

Abstract
Porphyrins are acknowledged for their efficacy as photosensitizers and show potential for the treatment of water contaminated with diverse dyes. This research emphasizes the use of meso-tetra(4-sulfonatophenyl)porphyrin (TPPS) as a photosensitizer for purifying water contaminated with rhodamine B. Investigations were conducted under homogeneous conditions using visible light irradiation, revealing the efficacy of the porphyrin in the decolorization of rhodamine B strongly depends on pH of the solution. This study demonstrated that within 120 min, the decolorization process rapidly removed about 95% of RhB at an initial pH of 3.0, while at pH 6.0, the removal rate was significantly lower, at only 12%. An extensive photophysical study of the TPPS was carried out at pH 6.0 and pH 3.0 including absorption and fluorescence spectra, fluorescence quantum yields, triplet absorption spectra, triplet lifetimes, triplet and singlet oxygen quantum yields in order to explain difference in the efficiency of RhB discoloration. A thorough investigation into mechanism revealed that neither reactive oxygen species nor singlet oxygen played a role in RhB decolorization within this system. Instead, the predominant route was found to be the electron transfer from photoexcited TPPS to RhB, followed by proton transfer at pH 3.0, leading to the generation of a colorless leuco form.
Keywords: Dye photodegradation, Photocatalysis, Anionic porphyrin, Rhodamine B, Time-resolved spectroscopy, Scavengers
Subject terms: Chemistry, Materials science
Introduction
The discharge of dyes from textile processes into the environment has emerged as a significant source of water pollution, contributing to the hazard of organic compounds. About 15% of the total world production of dyes (700,000 tons per year) is released in textile effluents during textile dyeing1. The discharge of highly colored waste can affect the aquatic organisms either directly due to the toxicity of most dyes or indirectly by preventing their biological processes by impeding light penetration. Rhodamine B (RhB), a widely utilized xanthene dye with applications spanning various industries, has garnered attention due to its widespread use and potential environmental impact2,3. The high photostability of RhB, attributed to the rigid structure of the rhodamine molecule 4, leads to its accumulation in the environment. Unlike other xanthene dyes (eosin Y, rose bengal), RhB has a very low efficiency in forming triplet states, which limits its application as a photocatalyst5. Several techniques have been developed, including chemical and biological purification, physical and chemical adsorption, photocatalytic degradation and combined methods to remove this carcinogenic and neurotoxic dye from wastewater. Photocatalytic decomposition is an effective as well as a low-cost degradation method6–8.
The standard method for assessing the photocatalytic RhB transformation is by tracking the fading of its distinct absorbance peak at 554 nm. However, deducing the specific mechanisms behind the disappearance of RhB absorbance solely from UV–VIS spectra is not feasible, as it could result from a variety of processes (Fig. 1). These include (i) photodiscoloration owing to adsorption phenomena, (ii) direct photolysis (iii) photodiscoloration owing to reduction into colorless leuco form (iv) dye self-sensitization effects, (v) genuine photodegradation leading to the formation of stable byproducts or (vi) complete mineralization, resulting in the production of CO2 and H2O, leaving no residual contaminants9,10. In order to design more efficient photosensitizers for RhB degradation it is important to understand the mechanism involved in the phototransformation of RhB.
Fig. 1.
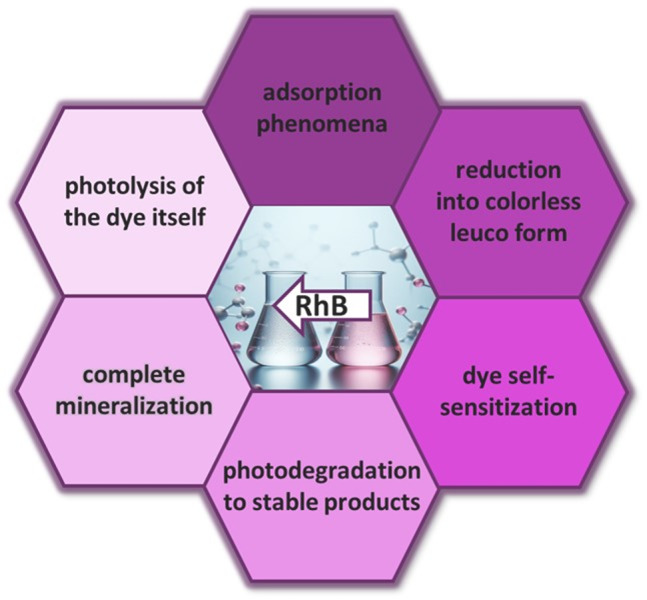
Various possible processes leading to the discoloration of rhodamine B.
For example, it is known that rhodamine B under irradiation with visible light in the presence of a photocatalyst, may undergo the N-deethylation process. This involves the subsequent detachment of ethyl groups from the amino nitrogen atoms, resulting in a hypsochromic shift of the absorption and emission maxima11. The process takes place when the photocatalyst is sensitized by RhB excited with visible light. Otherwise, when the photocatalyst is directly excited, RhB undergoes non-selective oxidation by generated reactive oxygen species, resulting in the chromophore decomposition. Additionally, a decrease in absorbance may also indicate formation of the leuco form of RhB, which is the product of a direct reduction of RhB with electrons12.
The use of organic photocatalysts, such as porphyrin-based catalysts, represents a promising approach for the remediation of dye pollutants due to their strong light absorption in the visible spectrum and their ability to act as powerful oxidants in excited states13. An organic photocatalyst can induce pollutant photooxidation via an initial electron transfer between its excited state and the contaminant (Type I mechanism). Alternatively, pollutant degradation can also be initiated by singlet oxygen generated by the photocatalyst (Type II mechanism)14. Organic homogeneous photocatalysis is primarily driven by organic dyes and transition metal complexes. However, in terms of sustainability, organic dyes are more economical, environmentally benign, and easy to use. A significant drawback of organic dyes is their inability to be recovered and reused. One strategy to address this issue is their heterogenization, such as immobilization on solid materials like silica or TiO₂, which improves reusability. 15–18 It is important to note that the adsorption of an organic dye onto a solid support may also affect the mechanism of pollutant degradation.
Among organic dyes, porphyrins have garnered significant attention as photocatalysts in the last years. 19–21 Anionic porphyrins, such as meso-tetra(4-sulfonatophenyl)porphyrin (TPPS), stand out due to their water solubility, stability, and photochemical reactivity, making them excellent candidates for harnessing light energy to drive the degradation of organic pollutants. Several studies have highlighted the successful application of TPPS in water treatment, showcasing its potential for addressing the challenges associated with organic pollutants22–25. While various studies have explored the photocatalytic capabilities of TPPS porphyrin, most of these investigations have concentrated on aggregated forms of TPPS porphyrin26,27. Our study, however, concentrates on the monomeric form of TPPS, which allows for a more detailed exploration of the mechanistic pathways involved. Homogeneous porphyrins, in contrast to their aggregates, are known to effectively form triplet which further can be involved in two possible mechanistic pathways in a photocatalytic process: energy transfer (to form singlet oxygen, Type II ) and electron transfer from the triplet excited state (Type I). An intriguing aspect that led us to investigate phototransformation of RhB by TPSS was the significant impact of pH on process efficiency, which proved to be a critical factor. The main functions and mechanisms of action for porphyrin type molecules are based on their photophysical properties, therefore by applying various spectroscopic methods we could identify parameters responsible for the enhanced activity under acidic pH.
