

Significance
SF3B4, a U2 spliceosome component, is required for identifying pre-mRNA branchpoint sequences. Pathogenic SF3B4 variants cause acrofacial dysostosis disorder with variable expressivity. Patients have distinct clinical presentations in the head, limb, and heart. In light of its ubiquitous expression, cause for the specificity of clinical presentation remains unknown. We generated a mouse model mimicking abnormalities and expressivity found in SF3B4 -related disorders. RNAseq revealed mis-splicing of transcription factors and histone modifiers important for craniofacial and heart development in Sf3b4 mutant samples. This was associated with increased proximal branch point usage and increased thymidine bases in the surrounding intronic sequence. Our findings suggest that the context of branchpoint sequences may predispose specific genes to splicing alterations, explaining tissue-specific defects in patients.
Keywords: Sf3b4, splicing, craniofacial, mouse model, RNAseq
Abstract
Pathogenic variants in SF3B4, a component of the U2 snRNP complex important for branchpoint sequence recognition and splicing, are responsible for the acrofacial disorders Nager and Rodriguez Syndrome, also known as SF3B4-related syndromes. Patients exhibit malformations in the head, face, limbs, vertebrae as well as the heart. To uncover the etiology of craniofacial malformations found in SF3B4-related syndromes, mutant mouse lines with homozygous deletion of Sf3b4 in neural crest cells (NCC) were generated. Like in human patients, these embryos had craniofacial and cardiac malformations with variable expressivity and penetrance. The severity and survival of Sf3b4 NCC mutants was modified by the level of Sf3b4 in neighboring non-NCC. RNA sequencing analysis of heads of embryos prior to morphological abnormalities revealed significant changes in expression of genes forming the NCC regulatory network, as well as an increase in exon skipping. Additionally, several key histone modifiers involved in craniofacial and cardiac development showed increased exon skipping. Increased exon skipping was also associated with use of a more proximal branch point, as well as an enrichment in thymidine bases in the 50 bp around the branch points. We propose that decrease in Sf3b4 causes changes in the expression and splicing of transcripts required for proper craniofacial and cardiac development, leading to abnormalities.
In humans, heterozygous pathogenic variants in SF3B4 are responsible for the most common type of acrofacial dysostosis, Nager syndrome (1, 2) (OMIM #154400) and Rodriguez syndrome (3) (OMIM#201170), here on referred to as SF3B4-related syndromes (4). Acrofacial Dysostosis and SF3B4-related syndromes are characterized by micrognathia and ear defects as well as limb defects. Craniofacial and preaxial limb defects common in SF3B4-related syndrome patients include malar and mandibular hypoplasia, cleft palate, downward slanted palpebral fissures, hearing loss, small or absent thumbs, and minor foot anomalies such as small metatarsals (1, 3, 5, 6). Additionally, some patients exhibit rib abnormalities and abnormal vertebral segmentation (1, 3), as well as ventricular septal defect, coarctation of the aorta, hypertrabeculation of the left ventricle and complex structural heart disease (1, 5, 6). To date, 41 distinct pathogenic variants have been identified in SF3B4 (1–14), and include whole gene deletion, nonsense, missense, frameshifts, and truncating changes. Since no genotype–phenotype correlation has been found (15), all mutations are predicted to be nonfunctional.
SF3B4 encodes for splicing associated protein (SAP49)/SF3B4 and is a core component of the SF3B complex found in both the U2 as well as the U12 small nuclear ribonucleoproteins (snRNP) (16). SF3B4 has two RNA recognition motifs (RRM) at its amino terminal that facilitates binding of U2 immediately upstream of the 3′ branch point adenosine during spliceosome assembly (17). The major or U2-dependent spliceosome catalyzes 99% of the RNA splicing reactions in humans (18), whereas the U12-dependent spliceosome (minor spliceosome) is responsible for splicing of 700 minor introns in 666 genes (19). The spliceosome is also crucial for alternative splicing, an important contributor to protein and genetic diversity (16, 20), that is indispensable for development and maintenance of complex organisms.
Our aim is to uncover the etiology of malformations seen in SF3B4-related syndromes. Previous work by our group and the group of Yamada et al. (21, 22), showed that mice with heterozygous mutation of Sf3b4 did not model craniofacial malformations seen in patients, but had growth retardation, microcephaly, as well as homeotic posteriorization of the axial skeleton. Additionally, though RNAseq analysis showed few changes in gene expression, a significant number of genes, including histone modifiers that regulate Hox expression, were abnormally spliced in heterozygous mouse embryos and somites (21).
Since structures affected in the face and head of patients are all derived from cranial neural crest cells (CNCC), herein, we use a mouse model with conditional mutation in Sf3b4 (21) and the Wnt1-Cre2 line to delete Sf3b4 in neural crest cells (NCC). Sf3b4 NCC mutant embryos exhibit craniofacial, heart, and vertebral abnormalities similar to those described in patients with SF3B4-related syndromes. These malformations were associated with decreased expression of NCC specifier genes, as well as aberrant splicing of histone modifiers important for craniofacial development. We identified sequence specific changes associated with abnormal branch point recognition and mis-splicing in Sf3b4 mutants. Overall, we show that Sf3b4 is necessary at the earliest stages of NCC differentiation and propose that transcripts with branch point sequences situated in thymine-rich contexts have an increased susceptibility to be mis-spliced when levels of SF3B4 are reduced.
Results
Homozygous Deletion of Sf3b4 with Wnt1-Cre2 Results in Craniofacial Malformations that Are Modified by the Genotype of Neighboring Cells.
Sf3b4 heterozygous mutant embryos on two genetic backgrounds showed vertebrae abnormalities but did not have craniofacial defects (21, 22). Here, to characterize the requirement for Sf3b4 during craniofacial development and the etiology of craniofacial malformations in SF3B4-related syndromes, Sf3b4 conditional mutant mice with LoxP sequences flanking exons 2–3 of Sf3b4 (Sf3b4L/+) (21), Wnt1-Cre2 (23), and Sf3b4 heterozygous with Wnt1-Cre2 transgene (Sf3b4+/–;Wnt1-Cre2 tg/+) mice were used to delete Sf3b4 in NCC (SI Appendix, Fig. S1 A and B), and generate Sf3b4L/L;Wnt1-Cre2 tg/+ (Sf3b4 ncc/ncc) or Sf3b4L/–;Wnt1-Cre2 tg/+ (Sf3b4 ncc/–) embryos. The Wnt1 promoter drives CRE expression and activity in the neuroepithelium and NCC that emerges from the forebrain and midbrain of E8.5 embryos with four or more somite (s) (23, 24).
We expect embryos with Sf3b4 mutant NCC to exhibit craniofacial defects by E9.5 and die before birth like Eftud2 and Snrpb mutants (25, 26). To determine whether this is the case, we analyzed Sf3b4 NCC mutant embryos on a mixed genetic background, generated as described in materials and methods section (SI Appendix, Fig. S1 A and B), from embryonic day (E) 8.5–E17.5. At E8.5, 72% (n = 34/47) of Sf3b4ncc/ncc embryos and 53% (n = 55/103) of Sf3b4 ncc/– mutants were normal, compared to 99% (n = 366/370) of somite-matched controls (Sf3b4+/+;Wnt1-Cre2 tg/+, Sf3b4+/–, and Sf3b4 ncc/+). Hypoplasia of the first pharyngeal arch and the fore- and mid-brain were the most common abnormalities found in 96% (n = 46/48) of Sf3b4 ncc/– and 54% (n = 7/13) of Sf3b4 ncc/ncc mutant embryos, though phenotypic differences were observed between these two genotypes. Specifically, 8% (n = 4/48) of Sf3b4ncc/– mutants exhibited hypoplasia of the heart, a phenotype not previously seen in spliceosomal mouse mutants, while Sf3b4 ncc/ncc mutants exhibited either hypoplasia of only the first pharyngeal arch [23% (n = 3/13)], or hypoplasia of only the midbrain [23% (n = 3/13)]. A day later, the majority of E9.5 Sf3b4 NCC mutants of both genotypes were abnormal (SI Appendix, Tables S1 and S2) and although 2% (n = 1/45) of Sf3b4 ncc/– mutants were undergoing resorption, all E9.5 Sf3b4 ncc/ncc mutants (n = 22) were alive. Thus, Sf3b4 NCC mutants can first be distinguished from their control littermates at E8.5, shortly after CRE expression.
