

Abstract
Orphan GPR52 is emerging as a promising neurotherapeutic target. Optimization of previously reported lead 4a employing an iterative drug design strategy led to the identification of a series of unique GPR52 agonists, such as 10a (PW0677), 15b (PW0729), and 24f (PW0866), with improved potency and efficacy. Intriguingly, compounds 10a and 24f showed greater bias for G protein/cAMP signaling and induced significantly less in vitro desensitization than parent compound 4a, indicating that reducing GPR52 β-arrestin activity with biased agonism results in sustained GPR52 activation. Further exploration of compounds 15b and 24f indicated improved potency and efficacy, and excellent target selectivity, but limited brain exposure warranting further optimization. These balanced and biased GPR52 agonists provide important pharmacological tools to study GPR52 activation, signaling bias, and therapeutic potential for neuropsychiatric and neurological diseases.
Graphical Abstract

INTRODUCTION
Orphan G protein-coupled receptors (oGPCRs; e.g., GPR6, GPR12, GPR37, GPR61, GPR88, GPR52, and GPR158) are emerging therapeutic targets for central nervous system (CNS) disorders, such as schizophrenia (SZ), Huntington’s disease (HD), Parkinson’s disease (PD), Alzheimer’s disease (AD), attention deficit hyperactivity disorder (ADHD) as well as metabolic disorders, obesity and cancer.1–9 GPR52 is an oGPCR that regulates diverse cAMP-mediated pathological and physiological processes.1 GPR52 was first identified as a novel oGPCR from the GenBank high-throughput genome database in 1999.10 Considerable effort has been made to determine the tissue distribution and expression of GPR52.11,12 In situ hybridization studies in rats and large-scale RNA sequencing approaches in humans indicate GPR52 is exclusively expressed in the brain, most abundantly in the striatum.11,12 Anatomical studies have demonstrated that GPR52 is specifically coexpressed with dopamine D2 receptors (D2R) in medium spiny neurons (MSNs) in the striatum.13 Activated GPR52 couples primarily to Gs or Golf protein that activates adenylyl cyclases to increase cellular cAMP. The Gs/cAMP signaling pathway of GPR52 may induce functional crosstalk with the Gi-coupled D2R,14 and modulate dopamine signaling in striatal neurons and in the broader corticostriatal brain circuitry.11,13 Recent human genome-wide association studies (GWAS) have revealed that GPR52 is also a schizophrenia (SZ) risk gene.15 GPR52-knockout mice display psychosis-related behaviors, whereas transgenic mice over-expressing GPR52 exhibit antipsychotic-like behaviors.11 These studies collectively suggest that GPR52 agonists may have antipsychotic-like activity. In addition, colocalization of GPR52 with Gs-coupled dopamine D1 receptors (D1R) in the medial prefrontal cortex suggests a functional role in attention, cognition, and memory.11,16,17 Based on its coexpression with D2R and D1R, activation of GPR52 could produce antipsychotic effects, improve cognition, and reduce the effects of psychostimulant drugs. Furthermore, GPR52 was reported to stabilize mutant huntingtin (mHTT) protein, exacerbating the cytotoxicity of mHTT in Huntington’s disease (HD).18–20 Interestingly, knockout of GPR52 or loss of GPR52 function suppresses the accumulation of mHTT in striatal neurons and improves HD-related movement impairments.18–20 Taken together, these findings indicate that GPR52 agonists could be beneficial for treating SZ, psychotic disorders, or substance use disorders (SUDs). However, GPR52 antagonists could be potential therapeutics for HD.
To date, the endogenous ligand of GPR52 has not been identified. The crystal structure of human GPR52 has been solved in three states: two ligand-free apo states (PBD codes: 6LI1 and 6LI2) and one active state in complex with agonist c17 (2) (PBD: 6LI0).21 In these structures, GPR52 displays an unprecedented phenomenon of self-activation as a result of binding its own extracellular loop 2 (ECL2) as an internal agonist to the classical orthosteric site.21,22 The lack of known endogenous GPR52 ligands and lack of close homology to known GPCRs poses challenges to fully understanding the functional roles of the receptor. Therefore, the development of surrogate ligands is highly desirable for probing the functional activity and therapeutical potential of GPR52.
So far, several surrogate ligands as GPR52 agonists have been identified (Figure 1). Setoh et al. identified the first GPR52 agonists, exemplified by compound 1 as a potent GPR52 agonist (EC50 = 30 nM) with poor aqueous solubility.23 Further optimization of compound 1 to improve the aqueous solubility led to the identification of compound 2 with high potency (EC50 = 21 nM) and better aqueous solubility (21 μg/mL at pH 6.8).24 In addition, compound 2 dose-dependently inhibited methamphetamine-induced hyper-locomotion in mice, a preclinical model for antipsychotic activity.24 Tokumaru et al. reported a series of 4-azolyl-benzamide analogues as GPR52 agonists, exemplified by compound 3 with high potency (EC50 = 75 nM).25 In animal models, compound 3 dose-dependently demonstrated antipsychotic activity and improved cognitive functions.26 Arena Pharmaceuticals disclosed a series of indoline-4-carboxamides as GPR52 agonists, exemplified by a representative compound 4a.27 Recently, Sosei Heptares reported an additional series of GPR52 agonists with a similar pharmacophore, exemplified by HTL0041178.28 Phase I clinical trials have begun with compound HTL0048149 (structure not disclosed) as the first-in-class GPR52 agonist from Sosei Heptares for the treatment of SZ positive, negative, and cognitive symptoms, potentially without the adverse effects accompanied by currently available antipsychotics.29 Some additional GPR52 modulators have been disclosed in recent patents.30,31
Figure 1.
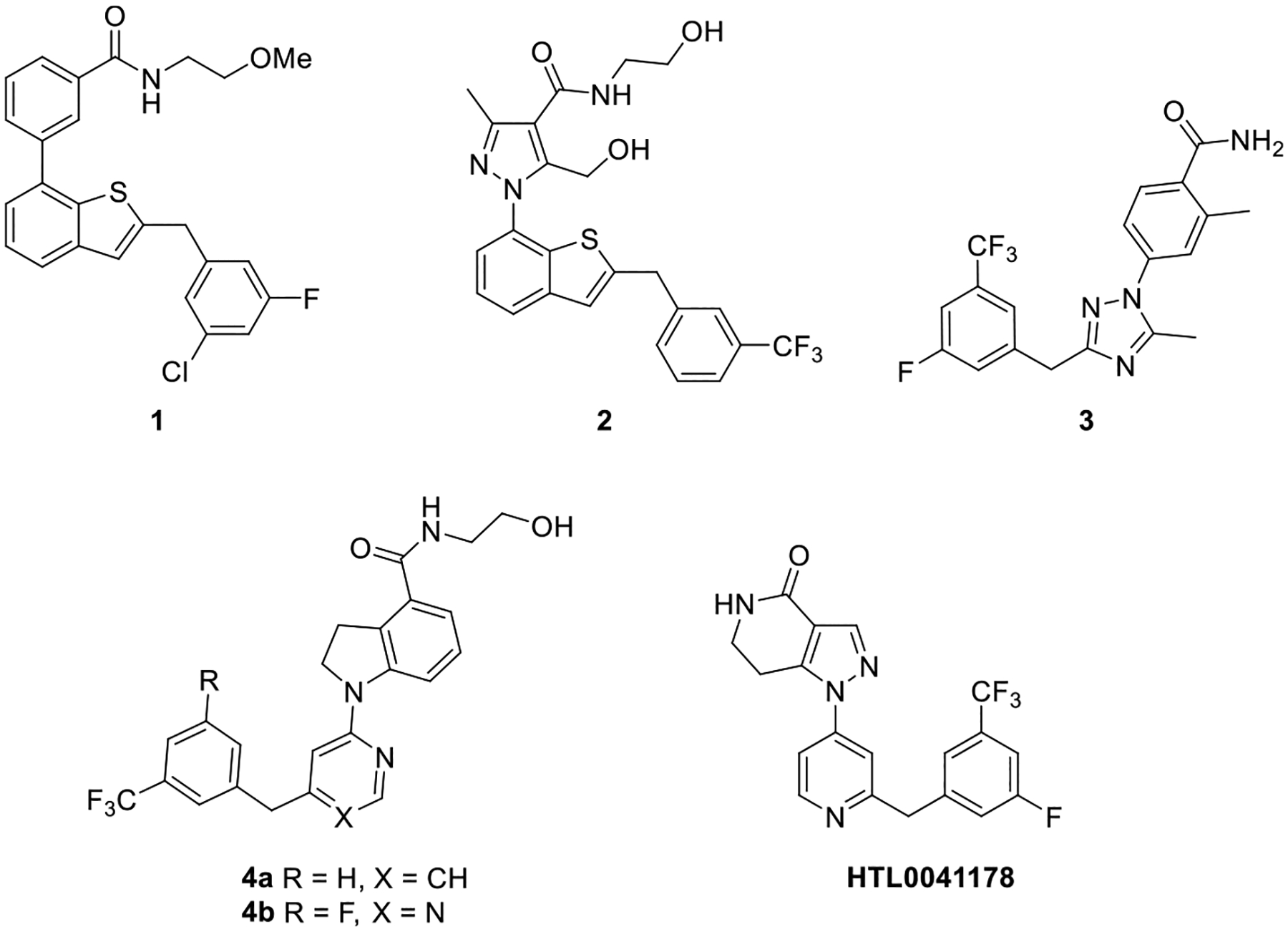
Chemical structures of representative GPR52 agonists.
Notably, prior SAR and pharmacology studies of these compounds strictly focused on GPR52 agonist activity for the canonical G protein Gs/olf cAMP pathway. However, most activated GPCRs also universally couple to the β-arrestin proteins, which control receptor trafficking and desensitization and may propagate receptor signaling pathways distinct from G proteins.32–34 Agonists that activate receptors to signal primarily via G proteins versus β-arrestin are known as biased agonists,35 which may improve agonist efficacy and/or reduce side effects induced by unbiased receptor ligands.36–39 Therefore, the development of potent and highly selective GPR52 agonists, including biased agonists, will provide useful pharmacological tools for elucidating GPR52 function as well as provide promising drug leads for the treatment of various CNS diseases.
Inspired by the understudied physiology of GPR52 and the potential neurotherapeutic applications of GPR52 agonists, we focused our drug discovery program on developing novel potent and selective GPR52 agonists. In our previous work, compound 4a was selected as the lead for further systematic SAR studies to identify more potent compounds.40,41 This prior optimization of compound 4a led to the identification of a series of 1-(pyrimidin-4-yl)indoline-4-carboxamides as selective GPR52 agonists, exemplified by compound 4b with improved efficacy, along with excellent target specificity and pharmacokinetic (PK) profiles. Further studies of amphetamine-induced hyperlocomotor behavior in mice revealed that compound 4b exhibited antipsychotic-like activity.40
In our continued research toward developing novel GPR52 agonists based on the lead 4a (Figure 2), we first opened the indoline ring moiety (highlighted in green) to enhance the flexibility of the molecules with a truncated scaffold and to expose an additional NH that could form new hydrogen bonds to improve the potency. In addition, we employed systematic medicinal chemistry around the aminoalcohol group (R1) and substituents on ring A and ring C (R2 and R3, respectively) of 4a (Figure 2; aminoalcohol side chain highlighted in blue, ring A highlighted in magenta, ring C highlighted in red). Remarkably, the opening of the indoline ring resulted in GPR52 biased agonism relative to parent compound 4a, with significantly increased G protein signaling potency and efficacy but decreased potency for β-arrestin recruitment. The most biased of these compounds also displayed reduced GPR52 agonist-induced desensitization, which allowed for sustained cAMP signaling. To our knowledge, these are among the first biased ligands for any oGPCR and the first biased agonists for GPR52.
Figure 2.

Proposed design strategy and structural modifications to 4a.
RESULTS AND DISCUSSION
Chemistry.
All compounds described herein were synthesized following the synthetic procedures depicted in Schemes 1–4. The intermediate 7 was prepared by Suzuki C−C coupling reaction using commercially available 1-(bromomethyl)-3-(trifluoromethyl)benzene 5 and (2-chloropyridin-4-yl)-boronic acid 6 in the presence of Pd(PPh3)4 (Scheme 1).27 The palladium-catalyzed C−N coupling reaction between intermediate 7 and ethyl 3-aminobenzoate provided intermediate 8, which subsequently underwent ester hydrolysis to yield intermediate 9. Compounds 10a−l were synthesized through the amide coupling between the intermediate 9 and corresponding amino derivatives.
Scheme 1.

Synthesis of Compounds 10a−la
aReagents and conditions: (a) Pd(PPh3)4, Na2CO3, toluene, EtOH, 85 °C, 1.5 h, 48%; (b) ethyl 3-aminobenzoate, Pd(OAc)2, XantPhos, Cs2CO3, 1,4-dioxane, 100 °C, 12 h, 76%; (c) (i) 4 N NaOH, EtOH, reflux, 2 h; (ii) 2 N HCl, 80%; (d) R1H, EDCI, DMAP, HOBt, DMF, rt, 12 h, 41−87%.
Scheme 4.

Synthesis of Compound 24a−ka
aReagents and conditions: (a) Pd(PPh3)4, Na2CO3, toluene, EtOH, 85 °C, 1.5 h, 53−93%; (b) methyl 3-amino-2-methylbenzoate, Pd(OAc)2, XantPhos, Cs2CO3, 1,4-dioxane, 100 °C, 12 h, 65−83%; (c) (i) 2 N NaOH, EtOH, reflux, 2 h; (ii) 2 N HCl, 73−93%; (d) R1H, EDCI, DMAP, HOBt, DMF, rt, 12 h, 38−93%.
Compounds 12d and 12e were produced via palladium-catalyzed cross-coupling reaction of 11a and 11b with cyclopropylboronic acid or furan-2-ylboronic acid, respectively (Scheme 2). The intermediates 13a−h were synthesized using intermediate 7 and a set of intermediates 12a−h following the synthetic procedure of intermediate 8. Ester hydrolysis of intermediates 13a−h led to compounds 14a−h, which subsequently coupled with 2-aminoethan-1-ol to provide the final compounds 15a−h. Compound 19 was prepared following similar synthetic procedures to compound 10a (Scheme 3). The C−N coupling reaction between intermediate 7 and methyl 4-aminobenzoate 16 afforded intermediate 17, which upon ester hydrolysis provided intermediate 18. Finally, condensation of intermediate 18 with 2-aminoethan-1-ol furnished the targeted compound 19.
Scheme 2.

Synthesis of Compounds 15a−ha
aReagents and conditions: (a) cyclopropylboronic acid or furan-2-ylboronic acid, Pd(dppf)2Cl2, K3PO4, 1,4-dioxnae, H2O, 85 °C, 12 h, 47% for 12d, 60% for 12e; (b) Pd(OAc)2, XantPhos, Cs2CO3, 1,4-dioxane, 100 °C, 12 h, 37–81%; (c) (i) 2 N NaOH, EtOH, reflux, 2 h; (ii) 2 N HCl, 79–94%; (d) NH2CH2CH2OH, EDCI, DMAP, HOBt, DMF, rt, 12 h, 38−86%.
Scheme 3.

Synthesis of Compound 19a
aReagents and conditions: (a) Pd(OAc)2, XantPhos, Cs2CO3, 1,4-dioxane, 100 °C, 12 h, 87%; (b) (i) 4 N NaOH, EtOH, reflux, 2 h; (ii) 2 N HCl, 91%; (c) NH2CH2CH2OH, EDCI, DMAP, HOBt, DMF, rt, 12 h, 77%.
The intermediates 21a−k were prepared following the synthetic procedure of intermediate 7 using commercially available 6 with 20a−k (Scheme 4). Following the synthetic route of intermediate 8, intermediates 22a−k were obtained using methyl 3-amino-2-methylbenzoate and 21a−k. Hydrolysis of methyl esters 22a−k led to the formation of corresponding acid 23a−k. The targeted compounds 24a−k were accessed following the same synthetic procedure as compound 10a, using intermediates 23a−k and 2-aminoethan-1-ol.
In Vitro Evaluation of GPR52 G Protein-Mediated cAMP Activities.
All the compounds were evaluated for their GPR52 agonist activity based on cAMP production using the GloSensor cAMP assay, and the results are summarized in Tables 1−3. Reported GPR52 agonist 4a was resynthesized and used as the parent scaffold and reference compound (EC50 = 119 nM, when screened in our assay in a 12-point concentration−response). Interestingly, GPR52 expression alone significantly elevated basal cAMP more than 20-fold over both empty vector control and Gs-coupled D1R, indicating that GPR52 has a high constitutive activity for cAMP signaling (Supplementary Figure S1A). The GloSensor cAMP assay used for GPR52 screening provided a consistent, robust agonist signal for all compounds described herein, although the limited aqueous solubility of compound 4a caused visible precipitation and decreased signal at high concentrations (Supplementary Figure S1B).
Table 1.
EC50 and Emax of Compounds 10a−l
| Compound | R1 | ClogPa | EC50(nM)b | Emax(%)b |
|---|---|---|---|---|
| 4a |

|
4.10 | 119 ± 18 | 100 ± 5 |
| 10a |

|
4.16 | 282 ± 46 | 197 ± 26 |
| 10b |

|
5.47 | 467 ± 60 | 131 ± 13 |
| 10c |

|
5.03 | 672 ± 134 | 99 ± 12 |
| 10d |
|
4.91 | 774 ± 124 | 93 ± 7 |
| 10e |

|
4.09 | 448 ± 59 | 164 ± 12 |
| 10f |

|
5.26 | 862 ± 157 | 188 ± 11 |
| 10g |

|
3.52 | 220 ± 39 | 214 ± 19 |
| 10h |

|
5.43 | 638 ± 137 | 152 ± 7 |
| 10i |

|
4.81 | 252 ± 54 | 131 ± 15 |
| 10j |

|
4.55 | 220 ± 43 | 129 ± 7 |
| 10k |
|
4.94 | 198 ± 37 | 129 ± 8 |
| 10l |

|
3.52 | 286 ± 3 | 268 ± 12 |

Clog P is calculated from a web calculator: http://biosig.unimelb.edu.au/pkcsm/prediction.
Values are mean ± SEM from n ≥ 3 independent experiments.
Emax (%) is the efficacy maximum of the compounds in the cAMP assay relative to compound 4a as 100% and DMSO as 0%.
Table 3.
EC50 and Emax of Compounds 24a−k

