

Abstract
Replicating poxviruses catalyze high-frequency recombination reactions by a process that is not well understood. Using transfected DNA substrates we show that these viruses probably use a single-strand annealing recombination mechanism. Plasmids carrying overlapping portions of a luciferase gene expression cassette and luciferase assays were first shown to provide an accurate method of assaying recombinant frequencies. We then transfected pairs of DNAs into virus-infected cells and monitored the efficiencies of linear-by-linear, linear-by-circle, and circle-by-circle recombination. These experiments showed that vaccinia virus recombination systems preferentially catalyze linear-by-linear reactions much more efficiently than circle-by-circle reactions and catalyze circle-by-circle reactions more efficiently than linear-by-circle reactions. Reactions involving linear substrates required surprisingly little sequence identity, with only 16-bp overlaps still permitting ∼4% recombinant production. Masking the homologies by adding unrelated DNA sequences to the ends of linear substrates inhibited recombination in a manner dependent upon the number of added sequences. Circular molecules were also recombined by replicating viruses but at frequencies 15- to 50-fold lower than are linear substrates. These results are consistent with mechanisms in which exonuclease or helicase processing of DNA ends permits the forming of recombinants through annealing of complementary single strands. Our data are not consistent with a model involving strand invasion reactions, because such reactions should favor mixtures of linear and circular substrates. We also noted that many of the reaction features seen in vivo were reproduced in a simple in vitro reaction requiring only purified vaccinia virus DNA polymerase, single-strand DNA binding protein, and pairs of linear substrates. The 3′-to-5′ exonuclease activity of poxviral DNA polymerases potentially catalyzes recombination in vivo.
Replicating poxviruses are subjected to extraordinarily high frequencies of homologous recombination while replicating in the cytoplasm of infected cells. When this fact is considered within our growing understanding of the links between repair, recombination, and replication (2, 5, 13), recombination reactions presumably provide viruses with several advantages. First, in the absence of any known poxvirus primase, recombination may play some role in priming DNA replication much as it does during T4 replication (8). Second, recombinational repair of double-stranded breaks is expected to enhance the survival of replicating viruses. Finally, recombinational processes provide a route whereby viruses might acquire new genes either by recombination with coinfecting viruses or by a strategy of gene capture from the host. This last process is poorly understood, yet it is presumed to have happened repeatedly during viral evolution, as evidenced by the still extensive similarities between many viral and host genes.
A number of studies have outlined the basic features of poxvirus recombination reactions using both viruses and DNAs transfected into virus-infected cells (1, 6, 19). Recombination occurs at such high frequencies that linkage is lost at distances exceeding ∼500 bp and exhibits the genetic phenomenon of high-negative interference due to the formation of large quantities of heteroduplex DNA (7, 15). Experiments using inhibitory drugs and conditional (temperature-sensitive) mutants have shown that virus recombination is absolutely dependent upon a functional virus-encoded DNA polymerase (6, 12, 24). These genetic studies have been more recently complemented by biochemical data showing that the DNA polymerase 3′-5′ exonuclease can catalyze joint molecule formation in vitro using a single-strand annealing (SSA) mechanism (25). While such a mechanism is fully consistent with all of the known features of poxvirus recombination in vivo, it remains to be proven that virus recombination depends partially or exclusively on such an exonuclease-based SSA process.
The recombination reaction catalyzed by vaccinia virus DNA polymerase in vitro assembles recombinants from linear duplex molecules sharing as little as 12 bp of end sequence identity (25). To test whether this reaction is of any relevance to the process occurring in virus-infected cells, we have examined what effect such double-stranded breaks and homology length have on the efficiency of poxvirus-mediated recombination reactions in vivo. Our results suggest that poxviral recombination systems can most efficiently utilize linear molecules capable of entering an SSA pathway of recombination while still being able to recombine circular DNAs by a second, and considerably less efficient, process. A striking difference between these two pathways is that recombination of linear molecules requires surprisingly little sequence homology. This observation may be of practical use when considering strategies for constructing recombinant poxviruses. It also provides new insights into the origins of spontaneous poxvirus mutations and into how poxviruses might periodically acquire new genes.
MATERIALS AND METHODS
Cells and virus culture.
Vaccinia virus (strain WR) was obtained from the American Type Culture Collection and cultured on BSC40 cells (E. Niles, State University of New York at Buffalo) in 60-mm-diameter dishes. Cells were cultured in Dulbecco's modified Eagle's medium (Gibco-BRL) supplemented with 5% fetal calf serum (Cansera), 1% nonessential amino acids, and antibiotic/antimycotic at 37°C in a 5% CO2 atmosphere.
DNA substrates.
The starting point for construction of the plasmids used in the experiments depicted by Fig. 1 was plasmid pRP406, which carries an intact firefly luciferase gene driven by a vaccinia virus P11K early/late promoter and two derivatives, pRP406Δ and pRP403Δ, which encode upstream and downstream portions of the gene, respectively (16). pRP406Δ and pRP403Δ share 366 bp of overlapping sequence identity in the center of the luciferase open reading frame which permits recombinational reconstruction of a functional gene. A series of upstream PCR primers in conjunction with a common downstream primer carrying the 3′ end of the luciferase gene, PCR, and a pRP406 template were used to clone downstream portions of the gene in pCR2.1 TOPO (Invitrogen). The resulting plasmids were named pXYT403Δ20, pXYT403Δ50, pXYT403Δ100, and pXYT403Δ200 to indicate they share 20 to 200 bp of identity with the 3′ end of the luciferase gene fragment in pRP406Δ. All of the primer sequences are listed in Table 1.
FIG. 1.