TPPS can exist in homogeneous aqueous solutions in various protonation states. The pK values of its first and second protonations are very close to 5.228. Therefore, at neutral pH, the TPPS molecule exists in the tetra-anionic form (TPPS4‒) and appears pinkish in solution. At pH 3.0, the solution turns green, indicating protonation at pyrrolic nitrogen within the porphyrin core (H2TPPS2‒) (Fig. S1). This paper aims to systematically probe the effect of pH on the photodegradation of RhB mediated by the anionic porphyrin TSPP. Through a combination of spectroscopic analyses, kinetic studies, and mechanistic investigations, we endeavor to unravel the pH-dependent intricacies governing the photochemical reactivity of this catalytic system. Our findings not only contribute to the fundamental understanding of the phototransformation process but also provide valuable insights for optimizing the efficiency of porphyrin-based catalysts in diverse environmental contexts.
Materials and methods
Reagents and materials
Meso-tetra(4-sulfonatophenyl)porphyrin (TPPS) (≥ 95%), tetraphenylporphyrin (TPP) (≥ 99%), meso-tetra(4-N-methylpyridyl)porphyrin (TMPyP) (≥ 97%), rhodamine B (RhB) (≥ 95% purity for HPLC), deuterium oxide (D2O) (99.9 atom %D), tert-butanol (t-BuOH) (suitable for HPLC, ≥ 99.5%), sodium iodide (NaI)(ACS reagent, ≥ 99.5%) 1,4-benzoquinone (BQ) (reagent grade, ≥ 98%), sodium azide (NaN3) (purum p.a., ≥ 99.0%) and ethanol (EtOH) (≥ 99.9% (GC), gradient grade, suitable for HPLC) were all purchased from Sigma-Aldrich and used with no further purifications. Meso-tetra(4-carboxyphenyl)porphyrin (TCPP) (≥ 97%) was purchased from Porphyrin Systems. All experiments were performed in Mili-Q water or deuterium oxide as a solvent. The pH of the solution was adjusted with an HCl solution. The stock solution of TPPS (8.6 µM) was prepared by dissolving the porphyrin in water. The stock solution of RhB (33 µM) was prepared by dissolving the compound in ultrapure water. The stock solutions were protected from light and stored at room temperature until use.
Experimental apparatus and methods
Cary 100 UV–Vis two-beam spectrometer was used to record UV–Vis absorption spectra in the range from 800 to 200 nm with 1 nm step, using quartz cells with various optical path length (2–10 mm). Fluorescence spectra were recorded in the range of 630‒800 nm on a JASCO FP-8550 spectrofluorometer for diluted solutions with an absorbance at the excitation wavelength lower than 0.1.
Fluorescence quantum yield of compounds TPPS have been determined by preparing series of dilution ranging in absorption from 0.04 to 0.10 and excited at 422 nm. TPP has been used as standard in experiments to determine fluorescence quantum yield29. The following equation have been applied to determine the quantum yield of fluorescence:
| 1 |
where ST stands for the standard, X stands for the respective TPPS at various pH, is the quantum yield of standard, is the quantum yield of TPPS, Grad is the gradient from the plot of integrated fluorescence intensity versus optical density. A solution of tetraphenylporphyrin (TPP) in ethanol was used as a standard sample (ΦF = 0.15)29.
The fluorescence lifetimes were measured on a Fluorescence Lifetime Spectrometer (FluoTime300 from PicoQuant) with a detection system based on time-correlated single-photon counting (TCSPC). The emission decay lifetimes were acquired using 437 nm diode laser as the excitation source. In addition, an instrument response function (IRF) was obtained using Ludox solution (colloidal silica). Singlet oxygen measurements were conducted via direct method. The direct measurements of singlet oxygen emission were carried out on a FluoTime 300 fluorescence spectrophotometer with an NIR PMT detector H10330–45 (Hamamatsu) equipped with a 1000 nm long-pass filter. The samples were excited at 437 nm using a high repetition rate 40 MHz picosecond laser diode (LDH-437 nm, PicoQuant). Data collection was performed using a computer-mounted PCI-board multichannel scaler (NanoHarp 250, PicoQuant). The TSPP absorbance was adjusted to be 0.2 at the excitation wavelength. The time-resolved measurements (decay traces at λ = 1270 nm) were collected using a so-called “burst mode”, in which the sample is first excited using multiple pulses of the laser to build up the population of singlet oxygen and then left to decay in the 500 μs time window. The experiment was performed in D2O. 10 mm × 10 mm quartz cells were used for all time-resolved emission measurements. The setup for the nanosecond laser flash photolysis (LFP) experiments and the data acquisition system have been previously described in detail30. LFP experiments employed a Nd:YAG laser (355 nm, 5 mJ, 7–9 ns) for excitation. Transient decays were recorded at individual wavelengths by the step-scan method with a step distance of 10 nm in the range of 380 to 800 nm as the mean of 8 probe pulses. Solutions for LFP were deoxygenated with high-purity argon for 45 min prior to the measurements. Experiments were performed in rectangular quartz cells (10 mm × 10 mm). Details of the method of determination of the quantum yields of intersystem crossing is described Supplementary Materials31.
The femtosecond transient absorption spectroscopy setup consisted of a short-pulse titanium sapphire oscillator (Mai Tai, Spectra Physics, 70 fs) followed by a high-energy titanium sapphire regenerative amplifier (Spitfire Ace, Spectra Physics, 100 fs). The 800 nm beam was split into two beams to generate: (1) a pump (λexc = 420 nm) from the optical parametric amplifier (Topas Prime with a NirVis frequency mixer) and (2) probe pulses in the UV–Vis range by using sapphire plate (Ultrafast Systems, Helios). The temporal resolution of the setup is about 200 fs. For transient UV–Vis measurements a quartz cell with 2 mm optical path of solution was used with the absorbance of about 0.4 at the excitation wavelength. The sample solution was stirred by a teflon-coated bar. Typical pump pulse energy was about 0.2 µJ. All experiments were performed at room temperature. Analysis of the transient absorption data was made using Surface Explorer software (Ultrafast Systems).
The acquired experimental data were analyzed using the commercial program OriginPro 2023b.
Phototransformation of RhB in aqueous solutions
For photodegradation experiments, the stock air-saturated aqueous solutions of RhB and TPPS were mixed to obtain a 3 mL solution with desired RhB and TPPS concentration by dilution with Milli-Q water. Solutions of RhB with the porphyrin were placed in quartz cuvette and irradiated using 150 W Xe lamp (Newport Corporation) with cut-off filter λ > 400 nm (spectral irradiance at 0.5 m from our Xe lamp is 20 mW m‒2nm‒1 at 420 nm) or diode laser 505 nm (Thorlabs Mounted LEDs, M505L4, 6.5 µW/mm2) or 420 nm (Thorlabs Mounted LEDs, M420L3, 6.5 µW/mm2. The mixture was irradiated for a certain time with continuous stirring at 800 rpm. All experiments were performed at room temperature. All experiments were performed on air-saturated solutions unless otherwise stated. At various time intervals the samples were analyzed by UV–Vis spectroscopy and the removal % was calculated from Eq. 2:
| 2 |
where Ao and At are the initial absorbance at 554 nm, the maximum absorbance for RhB and at time ‘t’ of irradiation, respectively.
For the data analysis of the RhB photodegradation in the presence of porphyrin first order kinetic Eq. (3) was used to fit the data:
| 3 |
where k is the first order rate constant, and t is the irradiation time.
