

Abstract
Different ligands stabilize specific conformations of the angiotensin II type 1 receptor (AT1R) that direct distinct signaling cascades mediated by heterotrimeric G proteins or β-arrestin. These different active conformations are thought to engage distinct intracellular transducers due to differential phosphorylation patterns in the receptor C-terminal tail (the “barcode” hypothesis). Here, we identified the AT1R barcodes for the endogenous agonist AngII, which stimulates both G protein activation and β-arrestin recruitment, and to a synthetic biased agonist that only stimulates β-arrestin recruitment. The endogenous and β-arrestin–biased agonists induced two different ensembles of phosphorylation sites along the C-terminal tail. The phosphorylation of eight serine and threonine residues in the proximal and middle portions of the tail was required for full β-arrestin functionality, whereas phosphorylation of the serine and threonine residues in the distal portion of the tail had little influence on β-arrestin function. Similarly, molecular dynamics simulations showed that the proximal and middle clusters of phosphorylated residues were critical for stable β-arrestin–receptor interactions. These findings demonstrate that ligands that stabilize different receptor conformations induce different phosphorylation clusters on the C-terminal tail as barcodes to evoke distinct receptor-transducer engagement, receptor trafficking, and signaling.
Introduction
The type 1 angiotensin II receptor (AT1R), which is stimulated by its endogenous ligand angiotensin II (AngII), is a G protein–coupled receptor (GPCR) that is a vital therapeutic target for various cardiovascular diseases[1]. Understanding the mechanism of GPCR activation and the resulting intracellular signaling is of fundamental importance considering that they represent the largest class of cell-surface receptors and are major therapeutic targets[2]. GPCRs are transmembrane proteins made of a single polypeptide chain that consists of unique extracellular and intracellular domains formed by seven transmembrane helices. They function as environmental sensors to transduce extracellular stimuli to activate intracellular signaling. The mechanism of GPCR activation involves ligand binding to the extracellular domain to induce a conformational change through movement of the transmembrane helices and engagement of signaling transducers such as heterotrimeric G proteins or β-arrestin to activate intracellular signaling cascades. Silencing or desensitization of G protein signaling following activation is largely achieved through phosphorylation of amino acid residues on the GPCR C-terminal tail by G protein–coupled receptor kinases (GRKs). Receptor tail phosphorylation allows tight interactions with β-arrestin, which terminates membrane G protein signaling by sterically blocking the binding site on the GPCR. In addition to its desensitization function, β-arrestin initiates β-arrestin-dependent signaling, receptor internalization, and sustained G protein signaling from internalized GPCRs [1, 3].
In addition to stimulating G protein signaling, ligand binding can also lead to the activation of β-arrestin–dependent signaling pathways. Biased signaling, the selective activation of some of these signaling pathways and not others by a GPCR, can be due to changes at the level of the ligand, receptor, or downstream transducers. The ability of different ligands to stabilize distinct active receptor conformations to promote biased signaling is referred to as biased agonism[4]. These structurally distinct, ligand-stabilized, active conformations recruit unique subsets of GRKs, leading to differential phosphorylation patterns on the C-terminal tail of the receptor, a concept referred to as the “barcode” hypothesis[5–7]. For example, the amount of GRK2 and GRK6 recruited to the β2-adrenergic receptor drives unique C-terminal tail phosphorylation following receptor activation with different ligands[7]. it is important of fully understand mechanisms of receptor phosphorylation because different phosphorylation “barcodes” appear to stabilize distinct active conformations of β-arrestin[8, 9], leading to the activation of unique signaling downstream cascades[10] and physiological responses[11]. Phosphorylation of the receptor C-terminal tail appears to determine β-arrestin affinity for the receptor, allowing β-arrestin to interact with a GPCR in a fully engaged high-affinity transmembrane core state or partially engaged low-affinity hanging state determined by the stability and duration of the GPCR–β-arrestin macromolecular complex[12–14]. Indeed, the pattern of C-terminal tail phosphorylation correlates with the arrestin-binding properties of the receptor[15, 16] suggesting that regional stoichiometry may determine the resulting activation of β-arrestin–dependent signaling pathways[17]. Although in vitro and molecular dynamics studies have demonstrated that different patterns of GPCR phosphorylation promote distinct receptor conformations[18, 19], there is a paucity of experimental evidence on which sites are actually phosphorylated in a GPCR C-terminal tail and their impact on β-arrestin conformation and activity.