Enzymatic detection of plasmid-by-plasmid recombination in vaccinia virus-infected cells. Plasmids carrying overlapping portions of a firefly luciferase gene under P11K promoter control were cotransfected into vaccinia virus-infected cells, and the recombinant frequency was calculated relative to the amount of luciferase detected in cells transfected with an intact luciferase gene (see Materials and Methods). Plasmids pXYT403Δ20 to pXYT403Δ200 share 0 to 200 bp of sequence identity with the promoter bearing the upstream portion of the luciferase gene in pRP406Δ, while pRP403Δ shares 366 bp of sequence identity with plasmid pRP406Δ. Less than 0.001% luciferase activity was detected in cells transfected with pRP406Δ or pRP403Δ alone relative to cells transfected with the intact gene. A β-galactosidase-encoding control plasmid was used in all transfections to normalize for dish-to-dish variations in transfection and infection efficiency. Error bars indicate standard deviations measured in five independent experiments.
TABLE 1.
Primers, plasmids, and PCR products used in this study
| Primer pair (5′-3′)a | PCR productb | Plasmid |
|---|---|---|
| GACCGCTTGAAGTCTTTAAT | 403Δ20 | pXYT403Δ20 |
| GGATCCGATTACAATAGCTAAG | ||
| TGGGACGAAGACGAACACTT | 403Δ50 | pXYT403Δ50 |
| GGATCCGATTACAATAGCTAAG | ||
| ACGCCTTGATTGACAAGGAT | 403Δ100 | pXYT403Δ100 |
| GGATCCGATTACAATAGCTAAG | ||
| CTGGATACCGGGAAACGCT | 403Δ200 | pXYT403Δ200 |
| GGATCCGATTACAATAGCTAAG | ||
| CTAAGGGTGTGGCCCTTCCG | LucM0 | |
| TAAAGACTTCAAGCGGTCAA | ||
| TGGGTTACCTAAGGGTGTGGCCTT | LucM8 | |
| TATTTAATAAAGACTTCAAGCGGTC | ||
| ACTGGGTTACCTAAGGGTGTGGCCC | LucM10 | |
| TGTATTTAATTAAAGACTTCAAGCGG | ||
| CTACTGGGTTACCTAAGGGTGTGGC | LucM12 | |
| TTTGTATTTAATTAAAGACTTCAAGC | ||
| ATCTACTGGGTTACCTAAGGGTGTG | LucM14 | |
| CCTTTGTATTTAATTAAAGACTTCAAGC | ||
| GGATCTACTGGGTTACCTAAGGGTG | LucM16 | |
| ATCCTTTGTATTTAATTAAAGACTTCAAGC | ||
| CTGGATCTACTGGGTTACCTAAGGG | LucM18 | |
| ATATCCTTTGTATTTAATTAAAGACTTCAAGC | ||
| CTCTGGATCTACTGGGTTACCTAAG | LucM20 | pXYTLucM20 |
| TGATATCCTTTGTATTTAATTAAAGACTTCAAGC | ||
| TGACAAAACAATTGCACTGATAATG | LucM50 | |
| AATATCGATTCCAATTCAGCGGGG | ||
| CTACCTCCCGGTTTTAATG | LucM100 | |
| CGTCGGGAAGACCTGCCACG | ||
| CGTGCAAAAAAAATTACCAATAA | LucM200 | |
| GTTGTTACTTGACTGGCGACG | ||
| GGCGCGTTATTTATCGGAGTTGCAG | LucM333 | pXYTLucM333 |
| TTTTACAATTTGGACTTTCCGCCCTTC | ||
| GGATCTACTGGGTTACCAGAAACAGGCATCTTACGCGTG | 826-bp SFV | pRP406-S800 |
| ATCCTTTGTATTTAATTAAACGGAAACGCCTTGGT | ||
| GGATCTACTGGGTTACCTCGTTGTCTGAGCTGCCG | 1,621-bp SFV | pRR406-S1600 |
| ATCCTTTGTATTTAATTAAACGGAAACGCCTTGG |
Underlined nucleotides overlap with the vector.
SFV, Shope fibroma virus.
For observation of recombination both in vivo and in vitro (see Fig. 3, 4, and 5) we constructed two additional sets of DNA substrates. A linear vector portion (pRP406/BstEII+PacI) was prepared by cutting pRP406 with BstEII and PacI to excise the middle portion of the luciferase gene followed by gel purification of the larger fragment. Linear inserts were prepared using PCR, a series of 12 primer pairs (Table 1), and a pRP406 template. These inserts spanned the BstEII-PacI gap in the luciferase gene, and primer placement ensured each end carried between 0 and 333 bp of terminal sequences identical to sequences located at the ends of the pRP406/BstEII+PacI substrate. Before use, all PCR products were gel purified and quantitated by UV spectrometry.
FIG. 3.

Recombination between linear molecules sharing different lengths of overlapping end sequence identity in vaccinia virus-infected cells. Each of 12 different PCR-amplified inserts (LucM0 to LucM333) plus a cut vector (pRP406/BstEII+PacI) were cotransfected into vaccinia virus-infected cells and the recombinant frequencies were calculated in two separate experiments as described in Materials and Methods. The cross is shown inset, with gray boxes indicating the luciferase open reading frame. Control experiments detected 0.055 and 0.035% luciferase activity in cells transfected with just LucM333 or pRP406/BstEII+PacI DNAs, respectively, relative to that of cells transfected with the intact gene. Results from separate replicate experiments are shown.
FIG. 4.

DNA polymerase catalyzed joining of linear molecules sharing different lengths of overlapping end homology. In vitro reactions contained PCR-amplified insert DNA (LucM0 to LucM333, labeled A), cut vector (pRP406/BstEII+PacI, labeled B), vaccinia virus single-strand DNA binding protein, and vaccinia virus DNA polymerase. After a 20-min incubation at 37°C the deproteinized reaction mixture was fractionated on a 0.8% agarose gel and the DNA was visualized by staining with ethidium bromide. Control reaction mixtures omitted the DNA polymerase (lane 2), the vector (lane 3), or the insert (lane 4). Size markers included circular (Form I and II) and linear (Form III) pRP406 species (lanes 17 and 18, respectively) and a 1-kbp DNA ladder. Shown inset are the proposed identities of various molecules.
FIG. 5.