Results
Photocatalytic transformation of RhB
Initially, the stability of RhB was tested by exposing the aqueous dye solution to visible light for 2 h. As shown in Fig. 2, in the control experiment without any photocatalyst added, only a minor degradation of RhB molecules (5% after 2 h) was observed. The dye demonstrated high stability, showing negligible degradation observed under illumination. High photostability of the RhB is attributed to the rigid structure of the rhodamine molecule4. Afterwards, samples of RhB in the presence of TPPS in aerated aqueous solutions were irradiated for 2 h under visible light with constant stirring. (Fig. 2a). The activity of the TPPS4‒ at pH 6.0 was quite modest, with only 12% RhB decomposition observed after 2 h of irradiation. Notably, the activity of H2TPPS2‒ at pH 3.0 was significantly higher, resulting in 95% RhB decomposition after the same time (Fig. 2b).
Fig. 2.

Photodegradation of RhB (7.5 µM) under visible irradiation with Xe lamp (λ > 400 nm) of aerated aqueous solutions with stirring at 800 rpm a) UV–Vis absorption spectra changes for RhB upon irradiation in the presence of H2TPPS2‒ (15 µM) at pH 3.0; inset: photograph of the solution before and after 2 h irradiation, b) photocatalytic activities for RhB degradation using TPPS4‒ (15 µM) at pH 6.0 (pink), at pH 3.0 (green), a control test without porphyrin at pH 6.0 (blue) and pH 3.0 (star).
The almost complete disappearance of RhB after 2 h of irradiation in the presence of H2TPPS2‒ at pH 3.0 was evidenced by the clear discoloration of the sample (inset in Fig. 2a). The control test, conducted in the dark with porphyrin and RhB present, showed no degradation. The calculated pseudo-first-order rate constants are k = 7.4 × 10‒3 min‒1 and k = 6.7 × 10‒4 min‒4 at pH 3.0 and pH 6.0, respectively, which is confirmed by the good fitting to Eq. (3) to the experimental data values (Supplementary Fig. S2). The calculated pseudo-first-order rate constant k for H2TPPS2‒ at pH 3.0 is one order of magnitude higher than for TPPS4‒ at pH 6.0.
To understand the impact of initial pH on the RhB degradation process by TPPS, experiments were conducted across a broader pH range: pH 2.0–8.0. The effect of the pH on the removal of RhB is illustrated in Fig. 3a. The results of these experiments revealed that the RhB removal decreased with increasing pH; the highest color removal efficiency was observed at the pH value of 3.0. Consequently, pH 3.0 is identified as the optimal pH for RhB degradation and selected for subsequent experiments. At higher pH values (pH > 6.0), the degradation of RhB was almost completely inhibited. Below pH 3.0, porphyrin tends to aggregate due to cooperative π-π electrostatic interactions between ionic substituent groups and protonated porphyrin core. 32,33 That could potentially also affect the efficiency of RhB photodegradation.
Fig. 3.

a) Effect of pH on photodegradation of RhB (7.5 µM) [TPPS]: 5.0 µM b) effect of the initial concentration of H2TPPS2‒ on photodegradation of RhB at pH 3.0. Conditions: irradiation with Xe lamp (λ > 400), aerated aqueous solutions, stirring at 800 rpm, T = 298 K.
To ascertain the correlation between the photodegradation of RhB at various pH and the properties of TPPS, control experiments were conducted. These experiments revealed negligible photodegradation of RhB in the absence of TPPS at the pH 3.0 and 6.0 (Supplementary Fig. S3). Additionally, comparative photodegradation experiments of RhB were conducted at pH 6.0 and pH 3.0 using another commonly used in photocatalysis anionic porphyrin, tetra-(4-carboxyphenyl)porphyrin (TCPP), instead of TPPS. However, RhB removal efficiency was negligible at both pH levels when TCPP was utilized (Supplementary Fig. S4). These findings underscore the unique properties of TPPS in facilitating the pH-dependent photodegradation of RhB. Figure 3b shows the effect of H2TPPS2‒ concentration on photodegradation at pH 3.0. Notably, the most efficient concentration of H2TPPS2‒ was 20 µM, resulting in complete RhB photodegradation after 1 h of irradiation.
Mechanistic evaluation
In our exploration of the photocatalytic removal of RhB using the anionic porphyrin TPPS, a striking increase in decolorization efficiency has been noted, particularly in acidic pH environments. This finding suggests a link between the protonation status of the TPPS core and its photocatalytic prowess. Nevertheless, to fully comprehend the underlying mechanism of RhB discoloration under these conditions, a more detailed study was warranted. Initially, we hypothesized that differences in TPPS’s activity toward RhB could be discerned through a comparative analysis of its spectroscopic properties at pH 3.0 and pH 6.0. Consequently, our study meticulously characterized TPPS across various states to shed light on this phenomenon.
Evaluation of TPPS spectroscopic properties
At neutral pH, the TPPS molecule exists in the tetra-anionic form TPPS4‒ and is pinkish in solution (Fig. S1). However, at pH 3.0, a notable color shift occurs, with the solution turning green, indicating protonation at pyrrolic nitrogen in the porphyrin core (Fig. 4). The absorption spectrum of H2TPPS2‒ in aqueous solution at pH 3.0 exhibits distinct bands at 434 nm (Soret band) and 550, 594, 645, and 708 nm (Q bands) whereas the TPPS4‒form at pH 6.0 displays bands centered at 414 nm (Soret band) and 516, 552, 580, and 634 nm (Q bands) (Fig. 4 and Table S1). The Soret band denotes the So‒S2 transition. In the visible region of the porphyrin spectrum there are four weak bands called the Q bands that correspond to So‒S1 transitions34. The considerable spectral changes as a function of pH stem from the protonation of the pyrrole nitrogen atoms in the center of the porphyrin ring. Notably, both forms of porphyrin exhibit comparable molar absorption coefficient values at the Soret band maximum (Table S1).
Fig. 4.
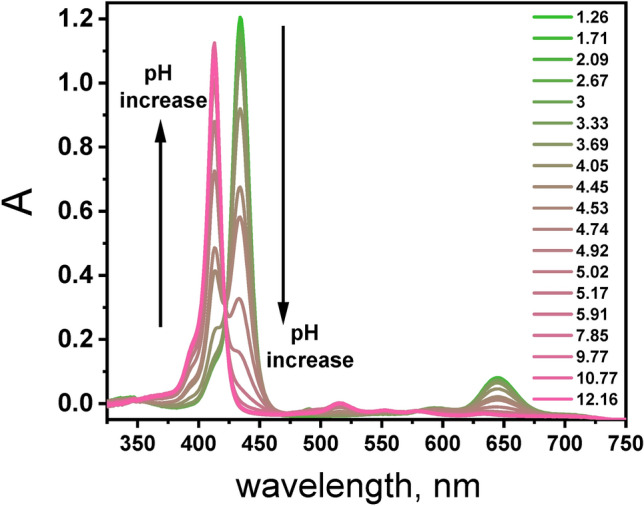
pH dependent absorption spectra of a porphyrin TPPS (3.1 µM) in aqueous solution.
From the perspective of photocatalysis, the stability of the compound is crucial. It was observed by UV–VIS spectroscopy that aqueous solution of both forms of porphyrin, H2TPPS2‒and TPPS4‒, remained stable throughout 24 h timeframe (Fig. S5). Extensive studies on porphyrins have shown that water-soluble derivatives are particularly prone to undergoing cooperative π-π electrostatic interactions between ionic substituent groups and the electron- rich central nitrogen atoms, leading to aggregation32,33. It is established that TSPP can aggregate under conditions of high ionic strength and low pH35. Given that this study focuses on RhB photodegradation under homogeneous conditions, it was imperative to ascertain the absence of TPPS aggregates. Although the formation of aggregates was deemed unlikely under the conditions utilized in this study – owing to relatively low porphyrin concentrations (< 20 μM) and low ionic strength (10‒3 ) – we carefully compared the UV–Vis spectra of TPPS solutions at pH 6.0 an pH 3.0 used for RhB degradation experiments, with the UV–Vis spectra of TPPS aggregates prepared according to ref.36 (Supplementary Fig. S6). TPPS aggregates are characterized by a distinctive band with maximum at 490 nm, which was not evident in our TPPS solutions (Supplementary Fig. S6). Based on that presence of TPPS aggregates under our experimental conditions was found to be highly unlikely.