Several lines of evidence point to the critical role of phosphorylation of specific amino acid residues in the C-terminal tail of the AT1R in the regulation of β-arrestin function. Truncation of the AT1R carboxyl terminus or mutation of phosphorylatable serine and threonine residues in the proximal and central regions of the carboxyl terminus (Thr332, Ser335, Thr336, Ser338) results in a weaker, less stable interaction with β-arrestin along with reduced receptor internalization[20, 21] and diminished β-arrestin–dependent activation of the mitogen-activated protein kinase (MAPK) extracellular signal–regulated kinase (ERK) [21]. In contrast, mutation of protein kinase C (PKC) phosphorylation sites (Ser331, Ser338, Ser348) or distal non-PKC phosphorylatable residues (Ser346, Ser347, Ser348) does not affect β-arrestin interaction or AT1R internalization[20]. In an investigation of the interaction between AT1R and β-arrestin following receptor stimulation using a mutated photoreactive receptor, the endogenous ligand AngII and the β-arrestin–biased ligand TRV027 elicited similar photo-crosslinking of AT1R residues but with varying intensity[22]. Moreover, important residues for β-arrestin binding were identified in intracellular loops 2 and 3 (ICL2 and ICL3), helix 8, and the proximal and middle regions the C-tail of the receptor[22].
The importance of GRK phosphorylation of the AT1R tail has been demonstrated with differential effects on β-arrestin recruitment, AT1R β-arrestin complex formation and β-arrestin–mediated ERK activation depending on whether the C-tail is phosphorylated by GRK2 or GRK3 (GRK2/3) or GRK5 or GRK6 (GRK5/6)[5, 23, 24]. Activation of the Gq heterotrimer appears to be critical for GRK2/3 recruitment to the AT1R[24]. As a result, only GRK5/6 contributes to AT1R phosphorylation[20, 25, 26], β-arrestin binding[20], and β-arrestin–mediated signaling that is promoted by β-arrestin–biased ligands of the AT1R, which do not to activate Gq[24].
Like other GPCRs, AT1R initiates intracellular signaling and physiological responses through both G protein and β-arrestin–dependent mechanisms, linking distinct GRK-mediated receptor phosphorylation events to cardiomyocyte survival and cardiac function[11, 27, 28]. At a structural level, the endogenous angiotensin II peptide engages the AT1R through an extensive ligand-binding cavity[29], thereby stimulating both G protein–mediated and β-arrestin–mediated responses, whereas synthetic biased ligands of different functional classes show distinct effects on the conformational heterogeneity of the receptor[30], resulting in preferential activation of G protein–mediated or β-arrestin–mediated responses.
Given the importance of understanding the fundamental mechanisms of AT1R signaling, we set out to determine whether an antagonist and both full and β-arrestin–biased AT1R ligands induce different receptor phosphorylation patterns and if these patterns correlate with unique downstream effects. We provide evidence that a broad ensemble of sites are differentially phosphorylated in the AT1R C-terminal tail in response to stimulation by endogenous and β-arrestin–biased ligands. We found that many of these sites play central roles in directing β-arrestin conformation and function, with some sites also directing G protein signaling.