In vivo recombination between circular DNAs bearing two different regions of luciferase homology. Vaccinia virus-infected cells were cotransfected with the indicated plasmids and recombinant frequencies were calculated as described in Materials and Methods. Plasmids pRP406-S800 and pRP406-S1600 differ in that the stuffer sequences separating the two N = 20-bp and two N = 333-bp homologies are 787 and 1,621 bp long, respectively. Results obtained from three separate experiments are plotted separately to illustrate the experimental variation.
Two of the PCR-amplified inserts, bearing 20 and 333 bp of terminal identity, were subsequently cloned using pCR2.1 TOPO vectors to create plasmids pXYTLucM20 and pXYTLucM333, respectively (see Fig. 5). These plasmids were used in conjunction with a third type of substrate produced using the cloning method that is the subject of this and previous investigations (25). Briefly, bipartite PCR primers (Table 1) were synthesized that were capable of amplifying 787- or 1,621-bp fragments of “stuffer” (Shope fibroma virus) DNA, but which added 16-bp 5′ ends homologous to the termini of BstEII-PacI-cut pRP406. The PCR-amplified inserts and cut vector were incubated with vaccinia virus polymerase at 37°C for 10 min (see below), and one tenth of the reaction mixture was transfected into Escherichia coli SURE cells (Invitrogen). The resulting recombinants were named pRP406-S800 and pRP406-S1600 (see Fig. 5 and 6). These and other plasmids were purified using polyethylene glycol and phenol (17) or commercial columns (Qiagen).
FIG. 6.

In vivo recombination between circular and linear DNAs bearing two different regions of luciferase homology. The experiments were identical to those shown in Fig. 5, except that linear DNA LucM20 or LucM333 was used instead of plasmid clones encoding these same DNAs (inset). The data are plotted on the same scale as that for Fig. 5 to facilitate direct comparison.
Sequence-tagged (mismatched) substrates.
Plasmid pBluescriptII-λ was constructed by cloning a 499-bp fragment of SalI-cut λ DNA into SalI-digested pBluescriptII KS (Invitrogen). The insert was excised with XhoI and HindIII, gel purified, and incubated with T4 DNA ligase, ATP, and a 10-fold molar excess of two mismatch-containing left and right adapter duplexes containing T · G and T · C mismatches (in bold), respectively: GCTTGGACCTCCACAGCCCCTpAGCTGGGAACGCGGTGCGGTCATC CGAACCTGGGGGTGTCGGGGAAGCTpCCCTTGCGCCCCGCCAGTAG
The resulting ligation products, or inserts, were gel purified and ethanol precipitated. Sequence-tagged vector molecules were prepared in a related manner. Plasmid pBluescriptII KS was digested with XhoI and XbaI, gel purified to remove the linker fragment, and incubated with T4 DNA ligase, ATP, and a 10-fold molar excess of A · C and A · G mismatch-containing (in bold) adapter duplex: pTCGAAGCTTGGACCACCACAGCCCCGGGAACGCGGAGCGGTCATCCT TCGAACCTGGCGGTGTCGGGGCCCTTGCGCCGCGCCAGTAGGAGATCp
The resulting circular ligation products were gel purified and cut with SmaI (underlined), and linear products were repurified by gel electrophoresis.
Vaccinia virus DNA polymerase and single-strand DNA binding protein.
Vaccinia virus DNA polymerase was prepared from cells infected with vTF7.5 and vTMPOL viruses (11, 24), and recombinant vaccinia virus gpI3L was prepared from E. coli (23).
In vitro recombination assay.
The reaction mixture for the in vitro recombination assay contained 30 mM Tris-HCl (pH 7.9), 5 mM MgCl2, 70 mM NaCl, 1.8 mM dithiothreitol, 80 μg of acetylated bovine serum albumin/ml, 250 ng (0.1 pmol) of vector DNA, 190 ng (0.4 pmol) of PCR-amplified insert DNA, 25 μg of vaccinia virus single-stranded DNA binding protein/ml, and 0.2 μg of vaccinia virus DNA polymerase in 20 μl (25). The reaction mixture was incubated at 37°C for 20 min and deproteinized by incubation in 0.1% sodium dodecyl sulfate, 0.2 mg of proteinase K/ml, and 50 mM EDTA at 37°C for 20 min. Joint molecules were separated using 0.8% agarose gels.
Transfection-based recombination assay.
The luciferase-based recombination assay has been described in detail elsewhere (16, 24). Briefly, 50-ng quantities of each DNA carrying different overlapping portions of the luciferase gene plus 50 ng of a plasmid encoding β-galactosidase (pRP7.5lacZ) were cotransfected with calcium phosphate, 2 h postinfection, into cells infected with vaccinia virus at a multiplicity of infection of 2. Protein extracts were prepared 5 h later. Luciferase and β-galactosidase levels were measured with a luminometer using luciferase (Promega) and luminescent β-galactosidase (Clontech) detection kits. Variations in infection and transfection efficiencies were corrected by normalizing luciferase expression levels against β-galactosidase levels, and the recombinant frequency (Rf) was calculated relative to the amount of luciferase detected in cells transfected in parallel with an intact luciferase gene (pRP406):
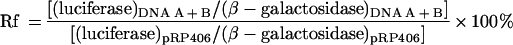 |
Each measurement was an average calculated from duplicate dishes, and the experiments were performed three to five times with essentially identical results.
To determine the sequence of recombinant junctions we used a method described in greater detail elsewhere (6, 25). Briefly, DNA was extracted from vaccinia virus-infected cells 20 h after transfection with a mixture containing 100 ng of each sequence-tagged (mismatched) linear DNA substrate. The resulting mixture of cellular, viral, and transfected DNA was cut with SacI and DpnI (to degrade unreplicated input DNA), recircularized at low DNA concentrations using ATP and T4 DNA ligase, and used to transform E. coli BMH71-18 mutS cells (Clontech) to ampicillin resistance. Recombinant plasmids were identified by restriction analysis, and both junction sequences were determined by automated sequencing.
Southern blotting.
