFSA FEEDAP Panel, 2018) |
Mandatory | Mandatory |
| Acquired antimicrobial resistance (AMR) genes of clinical relevance (EFSA, 2021c; EFSA BIOHAZ Panel, 2023b; EFSA FEEDAP Panel, 2018) | Mandatory for bacteria (regardless of QPS status) | Mandatory for bacteria (regardless of QPS status) |
| Assessment of the capacity of the microbial strain to produce antimicrobials of clinical relevance (EFSA FEEDAP Panel, 2018) | Applicable to TUs: | Applicable to TUs: |
|
|
|
|
|
|
|
|
|
| Toxigenicity and pathogenicity (EFSA, 2021c; EFSA FEEDAP Panel, 2018) | Applicable to TUs: | Applicable to TUs: |
|
|
|
|
|
|
| Purpose, characterisation and structure of the genetic modification(s) (EFSA FEEDAP Panel, 2018; EFSA, 2021c; EFSA GMO Panel, 2024) | Not applicable | Applicable to GM production strains |
| Whole genome sequence (WGS) data | According to the most up‐to‐date versions of the available and applicable EFSA scientific outputs (e.g. EFSA, 2021c) | According to the most up‐to‐date versions of the available and applicable EFSA scientific outputs (e.g. EFSA, 2021c) |
| Presence of viable cells in the novel food to be analysed (EFSA FEEDAP Panel, 2018) | Applicable to:
|
Applicable to:
|
| Presence of DNA in the novel food to be analysed (EFSA FEEDAP Panel, 2018) | Not applicable | Applicable to:
|
APPENDIX B.
B.1.
TABLE B.1.
Table with the input materials used in the production process of the novel food.
| Input material | Production process step(s) used at | Chemical identifier (CAS, EC number, etc.) | Specifications and/or certificates of analysis | Functional role | Regulatory status in EU (if applicable) |
|---|---|---|---|---|---|
| e.g. β‐cyclodextrin | Encapsulation | 7585‐39‐9 (hydrate) | Name of the document in the dossier | Emulsifier | Authorised food additive (E 459) |
APPENDIX C.
C.1.
Data on the protein quality of the novel food must be provided when the highest mean consumption of the novel food under the proposed conditions of use could substantially contribute to the average requirement (AR) for protein of one or more population groups (EFSA NDA Panel, 2012). For this purpose, a substantial contribution is defined as at least 15% of the population‐specific AR. For DietEx/FAIM age categories, the Panel proposes to use the AR for protein reported in Table C.1.
TABLE C.1.
Population‐specific average requirements for protein dietary intake.
| DietEx/FAIM category | Age range | AR (g/kg bw per day) a | AR (g/day) b |
|---|---|---|---|
| Infants | Up to and including 11 months | 1.12 | 10 |
| Young children | From 12 up to and including 35 months of age | 0.79 c | 10 |
| Other children | From 36 months up to and including 9 years of age | 0.72 d | 15 |
| Adolescents | From 10 up to and including 17 years of age | 0.71 e | 36 |
| Adults | From 18 years | 0.66 | 46 |
Mean AR for males and females (EFSA NDA Panel, 2012).
Reference body weights (bw): 8.9 kg for infants (average bw at 9 months), 12.4 kg for young children (average bw at 24 months), 21.3 kg for other children (average bw at 6 years) and 52.2 kg for adolescents (average bw at 14 years) (van Buuren et al., 2012); 70 kg for adults (EFSA Scientific Committee, 2012).
AR at 24 months (mid‐age of the category).
AR at 6 years (mid‐age of the category).
AR at 14 years (mid‐age of the category).
ANNEX A.
A.1.
The outcome of the public consultation which was open from 15 February 2024 until 14 April 2024 is presented in Annex A. Annex A is available under the Supporting Information section on the online version of the scientific output.
EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food Allergens) , Turck, D. , Bohn, T. , Castenmiller, J. , de Henauw, S. , Hirsch‐Ernst, K. I. , Maciuk, A. , Mangelsdorf, I. , McArdle, H. J. , Naska, A. , Pentieva, K. , Siani, A. , Thies, F. , Tsabouri, S. , Vinceti, M. , Aguilera Gómez, M. , Cubadda, F. , Frenzel, T. , Heinonen, M. , … Knutsen, H. K. (2024). Guidance on the scientific requirements for an application for authorisation of a novel food in the context of Regulation (EU) 2015/2283. EFSA Journal, 22(9), e8961. 10.2903/j.efsa.2024.8961
Adopted: 27 June 2024
Annex A is available under the Supporting Information section .
Notes
Regulation (EU) 2015/2283 of the European Parliament and of the Council on novel foods, amending Regulation (EU) No 1169/2011 of the European Parliament and of the Council and repealing Regulation (EC) No 258/97 of the European Parliament and of the Council and Commission Regulation (EC) No 1852/2001. OJ L 327, 11.12.2015, p. 1–22.
Commission Recommendation of 29 July 1997 concerning the scientific aspects and the presentation of information necessary to support applications for the placing on the market of novel foods and novel food ingredients and the preparation of initial assessment reports under Regulation (EC) No 258/97 of the European Parliament and of the Council. OJ L 253, 16.9.1997, p. 1–36.
Regulation (EC) No 258/97 of the European Parliament and of the Council of 27 January 1997 concerning novel foods and novel food ingredients. OJ L 43, 14.2.1997, p. 1–6. No longer in force, repealed by Regulation (EU) 2015/2283.
Regulation (EU) 2015/2283 of the European Parliament and of the Council on novel foods, amending Regulation (EU) No 1169/2011 of the European Parliament and of the Council and repealing Regulation (EC) No 258/97 of the European Parliament and of the Council and Commission Regulation (EC) No 1852/2001. OJ L 327, 11.12.2015, p. 1–22.
Regulation EU) 2019/1381 of the European Parliament and of the Council of 20 June 2019 on the transparency and sustainability of the EU risk assessment in the food chain and amending Regulations (EC) No 178/2002, (EC) No 1829/2003, (EC) No 1831/2003, (EC) No 2065/2003, (EC) No 1935/2004, (EC) No 1331/2008, (EC) No 1107/2009, (EU) 2015/2283 and Directive 2001/18/EC (OJ L 231, 6.9.2019, p. 1).
Commission Implementing Regulation (EU) 2017/2469 of 20 December 2017 laying down administrative and scientific requirements for applications referred to in Article 10 of Regulation (EU) 2015/2283 of the European Parliament and of the Council on novel foods. OJ L 351, 30.12.2017, p. 64.