RESULTS
Endogenous and biased AT1R agonists induce distinct patterns of phosphorylation of the C-terminal tail
To test the hypothesis that an endogenous (unbiased) AT1R agonist and a synthetic β-arrestin–biased AT1R agonist would induce distinct patterns of phosphorylation of the C-terminal tail, we used a mass spectrometry (MS)-based approach (Fig. 1). HEK293 cells that express high amounts of FLAG-tagged human AT1R (FLAG-AT1R) [30] were grown in suspension culture and left untreated (nonstimulated, NS) or treated with the endogenous AT1R ligand angiotensin II (AngII) or the β-arrestin–biased agonist TRV023, a peptide analogue of AngII, to induce receptor phosphorylation. An AT1R antagonist, telmisartan (Telm), was also included in the experiment protocol as a negative control. AT1R was enriched from cell lysates using FLAG-tag affinity chromatography and the purified receptor was processed for MS using conventional proteomic sample preparation techniques, including trypsin digestion and Tandem Mass Tag (TMT) reporter[31] labeling before analysis by liquid chromatography with tandem MS. After filtering the complete phosphoisomer dataset (table S2) for quality, we identified 49 unique phosphoisomers, 38 of which were found in the C-terminal tail (Fig. 2A).
Fig. 1. Schematic representation of LC-MS/MS sample preparation and analysis.
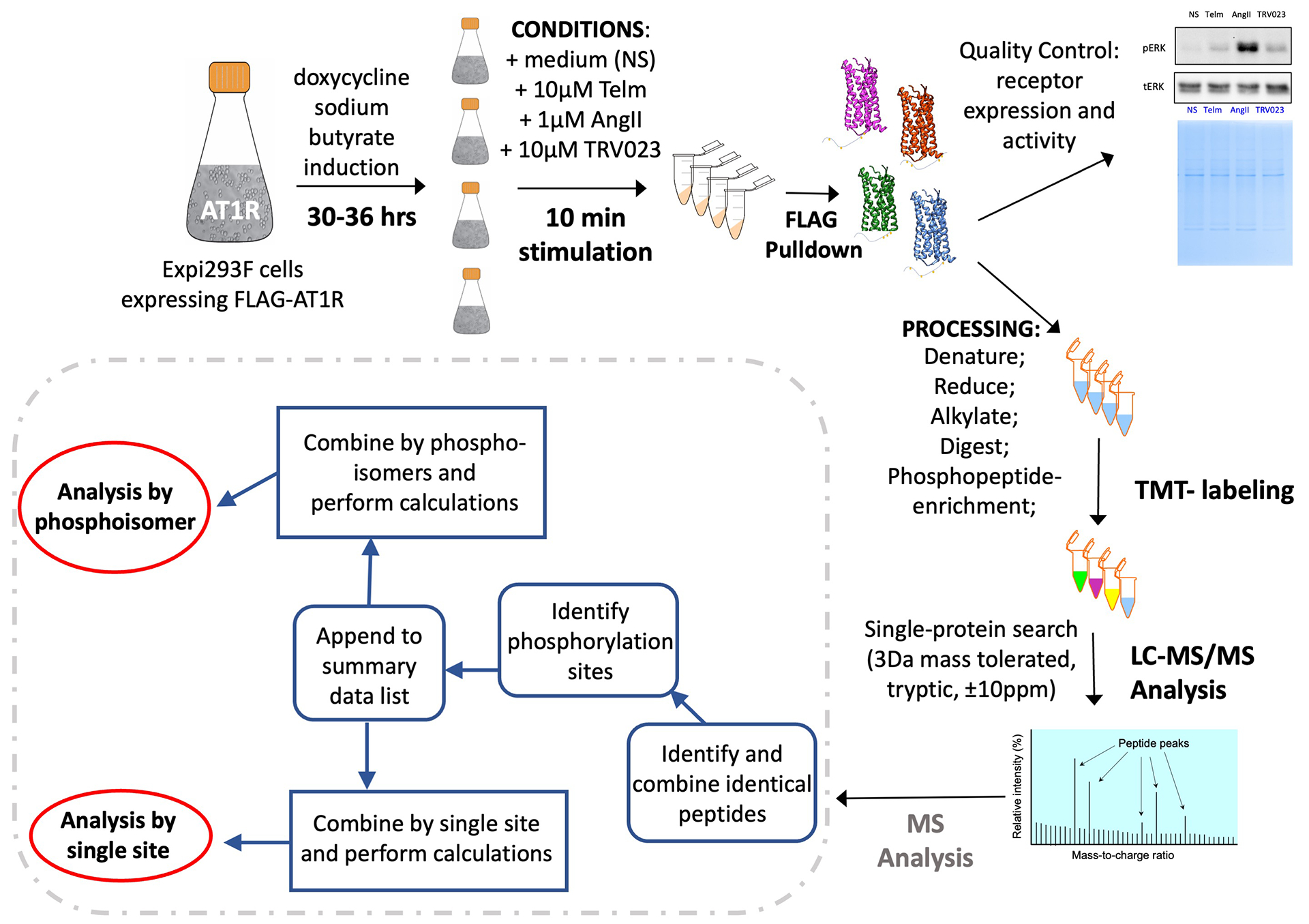