From E10.5 to E17.5, most Sf3b4 NCC mutants (n = 74, Sf3b4 ncc/ncc and 62 Sf3b4 ncc/–) were alive, as only 21% (n = 28/136) were undergoing resorption and did not have a heartbeat. Live mutant embryos weighed significantly less than their control littermates (SI Appendix, Fig. S1C) and exhibited a spectrum of defects which we grouped into three phenotypic Classes 1 to 3 (Fig. 1). Class 1 mutants appeared normal, and Class 2 mutants exhibited hypoplasia of the developing pharyngeal arches, or of the brain, or an open neural tube. E10.5–E12.5 Class 3 embryos exhibited hypoplasia of both the brain and pharyngeal arches, or reduced frontonasal mass, and/or mandible, while E13.5–E17.5 embryos in this Class showed anencephaly—an exposed forebrain, absent eyes and ears, as well as hypoplastic mandible. From a total of 136 Sf3b4 NCC mutants collected from E10.5–E17.5, Class 1 mutants were present at all stages with 20%, being Sf3b4 ncc/ncc, (n = 27/136) while 13% (n = 8/136) were Sf3b4 ncc/–. Similarly, whereas 10% (n = 14/136) of Sf3b4 NCC mutant embryos belonging to Class 2 were Sf3b4 ncc/ncc mutants, less than 1%, (n = 1/136) were Sf3b4 ncc/– mutants. In contrast, whereas 13% (n = 18/136) of Sf3b4 NCC Class 3 mutants were Sf3b4 ncc/ncc, 29% (n = 40/136) were Sf3b4 ncc/– mutants. Thus, homozygous mutation of Sf3b4 in NCCs, in the context of neighboring Sf3b4 heterozygous cells, results in a more penetrant and severe phenotype.
Fig. 1.

Neural crest–specific mutations in Sf3b4 in mouse embryos lead to craniofacial defects with variable expressivity. Representative images of embryos at E8.5–17.5 showing variable expressivity of craniofacial abnormalities observed in Sf3b4 NCC mutant embryos compared to wildtype. Embryo images of Class 1 and 2 are Sf3b4ncc/ncc. At E8.5 embryos 14 to 16 s are shown. All Class 3 embryo images are Sf3b4ncc/–. At E8.5 the white arrows point to hypoplastic structures. FB: forebrain, FL: forelimb, H: heart, HB: hindbrain, HL: hindlimb, M: mandible, MB: midbrain, N: nasal, OP: optic placode, PA: Pharyngeal arch. (Scale bar, E8.5–13.5, 500 μm and E17.5, 1,000 μm.)
Since Xenopus embryos with sf3b4 knockdown showed an expanded neural tube (27), the width of the left and the right neural epithelium of E12.5 Sf3b4 NCC mutants was measured at the level of the forelimb (SI Appendix, Fig. S1D, white line). No difference was found in the width of the neural folds of Class 1 and 2 Sf3b4 NCC mutants (n = 4), when compared to those of controls (n = 11). However, width of both the left and the right neural folds of Class 3 Sf3b4 NCC mutants (n = 11) was significantly reduced when compared to controls (t test, P < 0.0001) (SI Appendix, Fig. S1 F and G). Furthermore, 25% (n = 1/4) of Class 1 and 2, and 64% (n = 7/11) of Class 3 Sf3b4 NCC mutants had an open neural tube (SI Appendix, Fig. S1 D and E). Thus, deletion of mouse Sf3b4 in the dorsal neural tube results in a significant decrease in the size of the neural tube and open neural tube in a subset of embryos. When taken together, these data suggest that the reduced size of the neural epithelium may be responsible for the open neural tube found in Sf3b4 NCC mutants.
The morphological analysis above suggests that phenotypically normal-appearing embryos belonging to Class 1 may be born. To determine whether this was the case, all pups found from mating of Sf3b4L/+ males and Sf3b4L/+;Wnt-Cre2tg/+Wnttg/+ females (n = 68) or Sf3b4L/+ and Sf3b4+/–; Wnt1-Cre2tg/+ (n = 13) mice were genotyped. No Sf3b4 ncc/– pups were found from these matings (SI Appendix, Table S4). In contrast, 2 Sf3b4 ncc/ncc pups (SI Appendix, Table S3) were found at postnatal day (P) 21, when offspring were weaned. Both Sf3b4 ncc/ncc female mice were indistinguishable from their wildtype littermates and were euthanized (28) a year after their birth. Although a significant deviation from the expected frequency, (χ2 = 17.64, P = 0.0052), these data show that mice with homozygous deletion of Sf3b4 in NCC can be born and survive to adulthood. Additionally, we found no sex-specific difference in the expressivity and survival of E17.5 Sf3b4 NCC mutants (SI Appendix). These findings indicate that homozygous deletion of Sf3b4 in NCC is not fully penetrant when all other cells of the embryo are wildtype and that mutation of Sf3b4 in mouse NCC replicates the penetrance and expressivity of craniofacial malformations observed in human patients.
Neural Crest and Mesoderm-Derived Structures Are Abnormally Formed in Sf3b4 NCC Mutant Heads.
CNCC along with ectodermal placodes give rise to the cranial ganglia and their sensory neurons (29), and like previously described spliceosomal NCC mutants (25, 26), NCC-derived ganglia were reduced and failed to form in Sf3b4 NCC mutants (SI Appendix, Fig. S3).
CNCC also contribute to many of the cartilage and bones that are malformed in SF3B4-related syndrome patients. We stained E17.5 embryos with Alizarin red which binds to calcium, and Alcian blue, which binds to sulfated glycosaminoglycans and glycoproteins to visualize and characterize the craniofacial bones and cartilages, respectively. Our analysis revealed that one Class 1 Sf3b4 ncc/ncc mutant (n = 1/10) was normal and indistinguishable from its control littermates (n = 23), consistent with the fact that a subset of these mutant embryos is born and survive to adulthood. However, all remaining Sf3b4 NCC mutants [Class 1 (n = 9), Class 2 (n = 2), and Class 3 (n = 7), SI Appendix, Fig. S1H] exhibited at least one defect in the craniofacial region (SI Appendix, Table S6). These defects span a broad spectrum and were variable between individual embryos of the same genotype and Class. Nonetheless, Class 3 mutant embryos had the most severe and the highest burden of defects, exhibiting multiple malformations in the skull.
Malformations found in the cranium included the absence of CNCC and mesoderm-derived cartilage and bones ventral to the basisphenoid (n = 7/7) in Class 3 embryos (Fig. 2). At the other extreme, neural crest–derived structures, including the nasal cartilage, the palate, and the frontal bone were formed in the cranium of Class 1 and Class 2 mutants, but these bones had a smaller region of mineralization and were thus smaller than those of controls (Fig. 2). The basisphenoid which is derived from NCC and forms via endochondral ossification was present but abnormally shaped, while the pterygoid, also NCC-derived, was absent in a subset of Class 1, Class 2, and Class 3 mutants (Fig. 2). Furthermore, defects were also found in the palates of Class 1 and 2 mutants. A small subset of these embryos had cleft palate [Class 1 (n = 4/10) and Class 2 (n = 2/2)] while most were missing the palatal process of palatine (Fig. 2). The mandible or lower jaw, which uses Meckel’s cartilage as a template for its development, was present in all Class 1 and 2 Sf3b4 NCC mutants. However, mandibles of Class 2 mutants were less ossified than those of controls (SI Appendix, Fig. S2A). Furthermore, in Class 3 mutants, the mandibles were dysmorphic and showed no obvious distal and proximal patterning (Fig. 2).
Fig. 2.

Sf3b4 mutants at E17.5 have various skeletal craniofacial abnormalities. Craniofacial defects were seen in the cranium, palate, mandible, and middle ear structures in Sf3b4 NCC mutants. A schematic beside each row indicates orientation of the cranium. A: anterior, An: angular process, Bs: basisphenoids, Cn: condylar process, Cp: coronoid process, Hy: Hyoid, I: inferior, In: Incus, L: Left, Lp: lenticular process, M: malleus, Ma: mandible, MC: Meckel’s cartilage, Pbm: processus brevis malleus, P: posterior, Pl: palatine, Pppl: palatal process of palatine, Pppm: palatal process of premaxilla, Ptg: Pterygoid, R: Right, S: superior, St: stapes, Stp: styloid process. red * indicates dysmorphic middle ear cartilage. (Scale bar, 1,000 μm.)
The proximal portion of Meckel’s cartilage contributes to the malleus and incus—two of three ossicles of the middle ear. The third middle ear ossicle, the stapes, is derived from NCCs and mesoderm, and forms independently of Meckel’s cartilage. In Sf3b4 NCC mutants, the proximal portion of Meckel’s cartilage was bilaterally reduced in Class 1 mutants (n = 2/10) (Fig. 2) and missing in all Class 3 mutants. Similarly, middle ear ossicles formed from this cartilage, were malformed or missing. A subset of Class 1 and Class 2 mutants lacked the lateral process of the malleus and had hypoplasia of the manubrium and the lenticular process of the incus (n = 3/12, SI Appendix, Fig. S2B). Additionally, the stapes was absent in a subset of Class 1 and Class 2 mutants, but the most severe defects were seen in Class 3 mutants, which lacked all middle ear ossicles and had ectopic cartilages in their places (Fig. 2). Interestingly, the middle ear ossicles are derived from the first and second PA which were hypoplastic at earlier embryonic stages.