Subsequently, the excited-state properties of TPPS were evaluated by means of steady-state and time-resolved absorption and emission techniques. The measured excited-state properties of the porphyrin at pH 6.0 and pH 3.0 included the lifetimes of the lowest singlet excited state (S1) (τs) and triplet state (T1) (τT), the S1 → S0 fluorescence quantum yield (ΦF), and the yield of S1 → T1 intersystem crossing (ΦISC). The yield of S1 → S0 internal conversion was obtained by calculating the difference ΦIC = 1 – ΦF – ΦISC. Rate constants of the fluorescence (kF), intersystem crossing (kISC), and internal conversion (kIC) were obtained from the quantum yield of the respective process via the equation kP = ΦP/τS, where P = F, ISC or IC.
The fluorescence spectra of both forms of the TPPS porphyrin are shown in Fig. 5a, with select photophysical properties summarized in Table 1. The fluorescence quantum yield was determined following the method described by Williams et al.37, which involves the employing well-characterized standard samples with known ΦF values and measuring emission spectra for a series of concentrations of TPPS (Supplementary Fig. S7).
Fig. 5.

(a) Normalized emission spectra of aqueous solution of TPPS at pH 3.0 (green) and at pH 6.0 (pink) measured at excitation 422 nm (A422 nm = 0.05) (b) decay of TPPS fluorescence recorded in at pH 3.0 (green) and pH 6.0 (pink) prompt (grey); λex = 437 nm, λem = 660 nm for pH = 3.0 and λem = 640 nm for pH 6.0; black lines show the monoexponential decay fits.
Table 1.
Spectroscopic properties of singlet excited state S1: S1 lifetimes (τS), fluorescence (ΦF), triplet (ΦISC) and internal conversion (ΦIC) quantum yields and the rates constants of intersystem crossing (kISC), radiative (kIC) and internal conversion (kISC), Stokes shift (∆λStokes) and S1 energy level (ES1) of protonated and non-protonated TPPS in water.
| λem(nm) | τS (ns) | ΦF | ΦIC | ΦISC | kF(107s‒1) | kISC(107 s‒1) | kIC(107 s‒1) | ∆λStokes(cm‒1) | ES1(kJ/mol) | |
|---|---|---|---|---|---|---|---|---|---|---|
| TPPS4‒ | 639 | 10.05 | 0.18 | 0.30 | 0.52 | 1.8 | 4.9 | 2.9 | 147.9 | 187.3 |
| H2TPPS2‒ | 660 | 3.85 | 0.11 | 0.47 | 0.50 | 2.7 | 10.9 | 12.2 | 535.4 | 182.5 |
Upon excitation at 422 nm, TPSS at both pH values exhibited a broad fluorescence spectrum in the range of 625 to 750 nm. At pH 6.0, TPPS4‒ displayed a broad emission comprising two unresolved Q(0,0) and Q(0,1) bands at ca. 640 nm and 702 nm, respectively, with a quantum yield of 0.18. As shown in Fig. 5a protonation reduced the two fluorescence bands (Qx(0, 0) and Qx(0, 1)) to just a single broad structureless band (Q(0, 0)) with the maximum at 660 nm. Upon protonation of the TSPP molecule through the addition of HCl, the second emission band Q(0,1) diminished – a phenomenon well-documented for protonated porphyrins38. The fluorescence quantum yields ΦF also underwent changes upon macrocycle protonation, with the emission quantum yield at pH 3.0 found to be 0.11. Protonation or deprotonation events have the potential to alter non-radiative decay pathways, thereby influencing the interplay between radiative and non-radiative processes governing fluorescence emission.
The fluorescence lifetime calculated from the emission decay was 3.85 ns for TPPS in an acidic environment and 10.05 ns under pH 6.0 (Fig. 5b). Protonation shortened the fluorescence lifetime of the molecule. Considering the results obtained from both steady-state and time-resolved emission experiments, it can be concluded that the formation of the protonated species leads to an increase in the fluorescence probability kF. However, due to competition with enhanced radiationless deactivation rates, the fluorescence quantum yield ΦF decreases for the protonated form. One can point out the substantial quantum yield of the internal conversion in both forms of TPPS, with significantly higher value for the protonated TPPS. Smaller ΦF and ΦISC values for the protonated TPPS are mainly at the expense of the internal conversion.
Thus far, we have investigated the properties of the singlet excited states of TPPS. Nanosecond transient absorption spectroscopy served as an additional tool to get further insight into the excited-state photophysics of the triplet state of TPPS. This technique enabled the study of the triplet excited state lifetime and transient absorption spectra for TPPS. Nanosecond transient absorption difference spectra were acquired for deoxygenated TPSS solution in water at pH 6.0 and 3.0 (Fig. 6). The resulting difference spectra for H2TPPS2‒ exhibited an intense ground-state bleach at ∼430 nm and a less intense band at 650 nm (Fig. 6c). Both bands match the position of the ground-state absorption spectrum. The positive signal with a maximum at 500 nm can be attributed to the triplet state absorption as confirmed by its quenching in the presence of oxygen. In addition, a low-intensity signal in the region of 700–800 nm was also observed. Upon laser excitation at 355 nm, TPPS at pH 6.0 exhibited a strong bleaching peak, identical to the ground state peak at 410 nm and the triplet state absorption around 430–500 nm, consistent with the literature data (Fig. 6a)31,39. The triplet lifetimes for TPPS at pH 3.0 and pH 6.0 were obtained from monoexponential fits of the bleach recovery (435 nm and 415 nm) and transient decay (450 nm and 500 nm) (Fig. 6b, d, Table S2). Both kinetic traces were characterized by the same time constant for corresponding pH values. The TPPS triplet lifetime was found to be 146 µs and 320 µs at pH 3.0 and pH 6.0 respectively.
Fig. 6.

Transient absorption spectra (a and c) and decay profiles (b and d) obtained during laser flash photolysis (with excitation at 355 nm) of deoxygenated aqueous solutions of TPPS at a) pH 6.0 ([TPPS4-] = 6.5 µM); c) pH 3.0 ([H2TPPS2‒] = 7.7 µM); monoexponential decay fits shown as black lines.
The T1 quantum yield (ФISC) is one of the most important characteristics in the study of the photochemical properties of porphyrins. The ФISC values were determined by adopting the methods recently developed by Gonçalves et al.31 (see details in Supplementary). ФISC for TPPS4‒ and H2TPPS2‒ were found to be 0.52 and 0.50, respectively (Fig S8, Fig. S9, Table S2).
High values of ФISC indicates that the S1 → T1 intersystem crossing dominates the decay of the S1 excited state in both forms of TPPS. Since various oxygen forms could potentially play a role in the photodegradation of RhB in the presence of TPPS, it was of interest to determine the bimolecular rate constant for the quenching of triplet excited state of TPPS by O2. The triplet lifetimes of both TPPS forms were strongly reduced in the presence of oxygen under ambient conditions (Table S3, Supplementary Fig. S10). The bimolecular quenching rate constants of TPPS by oxygen (kq) in air-saturated solutions were obtained from Eq. (4):
| 4 |
where is the lifetime of triplet states in degassed solutions and is the lifetime of triplet state in air-saturated solutions. Assuming [O2] concentration of 1.22 mM (in water)40 we calculated kq based on Eq. (4) (Table S3). The bimolecular rate constant kq for the quenching of triplet states of TPPS by oxygen was found to be 5.0 × 108 M‒1 s‒1 and 5.3 × 108 M‒1 s‒1 for pH 3.0 and pH 6.0, respectively.