Expi293F™ cells were induced to express N-terminal FLAG-tagged human WT AT1R (FLAG-AT1R) for 30–36h. Separate cell suspensions were then stimulated for 10 mins with the endogenous ligand AngII, the β-arrestin–biased ligand TRV023, or pretreated with the AT1R antagonist Telm for 24h. A separate cell suspension was not stimulated (NS). The receptor was purified by FLAG pulldown, digested, and labeled with TMT probes. The peptides were enriched for the phosphopeptides by High-Select Fe-NTA Phosphopeptide Enrichment Kit and analyzed by liquid chromatography-tandem MS. n = 5 experiments for each treatment condition. The MS data have been combined and analyzed by phosphoisomers or single phosphorylation site.
Fig. 2. Identification of ligand-specific AT1R C-terminal tail phosphorylation patterns.
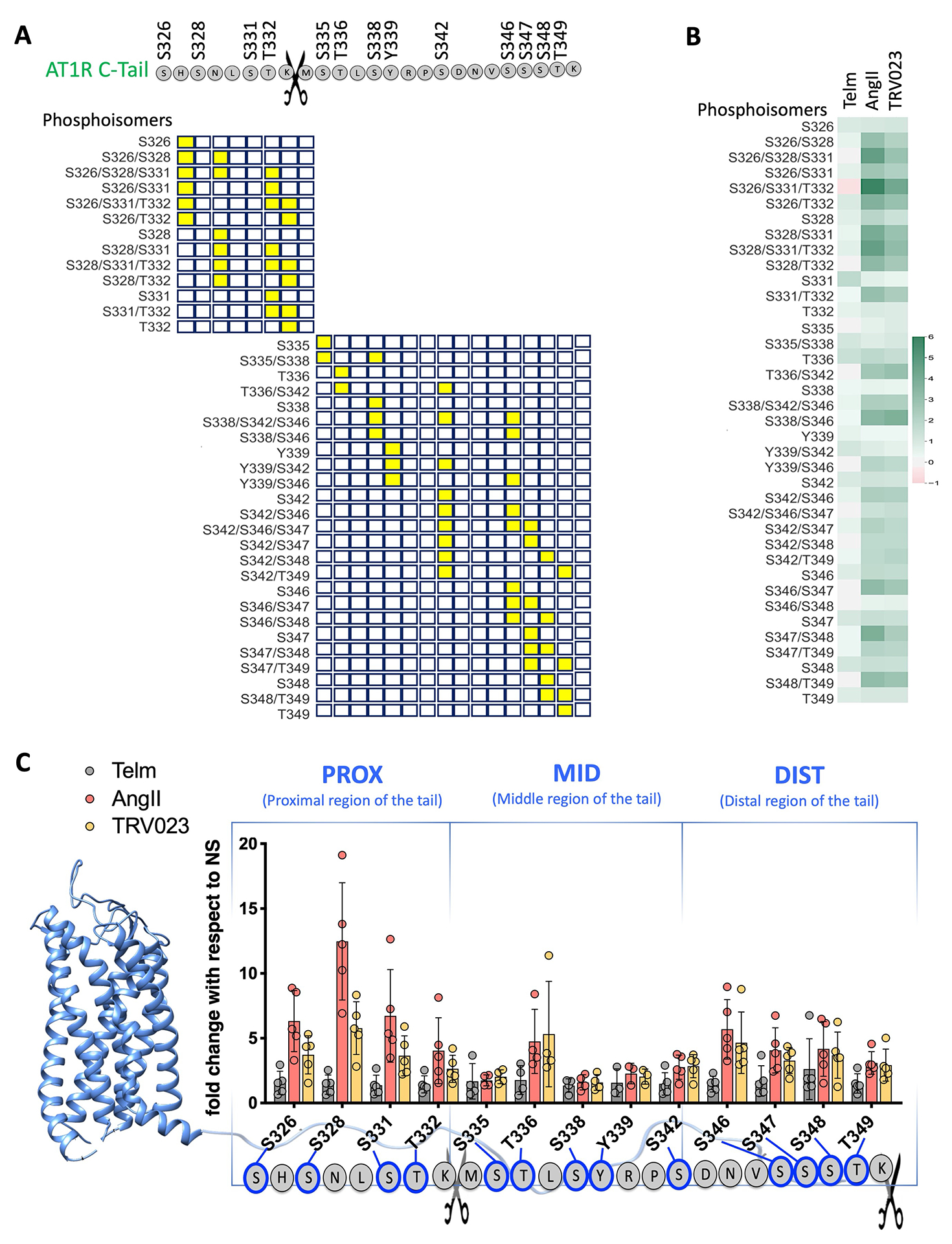
(A) Graphical representation of the location of phosphorylated sites (yellow squares) along the C-terminal tail peptides (open squares). Scissors indicate the trypsin cleavage site. (B) Heatmap showing the amounts of phosphorylation of the indicated AT1R C-terminal tail phosphoisomers in AT1R-expressing Expi293F™ cells stimulated the AT1R inhibitor Telm, the full AT1R agonist Angiotensin II (AngII), and the β-arrestin-biased AT1R agonist TRV023. Quantitative data were obtained by MS and are reported as fold change compared to cells that were not stimulated (NS). All of