Regulation (EC) No 178/2002 of the European Parliament and of the Council of 28 January 2002, laying down the general principles and requirements of food law, establishing the European Food Safety Authority, and laying down procedures in matters of food safety, as amended by Regulation (EU) 2019/1381 of the European Parliament and of the Council of 20 June 2019 on the transparency and sustainability of the EU risk assessment in the food chain and amending Regulations (EC) No 178/2002, (EC) No 1829/2003, (EC) No 1831/2003, (EC) No 2065/2003, (EC) No 1935/2004, (EC) No 1331/2008, (EC) No 1107/2009, (EU) 2015/2283 and Directive 2001/18/EC, PE/41/2019/REV/1. OJ L 231, 6.9.2019, p. 1–28.
Regulation (EC) No 1925/2006 of the European Parliament and of the Council of 20 December 2006 on the addition of vitamins and minerals and of certain other substances to foods. OJ L 404, 30.12.2006, p. 26–38.
Regulation (EU) No 609/2013 of the European Parliament and of the Council of 12 June 2013 on food intended for infants and young children, food for special medical purposes, and total diet replacement for weight control and repealing Council Directive 92/52/EEC, Commission Directives 96/8/EC, 1999/21/EC, 2006/125/EC and 2006/141/EC, Directive 2009/39/EC of the European Parliament and of the Council and Commission Regulations (EC) No 41/2009 and (EC) No 953/2009 OJ L 181, 29.6.2013, p. 35–56.
Directive 2002/46/EC of the European Parliament and of the Council of 10 June 2002 on the approximation of the laws of the Member States relating to food supplements OJ L 183, 12.7.2002, p. 51–57.
Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes, OJ L 276, 20.10.2010, p. 33.
In vitro, in silico and in chemico approaches are often referred to as New Approach Methodologies or NAMs. These methodologies may be complemented by physiologically based toxicokinetic (PBTK) modelling to simulate the concentration‐time profile in blood and at the target site, and in vitro to in vivo extrapolation (IVIVE) to translate in vitro bioactivity measurements into in vivo concentrations, doses or exposures.
Directive 2004/10/EC of the European Parliament and of the Council of 11 February 2004 on the harmonisation of laws, regulations and administrative provisions relating to the application of the principles of good laboratory practice and the verification of their applications for tests on chemical substances, OJ L 50, 20.2.2004, p. 44.
OECD Series on Principles of Good Laboratory Practice and Compliance Monitoring. Number 1. OECD Principles on Good Laboratory Practice (as revised in 1997) ENV/MC/CHEM(98)17.
Applicable when (EFSA Scientific Committee, 2021a) applies.
For the purpose of this guidance, proteins are not included in the definition of polymers.
Regulation (EC) No 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No 793/93 and Commission Regulation (EC) No 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC. OJ L 396, 30.12.2006, p. 1–849.
Regulation (EC) No 1272/2008 of the European Parliament and of the Council of 16 December 2008 on classification, labelling and packaging of substances and mixtures, amending and repealing Directives 67/548/EEC and 1999/45/EC, and amending Regulation (EC) No 1907/2006. OJ L 353, 31.12.2008, p. 1–1355.
These requirements are based on the EFSA Scientific Committee guidance on the safety assessment of botanicals and botanical preparations intended for use as ingredients in food supplements (EFSA Scientific Committee, 2009).
Plants of the World Online (https:/powo.science.kew.org/) facilitated by the Royal Botanic Gardens, Kew; The USDA‐ARS Germplasm Resources Information Network (GRIN) database (https://npgsweb.ars‐grin.gov/ gringlobal/taxon/taxonomysimple.aspx) in case Plants of the World Online does not provide the required information; The International Plant Names Index (https://www.ipni.org/) in case the two above sources do not provide the required information.
Mycobank (www.mycobank.org), Index fungorum (https://www.indexfungorum.org), Catalogue of Life (CoL), Integrated Taxonomic Information System (ITIS), Global Biodiversity Information Facility (GBIF), Encyclopedia of Life (EOL).
GMO refers to organisms obtained through the techniques covered by Directive 2001/18/EC of the European Parliament and of the Council of 12 March 2001 on the deliberate release into the environment of genetically modified organisms and repealing Council Directive 90/220/EEC.
Commission Regulation (EU) 2015/1162 of 15 July 2015 amending Annex V to Regulation (EC) No 999/2001 of the European Parliament and of the Council laying down rules for the prevention, control and eradication of certain transmissible spongiform encephalopathies.
Regulation (EU) 2017/625 of the European Parliament and of the Council of 15 March 2017 on official controls and other official activities performed to ensure the application of food and feed law, rules on animal health and welfare, plant health and plant protection products.
Regulation (EC) No 853/2004 of the European Parliament and of the Council of 29 April 2004 laying down specific hygiene rules for food of animal origin.
As defined in Regulation (EU) 2015/2283.
Quality of the input material can be proven for commercial products by the certificates of analysis of the purchased products or by specifications for non‐commercial products and certificates of analysis that prove the product complies with specifications.
Regulation (EC) No 1935/2004 of the European Parliament and of the Council of 27 October 2004 on materials and articles intended to come into contact with food and repealing Directives 80/590/EEC and 89/109/EEC, OJ L 338, 13.11.2004, p. 4.
The ratio of the concentration of a chemical substance in a product to the concentration of the same substance in the starting material (the source).
Regulation (EC) No 852/2004 of the European parliament and of the council of 29 April 2004 on the hygiene of foodstuffs, OJ L 139, 30.4.2004, p. 1.
Regulation (EC) No 1332/2008 of the European Parliament and of the Council of 16 December 2008 on food enzymes and amending Council Directive 83/417/EEC, Council Regulation (EC) No 1493/1999, Directive 2000/13/EC, Council Directive 2001/112/EC and Regulation (EC) No 258/97. OJ L 354, 31.12.2008, p. 7.
Regulation (EC) No 1333/2008 of the European Parliament and of the Council of 16 December 2008 on food additives (Text with EEA relevance). OJ L 354, 31.12.2008, p. 16–33.
Directive 2009/32/EC of the European Parliament and of the Council of 23 April 2009 on the approximation of the laws of the Member States on extraction solvents used in the production of foodstuffs and food ingredients (Recast) (Text with EEA relevance). OJ L 141, 6.6.2009, p. 3–11.
https://publications.jrc.ec.europa.eu/repository/handle/JRC59782; https://www.oecd‐ilibrary.org/environment/guidance‐document‐on‐good‐in‐vitro‐method‐practices‐givimp/good‐cell‐culture‐practice‐gccp_9789264304796‐16‐en.