Sf3b4 NCC mutants also had abnormalities in neural crest-derived cartilages and bones of the neck. The clavicles were asymmetric, shorter, and dysmorphic in Class 1 (n = 3/10), Class 2 (n = 1/2), and all Class 3 (n = 7/7) mutants (SI Appendix, Fig. S2C). In one Class 1 mutant, the body of the hyoid was not ossified, it was vertically positioned and abnormally attached to the cricoid cartilage (SI Appendix, Fig. S2D). In another, the hyoid was malformed and attached to the thyroid cartilage as two separate horn-like structures (SI Appendix, Fig. S2D). The hyoid was missing in all severely affected mutants. Furthermore, the thyroid cartilage was dysmorphic in Class 1 embryos (n = 1/10) (SI Appendix, Fig. S2D), and along with the tracheal rings were also missing in Class 1 (n = 1/10) (SI Appendix, Fig. S2D), Class 2 (n = 1/2) and all Class 3 (n = 7/7) mutants. Additionally, vertebral transformation and malformations previously described in Sf3b4+/– embryos (21) were also seen in Sf3b4 ncc/– embryos (SI Appendix). These analyses indicate that Sf3b4 NCC embryos, including normal-appearing Class 1 mutants, have mineralization, cartilage, and bone defects. Our data also show that mouse Sf3b4 is required for proper morphogenesis of NCC-derived cartilages and bones that are malformed in patients with SF3B4-related syndromes.
Sf3b4 Is Required for Normal Heart Development.
Cardiac NCCs migrate into the outflow tract of the developing heart and contribute to the aorticopulmonary septum and the endocardial cushion (30). By E12.5, septation of the distal outflow tract has completed (31). At this stage, we observed a large increase in the percentage of dead/reabsorbed Sf3b4 NCC mutants (SI Appendix, Tables S1 and S2). To determine whether heart malformations contribute to the significant loss of Sf3b4 mutants, E12.5 embryos were analyzed. No discernable differences were found in wholemount hearts of controls (n = 2) and Class 1 Sf3b4 ncc/ncc (n = 2) mutant embryos (Fig. 3 A and B). In contrast, a similar analysis of the hearts of Class 2 Sf3b4 ncc/ncc (n = 2) and Class 3 Sf3b4 ncc/– (n = 2) mutants showed that the right and left atria were hypoplastic and that the aorticopulmonary septum, which separates the aortic and the pulmonary trunks, was not present resulting in persistent truncus arteriosus (Fig. 3 C and D). However, histological analysis of frontal and sagittal sections of E12.5 embryos revealed heart defects in Sf3b4 NCC embryos of all Classes. In Class 1 mutant embryos (n = 2), the aorticopulmonary septum (Fig. 3 E ′ and F ′) was thicker than those of controls, whereas walls of both ventricles were thicker in mutants of all Classes (n = 6) (Fig. 3 F ′, G′, and H ′, black line). Additionally, the dorsal aorta was smaller than those of controls in Class 1 mutants (Fig. 3 E ″ and F ″), abnormally connected to the pulmonary trunk in Class 2 mutants (Fig. 3G″), and undetectable in Class 3 mutants (Fig. 3H″). Similarly, the aorticopulmonary trunk was not found in Class 2 and Class 3 mutants (Fig. 3 G ′ and H ′) leading to the presence of a common truncus. Additional heart defects seen in Class 3 Sf3b4 ncc/– mutants included a smaller heart, that did not completely occupy the mediastinum, small and abnormally positioned left and right atriums, and an absence of the aortic/pulmonary valves (Fig. 3H ′), when compared to wild type (Fig. 3E ′). Thus, Sf3b4 NCC mutants have several heart defects that have been reported in patients with SF3B4-related syndromes (5) and reveal a role for SF3B4 during septation of the outflow tracts as well as in the differentiation and growth of the atria and ventricles. Based on these findings, we infer that cardiac abnormalities may contribute to loss of Sf3b4 NCC mutants.
Fig. 3.

Heart defects were observed in E12.5 Sf3b4 mutants. (A–D) Whole mount heart of wild type and mutants. (A) Sf3b4+/+;Wnttg/+, (B) Class 1 Sf3b4ncc/ncc, (C) Class 2 Sf3b4ncc/ncc with a normal craniofacial region and hypoplastic mid- and hindbrain, (D) Class 3 Sf3b4ncc/–. A: atrium, L: left, OFT: outflow tract, the blue star indicates the visible aorticopulmonary septation in the outflow tract. (Scale bar, 1,000 μm.) (E′–H′) Frontal serial sections of the heat. For all frontal sections, orientation is indicated beside E′. (E′) wildtype, (F′) Class 1 Sf3b4ncc/ncc, (G′) Class 2 Sf3b4ncc/ncc, (H′) Class 3 Sf3b4ncc/–. (E″–H″) Sagittal, serial sections of the heart. For all sagittal sections, orientation is indicated beside E″. (E″) wildtype, (F″) Class 1 Sf3b4ncc/ncc, (G″) Class 2 Sf3b4ncc/ncc, (H″) Class 3 Sf3b4ncc/–. The black line shows thickness of the ventricle wall. C: caudal, D: distal, Dao: dorsal aorta, P: proximal, Ro: rostral. A: atrium, AT: aortic trunk, PT: pulmonary trunk, V: ventricle. (Scale bar, 100 μm.)
Fewer Sf3b4 Mutant NCC Are Found in the Developing Craniofacial Region.
Hypoplasia and absence of CNCC derivatives in Sf3b4 mutants could be due to reduced NCC generation, migration, or survival. To track CRE-expressing NCC, we used the RosamT/mG reporter, which marks these cells and their derivatives with GFP (32). At E8.5, the distribution of GFP+ NCC found in the head and in streams at the levels of the first and second pharyngeal arches of Sf3b4 ncc/ncc mutants (n = 4) and controls (n = 4) were comparable (Fig. 4A). However, the GFP signal appeared patchy and reduced in the frontonasal region of mutants (Fig. 4A, arrow). Furthermore, few GFP+ cells were found in the first pharyngeal arch and frontonasal prominences of a phenotypically normal E8.5 Sf3b4 ncc/– mutant (n = 3) with 8-somites (Fig. 4A), when compared to a somite-matched control (Fig. 4A and SI Appendix, Fig. S4A). A day later, significantly fewer GFP+ cells were found in the 1st pharyngeal arch of E9.5 Sf3b4 NCC mutant embryos (t test; Sf3b4 ncc/ncc P = 0.0006, Sf3b4 ncc/– P = 0.0001) (Fig. 5 A and C). We postulated that the reduced number of GFP+ (Sf3b4 homozygous mutant) cells found in the first pharyngeal arch and frontonasal prominence of E8.5 Sf3b4 NCC embryos is due to a reduction in NCC generation, as previously shown in Xenopus sf3b4 morphants (27), as well as reduced cell survival in the developing craniofacial region.
Fig. 4.

Sf3b4ncc/– mutants have increased cell death early in the neural tube. (A) Sf3b4+/+;Wnttg/+embryos with RosamT/mG have GFP expression in the head and first pharyngeal arch whereas decreased GFP expression is observed in the frontonasal region of Sf3b4ncc/ncc embryos, white arrow. And Sf3b4ncc/– embryos have decreased GFP in the head, white arrow, and no GFP in the first PA. Images are shown in brightfield followed by fluorescence. PA; pharyngeal arch. (Scale bar, 20 μm.) (B) Representative sagittal view images of TUNEL assay at E8.5 in whole wild type and mutant embryos. White arrow indicates TUNEL positive nuclei. H: heart. (Scale bar, 100 μm.)
Fig. 5.