Sixty-millimeter-diameter dishes of BSC40 cells were infected for 2 h at a multiplicity of infection of 5 and then were transfected with 200 ng of pRP406Δ plus 200 ng of pXYT403Δ20, pXYT403Δ50, pXYT403Δ100, pXYT403Δ200, or pRP403Δ. The DNA was extracted 20 h posttransfection (26), cleaved with SpeI, XhoI, and DpnI, sized on a 1.2% agarose gel, and transferred to a nylon membrane (Bio-Rad). The DNA was hybridized to an [α-32P]dCTP-labeled luciferase gene probe and exposed to film, and the band intensities were determined by densitometry.
RESULTS
Effect of homology length on vaccinia virus-mediated recombination.
Previous studies have shown that poxvirus-infected cells recombine transfected plasmids (6) in a reaction that is most easily monitored using a luciferase reporter gene (16). However, little is known about the homology requirements in these reactions beyond the fact that 366 bp of common sequence is sufficient to permit efficient recombination between plasmids bearing overlapping portions of the luciferase gene (16, 24). To better define the minimal length of sequence identity required by poxviral recombination systems, we prepared a series of additional plasmids encoding the C terminus of the luciferase protein and sharing 20, 50, 100, or 200 bp of sequence identity with a plasmid encoding the N terminus of the gene product (pRP406Δ). Supercoiled plasmids were then transfected into vaccinia virus-infected cells, and luciferase expression levels were used to estimate the frequency of recombination. The results of these experiments are shown in Fig. 1. These assays are very sensitive, making it possible to detect as little as 0.1% recombination when plasmids shared only 50 bp of homologous sequence in the overlapping portion of the luciferase gene. But the production of substantial numbers of recombinants needed more extensive homologies, with 366-bp overlaps being required to generate ∼8% recombinant production (Fig. 1).
There is always some concern that enzyme-based assays might not accurately reflect the frequencies of recombination at the DNA level. Therefore, we also extracted DNA from virus-infected cells and used Southern blots to search for the presence of recombinant restriction fragments. These experiments are not as sensitive as luciferase-based assays, but we still detected recombinant products among the DNAs extracted from cells transfected with molecules sharing ≥100 bp of sequence identity (Fig. 2, inset). We also detected comparable yields of recombinant fragments (within a factor of about two), showing that the enzyme-based assays provide a simpler yet still reliable measure of recombination frequency at the DNA level. Collectively these results showed that the frequency of plasmid-by-plasmid recombination reactions depends upon the length of homology between recombining molecules. Under these circumstances at least 50, and preferably over 100, base pairs of sequence identity are required to permit production of substantial amounts of recombinants.
FIG. 2.

Southern blot analysis of recombinant molecules recovered from vaccinia virus-infected cells. DNA was recovered from cells transfected with the luciferase-encoding plasmids indicated in the legend to Fig. 1, digested with SpeI, XhoI, and DpnI (to degrade unreplicated input DNA), and size fractionated. After transfer to a nylon membrane, plasmid sequences were detected with a 32P-labeled luciferase probe and the yield of 1.7-kbp recombinant molecules (indicated by the arrow) was determined by densitometry. Cells were also transfected with a plasmid carrying an intact luciferase gene (lane 8), and the recombinant frequencies were calculated relative to the intensity of the 1.7-kbp full-length luciferase-encoding fragment seen in that lane. No recombinant DNA was detected in cells transfected with pRP406Δ or pRP403Δ alone (lanes 1 and 2, respectively). Note that different vectors were used to construct pXYT403Δ20 to pXYT403Δ200 (lanes 3 through 6) versus those for pRP403Δ (lanes 2 and 7). Recombinant frequencies can still be calculated because the luciferase-encoding recombinant fragments are identical, but differences in flanking vector sequences produce other changes in the restriction patterns.
Poxvirus recombination systems most efficiently recombine linear molecules.
To examine the effect of double-stranded breaks on this recombination system we devised a second set of luciferase-based recombination substrates. Plasmid pRP406 was digested with BstEII and PacI to excise 718 bp of DNA from the middle of the luciferase open reading frame. Then a series of PCR products were prepared which spanned the gap and had ends extending variable distances into the DNA sequences flanking the BstEII and PacI cut sites. These DNAs were all gel purified and then used as recombination substrates both in vivo (Fig. 3) and in vitro (Fig. 4).
When these DNAs were separately transfected into virus-infected cells, no significant luciferase activity was detected (<0.05% of the luciferase measured in pRP406-transfected cells). However, cotransfecting the promoter-encoding vector plus different PCR-amplified inserts into infected cells produced substantial numbers of recombinants with an efficiency dependent upon the extent of homology. Very little sequence identity was needed to still permit recombinant formation under these conditions. Even with substrates sharing no apparent homologous end sequences, 0.5% recombinants were detected (∼10-fold over background), and this number increased to 2.5 or 4% when molecules overlapped by 14 or 16 bp, respectively (Fig. 3). Substrates sharing over 100 bp of end sequence identity produced recombinants at frequencies ranging from 34 to 61% (Fig. 3). There was some evidence for at least one peak in the frequency plot between 16 and 18 bp, followed by a substantial increase in yield when the overlap exceeded ∼50 bp.
We also tested whether these molecules could be used as substrates in an in vitro recombination reaction catalyzed by vaccinia virus DNA polymerase and single-strand DNA binding protein (24). Gel electrophoresis was used to monitor the yield of joint molecules (Fig. 4). We saw a good correlation between the efficiency of joint molecule formation in vitro and the yield of recombinants detected in vivo, particularly with molecules sharing shorter overlaps (0 to 100 bp). Little or no joint molecule formation was detected with molecules sharing less than 10 bp of end sequence identity (Fig. 4, lanes 5 to 7). Molecules sharing 12 or 14 bp of sequence identity produced linear dimers, those sharing 16 bp homologies were fused into a mixture of linear dimers and dimeric circles, and those sharing 18 to 100 bp of sequence identity were converted primarily into nicked circles (Fig. 4, lanes 8 to 14). The situation was more complicated with substrates sharing 200- to 333-bp homologies. A mixture of what appeared to be linear and circular dimers were formed in reactions containing the 200- and 333-bp overlap substrates (lanes 15 and 16), which seemed to migrate slightly slower than what should be identical products seen in other reactions (e.g., lanes 8 and 10). Generally the results show that the minimal extent of homology required to produce recombinants in vitro is about the same as that seen in vivo (∼12 bp). Moreover, the local yield optimum seen with substrates sharing 16 to 18 bp of sequence identity in vivo (Fig. 3) correlates well with high-efficiency production of circular molecules in vitro (Fig. 4, lane 11). The in vitro system did not reproduce the in vivo situation well when molecules shared long patches of sequence identity.