As defined by the EFSA Scientific Committee (2011a).
Matrix accreditation evaluates a laboratory's competence in accurately analysing specific food samples, ensuring quality and reliability in testing diverse and complex matrices. Such accreditation is crucial as validated analytical methods for one product may not be applicable to other types of products.
Employed to analyse another type of food matrix.
As defined in Section 1.
Batch as defined in Directive 2004/10/EC of the European Parliament and of the Council of 11 February 2004 on the harmonisation of laws, regulations and administrative provisions relating to the application of the principles of good laboratory practice and the verification of their applications for tests on chemical substances. OJ L 050, 20.2.2004, p. 44.
i.e. the protein content contributes to at least 12% of the energy content of the novel food [Regulation (EC) No 1924/2006 of the european parliament and of the council of 20 December 2006 on nutrition and health claims made on foods. OJ L 404, 30.12.2006, p. 9.
Solubility and dissolution rate can be used to demonstrate that under the anticipated use conditions the material will be fully dissolved in the marketed product, in food or, following ingestion, during the gastrointestinal tract processes, and that consumers will not be exposed to particles after food consumption.
Antinutrients are compounds that can interfere with the absorption of essential nutrients. Novel foods can contain antinutrients such as tannins, lectins, trypsin inhibitors, amylase inhibitors, phytic acid and phytates, oxalates or saponins, among others.
‘Dietary fibre’ in the context of this Guidance is understood as defined by EFSA NDA Panel, 2010c, i.e. all non‐digestible carbohydrates, and not as defined by Regulation (EU) 1169/2011, which sets the additional requirement of having a beneficial physiological effect for edible carbohydrate polymers obtained from food raw material by physical, enzymatic or chemical means, and for edible synthetic carbohydrate polymers.
Commission Implementing Regulation (EU) 2017/2470 of 20 December 2017 establishing the Union list of novel foods in accordance with Regulation (EU) 2015/2283 of the European Parliament and of the Council on novel foods. OJ L 351, 30.12.2017, p. 72–201.
Regulation (EC) No 1925/2006 of the European Parliament and of the Council of 20 December 2006 on the addition of vitamins and minerals and of certain other substances to foods. OJ L 404, 30.12.2006, p. 26–38.
Regulation (EU) No 609/2013 of the European Parliament and of the Council of 12 June 2013 on food intended for infants and young children, food for special medical purposes, and total diet replacement for weight control and repealing Council Directive 92/52/EEC, Commission Directives 96/8/EC, 1999/21/EC, 2006/125/EC and 2006/141/EC, Directive 2009/39/EC of the European Parliament and of the Council and Commission Regulations (EC) No 41/2009 and (EC) No 953/2009. OJ L 181, 29.6.2013, p. 35–56.
Directive 2002/46/EC of the European Parliament and of the Council of 10 June 2002 on the approximation of the laws of the Member States relating to food supplements. OJ L 183, 12.7.2002, p. 51–57.
The FAIM tool was developed by EFSA for estimating chronic exposure to food additives in the regulatory framework of food additives Regulation (EU) 1333/2008. Considering that the exposure assessment of food additives and intake assessment of novel foods share common principles, the FAIM tool can also be used for the intake assessment of novel food. Refer to Section 6.3 for the use of the FAIM tool to estimate the intake of the novel food. FAIM tool is: https://www.efsa.europa.eu/en/applications/food‐improvement‐agents/tools.
The DietEx tool was developed by EFSA for estimating chronic exposure to substance present in food, naturally present or intentionally added to foods like Novel Foods. Refer to Section 6.3 for the use of DietEx tool to estimate the intake of the novel food. DietEx tool is: https://www.efsa.europa.eu/en/science/tools‐and‐resources/dietex.
Commission Directive 96/8/EC of 26 February 1996 on foods intended for use in energy‐restricted diets for weight reduction. OJ L 55, 6.3.1996, p. 22–26. https://eur‐lex.europa.eu/eli/dir/1996/8/oj.
This approach takes into consideration animal welfare by adopting animal testing strategies in line with the Rs (replacement, reduction and refinement) to allow for fewer tests to be conducted when the information available for a risk assessment is sufficient (Directive 2010/63/EU).
Nutritional status can be defined as an individual's ability to meet the physiological requirements for a particular nutrient.
Please refer to the ‘Overview on Tolerable Upper Intake Levels as derived by the Scientific Committee on Food (SCF) and the EFSA Panel on Dietetic Products, Nutrition and Allergies (NDA) on Dietary reference values’ which is updated any time a new opinion is published and is available at this link: https://www.efsa.europa.eu/sites/default/files/2024‐05/ul‐summary‐report.pdf.
When specifications are not established for a nutrient, the maximum concentration reported in batch‐to‐batch analysis is to be considered.
Average and 95th percentile estimated intake of nutrients in European populations can be found in EFSA's opinions on Dietary Reference Values for nutrients. https://efsa.onlinelibrary.wiley.com/doi/toc/10.1002/(ISSN)1831‐4732.021217.
A series of consensus documents relevant for the safety assessment of specific foods have been developed by the Organization for Economic Co‐operation and Development (OECD) task force for the safety of novel foods and feeds. These documents address compositional considerations, including both nutrient and antinutrients, and should be used by the applicants as reference. https://www.oecd.org/science/biotrack/consensus‐document‐for‐work‐on‐safety‐novel‐and‐foods‐feeds‐plants.htm.
Regulation (EU) No 1169/2011 of the European Parliament and of the Council of 25 October 2011 on the provision of food information to consumers, amending Regulations (EC) No 1924/2006 and (EC) No 1925/2006 of the European Parliament and of the Council, and repealing Commission Directive 87/250/EEC, Council Directive 90/496/EEC, Commission Directive 1999/10/EC, Directive 2000/13/EC of the European Parliament and of the Council, Commission Directives 2002/67/EC and 2008/5/EC and Commission Regulation (EC) No 608/2004 Text with EEA relevance. OJ L 304, 22.11.2011, p. 18–63.
Regulation (EC) No 1925/2006 of the European Parliament and of the Council of 20 December 2006 on the addition of vitamins and minerals and of certain other substances to foods. OJ L 404, 30.12.2006, p. 26–38.