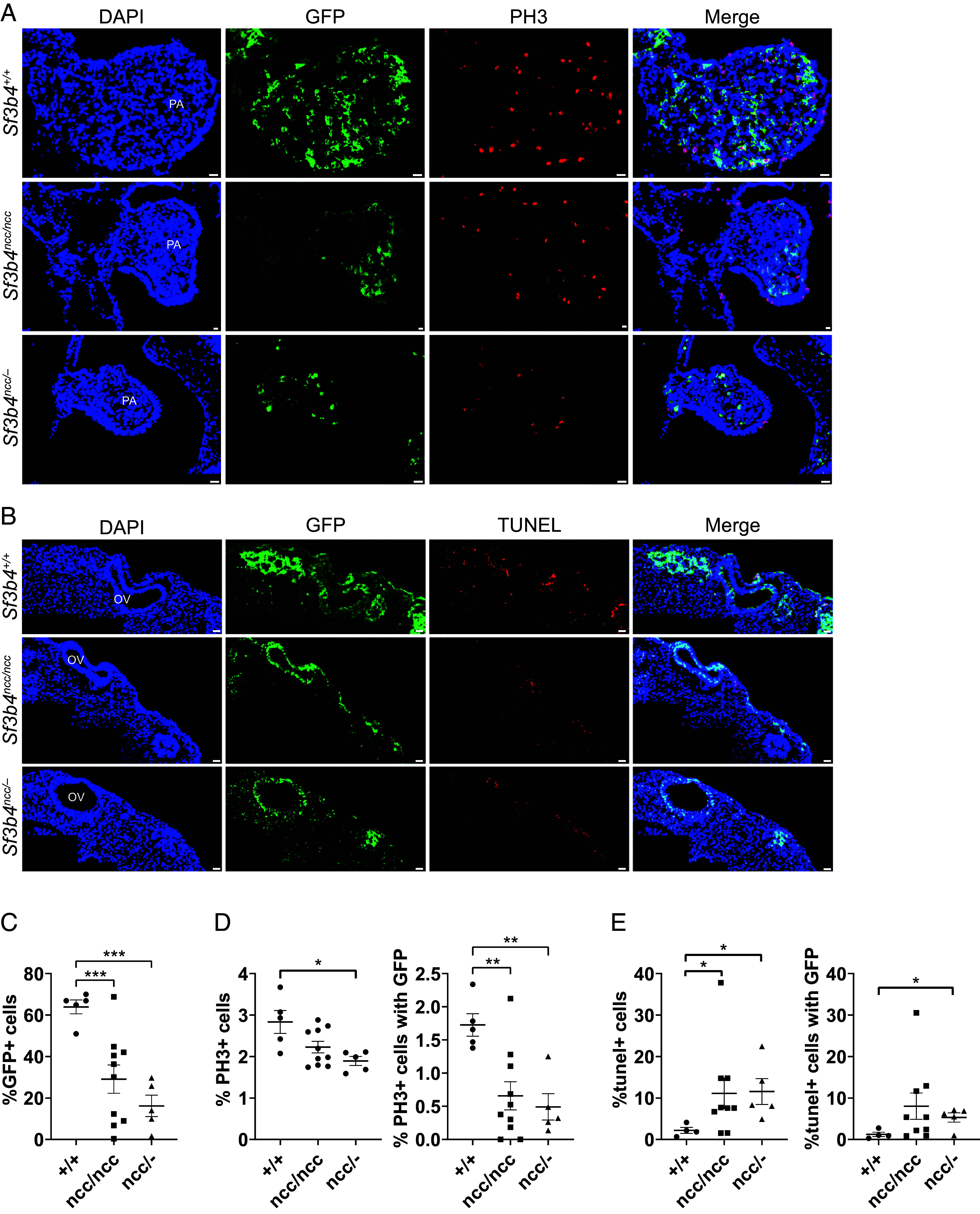
NCCs with Sf3b4 mutations have decreased proliferation and increased cell death. (A) Representative images of first pharyngeal arch sections of wild type and mutant embryos stained with PH3 to look at proliferation. PA; pharyngeal arch. (Scale bar, 20 μm.) (B) Representative images of TUNEL assay on sections of hindbrain with otic vesicle in wild type and mutant embryos. OV; otic vesicle. (Scale bar, 20 μm.) (C) Quantification of GFP expression in the first PA at E9.5 with a significant decrease in Sf3b4 mutants. Each data point represents the average of four sections from one embryo, error bars are ±SEM, ***P < 0.001. (D) Graph showing quantification of PH3+ cells in the pharyngeal arch with a significant decrease in the mutants. Each data point represents the average of four sections from one embryo, error bars are ±SEM, *P < 0.05, **P < 0.005. (E) Graph showing quantification of TUNEL+ cells in the hindbrain and otic vesicle region with a significant increase in the mutants. Each data point represents the average of 3/4 sections from one embryo, error bars are ±SEM, *P < 0.05, **P < 0.005.
Normal Levels of Sf3b4 Are Required for NCC Survival and Proliferation.
To determine whether cell death is increased in Sf3b4 mutant NCCs we used TUNEL to label cell death in E8.5 embryos with 5 to 7 s, before phenotypic differences are seen. We found that cell death was increased in the neural tube and region underlying the neural tube of E8.5 Sf3b4 ncc/– embryos (n = 3) when compared to wild-type (n = 3) or Sf3b4 ncc/ncc embryos (n = 2) (Fig. 4B and SI Appendix, Fig. S4G).
To examine the fate of Sf3b4 mutant cells at E9.5 when 96% of Sf3b4 ncc/– and approximately 50% of Sf3b 4 ncc/ncc mutants are phenotypically abnormal, cell death and proliferation were measured in E9.5 embryos. In the region immediately surrounding the hindbrain and otic vesicle, where the NCC emerge, the proportion of histone H3 phosphorylation on serine-10 (PH3)-positive (mitotic) cells found in wild type and Sf3b4 NCC mutants was comparable (SI Appendix, Fig. S4 B and C). In contrast, the proportion of GFP+ cells that were also TUNEL positive was significantly increased in Sf3b4 ncc/– mutants (t-test, P = 0.018) (Fig. 5 B and E). Additionally, the total number of all TUNEL positive cells (GFP+ and GFP–) was significantly increased in Sf3b4 NCC mutants when compared to controls (t test, Sf3b4 ncc/ncc P = 0.043 and Sf3b4 ncc/– P = 0.037) (Fig. 5 B and E). In contrast, a similar analysis in the first pharyngeal arch of controls (n = 5), and Sf3b4 NCC mutants [Sf3b4 ncc/ncc(n = 10) and Sf3b4 ncc/– (n = 5)] revealed no difference in the proportion of cleaved caspase-3 positive cells, an apoptotic marker (SI Appendix, Fig. S4F), or in TUNEL-positive cells in the 1st pharyngeal arch of controls (n = 4), Sf3b4 ncc/ncc (n = 9), and Sf3b4 ncc/– mutants (n = 5, SI Appendix, Fig. S4 D and S4 E). Additionally, the proportion of GFP+ cells stained with PH3 was significantly reduced in the first pharyngeal arch of Sf3b4 ncc/ncc (n = 10, 5 each from Class 1 and Class 3) and Sf3b4 ncc/– (n = 5, all Class 3) mutants, when compared to controls (n = 5) (t test, P = 0.0018 and P = 0.0016, respectively) (Fig. 5 A and D). Although the proportion of all cells (GFP+ and GFP–) positive for PH3 was only significantly reduced in the first pharyngeal arches of Sf3b4 ncc/– mutants, when compared to controls (t test, P = 0.0233) (Fig. 5 A and D). Our data show that increased cell death in the neuroepithelium contributes to the decreased number of NCC found in E8.5 Sf3b4 ncc/– mutants and reveals that it is reduced proliferation which underlies the reduced number of NCC found in the first pharyngeal arch of Sf3b4 NCC mutants of both genotype.
To further investigate the impact of reduced Sf3b4 specifically to NCC proliferation and survival, we investigated Sf3b4 knock-down in O9-1 cells, a mouse NCC line. We found that the proportion of BrdU and PH3 positive O9-1 cells was significantly reduced in cells transfected with Sf3b4 siRNA compared to control transfections (SI Appendix, Fig. S4 H and I). Additionally, to investigate NCC death after Sf3b4 knockdown, O9-1 cells were stained with a fluorophore-conjugated Annexin V 24 h posttransfection. Annexin V binds to membrane phosphatidylserines that are exposed to the extracellular environment during the early stages of apoptosis (33). The Annexin V assay revealed that O9-1 cells transfected with Sf3b4 siRNAs fluoresce at significantly higher levels relative to the control condition (SI Appendix, Fig. S4J). These data indicate that normal levels of Sf3b4 are necessary for NCC proliferation and survival both in vivo and in vitro.
Homozygous Deletion of Sf3b4 Disrupts the Transcriptomic Landscape Before the Onset of Morphological Defects.
Sf3b4 is important for the U2 snRNP to attach upstream of the branch point site (17), we next compared the transcriptome of E8.5 normal looking Sf3b4 ncc/– and wild type embryos between 8 and 10 s using RNAseq. Previously, a similar analysis of E9.5 Sf3b4+/– embryos and somite showed an increase in exon skipping associated differential splicing events (DSE) and few differentially expressed genes (21). In contrast, RNAseq analysis of E8.5 Sf3b4 NCC mutants revealed 733 differentially expressed genes, of which 330 genes were significantly up-regulated, and 403 genes were significantly down-regulated (SI Appendix, Fig. S5A). Additionally, a greater than fivefold increase in skipped exon (SE) events was found in Sf3b4 NCC mutants, when compared to controls; 315 SEs were found in 246 genes in control, versus 1,620 SEs in 1,020 genes in mutants (SI Appendix, Figs. S5C and S6A). To determine whether DSEs found in Sf3b4 ncc/– mutants were associated with any molecular function or biological processes, gene ontology and KEGG pathway were analyzed. No enrichment was found for any splice events except for one term, H4K20 histone methyltransferase activity, for alternative 5′ splice site (A5SS) events (SI Appendix, Fig. S6B). Thus, homozygous deletion of Sf3b4 in NCC alters the transcriptomic landscape of mutant embryos prior to any morphological defects.
Splicing and Level of Genes in the NCC Regulatory Network Are Aberrant in Sf3b4 NCC Mutants.
Transcripts in the NCC gene regulatory network (34) important for NCC development were mostly decreased in Sf3b4 NCC mutants. Specifically, we found significant reduction in expression of Tfap2b part of the neural plate border module, and in several Sox genes important in NCC specification and migration: Sox5, Sox6, Sox9, and Sox10 (SI Appendix, Table S7). Several other genes important for neural crest generation were also significantly reduced in mutants, including: Ets1, En2 and Grhl3, Dnmt3b, Dnmt1, and Nes (SI Appendix, Table S7). In contrast, E-cadherin levels were significantly increased in these mutants. E-cadherin is down-regulated when cells undergo epithelial–mesenchymal transition (EMT), an essential first step in neural crest migration from the hindbrain. Though splicing changes were not found in most transcripts, a significant increase in SE events was found for exon 4 of Sox5, exon 6 of Tfap2a, exon 9 or 4 of Dnmt3a and exon 4 of Dnmt3b. In addition, increased usage of alternative 3’SS was also found in exon 4 of Nes (SI Appendix, Table S8). Furthermore, HCR revealed reduced levels of Tfap2b and Sox9 along with discontinuous expression of Sox9 in the neural plate of morphologically normal E8.5 Sf3b4 ncc/ncc mutants (n = 3) with 7/8 s when compared to somite-matched controls (SI Appendix, Fig. S6C).