Finally, to quantitatively evaluate the ability of TPPS to produce singlet oxygen, singlet oxygen generation was monitored in real-time through time-resolved measurements of its 1O2 phosphorescence41. Figure 7 displays the tail of the time-resolved singlet oxygen decay at λ = 1270 nm for TPPS at two pH values. The singlet oxygen decay was fitted to a lifetime of 65 µs. This lifetime value aligns well with the published literature values for the lifetime of singlet oxygen in D2O42. Based on the comparison of the signal amplitude with the reference compound TMPyP (Φ∆ = 0.74)43, it was calculated that the quantum yields of singlet oxygen generation was 0.58 and 0.59 for TPPS in pH 3.0 and pH 6.0 respectively. High value of singlet oxygen quantum yield for TPPS form suggests that it might find application in photodegradation of organic dyes.
Fig. 7.

Decay curves of the singlet oxygen generated by air-saturated solutions of TPPS at pH 3.0 (green) and pH 6.0 (pink), TMPyP as reference (black) in D2O (λexc = 437 nm, Abs437nm = 0.2, λem = 1270 nm, collection time: 60 min).
In summary, both forms of porphyrin, TPPS4‒ and H2TPPS2‒ exhibited significant absorption in the visible range with similar molar absorption coefficients, nearly identical intersystem crossing efficiency, long-lived triplet state, and comparable singlet oxygen generation efficiency. The strikingly similar properties of both porphyrin forms, crucial from the perspective of photocatalysis, do not provide a clear explanation for the substantial increase in activity towards RhB photodegradation at pH 3.0. This indicates the necessity for further research to understand the mechanisms underlying this phenomenon.
Discussion on possible mechanistic pathways
As discussed in the Introduction, the phototransformation of RhB manifested by the decrease in the characteristic absorbance peak at 554 nm can occur through various pathways (Fig. 1). These pathways include: (i) photodiscoloration due to adsorption phenomena, (ii) photodiscoloration due to reduction into a colorless leuco form (iii) RhB self-sensitization effects, (iv) photodegradation leading to the formation of stable organic products, and (v) complete mineralization, resulting in the production of CO2 and H2O with no residual contaminants. In our homogeneous system, the adsorption of RhB on the photocatalyst can be disregarded. Similarly, the possibility of photocatalytic transformations of RhB through N-deethylation can be ruled out, as the anticipated hypsochromic shift in absorption, indicative of N-deethylation, was not observed44.
The remaining processes were investigated through the following methods: 1) photolysis of RhB in the presence of TPPS, with radical and oxygen singlet traps 2) photolysis of RhB in the presence of TPPS using LED that can selectively excite RhB, 3) emission and femtosecond transient absorption spectroscopy studies of TPPS in the presence of RhB.
Although reactive oxygen species (ROS) are frequently associated with the photodegradation of organic compounds,45–47 their central role is not guaranteed. Therefore, it’s essential to carry out thorough trapping experiments to precisely determine the mechanisms at play and pinpoint the true agents of the degradation pathway. According to previous reports, the •OH, O2•‒ and 1O2 were assumed to be the major active species responsible for photocatalytic reaction23,24,45–47. Therefore, a series of additional experiments were conducted to probe the mechanism involved in the visible light-induced RhB photodegradation by H2TPPS2‒ at pH 3.0 (Fig. 8). The discrimination among the produced ROS can be obtained by introducing radical-trapping agents during light exposure. These scavenger molecules by interacting with photogenerated radicals, inhibit their reaction with the organic dye. In particular, sodium iodide NaI, t-BuOH, and 1,4-benzoquinone (BQ) can be efficiently used to scavenge photogenerated holes, hydroxyl radical (•OH) , superoxide radical O2•‒ , respectively18,48. In addition NaN3 is known to acts as singlet oxygen (1O2) scavenger49. The results obtained (Fig. 8) show that RhB degradation in the presence of t-BuOH follows almost the same kinetics as in the absence of the scavenger, as confirmed by the coincidence of the experimental points with the pseudo-first order kinetic curve in the absence of the scavenger. Therefore •OH are not the main oxidizing species in the process of RhB degradation.
Fig. 8.
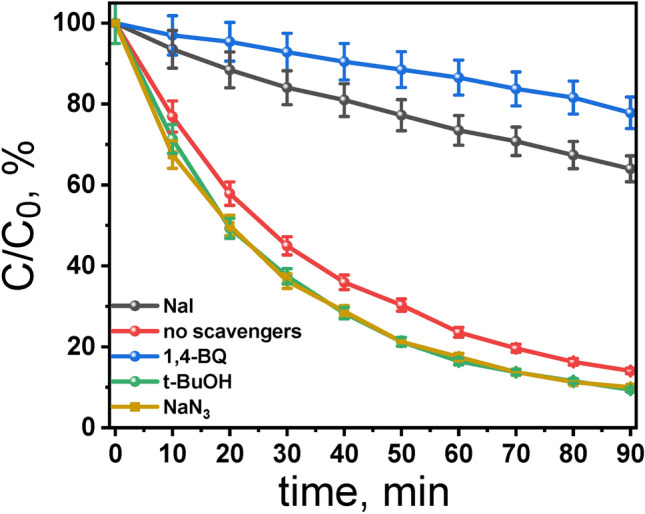
Effect of radical-hole scavengers on the degradation rate of RhB (7.5 µM) by using TPPS (5.14 µM) at pH 3.0: blue – 1,4-benzoquinone, black – NaI, red – no scavenger, green – t-BuOH; orange – NaN3. Conditions: irradiation with Xe lamp (λ > 400), aerated aqueous solutions, stirring at 800 rpm, T = 298 K, Scavengers concentration: 10 mmol L‒1.
It has been suggested that the reactive species responsible for the photooxidation of substrates by oxygen using visible light activation of porphyrin involve singlet oxygen (1O2), generated by triplet–triplet energy transfer from the excited state of porphyrin to oxygen. TPPS forms 1O2 in high quantum yield (Table S3). Therefore, to identify whether 1O2 is involved in the photodegradation of RhB, we investigated the influence of NaN3 on the photodegradation process. The results show that the degree of RhB photodegradation remains unchanged in the presence of NaN3. Thus, it is inferred that the mechanism for the photodegradation of RhB by H2TPPS2‒ does not to involve singlet oxygen processes.
However, a different situation was observed in the presence of BQ, where the RhB concentration decreased by only 36% after 90 min of irradiation. The significant influence of BQ on photodegradation strongly suggests that the dye photodegradation is largely caused by O2•‒‒. It has been previously reported that the superoxide anion radical (O2•‒‒) plays a role in the photodegradation of pollutants9,50,51. Surprisingly, experiment involving irradiation of argon-saturated solution led to the decrease in the absorbance of RhB which was comparable to that observed in air-saturated sample (Supplementary Fig. S11). Therefore, the possibility of direct interaction between the photoexcited H2TPPS2‒ and BQ has been examined. It was found that both the singlet and triplet excited states of H2TPPS2‒ can be quenched by BQ (details in Supplementary Material and Fig. S12). This discovery suggests that conclusions drawn from trapping experiments using BQ must be approached cautiously, as there is a risk of BQ directly interacting with the excited state of photocatalysts, thereby reducing the efficiency of the photocatalytic process.