| ||||
|---|---|---|---|---|
| compound | R3 | Clog Pa | EC50 (nM)b | Emax (%)b |
| 4a | 3-CF3 | 4.10 | 119 ± 16 | 100 ± 5 |
| 15b | 3-CF3 | 4.47 | 47 ± 8 | 143 ± 5 |
| 24a | 3,5-di-CF3 | 5.48 | 211 ± 51 | 127 ± 11 |
| 24b | 2,4-di-CF3 | 5.48 | 52 ± 11 | 150 ± 12 |
| 24c | 4-CF3 | 4.47 | 220 ± 58 | 120 ± 9 |
| 24d | 2-CF3 | 4.47 | 143 ± 33 | 122 ± 13 |
| 24e | 3-Cl, 5-F | 4.24 | 46 ± 8 | 135 ± 9 |
| 24f | 3-OMe | 3.46 | 53 ± 10 | 133 ± 10 |
| 24g | H | 3.45 | 278 ± 48 | 139 ± 9 |
| 24h | 4-Cl, 2-F | 4.24 | 235 ± 47 | 131 ± 8 |
| 24i | 3-F | 3.59 | 136 ± 33 | 137 ± 7 |
| 24j | 4-F | 3.59 | 90 ± 17 | 146 ± 9 |
| 24k | 3-OCF3 | 4.35 | 30 ± 8 | 137 ± 7 |
Clog P is calculated from a web calculator: http://biosig.unimelb.edu.au/pkcsm/prediction.
Values are mean ± SEM from n ≥ 3 independent experiments.
Emax (%) is the efficacy maximum of the compounds in the cAMP assay relative to compound 4a as 100% and DMSO as 0%.
We embarked on a systematic structure−activity relationship (SAR) study, at the outset exploring the indoline ring moiety of compound 4a (Figure 2, highlighted in green) (Table 1). Opening of the indoline ring led to the formation of compound 10a (EC50 = 282 nM, Emax = 197%), which demonstrated a 2.4-fold decrease in potency but a significant increase in efficacy compared to reference 4a. To probe the flexibility of the side chain (R1), the aminoalcohol was replaced with a set of cycloalkanes 10b−d or appended with substituents on the aminoalcohol side chain 10e−g. Compounds 10b−g displayed reduced potency in comparison to 4a, ranging from a 1.8- to 7.2-fold shift. Incorporation of fluorine or a group of fluorine atoms in a molecule may increase the potency and the permeability, reduce the pKa and clearance, and restrict the conformation.42,43 Therefore, we leveraged the application of fluorine in our medicinal chemistry campaign. Replacing the hydroxy group of the aminoalcohol of 10a with difluoro afforded compound 10h. In contrast to our expectation, compound 10h displayed a 2.3-fold potency loss and a slight decrease in efficacy compared to 10a. Swapping the hydroxy group of the aminoalcohol of 10a with a methoxy group yielded compound 10i (EC50 = 252 nM), which showed a slight decrease in potency versus 4a. Extending the alkyl chain of the aminoalcohol of 10a led to compounds 10j−l, which exhibited similar potency to 10a. Collectively, none of compounds 10a−l displayed improved potency over 4a. Therefore, we decided to use the aminoalcohol as the optimal side chain for further modifications. This is consistent with prior SAR studies indicating ethanolamine is critical for GPR52 potency.40
Having optimized the side chain, we next turned our attention to the role of substituents on ring A of 10a, and the results are summarized in Table 2. Appending substitutions at the 2-position led to compounds 15a−e, which demonstrated increased potency, except 15a, but slightly reduced efficacy relative to 10a. Notably, 15b (EC50 = 47 nM, Emax = 143%) and 15d (EC50 = 30 nM, Emax = 146%) displayed the best improvement in potency. The addition of substituents at the 4-position of ring A provided compounds 15f and 15g. However, substitution at the 4-position of ring A was not tolerated and led to a significant decrease in potency. In addition, the introduction of fluorine at the 6-position of ring A afforded compound 15h, which significantly lost efficacy compared to 10a (Emax = 126 and 197%, respectively) but had minimal effect on potency. Transposition of amide from the 1-position to the 6-position on ring A yielded compound 19 that exhibited a substantial loss in potency relative to both 4a and 10a, though it still retained efficacy comparable to 4a. Taken together, small alkyl groups (e.g., methyl, cyclopropyl) on the 2-position of ring A were found to improve potency over both 4a and 10a, while maintaining the increased efficacy of the open-ring 10a for GPR52 cAMP signaling.
Table 2.
EC50 and Emax of Compounds 15a−h and 19
| Compound | R2 | ClogPa | EC50(nM)b | Emax(%)b |
|---|---|---|---|---|
| 4a | Indoline | 4.10 | 119 ± 18 | 100 ± 5 |
| 10a | H | 4.16 | 282 ± 46 | 197 ± 26 |
| 15a | 2-F | 4.30 | 342 ± 78 | 134 ± 2 |
| 15b | 2-Me | 4.47 | 47 ± 8 | 143 ± 5 |
| 15c | 2-OMe | 4.17 | 64 ± 19 | 149 ± 8 |
| 15d | 2-cyclopropyl | 5.03 | 30 ± 9 | 146 ± 9 |
| 15e | 2-(furan-2-yl) | 5.42 | 84 ± 12 | 147 ± 6 |
| 15f | 4-Me | 4.47 | 984 ± 18 | 94 ± 6 |
| I5g | 4-OMe | 4.17 | 1432 ± 207 | 111 ± 7 |
| I5h | 6-F | 4.30 | 229 ± 38 | 126 ± 9 |
| 19 |

|
4.16 | 1086 ± 85 | 115 ± 10 |

Clog P is calculated from a web calculator: http://biosig.unimelb.edu.au/pkcsm/prediction.
Values are mean ± SEM from n ≥ 3 independent experiments.
Emax (%) is the efficacy maximum of the compounds in the cAMP assay relative to compound 4a as 100% and DMSO as 0%.
By taking advantage of the results presented in Tables 1 and 2, we established the aminoalcohol as a privileged side chain and the methyl group at the 2-position of ring A as a principal substituent to further investigate the effects of substitutions on ring C of 15b (Table 3). The introduction of an additional CF3 group led to compound 24a (3,5-di-CF3), which displayed a 4-fold loss in potency (EC50 = 211 nM) compared to 15b. Conversely, compound 24b (2,4-di-CF3) maintained potency (EC50 = 52 nM). Transposition of the CF3 group from the 3-position to the 2- or 4-position led to compounds 24c and 24d, respectively. Interestingly, these mono-CF3 substitutions in 24c and 24d showed a 3-fold decrease in potency compared to 24b with both additions. Installation of Cl at the 3-position and F at the 5-position on ring C provided compound 24e that exhibited similar activity (EC50 = 46 nM) to 15b. In contrast, compound 24h containing Cl at the 4-position and F at the 2-position on ring C demonstrated a reduction in potency (EC50 = 235 nM). Furthermore, incorporating F at the 4-position on ring C led to the formation of 24j, which demonstrated a slight loss in potency (EC50 = 90 nM) compared to 15b. To investigate the effects of steric hindrance caused by the substituents at the 3-position of ring C, compounds 24f, 24g, 24i, and 24k were designed and synthesized, which mainly displayed good potency in the order OCF3 > CF3 > OMe > F > H, with no significant alterations to efficacy among the compounds. Among these, compound 24k (EC50 = 30 nM) was not only more potent than 4a but also more potent than 15b.
Notably, the halogenation of 24e and 24h had opposite effects compared to the analogous compounds 24a and 24b, respectively, with bulkier di-CF3 additions at the same positions. Double substitution at the 3,5-positions appeared to be more favorable with the smaller F and Cl additions, whereas larger CF3 additions at the 2,4-positions were more potent. However, this observation only held for the activity of compounds with double additions to ring C, with single substituents at the 3-position favoring more sterically hindering groups (as discussed above). Collectively, the results discussed above suggest that ring C can tolerate either electron-donating or electron-withdrawing substituents. In addition, compounds with large groups (e.g., OCF3 and CF3) at the 3-position of ring C showed better GPR52 agonist efficacy.
Overall, opening the indoline ring of 4a led to an increase in efficacy for GPR52 activation of G protein-mediated cAMP signaling. The rationale for the improved efficacy of these compounds can presumably be attributed to the flexibility of the scaffold. Modification to the aminoalcohol moiety generally had no effect or reduced potency and efficacy compared to 10a (Figure 3A). The addition of substituents to the 2-position of ring A is essential to improve potency over 4a and the open-ring scaffold 10a (Figure 3B). Ring C is amenable to a wide array of modifications, with bulky substitutions at the 3-position being optimal for improving potency (Figure 3C).
Figure 3.

Concentration−responses of GPR52 agonists for cAMP signaling in HEK293 cells expressing human GPR52 and the GloSensor cAMP reporter. (A) Opening the indoline ring of 4a to yield 10a substantially increased efficacy but reduced potency. Further modification around the aminoalcohol moiety (blue) generally had minimal effects or reduced potency and efficacy compared to 10a. (B) Addition of substituents around ring A (magenta), particularly at the 2-position, improved both potency and efficacy over 4a but did not achieve the efficacy of 10a. (C) Substitutions to ring C (red) of 15b generally lost potency while maintaining the increased efficacy relative to 4a. Results are mean ± SEM from triplicate testing in a representative experiment, with similar results observed in n ≥ 3 experiments.
In Vitro Evaluation of GPR52 β-Arrestin Recruitment and Ligand Bias.
In addition to canonical G protein signaling, most agonist-activated GPCRs couple to β-arrestin, a multifunctional scaffolding protein that controls receptor trafficking and activation of various kinases.32,44 However, to the best of our knowledge, GPR52 recruitment of β-arrestin has not been previously reported. Therefore, we utilized a robust method of measuring the recruitment of β-arrestin2 to human GPR52, using the transcriptional activation following arrestin translocation (Tango) assay. The Tango assay is a scalable, gene reporter-based assay in which GPCR recruitment of β-arrestin results in transcription of luciferase.45,46 For the Tango assay, GPR52 is modified to include a portion of the vasopressin V2 receptor C-terminus followed by a TEV protease cleavage site and the Tta protein sequence. The V2R tail has been shown to increase β-arrestin recruitment47,48 and was added to enhance Tango assay performance.46 As an initial control, we compared human GPR52 wildtype with human GPR52 Tango testing 4a concentration responses using the GloSensor cAMP assay (Supplementary Figure S2). No significant differences were observed for potency or efficacy of 4a, suggesting that agonist pharmacology is similar between the wildtype and Tango versions of human GPR52. In further validation studies, GPR52 displayed high constitutive activity to recruit β-arrestin, significantly elevated above that of another Gs-coupled receptor, D1R (Supplementary Figure S3A). The concentration-response of the positive control agonist 4a produced robust β-arrestin recruitment to GPR52, approximately 25-fold over basal. This response was specific to GPR52, with no response observed when tested against the D1R (Supplementary Figure S3B). As a quantitative assessment of assay quality, a z-factor of 0.812 was determined testing 4a agonism at GPR52, indicating the assay had excellent signal-to-noise with low error.49
Alongside 4a, 12 of the most potent and efficacious GPR52 ligands for G protein/cAMP signaling were selected for concentration−response screening in the Tango β-arrestin assay. The results are summarized in Table 4, along with calculated bias factors for activation of the G protein/cAMP or β-arrestin pathways. These bias factors provide a scale to compare agonist activity for GPR52 signaling in one pathway versus the other, relative to the parent compound 4a.50 All of the tested compounds displayed some degree of GPR52 G protein/cAMP bias compared to 4a: the most biased being 24f (G protein bias factor = 81.9) and the least biased being 24b (G protein bias factor = 2.41).
Table 4.
β-Arrestin Recruitment EC50 and Emax and Bias Factors of Selected Compounds
| compound | EC50 (nM)a | Emax (%)a | G protein bias factor | β-arrestin bias factor |
|---|---|---|---|---|
| 4a | 22 ± 5 | 100 ± 10 | 1.00 | 1.00 |
| 10a | 483 ± 46 | 70 ± 6 | 25.8 | 0.039 |
| 10g | 1585 ± 250 | 125 ± 12 | 66.3 | 0.015 |
| 10l | 1582 ± 149 | 127 ± 7 | 63.2 | 0.016 |
| 15b | 110 ± 11 | 109 ± 3 | 16.4 | 0.061 |
| 15c | 247 ± 62 | 100 ± 7 | 31.2 | 0.032 |
| 15d | 14 ± 4 | 92 ± 2 | 4.10 | 0.244 |
| 15e | 335 ± 69 | 118 ± 5 | 26.9 | 0.037 |
| 24b | 17 ± 3 | 109 ± 6 | 2.41 | 0.415 |
| 24e | 89 ± 12 | 120 ± 3 | 11.7 | 0.085 |
| 24f | 1036 ± 136 | 172 ± 7 | 81.9 | 0.012 |
| 24j | 563 ± 28 | 142 ± 6 | 34.7 | 0.029 |
| 24k | 349 ± 25 | 131 ± 4 | 64.9 | 0.015 |
Values are mean ± SEM from n ≥ 3 independent experiments.
Emax (%) is the efficacy maximum of the compounds in the β-arrestin recruitment assay relative to compound 4a as 100% and DMSO as 0%.
The opening of the indoline ring of 4a to yield 10a resulted in a profound 20-fold loss in potency for GPR52 β-arrestin activation and a 30% reduction in the maximum response (Figure 4A). The introduction of additional hydroxy groups to the side chain in 10g and 10l further decreased β-arrestin potency by more than 70-fold but increased the efficacy relative to both 4a and 10a. Substitution of small alkyl groups at the 2-position of ring A (15b−e) rescued both potency and efficacy for β-arrestin recruitment that was lost in 10a (Figure 4B). This increase in β-arrestin potency, along with the identical rank order in potency, was similarly observed in the cAMP assay, suggesting a significant role of the 2-position of ring A in the ligand−receptor interaction with GPR52. Substitution to ring C of 15b generally demonstrated limited influence on GPR52 β-arrestin efficacy but had variant effects on potency for β-arrestin activation (Figure 4C). A structure−activity trend for β-arrestin recruitment was not clear with ring C modifications. As shown in the cAMP signaling assays, however, ring C is amenable to a wide array of modifications that also maintained activity for β-arrestin recruitment.
Figure 4.

Concentration−responses of GPR52 agonists for β-arrestin recruitment in HTLA cells expressing the human GPR52 Tango construct. (A) The open-ring derivatives of 4a had greatly reduced compound potency and efficacy, and modification of the terminal aminoalcohol moiety (blue) further reduced potency but rescued efficacy. (B) Modifications around ring A (magenta) generally resulted in loss of potency and similar maximal efficacy compared to 4a. (C) Substitutions to ring C (red) of 15b generally increased maximal β-arrestin recruitment. Results are mean ± SEM from triplicate testing in a representative experiment, with similar results observed in n ≥ 3 experiments.
Interestingly, the parent compound 4a was more active in the β-arrestin pathway, with roughly 5-fold greater potency for β-arrestin activation than for G protein/cAMP activation (Figure 5A). Simply removing two carbons of the 4a indoline ring, yielding 10a, both sharply increased efficacy for cAMP signaling and sharply decreased potency and efficacy for β-arrestin, suggesting that interactions with these carbons may be a driving factor for agonist recruitment of β-arrestin to GPR52 (Figure 5B). Alkyl substitutions at the 2-position of ring A largely recovered this lost β-arrestin activity. This can be seen with the methyl substitution in 15b, which provided a more balanced profile between the G protein and β-arrestin pathways (Figure 5C). These alkyl substitutions at the 2-position of ring A (15b, 15c, 15d, 15e) all enhanced potency both for β-arrestin and G protein activation. These results indicate that the receptor interactions, or possibly the steric hindrance, afforded by substituents at this position are valuable for ligand−receptor binding, rather than for introducing bias for any pathway. Taken together, these pharmacological studies clearly establish that opening the indoline of 4a, creating more structurally flexible GPR52 agonists, promotes bias for the G protein/cAMP pathway over the β-arrestin pathway. These studies provide a structural basis for agonist tools that can fine-tune GPR52 signaling toward β-arrestin or G protein/cAMP.
Figure 5.

Comparison of concentration−responses of select GPR52 agonists for both cAMP signaling and β-arrestin recruitment. (A) Reference compound 4a showed ~5-fold greater potency for β-arrestin than for cAMP signaling. (B) The removal of the indoline ring in 10a greatly reduced activity for the β-arrestin pathway but nearly doubled the maximum efficacy for cAMP production. (C) The methyl substitution at ring A in 15b rescued β-arrestin activity and increased potency for cAMP signaling. (D) 10g showed similar cAMP signaling activity to 10a, with greater efficacy for β-arrestin recruitment. (E,F) 24f and 24j had similar cAMP signaling activity to 15b, although the substitutions at ring C reduced potency for β-arrestin activation. Results are mean ± SEM from triplicate testing in a representative experiment, with similar results observed in n ≥ 3 experiments.
Evaluation of G Protein-Biased and Unbiased Agonists on GPR52 Desensitization.
Functional selectivity of agonists for GPCR signaling pathways may provide greater therapeutic efficacy as well as reduced side effect profiles.33,35,36,38,39 GPCR β-arrestin activation has historically been conceptualized to interrupt G protein signaling and to induce receptor internalization.34,44,51 Prolonged recruitment of β-arrestin to GPCRs can desensitize receptor signaling responses to agonists,37,51 potentially leading to tachyphylaxis.33,34,37,39 To evaluate the functional role of bias for G protein activation over β-arrestin recruitment with our GPR52 agonists, we examined the effect of extended treatment with these compounds on GPR52 desensitization. A schematic shown in Figure 6A outlines agonist-induced recruitment of β-arrestin to GPR52 to inhibit G protein coupling, causing desensitization of GPR52 G protein signaling. Continuous recruitment of β-arrestin is expected to decrease the pool of available receptors for G protein signaling.
Figure 6.

Desensitization studies of G protein-biased and balanced GPR52 agonists. (A) GPR52 agonists recruit β-arrestin to the receptor, inhibiting G protein activation and cAMP production. (B) HEK293 cells were pretreated with cAMP assay EC50 concentrations of GPR52 agonists, washed, then challenged with 4a (1 μM). (C) Desensitization to the 4a cAMP response was greatest for 24b and 4a, which were the most potent agonists for GPR52 β-arrestin recruitment. Notably, the G protein-biased compounds 10a and 24f induced significantly less GPR52 desensitization than 2 h treatments with 4a. (D) Pearson’s correlation analysis reported a significant, positive correlation between compound potency for GPR52 β-arrestin recruitment and the GPR52 cAMP signaling remaining after 2 h pretreatment with compounds. Compounds with greater potency for β-arrestin recruitment showed a greater reduction in GPR52 G protein cAMP signaling. Results are mean ± SEM from n = 3 independent experiments. A, p < 0.05 vs DMSO; b, p < 0.05 vs 4a; one-way ANOVA with Tukey’s posthoc test.
To assess potential agonist desensitization, five compounds with a wide range of calculated bias factors were tested for their activity to desensitize GPR52 G protein/cAMP signaling. For this, cells expressing human GPR52 were treated with vehicle (control) or agonists for 2 h with an EC50 concentration previously determined in the cAMP assay. Following compound washout, cells were then challenged with a saturating concentration (1 μM) of 4a, and the cAMP response was measured (Figure 6B). Treatment of cells for 2 h with 4a, 24b, and 15b significantly decreased cAMP responses, whereas the highly G protein-biased agonists 10a and 24f showed no significant difference from the vehicle-treated cells. The percent desensitization of GPR52 displayed a similar trend (Figure 6C), with the highest level of desensitization caused by 4a, 24b, and 15b. Notably, the highly G protein-biased agonists 10a and 24f showed significantly less desensitization than parent compound 4a. The trend in agonist-induced GPR52 desensitization generally follows the trend observed for the G protein bias factor, with less receptor desensitization as the G protein bias factor increases. 10a showed very low receptor desensitization, likely due to both the reduced potency and partial efficacy for β-arrestin recruitment (see Table 4). Together, these findings suggest the functional bias toward or away from β-arrestin correlates with GPR52 desensitization. To test this, a Pearson’s correlation analysis (Figure 6D) was performed comparing the GPR52 cAMP response after agonist pretreatments (see Figure 6B) versus the agonist EC50 for GPR52 β-arrestin recruitment (see Table 4). A significant, positive correlation (p = 0.0358) was determined. Prolonged treatment of compounds with greater potency for the GPR52 β-arrestin pathway correlated with significantly less G protein-activated cAMP signaling. These results demonstrate that the highly G protein-biased GPR52 agonists, such as 10a and 24f, are valuable chemical probes to further investigate the effects of GPR52 signaling bias. These G protein-biased probes could provide sustained agonist activation of GPR52 with reduced desensitization and could reduce the potential for agonist tolerance and tachyphylaxis.
Molecular Docking of Compounds 4a, 24f, and 15b with GPR52.
Molecular docking provides significant information about the potential binding mode and receptor−ligand interactions that could assist in structure-based drug design. Recent cocrystal structural analysis of GPR52 bound to compound 2 (PDB code: 6LI0) revealed binding at a narrow side pocket between extracellular loop2 (ECL2), N-terminus, TM1, TM2, and TM7.21 To ascertain comprehensive docking poses of our compounds with GPR52, Schrödinger Drug Discovery Suite was employed for modeling GPR52 and selected compounds based on the cocrystal structure. For the docking studies, we selected representative compounds 4a (parent compound) and 24f (which showed the largest G protein bias factor) to find distinct binding modes that could potentially explain the observed differences in bias. Due to its increased potency and efficacy, compound 15b was also chosen to further examine the molecular interactions with GPR52. 4a, 15b, and 24f demonstrated energetically favorable interactions and occupied similar binding poses to 2 between TM1, TM2, TM7, N-terminus, and ECL2 (Figure 7 and Supplementary Figure S4).
Figure 7.