To identify pathways dysregulated in Sf3b4 NCC mutants, gene ontology analysis using all significantly differentially expressed genes was performed. Several pathways were enriched in these mutants, including intrinsic apoptotic signaling pathway by P53 among up-regulated genes, and neurogenesis among the most down-regulated (SI Appendix, Fig. S5B). Thus, we concluded that abnormal splicing of one or more genes, yet to be identified, results in abnormal expression of genes important for NCC development, and hypothesized that NCC with reduced Sf3b4 levels may arrest during the early stages of EMT and undergo P53-mediated cell death.
Reducing Levels of Trp53 Did Not Rescue Malformations or Viability of Sf3b4 NCC Mutant Embryos.
Increased P53 activity contributes to craniofacial malformations in several mouse models of neurocristopathies and spliceosomopathies (25, 26, 35, 36), while perturbation of global molecular processes that activate P53 contributes to NCC death in human cell lines and in mouse models (35). Therefore, we next sought to determine whether P53-activity was increased in cells with reduced levels of Sf3b4. RNAseq analysis showed significant enrichment of the P53 pathway, and significant increases in the levels of P53-downstream targets: Mdm2 (1.73-fold), Phlda3 (21.75-fold), Trp53inp1 (7.41), Ccng1 (6.35-fold), Cdkn1a (20.96-fold), and Eda2r (17.04-fold). In addition, when P53 activity was analyzed in O9-1 cells transfected with Sf3b4 siRNA, we found a remarkable increase in activated P53 (SI Appendix, Fig. S7A). To evaluate the extent of P53 activation upon Sf3b4 knockdown in O9-1 cells, the expression of cell cycle genes that are directly and indirectly regulated by P53 was quantified by RT-qPCR. This analysis revealed a significant upregulation of Ccng1 (2.63-fold) and Cdkn1a (3.23-fold) in knockdown samples relative to control (SI Appendix, Fig. S7B). These data reveal that Ccng1 and Cdkn1a, two important mediators of the G1-S phase transition (37–39) that are transcriptionally regulated by P53 are significantly overexpressed in knockdown samples compared to control. We further evaluated whether Sf3b4 knockdown leads to activation of the P53 apoptosis pathway. RT-qPCR data showed a significant upregulation of P53-regulated genes, including Mdm2 (5.61-fold), Phlda3 (2.78-fold), Puma (3.72-fold), and Trp53inp1 (11.73-fold) in Sf3b4 knockdown cells compared to control (SI Appendix, Fig. S7C), suggesting a role for P53 mediated apoptosis in NCC with loss of Sf3b4.
Sakai and colleagues showed that neuroepithelial cells in E8.5 mouse embryos have higher levels of ROS relative to other cells and that exposure of wild-type mouse embryos to a ROS generator drug (3-NP) leads to a substantial increase in neuroepithelial cell death (40). The marked upregulation of Trp53inp1, a gene with known antioxidant function, in Sf3b4 knockdown cells (SI Appendix, Fig. S7C) led to the consideration of the cells’ oxidative state as a factor that might influence the observed increase in apoptosis. To evaluate this hypothesis, the levels of ROS and Superoxide were assayed in Sf3b4 knockdown and control O9-1 cells. Interestingly, while the levels of both ROS and Superoxide for the control wells clustered together, the levels for the knockdown wells were much more variable (SI Appendix, Fig. S7 D and E). To further evaluate the role of oxidative stress in Sf3b4-mediated apoptosis, the expression of key antioxidant genes Sod1, catalase, and Nqo1 (41) was also assayed in Sf3b4 knockdown and control cells (SI Appendix, Fig. S7 F and G). Although the group means for each gene were not significantly different, their reference-normalized expression (ΔCT) for the control condition exhibited higher variance relative to the knockdown condition. Taken together, our data suggest that Sf3b4 knockdown increased susceptibility to stress in NCCs.
Reducing levels of P53 has variable impact on craniofacial development and survivability of mouse models of craniofacial spliceosomopathies. For Snrpb NCC mutants, reducing levels of P53 resulted in increased survival but failed to rescue patterning defects in the mandible (25). In contrast, reducing levels of P53 alone, proved insufficient to improve craniofacial development or survival of Eftud2 NCC mutants (25, 42). To test whether decreasing P53 rescues the craniofacial malformations seen in Sf3b4 ncc/– mutants, we mated Sf3b4+/–;Trp53L/+;Wnt1-cre2 tg/+ mice with Sf3b4L/+;Trp53L/L mice to generate pups that have NCCs with homozygous deletions for both Sf3b4 and P53. We followed pups from 4 litters from P0 to P21. Of 42 pups born from these matings, four died at P1 and could not be followed to weaning. However, genotyping of carcasses collected from these pups revealed that two were conditionally heterozygous for Sf3b4 and Trp53 (Sf3b4L/+;Trp53L/+;Wnt1-Cre2tg/+) and 2 were heterozygous for the conditional alleles but did not carry CRE (Sf3b4L/+;Trp53L/+). Furthermore, zero Sf3b4 and Trp53 double homozygous NCC mutants were found among the 38 pups weaned at P21. Additionally, E9.5 Sf3b4 and Trp53 double homozygous NCC mutants were indistinguishable from Class 3 E9.5 Sf3b4 NCC mutants (SI Appendix, Fig. S7H). Thus, we conclude that increased P53 activity is not sufficient to explain the observed malformations and reduced survival associated with loss of Sf3b4 in NCC.
Mis-Splicing of Chromatin Remodelers Contributes to Malformations in Sf3b4 Mutants.
Previously, histone modifiers were found to be differentially spliced in Sf3b4+/– embryos when compared to controls (21). Consequently, a list of genes with significant increases in SE with FDR < 0.05 was used to query MGI for gene ontology terms related to histone modifier and chromatin remodeler, and craniofacial and heart development. Six genes: Tcf7l2, Kdm6a, Kdm5c, Setd5, Carm1, and Dnmt3b were identified from this analysis (SI Appendix, Table S9). Next, RT-PCR was used to show that SE events predicted in Tcf7l2 and Kdm6a were also seen in RNA collected from additional pools of Sf3b4 mutants (SI Appendix, Fig. S5 D and E). In addition, RT-qPCR revealed an increase in levels of Kdm5c, Carm1, and Setd5 in E8.5 Sf3b4 mutant samples, when compared to controls although not significant (SI Appendix, Fig. S6D). Thus, we concluded that reducing Sf3b4 levels in NCC affects splicing of transcription factors and histone modifiers involved in normal craniofacial development.
Short AT-Rich and GC-Poor Exons in Short Genes Are More Skipped in Sf3b4 Mutants.
To investigate whether aberrant splicing events were characterized by specific sequences, we compared alternative events preferentially found in mutant samples, wild-type samples, and a set of 1,000 randomly chosen alternative splicing events. No difference was found for 5′ splice site (SS) and 3′SS strengths of any alternative splicing events [skipped exons (SE), retained introns (RI), mutually exclusive exons (MXE), alternative 3′ splice sites (A3SS) and alternative 5′ splice sites (A5SS)], although a trend toward weaker 5′SS strength was seen for RI and A3SS events found in mutants (SI Appendix, Fig. S8 C and D).
To further characterize context and features of exons with skipped events we analyzed the length and the nucleotide composition of genes with skipped exons, the exons skipped, and the intronic sequences flanking them in wild type and mutant samples. Our analysis revealed that genes with SE events were significantly shorter in Sf3b4 mutants (P = 0.00031) and showed no significant difference in overall nucleotide composition, when compared to genes with SE events in wild type samples (Fig. 6A and SI Appendix, Fig. S8B). Furthermore, skipped exons and their upstream introns were also significantly shorter in mutant embryos (P = 0.004 and P = 0.0091 respectively), when compared to skipped exons and their upstream introns in wild type controls (Fig. 6 B and C). Additionally, the nucleotide composition of both upstream and downstream introns was significantly AT-rich and GC-poor in Sf3b4 mutants, when compared to intronic sequences in wild type embryos (Fig. 6 D–G). Similarly, the SE and MXE of Sf3b4 mutants were also GC poor, when compared to similar events found in controls (SI Appendix, Fig. S8 E and F). Altogether these analyses indicate AT-rich short exons found in a subset of genes are more likely to be skipped when levels of Sf3b4 are reduced.
Fig. 6.

Altered recognition of branch point features in skipped exon events contribute to increased alternative splicing in Sf3b4 NCC mutants. (A) Violin plot showing that the length of gene in which the skipped exon event is observed is significantly shorter in mutants. (B) Violin plot showing a significant decrease in the length of skipped exons in mutant compared to wildtype. (C) Violin plot showing that the length of the intron upstream of the skipped exon is significantly shorter in mutants than in wildtype. (D) Violin plot showing significantly increased proportions of AT present in the intron downstream from the skipped exon. (E) Violin plot showing significantly decreased proportions of GC present in the intron downstream from the skipped exon. (F) Violin plot showing significantly increased proportions of AT present in the intron upstream from the skipped exon. (G) Violin plot showing significantly decreased proportions of GC present in the intron upstream from the skipped exon. (H) Violin plot showing predicted branch point signals for SE events with a significant difference in preference for branch point at specific distance from 3′SS in wildtype (28 bp) and mutant (26 bp). (I) Predicted sequence composition 50 bp around the branch point showing an increase in thymine bases surrounding the branch-point A in mutants.