All our trapping experiments revealed that when the photocatalyst is directly excited, RhB does not undergo non-selective oxidation by generated reactive oxygen species or react with singlet oxygen. Additionally, under LED 505 nm irradiation, selective excitation of RhB did not induce a notable decrease in its absorbance (Supplementary Fig. S13). Hence, the excitation of RhB followed by oxidation in the presence of TPPS does not account for the observed transformation of RhB. This mechanism of RhB sensitization has been previously proposed, for example, for the TiO2 photocatalyst44.
It was found that only selective excitation of H2TPPS2‒ with 420-nm LED leads to efficient color removal of the RhB (Supplementary Fig. S13). After excluding the action of ROS and N-deethylation process, the decrease in absorbance may indicate the formation of the colorless leuco form of RhB, which is the product of the direct reduction of RhB. It was reported that the formation of the leuco form of RhB can occur through a one-step hydrogen atom transfer [H• = H+ + e‒) or a two-step process involving electron transfer followed by proton transfer52,53. It has been postulated that the H• forms a σ-bond with the π-electron of C-9 carbon (marked with an asterisk in Fig. 9) in RhB, which completely disrupts the conjugated structure and is the main cause of the disappearance of color during the reduction process52,53. The mechanism of RhB photodiscoloration involving two-step process of electron transfer followed by proton transfer would explain the enhanced removal of RhB color under acidic pH54. This catalytic reduction technique has received significant attention due to its ease of application, cost-effectiveness, cleanliness, and ability to rapidly purify contaminated water from colorful organic dyes2. For example, silver nanoparticles on various supports in the presence of reductant such as NaBH4 were reported to efficiently remove RhB by reduction to leuco for 55–57. To validate the proposed reaction mechanism, involving the direct reduction of RhB by the excited state of TPPS we investigated the interaction between the excited state of TPPS and RhB, by monitoring the emission decay profiles of TPPS at pH 3.0 and pH 6.0 in the presence of varying RhB concentrations (Fig. S14). On the basis of the Stern–Volmer plot (the inset in Supplementary Fig. S14a), the quenching rate constant (kq) calculated from the equation τ0/τ = 1 + kqτ0[RhB] is 7.3 × 1011 M‒1 s‒1 for pH 3.0. The value is reaching the diffusion control limit. It’s noteworthy that a very similar value for the quenching rate constant was obtained for TPPS at pH 6.0 (Supplementary Fig. S14b), indicating that the second step in the discoloration of RhB, namely proton transfer, is crucial for the overall process.
Fig. 9.

Mechanistic scheme for the photodiscoloration of RhB in the presence of TPPS under acidic pH.
Femtosecond transient absorption measurements were carried out to further confirm deactivation of the singlet excited state of TPPS by RhB, as expected from the emission experiments. The transient absorption spectra recorded shortly after a 425 nm laser pulse excitation of TPPS exhibited absorption band with the characteristic spectral shape of singlet excited states of porphyrins (Fig. S15)58–60. TPPS in its singlet excited state has a broad, intense absorption band with a maximum at 460 nm and a Q-band bleach with maximum at 650 nm that matches with the Q-band position observed in stationary UV–Vis absorption spectra. TA spectra recorded in the presence of RhB are disturbed in the regions 500–600 nm due to significant absorption of probe light by RhB in this region but besides this the TA obtained in the presence and absence of RhB show shape similarity. Kinetic profiles at 460 nm recorded for TPPS in the absence and presence of RhB are depicted in Figure S16. It is clear that within the same time observation window (3 ns), a significant difference can be seen between the two investigated samples. Undoubtedly the kinetic profile at 480 nm for TPPS in the presence of RhB showed faster decay dynamics compared with the TPSS in the absence of RhB.
The discoloration reaction of RhB was found to be catalytic, as under air-saturated conditions, the decrease of the RhB concentration was nearly six time higher that the corresponding depletion of H2TPPS2‒. Interestingly, the depletion of H2TPPS2‒ was significantly more pronounced when visible light irradiation of RhB in the presence of H2TPPS2‒ was performed under argon-saturated condition (Supplementary Fig. S17).
Based on the obtained results, the mechanism for the discoloration of RhB in the presence of H2TPPS2‒ was proposed (Fig. 9). As discussed earlier, the excited TPPS may undergo a series of energy-transfer and electron-transfer reactions generating reactive oxygen species (ROS) such as singlet oxygen or superoxide radical. Generally, the degradation of organic compounds can be initiated indirectly by ROS or directly by interaction with the excited state of TPPS. Initially, by applying scavengers, we ruled out ROS and singlet oxygen as reagents involved in the RhB discoloration. Interestingly, the absence of O2 (under argon saturation condition) did not inhibit the degradation of RhB. This implies that the degradation RhB under acidic pH is initiated by the direct electron transfer between photoexcited TPPS and RhB followed by proton transfer that leads to the formation of colorless leuco form (Fig. 9, reaction 4). Although this reaction does not involve O2, the dissolved oxygen is essentially needed to recycle TPPS (Fig. 9, reaction 5). In the presence of O2 the superoxide radical is formed which can react with the TPPS radical cation. In the absence of O2, TPPS radical cation accumulates as the reaction proceeds and the overall activity should decrease gradually. Our study has shown that color removal is not equivalent to dye mineralization; indeed, total color removal can be achieved without mineralization.
Conclusions
This study underscores the potential of meso-tetra(4-sulfonatophenyl)porphyrin (TPPS) as a catalyst for the decolorization of water contaminated with RhB under visible light irradiation in homogeneous conditions. Our findings highlight the critical role of pH in determining the efficacy of TPPS in RhB decolorization. Specifically, rapid and efficient removal of RhB, with approximately 95% decolorization achieved within 120 min, was observed at an initial pH of 3.0. In contrast, the removal rate at pH 6.0 was notably lower, at only 12%. A comprehensive investigation into the photophysical properties of TPPS at different pH levels elucidated key factors influencing RhB discoloration. Our mechanistic studies revealed that the degradation of RhB in the presence of TPPS occurs primarily through a direct electron transfer mechanism, followed by proton transfer at acidic pH conditions. Interestingly, reactive oxygen species (ROS) and singlet oxygen were found to play negligible roles in the decolorization process within this system. Instead, the dissolved oxygen was found to be essential for recycling TPPS. Importantly, our study demonstrates that color removal does not necessarily equate to dye mineralization, as total color removal can be achieved without permanent damage to the chromophore within the RhB structure. This distinction emphasizes the need for careful mechanistic investigations to properly interpret experimental data and understand the underlying processes involved in dye degradation.
Based on our findings, H2TPPS2‒ emerges as a promising sensitizer for immobilization onto various supports, such as TiO2, silica or 2D materials. The insights gained from this study contribute to the development of efficient and environmentally sustainable strategies for water purification and pollutant remediation.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
A.L thanks for funding to program under the project at AMU "Initiative of Excellence—Research University" (proposal no. 054/13/SNS/0025).
Author contributions
A.L: experimentation, data analysis, writing the manuscript; A.L-A: conceptualization, interpretation of data, revising and writing the manuscript.
Funding
Initiative of Excellence—Research University,054/13/SNS/0025
Data availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information file).
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-024-73586-3.
References
- 1.Agarwal, J. & Sonia. 13 - Dyes and dyeing processes for natural textiles and their key sustainability issues. in Fundamentals of Natural Fibres and Textiles (ed. Mondal, Md. I. H.) 439–472 (Woodhead Publishing, 2021). 10.1016/B978-0-12-821483-1.00013-9.