Predicted binding mode and molecular docking of 4a and 24f with GPR52. (A) Compound 4a is shown in green docking into the GPR52 binding pocket surrounded by N-terminus, TM1, TM2, TM7, and ECL2 based on the GPR52 crystal structure (PBD: 6LI0). Key interacting residues in the binding site are drawn as gray sticks. Hydrogen bonds are shown as magenta dashed lines, H−F bond is shown as an orange dashed line, and π−π stacking is shown as a cyan dashed line. (B) Interaction diagram of 4a docking into GPR52. Hydrogen bonds are shown as magenta lines, and π−π stacking interaction is shown as a green line. (C) Compound 24f is shown in magenta docking into the binding pocket of GPR52. (D) Interaction diagram of 24f docking into GPR52.
There were three pairs of critical hydrogen bonds between each docked compound and residues within the receptor: the hydroxy group of Ser35 in the N-terminus, the carbonyl group of the amide in Ile189 at ECL2, and the amine group of the amide in Cys40 in TM1. The flexible aminoalcohol side chain may also form a hydrogen bond with the amine group of the amide in Glu191 in ECL2, rather than with Ser35, with both positions predicted to be equally energetically favorable (Supplementary Figure S4). This binding model may explain the significant activity loss when changing the aminoalcohol moiety to piperidine or morpholine, disrupting hydrogen bonding interactions. The pyridine ring (ring B) of each compound formed a π−π stacking interaction with Phe300 of TM7. Ring C of all three compounds pointed into a hydrophobic pocket and formed hydrophobic interactions with residues Ile47, Cys94, Phe117, Ile121, Tyr185, Trp304, and Ile307. This may explain why the ring C could tolerate such varied substitutions and why hydrophobic groups (e.g., OCF3 and CF3) were more favorable for increasing potency. Notably, each of these docked compounds interacted closely with the ECL2 domain through both hydrogen bonds (Ile189 and Glu191) and hydrophobic interactions (Tyr185, His186, Asp188, Ile189, Phe190, and Glu191).
The unusual orientation of ECL2 deep in the canonical class A GPCR orthosteric binding pocket has led to the suggestion that ECL2 acts as a self-agonist for GPR52.21 The extensive interaction between GPR52 agonists and ECL2 indicates that the ligands may act in an allosteric manner to stabilize receptor self-activation by the ECL2. Based on the results from the cAMP signaling and β-arrestin recruitment assays, the molecular region around ring B appears to be highly important for agonist potency (Figures 3B and 4B). However, no significant receptor interactions with this region were observed in the docking models. Altogether, 4a, 15b, and 24f all showed nearly identical binding modes with this docking exploration, but no obvious changes to account for the variations in signaling bias for G protein or β-arrestin were revealed. However, docking into the GPR52 crystal structure is a structurally rigid, static model, and it does not provide a wide range of receptor conformations that likely occur for GPR52, limiting these interpretations. More advanced dynamic computational methods may assist in elucidating the exact mechanisms and conformational changes underlying the unique agonist bias for 24f versus 4a. For example, the increased flexibility of 24f between rings A and B, due to the opening of the indoline ring in 4a, may allow unique interactions that can only be revealed in molecular dynamic simulations. While there were no apparent differences in the docking model, these GPR52 agonists may also have differences in binding kinetics that could affect signaling bias for the G protein or β-arrestin pathways.52,53 In addition to docking with the GPR52-c17 cocrystal structure (PBD: 6LI0), a similar docking of 24f and 4a with the mini-Gs-coupled active state cryo-EM structure of GPR52 (PDB: 8HMP)54 was performed, that yielded a similar docking pose and similar ligand interactions (data not shown).
Counter Screening of Compound 15b.
Considering the in vitro activity for GPR52 activation, compound 15b was chosen as a representative probe out of the active scaffolds for further pharmacological evaluation. To ascertain the selectivity of GPR52 agonist 15b against other GPCRs and off-targets, broad-panel counter-screening was performed by the National Institute of Mental Health Psychoactive Drug Screening Program (NIMH-PDSP).55 The results revealed that 15b, at 10 μM concentration, displayed no significant binding affinity (Ki) at over 30 GPCRs, transporters, and ion channels but showed a moderate interaction with 5-HT2B (Table 5). In addition, 15b did not show inhibition or binding affinity toward the human ether-a-go-go-related gene (hERG) potassium channel, indicating a low risk of cardiovascular toxicity.56,57 These results are comparable to the lack of off-target activity reported for the structurally related compound 4b.40
Table 5.
Broad-Panel Counter Screening of Compound 15b against Other GPCRs, Ion Channels, and Transporters
| GPCRs, transporters, ion channels | % inhibition (10 μM)a | Ki (nM)a | GPCRs, transporters, ion channels | % inhibition (10 μM) | Ki (nM)a |
|---|---|---|---|---|---|
| 5-HT1A | 35.04 | ND | D1 | 7.92 | ND |
| 5-HT1B | 11.55 | ND | D2 | −0.64 | ND |
| 5-HT1D | 29.09 | ND | D3 | 10.45 | ND |
| 5-HT1E | 18.55 | ND | D4 | 20.65 | ND |
| 5-HT2A | 25.21 | ND | D5 | 8.29 | ND |
| 5-HT2B | 91.67 | 425.4 ± 81.4 | DAT | 15.46 | ND |
| 5-HT2C | 41.39 | ND | GABAA | −17.94 | ND |
| 5-HT3 | 22.6 | ND | H1 | 15.7 | ND |
| 5-HT5A | 20.55 | ND | H2 | 25.13 | ND |
| 5-HT6 | 15.34 | ND | KOR | 18.73 | ND |
| 5-HT7A | 20.68 | ND | M1 | −1.71 | ND |
| α 1A | 16.45 | ND | M2 | 3.15 | ND |
| α 1B | −5.51 | ND | M3 | 39.69 | ND |
| α 1D | −10.52 | ND | M4 | 28.71 | ND |
| α 2A | 23 | ND | M5 | −1.37 | ND |
| β 1 | 10.39 | ND | MOR | 20.45 | ND |
| β 2 | −7.69 | ND | hERG | 11.04 | ND |
Values are mean ± SEM of at least three independent experiments.
SEM < 20% where not listed. “ND” means that a Ki binding affinity was not determined because off-target inhibition at 10 μM did not reach 50%.
In Vivo PK Profiles of Compounds 15b and 24f.
Compound 15b (based on enhanced GPR52 potency and efficacy) and 24f (based on bias toward G protein signaling pathway) were chosen for further in vivo PK profiling. Both compounds were evaluated in rats after a single dose of 20 mg/kg by oral (PO) or 10 mg/kg by intravenous (IV) administration, and the results are summarized in Table 6. 15b and 24f displayed acceptable plasma exposure over time after IV administration (AUC0−∞ = 4679 and 4746 ng h/mL, respectively), high maximum serum concentration (Cmax = 6456 and 13733 ng/mL, respectively), and good volume of distribution (Vss = 1.85 and 0.55 L/kg, respectively). 15b exhibited good PK parameters after PO (AUC0−∞ = 3535 ng h/mL, Cmax = 2212 ng/mL, Vss = 9.6 L/kg), whereas 24f showed low plasma exposure (AUC0−∞ = 662 ng h/mL, Cmax = 192 ng/mL) after PO. This difference appears to be largely due to the reduced oral bioavailability of 24f compared to 15b (F = 7.0 and 37.6%, respectively).
Table 6.
In Vivo PK Profiles of Compounds 15b or 24f Following 20 mg/kg Oral and 10 mg/kg Intravenous Dosing in Ratsa
| route | AUC0−∞ (ng h/mL) | t1/2 (h) | Tmax (h) | Cmax (ng/mL) | CL (L/h/kg) | Vss (L/kg) | F (%) |
|---|---|---|---|---|---|---|---|
| 15b PO | 3535 ± 1411 | 1.1 ± 0.02 | 0.5 | 2212 ± 587 | 6.2 ± 2.1 | 9.6 ± 3.2 | 37.6 ± 15 |
| 15b IV | 4679 ± 1280 | 0.6 ± 0.2 | 6456 ± 988 | 2.3 ± 0.7 | 1.8 ± 0.1 | ||
| 24f PO | 662 ± 191 | 3.3 ± 1.5 | 1.0 | 192 ± 109 | nd | nd | 7.0 ± 2.0 |
| 24f IV | 4746 ± 268 | 0.7 ± 0.3 | 13733 ± 1358 | 35.2 ± 2.0 | 0.55 ± 0.08 |
Values are mean ± SD from n = 3 biological replicates.
Cmax, maximum concentration of drug in plasma; Tmax, time to maximum concentration of drug in plasma; AUC, area under the curve (t = 0 to 24 h); Vss, volume of distribution at steady state; CL, plasma clearance; t1/2, terminal half-life; F, absolute oral bioavailability; nd, not determined. Formulation for PO and IV: DMSO:20% HP-β-CD in saline = 1:9.
In addition, 24f showed a high plasma clearance rate (CL = 35.2 L/h/kg). 15b and 24f demonstrated limited brain exposure (B/P ratio = 0.06 and 0.02, respectively) despite a substantial concentration of the ligand in the brain after the blood-brain barrier (BBB) penetration (Table 7). Therefore, PK profiles of 15b and 24f require further optimization to improve oral bioavailability, reduce plasma clearance, and enhance BBB permeability prior to their application as in vivo probes.
Table 7.
Brain Penetration Analysisa
| route | time (h) | brain conc. (ng/g) | plasma conc. (ng/g) | brain/plasma ratio |
|---|---|---|---|---|
| 15b IV | 0.25 | 198 ± 32 | 3668 ± 845 | 0.05 ± 0.01 |
| 1 | 62 ± 20 | 1175 ± 526 | 0.06 ± 0.04 | |
| 24f IV | 0.25 | 124 ± 26 | 6103 ± 265 | 0.02 ± 0.003 |
| 1 | 9.0 ± 0.8 | 376 ± 26 | 0.02 ± 0.001 |
Values are mean ± SD from n = 3 biological replicates.
Concentrations of compounds 15b and 24f in the brain and plasma were determined at 0.25 and 1 h after a single dose of 10 mg/kg by IV. Formulation: DMSO:20% HP-β-CD in saline = 1:9.
CONCLUSIONS
We conducted a structural optimization of compound 4a, employing an iterative drug design strategy that led to the discovery of a series of potent GPR52 agonists. Compounds 15b, 15c, 15d, 24b, 24e, 24f, and 24k were found to be more potent for cAMP signaling than both 4a and our previously optimized compound 4b.40 Our studies into β-arrestin recruitment of this series revealed that the parent compound 4a was unexpectedly highly potent for GPR52 β-arrestin recruitment (EC50,β-arrestin = 22 nM). Opening the indoline ring system to create 10a profoundly reduced GPR52 β-arrestin recruitment potency and efficacy (EC50,β-arrestin = 483 nM, Emax,β-arrestin = 70%). This finding provides a structural basis to generate GPR52 G protein-biased agonist probes and indicates the indoline ring system is important for imparting potent β-arrestin recruitment. To the best of our knowledge, these are the first G protein-biased GPR52 agonists. 10a and 24f, with a greater bias for G protein/cAMP signaling compared to 4a, also induced significantly less in vitro desensitization than 4a. This bias may be relevant for improving in vivo pharmacodynamics by increasing the duration of GPR52 activation and reducing receptor internalization and tachyphylaxis. Molecular docking studies of compounds 4a, 15b, and 24f with GPR52 revealed a conserved binding mode with three pairs of hydrogen bonds, π−π stacking, and hydrophobic interactions, providing insights for future structure-based drug design. Compound 15b was highly selective for GPR52 agonism with no significant off-target effects at other brain GPCRs. 15b and 24f are also bioavailable but have less than optimal brain permeability, requiring further optimization. These balanced and biased GPR52 agonists provide important pharmacological probes to determine the impact of biased agonism on GPR52 function. In addition, these compounds provide an innovative conceptual framework for evaluating reduced β-arrestin-mediated desensitization of GPR52, which may provide sustained agonist efficacy for therapeutic applications.
EXPERIMENTAL SECTION
Chemistry.
All commercially available starting materials and solvents were reagent grade and used without further purification. Reactions were performed under a nitrogen atmosphere in dry glassware with magnetic stirring. Preparative column chromatography was performed using silica gel 60, particle size 0.063−0.200 mm (70−230 mesh, flash). Analytical TLC was carried out employing silica gel 60 F254 plates (Merck, Darmstadt). Visualization of the developed chromatograms was performed with detection by UV (254 nm). NMR spectra were recorded on a Bruker-600 (1H, 300 MHz; 13C, 75 MHz) spectrometer. 1H and 13C NMR spectra were recorded with TMS as an internal reference. Chemical shifts downfield from TMS were expressed in ppm, and J values were given in Hz. High-resolution mass spectra (HRMS) were obtained from Thermo Fisher LTQ Orbitrap Elite mass spectrometer. Parameters include the following: the nano ESI spray voltage was 1.8 kV, the capillary temperature was 275 °C, and the resolution was 60000; ionization was achieved by positive mode. The purity of the final compounds was determined by analytical HPLC, which was carried out on a Shimadzu HPLC system (model: CBM-20A LC-20AD SPD-20A UV/vis). HPLC analysis conditions: Waters μBondapak C18 (300 × 3.9 mm), flow rate 0.5 mL/min, UV detection at 270 and 254 nm, linear gradient from 10% acetonitrile in water (0.1% TFA) to 100% acetonitrile (0.1% TFA) in 20 min, followed by 30 min of the last-named solvent. All biologically evaluated compounds are >95% pure.
2-Chloro-4-(3-(trifluoromethyl)benzyl)pyridine (7).
A mixture of 1-(bromomethyl)-3-(trifluoromethyl)benzene 5 (287 mg, 1.2 mmol), Na2CO3 (424 mg, 4 mmol), Pd(PPh3)4 (115 mg, 0.05 mmol) and (2-chloropyridin-4-yl)boronic acid 6 (157 mg, 1 mmol) in toluene (5 mL) and ethanol (2.5 mL) was heated to 85 °C for 1.5 h under nitrogen atmosphere. After completion of the reaction, it was cooled to room temperature, poured into water, and then extracted with EtOAc (20 mL × 3). The organic phase was washed with brine, dried over anhydrous Na2SO4, and subsequently concentrated under reduced pressure. The residue was purified by silica gel chromatography (Gradient: 10−20% EtOAc in hexane) provided compound 7 as a light-yellow oil (131 mg, 48%). 1H NMR (300 MHz, Chloroform-d) δ 8.32 (d, J = 5.1 Hz, 1H), 7.56 (d, J = 7.8 Hz, 1H), 7.52−7.42 (m, 2H), 7.36 (d, J = 7.7 Hz, 1H), 7.16 (s, 1H), 7.04 (d, J = 5.0 Hz, 1H), 4.04 (s, 2H).
Ethyl 3-((4-(3-(Trifluoromethyl)benzyl)pyridin-2-yl)amino)-benzoate (8).
A mixture of 7 (542 mg, 2 mmol), Pd(OAc)2 (44 mg, 0.2 mmol), XantPhos (231 mg, 0.4 mmol), Cs2CO3 (1.3 g, 4 mmol) and ethyl 3-aminobenzoate (330 mg, 2 mmol) in 1,4-dioxane (10 mL) was subjected to three rounds of vacuum evacuation followed by introduction of nitrogen. The reaction mixture was then stirred at 100 °C overnight. The reaction was cooled to room temperature, poured in water, and then extracted with EtOAc (20 mL × 3). The organic phase was washed with brine, dried over anhydrous Na2SO4, and then concentrated under reduced pressure. The residue was purified by silica gel chromatography (Gradient: 10−20% EtOAc in hexane) to provide product 8 as a colorless oil (610 mg, 76%). 1H NMR (300 MHz, Chloroform-d) δ 8.16 (d, J = 5.1 Hz, 1H), 7.97 (t, J = 1.9 Hz, 1H), 7.75−7.61 (m, 2H), 7.56−7.35 (m, 6H), 6.72 (s, 1H), 6.66−6.58 (m, 2H), 4.39 (q, J = 7.0 Hz, 3H), 3.96 (s, 2H), 1.43−1.38 (m, 3H).
3-((4-(3-(Trifluoromethyl)benzyl)pyridin-2-yl)amino)benzoic Acid (9).