Sf3b4 Is Necessary for Branchpoint Recognition.
Since, we previously showed a nonsignificant preference for usage of a branch point that was closer to the 3’SS in mutant-specific A3SS events (21), we analyzed position of predicted branch point adenosine (BPA) associated with SE events. This analysis indicated that BPA associated with SE events in Sf3b4 mutants were closer to the 3′ splice site (26 bp in mutants compared to 28 bp in controls, P = 0.007) (Fig. 6H) when compared to BPAs associated with SE events in wild type embryos. In addition, nucleotide composition analysis of 50 bp flanking the BP sequence showed an increase in thymine bases surrounding the branch-point adenosine, when compared to controls (Fig. 6I). Our data further support a requirement for normal levels of SF3B4 to prevent skipping of short AT-rich exons with AT-rich branch points that are closer to the 3′ splice site.
Discussion
Craniofacial spliceosomopathies, like SF3B4-related syndromes and MFDM which is caused by pathogenic variants in EFTUD2, are also termed neurocristopathies since craniofacial defects are predominantly found in structures derived from NCCs. In this study, we showed that homozygous deletion of Sf3b4 in NCCs results in craniofacial defects with variable expressivity mimicking those seen in patients. Loss of Sf3b4 in NCCs results in mis-splicing and reduced expression of NCC specifier genes and histone modifiers as well as reduced survival of mutant NCCs and surrounding cells. Furthermore, though the P53 pathway was significantly up-regulated by loss of Sf3b4 in NCCs, craniofacial defects and survival of Sf3b4 mutants were not solely dependent on P53, like Eftud2 NCC mutants (42). Additionally, aberrant splicing and altered branch point usage were predominantly in AT-rich exons of shorter genes. Overall, we show that craniofacial malformations like those found in patients with SF3B4-related syndromes were present in the majority of Sf3b4 NCC mutants. In addition to craniofacial defects, Sf3b4 ncc/– embryos also had vertebral abnormalities like those seen in Sf3b4+/– embryos (21, 22). Therefore, the Sf3b4 NCC mutant embryos described herein model the spectrum of malformations and expressivity found from Nager to Rodriguez syndrome (1, 3).
The penetrance and expressivity of Sf3b4-associated malformations seen in Sf3b4 NCC mutants were modified by the number of wild type Sf3b4 alleles present in cells that neighbored homozygous mutant NCCs. Thus, we conclude that wild type level of SF3B4 in neural crest–derived mesenchyme and their neighboring cells is required for normal craniofacial development. We predict that expression of one, or more transcript that protects Sf3b4 homozygous mutant NCCs from death are reduced or missing in heterozygous nonneural crest–derived mesenchyme. Hence, explaining the more severe phenotype seen in Sf3b4 ncc/– embryos, when compared to Sf3b4 ncc/ncc. We postulate that normal variance in the expression level of SF3B4 in non neural crest–derived cells versus neural crest–derived mesenchyme, may lead to differential splicing of similar transcripts in humans explaining why patients with the same pathogenic variants may present with Nager syndrome or the more severe Rodriguez syndrome. Single-cell analysis of the developing craniofacial region of Sf3b4 NCC mutants in the context of wild type and heterozygous neighbors will be necessary to identify these targets in the mouse.
Several malformations found may contribute to the reduced numbers of Sf3b4 NCC mutants born, including a role for Sf3b4 in heart development. We propose that Sf3b4 NCC mutants of all Classes die due to palatal and/or heart defects, similar to other mouse models of craniofacial spliceosomopathies (25, 26). Since a subset of Nager Syndrome patients show heart defects, including coarctation of the dorsal aorta and complex heart defects, the role of SF3B4 in cardiac NCCs and heart mesoderm needs further investigation. Finally, our data support a role for Sf3b4 in cartilage and bone differentiation, as proposed by Marques et al (13), and also reveals a requirement for this gene in patterning of the lower jaw.
In addition, using RNAseq, mis-splicing was found in several transcripts important for NCC development, including Tcf7l2/TCF4 a transcription factor that together with β-catenin binds to and activates expression of WNT targets (43, 44). Intriguingly, increased skipping of exon 4 of Tcf7l2 was reported in TXNL4A patient iNCCs (45) and was predicted to result in loss of the CTNNB1 domain of TCF4 reducing WNT transcriptional response (44), suggesting that this particular splicing event is shared between the mouse and human. Thus, we propose that reduced WNT signaling during NCC development underlies shared craniofacial defects seen in Burn-Mckeown and SF3B4-related syndromes, and possibly other spliceosomopathies. Additional mis-splicing events in key NCC genes like Tfap2a (34) and Sox9 (46–48), which are predicted to introduce PTCs and reduce level of these genes, may further contribute to the reduced amount of NCC generated in Sf3b4 NCC mutants (Fig. 7).
Fig. 7.

Model of how loss of Sf3b4 leads to abnormalities in Sf3b4 NCC mutants. Our data indicate that SF3B4 is required for proper splicing of short genes, with BPAs that are closer to the 3′SS in AT-rich and GC-poor introns flanking exons that are themselves AT-rich and GC-poor. In Sf3b4 homozygous mutant NCC there is increased skipping of these exons in NCC GRN specifiers and histone modifiers important for neural crest development.
Sf3b4 NCC mutants also exhibited abnormal expression of genes important for NCC specification and migration. Specifically, reduced levels of Sox10, which is switched on when NCCs begin EMT (49–51), and increased expression of epithelial markers such as Grhl3, which is required for neural tube closure (52) and E-cad, which is normally down-regulated in cells undergoing EMT (53, 54), indicate that Sf3b4 mutant cells may be blocked in exiting the neural tube. Furthermore, reduced levels of Tfap2b and Sox9 in Sf3b4 ncc/ncc mutants support a model whereby reduced levels of specific transcription factors, for example, Tfap2b, Sox5, and Sox9 directly contribute to a delay, or block, in EMT of Sf3b4 mutant NCCs. Our findings are aligned with those of Devota et al. (27) who also found reduced NCC migration when sf3b4 was knocked down in Xenopus. However, unlike Xenopus embryos with sf3b4 knockdown, Sf3b4 NCC mutant mouse embryos do not show an expanded neural tube, likely due to the increased cell death of NCC precursors in the neural tube. In the pharyngeal arches, increased cell death of NCCs carrying mutations in core components of the spliceosome appears to be shared (25, 26). However, in Sf3b4 mutants reduced proliferation also contributes to reduced NCC survival in the pharyngeal region. Though P53-mediated cell death likely contributes to reduced survival of Sf3b4 mutant NCCs, genetic deletion of Trp53 did not rescue or improve craniofacial development in these mutant embryos. Hence, additional mechanism underlying cell death and the proliferation-arrest of Sf3b4 mutant NCCs remains to be identified.
We showed that heterozygous loss of Sf3b4 results in increased exon skipping and intron retention of histone modifiers required for development and patterning of the vertebrae (21). Hence, we postulated that reduction of Sf3b4 in NCCs would also result in aberrant splicing of histone modifiers required for craniofacial development. In fact, homozygous loss of Sf3b4 in NCCs had a much greater impact on splicing and gene ontology enrichment and only showed enrichment for H4-K20 methyltransferase in A5SS events. H4K20 methylation is associated with chromatin remodeling, cell cycle regulation, and DNA repair (55–57). Thus, abnormal splicing of enzymes important for H4K20 methylation may contribute to dysregulation of these cellular processes in Sf3b4 NCC mutants, or alternatively, dysregulation in these processes may contribute to abnormal H4K20 methylation. Since the current study does not indicate the direction of this interaction, future studies will need to explore this finding. Nonetheless, we did find increased SE in several histone modifiers important for craniofacial and heart development, including exon 24 of Kdm5c and exon 11 of Kdm6a. SE events in Kdm5c and Kdm6a are predicted to disrupt their protein interaction, and although Kdm5c and Kdm6a are X-linked, we do not see a sex-specific modification of expressivity or penetrance to Sf3b4 loss in mice. Also, like in this study, a large increase in SE was found in E9.5 whole embryos and isolated somites, and an increase in RI in embryos only (21), suggesting that there may be tissue-specificity in intron retention events. Differences in the number of RI events found between whole embryo and isolated tissues may reflect tissue-specificity in intron retention or may be a consequence of the larger number of transcripts present in the whole embryo. Our current analysis does not allow us to distinguish between these two possibilities.