the phosphoisomers identified by MS after post-database searching analysis are included in the heatmap. N=5 independent experiments per treatment group. (C) Intensity of each single phosphosite in the proximal (PROX), middle (MID), and distal (DIST) portions of the AT1R C-terminal tail after stimulation with Telm, AngII, or TRV023, as determined from the single-site analysis (see Materials and Methods). N=5 independent experiments per treatment group.
We identified 13 sites of phosphorylation in the C-terminal tail: Ser326, Ser328, Ser331, Thr332, Ser335, Thr336, Ser338, Tyr339, Ser342, Ser346, Ser347, Ser348, Thr349 (Fig. 2A). Both ligands increased phosphorylation of the C-terminal tail overall and substantially increasing the quantity of multiply phosphorylated peptides, indicating phosphorylation clusters or ensembles. The magnitude of increase was greatest in the proximal region of the tail containing the phosphorylation sites Ser326, Ser328, Ser331, and Thr332 following receptor activation by AngII and less so by TRV023 (Fig. 2B). Specifically, triply phosphorylated peptides Ser326/Ser328/Ser331 and Ser326/Ser331/The332 and doubly phosphorylated peptides, such as Ser328/Ser331 and Ser331/Thr332, showed increases over NS up to 6-fold for AngII and 4.2-fold for TRV023 (Fig. 2B). As expected, the phosphorylation amounts in response to Telm were similar to those in unstimulated cells.