To a mixture of 8 (600 mg, 1.5 mmol) in MeOH (4 mL) was added a 4 N solution of NaOH (2 mL), and then the resultant mixture was refluxed for 2 h. Adjusting the pH of the reaction mixture to pH 1 with 2 N HCl solution resulted in the formation of a white solid precipitate out of the solution. The white solid was filtered and washed with water, the cake was collected and dried to afford product 9 as a white solid (445 mg, 80%). 1H NMR (300 MHz, DMSO-d6) δ 12.77 (s, 1H), 9.20 (s, 1H), 8.26 (t, J = 1.9 Hz, 1H), 8.09 (d, J = 5.2 Hz, 1H), 7.93 (dd, J = 7.8, 2.3 Hz, 1H), 7.70−7.51 (m, 4H), 7.45 (d, J = 7.6 Hz, 1H), 7.34 (t, J = 7.9 Hz, 1H), 6.70 (d, J = 5.2 Hz, 1H), 6.63 (s, 1H), 4.03 (s, 2H).
N-(2-Hydroxyethyl)-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)amino)benzamide (10a).
Compound 9 (74 mg, 0.2 mmol) and 2-aminoethan-1-ol (25 mg, 0.4 mmol) were dissolved in DMF (5 mL) and the resultant mixture was cooled to 0 °C using an ice bath. HOBt (28 mg, 0.2 mmol), EDCI (39 mg, 0.2 mmol), and DMAP (48 mg, 0.4 mmol) were sequentially added to the reaction mixture at 0 °C. Thereafter, the ice bath was removed and the reaction was allowed to stir at room temperature overnight. After completion of the reaction (detected by TLC), the reaction was quenched by water and then extracted with EtOAc (20 mL × 3). The combined organic layers were washed with brine, dried anhydrous Na2SO4, and the solvent was removed using a rotary evaporator to afford a yellow oil. This material was further purified by preparative TLC plates using CH2Cl2/MeOH = 50:1 as the eluent to yield 10a as a colorless oil (71 mg, 87%). 1H NMR (300 MHz, Chloroform-d) δ 8.03 (d, J = 5.2 Hz, 1H), 7.68 (d, J = 2.0 Hz, 1H), 7.60−7.45 (m, 2H), 7.44−7.29 (m, 4H), 7.28−7.13 (m, 3H), 6.60−6.47 (m, 2H), 3.86 (s, 3H), 3.69 (t, J = 5.0 Hz, 2H), 3.50 (t, J = 5.2 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 168.7, 155.9, 150.8, 147.9, 141.0, 139.9, 135.2, 132.4, 130.9 (d, J = 32.1 Hz), 129.2, 129.1, 125.8, 125.6 (q, J = 3.8 Hz), 123.5 (q, J = 3.7 Hz), 122.2, 120.4, 118.3, 116.1, 109.9, 61.6, 42.8, 40.9; HRMS (ESI) calcd for C22H21F3N3O2 416.1580 (M + H)+, found 416.1578.
N-((1R,4R)-4-Hydroxycyclohexyl)-3-((4-(3-(trifluoromethyl)-benzyl)pyridin-2-yl)amino)benzamide (10b).
Compound 10b (41 mg, 65%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 9.09 (s, 1H), 8.05 (dd, J = 9.1, 6.4 Hz, 2H), 7.92 (d, J = 6.2 Hz, 2H), 7.68−7.53 (m, 4H), 7.28 (d, J = 4.6 Hz, 2H), 6.73−6.59 (m, 2H), 4.53 (d, J = 4.4 Hz, 1H), 4.01 (s, 2H), 3.77−3.60 (m, 1H), 3.45−3.35 (m, 1H), 1.92−1.74 (m, 4H), 1.30 (dt, J = 24.8, 12.5 Hz, 4H); 13C NMR (75 MHz, DMSO) δ 166.5, 156.5, 150.8, 147.8, 142.2, 141.6, 136.0, 133.6, 130.1, 128.7, 126.0−125.6 (m), 123.8−123.4 (m), 120.7, 119.2, 117.8, 115.8, 110.8, 68.8, 48.3, 34.7, 30.7; HRMS (ESI) calcd for C26H27F3N3O2 470.2049 (M + H)+, found 470.2048.
(4-Hydroxypiperidin-1-yl)(3-((4-(3-(trifluoromethyl)benzyl)-pyridin-2-yl)amino)phenyl)methanone (10c).
Compound 10c (40 mg, 67%) was synthesized following the synthetic procedure of compound 10a as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.09 (d, J = 5.2 Hz, 1H), 7.53−7.38 (m, 5H), 7.37−7.25 (m, 2H), 7.21 (s, 1H), 6.96 (d, J = 7.5 Hz, 1H), 6.61 (s, 1H), 6.55 (d, J = 5.2 Hz, 1H), 4.17 (s, 1H), 3.91 (s, 3H), 3.69 (s, 1H), 3.24 (d, J = 37.9 Hz, 2H), 2.75 (s, 1H), 1.85 (d, J = 32.6 Hz, 2H), 1.55 (d, J = 22.3 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 170.3, 155.9, 150.6, 148.2, 141.0, 140.0, 136.8, 132.4, 131.1, 130.9 (d, J = 32.2 Hz), 130.7, 129.2, 129.1, 125.6 (d, J = 3.8 Hz), 123.5 (d, J = 3.8 Hz), 120.3, 120.1, 117.7, 116.1, 109.6, 66.9, 45.0, 41.0, 39.5, 34.6, 33.9; HRMS (ESI) calcd for C25H25F3N3O2 456.1893 (M + H)+, found 456.1890.
Morpholino(3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)-amino)phenyl)methanone (10d).
Compound 10d (46 mg, 78%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 8.10 (d, J = 5.2 Hz, 1H), 7.51 (d, J = 7.3 Hz, 2H), 7.47−7.39 (m, 3H), 7.38−7.26 (m, 2H), 7.23 (s, 1H), 6.99 (d, J = 7.5 Hz, 1H), 6.63−6.54 (m, 2H), 3.92 (s, 2H), 3.80−3.41 (m, 8H); 13C NMR (75 MHz, CDCl3) δ 170.3, 155.8, 150.6, 148.2, 141.1, 140.0, 136.0, 132.4, 130.9 (d, J = 31.9 Hz), 129.3, 129.1, 125.6 (q, J = 3.8 Hz), 123.5 (q, J = 3.8 Hz)., 120.5, 120.4, 118.0, 116.2, 109.6, 77.5, 77.1, 76.7, 66.9, 41.0; HRMS (ESI) calcd for C24H23F3N3O2 442.1737 (M + H)+, found 442.1734.
Methyl (3-((4-(3-(Trifluoromethyl)benzyl)pyridin-2-yl)amino)-benzoyl)-D-allothreoninate (10e).
Compound 10e (47 mg, 72%) was synthesized following the synthetic procedure of compound 10a as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.04 (d, J = 5.2 Hz, 1H), 7.73 (s, 1H), 7.59 (d, J = 8.1 Hz, 1H), 7.53−7.29 (m, 7H), 7.22 (t, J = 7.9 Hz, 1H), 6.61 (s, 1H), 6.53 (d, J = 5.2 Hz, 1H), 4.77 (dd, J = 8.7, 2.8 Hz, 1H), 4.41 (dd, J = 6.6, 2.9 Hz, 1H), 3.88 (s, 2H), 3.69 (s, 4H), 1.23 (d, J = 6.4 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 171.8, 168.3, 155.8, 150.9, 147.9, 141.0, 139.9, 134.7, 132.4, 131.1, 129.3, 129.1, 125.7−125.4 (m), 123.5 (q, J = 3.8 Hz), 122.5, 120.6, 118.4, 116.1, 109.6, 68.0, 58.3, 52.5, 41.0, 20.1; HRMS (ESI) calcd for C25H25F3N3O4 488.1791 (M + H)+, found 488.1792.
N-((1S)-1,3-Dihydroxy-1-phenylpropan-2-yl)-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)amino)benzamide (10f).
Compound 10f (38 mg, 55%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 9.12 (s, 1H), 8.09 (d, J = 5.3 Hz, 1H), 7.98 (s, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.67−7.55 (m, 4H), 7.49 (d, J = 8.7 Hz, 1H), 7.37 (d, J = 7.5 Hz, 2H), 7.24 (dt, J = 21.9, 7.2 Hz, 5H), 6.69 (d, J = 5.3 Hz, 1H), 6.63 (s, 1H), 5.55 (d, J = 5.3 Hz, 1H), 4.96 (t, J = 4.5 Hz, 1H), 4.79 (t, J = 5.6 Hz, 1H), 4.13 (s, 1H), 4.02 (s, 2H), 3.60 (dt, J = 12.5, 6.7 Hz, 1H), 3.45−3.36 (m, 1H); 13C NMR (75 MHz, DMSO-d6) δ 166.9, 156.5, 150.8, 147.8, 144.1, 142.3, 141.6, 135.7, 133.6, 130.1, 128.8, 128.2, 127.1, 126.6, 125.8 (d, J = 4.2 Hz), 123.6 (d, J = 4.1 Hz), 120.8, 119.2, 117.3, 115.9, 110.9, 70.6, 61.0, 57.5; HRMS (ESI) calcd for C29H27F3N3O3 522.1999 (M + H)+, found 522.2001.
N-(1,3-Dihydroxypropan-2-yl)-3-((4-(3-(trifluoromethyl)benzyl)-pyridin-2-yl)amino)benzamide (10g).
Compound 10g (36 mg, 60%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 8.03 (d, J = 5.4 Hz, 1H), 7.66 (d, J = 16.9 Hz, 1H), 7.57−7.21 (m, 9H), 6.64−6.44 (m, 2H), 3.81 (d, J = 50.4 Hz, 9H); 13C NMR (75 MHz, CDCl3) δ 168.7, 155.8, 151.1, 147.6, 140.8, 139.8, 135.0, 132.4, 129.2, 129.1, 125.6 (d, J = 3.8 Hz), 123.5 (d, J = 3.7 Hz), 122.3, 120.6, 118.2, 116.0, 109.9, 61.9, 53.1, 40.9; HRMS (ESI) calcd for C23H23F3N3O3 446.1682 (M + H)+, found 446.1683.
N-(2,2-Difluoroethyl)-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)amino)benzamide (10h).
Compound 10h (34 mg, 62%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 8.12 (d, J = 5.1 Hz, 1H), 7.86 (t, J = 1.8 Hz, 1H), 7.60−7.48 (m, 2H), 7.48−7.40 (m, 2H), 7.39−7.29 (m, 3H), 7.00 (s, 1H), 6.78 (t, J = 6.2 Hz, 1H), 6.65−6.53 (m, 2H), 5.96 (tt, J = 56.1, 4.1 Hz, 1H), 3.94 (s, 2H), 3.81 (tdd, J = 14.8, 6.2, 4.1 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 168.1, 166.6, 155.7, 150.8, 148.2, 141.2, 139.9, 134.5, 132.4, 131.2, 130.8, 129.4, 129.2, 125.8, 125.6 (d, J = 3.8 Hz), 123.6 (d, J = 3.8 Hz), 122.6, 120.3, 118.2, 116.9, 116.4, 113.7 (t, J = 241.4 Hz), 110.5, 109.6, 42.23 (t, J = 26.7 Hz), 41.0; HRMS (ESI) calcd for C22H19F5N3O 436.1443 (M + H)+, found 436.1439.
N-(2-Methoxyethyl)-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)amino)benzamide (10i).
Compound 10i (38 mg, 66%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 8.13 (d, J = 5.2 Hz, 1H), 7.82 (t, J = 1.9 Hz, 1H), 7.59 (dt, J = 7.0, 2.2 Hz, 1H), 7.53−7.39 (m, 3H), 7.34 (q, J = 7.4 Hz, 3H), 7.07 (s, 1H), 6.72 (t, J = 5.5 Hz, 1H), 6.65−6.53 (m, 2H), 3.93 (s, 2H), 3.65 (q, J = 5.1 Hz, 2H), 3.56 (t, J = 4.9 Hz, 2H), 3.39 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 167.5, 155.9, 150.7, 148.3, 141.1, 140.0, 135.6, 132.4, 129.3, 129.1, 125.6 (d, J = 3.7 Hz), 123.5 (d, J = 3.9 Hz), 122.3, 120.3, 118.4, 116.2, 109.3, 71.2, 58.8, 41.0, 39.7; HRMS (ESI) calcd for C23H23F3N3O2 430.1737 (M + H)+, found 430.1735.
N-(3-Hydroxypropyl)-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)amino)benzamide (10j).
Compound 10j (37 mg, 64%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 8.04 (d, J = 5.4 Hz, 1H), 7.74 (s, 1H), 7.56 (d, J = 6.7 Hz, 1H), 7.38 (dt, J = 33.9, 12.4 Hz, 7H), 7.25−7.17 (m, 1H), 6.55 (d, J = 13.4 Hz, 2H), 3.87 (s, 2H), 3.63 (d, J = 5.8 Hz, 2H), 3.50 (t, J = 6.1 Hz, 3H), 1.71 (q, J = 5.8 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 168.7, 166.6, 155.8, 150.9, 147.7, 141.0, 139.9, 135.2, 132.4, 131.1, 129.2, 129.1, 125.6 (d, J = 4.1 Hz), 123.5 (d, J = 3.8 Hz), 122.2, 120.4, 118.2, 116.1, 109.9, 59.8, 41.0, 37.4, 31.8; HRMS (ESI) calcd for C23H23F3N3O2 430.1737 (M + H)+, found 430.1736.
N-(4-Hydroxybutyl)-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)amino)benzamide (10k).
Compound 10k (41 mg, 69%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 8.02 (d, J = 5.1 Hz, 1H), 7.75 (s, 1H), 7.40 (ddt, J = 45.2, 31.4, 12.5 Hz, 9H), 6.62−6.42 (m, 2H), 3.86 (d, J = 4.0 Hz, 2H), 3.58 (q, J = 5.4 Hz, 2H), 3.35 (t, J = 5.8 Hz, 2H), 3.21 (s, 1H), 1.58 (dt, J = 12.7, 6.1 Hz, 4H); 13C NMR (75 MHz, CDCl3) δ 168.1, 155.9, 150.8, 147.8, 141.0, 139.9, 135.7, 132.4, 130.87 (d, J = 32.5 Hz), 129.1, 125.6 (d, J = 3.7 Hz), 123.5 (d, J = 3.4 Hz), 122.0, 120.4, 118.1, 116.0, 110.0, 62.0, 40.9, 39.9, 29.8, 26.1; HRMS (ESI) calcd for C24H25F3N3O2 444.1893 (M + H)+, found 444.1889.
N-(2,3-Dihydroxypropyl)-3-((4-(3-(trifluoromethyl)benzyl)-pyridin-2-yl)amino)benzamide (10l).
Compound 10l (40 mg, 67%) was synthesized following the synthetic procedure of compound 10a as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 7.92−7.72 (m, 2H), 7.60−7.49 (m, 2H), 7.29 (ddd, J = 37.2, 22.4, 7.8 Hz, 5H), 7.02 (t, J = 7.4 Hz, 1H), 6.48 (s, 1H), 6.36 (d, J = 4.9 Hz, 1H), 4.43 (s, 2H), 4.10 (s, 1H), 3.75 (d, J = 26.6 Hz, 4H), 3.57−3.19 (m, 3H); 13C NMR (75 MHz, CDCl3) δ 169.2, 168.8, 155.7, 150.9, 147.5, 141.0, 139.9, 134.9, 134.7, 132.3, 131.0, 130.6, 129.0, 125.8, 125.5 (d, J = 3.6 Hz), 123.4 (d, J = 3.7 Hz), 122.2, 122.0, 120.4, 118.4, 115.9, 110.0, 70.9, 63.9, 61.5, 42.8, 40.8; HRMS (ESI) calcd for C23H23F3N3O3 446.1686 (M + H)+, found 446.1683.
Ethyl 3-Amino-2-cyclopropylbenzoate (12d).
A mixture of ethyl 3-amino-2-bromobenzoate 11a (260 mg, 1.0 mmol), K3PO4 (636 mg, 3 mmol), Pd(dppf)Cl2 (141 mg, 0.05 mmol) and cyclopropylboronic acid (103 mg, 1.2 mmol) in 1,4-dioxane (2 mL) and water (0.5 mL) was heated to 85 °C for 12 h under nitrogen atmosphere. After completion of the reaction, it was cooled to room temperature, poured in water, and then extracted with EtOAc (20 mL × 3). The organic phase was washed with brine, dried over anhydrous Na2SO4, and then concentrated under reduced pressure. The residue was purified by silica gel chromatography (Gradient: 10−20% EtOAc in hexane) to provide compound 12d as a light yellow oil (96 mg, 47%). 1H NMR (300 MHz, Chloroform-d) δ 7.08 (dt, J = 9.9, 4.9 Hz, 1H), 6.97 (d, J = 4.7 Hz, 1H), 6.77 (t, J = 5.6 Hz, 1H), 4.47−4.29 (m, 3H), 4.15 (s, 2H), 1.91−1.74 (m, 1H), 1.03−0.93 (m, 2H), 0.47 (t, J = 5.2 Hz, 2H).
Methyl 3-Amino-2-(furan-2-yl)benzoate (12e).
Compound 12e (130 mg, 60%) was synthesized following the synthetic procedure of compound 12d as a yellow oil. 1H NMR (300 MHz, Chloroform-d) δ 7.54 (d, J = 1.8 Hz, 1H), 7.26−7.15 (m, 2H), 6.89 (dd, J = 7.7, 1.6 Hz, 1H), 6.59−6.44 (m, 2H), 4.10 (s, 2H), 3.73 (s, 3H).
Methyl 2-Fluoro-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)-amino)benzoate (13a).
Compound 13a (154 mg, 38%) was synthesized following the synthetic procedure of compound 8 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.42 (t, J = 8.0 Hz, 1H), 8.19 (d, J = 5.2 Hz, 1H), 7.58−7.36 (m, 5H), 7.17 (t, J = 8.0 Hz, 1H), 6.73−6.52 (m, 3H), 3.99 (s, 2H), 3.95 (s, 3H).
Methyl 2-Methyl-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)-amino)benzoate (13b).
Compound 13b (242 mg, 61%) was synthesized following the synthetic procedure of compound 8 as a light-yellow oil. 1H NMR (300 MHz, Chloroform-d) δ 8.10 (d, J = 5.2 Hz, 1H), 7.64 (d, J = 7.8 Hz, 1H), 7.53 (dd, J = 14.0, 7.7 Hz, 2H), 7.44 (d, J = 7.0 Hz, 2H), 7.34 (d, J = 7.7 Hz, 1H), 7.24 (t, J = 7.9 Hz, 1H), 6.55 (d, J = 5.2 Hz, 1H), 6.37 (s, 2H), 3.90 (s, 2H), 2.46 (s, 3H).
Methyl 2-Methoxy-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)-amino)benzoate (13c).
Compound 13c (157 mg, 75%) was synthesized following the synthetic procedure of compound 8 as a light-yellow oil. 1H NMR (300 MHz, Chloroform-d) δ 8.36 (dd, J = 8.2, 1.6 Hz, 1H), 8.22−8.16 (m, 1H), 7.57−7.40 (m, 5H), 7.14 (t, J = 8.0 Hz, 1H), 7.06 (s, 1H), 6.64 (d, J = 4.3 Hz, 2H), 3.99 (s, 2H), 3.95 (s, 3H), 3.90 (s, 3H).
Ethyl 2-Cyclopropyl-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)amino)benzoate (13d).
Compound 13d (154 mg, 70%) was synthesized following the synthetic procedure of compound 8 as a light-yellow oil. 1H NMR (300 MHz, Chloroform-d) δ 8.17 (d, J = 5.2 Hz, 1H), 7.84 (dd, J = 5.5, 4.1 Hz, 1H), 7.53 (d, J = 8.0 Hz, 1H), 7.47 (d, J = 6.9 Hz, 2H), 7.38 (d, J = 7.5 Hz, 1H), 7.27−7.19 (m, 2H), 7.07 (s, 1H), 6.68 (s, 1H), 6.61 (dd, J = 5.2, 1.4 Hz, 1H), 4.39 (s, 2H), 3.97 (s, 2H), 1.97−1.86 (m, 1H), 1.42 (s, 3H), 1.11−1.01 (m, 2H), 0.55−0.45 (m, 2H).
Methyl 2-(Furan-2-yl)-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)amino)benzoate (13e).
Compound 13e (83 mg, 37%) was synthesized following the synthetic procedure of compound 8 as a light-yellow oil. 1H NMR (300 MHz, Chloroform-d) δ 8.21−8.07 (m, 2H), 7.63−7.50 (m, 2H), 7.40 (dt, J = 21.8, 7.4 Hz, 5H), 6.88 (s, 1H), 6.65−6.46 (m, 4H), 3.94 (s, 2H), 3.74 (s, 3H).
Methyl 4-Methyl-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)-amino)benzoate (13f).
Compound 13f (270 mg, 68%) was synthesized following the synthetic procedure of compound 8 as a light-yellow oil. 1H NMR (300 MHz, Chloroform-d) δ 8.23−8.02 (m, 2H), 7.73 (dd, J = 7.8, 1.7 Hz, 1H), 7.55−7.29 (m, 5H), 6.58 (d, J = 5.3 Hz, 1H), 6.49 (s, 1H), 6.28 (s, 1H), 3.93 (s, 2H), 2.19 (s, 2H).
Methyl 4-Methoxy-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)-amino)benzoate (13g).
Compound 13g (313 mg, 75%) was synthesized following the synthetic procedure of compound 8 as a light-yellow oil. 1H NMR (300 MHz, Chloroform-d) δ 8.79 (d, J = 2.1 Hz, 1H), 8.22 (d, J = 5.2 Hz, 1H), 7.71 (dd, J = 8.5, 2.1 Hz, 1H), 7.46 (dq, J = 21.3, 7.8 Hz, 4H), 6.92 (d, J = 8.6 Hz, 2H), 6.71−6.57 (m, 2H), 3.97 (d, J = 5.9 Hz, 5H), 3.91 (s, 3H).
Methyl 2-Fluoro-5-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)-amino)benzoate (13h).
Compound 13h (163 mg, 81%) was synthesized following the synthetic procedure of compound 8 as a light-yellow oil. 1H NMR (300 MHz, Chloroform-d) δ 8.14 (d, J = 5.2 Hz, 1H), 7.87 (dd, J = 6.1, 2.9 Hz, 1H), 7.64 (ddd, J = 8.9, 4.1, 3.0 Hz, 1H), 7.53 (d, J = 7.6 Hz, 1H), 7.48−7.44 (m, 2H), 7.38 (d, J = 7.7 Hz, 1H), 7.11 (dd, J = 10.1, 8.9 Hz, 1H), 6.62 (dd, J = 5.2, 1.4 Hz, 1H), 6.50 (d, J = 1.3 Hz, 1H), 6.45 (s, 1H), 3.95 (d, J = 3.4 Hz, 5H).
2-Fluoro-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)amino)-benzoic Acid (14a).