Regulation (EU) No 609/2013 of the European Parliament and of the Council of 12 June 2013 on food intended for infants and young children, food for special medical purposes, and total diet replacement for weight control and repealing Council Directive 92/52/EEC, Commission Directives 96/8/EC, 1999/21/EC, 2006/125/EC and 2006/141/EC, Directive 2009/39/EC of the European Parliament and of the Council and Commission Regulations (EC) No 41/2009 and (EC) No 953/2009. OJ L 181, 29.6.2013, p. 35–56.
Directive 2002/46/EC of the European Parliament and of the Council of 10 June 2002 on the approximation of the laws of the Member States relating to food supplements. OJ L 183, 12.7.2002, p. 51–57.
As defined in Regulation (EC) No 609/2013 on food intended for infants and young children, food for special medical purposes, and total diet replacement for weight control and repealing Council Directive 92/52/EEC, Commission Directives 96/8/EC, 1999/21/EC, 2006/125/EC and 2006/141/EC, Directive 2009/39/EC of the European Parliament and of the Council and Commission Regulations (EC) No 41/2009 and (EC) No 953/2009 Text with EEA relevance. OJ L 181, 29.6.2013, p. 35–56.
Commission Delegated Regulation (EU) 2017/1798 of 2 June 2017 supplementing Regulation (EU) No 609/2013 of the European Parliament and of the Council as regards the specific compositional and information requirements for total diet replacement for weight control; Commission Delegated Regulation (EU) 2016/127 of 25 September 2015 supplementing Regulation (EU) No 609/2013 of the European Parliament and of the Council as regards the specific compositional and information requirements for infant formula and follow‐on formula and as regards requirements on information relating to infant and young child feeding.
REFERENCES
- EFSA (European Food Safety Authority) . (2010). Application of systematic review methodology to food and feed safety assessments to support decision making. EFSA Journal, 8(6), 1637. 10.2903/j.efsa.2010.1637 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority) . (2012). Compendium of botanicals reported to contain naturally occurring substances of possible concern for human health when used in food and food supplements. EFSA Journal, 10(5), 2663. 10.2903/j.efsa.2012.2663 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority) . (2017). Dietary Reference Values for nutrients. Summary Report. EFSA Supporting Publications, 14(2), e15121. 10.2903/sp.efsa.2017.e15121 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority) . (2021a). Administrative guidance for the processing of applications for regulated products (update 2021). EFSA Supporting Publications, 18(3), 6471E. 10.2903/sp.efsa.2021.EN-6471 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority) . (2021b). EFSA's catalogue of support initiatives during the life‐cycle of applications for regulated products. EFSA Supporting Publications, 18(3), 6472E. 10.2903/sp.efsa.2021.EN-6472 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority) . (2021c). EFSA statement on the requirements for whole genome sequence analysis of microorganisms intentionally used in the food chain. EFSA Journal, 19(7), 6506. 10.2903/j.efsa.2021.6506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority) . (2021d). Decision of the Executive Director of the European Food Safety Authority Laying down practical arrangements concerning transparency and confidentiality. https://www.efsa.europa.eu/sites/default/files/corporate_publications/files/210111‐PAs‐transparency‐and‐confidentiality.pdf
- EFSA (European Food Safety Authority) , Hart, A. , Maxim, L. , Siegrist, M. , Von Goetz, N. , da Cruz, C. , Merten, C. , Mosbach‐Schulz, O. , Lahaniatis, M. , Smith, A. , & Hardy, A. (2019). Guidance on Communication of uncertainty in scientific assessments. EFSA Journal, 17(1), 5520. 10.2903/j.efsa.2019.5520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority) . (2024). Administrative guidance for the preparation of novel food applications in the context of Article 10 of Regulation (EU) 2015/2283. 10.2903/sp.efsa.2024.EN-9041 In press. Publication scheduled in October 2024 [DOI]
- EFSA BIOHAZ Panel (EFSA Panel on Biological Hazards) , Koutsoumanis, K. , Allende, A. , Alvarez‐Ordóñez, A. , Bolton, D. , Bover‐Cid, S. , Chemaly, M. , De Cesare, A. , Hilbert, F. , Lindqvist, R. , Nauta, M. , Nonno, R. , Peixe, L. , Ru, G. , Simmons, M. , Skandamis, P. , Suffredini, E. , Cocconcelli, P. S. , Suarez, J. E. , … Herman, L. (2023a). Statement on how to interpret the QPS qualification on ‘acquired antimicrobial resistance genes’. EFSA Journal, 21(10), 8323. 10.2903/j.efsa.2023.8323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA BIOHAZ Panel (EFSA Panel on Biological Hazards) , Koutsoumanis, K. , Allende, A. , Álvarez‐Ordóñez, A. , Bolton, D. , Bover‐Cid, S. , Chemaly, M. , de Cesare, A. , Hilbert, F. , Lindqvist, R. , Nauta, M. , Peixe, L. , Ru, G. , Simmons, M. , Skandamis, P. , Suffredini, E. , Cocconcelli, P. S. , Fernández Escámez, P. S. , Maradona, M. P. , … Herman, L. (2023b). Update of the list of qualified presumption of safety (QPS) recommended microorganisms intentionally added to food or feed as notified to EFSA. EFSA Journal, 21(1), 7747. 10.2903/j.efsa.2023.7747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA CEP Panel (EFSA Panel on Food Contact Materials, Enzymes and Processing Aids) , Lambré, C. , Barat Baviera, J. M. , Bolognesi, C. , Cocconcelli, P. S. , Crebelli, R. , Gott, D. M. , Grob, K. , Lampi, E. , Mengelers, M. , Mortensen, A. , Rivière, G. , Steffensen, I.‐L. , Tlustos, C. , Van Loveren, H. , Vernis, L. , Zorn, H. , Glandorf, B. , … Chesson, A. (2021). Scientific guidance for the submission of dossiers on food enzymes. EFSA Journal, 19(10), 6851. 10.2903/j.efsa.2021.6851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA FEEDAP Panel (EFSA Panel on Additives Products or Substances used in Animal Feed) , Rychen, G. , Aquilina, G. , Azimonti, G. , Bampidis, V. , Bastos, M. D. L. , Bories, G. , Chesson, A. , Cocconcelli, P. S. , Flachowsky, G. , Gropp, J. , Kolar, B. , Kouba, M. , López‐Alonso, M. , López Puente, S. , Mantovani, A. , Mayo, B. , Ramos, F. , … Galobart, J. (2018). Guidance on the characterisation of microorganisms used as feed additives or as production organisms. EFSA Journal, 16(3), 5206. 10.2903/j.efsa.2018.5206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA GMO Panel (EFSA Panel on Genetically Modified Organisms) . (2010). Scientific Opinion on the assessment of allergenicity of GM plants and microorganisms and derived food and feed. EFSA Journal, 8(7), 1700. 10.2903/j.efsa.10.1700 [DOI] [Google Scholar]