Dividing the AT1R C-terminal tail into three regions based on the proximity to the beginning of the tail [proximal (PROX), middle (MID), and distal (DIST)], we found that in response to AngII the greatest increase in receptor phosphorylation occurred in the PROX region, whereas in response to TRV023 the PROX region was phosphorylated to much lesser degree (Fig. 2C). The MID region showed a substantial change in phosphorylation at Thr336, especially in combination with Ser342 (Fig. 2,B and C). Furthermore, we observed that both AngII and TRV023 induced comparable amounts of phosphorylation in the DIST region. These data suggest that AngII and TRV023 induce distinct patterns of phosphorylation clusters along the C-terminal tail. Although both ligands induced similar amounts of phosphorylation in the middle and distal regions of the C-terminal tail, AngII induced more robust phosphorylation of the proximal tail compared to TRV023.
Phosphorylation of both proximal and middle C-terminal tail residues in AT1R is required for β-arrestin recruitment and receptor internalization
To determine the cluster of phosphorylated residues on the C-terminal tail that is required for ligand-induced β-arrestin recruitment, we generated AT1R mutants by using site-directed mutagenesis to replace different clusters of phosphorylated Ser and Thr residues with Ala (Fig. 3, A and B). The proximal residues Ser326, Ser328, Ser331, and Thr332 were replaced in the AT1R-PROX mutant protein; middle residues Ser335, Thr336, Ser338, and Ser342 were replaced in AT1R-MID; distal residues Ser346, Ser347, Ser348, and Thr349 were replaced in AT1R-DIST and all 12 phosphorylatable Ser and Thr amino acids in the C-terminal tail were replaced in AT1R-NULL. Additional mutation combinations were also generated (Fig. 3C).