- 2.Sudarshan, S., Bharti, V. S., Harikrishnan, S., Shukla, S. P. & RathiBhuvaneswari, G. Eco-toxicological effect of a commercial dye Rhodamine B on freshwater microalgae Chlorella vulgaris. Arch Microbiol204, 658 (2022). [DOI] [PubMed] [Google Scholar]
- 3.Sharma, J., Sharma, S., Bhatt, U. & Soni, V. Toxic effects of Rhodamine B on antioxidant system and photosynthesis of Hydrilla verticillata. Journal of Hazardous Materials Letters3, 100069 (2022). [Google Scholar]
- 4.Liu, Y. et al. Insight into the relationship between the photostability and molecular structure of rhodamine dyes. Tetrahedron149, 133664 (2023). [Google Scholar]
- 5.Menzel, R. & Thiel, E. Intersystem crossing rate constants of rhodamine dyes: influence of the amino-group substitution. Chem Phys Lett291, 237–243 (1998). [Google Scholar]
- 6.Katheresan, V., Kansedo, J. & Lau, S. Y. Efficiency of various recent wastewater dye removal methods: A review. J Environ Chem Eng6, 4676–4697 (2018). [Google Scholar]
- 7.Bal, G. & Thakur, A. Distinct approaches of removal of dyes from wastewater: A review. Mater Today Proc50, 1575–1579 (2022). [Google Scholar]
- 8.Roy, M. & Saha, R. 6 - Dyes and their removal technologies from wastewater: A critical review. in Intelligent Environmental Data Monitoring for Pollution Management (eds. Bhattacharyya, S., Mondal, N. K., Platos, J., Snášel, V. & Krömer, P.) 127–160 (Academic Press, 2021). 10.1016/B978-0-12-819671-7.00006-3.
- 9.Koe, W. S., Lee, J. W., Chong, W. C., Pang, Y. L. & Sim, L. C. An overview of photocatalytic degradation: photocatalysts, mechanisms, and development of photocatalytic membrane. Environmental Science and Pollution Research27, 2522–2565 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Gupta, G. K. & Mondal, M. K. 20 - Fundamentals and mechanistic pathways of dye degradation using photocatalysts. in Photocatalytic Degradation of Dyes (eds. Shah, M., Dave, S. & Das, J.) 527–545 (Elsevier, 2021). 10.1016/B978-0-12-823876-9.00007-X.
- 11.Jakimińska, A., Pawlicki, M. & Macyk, W. Photocatalytic transformation of Rhodamine B to Rhodamine-110 – The mechanism revisited. J Photochem Photobiol A Chem433, (2022).
- 12.Collins, A. M., Zhang, X., Scragg, J. J., Blanchard, G. J. & Marken, F. Triple Phase Boundary Photovoltammetry: Resolving Rhodamine B Reactivity in 4-(3-Phenylpropyl)-Pyridine Microdroplets. ChemPhysChem11, 2862–2870 (2010). [DOI] [PubMed] [Google Scholar]
- 13.Gkika, D. A., Ladomenou, K., Bououdina, M., Mitropoulos, A. C. & Kyzas, G. Z. Adsorption and photocatalytic applications of porphyrin-based materials for environmental separation processes: A review. Science of The Total Environment908, 168293 (2024). [DOI] [PubMed] [Google Scholar]
- 14.Blázquez-Moraleja, A. et al.Organic photoredox catalysts: tuning the operating mechanisms in the degradation of pollutants.95, 899–912 (2023). [Google Scholar]
- 15.Silvestri, S., Fajardo, A. R. & Iglesias, B. A. Supported porphyrins for the photocatalytic degradation of organic contaminants in water: a review. Environmental Chemistry Letters vol. 20 731–771 Preprint at 10.1007/s10311-021-01344-2 (2022).
- 16.Chen, Y., Li, A., Huang, Z. H., Wang, L. N. & Kang, F. Porphyrin-based nanostructures for photocatalytic applications. Nanomaterials vol. 6 Preprint at 10.3390/nano6030051 (2016). [DOI] [PMC free article] [PubMed]
- 17.La, D. D., Rananaware, A., Salimimarand, M. & Bhosale, S. V. Well–dispersed assembled porphyrin nanorods on graphene for the enhanced photocatalytic performance. ChemistrySelect1, 4430–4434 (2016). [Google Scholar]
- 18.Ussia, M. et al. Hybrid nickel-free graphene/porphyrin rings for photodegradation of emerging pollutants in water. RSC Adv9, 30182–30194 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fang, H. et al. Electrostatic Assembly of Porphyrin-Functionalized Porous Membrane toward Biomimetic Photocatalytic Degradation Dyes. ACS Omega5, 8707–8720 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sułek, A., Pucelik, B., Kuncewicz, J., Dubin, G. & Dąbrowski, J. M. Sensitization of TiO2 by halogenated porphyrin derivatives for visible light biomedical and environmental photocatalysis. Catal Today335, 538–549 (2019). [Google Scholar]
- 21.Rebelo, S. L. H. et al. Photodegradation of atrazine and ametryn with visible light using water soluble porphyrins as sensitizers. Environ Chem Lett5, 29–33 (2007). [Google Scholar]
- 22.Tsolekile, N. et al. Synthesis of meso-tetra-(4-sulfonatophenyl) porphyrin (TPPS4) - CuInS/ZnS quantum dots conjugate as an improved photosensitizer. Int J Nanomedicine14, 7065–7078 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jadhav, R. W., La, D. D., Nguyen, C. Q. & Bhosale, S. V. Supramolecular nanoarchitectonics with TPPS porphyrin as a fluorescent probe for detection of aminoglycoside antibiotics and their photocatalytic applications for the degradation of rhodamine B dye. J Photochem Photobiol A Chem437, (2023).
- 24.Cai, J. H. et al. Photodegradation of 1,5-dihydroxynaphthalene catalyzed by meso-tetra(4-sulfonatophenyl)porphyrin in aerated aqueous solution. J Mol Catal A Chem292, 49–53 (2008). [Google Scholar]
- 25.Gmurek, M. TPPS4—Sensitized Photooxidation of Micropollutants—Singlet Molecular Oxygen Kinetic Study. Molecules27, (2022). [DOI] [PMC free article] [PubMed]
- 26.Teixeira, R. et al. Fluorescence spectroscopy of porphyrins and phthalocyanines: Some insights into supramolecular self-assembly, microencapsulation, and imaging microscopy. Molecules vol. 26 Preprint at 10.3390/molecules26144264 (2021). [DOI] [PMC free article] [PubMed]
- 27.Kim, T., Ham, S., Lee, S. H., Hong, Y. & Kim, D. Enhancement of exciton transport in porphyrin aggregate nanostructures by controlling the hierarchical self-assembly. Nanoscale10, 16438–16446 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Yushmanov, V. E., Imasato, H., Tominaga, T. T. & Tabak, M. ’H NMR and Electronic Absorption Spectroscopy of Paramagnetic Water-Soluble Meso-Tetraarylsubstituted Cationic and Anionic Metalloporphyrins. [DOI] [PubMed]
- 29.Taniguchi, M., Lindsey, J. S., Bocian, D. F. & Holten, D. Comprehensive review of photophysical parameters (ε, Φf, τs) of tetraphenylporphyrin (H2TPP) and zinc tetraphenylporphyrin (ZnTPP) – Critical benchmark molecules in photochemistry and photosynthesis. Journal of Photochemistry and Photobiology C: Photochemistry Reviews vol. 46 Preprint at 10.1016/j.jphotochemrev.2020.100401 (2021).
- 30.Pedzinski, T., Markiewicz, A. & Marciniak, B. Photosensitized oxidation of methionine derivatives. Laser flash photolysis studies. Research on Chemical Intermediates35, 497–506 (2009).