- EFSA GMO Panel (EFSA Panel on Genetically Modified Organisms) . (2011). Guidance on the risk assessment of genetically modified microorganisms and their products intended for food and feed use. EFSA Journal, 9(6), 2193. 10.2903/j.efsa.2011.2193 [DOI] [Google Scholar]
- EFSA GMO Panel (EFSA Panel on Genetically Modified Organisms) , Naegeli, H. , Birch, A. N. , Casacuberta, J. , De Schrijver, A. , Gralak, M. A. , Guerche, P. , Jones, H. , Manachini, B. , Messéan, A. , Nielsen, E. E. , Nogué, F. , Robaglia, C. , Rostoks, N. , Sweet, J. , Tebbe, C. , Visioli, F. , Wal, J.‐M. , Eigenmann, P. , … Fernandez Dumont, A. (2017). Guidance on allergenicity assessment of genetically modified plants. EFSA Journal, 15(6), 4862. 10.2903/j.efsa.2017.4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA GMO Panel (EFSA Panel on Genetically Modified Organisms) , Naegeli, H. , Bresson, J.‐L. , Dalmay, T. , Dewhurst, I. C. , Epstein, M. M. , Firbank, L. G. , Guerche, P. , Hejatko, J. , Moreno, F. J. , Mullins, E. , Nogué, F. , Rostoks, N. , Sánchez Serrano, J. J. , Savoini, G. , Veromann, E. , Veronesi, F. , & Dumont, A. F. (2021). Statement on in vitro protein digestibility tests in allergenicity and protein safety assessment of genetically modified plants. EFSA Journal, 19(1), 6350. 10.2903/j.efsa.2021.6350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA GMO Panel (EFSA Panel on Genetically Modified Organisms) , Mullins, E. , Bresson, J.‐L. , Dalmay, T. , Dewhurst, I. C. , Epstein, M. M. , George Firbank, L. , Guerche, P. , Hejatko, J. , Naegeli, H. , Nogué, F. , Rostoks, N. , Sánchez Serrano, J. J. , Savoini, G. , Veromann, E. , Veronesi, F. , Fernandez Dumont, A. , & Moreno, F. J. (2022). Scientific Opinion on development needs for the allergenicity and protein safety assessment of food and feed products derived from biotechnology. EFSA Journal, 20(1), 7044. 10.2903/j.efsa.2022.7044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA GMO Panel (EFSA Panel on Genetically Modified Organisms) , Mullins, E. , Bresson, J.‐L. , Dewhurst, I. C. , Epstein, M. M. , Firbank, L. G. , Guerche, P. , Hejatko, J. , Moreno, F. J. , Naegeli, H. , Nogué, F. , Rostoks, N. , Sánchez Serrano, J. J. , Savoini, G. , Veromann, E. , Veronesi, F. , Cocconcelli, P. S. , Glandorf, D. , Herman, L. , … Dalmay, T. (2024). New developments in biotechnology applied to microorganisms. EFSA Journal, 22(7), e8895. 10.2903/j.efsa.2024.8895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies) . (2008). Safety of synthetic lycopene ‐ scientific opinion of the panel on scientific panel on dietetic products, nutrition and allergies. EFSA Journal, 6(4), 676. 10.2903/j.efsa.2008.676 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies) . (2010a). Scientific Opinion on the safety of ‘conjugated linoleic acid (CLA)‐rich oil’ (Clarinol®) as a novel food ingredient. EFSA Journal, 8(5), 1601. 10.2903/j.efsa.2010.1601 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies) . (2010b). Scientific Opinion on the safety of ‘conjugated linoleic acid (CLA)‐rich oil’ (Tonalin® TG 80) as a novel food ingredient. EFSA Journal, 8(5), 1600. 10.2903/j.efsa.2010.1600 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies) . (2010c). Scientific Opinion on Dietary Reference Values for carbohydrates and dietary fibre. EFSA Journal 2010, 8(3), 1462. 10.2903/j.efsa.2010.1462 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies) . (2012). Scientific opinion on dietary reference values for protein. EFSA Journal, 10(2), 2557. 10.2903/j.efsa.2012.2557 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies) . (2016). Safety of UV‐treated milk as a novel food pursuant to regulation (EC) No 258/97. EFSA Journal, 14(1), 4370. 10.2903/j.efsa.2016.4370 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food Allergens) , Turck, D. , Bohn, T. , Castenmiller, J. , De Henauw, S. , Hirsch‐Ernst, K. I. , Maciuk, A. , Mangelsdorf, I. , McArdle, H. J. , Naska, A. , Pelaez, C. , Pentieva, K. , Siani, A. , Thies, F. , Tsabouri, S. , Vinceti, M. , Cubadda, F. , Frenzel, T. , … Knutsen, H. K. (2022a). Safety of frozen and freeze‐dried formulations of the lesser mealworm (Alphitobius diaperinus larva) as a novel food pursuant to regulation (EU) 2015/2283. EFSA Journal, 20(7), 7325. 10.2903/j.efsa.2022.7325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food Allergens) , Turck, D. , Bohn, T. , Castenmiller, J. , De Henauw, S. , Hirsch‐Ernst, K. I. , Maciuk, A. , Mangelsdorf, I. , McArdle, H. J. , Naska, A. , Pelaez, C. , Pentieva, K. , Siani, A. , Thies, F. , Tsabouri, S. , Vinceti, M. , Cubadda, F. , Frenzel, T. , … Knutsen, H. K. (2022b). Safety of hydrothermally treated kernels from edible Jatropha curcas L. (Chuta) as a novel food pursuant to Regulation (EU) 2015/2283. EFSA Journal, 20(1), 6998. 10.2903/j.efsa.2022.6998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food Allergens) , Turck, D. , Bohn, T. , Castenmiller, J. , De Henauw, S. , Hirsch‐Ernst, K. I. , Maciuk, A. , Mangelsdorf, I. , McArdle, H. J. , Naska, A. , Pelaez, C. , Pentieva, K. , Siani, A. , Thies, F. , Tsabouri, S. , Vinceti, M. , Cubadda, F. , Frenzel, T. , … Knutsen, H. K. (2022c). Safety of vitamin D2 mushroom powder as a novel food pursuant to regulation (EU) 2015/2283 (NF 2019/1471). EFSA Journal, 20(6), 7326. 