Though reduced levels of Sf3b4 in NCCs resulted in an increase in DSE and in DEG, like previously described spliceosomal NCC mutants (25, 26), the large number of SEs found allowed us to detect that genes and exons more effected were shorter than those found in wildtype, and along with their upstream introns were AT-rich and GC-poor (Fig. 7). Additionally, these SE were also associated with an increase of thymidine bases surrounding the branch point. To date these signatures are specific to Sf3b4, although SE events in TXNL4A iNCC are also in shorter exons, the associated upstream introns are longer (45). Furthermore, though branch point context was not reported for TXNL4A, usage of branch point sequences closer to the 3′SS, was not seen in Eftud2 and Snrpb NCC mutants, which may suggest that this is specific for reduced levels of U2 components (58). Hence, we predict that variations in splice site characteristics found in these mutants combine to contribute to differential splicing in specific sets of transcripts in a tissue-specific manner. Future studies using single-cell RNAseq and spatial transcriptomics will shed light on this hypothesis. Altogether, our studies are starting to reveal the specific features that are altered by mutation of core splicing factors and a level of splicing regulation that has remained an enigma.
Materials and Methods
O9-1 Mouse CNCC Culture and Transfection.
O9-1 cells were purchased from Millipore Sigma (SCC0049). The cells were cultured on Matrigel-coated plates in conditioned media (SI Appendix). The Lipofectamine® RNAiMAX transfection reagent was used to perform siRNA (10 pmol/μL) transfections (SI Appendix). Transfected cell cultures were used for Annexin V, reactive oxygen species (ROS), and superoxide staining (SI Appendix).
Generation of Neural Crest–Specific Deletion Mutation in Sf3b4.
Sf3b4L/+;Wnt1-Cre2tg/+ mice were mated to Sf3b4L/+ mice, to generate Sf3b4L/L;Wnt1-Cre2tg/+ (Sf3b4ncc/ncc) embryos. Alternatively, Sf3b4+/–;Wnt1-Cre2tg/+ mice were mated to Sf3b4L/+ mice to generate embryos with Sf3b4L/–;Wnt1-Cre2tg/+ (Sf3b4ncc/–). All procedures and experiments were performed according to the guidelines of the Canadian Council on Animal Care and approved by the Animal Care Committee of the McGill University Health Centre Research Institute (AUP#5112). For further details on generation of these lines, genotyping, and a list of all mouse lines used in this study, see SI Appendix.
Generation of Neural Crest–Specific Mutation in Sf3b4 and Trp53.
Sf3b4L/+;Trp53L/L mice were mated to Sf3b4+/-;Trp53L/L;Wnt1-Cre2tg/+ mice to generate pups as well as embryos with NCC-specific deletion in both Sf3b4 and Trp53. Pups were observed from P0 to weaning (P21) and embryos were collected at E9.5.
Collection of Embryos.
Females were placed with a male overnight and checked for vaginal plug in the morning. The day of the plug was considered embryonic day (E)0.5. Dissections were carried out under a Lecia stereomicroscope (Lecia MZ6). Extraembryonic membranes were removed, yolk sac was collected for DNA extraction and genotyping. For embryos collected from E8.5 to E10.5, the pairs of somite were counted. Embryos were collected in 1% or 4% PFA (unless otherwise stated).
Skeletal Preparation.
E17.5 embryos were eviscerated and fixed in 100% EtOH overnight. Skeletal preparations were carried out as previously described by staining with 0.3% Alcian blue/0.1% Alizarin red (21). Skeletal preparations were imaged and analyzed using the Lecia stereomicroscope (Lecia MZ6 Infinity1).
Preparation of Embryos for Embedding and Histology.
For cryoembedding, fixed embryos were cryoprotected in 30% sucrose overnight, embedded in cryomatrix, and sectioned sagittally at 10 μm thickness. For embedding in paraffin, fixed embryos were washed in 1xPBS then dehydrated to 100% EtOH and embedded using paraffin. Embedded embryos were sectioned at 10 μm thickness on a Lecia RM2155 microtome and mounted on positively charged slides for further staining with hematoxylin and eosin.
Immunofluorescence (IF) and TUNEL Assay.
Freshly collected embryos or cryosections were used for IF and TUNEL assay. IF experiments were performed as previously described (26, 59). TUNEL assay was carried out using a Cell Death Detection Kit, TMR Red (12156792910, Roche) as per the manufacturer’s protocol. For details on the antibodies used and analysis, including for O9-1 cells, see SI Appendix.
RNA Isolation.
For RNA isolation of embryos, head from E8.5 embryos of 8 to 10 somite were collected in 250 µL of RNAlater (Invitrogen AM7020). For each pool of RNA extraction, heads from three embryos were pooled. RNA extraction was carried out using the Qiagen RNeasy micro kit per the manufacturer’s instruction (Qiagen 74104). For RNA sequencing, three wild type and three mutant pools were generated with each pool containing heads from three embryos. Total RNA was then used for RNA sequencing and RT- and qPCR (SI Appendix). For details on RNA extraction of O9-1 cells see SI Appendix.
RNA Sequencing Analysis.
Sequencing libraries were prepared by the McGill Genome Centre (Montreal, Canada), using the TruSeq Stranded Total RNA Sample Preparation Kit (Illumina TS-122-2301) as previously described (25). Detailed description of how differential expression analysis as well as analysis for differentially spliced events and enrichment analysis was carried out is given in SI Appendix.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We would like to thank Dr Mitra Cowan, Platform Manager, McGill Integrated Core of Animal Modeling for performing the microinjection experiments. We would like to thank the McGill Genome Centre for running the RNAseq. We also thank members of the Majewska laboratory for their helpful comments on the manuscript. We thank the Research Institute of McGill University Health Centre for supporting S.K. We would also like to acknowledge the professional and technical support from the Animal Resource Division of Reseach Institute - McGill University Health Centre for maintaining our mice colonies. This project was funded by the Canadian Institutes of Health Research Grant (202203PJT-480346-DEV-CFAC-157303) and by an Award by the Azrieli Foundation to Loydie Jerome-Majewska. The funders had no role in the study design, data collection and analyses, decision to publish, or preparation of the manuscript.
Author contributions
J.L.F., J.M., and L.A.J.-M. designed research; S.K., J.L., F.M., E.E.S., and S.M. performed research; S.K., E.B., J.L., E.C., F.M., E.E.S., and S.M. analyzed data; and S.K. and L.A.J.-M. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
RNAseq data have been deposited in the GEO (GSE260990) repository and are publicly available as of the date of publication (60).
Supporting Information
References
- 1.Bernier F. P., et al. , Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am. J. Hum. Genet. 90, 925–933 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petit F., et al. , Nager syndrome: Confirmation of SF3B4 haploinsufficiency as the major cause. Clin. Genet. 86, 246–251 (2014). [DOI] [PubMed] [Google Scholar]
- 3.McPherson E., Zaleski C., Ye Z., Lin S., Rodriguez syndrome with SF3B4 mutation: A severe form of Nager syndrome? Am. J. Med. Genet. A 164A, 1841–1845 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Cadieux-Dion M., Hughes S., Engleman K., Rush E. T., Saunders C., Nager syndrome in patient lacking acrofacial dysostosis: Expanding the phenotypic spectrum of SF3B4-related disease. Am. J. Med. Genet. A 185, 1515–1518 (2021). [DOI] [PubMed] [Google Scholar]
- 5.Czeschik J. C., et al. , Clinical and mutation data in 12 patients with the clinical diagnosis of Nager syndrome. Hum. Genet. 132, 885–898 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Drivas T. G., Taylor J. A., Zackai E. H., The final demise of Rodriguez lethal acrofacial dysostosis: A case report and review of the literature. Am. J. Med. Genet. A 179, 1063–1068 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Cassina M., et al. , A synonymous splicing mutation in the SF3B4 gene segregates in a family with highly variable Nager syndrome. Eur. J. Hum. Genet. 25, 371–375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Castori M., et al. , A 22-week-old fetus with Nager syndrome and congenital diaphragmatic hernia due to a novel SF3B4 mutation. Mol. Syndromol. 5, 241–244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drozniewska M., et al. , Second-trimester prenatal diagnosis of Nager syndrome with a deletion including SF3B4 detected by chromosomal microarray. Clin. Case Rep. 8, 508–511 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayata K., et al. , A case of Nager syndrome diagnosed before birth. Acta Med. Okayama 73, 273–277 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Irving M. D., et al. , Rodriguez acrofacial dysostosis is caused by apparently de novo heterozygous mutations in the SF3B4 gene. Am. J. Med. Genet. A 170, 3133–3137 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Lund I. C., Vestergaard E. M., Christensen R., Uldbjerg N., Becher N., Prenatal diagnosis of Nager syndrome in a 12-week-old fetus with a whole gene deletion of SF3B4 by chromosomal microarray. Eur. J. Med. Genet. 59, 48–51 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Marques F., et al. , Altered mRNA splicing, chondrocyte gene expression and abnormal skeletal development due to SF3B4 mutations in Rodriguez acrofacial dysostosis. PLoS Genet. 12, e1006307 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mohapatra A. P., Satpathy A., Athulya P. U., Das L., Mohapatra I., Nager Syndrome co-harboring mutation consistent with Stickler syndrome: A rare case report. Asian J. Pediatr. Res. 13, 75–80 (2023). [Google Scholar]
- 15.Ulhaq Z. S., Soraya G. V., Istifiani L. A., Pamungkas S. A., Tse W. K. F., SF3B4 frameshift variants represented a more severe clinical manifestation in nager syndrome. Cleft Palate Craniofacial J. 60, 1041–1047 (2023). [DOI] [PubMed] [Google Scholar]
- 16.Will C. L., Luhrmann R., Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 3, a003707 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Champion-Arnaud P., Reed R., The prespliceosome components SAP 49 and SAP 145 interact in a complex implicated in tethering U2 snRNP to the branch site. Genes Dev. 8, 1974–1983 (1994). [DOI] [PubMed] [Google Scholar]
- 18.Wickramasinghe V. O., et al. , Regulation of constitutive and alternative mRNA splicing across the human transcriptome by PRPF8 is determined by 5’ splice site strength. Genome Biol. 16, 201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olthof A. M., Hyatt K. C., Kanadia R. N., Minor intron splicing revisited: Identification of new minor intron-containing genes and tissue-dependent retention and alternative splicing of minor introns. BMC Genomics 20, 686 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ule J., Blencowe B. J., Alternative splicing regulatory networks: Functions, mechanisms, and evolution. Mol. Cell 76, 329–345 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Kumar S., et al. , Sf3b4 regulates chromatin remodeler splicing and Hox expression. Differentiation 131, 59–73 (2023). [DOI] [PubMed] [Google Scholar]
- 22.Yamada T., et al. , Heterozygous mutation of the splicing factor Sf3b4 affects development of the axial skeleton and forebrain in mouse. Dev. Dyn. 249, 622–635 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Lewis A. E., Vasudevan H. N., O’Neill A. K., Soriano P., Bush J. O., The widely used Wnt1-Cre transgene causes developmental phenotypes by ectopic activation of Wnt signaling. Dev. Biol. 379, 229–234 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen G., et al. , S pecific and spatial labeling of P0-Cre versus Wnt1-Cre in cranial neural crest in early mouse embryos. Genesis 55, e23034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alam S. S., et al. , Snrpb is required in murine neural crest cells for proper splicing and craniofacial morphogenesis. Dis. Model. Mech. 15 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beauchamp M. C., et al. , Mutation in Eftud2 causes craniofacial defects in mice via mis-splicing of Mdm2 and increased P53. Hum. Mol. Genet. 30, 739–757 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Devotta A., Juraver-Geslin H., Gonzalez J. A., Hong C. S., Saint-Jeannet J. P., Sf3b4-depleted Xenopus embryos: A model to study the pathogenesis of craniofacial defects in Nager syndrome. Dev. Biol. 415, 371–382 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGill, Standard Operating Procedure 301.05, Rodent Euthanasia. https://www.mcgill.ca/research/files/research/301-rodent_euthanasia-2024-06-17.pdf. Accessed 17 June 2024.