Other effects of DNA structure on recombination frequency.
The preceding experiments illustrate a profound difference in the recombinogenicity of linear versus circular molecules in vaccinia virus-infected cells. For example, even though the events illustrated in Fig. 3 require two exchanges to produce a recombinant molecule, they are up to 30 times more frequent (with 200-bp sequence identity) than when circular molecules requiring only a single exchange are being recombined. However, these differences in the number of required exchanges complicates the interpretation of such experiments. To address this concern we created circular versions of the pRP406/BstEII+PacI and PCR-amplified substrates (Fig. 5) and then examined how plasmids requiring two exchanges would be recombined in virus-infected cells. Because it was unclear what effect sequence context and spatial geometry might have on this reaction, the pRP406/BstEII+PacI substrate was converted to circular molecules by inserting 787- or 1,621-bp nonhomologous stuffer fragments into the 718-bp interval once occupied by luciferase gene sequences. These plasmids were designated pRP406-S800 and pRP406-S1600, respectively. PCR products sharing 20 or 333 bp of sequence identity with portions of the luciferase gene remaining upstream of the BstEII and downstream of the PacI sites were circularized and propagated by being cloned into a commercial vector (pCR2.1 Topo). The resulting plasmids were designated pXYTLucM20 and pXYTLucM333.
In the first of these new experiments we examined the frequency of recombination between circular molecules (Fig. 5). Molecules sharing only 20 bp of sequence identity on each side of the stuffer sequence were essentially unreactive under these conditions, generating no significant luciferase activity above a background measured using separately transfected plasmids (0.0 to 0.1% recombination). However, increasing the length of sequence identity to 333 bp on each side of the insert substantially increased the recombination frequency. We detected 1.0% ± 0.1% recombination with homologies separated by a 1,621-bp stuffer insert and 3.1% ± 0.8% recombination with a 787-bp insert. Estimating that just one 333-bp patch of homology should permit about 6.6% recombination (from a polynomial curve fit to the results shown in Fig. 1, r2 = 1.0), the value of 3.1% ± 0.8% is significantly higher than one would have predicted on the assumption that recombinants are a product of two independent events (6.6% × 6.6% = 0.4%). When the distance between the two patches of sequence identity was increased from 787 (pRP406-S800) to 1,621 bp (pRP406-S1600), the recombination frequency decreased to a value much closer to that predicted assuming circle-by-circle recombinants are being produced by two independent recombination reactions.
We repeated these experiments using one circular and one linear substrate (Fig. 6). The recombining circular DNAs remained the same as those used in the experiment illustrated by Fig. 5 (pRP406-S800 and pRP406-S1600), while the linear DNAs were purified PCR products unencumbered by flanking vector sequences. When the linear substrates bore 333 nucleotides (nt) of luciferase homology at each end of the molecule (Table 1, LucM333), we noted a sixfold reduction in the frequency of recombination with pRP406-S800 versus that observed using pXYTLucM333-by-pRP406-S800. The reduction is even more substantial (∼100-fold) when compared with the 49% recombination frequency observed using LucM333 and pRP406/BstEII+PacI (Fig. 4). A smaller but still substantial reduction of 2.5-fold was observed using the LucM333 linear product and pRP406-S1600 relative to the frequency of pXYTLucM333-by-pRP406-S1600 recombination. The very low frequencies of recombination observed using 20-nt homologies prevents us from quantifying the effect of using one linear substrate on recombination frequencies. It was nevertheless clear that recombination is not stimulated under these conditions. Poxviruses catalyze circle-by-circle, linear-by-linear, and circle-by-linear recombination, but the last type of event is the least favored.
Effect of nonhomologous border sequences on linear-by-linear recombination.
Digesting the plasmids pRP406-S800 and pRP406-S1600 with MluI generates linear substrates in which the luciferase homology is embedded within various amounts of unrelated border sequences. For example, cutting pRP406-S800 with MluI leaves the upstream end of the luciferase gene homology only 17 bp from the closest end of the DNA, while the 3′ end of the luciferase gene remains buried 780 bp from the MluI cut site. Cutting pRP406-S1600 with MluI leaves the upstream and downstream portions of the luciferase gene homology 811 and 780 bp from the cut site, respectively. We transfected these MluI-cut DNAs into virus-infected cells along with the linear substrates LucM20 or LucM333 and measured the recombinant frequency. The results are shown in Fig. 7. For comparison purposes we have also replotted the recombinant frequencies that were seen when the luciferase homologies were located immediately adjacent to the two vector ends in pRP406 cut with BstEII and PacI (Fig. 3). Under these circumstances, the only nonhomologous sequences would be some single 3′-terminal deoxyadenosines added by Taq DNA polymerase.
FIG. 7.

In vivo recombination between linear molecules bearing nonhomologous termini. Vaccinia virus-infected cells were cotransfected with the indicated DNAs and the recombinant frequencies were calculated as described in Materials and Methods. Plasmids pRP406-S800 and pRP406-S1600 were cut with MluI. This leaves one or two ∼800-bp patches of sequence at the DNA end(s) which is unrelated to the luciferase gene (inset). The added DNA partially or completely sequesters the 20 (closed circles)- and 333 (open circles)-bp sequences which are homologous to the luciferase gene fragments carried by LucM20 and LucM333, respectively. Error bars indicate standard deviations measured in three independent experiments.