10.2903/j.efsa.2022.7326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food Allergens) , Turck, D. , Bohn, T. , Castenmiller, J. , de Henauw, S. , Hirsch‐Ernst, K. I. , Knutsen, H. K. , Maciuk, A. , Mangelsdorf, I. , McArdle, H. J. , Naska, A. , Peláez, C. , Pentieva, K. , Thies, F. , Tsabouri, S. , Vinceti, M. , Bresson, J.‐L. , & Siani, A. (2022d). Scientific advice related to nutrient profiling for the development of harmonised mandatory front‐of‐pack nutrition labelling and the setting of nutrient profiles for restricting nutrition and health claims on foods. EFSA Journal, 20(4), 7259. 10.2903/j.efsa.2022.7259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel . (2022e). Guidance for establishing and applying tolerable upper intake levels for vitamins and essential minerals. Draft for internal testing. EFSA Journal, 20(1), e200102, 27 pp. 10.2903/j.efsa.2022.e200102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food Allergens) , Turck, D. , Bohn, T. , Castenmiller, J. , De Henauw, S. , Hirsch‐Ernst, K. I. , Maciuk, A. , Mangelsdorf, I. , McArdle, H. J. , Naska, A. , Pelaez, C. , Pentieva, K. , Siani, A. , Thies, F. , Tsabouri, S. , Vinceti, M. , Aguilera Gómez, M. , Cubadda, F. , … Knutsen, H. K. (2023a). Safety of UV‐treated powder of whole yellow mealworm (Tenebrio molitor larva) as a novel food pursuant to regulation (EU) 2015/2283. EFSA Journal, 21(5), 8009. 10.2903/j.efsa.2023.8009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food Allergens) , Turck, D. , Bohn, T. , Castenmiller, J. , De Henauw, S. , Hirsch‐Ernst, K. I. , Maciuk, A. , Mangelsdorf, I. , McArdle, H. J. , Naska, A. , Pelaez, C. , Pentieva, K. , Siani, A. , Thies, F. , Tsabouri, S. , Vinceti, M. , Aguilera Gómez, M. , Cubadda, F. , … Knutsen, H. K. (2023b). Safety of paramylon as a novel food pursuant to regulation (EU) 2015/2283. EFSA Journal, 21(5), 7995. 10.2903/j.efsa.2023.7995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food Allergens) , Turck, D. , Bohn, T. , Castenmiller, J. , De Henauw, S. , Hirsch‐Ernst, K. I. , Maciuk, A. , Mangelsdorf, I. , McArdle, H. J. , Naska, A. , Pelaez, C. , Pentieva, K. , Siani, A. , Thies, F. , Tsabouri, S. , Vinceti, M. , Aguilera Gómez, M. , Cubadda, F. , … Knutsen, H. K. (2023c). Safety of partially hydrolysed protein from spent barley (Hordeum vulgare) and rice (Oryza sativa) as a novel food pursuant to regulation (EU) 2015/2283. EFSA Journal, 21(9), 8064. 10.2903/j.efsa.2023.8064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food Allergens) , Turck, D. , Bohn, T. , Castenmiller, J. , De Henau, S. , Engel, K.‐H. , Hirsch‐Ernst, K. I. , Kearney, J. , Knutsen, H. K. , Maciuk, A. , Mangelsdorf, I. , McArdle, H. J. , Naska, A. , Pentieva, K. , Siani, A. , Thies, F. , Tsabouri, S. , Vincenti, M. , Peláez, C. , … Turla, E. (2024a). Guidance on the scientific requirements for a notification and application for authorisation of traditional foods from third countries in the context of Regulation (EU) 2015/2283. EFSA Journal, 22(8), e8966. 10.2903/j.efsa.2024.8966 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food Allergens) , Turck, D. , Bohn, T. , Castenmiller, J. , de Henauw, S. , Hirsch‐Ernst, K.‐I. , Knutsen, H. K. , Maciuk, A. , Mangelsdorf, I. , McArdle, H. J. , Pentieva, K. , Siani, A. , Thies, F. , Tsabouri, S. , Vinceti, M. , Kass, G. , Heng, L. , Sofroniou, A. , Ververis, E. , … Naska, A. (2024b). Guidance on scientific principles and data requirements for the safety and relative bioavailability assessment of new micronutrient sources. EFSA Journal, 22(8), e8946. 10.2903/j.efsa.2024.8946 [DOI] [Google Scholar]
- EFSA PPR Panel (EFSA Panel on Plant Protection Products and their Residues) , Hernandez‐Jerez, A. F. , Adriaanse, P. , Aldrich, A. , Berny, P. , Coja, T. , Duquesne, S. , Focks, A. , Marinovich, M. , Millet, M. , Pelkonen, O. , Pieper, S. , Tiktak, A. , Topping, C. J. , Widenfalk, A. , Wilks, M. , Wolterink, G. , Gundert‐Remy, U. , … Parra Morte, J. M. (2021). Scientific opinion of the scientific panel on plant protection products and their residues (PPR panel) on testing and interpretation of comparative in vitro metabolism studies. EFSA Journal, 19(12), 6970. 10.2903/j.efsa.2021.6970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee . (2009). Guidance on safety assessment of botanicals and botanical preparations intended for use as ingredients in food supplements. EFSA Journal, 7(9), 1249. 10.2903/j.efsa.2009.1249 [DOI] [Google Scholar]
- EFSA Scientific Committee . (2011a). Guidance on conducting repeated‐dose 90‐day oral toxicity study in rodents on whole food/feed. EFSA Journal, 9(12), 2438. 10.2903/j.efsa.2011.2438 [DOI] [Google Scholar]
- EFSA Scientific Committee . (2011b). Scientific opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA Journal, 9(9), 2379. 10.2903/j.efsa.2011.2379 [DOI] [Google Scholar]
- EFSA Scientific Committee . (2012a). Guidance on selected default values to be used by the EFSA scientific committee, scientific panels and units in the absence of actual measured data. EFSA Journal, 10(3), 2579. 10.2903/j.efsa.2012.2579 [DOI] [Google Scholar]
- EFSA Scientific Committee . (2012b). Scientific Opinion on exploring options for providing advice about possible human health risks based on the concept of threshold of toxicological concern (TTC). EFSA Journal, 10(7), 2750. 10.2903/j.efsa.2012.2750 [DOI] [Google Scholar]
- EFSA Scientific Committee . (2015). Risk profile related to production and consumption of insects as food and feed. EFSA Journal, 13(10), 4257. 10.2903/j.efsa.2015.4257 [DOI] [Google Scholar]