- 29.Méndez-Maldonado K., Vega-López G. A., Aybar M. J., Velasco I., Neurogenesis from neural crest cells: Molecular mechanisms in the formation of cranial nerves and ganglia. Front. Cell Dev. Biol. 8, 635 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kirby M. L., Waldo K. L., Neural crest and cardiovascular patterning. Circ. Res. 77, 211–215 (1995). [DOI] [PubMed] [Google Scholar]
- 31.Savolainen S. M., Foley J. F., Elmore S. A., Histology atlas of the developing mouse heart with emphasis on E11.5 to E18.5. Toxicol. Pathol. 37, 395–414 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muzumdar M. D., Tasic B., Miyamichi K., Li L., Luo L., A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 (2007). [DOI] [PubMed] [Google Scholar]
- 33.Schutte B., Nuydens R., Geerts H., Ramaekers F., Annexin V binding assay as a tool to measure apoptosis in differentiated neuronal cells. J. Neurosci. Methods 86, 63–69 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Simoes-Costa M., Bronner M. E., Establishing neural crest identity: A gene regulatory recipe. Development 142, 242–257 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bowen M. E., Attardi L. D., The role of p53 in developmental syndromes. J. Mol. Cell Biol. 11, 200–211 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calo E., et al. , Tissue-selective effects of nucleolar stress and rDNA damage in developmental disorders. Nature 554, 112–117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harper J. W., Adami G. R., Wei N., Keyomarsi K., Elledge S. J., The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75, 805–816 (1993). [DOI] [PubMed] [Google Scholar]
- 38.Tomasini R., et al. , TP53INP1s and homeodomain-interacting protein kinase-2 (HIPK2) are partners in regulating p53 activity. J. Biol. Chem. 278, 37722–37729 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Zhao L., et al. , Cyclin G1 has growth inhibitory activity linked to the ARF-Mdm2-p53 and pRb tumor suppressor pathways. Mol. Cancer Res. 1, 195–206 (2003). [PubMed] [Google Scholar]
- 40.Sakai D., Trainor P. A., Face off against ROS: Tcof1/Treacle safeguards neuroepithelial cells and progenitor neural crest cells from oxidative stress during craniofacial development. Dev., Growth Differ. 58, 577–585 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong J., Sulik K. K., Chen S.-Y., Nrf2-mediated transcriptional induction of antioxidant response in mouse embryos exposed to ethanol in vivo: Implications for the prevention of fetal alcohol spectrum disorders. Antioxidants Redox Signal. 10, 2023–2033 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beauchamp M. C., Boucher A., Dong Y., Aber R., Jerome-Majewska L. A., Craniofacial defects in embryos with homozygous deletion of Eftud2 in their neural crest cells are not rescued by Trp53 deletion. Int. J. Mol. Sci. 23, 9033 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cho E. A., Dressler G. R., TCF-4 binds β-catenin and is expressed in distinct regions of the embryonic brain and limbs. Mech. Dev. 77, 9–18 (1998). [DOI] [PubMed] [Google Scholar]
- 44.Weise A., et al. , Alternative splicing of Tcf7l2 transcripts generates protein variants with differential promoter-binding and transcriptional activation properties at Wnt/beta-catenin targets. Nucleic Acids Res. 38, 1964–1981 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wood K. A., et al. , Modelling the developmental spliceosomal craniofacial disorder Burn-McKeown syndrome using induced pluripotent stem cells. PLoS ONE 15, e0233582 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee Y. H., Saint-Jeannet J. P., Sox9 function in craniofacial development and disease. Genesis 49, 200–208 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ng L.-J., et al. , SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev. Biol. 183, 108–121 (1997). [DOI] [PubMed] [Google Scholar]
- 48.Zhao Q., Eberspaecher H., Lefebvre V., de Crombrugghe B., Parallel expression of Sox9 and Col2a1 in cells undergoing chondrogenesis. Dev. Dyn. 209, 377–386 (1997). [DOI] [PubMed] [Google Scholar]
- 49.McKeown S. J., Lee V. M., Bronner-Fraser M., Newgreen D. F., Farlie P. G., Sox10 overexpression induces neural crest-like cells from all dorsoventral levels of the neural tube but inhibits differentiation. Dev. Dyn. 233, 430–444 (2005). [DOI] [PubMed] [Google Scholar]
- 50.Pusch C., et al. , The SOX10/Sox10 gene from human and mouse: Sequence, expression, and transactivation by the encoded HMG domain transcription factor. Hum. Genet. 103, 115–123 (1998). [DOI] [PubMed] [Google Scholar]
- 51.Southard-Smith E. M., Kos L., Pavan W. J., Sox10 mutation disrupts neural crest development in Dom Hirschsprung mouse model. Nat. Genet. 18, 60–64 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Sundararajan V., Pang Q. Y., Choolani M., Huang R.Y.-J., Spotlight on the granules (Grainyhead-like proteins)—From an evolutionary conserved controller of epithelial trait to pioneering the chromatin landscape. Front. Mol. Biosci. 7, 213 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rogers C. D., Sorrells L. K., Bronner M. E., A catenin-dependent balance between N-cadherin and E-cadherin controls neuroectodermal cell fate choices. Mech. Dev. 152, 44–56 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Scarpa E., et al. , Cadherin switch during EMT in neural crest cells leads to contact inhibition of locomotion via repolarization of forces. Dev. Cell 34, 421–434 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Corvalan A. Z., Coller H. A., Methylation of histone 4’s lysine 20: A critical analysis of the state of the field. Physiol. Genomics 53, 22–32 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Evertts A. G., et al. , H4K20 methylation regulates quiescence and chromatin compaction. Mol. Biol. Cell 24, 3025–3037 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shoaib M., et al. , Histone H4 lysine 20 mono-methylation directly facilitates chromatin openness and promotes transcription of housekeeping genes. Nat. Commun. 12, 4800 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Taggart A. J., et al. , Large-scale analysis of branchpoint usage across species and cell lines. Genome Res. 27, 639–649 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zakariyah A., Hou W., Slim R., Jerome-Majewska L., TMED2/p24β1 is expressed in all gestational stages of human placentas and in choriocarcinoma cell lines. Placenta 33, 214–219 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Kumar S., Bareke E., Majewski J., Jerome-Majewska L., Data from “Investigating the Etiology of Craniofacial and Cardiac Malformations in a Mouse Model of SF3B4-Related Syndromes.” NCBI Gene Expression Omnibus. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE260990. Deposited 6 March 2004. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
RNAseq data have been deposited in the GEO (GSE260990) repository and are publicly available as of the date of publication (60).