The results show that embedding homologous DNAs within extensive regions of sequence nonhomology greatly inhibits linear-by-linear recombination. When the luciferase homology spanned 333 bp on each side of the break, a single 780-bp block of DNA inhibited recombination 4.4-fold and inhibited two of these blocks 16-fold relative to the frequency measured with no interfering sequences. Similar inhibitory effects seem to be seen when only 20-base homologies are available for recombination, being reduced from 3.7 to 0.25% recombination by a single 780-bp block of DNA (∼15-fold) and becoming undetectable by our methods (<0.05%) when the 20-base homologies are masked by two such blocks of DNA. The high frequency of recombination that is permitted when recombining sequences are located at the ends of DNA would seem to be inhibited by the presence of masking stretches of nonhomologous DNA.
Preferential loss of 3′ nucleotides accompanies linear-by-linear recombination.
The preceding observations are most simply explained by schemes in which poxviruses use the DNA polymerase-encoded 3′ to 5′ exonuclease to promote SSA reactions (see Discussion). This would predict that, whereas all other SSA reactions described to date involve 5′-to-3′ DNA degradation, poxvirus reactions should exhibit the opposite polarity. That is, sequences located on the 3′ ends of linear substrates should be preferentially lost during recombination. To test this novel prediction, we ligated mismatch-containing 20-mer oligonucleotide duplexes to the ends of linear DNAs and purified the resulting products. Both of the inserts and both of the vector ends were modified in this manner. This process tags the four strands located at each duplex end with homologous sequences that can all be differentiated by a single base substitution (Table 2). Plasmids were recovered from bacteria transformed with in vitro- or in vivo-generated recombinant molecules and sequenced to determine which end(s) survived these reactions. Control experiments showed that 100% (12 out of 12) of the recombinant junctions formed in vitro retained sequences originally located on the 5′-ended strands (3′ to 5′ degradation as first shown using 32P labels [25]), confirming the validity of the approach. Such an extreme bias was not detected in molecules recovered from transfected cells, but a strong preference for 3′ to 5′ degradation was nevertheless noted. Of 32 sequence-tagged junctions analyzed in 16 recombinant plasmids (mismatches were located at both recombining ends), 75% of junction sequences were those predicted to arise through reactions involving 3′ to 5′ degradation of duplex ends (Table 2). Chi-squared analysis showed that these results reflect a statistically significant bias in favor of 3′ to 5′ events. (The probability that the observed ratio reflects an underlying 1:1 ratio is less than 1%.) Further experiments are required to investigate the possible impact of hypothetical cytoplasmic mismatch repair systems, but the peculiar polarity of poxviral recombination reactions lends credence to the hypothesis that at least some of the recombinants formed in vivo arise through resection of 3′ ends.
TABLE 2.
Sequence of recombinant junctions in molecules recovered from cells transfected with linear substrates encoding sequence-tagged (mismatched) terminia
| Sequence-tagged mismatches at homologous endsb | Fraction of recovered molecules with sequence Xc for nucleotide:
|
Predicted effect of resection polarity on junction sequence
|
||||
|---|---|---|---|---|---|---|
| G | A | T | C | 3′ to 5′ | 5′ to 3′ | |
| Left end | ||||||
 |
5/16 | 3/16 | 6/16 | 2/16 | G or T | A or C |
| Right end | ||||||
 |
5/16 | 8/16 | 1/16 | 2/16 | G or A | T or C |
Duplex oligonucleotides were ligated to all four ends of two linear recombination substrates, adding 20 nt of sequence identity to each end interrupted by a single, central, mismatched base pair. These substrates were transfected into virus-infected cells (Fig. 3), and recombinants were recovered in E. coli (6) and sequenced (25).
Diagrams illustrate the base substitutions used to differentiate all eight 3′- and 5′-ended strands. The left end of the linear insert is identical to the vector end shown immediately below it, except that T · G and A · C mismatches differentiate insert from vector ends. T · C and A · G mismatches similarly differentiate the right end of the insert from its vector target. See Materials and Methods for sequence details.
Top-strand sequence (as drawn) at variant site.
DISCUSSION
Our data suggest that a poxviral recombination pathway exists which can very efficiently recombine pairs of linear substrates but has great difficulty recombining pairs of circular molecules and even more problems using one linear and one circular partner.
The recombination of linear molecules bearing shared end homologies is readily explained using an SSA recombination model (Fig. 8). This is a well-documented process that has been detected in many biological systems, including phage-infected E. coli (20, 22), yeast (21), and human cells (9, 10). As the name suggests, SSA reactions generate recombinants through annealing of complementary single-stranded DNAs. These single-stranded ends can hybridize spontaneously or in protein-enhanced reactions and are most simply generated by helicases or exonucleases acting upon DNA ends. That vaccinia virus recombination systems require duplex ends is suggested by the observations that circles are poor substrates and that adding ∼800 bp of nonhomologous sequence to one or two ends inhibited recombination 4- and 16-fold, respectively (Fig. 7). Such terminal sequences would interfere with exonucleases or helicases attempting to expose complementary sequences on two interacting molecules. While SSA is not an unusual reaction, it is remarkable how little sequence identity is required by poxviral recombination systems in vivo. The SSA pathway in Saccharomyces cerevisiae repairs a single double-stranded break flanked by 29-bp homologies with ∼0.2% efficiency, and repair efficiency increases linearly up to 100% with 415-bp homologies (21). This is bettered by vaccinia virus-infected cells, where repair of two such breaks can still be detected with substrates sharing as little as 12 to 14 bp of sequence identity on each end of the DNA (0.8 to 2.5%), and only 18 bp of sequence identity permits up to 8% recombinant production.
FIG. 8.