Fig. 3. AT1R C-terminal tail mutants generated in this study.
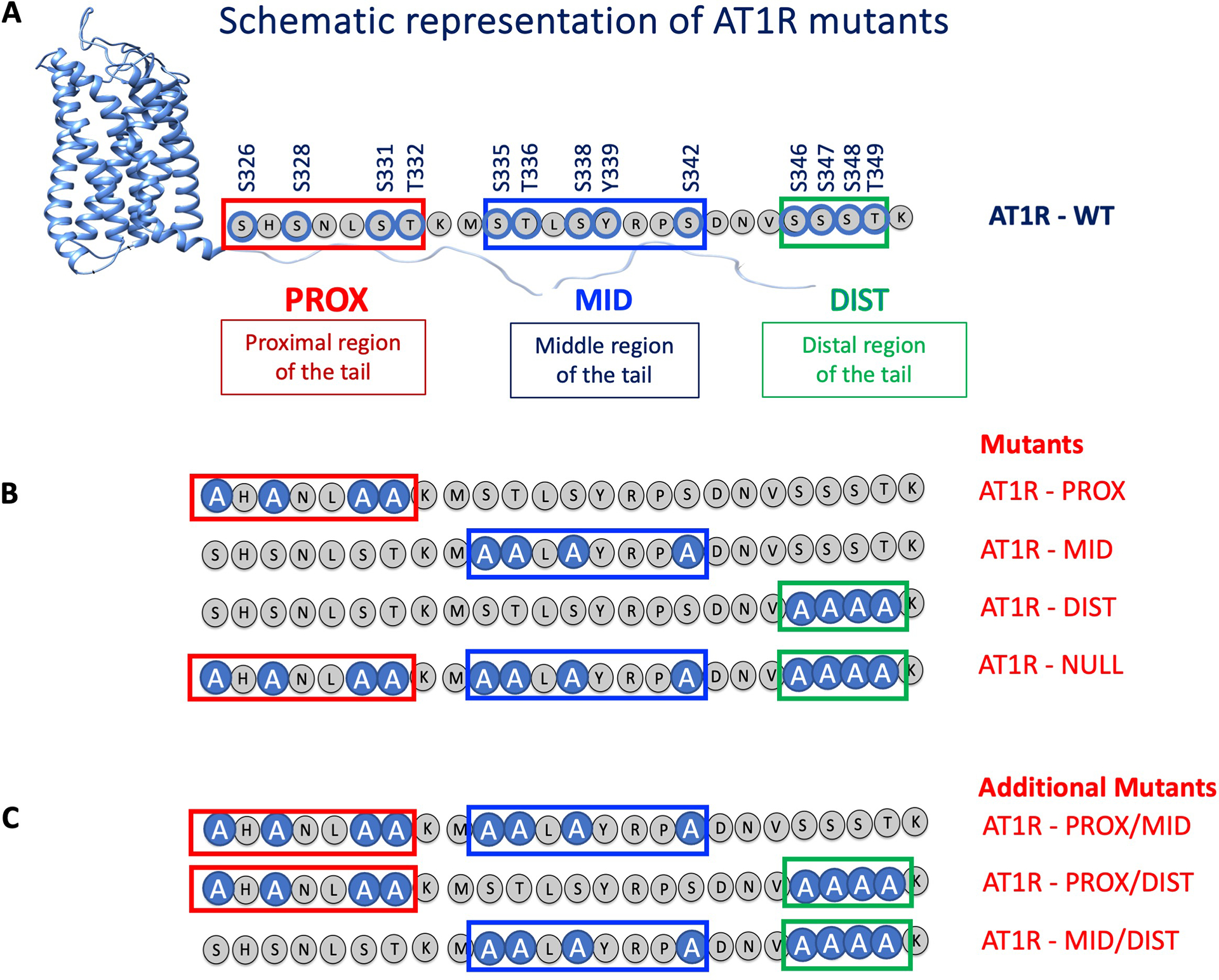
(A) Schematic showing the serine, threonine, and tyrosine residues in the proximal (PROX), middle (MID), and distal (DIST) regions of the C-terminal tail of AT1R that were phosphorylated upon receptor stimulation. (B) All of the phosphorylatable serine and threonine residues within a region were substituted with alanine in the AT1R-PROX, AT1R-MID and AT1R-DIST mutant proteins. All the phosphorylatable serine and threonine sites were changed to alanine in the AT1R-NULL mutant protein. (C) Additional AT1R mutants with a combination of substitutions in two regions were also generated, AT1R-PROX/MID, AT1R-PROX/DIST, and AT1R-MID/DIST.
We measured β-arrestin recruitment by bioluminescence resonance energy transfer (BRET) in response to ligand activation in HEK293 cells cotransfected with wild-type (WT) or mutant AT1R fused to RLuc8 and β-arrestin fused to enhanced green fluorescent protein (eGFP) (Fig. 4A). AngII induced robust β-arrestin recruitment to WT AT1R and the AT1R-DIST mutant, but its recruitment was reduced by 50% if either the PROX sites or MID sites were mutated (Fig. 4B). A similar pattern was observed for TRV023, although the overall response was less robust than that for AngII (Fig. 4B). Mutation of all 12 Ser and Thr phosphorylation sites (AT1R-NULL) largely abrogated β-arrestin recruitment by either ligand (Fig. 4B). Additional mutation combinations showed results similar to those with the selective cluster mutants (fig. S1, A to F).
Fig. 4. β-arrestin recruitment to AT1R C-terminal tail PROX and MID mutants is reduced.