Compound 14a (140 mg, 90%) was synthesized following the synthetic procedure of compound 9 as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.11 (s, 1H), 8.74 (s, 1H), 8.36 (t, J = 8.0 Hz, 1H), 8.04 (d, J = 5.2 Hz, 1H), 7.69−7.53 (m, 4H), 7.39 (t, J = 6.7 Hz, 1H), 7.19 (q, J = 8.7, 8.0 Hz, 1H), 6.84 (s, 1H), 6.72 (d, J = 5.1 Hz, 1H), 4.01 (s, 2H).
2-Methyl-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)amino)-benzoic Acid (14b).
Compound 14b (230 mg, 91%) was synthesized following the synthetic procedure of compound 9 as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 10.45 (s, 1H), 7.90 (d, J = 6.4 Hz, 1H), 7.81−7.68 (m, 2H), 7.59 (dq, J = 17.1, 7.7 Hz, 4H), 7.38 (t, J = 7.8 Hz, 1H), 7.01−6.76 (m, 2H), 4.16 (s, 2H), 2.37 (s, 3H).
2-Methoxy-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)amino)-benzoic Acid (14c).
Compound 14c (179 mg, 89%) was synthesized following the synthetic procedure of compound 9 as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 13.04 (s, 1H), 10.49 (s, 1H), 7.95 (d, J = 6.4 Hz, 1H), 7.78−7.54 (m, 6H), 7.28 (t, J = 7.8 Hz, 1H), 7.10 (d, J = 1.4 Hz, 1H), 6.94 (dd, J = 6.5, 1.5 Hz, 1H), 4.19 (s, 2H), 3.70 (s, 3H).
2-Cyclopropyl-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)-amino)benzoic Acid (14d).
Compound 14d (194 mg, 94%) was synthesized following the synthetic procedure of compound 9 as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 10.50 (s, 1H), 7.92 (dd, J = 6.5, 4.6 Hz, 1H), 7.72 (s, 1H), 7.63 (q, J = 5.3, 4.4 Hz, 3H), 7.56−7.41 (m, 3H), 7.01 (s, 1H), 6.93 (dt, J = 6.5, 1.7 Hz, 1H), 4.19 (s, 2H), 1.89−1.74 (m, 1H), 0.78 (d, J = 8.2 Hz, 2H), 0.40−0.22 (m, 2H).
2-(Furan-2-yl)-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)-amino)benzoic Acid (14e).
Compound 14e (63 mg, 79%) was synthesized following the synthetic procedure of compound 9 as a light yellow solid. 1H NMR (300 MHz, Chloroform-d) δ 10.34 (s, 1H), 7.81−7.61 (m, 2H), 7.56−7.38 (m, 4H), 7.24 (t, J = 10.9 Hz, 3H), 6.68−6.15 (m, 4H), 3.84 (s, 2H).
4-Methyl-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)amino)-benzoic Acid (14f).
Compound 14f (240 mg, 92%) was synthesized following the synthetic procedure of compound 9 as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 10.49 (s, 1H), 7.97−7.87 (m, 2H), 7.83 (d, J = 8.0 Hz, 1H), 7.72 (s, 1H), 7.61 (q, J = 7.4, 5.3 Hz, 3H), 7.49 (d, J = 8.0 Hz, 1H), 7.05−6.74 (m, 2H), 4.16 (s, 2H), 2.26 (s, 3H).
4-Methoxy-3-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)amino)-benzoic Acid (14g).
Compound 14g (254 mg, 81%) was synthesized following the synthetic procedure of compound 9 as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 10.00 (s, 1H), 8.06 (s, 1H), 7.90 (t, J = 7.7 Hz, 2H), 7.77−7.53 (m, 4H), 7.26 (d, J = 8.7 Hz, 1H), 7.00−6.82 (m, 2H), 4.15 (s, 2H), 3.85 (s, 3H).
2-Fluoro-5-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)amino)-benzoic Acid (14h).
Compound 14h (181 mg, 93%) was synthesized following the synthetic procedure of compound 9 as a light yellow solid. 1H NMR (300 MHz, DMSO-d6) δ 10.27 (s, 1H), 8.03−7.91 (m, 2H), 7.77−7.69 (m, 2H), 7.62 (d, J = 5.7 Hz, 3H), 7.36 (dd, J = 10.5, 8.7 Hz, 1H), 6.95−6.82 (m, 2H), 4.13 (s, 2H).
2-Fluoro-N-(2-hydroxyethyl)-3-((4-(3-(trifluoromethyl)benzyl)-pyridin-2-yl)amino)benzamide (15a).
Compound 15a (33 mg, 38%) was synthesized following the synthetic procedure of compound 10a as a colorless oil (33 mg, 38%). 1H NMR (300 MHz, Chloroform-d) δ 8.09 (q, J = 7.1, 5.8 Hz, 2H), 7.62−7.30 (m, 5H), 7.20 (dt, J = 10.4, 5.6 Hz, 1H), 7.07 (t, J = 8.0 Hz, 1H), 6.97 (s, 1H), 6.69−6.47 (m, 2H), 3.92 (s, 2H), 3.75 (t, J = 5.0 Hz, 2H), 3.57 (q, J = 5.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 164.6, 164.6, 155.4, 152.8, 150.9, 148.0, 139.8, 132.4, 129.4, 129.2, 125.6 (d, J = 3.6 Hz), 124.3 (d, J = 4.2 Hz), 123.9, 123.6 (d, J = 3.9 Hz), 123.4, 121.7 (d, J = 11.1 Hz), 116.7, 110.1, 61.5, 42.7, 40.9; HRMS (ESI) calcd for C22H20F4N3O2 434.1486 (M + H)+, found 434.1481.
N-(2-Hydroxyethyl)-2-methyl-3-((4-(3-(trifluoromethyl)benzyl)-pyridin-2-yl)amino)benzamide (15b).
Compound 15b (33 mg, 77%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 8.03 (d, J = 5.4 Hz, 1H), 7.43 (td, J = 24.9, 22.2, 7.7 Hz, 5H), 7.24−7.09 (m, 2H), 6.86 (s, 1H), 6.62−6.38 (m, 3H), 4.16 (s, 1H), 3.91 (s, 2H), 3.79 (t, J = 5.0 Hz, 2H), 3.57 (q, J = 5.3 Hz, 2H), 2.30 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 171.0, 166.6, 156.8, 151.2, 147.9, 139.9, 139.1, 138.5, 132.3, 129.3, 129.1, 126.5, 124.6, 122.9, 115.6, 108.2, 62.1, 42.6, 41.1, 14.7; HRMS (ESI) calcd for C23H23F3N3O2 430.1737 (M + H)+, found 430.1734.
N-(2-Hydroxyethyl)-2-methoxy-3-((4-(3-(trifluoromethyl)benzyl)-pyridin-2-yl)amino)benzamide (15c).
Compound 15c (36 mg, 60%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 8.16 (d, J = 5.2 Hz, 1H), 8.05 (dt, J = 8.1, 3.1 Hz, 2H), 7.61 (dd, J = 7.9, 1.6 Hz, 1H), 7.54−7.37 (m, 4H), 7.16 (t, J = 8.0 Hz, 1H), 6.96 (s, 1H), 6.63 (dd, J = 6.7, 1.4 Hz, 2H), 3.97 (s, 2H), 3.82 (d, J = 8.3 Hz, 5H), 3.65 (t, J = 5.0 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 166.4, 155.6, 150.9, 148.4, 147.9, 139.9, 134.2, 132.4, 129.2, 126.6, 125.6 (d, J = 3.7 Hz), 124.9, 123.9, 123.6 (d, J = 3.8 Hz), 122.7, 116.5, 109.6, 62.2, 61.7, 42.7, 41.0; HRMS (ESI) calcd for C23H23F3N3O3 446.1686 (M + H)+, found 446.1682.
2-Cyclopropyl-N-(2-hydroxyethyl)-3-((4-(3-(trifluoromethyl)-benzyl)pyridin-2-yl)amino)benzamide (15d).
Compound 15d (42 mg, 69%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 8.13 (d, J = 5.2 Hz, 1H), 7.74 (dd, J = 8.2, 1.3 Hz, 1H), 7.53 (d, J = 7.7 Hz, 1H), 7.48−7.42 (m, 2H), 7.38 (d, J = 7.6 Hz, 1H), 7.19 (t, J = 7.9 Hz, 1H), 7.09−6.99 (m, 2H), 6.66 (d, J = 5.1 Hz, 1H), 6.61 (dd, J = 5.2, 1.4 Hz, 1H), 6.52 (t, J = 5.6 Hz, 1H), 3.97 (d, J = 5.5 Hz, 2H), 3.80 (t, J = 4.9 Hz, 2H), 3.61−3.54 (m, 2H), 1.87−1.73 (m, 1H), 1.03 (dd, J = 8.3, 1.8 Hz, 2H), 0.62−0.47 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 171.2, 155.8, 150.8, 148.4, 141.3, 139.9, 139.5, 132.4, 129.2, 128.6, 127.3, 125.6, 123.5, 121.2, 120.6, 116.3, 109.3, 62.1, 42.9, 41.1, 10.4, 7.2; HRMS (ESI) calcd for C25H25F3N3O2 456.1893 (M + H)+, found 456.1890.
2-(Furan-2-yl)-N-(2-hydroxyethyl)-3-((4-(3-(trifluoromethyl)-benzyl)pyridin-2-yl)amino)benzamide (15e).
Compound 15e (26 mg, 54%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 8.06 (d, J = 5.1 Hz, 1H), 7.86 (d, J = 8.2 Hz, 1H), 7.56−7.39 (m, 4H), 7.31 (dd, J = 19.7, 7.8 Hz, 2H), 7.14 (d, J = 7.6 Hz, 1H), 6.93 (s, 1H), 6.63−6.41 (m, 4H), 6.33 (t, J = 5.8 Hz, 1H), 3.91 (s, 2H), 3.52 (d, J = 5.0 Hz, 2H), 3.31 (d, J = 5.2 Hz, 2H), 3.16 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 170.3, 155.6, 150.8, 148.4, 148.2, 143.2, 139.9, 139.3, 138.4, 131.0 (d, J = 31.9 Hz), 129.2, 125.7−125.5 (m), 123.5 (d, J = 4.0 Hz), 121.6, 121.3, 118.8, 116.6, 111.5, 111.0, 109.7, 61.6, 42.7, 41.0; HRMS (ESI) calcd for C26H23F3N3O3 482.1686 (M + H)+, found 482.1686.
N-(2-Hydroxyethyl)-4-methyl-3-((4-(3-(trifluoromethyl)benzyl)-pyridin-2-yl)amino)benzamide (15f).
Compound 15f (37 mg, 86%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 8.04 (d, J = 5.3 Hz, 1H), 7.88 (d, J = 1.9 Hz, 1H), 7.57−7.30 (m, 5H), 7.20 (d, J = 7.9 Hz, 1H), 6.98 (t, J = 5.6 Hz, 1H), 6.72 (s, 1H), 6.53 (d, J = 5.3 Hz, 1H), 6.41 (s, 1H), 4.31 (s, 1H), 3.89 (s, 2H), 3.75 (t, J = 5.0 Hz, 2H), 3.55 (q, J = 5.2 Hz, 2H), 2.23 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 168.3, 156.6, 151.1, 148.0, 139.9, 138.5, 134.9, 133.1, 132.3, 131.1, 129.1, 125.6 (d, J = 3.9 Hz), 123.5 (d, J = 3.7 Hz), 122.7, 121.4, 115.8, 108.6, 62.0, 42.9, 41.1, 18.0; HRMS (ESI) calcd for C23H23F3N3O2 430.1737 (M + H)+, found 430.1734.
N-(2-Hydroxyethyl)-4-methoxy-3-((4-(3-(trifluoromethyl)benzyl)-pyridin-2-yl)amino)benzamide (15g).
Compound 15g (36 mg, 41%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 8.56 (d, J = 2.2 Hz, 1H), 8.10 (d, J = 5.2 Hz, 1H), 7.56−7.32 (m, 5H), 7.22 (t, J = 5.6 Hz, 1H), 7.02 (s, 1H), 6.81 (d, J = 8.5 Hz, 1H), 6.56 (d, J = 5.3 Hz, 2H), 3.92 (s, 3H), 3.85 (s, 3H), 3.75 (t, J = 4.9 Hz, 2H), 3.54 (d, J = 5.1 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 168.7, 155.5, 150.7, 147.8, 139.9, 132.4, 130.9 (d, J = 32.4 Hz) 129.8, 129.1, 126.9, 125.6 (d, J = 3.8 Hz), 123.5 (d, J = 3.8 Hz), 121.2, 116.5, 116.2, 110.6, 109.6, 62.1, 55.8, 43.0, 41.0; HRMS (ESI) calcd for C23H23F3N3O3 446.1686 (M + H)+, found 446.1683.
2-Fluoro-N-(2-hydroxyethyl)-5-((4-(3-(trifluoromethyl)benzyl)-pyridin-2-yl)amino)benzamide (15h).
Compound 15h (37 mg, 64%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d and MeOD) δ 8.07 (d, J = 5.6 Hz, 1H), 7.88 (dd, J = 6.6, 2.9 Hz, 1H), 7.69 (ddd, J = 8.8, 4.3, 2.9 Hz, 1H), 7.50 (d, J = 7.7 Hz, 1H), 7.46−7.39 (m, 2H), 7.35 (d, J = 7.8 Hz, 1H), 7.31−7.20 (m, 2H), 7.00 (dd, J = 11.3, 8.9 Hz, 1H), 6.56 (dd, J = 3.9, 1.6 Hz, 2H), 3.91 (s, 2H), 3.80 (t, J = 5.1 Hz, 2H), 3.63 (d, J = 5.3 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 164.4, 157.4, 156.0, 154.2, 150.8, 148.1, 139.9, 137.5, 132.4, 131.1, 129.1, 125.6 (d, J = 3.9 Hz), 124.5 (d, J = 8.6 Hz), 123.5 (d, J = 3.8 Hz), 122.6, 121.1 (d, J = 12.5 Hz), 116.7, 116.3, 116.1, 109.2, 61.8, 42.9, 41.0; HRMS (ESI) calcd for C22H20F4N3O2 434.1486 (M + H)+, found 434.1483.
Methyl 4-((4-(3-(Trifluoromethyl)benzyl)pyridin-2-yl)amino)-benzoate (17).
Compound 17 (168 mg, 87%) was synthesized following the synthetic procedure of compound 8 as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 8.26−8.17 (m, 1H), 8.04−7.94 (m, 2H), 7.61−7.33 (m, 6H), 6.80−6.64 (m, 3H), 4.00 (s, 2H), 3.91 (s, 3H).
4-((4-(3-(Trifluoromethyl)benzyl)pyridin-2-yl)amino)benzoic Acid (18).
Compound 18 (169 mg, 91%) was synthesized following the synthetic procedure of compound 9 as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 10.03 (s, 1H), 8.10 (d, J = 5.7 Hz, 1H), 7.96−7.84 (m, 2H), 7.76−7.50 (m, 6H), 6.89 (dd, J = 7.5, 2.0 Hz, 2H), 4.11 (s, 2H).
N-(2-Hydroxyethyl)-4-((4-(3-(trifluoromethyl)benzyl)pyridin-2-yl)amino)benzamide (19).
Compound 19 (43 mg, 77%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d and MeoD) δ 8.03 (dd, J = 5.1, 2.3 Hz, 1H), 7.67 (dd, J = 8.8, 2.5 Hz, 2H), 7.50−7.25 (m, 7H), 6.71−6.50 (m, 2H), 3.90 (d, J = 2.3 Hz, 2H), 3.68 (d, J = 2.4 Hz, 2H), 3.50−3.44 (m, 2H); 13C NMR (75 MHz, CDCl3 and MeoD) δ 168.6, 155.3, 151.1, 147.8, 144.0, 139.8, 132.4, 129.1, 128.3, 126.5, 125.5 (d, J = 3.8 Hz), 123.5 (d, J = 3.5 Hz), 117.7, 116.5, 110.4, 61.3, 42.5, 40.9; HRMS (ESI) calcd for C22H21F3N3O2 416.1580 (M + H)+, found 416.1576.
4-(3,5-Bis(trifluoromethyl)benzyl)-2-chloropyridine (21a).
Compound 21a (540 mg, 80%) was synthesized following the synthetic procedure of compound 7 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.37 (d, J = 5.1 Hz, 1H), 7.83 (s, 1H), 7.64 (s, 2H), 7.17 (s, 1H), 7.08−6.96 (m, 1H), 4.11 (s, 2H).
4-(2,4-Bis(trifluoromethyl)benzyl)-2-chloropyridine (21b).
Compound 21b (260 mg, 77%) was synthesized following the synthetic procedure of compound 7 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.34 (d, J = 5.1 Hz, 1H), 8.00 (s, 1H), 7.80 (d, J = 8.1 Hz, 1H), 7.37 (d, J = 8.1 Hz, 1H), 7.10 (s, 1H), 6.99 (d, J = 5.1 Hz, 1H), 4.24 (s, 2H).
2-Chloro-4-(4-(trifluoromethyl)benzyl)pyridine (21c).
Compound 21c (81 mg, 75%) was synthesized following the synthetic procedure of compound 7 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.34−8.27 (m, 1H), 7.61 (d, J = 8.0 Hz, 2H), 7.31 (d, J = 8.0 Hz, 2H), 7.15 (dd, J = 1.6, 0.7 Hz, 1H), 7.08−7.00 (m, 1H), 4.04 (s, 2H).
2-Chloro-4-(2-(trifluoromethyl)benzyl)pyridine (21d).
Compound 21d (64 mg, 79%) was synthesized following the synthetic procedure of compound 7 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.29 (d, J = 5.1 Hz, 1H), 7.73 (dd, J = 7.9, 1.4 Hz, 1H), 7.54 (td, J = 7.6, 1.4 Hz, 1H), 7.42 (t, J = 7.6 Hz, 1H), 7.22 (d, J = 7.7 Hz, 1H), 7.12−7.05 (m, 1H), 6.99 (dd, J = 5.1, 1.5 Hz, 1H), 4.18 (s, 2H).
2-Chloro-4-(3-chloro-5-fluorobenzyl)pyridine (21e).
Compound 21e (70 mg, 91%) was synthesized following the synthetic procedure of compound 7 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.33 (d, J = 5.1 Hz, 1H), 7.14 (dd, J = 1.6, 0.7 Hz, 1H), 7.08−6.94 (m, 3H), 6.79 (dt, J = 8.9, 1.9 Hz, 1H), 3.93 (s, 2H).
2-Chloro-4-(3-methoxybenzyl)pyridine (21f).
Compound 21f (60 mg, 86%) was synthesized following the synthetic procedure of compound 7 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.28 (d, J = 5.1 Hz, 1H), 7.32−7.25 (m, 1H), 7.20−7.14 (m, 1H), 7.05 (dd, J = 5.1, 1.4 Hz, 1H), 6.90−6.68 (m, 3H), 3.94 (s, 2H), 3.81 (s, 3H).
4-Benzyl-2-chloropyridine (21g).
Compound 21g (53 mg, 87%) was synthesized following the synthetic procedure of compound 7 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.28 (d, J = 5.1 Hz, 1H), 7.40−7.26 (m, 3H), 7.23−7.12 (m, 3H), 7.05 (dd, J = 5.1, 1.4 Hz, 1H), 3.97 (s, 2H).
2-Chloro-4-(4-chloro-2-fluorobenzyl)pyridine (21h).
Compound 21h (59 mg, 77%) was synthesized following the synthetic procedure of compound 7 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.29 (d, J = 5.1 Hz, 1H), 7.18−7.09 (m, 4H), 7.04 (dd, J = 5.1, 1.4 Hz, 1H), 3.95 (d, J = 1.3 Hz, 2H).
2-Chloro-4-(3-fluorobenzyl)pyridine (21i).
Compound 21i (54 mg, 80%) was synthesized following the synthetic procedure of compound 7 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.30 (d, J = 5.1 Hz, 1H), 7.37−7.27 (m, 1H), 7.17−7.12 (m, 1H), 7.08−6.93 (m, 3H), 6.88 (dt, J = 9.6, 2.1 Hz, 1H), 3.96 (s, 2H).
2-Chloro-4-(4-fluorobenzyl)pyridine (21j).
Compound 21j (35 mg, 53%) was synthesized following the synthetic procedure of compound 7 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.29 (d, J = 5.1 Hz, 1H), 7.20−7.10 (m, 3H), 7.09−6.98 (m, 3H), 3.95 (s, 2H).
2-Chloro-4-(3-(trifluoromethoxy)benzyl)pyridine (21k).
Compound 21k (80 mg, 93%) was synthesized following the synthetic procedure of compound 7 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.31 (d, J = 5.1 Hz, 1H), 7.38 (t, J = 7.9 Hz, 1H), 7.19−7.08 (m, 3H), 7.04 (dd, J = 5.1, 1.5 Hz, 2H), 3.99 (s, 2H).
Methyl 3-((4-(3,5-Bis(trifluoromethyl)benzyl)pyridin-2-yl)amino)-2-methylbenzoate (22a).
Compound 22a (110 mg, 79%) was synthesized following the synthetic procedure of compound 8 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.15 (d, J = 5.2 Hz, 1H), 7.77 (s, 1H), 7.69 (s, 1H), 7.62 (s, 2H), 7.55 (d, J = 8.0 Hz, 1H), 7.24 (d, J = 8.1 Hz, 1H), 6.54 (d, J = 5.3 Hz, 1H), 6.34 (s, 2H), 3.97 (s, 2H), 3.93 (s, 3H), 2.47 (s, 3H).
Methyl 3-((4-(2,4-Bis(trifluoromethyl)benzyl)pyridin-2-yl)amino)-2-methylbenzoate (22b).
Compound 22b (91 mg, 65%) was synthesized following the synthetic procedure of compound 8 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.13 (d, J = 5.2 Hz, 1H), 7.66 (dd, J = 7.8, 1.3 Hz, 1H), 7.56 (dd, J = 8.2, 1.3 Hz, 1H), 7.26−7.20 (m, 3H), 7.05 (d, J = 11.4 Hz, 1H), 6.55 (dd, J = 5.2, 1.4 Hz, 1H), 6.34 (d, J = 5.0 Hz, 2H), 3.93 (s, 3H), 3.90 (s, 2H), 2.47 (s, 3H).
Methyl 2-Methyl-3-((4-(4-(trifluoromethyl)benzyl)pyridin-2-yl)-amino)benzoate (22c).