Recombination reactions catalyzed by vaccinia virus in vivo and in vitro. Pathway A efficiently joins linear molecules sharing ∼12 bp of end sequence identity and can be detected both in vivo (Fig. 3) and in vitro (Fig. 4). It probably involves a simple exonuclease-catalyzed SSA reaction which may well be catalyzed in vivo as it is in vitro by vaccinia virus DNA polymerase. Pathway B could catalyze circle-by-circle recombination reactions if the substrates share >50-bp homologies (Fig. 1 and 5) and is based on the proposal that viruses use a rolling-hairpin replication scheme (14). Pathway C illustrates a strand invasion reaction. Strand invasion reactions efficiently catalyze linear-by-circular recombination, but because this reaction is so inefficient in transfected cells (Fig. 6) we suggest that pathway C is not used by replicating poxviruses.
A characteristic property of yeast SSA reactions is that they promote deletions of sequences flanked by direct repeats. That this is a feature of poxvirus mutational processes has been suggested by several earlier studies. In particular, Shchelkunov and Totmenin (18) compared the DNA sequences of vaccinia and variola viruses and noted that deletion mutations seem to preferentially occur at sites encoding 3- to 21-bp direct repeats. They further suggested that a virus-encoded enzyme might be catalyzing these events. The reactions we have characterized in this study are clearly compatible with their observations garnered through a bioinformatics-based approach. Moreover, their work suggests that the reactions which we have characterized using transfected DNAs can probably also modify viral genomes.
Whether these in vivo reactions are catalyzed by vaccinia virus DNA polymerase cannot be established with certainty by our experiments (Fig. 3 and 4 and Table 2). Genetic studies have clearly established that recombination requires a functional polymerase (24), and it is noteworthy that the minimal amount of sequence identity required to produce joint molecules in vitro is almost the same as that seen in vivo (≥12 bp). There is also a local reaction optimum in vivo at ∼18 bp (Fig. 3), which is again nearly the same as the in vitro optima of 16 (reported previously using different DNAs [25]) or 18 bp (Fig. 4). However, these features may have more to do with DNA stability and annealing kinetics than reflecting commonalities between the enzymes used both in vivo and in vitro. We also noted that the in vitro reaction rather poorly joined substrates bearing 333-bp homologies, while such molecules are efficiently recombined in vivo (Fig. 3 and 4). This discrepancy could be a simple in vitro artifact caused by the fact that a 20-min reaction might not provide enough time to expose complementary sequences, or it may be evidence that another enzyme(s) joins molecules in vivo. If another enzyme(s) does catalyze viral recombination, the fact that 75% of the recombinants recovered from cells transfected with sequence-tagged molecules showed evidence of 3′-end resection (Table 2) suggests that such enzymes would, like the polymerase, exhibit 3′ to 5′ exonuclease activities.
Recombination of circular molecules in virus-infected cells cannot be explained by a simple exonuclease or helicase catalyzed reaction. However, a variant of the SSA process could still account for our observations if it is remembered that circular molecules are also being replicated in trans in poxvirus-infected cells (3). It is not clear how poxviruses do this, but the rolling-hairpin reactions that probably replicate viral genomes (14) would, using plasmid templates, create rolling circles. SSA reactions could hybridize the displaced complementary strands which might then be processed into mature recombinants (Fig. 8). Note that linear molecules might not effectively exploit such a recombinational pathway, because only one single-stranded molecule would be produced per initiation event, whereas multiple DNA copies are produced by rolling circle reactions. The fact that circle-by-circle recombination reactions are significantly less efficient and require longer homologies than do linear-by-linear reactions may reflect difficulties associated with initiating replication on nonviral templates (4). There may also be difficulties caused by competition between recombination reactions (which require single-stranded substrates) and the replication reactions which sequester single-stranded DNA in a double-stranded form. Some support for the latter idea can be drawn from the behavior of homologous DNAs separated by stuffer sequences. Sequences separated by 787-bp inserts recombined at frequencies eightfold higher than those predicted if recombinants are a product of two independent events, whereas 1,621 bp seems to be approaching a distance that permits independent recombination. One interpretation of this observation might be that ∼800 nt of separation is short enough to allow formation of hybrid duplexes spanning both homologies (thus initiating formation of a recombinant in a single step), while greater distances provide sufficient time for replication to sequester one of the homologies as duplex DNA.
In both situations we favor an SSA mechanism of recombination in preference to a strand invasion reaction of the type catalyzed by RecA-like proteins. Certainly no such enzyme seems to be encoded by any known poxvirus. Furthermore, mixtures of linear and circular DNAs are the most poorly recombined substrates of the three structural combinations tested (Fig. 6), while this combination of DNAs provides good substrates where invasive recombination reactions are active (for example, see reference 20).
One last point seems worthy of note, and that is the remarkable ability of poxviral enzymes to generate recombinants under seemingly unfavorable circumstances. As little as 12 bp of sequence identity on each side of a double-stranded break was sufficient to permit 0.8% recombination between linear molecules (Fig. 3), and even when one of two 20-bp patches of sequence identity was obscured by an ∼800-bp nonhomology, we still detected 0.3% recombination (Fig. 7). This suggests a simple scenario that might explain how replicating poxviruses have acquired host gene homologs. Reverse-transcribed host cDNAs would often bear a poly(dA)-poly(dT) end homologous to the sequences comprising poxviral promoters, and this could provide one of the two homologies needed for recombination with an accidentally broken virus genome. Such events would very likely disrupt existing genes, and this might also explain why host- and virus-specific pathogenes seem to be acquired and/or located within terminal-inverted (i.e., diploid) repeats. We are presently examining the effects of nucleotide substitutions on linear-by-linear recombination, because 16- to 20-bp patches of perfect sequence identity are still statistically rare features and mismatches must be accommodated if this scheme is to work. The experiments outlined in Table 2 showed that vaccinia virus recombination systems tolerate single base substitutions within 20-bp-long end homologies, and other experiments show that two mismatches can also be accommodated by these systems (X.-D. Yao, unpublished data). This accumulating evidence suggests that even base mismatches may not suffice to inhibit poxvirus gene acquisition strategies.
ACKNOWLEDGMENTS
We thank A. Hilliker for his advice on statistics.
This work was supported by an award from the Canadian Institutes for Health Research and salary support from the Guelph Food System Biotechnology Center and the Natural Sciences and Engineering Research Council (NSERC).