- EFSA Scientific Committee , Hardy, A. , Benford, D. , Halldorsson, T. , Jeger, M. J. , Knutsen, H. K. , More, S. , Naegeli, H. , Noteborn, H. , Ockleford, C. , Ricci, A. , Rychen, G. , Schlatter, J. R. , Silano, V. , Solecki, R. , Turck, D. , Benfenati, E. , Chaudhry, Q. M. , Craig, P. , … Younes, M. (2017a). Scientific Opinion on the guidance on the use of the weight of evidence approach in scientific assessments. EFSA Journal, 15(8), 4971. 10.2903/j.efsa.2017.4971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , Hardy, A. , Benford, D. , Halldorsson, T. , Jeger, M. J. , Knutsen, H. K. , More, S. , Naegeli, H. , Noteborn, H. , Ockleford, C. , Ricci, A. , Rychen, G. , Schlatter, J. R. , Silano, V. , Solecki, R. , Turck, D. , Bresson, J.‐L. , Dusemund, B. , Gundert‐Remy, U. , … Mortensen, A. (2017b). Guidance on the risk assessment of substances present in food intended for infants below 16 weeks of age. EFSA Journal, 15(5), 4849. 10.2903/j.efsa.2017.4849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , Hardy, A. , Benford, D. , Halldorsson, T. , Jeger, M. J. , Knutsen, H. K. , More, S. , Naegeli, H. , Noteborn, H. , Ockleford, C. , Ricci, A. , Rychen, G. , Schlatter, J. R. , Silano, V. , Solecki, R. , Turck, D. , Younes, M. , Bresson, J.‐L. , Griffin, J. , … Alexander, J. (2017c). Guidance on the assessment of the biological relevance of data in scientific assessments. EFSA Journal, 15(8), 4970. 10.2903/j.efsa.2017.4970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , More, S. , Bampidis, V. , Benford, D. , Boesten, J. , Bragard, C. , Halldorsson, T. , Hernandez‐Jerez, A. , Hougaard‐Bennekou, S. , Koutsoumanis, K. , Naegeli, H. , Nielsen, S. S. , Schrenk, D. , Silano, V. , Turck, D. , Younes, M. , Aquilina, G. , Crebelli, R. , Gürtler, R. , … Schlatter, J. (2019a). Genotoxicity assessment of chemical mixtures. EFSA Journal, 17(1), 5519. 10.2903/j.efsa.2019.5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , More, S. J. , Bampidis, V. , Benford, D. , Bragard, C. , Halldorsson, T. I. , Hernández‐Jerez, A. F. , Hougaard Bennekou, S. , Koutsoumanis, K. P. , Machera, K. , Naegeli, H. , Nielsen, S. S. , Schlatter, J. R. , Schrenk, D. , Silano, V. , Turck, D. , Younes, M. , Gundert‐Remy, U. , Kass, G. E. N. , … Wallace, H. M. (2019b). Guidance on the use of the threshold of toxicological concern approach in food safety assessment. EFSA Journal, 17(6), 5708. 10.2903/j.efsa.2019.5708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , More, S. , Bampidis, V. , Benford, D. , Bragard, C. , Halldorsson, T. , Hernández‐Jerez, A. , Bennekou, S. H. , Koutsoumanis, K. , Lambré, C. , Machera, K. , Naegeli, H. , Nielsen, S. , Schlatter, J. , Schrenk, D. , Silano, V. , Turck, D. , Younes, M. , Castenmiller, J. , … Schoonjans, R. (2021a). Guidance on technical requirements for regulated food and feed product applications to establish the presence of small particles including nanoparticles. EFSA Journal, 19(8), 6769. 10.2903/j.efsa.2021.6769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , More, S. , Bampidis, V. , Benford, D. , Bragard, C. , Halldorsson, T. , Hernández‐Jerez, A. , Hougaard Bennekou, S. , Koutsoumanis, K. , Lambré, C. , Machera, K. , Naegeli, H. , Nielsen, S. , Schlatter, J. , Schrenk, D. , Silano, V. , Turck, D. , Younes, M. , Castenmiller, J. , … Schoonjans, R. (2021b). Guidance on risk assessment of nanomaterials to be applied in the food and feed chain: Human and animal health. EFSA Journal, 19(8), 6768. 10.2903/j.efsa.2021.6768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , More, S. , Bampidis, V. , Benford, D. , Bragard, C. , Halldorsson, T. , Hougaard Bennekou, S. , Koutsoumanis, K. , Machera, K. , Naegeli, H. , Nielsen, S. , Schlatter, J. , Schrenk, D. , Silano, V. , Turck, D. , Younes, M. , Aggett, P. , Castenmiller, J. , Giarola, A. , … Hernandez‐Jerez, A. (2021c). Statement on the derivation of Health‐Based Guidance Values (HBGVs) for regulated products that are also nutrients. EFSA Journal, 19(3), 6479. 10.2903/j.efsa.2021.6479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , More, S. , Bampidis, V. , Benford, D. , Bragard, C. , Halldorsson, T. , Hernández‐Jerez, A. , Bennekou, S. H. , Koutsoumanis, K. , Lambré, C. , Machera, K. , Mullins, E. , Nielsen, S. , Schlatter, J. , Schrenk, D. , Turck, D. , Younes, M. , Herman, L. , Pelaez, C. , … Cocconcelli, P. S. (2022a). Scientific opinion on the evaluation of existing guidelines for their adequacy for the food and feed risk assessment of microorganisms obtained through synthetic biology. EFSA Journal, 20(8), 7479. 10.2903/j.efsa.2022.7479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , More, S. J. , Bampidis, V. , Benford, D. , Bragard, C. , Halldorsson, T. I. , Hernández‐Jerez, A. F. , Bennekou, S. H. , Koutsoumanis, K. , Lambré, C. , Machera, K. , Mennes, W. , Mullins, E. , Nielsen, S. S. , Schrenk, D. , Turck, D. , Younes, M. , Aerts, M. , Edler, L. , … Schlatter, J. (2022b). Guidance on the use of the benchmark dose approach in risk assessment. EFSA Journal, 20(10), 7584. 10.2903/j.efsa.2022.7584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , More, S. J. , Bampidis, V. , Benford, D. , Bragard, C. , Halldorsson, T. I. , Hernández‐Jerez, A. F. , Bennekou, S. H. , Koutsoumanis, K. , Lambré, C. , Machera, K. , Mullins, E. , Nielsen, S. S. , Schlatter, J. R. , Schrenk, D. , Turck, D. , Younes, M. , Boon, P. , Ferns, G. A. , … Leblanc, J.‐C. (2023). Re‐evaluation of the existing health‐based guidance values for copper and exposure assessment from all sources. EFSA Journal, 21(1), 7728. 10.2903/j.efsa.2023.7728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EUCAST (The European Committee on Antimicrobial Susceptibility Testing) . (2024). Breakpoint tables for interpretation of MICs and zone diameters. Version 14.0. Available online: http://www.eucast.org
- FAO . (2013). Dietary protein quality evaluation in human nutrition. Report of an FAQ expert consultation. FAO Food and Nutrition Paper, 92, 1–66. https://www.fao.org/ag/humannutrition/35978‐02317b979a686a57aa4593304ffc17f06.pdf [PubMed] [Google Scholar]
- FAO & IAEA . (2024). Development of a protein database and the way forward for reviewing protein requirements – Report of a joint FAO/IAEA technical meeting in Vienna, 10–13 October 2022. Rome. 10.4060/cd1021en [DOI]