Compound 22c (88 mg, 73%) was synthesized following the synthetic procedure of compound 8 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.11 (d, J = 5.2 Hz, 1H), 7.65 (dd, J = 7.8, 1.3 Hz, 1H), 7.61−7.52 (m, 3H), 7.34−7.20 (m, 4H), 6.55 (dd, J = 5.2, 1.4 Hz, 1H), 6.36 (s, 1H), 6.32 (s, 1H), 3.93 (s, 3H), 3.91 (s, 2H), 2.47 (s, 3H).
Methyl 2-Methyl-3-((4-(2-(trifluoromethyl)benzyl)pyridin-2-yl)-amino)benzoate (22d).
Compound 22d (86 mg, 72%) was synthesized following the synthetic procedure of compound 8 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.10 (d, J = 5.2 Hz, 1H), 7.73−7.55 (m, 3H), 7.47 (d, J = 7.5 Hz, 1H), 7.37 (d, J = 7.6 Hz, 1H), 7.26−7.17 (m, 2H), 6.52 (d, J = 5.2 Hz, 1H), 6.37 (s, 1H), 6.27 (s, 1H), 4.07 (s, 2H), 3.93 (s, 3H), 2.47 (s, 3H).
Methyl 3-((4-(3-Chloro-5-fluorobenzyl)pyridin-2-yl)amino)-2-methylbenzoate (22e).
Compound 22e (85 mg, 74%) was synthesized following the synthetic procedure of compound 8 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.12 (d, J = 5.2 Hz, 1H), 7.66 (dd, J = 7.8, 1.3 Hz, 1H), 7.58 (dd, J = 8.0, 1.3 Hz, 1H), 7.28 (d, J = 4.1 Hz, 1H), 6.98 (dd, J = 7.6, 2.0 Hz, 2H), 6.77 (dt, J = 9.4, 2.0 Hz, 1H), 6.55 (dd, J = 5.2, 1.4 Hz, 1H), 6.40−6.28 (m, 2H), 3.93 (s, 3H), 3.81 (s, 2H), 2.48 (s, 3H).
Methyl 3-((4-(3-Methoxybenzyl)pyridin-2-yl)amino)-2-methylbenzoate (22f).
Compound 22f (48 mg, 67%) was synthesized following the synthetic procedure of compound 8 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.09 (d, J = 5.2 Hz, 1H), 7.61 (td, J = 8.2, 1.3 Hz, 2H), 7.24 (td, J = 7.8, 3.9 Hz, 2H), 6.78 (ddd, J = 7.4, 5.6, 2.0 Hz, 2H), 6.71 (t, J = 2.1 Hz, 1H), 6.59 (dd, J = 5.2, 1.4 Hz, 1H), 6.43 (s, 1H), 6.28 (s, 1H), 3.93 (s, 3H), 3.83 (s, 2H), 3.80 (s, 3H), 2.47 (s, 3H).
Methyl 3-((4-Benzylpyridin-2-yl)amino)-2-methylbenzoate (22g).
Compound 22g (72 mg, 72%) was synthesized following the synthetic procedure of compound 8 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.09 (d, J = 5.2 Hz, 1H), 7.61 (ddd, J = 10.4, 8.1, 1.3 Hz, 2H), 7.37−7.21 (m, 4H), 7.20−7.13 (m, 2H), 6.58 (dd, J = 5.2, 1.3 Hz, 1H), 6.46−6.39 (m, 1H), 6.28 (s, 1H), 3.93 (s, 3H), 3.86 (s, 2H), 2.47 (s, 3H).
Methyl 3-((4-(4-Chloro-2-fluorobenzyl)pyridin-2-yl)amino)-2-methylbenzoate (22h).
Compound 22h (61 mg, 80%) was synthesized following the synthetic procedure of compound 8 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.10 (d, J = 5.2 Hz, 1H), 7.65 (dd, J = 7.7, 1.4 Hz, 1H), 7.58 (dd, J = 8.0, 1.3 Hz, 1H), 7.24 (d, J = 7.9 Hz, 1H), 7.11−7.03 (m, 3H), 6.56 (dd, J = 5.2, 1.4 Hz, 1H), 6.40 (s, 1H), 6.30 (s, 1H), 3.93 (s, 3H), 3.83 (s, 2H), 2.47 (s, 3H).
Methyl 3-((4-(3-Fluorobenzyl)pyridin-2-yl)amino)-2-methylbenzoate (22i).
Compound 22i (54 mg, 77%) was synthesized following the synthetic procedure of compound 8 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.11 (d, J = 5.2 Hz, 1H), 7.64 (dd, J = 7.9, 1.3 Hz, 1H), 7.61−7.56 (m, 1H), 7.28−7.21 (m, 2H), 6.95 (dd, J = 8.0, 2.0 Hz, 2H), 6.87 (d, J = 9.6 Hz, 1H), 6.57 (dd, J = 5.2, 1.4 Hz, 1H), 6.40 (s, 1H), 6.29 (s, 1H), 3.93 (s, 3H), 3.84 (s, 2H), 2.47 (s, 3H).
Methyl 3-((4-(4-Fluorobenzyl)pyridin-2-yl)amino)-2-methylbenzoate (22j).
Compound 22j (58 mg, 83%) was synthesized following the synthetic procedure of compound 8 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.09 (d, J = 5.2 Hz, 1H), 7.64 (dd, J = 7.8, 1.3 Hz, 1H), 7.57 (dd, J = 8.0, 1.3 Hz, 1H), 7.24 (d, J = 7.8 Hz, 1H), 7.12 (dd, J = 8.5, 5.5 Hz, 2H), 7.00 (t, J = 8.7 Hz, 2H), 6.55 (dd, J = 5.2, 1.4 Hz, 1H), 6.36 (d, J = 8.6 Hz, 2H), 3.93 (s, 3H), 3.82 (s, 2H), 2.46 (s, 3H).
Methyl 2-Methyl-3-((4-(3-(trifluoromethoxy)benzyl)pyridin-2-yl)-amino)benzoate (22k).
Compound 22k (68 mg, 82%) was synthesized following the synthetic procedure of compound 8 as a colorless oil. 1H NMR (300 MHz, Chloroform-d) δ 8.09 (d, J = 5.2 Hz, 1H), 7.64 (dd, J = 7.8, 1.3 Hz, 1H), 7.56 (dd, J = 7.9, 1.4 Hz, 1H), 7.32 (dd, J = 7.8, 3.6 Hz, 1H), 7.23 (d, J = 7.7 Hz, 1H), 7.11− 7.07 (m, 2H), 7.02 (s, 1H), 6.55 (dd, J = 5.3, 1.4 Hz, 2H), 6.37 (s, 1H), 3.92 (s, 3H), 3.86 (s, 2H), 2.46 (s, 3H).
3-((4-(3,5-Bis(trifluoromethyl)benzyl)pyridin-2-yl)amino)-2-methylbenzoic Acid (23a).
Compound 23a (76 mg, 84%) was synthesized following the synthetic procedure of compound 9 as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 10.55 (s, 1H), 8.01 (d, J = 11.6 Hz, 2H), 7.92−7.88 (m, 1H), 7.82−7.78 (m, 1H), 7.57−7.52 (m, 2H), 7.42 (d, J = 7.7 Hz, 1H), 6.98−6.93 (m, 2H), 4.29 (s, 2H), 2.36 (s, 3H).
3-((4-(2,4-Bis(trifluoromethyl)benzyl)pyridin-2-yl)amino)-2-methylbenzoic Acid (23b).
Compound 23b (78 mg, 86%) was synthesized following the synthetic procedure of compound 9 as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 10.69 (d, J = 4.5 Hz, 1H), 7.91 (dd, J = 6.5, 4.5 Hz, 1H), 7.80 (ddd, J = 7.8, 4.1, 1.4 Hz, 1H), 7.66−7.54 (m, 4H), 7.47−7.40 (m, 1H), 7.00−6.88 (m, 2H), 4.19 (s, 2H), 2.38 (s, 3H).
2-Methyl-3-((4-(4-(trifluoromethyl)benzyl)pyridin-2-yl)amino)-benzoic Acid (23c).
Compound 23c (70 mg, 91%) was synthesized following the synthetic procedure of compound 9 as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.14 (s, 1H), 10.63 (s, 1H), 7.89 (dd, J = 6.5, 4.3 Hz, 1H), 7.83−7.69 (m, 3H), 7.56 (ddd, J = 13.2, 8.0, 1.4 Hz, 3H), 7.42 (q, J = 7.5 Hz, 1H), 7.00−6.83 (m, 2H), 4.17 (s, 2H), 2.37 (s, 3H).
2-Methyl-3-((4-(2-(trifluoromethyl)benzyl)pyridin-2-yl)amino)-benzoic Acid (23d).
Compound 23d (59 mg, 76%) was synthesized following the synthetic procedure of compound 9 as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.04 (s, 1H), 10.27 (s, 1H), 7.87 (d, J = 6.4 Hz, 1H), 7.83−7.66 (m, 3H), 7.61−7.47 (m, 3H), 7.39 (t, J = 7.8 Hz, 1H), 6.79 (dd, J = 6.5, 1.6 Hz, 1H), 6.68 (s, 1H), 4.26 (s, 2H), 2.34 (s, 3H).
3-((4-(3-Chloro-5-fluorobenzyl)pyridin-2-yl)amino)-2-methylbenzoic Acid (23e).
Compound 23e (58 mg, 78%) was synthesized following the synthetic procedure of compound 9 as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.11 (s, 1H), 10.51 (s, 1H), 7.88 (dd, J = 6.5, 4.5 Hz, 1H), 7.80 (dd, J = 7.8, 1.4 Hz, 1H), 7.56 (dd, J = 7.9, 1.4 Hz, 1H), 7.47−7.29 (m, 3H), 7.24 (dt, J = 9.6, 1.8 Hz, 1H), 6.98−6.85 (m, 2H), 4.08 (s, 2H), 2.38 (s, 3H).
3-((4-(3-Methoxybenzyl)pyridin-2-yl)amino)-2-methylbenzoic Acid (23f).
Compound 23f (58 mg, 83%) was synthesized following the synthetic procedure of compound 9 as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 10.58 (s, 1H), 7.83 (td, J = 7.6, 7.1, 2.1 Hz, 2H), 7.54 (dd, J = 7.9, 1.5 Hz, 1H), 7.42 (t, J = 7.8 Hz, 1H), 7.27 (t, J = 7.8 Hz, 1H), 6.95−6.78 (m, 4H), 6.68 (dd, J = 4.5, 1.9 Hz, 1H), 4.02 (s, 2H), 3.75 (s, 3H), 2.37 (s, 3H).
3-((4-Benzylpyridin-2-yl)amino)-2-methylbenzoic Acid (23g).
Compound 23g (59 mg, 93%) was synthesized following the synthetic procedure of compound 9 as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.02 (s, 1H), 10.65 (s, 1H), 7.87 (dd, J = 6.5, 4.0 Hz, 1H), 7.79 (ddd, J = 6.1, 4.8, 1.4 Hz, 1H), 7.61−7.52 (m, 1H), 7.44−7.25 (m, 6H), 6.95 (d, J = 1.6 Hz, 1H), 6.87 (dt, J = 6.5, 1.7 Hz, 1H), 4.05 (s, 2H), 2.37 (s, 3H).
3-((4-(4-Chloro-2-fluorobenzyl)pyridin-2-yl)amino)-2-methylbenzoic Acid (23h).
Compound 23h (54 mg, 73%) was synthesized following the synthetic procedure of compound 9 as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.14 (s, 1H), 10.29 (s, 1H), 7.82 (dd, J = 18.5, 7.1 Hz, 2H), 7.57−7.32 (m, 5H), 6.91−6.77 (m, 2H), 4.09 (s, 2H), 2.36 (s, 3H).
3-((4-(3-Fluorobenzyl)pyridin-2-yl)amino)-2-methylbenzoic Acid (23i).
Compound 23i (59 mg, 88%) was synthesized following the synthetic procedure of compound 9 as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.06 (s, 1H), 10.52 (s, 1H), 7.92−7.76 (m, 2H), 7.55 (dd, J = 7.9, 1.4 Hz, 1H), 7.47−7.35 (m, 2H), 7.24−7.06 (m, 3H), 6.97−6.83 (m, 2H), 4.08 (s, 2H), 2.37 (s, 3H).
3-((4-(4-Fluorobenzyl)pyridin-2-yl)amino)-2-methylbenzoic Acid (23j).
Compound 23j (53 mg, 79%) was synthesized following the synthetic procedure of compound 9 as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.00 (s, 1H), 10.44 (s, 1H), 7.92−7.73 (m, 2H), 7.54 (dd, J = 7.9, 1.4 Hz, 1H), 7.46−7.32 (m, 3H), 7.24−7.13 (m, 2H), 6.87 (d, J = 7.3 Hz, 2H), 4.05 (s, 2H), 2.36 (s, 3H).
2-Methyl-3-((4-(3-(trifluoromethoxy)benzyl)pyridin-2-yl)amino)-benzoic Acid (23k).
Compound 23k (68 mg, 85%) was synthesized following the synthetic procedure of compound 9 as a white solid. 1H NMR (300 MHz, DMSO-d6) δ 13.04 (s, 1H), 10.42 (s, 1H), 7.90−7.77 (m, 2H), 7.58−7.47 (m, 2H), 7.43−7.34 (m, 3H), 7.28 (d, J = 8.2 Hz, 1H), 6.90 (d, J = 6.5 Hz, 2H), 4.13 (s, 2H), 2.37 (s, 3H).
3-((4-(3,5-Bis(trifluoromethyl)benzyl)pyridin-2-yl)amino)-N-(2-hydroxyethyl)-2-methylbenzamide (24a).
Compound 24a (68 mg, 68%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d and MeOD) δ 7.98 (d, J = 5.3 Hz, 1H), 7.75 (s, 1H), 7.61 (s, 2H), 7.34 (t, J = 4.7 Hz, 1H), 7.15 (d, J = 4.6 Hz, 2H), 6.49 (dd, J = 5.3, 1.4 Hz, 1H), 6.44 (s, 1H), 3.95 (s, 2H), 3.71 (t, J = 5.1 Hz, 2H), 3.50 (t, J = 5.3 Hz, 2H), 2.23 (s, 3H); 13C NMR (75 MHz, CDCl3 and MeOD) δ 171.4, 171.3, 166.6, 157.1, 149.9, 148.3, 141.5, 138.8, 138.5, 132.1, 131.6, 130.0, 129.3, 129.0, 126.4, 125.2, 125.0, 123.3, 121.4, 120.7, 115.2, 108.4, 61.3, 42.4, 40.8, 14.7; HRMS (ESI) calcd for C24H22F6N3O2 498.1611 (M + H)+, found 498.1609.
3-((4-(2,4-Bis(trifluoromethyl)benzyl)pyridin-2-yl)amino)-N-(2-hydroxyethyl)-2-methylbenzamide (24b).
Compound 24b (50 mg, 75%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 7.99 (d, J = 5.2 Hz, 1H), 7.37 (dd, J = 6.7, 2.6 Hz, 1H), 7.26−7.16 (m, 2H), 7.14−7.00 (m, 3H), 6.90 (t, J = 5.6 Hz, 1H), 6.71 (s, 1H), 6.50 (dd, J = 5.3, 1.4 Hz, 1H), 6.44 (s, 1H), 3.87 (s, 2H), 3.69 (t, J = 5.0 Hz, 2H), 3.48 (d, J = 5.2 Hz, 2H), 3.23 (s, 1H), 2.21 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 171.1, 164.2, 160.9, 157.0, 150.1, 148.3, 142.8 (d, J = 7.4 Hz), 139.0, 138.5, 129.4, 126.4, 124.7, 123.0, 121.5−121.4 (m), 119.4 (d, J = 20.9 Hz), 115.5, 111.10 (d, J = 24.7 Hz), 108.4, 61.5, 42.5, 40.8, 14.7; HRMS (ESI) calcd for C24H22F6N3O2 498.1611 (M + H)+, found 498.1609.
N-(2-Hydroxyethyl)-2-methyl-3-((4-(4-(trifluoromethyl)benzyl)-pyridin-2-yl)amino)benzamide (24c).
Compound 24c (54 mg, 93%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d and MeOD) δ 7.92 (d, J = 5.3 Hz, 1H), 7.53 (d, J = 8.0 Hz, 2H), 7.34−7.23 (m, 3H), 7.17−7.07 (m, 2H), 6.49 (dd, J = 5.3, 1.4 Hz, 1H), 6.43 (s, 1H), 3.87 (s, 2H), 3.69 (t, J = 5.2 Hz, 2H), 3.48 (t, J = 5.1 Hz, 2H), 2.21 (s, 3H); 13C NMR (75 MHz, CDCl3 and MeOD) δ 171.4, 156.9, 151.2, 147.8, 143.1, 138.9, 138.4, 129.8, 129.3, 129.1, 128.7, 126.3, 125.9, 125.5 (q, J = 3.8 Hz), 125.1, 123.2, 115.5, 108.6, 61.2, 42.5, 42.3, 41.0, 14.7; HRMS (ESI) calcd for C23H23F3N3O2 430.1737 (M + H)+, found 430.1735.
N-(2-Hydroxyethyl)-2-methyl-3-((4-(2-(trifluoromethyl)benzyl)-pyridin-2-yl)amino)benzamide (24d).
Compound 24d (67 mg, 78%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 7.93 (d, J = 5.3 Hz, 1H), 7.66 (dd, J = 7.9, 1.4 Hz, 1H), 7.50−7.42 (m, 1H), 7.34 (dd, J = 8.5, 6.2 Hz, 2H), 7.19 (d, J = 7.7 Hz, 1H), 7.11−6.96 (m, 3H), 6.72 (s, 1H), 6.50−6.37 (m, 2H), 4.03 (s, 2H), 3.64 (t, J = 5.1 Hz, 3H), 3.44 (t, J = 5.2 Hz, 2H), 2.17 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 171.2, 156.7, 151.1, 147.7, 139.1, 138.4, 137.1, 132.0, 131.8, 129.0, 128.6, 126.8, 126.3, 126.2, 126.1 (q, J = 5.8, 5.3 Hz), 124.3, 122.7, 115.7, 108.9, 61.4, 42.5, 37.5, 37.4, 14.7; HRMS (ESI) calcd for C23H23F3N3O2 430.1737 (M + H)+, found 430.1735.
3-((4-(3-Chloro-5-fluorobenzyl)pyridin-2-yl)amino)-N-(2-hydroxyethyl)-2-methylbenzamide (24e).
Compound 24e (55 mg, 66%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 7.94 (d, J = 5.3 Hz, 1H), 7.34 (dd, J = 7.0, 2.3 Hz, 1H), 7.14−7.02 (m, 3H), 6.94 (dp, J = 4.4, 2.0 Hz, 2H), 6.87−6.70 (m, 2H), 6.52−6.39 (m, 2H), 3.76 (s, 2H), 3.64 (t, J = 5.1 Hz, 3H), 3.43 (q, J = 5.0 Hz, 2H), 2.18 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 171.2, 166.6, 164.3, 161.0, 156.9, 150.4, 148.1, 142.7 (d, J = 8.2 Hz), 139.1, 138.4, 135.0 (d, J = 10.8 Hz), 129.4, 126.4, 125.0 (d, J = 3.1 Hz), 124.7, 122.9, 115.5, 114.6 (d, J = 6.4 Hz), 114.3 (d, J = 3.2 Hz), 108.6, 61.3, 42.5, 40.7, 14.8; HRMS (ESI) calcd for C22H22ClFN3O2 414.1379 (M + H)+, found 414.1377.
N-(2-Hydroxyethyl)-3-((4-(3-methoxybenzyl)pyridin-2-yl)amino)-2-methylbenzamide (24f).
Compound 24f (20 mg, 38%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 8.00 (s, 1H), 7.41 (t, J = 5.4 Hz, 1H), 7.31−6.92 (m, 4H), 6.80−6.41 (m, 6H), 3.97−3.62 (m, 7H), 3.52 (t, J = 5.2 Hz, 2H), 3.05 (s, 1H), 2.24 (d, J = 3.9 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 171.1, 159.8, 156.5, 152.1, 147.8, 140.5, 139.2, 138.4, 129.6, 128.9, 126.4, 124.2, 122.6, 121.4, 116.0, 114.9, 111.7, 108.6, 61.9, 55.2, 42.6, 41.4, 14.7; HRMS (ESI) calcd for C23H26N3O3 392.1969 (M + H)+, found 392.1964.
3-((4-Benzylpyridin-2-yl)amino)-N-(2-hydroxyethyl)-2-methylbenzamide (24g).
Compound 24g (37 mg, 77%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 7.94 (d, J = 5.3 Hz, 1H), 7.35 (dd, J = 7.1, 2.2 Hz, 1H), 7.33−7.27 (m, 2H), 7.26−7.20 (m, 1H), 7.19−7.13 (m, 2H), 7.12−7.04 (m, 2H), 6.96 (t, J = 5.6 Hz, 1H), 6.54 (dd, J = 5.3, 1.3 Hz, 2H), 6.47 (s, 1H), 3.83 (s, 2H), 3.66 (t, J = 5.0 Hz, 2H), 3.45 (d, J = 5.2 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 171.2, 156.6, 152.2, 147.7, 139.2, 139.0, 138.4, 129.0, 129.0, 128.6, 126.6, 126.3, 124.4, 122.7, 115.9, 108.8, 61.5, 42.5, 41.4, 14.8; HRMS (ESI) calcd for C22H24N3O2 362.1863 (M + H)+, found 362.1862.
3-((4-(4-Chloro-2-fluorobenzyl)pyridin-2-yl)amino)-N-(2-hydroxyethyl)-2-methylbenzamide (24h).
Compound 24h (36 mg, 65%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 7.96 (d, J = 5.3 Hz, 1H), 7.37 (dd, J = 7.2, 2.1 Hz, 1H), 7.18−7.03 (m, 5H), 6.87 (t, J = 5.7 Hz, 1H), 6.72−6.50 (m, 2H), 6.46 (s, 1H), 3.81 (s, 2H), 3.69 (t, J = 5.0 Hz, 2H), 3.48 (q, J = 5.2 Hz, 2H), 3.20 (s, 1H), 2.20 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 171.1, 159.0, 156.7, 150.3, 148.0, 138.8 (d, J = 53.6 Hz), 133.4 (d, J = 10.0 Hz), 131.7 (d, J = 5.3 Hz), 129.1, 126.4, 124.9, 124.7 (d, J = 3.5 Hz), 124.4, 122.7, 116.3 (d, J = 25.6 Hz), 115.4, 108.4, 61.6, 42.6, 34.0, 34.0, 14.7; HRMS (ESI) calcd for C22H22ClFN3O2 414.1379 (M + H)+, found 414.1377.
3-((4-(3-Fluorobenzyl)pyridin-2-yl)amino)-N-(2-hydroxyethyl)-2-methylbenzamide (24i).
Compound 24i (33 mg, 65%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 7.93 (s, 1H), 7.33 (dd, J = 6.7, 2.7 Hz, 1H), 7.24 (td, J = 8.2, 6.2 Hz, 1H), 7.11− 7.03 (m, 3H), 6.96−6.89 (m, 2H), 6.88−6.74 (m, 2H), 6.55−6.38 (m, 2H), 3.80 (s, 2H), 3.64 (t, J = 5.1 Hz, 3H), 3.43 (q, J = 5.2 Hz, 2H), 2.18 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 171.2, 164.5, 161.3, 156.8, 151.4, 147.8, 141.4 (d, J = 7.2 Hz), 139.1, 138.4, 130.1 (d, J = 8.4 Hz), 129.3, 126.3, 124.7, 124.6, 122.8, 115.8 (d, J = 21.3 Hz), 113.5 (d, J = 21.0 Hz), 108.7, 61.3, 42.5, 41.0, 14.8; HRMS (ESI) calcd for C22H23FN3O2 380.1769 (M + H)+, found 380.1765.