- 31.Borissevitch, I. E. et al. An Alternative Method to Determine the Quantum Yield of the Excited Triplet State Using Laser Flash Photolysis. Photonics10, 409 (2023). [Google Scholar]
- 32.De Napoli, M. et al. Hierarchical Porphyrin Self-Assembly in Aqueous Solution. J Am Chem Soc126, 5934–5935 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Snitka, V., Rackaitis, M. & Rodaite, R. Assemblies of TPPS4 porphyrin investigated by TEM, SPM and UV–vis spectroscopy. Sens Actuators B Chem109, 159–166 (2005). [Google Scholar]
- 34.Simpson, M. C. & Novikova, N. I. Porphyrins: Electronic Structure and Ultraviolet/Visible Absorption Spectroscopy. in Fundamentals of Porphyrin Chemistry 505–586 (2022). 10.1002/9781119129301.ch11.
- 35.Hanyż, I. & Wróbel, D. The influence of pH on charged porphyrins studied by fluorescence and photoacoustic spectroscopy. Photochemical & Photobiological Sciences1, 126–132 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Kim, T., Ham, S., Lee, S. H., Hong, Y. & Kim, D. Supporting Information for Enhancement of Exciton Transport in Porphyrin Aggregate Nanostructures by Controlling Hierarchical Self-Assembly. (2018). [DOI] [PubMed]
- 37.Williams, A. T. R., Winfield, S. A. & Miller, J. N. Relative fluorescence quantum yields using a computer-controlled luminescence spectrometer. Analyst108, 1067–1071 (1983). [Google Scholar]
- 38.Uttamlal, M. & Sheila Holmes-Smith, A. The excitation wavelength dependent fluorescence of porphyrins. Chem Phys Lett454, 223–228 (2008).
- 39.Parra, G. G. et al. Effects of meso-tetrakis (4-sulfonatophenyl) porphyrin (TPPS4) aggregation on its spectral and kinetic characteristics and singlet oxygen production. Spectrochim Acta A Mol Biomol Spectrosc261, 120063 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Lakowicz, J. R. & Weber, G. Quenching of protein fluorescence by oxygen. Detection of structural fluctuations in proteins on the nanosecond time scale. Biochemistry12, 4171–4179 (1973). [DOI] [PMC free article] [PubMed]
- 41.Jiménez-Banzo, A., Ragàs, X., Kapusta, P. & Nonell, S. Time-resolved methods in biophysics. 7. Photon counting vs. analog time-resolved singlet oxygen phosphorescence detection. Photochemical & Photobiological Sciences7, 1003–1010 (2008). [DOI] [PubMed]
- 42.Westberg, M., Bregnhøj, M., Etzerodt, M. & Ogilby, P. R. No Photon Wasted: An Efficient and Selective Singlet Oxygen Photosensitizing Protein. J Phys Chem B121, 9366–9371 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Lei, W., Jiang, G., Zhou, Q., Zhang, B. & Wang, X. Greatly enhanced binding of a cationic porphyrin towards bovine serum albumin by cucurbit[8]uril. Physical Chemistry Chemical Physics12, 13255–13260 (2010). [DOI] [PubMed] [Google Scholar]
- 44.Jakimińska, A., Pawlicki, M. & Macyk, W. Photocatalytic transformation of Rhodamine B to Rhodamine-110 – The mechanism revisited. J Photochem Photobiol A Chem433, 114176 (2022). [Google Scholar]
- 45.Kumari, H. et al. A Review on Photocatalysis Used For Wastewater Treatment: Dye Degradation. Water, Air, and Soil Pollution vol. 234 Preprint at 10.1007/s11270-023-06359-9 (2023). [DOI] [PMC free article] [PubMed]
- 46.Lanjwani, M. F., Tuzen, M., Khuhawar, M. Y. & Saleh, T. A. Trends in photocatalytic degradation of organic dye pollutants using nanoparticles: A review. Inorg Chem Commun159, 111613 (2024). [Google Scholar]
- 47.Parul, Kaur, K., Badru, R., Singh, P. P. & Kaushal, S. Photodegradation of organic pollutants using heterojunctions: A review. J Environ Chem Eng8, 103666 (2020).
- 48.Yin, M., Li, Z., Kou, J. & Zou, Z. Mechanism investigation of visible light-induced degradation in a heterogeneous TiO2/eosin Y/rhodamine B system. Environ Sci Technol43, 8361–8366 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Sun, Z., Zhao, M. & Zhou, X. Reliability analysis of NaN3 as quencher of 1O2 in heterogeneous persulfate catalytic oxidation system. Catal Commun183, 106774 (2023). [Google Scholar]
- 50.Khajone, V. B. & Bhagat, P. R. Synthesis of polymer-supported Brønsted acid-functionalized Zn–porphyrin complex, knotted with benzimidazolium moiety for photodegradation of azo dyes under visible-light irradiation. Research on Chemical Intermediates46, 783–802 (2020). [Google Scholar]
- 51.Rilda, Y. et al. The use of the low-temperature sol–gel method for ZnO-TiO2 nanorods synthesis: structural analysis, morphology and photodegradation properties of methyl orange dye with benzoquinone scavenger. Journal of the Iranian Chemical Society19, 2023–2030 (2022). [Google Scholar]
- 52.Harada, H., Hosoki, C. & Kudo, A. Overall water splitting by sonophotocatalytic reaction: the role of powdered photocatalyst and an attempt to decompose water using a visible-light sensitive photocatalyst. J Photochem Photobiol A Chem141, 219–224 (2001). [Google Scholar]
- 53.Choudhary, M., Brink, R., Nandi, D., Siwal, S. & Mallick, K. Gold nanoparticle within the polymer chain, a multi-functional composite material, for the electrochemical detection of dopamine and the hydrogen atom-mediated reduction of Rhodamine-B, a mechanistic approach. J Mater Sci52, 770–781 (2017). [Google Scholar]
- 54.Hernández-Gordillo, A. et al. Good practices for reporting the photocatalytic evaluation of a visible-light active semiconductor: Bi2O3, a case study. Catal Sci Technol9, 1476–1496 (2019). [Google Scholar]
- 55.Ecer, Ü., Şahan, T., Zengin, A. & Gubbuk, İH. Decolorization of Rhodamine B by silver nanoparticle–loaded magnetic sporopollenin: characterization and process optimization. Environmental Science and Pollution Research29, 79375–79387 (2022). [DOI] [PubMed] [Google Scholar]
- 56.He, K. et al. Fabrication of ploydopamine–kaolin supported Ag nanoparticles as effective catalyst for rapid dye decoloration. Chemosphere219, 400–408 (2019). [DOI] [PubMed] [Google Scholar]
- 57.Alamier, W. M. et al. Green Synthesis of Silver Nanoparticles Using Acacia ehrenbergiana Plant Cortex Extract for Efficient Removal of Rhodamine B Cationic Dye from Wastewater and the Evaluation of Antimicrobial Activity. ACS Omega8, 18901–18914 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gacka, E. et al. Noncovalent Porphyrin-Graphene Oxide Nanohybrids: The pH-Dependent Behavior. The Journal of Physical Chemistry C123, 3368–3380 (2019). [Google Scholar]
- 59.Wojcik, A. & Kamat, P. V. Reduced Graphene Oxide and Porphyrin. An Interactive Affair in 2-D. ACS Nano4, 6697–6706 (2010). [DOI] [PubMed]
- 60.Aly, S. M., Parida, M. R., Alarousu, E. & Mohammed, O. F. Ultrafast electron injection at the cationic porphyrin–graphene interface assisted by molecular flattening. Chemical Communications50, 10452–10455 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its Supplementary Information file).