- FAO/WHO (Food and Agriculture Organization of the United Nations and World Health Organization) . (2009). Principles and methods for the risk assessment of chemicals in food. Environmental health criteria 240. WHO. Available online: https://www.who.int/publications/i/item/9789241572408 [Google Scholar]
- ICH (International Council for Harmonisation of Technical Requirements For Pharmaceuticals For Human Use) . (2024). Harmonised Guideline on Drug Interaction Studies – M12. https://database.ich.org/sites/default/files/ICH_M12_Step4_Guideline_2024_0521_0.pdf
- IUPAC (International Union of Applied and Pure Chemistry) . (2020). Publication of this document by any means is permitted on condition that it is whole and unchanged. Pure Applied Chemistry. [Google Scholar]
- IUPAC (International Union of Applied and Pure Chemistry) . (2015). Publication of this document by any means is permitted on condition that it is whole and unchanged. Copyright © IUPAC & De Gruyter 2015. Pure Applied Chemistry, 87, 1039—1049. [Google Scholar]
- IUPAC (International Union of Applied and Pure Chemistry) . (2012). Publication of this document by any means is permitted on condition that it is whole and unchanged. Copyright © IUPAC 2012. Pure Applied Chemistry, 84, 2167—2169. [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2001). Test no. 416: Two‐generation reproduction toxicity. In OECD guidelines for the testing of chemicals, section 4. OECD Publishing. Available online: 10.1787/9789264070868-en [DOI] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2007a). Test no. 426: Developmental neurotoxicity study. In OECD guidelines for the testing of chemicals, section 4. OECD Publishing. Available online: 10.1787/9789264067394-en [DOI] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2007b). Test no. 440: Uterotrophic bioassay in rodents: A short‐term screening test for oestrogenic properties. In OECD guidelines for the testing of chemicals, section 4. OECD Publishing. Available online: 10.1787/9789264067417-en [DOI] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2009). Test no. 441: Hershberger bioassay in rats: A short‐term screening assay for (Anti) androgenic properties. In OECD guidelines for the testing of chemicals, section 4. OECD Publishing. Available online: 10.1787/9789264076334-en [DOI] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2010). Test no. 417: Toxicokinetics. In OECD guidelines for testing of chemicals, section 4. OECD Publishing. Available online: 10.1787/9789264070882-en [DOI] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2016). Test no. 421: Reproduction/Developmental toxicity screening test. In OECD guidelines for the testing of chemicals, section 4. OECD Publishing. Available online: 10.1787/9789264264380-en [DOI] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2018a). Test no. 408: Repeated dose 90‐day oral toxicity study in rodents. In OECD guidelines for the testing of chemicals, section 4. OECD Publishing. 10.1787/9789264070707-en [DOI] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2018b). Test no. 414: Prenatal developmental toxicity study. In OECD g for the testing of chemicals, section 4. OECD Publishing. Available online: 10.1787/9789264070820-en [DOI] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2018c). Test no. 443: Extended one‐generation reproductive toxicity study. In OECD guidelines for the testing of chemicals, section 4. OECD Publishing. Available online: 10.1787/9789264185371-en [DOI] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2018d). Test no. 451: Carcinogenicity studies. In OECD guidelines for the testing of chemicals, section 4. OECD Publishing. Available online: 10.1787/9789264071186-en [DOI] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2018e). Test no. 452: Chronic toxicity studies. In OECD guidelines for the testing of chemicals, section 4. OECD Publishing. Available online: 10.1787/9789264071209-en [DOI] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2018f). Test no. 453: Combined chronic toxicity/carcinogenicity studies. In OECD guidelines for the testing of chemicals, section 4. OECD Publishing. Available online: 10.1787/9789264071223-en [DOI] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2020). Test no. 471: Bacterial reverse mutation test. In OECD guidelines for the testing of chemicals, section 4. OECD Publishing. Available online: 10.1787/9789264071247-en [DOI] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2021). Guidance document on the characterisation, validation and reporting of physiologically based kinetic (PBK) models for regulatory purposes, OECD series on testing and assessment, No. 331, Environment,Health and Safety, Environment directorate. OECD. [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2023a). Test no. 487: In vitro mammalian cell micronucleus test. In OECD guidelines for the testing of chemicals, section 4. OECD Publishing. Available online: 10.1787/9789264264861-en [DOI] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2023b). Test no. 456: H295R Steroidogenesis assay. In OECD guidelines for the testing of chemicals, section 4. OECD Publishing. Available online: 10.1787/9789264122642-en [DOI] [Google Scholar]
- OECD (Organisation for Economic Co‐operation and Development) . (2024). Test No. 493: Performance‐based test guideline for human recombinant estrogen receptor (hrER). In Vitro assays to detect chemicals with ER binding affinity, OECD guidelines for the testing of chemicals, section 4. OECD Publishing. Available online: 10.1787/9789264242623-en [DOI] [Google Scholar]
- van Buuren, S. , Schönbeck, Y. , & van Dommelen, P. (2012). Collection, collation and analysis of data in relation to reference heights and reference weights for female and male children and adolescents (0–18 years) in the EU, as well as in relation to the age of onset of puberty and the age at which different stages of puberty are reached in adolescents in the EU. EFSA Supporting Publications, 9(3), 255E. 10.2903/sp.efsa.2012.EN-255 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Outcome of Public Consultation