3-((4-(4-Fluorobenzyl)pyridin-2-yl)amino)-N-(2-hydroxyethyl)-2-methylbenzamide (24j).
Compound 24j (27 mg, 71%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 7.97 (d, J = 5.2 Hz, 1H), 7.38 (dd, J = 7.2, 2.2 Hz, 1H), 7.16−7.04 (m, 4H), 7.03− 6.92 (m, 2H), 6.83 (t, J = 5.6 Hz, 1H), 6.64−6.47 (m, 2H), 6.44 (s, 1H), 3.81 (s, 2H), 3.70 (t, J = 5.0 Hz, 2H), 3.49 (q, J = 5.3 Hz, 2H), 3.00 (s, 1H), 2.21 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 171.2, 166.6, 163.2, 160.0, 156.7, 152.0, 147.9, 139.2, 138.4, 134.7 (d, J = 3.2 Hz), 130.4 (d, J = 8.0 Hz), 129.1, 126.4, 124.4, 122.7, 115.7, 115.6, 115.3, 108.5, 61.6, 42.6, 40.5, 14.7; HRMS (ESI) calcd for C22H23FN3O2 380.1769 (M + H)+, found 380.1765.
N-(2-Hydroxyethyl)-2-methyl-3-((4-(3-(trifluoromethoxy)benzyl)-pyridin-2-yl)amino)benzamide (24k).
Compound 24k (47 mg, 78%) was synthesized following the synthetic procedure of compound 10a as a white solid. 1H NMR (300 MHz, Chloroform-d) δ 8.01 (d, J = 4.5 Hz, 1H), 7.44−7.25 (m, 2H), 7.20−6.97 (m, 5H), 6.78 (d, J = 5.1 Hz, 1H), 6.65−6.37 (m, 3H), 3.85 (d, J = 3.6 Hz, 2H), 3.71 (d, J = 5.1 Hz, 2H), 3.50 (t, J = 5.1 Hz, 2H), 3.26 (s, 1H), 2.23 (t, J = 2.8 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 171.1, 166.6, 156.8, 151.1, 149.4, 148.1, 141.3, 139.1, 138.4, 129.9, 129.1, 127.4, 126.4, 124.4, 122.8, 121.4, 119.0, 115.7, 108.5, 61.7, 42.6, 41.0, 14.7; HRMS (ESI) calcd for C23H23F3N3O3 446.1686 (M + H)+, found 446.1682.
Molecular Docking.
The molecular docking study was performed using the Schrödinger Small-Molecule Drug Discovery Suite (Schrödinger, LLC, New York, NY, 2020). The cocrystal structure of GPR52 bound with compound 2 (PDB code: 6LI0) was downloaded from the RCS PDB bank. The cocrystal structure was preprocessed and minimized with Schrödinger Protein Preparation Wizard using default settings. The grid center was chosen on the centroid of the existing ligand and the size of the grid box was set to 30 Å on each side. The 3D structure of ligands 4a, 15b, and 24f were created using Schrödinger Maestro and a low energy conformation was calculated using LigPrep. Docking was employed with Glide using the SP precision. Docked poses were incorporated into Schrödinger Maestro for visualization and analysis of binding site interactions.
Cell Culture and Plasmids.
Wildtype (WT) human embryonic kidney 293 (HEK293) cells were a generous gift from Dr. Asuka Inoue, Tohoku University.58 The wild-type HEK293 cells were cultured in DMEM (Gibco, Carlsbad, CA) with 10% FBS (Omega Scientific, Tarzana, CA) and 1% penicillin/streptomycin (Gibco, Carlsbad, CA) in a humidified incubator at 37 °C in 5% CO2. HTLA cells (HEK293 cells stably expressing tTA-dependent luciferase reporter and a β-arrestin2-TEV fusion gene) were a generous gift from Dr. Bryan Roth, University of North Carolina at Chapel Hill.46 The HTLA cells were cultured in DMEM with 10% FBS, 1% penicillin/streptomycin, 100 μg/mL hygromycin (Thermofisher, Waltham, MA), and 2 μg/mL puromycin (Gibco/Fisher Scientific, Hampton, NH) in a humidified incubator at 37 °C in 5% CO2. The human GPR52 Tango plasmid was purchased from Addgene (Watertown, MA). A human GPR52 WT construct was generated from the GPR52 Tango plasmid via placement of a stop codon following the receptor coding sequence, as previously described.40
Transfection.
For the cAMP GloSensor assays, 6.5 × 105 wildtype HEK293 cells were plated in 6-well plates (Corning, Oneonta, NY) 24 h before transfection to achieve 70−80% confluency the following day. Each well of wildtype HEK293 cells was transiently transfected with 0.10 μg of wild-type human GPR52 WT plasmid and 1 μg of 22F GloSensor plasmid (Promega, Madison, WI) using 10 μL of Lipofectamine 2000 (Invitrogen, Waltham, MA) in 0.4 mL of Opti-MEM (Gibco, Carlsbad, CA) and 2 mL of growth media. For the Tango β-arrestin recruitment assays, 6.5 × 105 HTLA cells were plated in 6-well plates 24 h before transfection to achieve 70−80% confluency the following day. Each well was transiently transfected with 0.25 μg of GPR52 Tango plasmid using 10 μL of Lipofectamine 2000 in 0.4 mL of Opti-MEM and 2 mL of growth media.
cAMP GloSensor Assay.
Eighteen h after transfection, wildtype HEK293 cells with GPR52 and GloSensor plasmids (see transfections above) were seeded at 50,000 cells/well in growth media into poly-L-lysine (Trevigen, Gaithersburg, MD) coated 96-well white clear-bottomed cell culture plates (Greiner Bio-One, Monroe, NC). Four h later, complete media was aspirated and replaced with 90 μL/well of 1% GloSensor reagent (Promega, Madison, WI) in Hanks balanced salt solution (HBSS, Gibco/Fisher Scientific, Hampton, NH) and 20 mM HEPES. Cells were serum starved for 2 h at room temperature in the dark. Test compounds were weighed and diluted to 2 mM in DMSO. Serial dilutions of each test compound were prepared in 1% GloSensor reagent/20 mM HEPES in HBSS and were transferred to a 96-well source plate. Cells were incubated with compound solutions for 15 min. Luminescence in light counts per second (lcps) was recorded on a MicroBeta2 Microplate counter (PerkinElmer, Waltham, MA). Data from at least three independent experiments (n = 3−24) conducted in technical triplicate are presented as a percentage of compound 4a response (compound 4a response set to 100%). All in vitro pharmacological data were analyzed using GraphPad Prism 8.4.3 software (La Jolla, CA). Data from cAMP ligand concentration−response assays are presented as the half-maximum (EC50, nM) and maximum effect (Emax) (means ± SEM) as computed by GraphPad using a four-parameter nonlinear regression curve-fitting algorithm. For compounds that did not show saturating concentration−responses in the GloSensor assay, the curve-fitting algorithm in GraphPad Prism was constrained by defining the maximum efficacy as the luminescence observed at the highest tested concentration.
Tango β-Arrestin Recruitment Assay.
Eighteen h after transfection, HTLA cells expressing GPR52 Tango plasmid (see transfections above) were seeded at 80,000 cells/well in 100 μL/well growth media into poly-L-lysine coated 96-well white clear-bottomed cell culture plates. Test compounds were weighed and diluted to 2 mM in DMSO. Serial dilutions of each compound were prepared in growth media in a 96-well source plate. At 24 h post-transfection, cells were treated with compound solutions and returned to the incubator. After 20 h incubation, growth media was aspirated and the cells were lysed with 50 μL/well of 40-fold diluted Bright-Glo luciferase substrate (Promega, Madison, WI) in HBSS for 20 min at room temperature. Luminescence in light counts per second (lcps) was recorded on a MicroBeta2 Microplate counter. Data from at least three independent experiments (n = 3−6) conducted in technical triplicate are presented as a percentage of compound 4a response (compound 4a response set to 100%). All data were analyzed using GraphPad Prism 8.4.3 software. Data from β-arrestin recruitment concentration−response assays are presented as the half-maximum (EC50, nM) and maximum effect (Emax) (means ± SEM) as computed by GraphPad using a four-parameter nonlinear regression curve-fitting algorithm. Notably, some compounds when tested for β-arrestin recruitment did not show saturating concentration−responses; in these cases, the curve-fitting algorithm in GraphPad Prism was constrained by defining the maximum efficacy as the luminescence observed at the highest tested concentration.
GPR52 Desensitization Studies.
Eighteen h after transfection of wildtype HEK293 cells with GPR52 and GloSensor plasmids (see transfections above), cells were seeded at 50,000 cells/well into poly-L-lysine coated 96-well white clear-bottomed cell culture plates in growth media (Greiner Bio-One, Monroe, NC). Four h later, complete media was aspirated and replaced with 90 μL/well serum-free DMEM media. Compounds were diluted in serum-free media to respective EC50 concentrations determined from the cAMP concentration−response assays (see above). Cells were incubated at room temperature in the dark with the EC50 concentration (determined in cAMP assays) of compounds for 2 h to induce GPR52 desensitization. During the final 60 min of compound pretreatment, cells were also incubated in 1% GloSensor reagent/20 mM HEPES in HBSS to load cells with substrate for the cAMP assay. Following the 2 h pretreatments, the buffer was aspirated, and wells were washed for 5 min intervals three times with 20 mM HEPES in HBSS. After washing, cells were challenged with 1 μM compound 4a for 15 min to determine the remaining GPR52 G protein/cAMP signaling activity. Luminescence in light counts per second (lcps) was recorded on a MicroBeta2 Microplate counter. Data from three independent experiments (n = 3) conducted in technical quad-ruplicate are presented as raw counts (lcps) or percent desensitization (normalized to 100% DMSO control response). All data were analyzed using GraphPad Prism 8.4.3 software. Statistically significant differences were determined by one-way ANOVA with Tukey’s posthoc test. A Pearson’s correlation analysis was used to compare the GPR52 cAMP response after agonist pretreatments versus the agonist EC50 for GPR52 β-arrestin recruitment. The resulting correlation p-value and a coefficient of determination (R2) were determined.
Bias Factor Calculations.
Mean efficacy (Emax) and potency (EC50) were taken from at least three independent experiments for all compounds tested in both the cAMP and β-arrestin recruitment assays. Formulas for bias factor calculations were adopted from Kenakin.50 The bias factor calculation method requires an agonist to have an Emax > 35% compared to the reference agonist, which all tested compounds displayed. Emax and EC50 values for each compound were entered into the equation log(Emax/EC50) for both cAMP and β-arrestin recruitment assays, then subtracted from the respective log(Emax/EC50) for the reference compound 4a to obtain a Δlog(Emax/EC50) for each compound. A ΔΔlog(Emax/EC50) value for each compound was calculated by subtraction of Δlog(Emax/EC50)β-arrestin from Δlog(Emax/EC50)cAMP. The inverse log of this ΔΔlog(Emax/EC50) value is the G protein bias factor. Conversely, the β-arrestin bias factor is calculated from the inverse log of ΔΔlog(Emax/EC50) when Δlog(Emax/EC50)cAMP is subtracted from Δlog(Emax/EC50)β-arrestin. The calculated bias factors represent the relative propensity to activate one pathway over another in reference to compound 4a.
In Vivo PK Studies.
Male Sprague−Dawley rats (n = 3 per treatment group; Beijing Vital River Laboratory, Animal Technology Co., Ltd., Beijing, China) weighing 200−250 g at the beginning of the experiment were housed three per cage in a pathogen-free, temperature-controlled (20−26 °C), and humidity-controlled (40−70%) environment with a 12 h light−dark cycle and ad libitum access to food and filtered water. Rats were randomly assigned to treatment groups. Vehicle (10% dimethyl sulfoxide (DMSO) and 90% 2-hydroxypropyl-β-cyclodextrin (HP-β-CD); Cyclodextrin Technologies Development, Inc., High Springs, FL, USA) or compound 15b/24f dissolved in the vehicle was administered to rats IV at 10 mg/kg or PO at 20 mg/kg. The rat was restrained manually at the designated time points (0.08, 0.25, 0.5, 1.0, 2.0, 4.0, 8.0, and 24 h postdosing for IV; 0.25, 0.5, 1.0, 2.0, 4.0, 8.0, and 24 h postdosing for PO). 300 μL of blood (yielding ~50 μL of plasma) samples are taken from the animal’s jugular vein and placed into micro K2EDTA tubes. Blood samples were placed on ice and centrifuged at 6000 rpm for 5 min at 4 °C to generate plasma samples within 0.5 h of collection. Brain samples were collected at 0.25 and 1 h postdosing. All samples were stored at −80 °C. The concentration of 15b/24f in each sample was analyzed by Sundia MediTech Co., Ltd. The study and the related standard operating procedures were reviewed and approved by the Sundia Institutional Animal Care and Use Committee. The Sundia animal facility is approved with yearly inspection by the Shanghai Laboratory Animal Management Committee. All treatment assignments were blinded to investigators who performed PK assays and end-point statistical analyses.
The PK parameters of compounds 15b and 24f were calculated according to a noncompartmental model using WinNonlin (Pharsight Corporation, ver 8.1, Mountain View, CA, USA). The peak concentration (Cmax) was directly obtained by visual inspection of the plasma concentration−time profile. The elimination rate constant (λ) was obtained by the least-squares fitted terminal log−linear portion of the slope of the plasma concentration−time profile. The elimination half-life (t1/2) was evaluated according to 0.693/λ. The area under the plasma concentration−time curve from 0 to time t (AUC0−t) was evaluated using the linear trapezoidal rule and further extrapolated to infinity (AUC0−∞) according to the following equation: AUC0−∞ = AUC0−t + Clast/λ. The pharmacokinetic parameters are presented as mean ± SEM.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants U18 DA052543 (J.A.A. and J.Z.) and T32 DA007287 (H.R.S. and D.E.F.), a 2022 Predoctoral Fellowship from the PhRMA Foundation (R.E.M.), and the Center for Addiction Sciences and Therapeutics at the University of Texas Medical Branch (UTMB). J.Z. is also partially supported by the John D. Stobo, M. D., Distinguished Chair Endowment, and the Edith and Robert Zinn Chair Endowment in Drug Discovery. The authors would also like to acknowledge Dr. Asuka Inoue, Tohoku University, for the generous gift of the HEK cell line, Dr. Bryan Roth, UNC-Chapel Hill, for the generous gift of the HTLA cell line, Dr. Lawrence C. Sowers at the UTMB Department of Pharmacology and Toxicology as well as Dr. Tianzhi Wang at the NMR core facility of UTMB for the NMR spectroscopy assistance, and Dr. Xuemei Luo at UTMB Mass Spectrometry Core with funding support from University of Texas System Proteomics Network for the HRMS analysis.
ABBREVIATIONS
- cAMP
cyclic adenosine monophosphate
- CNS
central nervous system
- DMAP
4-dimethylaminopyridine
- DMF
N-dimethylformamide
- EC50
half maximal effective concentration
- ECL2
extracellular loop 2
- EDCI
N-(3-(dimethylamino)propyl)-N′-ethylcarbo-diimide hydrochloride
- GPCRs
G-protein-coupled receptors
- HD
Huntington’s disease
- HEK293
human embryonic kidney 293
- hERG
human ether-a-go-go-related gene
- HOBt
1-hydroxybenzotriazole
- IV
intravenous
- mHTT
mutant huntingtin
- MSNs
medium spiny neurons
- NAc
nucleus accumbens
- NIMH
national institute of mental health
- Pd(dppf)2Cl2
1,1′-bis(diphenylphosphino)ferrocene-palladium(II)dichloride dichloromethane complex
- PDSP
psychoactive drug screening program
- PK
pharmacokinetic
- PO
oral
- SAR
structure−activity relationship
- SUD
substance-use disorder
- SZ
schizophrenia
- TM
transmembrane helix
- WT
Wildtype
- XantPhos
4,5-bis(diphenylphosphino)-9,9-dimethylxanthene
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.4c00856.