REFERENCES
- 1.Ball L A. High-frequency homologous recombination in vaccinia virus DNA. J Virol. 1987;61:1788–1795. doi: 10.1128/jvi.61.6.1788-1795.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cox M M, Goodman M F, Kreuzer K N, Sherratt D J, Sandler S J, Marians K J. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- 3.DeLange A M, McFadden G. Sequence-nonspecific replication of transfected plasmid DNA in poxvirus-infected cells. Proc Natl Acad Sci USA. 1986;83:614–618. doi: 10.1073/pnas.83.3.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Du S, Traktman P. Vaccinia virus DNA replication: two hundred base pairs of telomeric sequence confer optimal replication efficiency on minichromosome templates. Proc Natl Acad Sci USA. 1996;93:9693–9698. doi: 10.1073/pnas.93.18.9693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Egelman E. A common structural core in proteins active in DNA recombination and replication. Trends Biochem Sci. 2000;25:179–182. doi: 10.1016/s0968-0004(00)01555-8. [DOI] [PubMed] [Google Scholar]
- 6.Evans D H, Stuart D, McFadden G. High levels of genetic recombination among cotransfected plasmid DNAs in poxvirus-infected mammalian cells. J Virol. 1988;62:367–375. doi: 10.1128/jvi.62.2.367-375.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fisher C, Parks R J, Lauzon M L, Evans D H. Heteroduplex DNA formation is associated with replication and recombination in poxvirus-infected cells. Genetics. 1991;129:7–18. doi: 10.1093/genetics/129.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kreuzer K N. Recombination-dependent DNA replication in phage T4. Trends Biochem Sci. 2000;25:165–173. doi: 10.1016/s0968-0004(00)01559-0. [DOI] [PubMed] [Google Scholar]
- 9.Lin F L, Sperle K, Sternberg N. Model for homologous recombination during transfer of DNA into mouse L cells: role for DNA ends in the recombination process. Mol Cell Biol. 1984;4:1020–1034. doi: 10.1128/mcb.4.6.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin F L, Sperle K, Sternberg N. Repair of double-stranded DNA breaks by homologous DNA fragments during transfer of DNA into mouse L cells. Mol Cell Biol. 1990;10:113–119. doi: 10.1128/mcb.10.1.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McDonald W F, Traktman P. Overexpression and purification of the vaccinia virus DNA polymerase. Protein Expr Purif. 1994;5:409–421. doi: 10.1006/prep.1994.1059. [DOI] [PubMed] [Google Scholar]
- 12.Merchlinsky M. Intramolecular homologous recombination in cells infected with temperature-sensitive mutants of vaccinia virus. J Virol. 1989;63:2030–2035. doi: 10.1128/jvi.63.5.2030-2035.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Michel B. Replication fork arrest and DNA recombination. Trends Biochem Sci. 2000;25:173–178. doi: 10.1016/s0968-0004(00)01560-7. [DOI] [PubMed] [Google Scholar]
- 14.Moyer R W, Graves R L. The mechanism of cytoplasmic orthopoxvirus DNA replication. Cell. 1981;27:391–401. doi: 10.1016/0092-8674(81)90422-0. [DOI] [PubMed] [Google Scholar]
- 15.Parks R J, Evans D H. Effect of marker distance and orientation on recombinant formation in poxvirus-infected cells. J Virol. 1991;65:1263–1272. doi: 10.1128/jvi.65.3.1263-1272.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parks R J, Winchcombe-Forhan C, DeLange A M, Xing X, Evans D H. DNA ligase gene disruptions can depress viral growth and replication in poxvirus-infected cells. Virus Res. 1998;56:135–147. doi: 10.1016/s0168-1702(98)00055-0. [DOI] [PubMed] [Google Scholar]
- 17.Sambrook J, Fritsch E F, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- 18.Shchelkunov S N, Totmenin A V. Two types of deletions in orthopoxvirus genomes. Virus Genes. 1995;9:231–245. doi: 10.1007/BF01702879. [DOI] [PubMed] [Google Scholar]
- 19.Spyropoulos D D, Roberts B E, Panicali D L, Cohen L K. Delineation of the viral products of recombination in vaccinia virus-infected cells. J Virol. 1988;62:1046–1054. doi: 10.1128/jvi.62.3.1046-1054.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stahl M M, Thomason L, Poteete A R, Tarkowski T, Kuzminov A, Stahl F W. Annealing vs. invasion in phage lambda recombination. Genetics. 1997;147:961–977. doi: 10.1093/genetics/147.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sugawara N, Ira G, Haber J E. DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. Mol Cell Biol. 2000;20:5300–5309. doi: 10.1128/mcb.20.14.5300-5309.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tomso D J, Kreuzer K N. Double-strand break repair in tandem repeats during bacteriophage T4 infection. Genetics. 2000;155:1493–1504. doi: 10.1093/genetics/155.4.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tseng M, Palaniyar N, Zhang W, Evans D H. DNA binding, aggregation, and annealing properties of the vaccinia virus I3L gene product. J Biol Chem. 1999;274:21637–21644. doi: 10.1074/jbc.274.31.21637. [DOI] [PubMed] [Google Scholar]
- 24.Willer D O, Mann M J, Zhang W, Evans D H. Vaccinia virus DNA polymerase promotes DNA pairing and strand-transfer reactions. Virology. 1999;257:511–523. doi: 10.1006/viro.1999.9705. [DOI] [PubMed] [Google Scholar]
- 25.Willer D O, Yao X-D, Mann M J, Evans D H. In vitro concatemer formation catalyzed by vaccinia virus DNA polymerase. Virology. 2000;278:562–569. doi: 10.1006/viro.2000.0686. [DOI] [PubMed] [Google Scholar]
- 26.Yao X-D, Matecic M, Elias P. Direct repeats of the herpes simplex virus a sequence promote nonconservative homologous recombination that is not dependent on XPF/ERCC4. J Virol. 1997;71:6842–6849. doi: 10.1128/jvi.71.9.6842-6849.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]