Supplementary Figures S1−S4, HPLC analysis spectra of representative compounds, 1H and 13C NMR spectra of all new compounds (PDF)
Molecular formula strings and data (CSV)
Docking with GPR52 (PDB)
Docking of 4a with GPR52 (PDB)
Docking of 15b with GPR52 (PDB)
Docking of 24f with GPR52 (PDB)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.4c00856
The authors declare the following competing financial interest(s): J.A.A. and J.Z., as well as their teams, were partially supported by MapLight Therapeutics, Inc. through an industry-funded collaborative research project, which was generally related to this manuscript.
Contributor Information
Ryan E. Murphy, Center for Addiction Sciences and Therapeutics, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas 77555, United States;.
Pingyuan Wang, Center for Addiction Sciences and Therapeutics, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas 77555, United States; Present Address: Ocean University of China, Qingdao, Shangdong 266003, China;.
Saghir Ali, Center for Addiction Sciences and Therapeutics, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas 77555, United States.
Hudson R. Smith, Center for Addiction Sciences and Therapeutics, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas 77555, United States
Daniel E. Felsing, Center for Addiction Sciences and Therapeutics, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas 77555, United States; Present Address: Neurocrine Biosciences, San Diego, California 92130, United States.
Haiying Chen, Center for Addiction Sciences and Therapeutics, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas 77555, United States.
Jia Zhou, Center for Addiction Sciences and Therapeutics, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas 77555, United States;.
John A. Allen, Center for Addiction Sciences and Therapeutics, Department of Pharmacology and Toxicology, University of Texas Medical Branch, Galveston, Texas 77555, United States;.
REFERENCES
- (1).Ali S; Wang P; Murphy RE; Allen JA; Zhou J Orphan GPR52 as an emerging neurotherapeutic target. Drug Discovery Today 2024, 29, No. 103922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Wang P; Lv L; Li H; Wang CY; Zhou J Opportunities and challenges in drug discovery targeting the orphan receptor GPR12. Drug Discovery Today 2023, 28, No. 103698. [DOI] [PubMed] [Google Scholar]
- (3).Bolinger AA; Frazier A; La JH; Allen JA; Zhou J Orphan G Protein-Coupled Receptor GPR37 as an Emerging Therapeutic Target. ACS Chem. Neurosci 2023, 14, 3318–3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ye N; Li B; Mao Q; Wold EA; Tian S; Allen JA; Zhou J Orphan Receptor GPR88 as an Emerging Neurotherapeutic Target. ACS Chem. Neurosci 2019, 10, 190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Alavi MS; Shamsizadeh A; Azhdari-Zarmehri H; Roohbakhsh A Orphan G protein-coupled receptors: The role in CNS disorders. Biomed. Pharmacother 2018, 98, 222–232. [DOI] [PubMed] [Google Scholar]
- (6).Sutton LP; Orlandi C; Song C; Oh WC; Muntean BS; Xie K; Filippini A; Xie X; Satterfield R; Yaeger JDW; et al. Orphan receptor GPR158 controls stress-induced depression. eLife 2018, 7, No. e33273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jeong E; Kim Y; Jeong J; Cho Y Structure of the class C orphan GPCR GPR158 in complex with RGS7-Gβ5. Nat. Commun 2021, 12, 6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lees JA; Dias JM; Rajamohan F; Fortin JP; O’Connor R; Kong JX; Hughes EAG; Fisher EL; Tuttle JB; Lovett G; et al. An inverse agonist of orphan receptor GPR61 acts by a G protein-competitive allosteric mechanism. Nat. Commun 2023, 14, 5938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Margolin DH; Brice NL; Davidson AM; Matthews KL; Carlton MBL A Phase I, First-in-Human, Healthy Volunteer Study to Investigate the Safety, Tolerability, and Pharmacokinetics of CVN424, a Novel G Protein-Coupled Receptor 6 Inverse Agonist for Parkinson’s Disease. J. Pharmacol. Exp. Ther 2022, 381, 33–41. [DOI] [PubMed] [Google Scholar]
- (10).Sawzdargo M; Nguyen T; Lee DK; Lynch KR; Cheng R; Heng HH; George SR; O’Dowd BF Identification and cloning of three novel human G protein-coupled receptor genes GPR52, ΨGPR53 and GPR55: GPR55 is extensively expressed in human brain. Mol. Brain Res 1999, 64, 193–198. [DOI] [PubMed] [Google Scholar]
- (11).Komatsu H; Maruyama M; Yao S; Shinohara T; Sakuma K; Imaichi S; Chikatsu T; Kuniyeda K; Siu FK; Peng LS; et al. Anatomical transcriptome of G protein-coupled receptors leads to the identification of a novel therapeutic candidate GPR52 for psychiatric disorders. PLoS One 2014, 9, No. e90134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Yang RY; Quan J; Sodaei R; Aguet F; Segrè AV; Allen JA; Lanz TA; Reinhart V; Crawford M; Hasson S; et al. A systematic survey of human tissue-specific gene expression and splicing reveals new opportunities for therapeutic target identification and evaluation. bioRxiv 2018, No. 311563. [Google Scholar]
- (13).Spark DL; Mao M; Ma S; Sarwar M; Nowell CJ; Shackleford DM; Sexton PM; Nithianantharajah J; Stewart GD; Langmead CJ In the Loop: Extrastriatal Regulation of Spiny Projection Neurons by GPR52 . ACS Chem. Neurosci 2020, 11, 2066–2076. [DOI] [PubMed] [Google Scholar]
- (14).Murphy RE; Felsing DE; Allen JA Bi-directional cAMP signaling crosstalk between dopamine D2 receptors and the constitutively active orphan receptor GPR52. FASEB J. 2021, 35 (S1), No. 04255. [Google Scholar]
- (15).Pardinas AF; Holmans P; Pocklington AJ; Escott-Price V; Ripke S; Carrera N; Legge SE; Bishop S; Cameron D; Hamshere ML; et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet 2018, 50, 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Felsing DE; Jain MK; Allen JA Advances in Dopamine D1 Receptor Ligands for Neurotherapeutics. Curr. Top. Med. Chem 2019, 19, 1365–1380. [DOI] [PubMed] [Google Scholar]
- (17).Surmeier DJ; Ding J; Day M; Wang Z; Shen W D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007, 30, 228–235. [DOI] [PubMed] [Google Scholar]
- (18).Yao Y; Cui X; Al-Ramahi I; Sun X; Li B; Hou J; Difiglia M; Palacino J; Wu ZY; Ma L; et al. A striatal-enriched intronic GPCR modulates huntingtin levels and toxicity. eLife 2015, 4, No. e05449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Li G; Marlin MC Rab family of GTPases. Methods Mol. Biol 2015, 1298, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Song H; Li H; Guo S; Pan Y; Fu Y; Zhou Z; Li Z; Wen X; Sun X; He B; et al. Targeting Gpr52 lowers mutant HTT levels and rescues Huntington’s disease-associated phenotypes. Brain 2018, 141, 1782–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lin X; Li M; Wang N; Wu Y; Luo Z; Guo S; Han GW; Li S; Yue Y; Wei X; et al. Structural basis of ligand recognition and self-activation of orphan GPR52. Nature 2020, 579, 152–157. [DOI] [PubMed] [Google Scholar]
- (22).Krumm B; Roth BL A self-activating orphan receptor. Nature 2020, 579, 35–36. [DOI] [PubMed] [Google Scholar]
- (23).Setoh M; Ishii N; Kono M; Miyanohana Y; Shiraishi E; Harasawa T; Ota H; Odani T; Kanzaki N; Aoyama K; et al. Discovery of the first potent and orally available agonist of the orphan G-protein-coupled receptor 52. J. Med. Chem 2014, 57, 5226–5237. [DOI] [PubMed] [Google Scholar]
- (24).Nakahata T; Tokumaru K; Ito Y; Ishii N; Setoh M; Shimizu Y; Harasawa T; Aoyama K; Hamada T; Kori M; et al. Design and synthesis of 1-(1-benzothiophen-7-yl)-1H-pyrazole, a novel series of G protein-coupled receptor 52 (GPR52) agonists. Bioorg. Med. Chem 2018, 26, 1598–1608. [DOI] [PubMed] [Google Scholar]
- (25).Tokumaru K; Ito Y; Nomura I; Nakahata T; Shimizu Y; Kurimoto E; Aoyama K; Aso K Design, synthesis, and pharmacological evaluation of 4-azolyl-benzamide derivatives as novel GPR52 agonists. Bioorg. Med. Chem 2017, 25, 3098–3115. [DOI] [PubMed] [Google Scholar]
- (26).Nishiyama K; Suzuki H; Harasawa T; Suzuki N; Kurimoto E; Kawai T; Maruyama M; Komatsu H; Sakuma K; Shimizu Y; et al. FTBMT, a novel and selective GPR52 agonist, demonstrates antipsychotic-like and procognitive effects in rodents, revealing a potential therapeutic agent for schizophrenia. J. Pharmacol. Exp. Ther 2017, 363, 253–264. [DOI] [PubMed] [Google Scholar]
- (27).Xiong Y, Grottick AJ; Korman H; Lehmann J; Ren AS; Semple G 1-Heteroaryl-indoline-4-carboxamines as modulators of GPR52 usedul for the treatment or prevention of disorders related thereto. WO2016176571 A1, 2016.
- (28).Poulter S; Austin N; Armstrong R; Barnes M; Bucknell SJ; Higueruelo A; Banerjee J; Mead A; Mould R; MacSweeney C; et al. The Identification of GPR52 Agonist HTL0041178, a Potential Therapy for Schizophrenia and Related Psychiatric Disorders. ACS Med. Chem. Lett 2023, 14, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sosei Heptares Doses First Subject in Phase I Trial with HTL0048149, a First-in-Class GPR52 Agonist for Schizophrenia, 2023. https://soseiheptares.com/news/851/129/Sosei-Heptares-Doses-First-Subject-in-Phase-I-Trial-with-HTL0048149-a-First-in-Class-GPR52-Agonist-for-Schizophrenia.html.
- (30).Watson SP; Swain NA; O’Brien MA GPR52 modulator compounds. WO 2023/041909 A1, 2023.
- (31).Gerlach K; Berta DG; Ferrara M; Gutsche K; Mueller-Vieira U; Hobson S; Runge F; Semple G; Vintonyak V; Xiong Y 3-Phenoxyazetidin-1-yl-heteroaryl pyrrolidine derivatives and the use thereof as medicament. WO 2023/041432 A1, 2023.
- (32).DeWire SM; Ahn S; Lefkowitz RJ; Shenoy SK β-Arrestins and Cell Signaling. Annu. Rev. Physiol 2007, 69, 483–510. [DOI] [PubMed] [Google Scholar]
- (33).Peterson YK; Luttrell LM The Diverse Roles of Arrestin Scaffolds in G Protein−Coupled Receptor Signaling. Pharmacol. Rev 2017, 69, 256–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Wess J; Oteng A-B; Rivera-Gonzalez O; Gurevich EV; Gurevich VV β−Arrestins: Structure, Function, Physiology, and Pharmacological Perspectives. Pharmacol. Rev 2023, 75, 854–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Urban JD; Clarke WP; von Zastrow M; Nichols DE; Kobilka B; Weinstein H; Javitch JA; Roth BL; Christopoulos A; Sexton PM; et al. Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther 2007, 320, 1–13. [DOI] [PubMed] [Google Scholar]
- (36).Schmid CL; Kennedy NM; Ross NC; Lovell KM; Yue Z; Morgenweck J; Cameron MD; Bannister TD; Bohn LM Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Gray DL; Allen JA; Mente S; O’Connor RE; DeMarco GJ; Efremov I; Tierney P; Volfson D; Davoren J; Guilmette E; Salafia M; et al. Impaired β-arrestin recruitment and reduced desensitization by non-catechol agonists of the D1 dopamine receptor. Nat. Commun 2018, 9, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Rajagopal S; Rajagopal K; Lefkowitz RJ Teaching old receptors new tricks: biasing seven-transmembrane receptors. Nat. Rev. Drug Discovery 2010, 9, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Rankovic Z; Brust TF; Bohn LM Biased agonism: An emerging paradigm in GPCR drug discovery. Bioorg. Med. Chem. Lett 2016, 26, 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wang P; Felsing DE; Chen H; Stutz SJ; Murphy RE; Cunningham KA; Allen JA; Zhou J Discovery of Potent and Brain-Penetrant GPR52 Agonist that Suppresses Psychostimulant Behavior. J. Med. Chem 2020, 63, 13951–13972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhou J; Allen JA; Wang P; Felsing DE Heterocyclic G-protein-coupled receptor 52 (GPR52) agonists. US 11,773,065 B2, 2023.
- (42).Ali S; Zhou J Highlights on U.S. FDA-approved fluorinated drugs over the past five years (2018−2022). Eur. J. Med. Chem 2023, 256, No. 115476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Ali S; Bolinger AA; Zhou J Highlights on Fluorine-Containing Drugs Approved by U.S. FDA in 2023. Curr. Top. Med. Chem 2024, 24, 843–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Ferguson SS; Downey WE 3rd; Colapietro AM; Barak LS; Menard L; Caron MG Role of β-Arrestin in Mediating Agonist-Promoted G Protein-Coupled Receptor Internalization. Science 1996, 271, 363–366. [DOI] [PubMed] [Google Scholar]
- (45).Kroeze WK; Sassano MF; Huang XP; Lansu K; McCorvy JD; Giguere PM; Sciaky N; Roth BL PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol 2015, 22, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Barnea G; Strapps W; Herrada G; Berman Y; Ong J; Kloss B; Axel R; Lee KJ The genetic design of signaling cascades to record receptor activation. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Kim K-M; Caron MG Complementary roles of the DRY motif and C-terminus tail of GPCRS for G protein coupling and β-arrestin interaction. Biochem. Biophys. Res. Commun 2008, 366, 42–47. [DOI] [PubMed] [Google Scholar]
- (48).Tohgo A; Choy EW; Gesty-Palmer D; Pierce KL; Laporte S; Oakley RH; Caron MG; Lefkowitz RJ; Luttrell LM The Stability of the G Protein-coupled Receptor-β-Arrestin Interaction Determines the Mechanism and Functional Consequence of ERK Activation. J. Biol. Chem 2003, 278, 6258–6267. [DOI] [PubMed] [Google Scholar]
- (49).Zhang JH; Chung TD; Oldenburg KR A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen 1999, 4, 67–73. [DOI] [PubMed] [Google Scholar]
- (50).Kenakin T A Scale of Agonism and Allosteric Modulation for Assessment of Selectivity, Bias, and Receptor Mutation. Mol. Pharmacol 2017, 92, 414–424. [DOI] [PubMed] [Google Scholar]
- (51).Lohse MJ; Benovic JL; Codina J; Caron MG; Lefkowitz RJ β-Arrestin: a Protein that Regulates β-adrenergic Receptor Function. Science 1990, 248, 1547–1550. [DOI] [PubMed] [Google Scholar]
- (52).Klein Herenbrink C; Sykes DA; Donthamsetti P; Canals M; Coudrat T; Shonberg J; Scammells PJ; Capuano B; Sexton PM; Charlton SJ; et al. The role of kinetic context in apparent biased agonism at GPCRs. Nat. Commun 2016, 7, 10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Wacker D; Wang S; McCorvy JD; Betz RM; Venkatakrishnan AJ; Levit A; Lansu K; Schools ZL; Che T; Nichols DE; et al. Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell 2017, 168, 377–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Fan Y; Lin X; Pan B; Chen B; Liu D; Wuthrich K; Xu F Allosteric coupling between G-protein binding and extracellular ligand binding sites in GPR52 revealed by (19)F-NMR and cryo-electron microscopy. MedComm 2023, 4, No. e260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang XP; Norval S; Sassano MF; Shin AI; Webster LA; et al. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Kalyaanamoorthy S; Barakat KH Development of Safe Drugs: The hERG Challenge. Med. Res. Rev 2018, 38, 525–555. [DOI] [PubMed] [Google Scholar]
- (57).Garrido A; Lepailleur A; Mignani SM; Dallemagne P; Rochais C hERG toxicity assessment: Useful guidelines for drug design. Eur. J. Med. Chem 2020, 195, No. 112290. [DOI] [PubMed] [Google Scholar]
- (58).Grundmann M; Merten N; Malfacini D; Inoue A; Preis P; Simon K; Rüttiger N; Ziegler N; Benkel T; Schmitt NK; et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun 2018, 9, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.