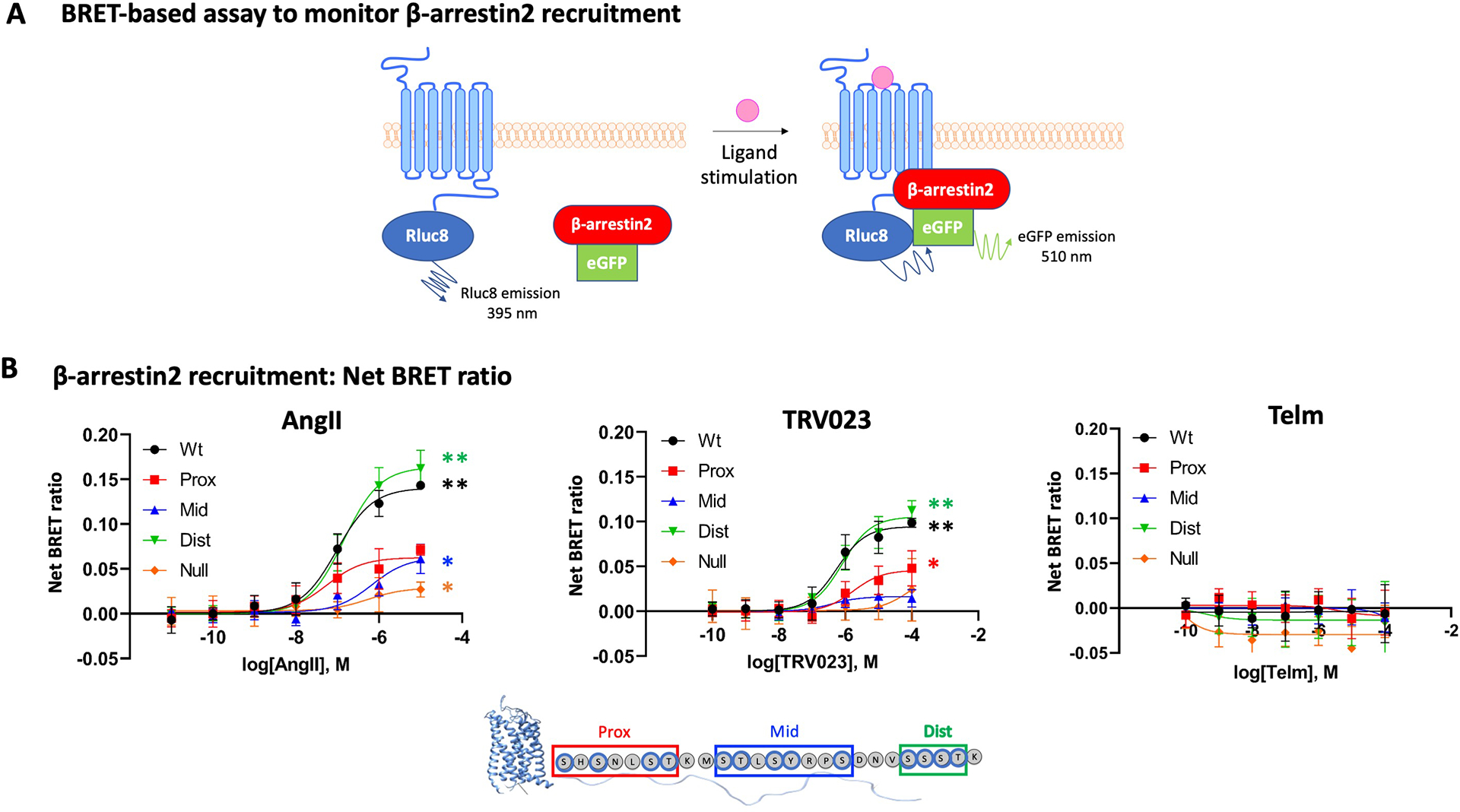
(A) Illustration of the BRET-based assay to monitor β-arrestin recruitment. RLuc8-tagged AT1R is the energy donor, and eGFP-tagged β-arrestin is the energy acceptor. Ligand-induced recruitment of β-arrestin to AT1R increases the BRET signal. (B) HEK293 cells were transfected with RLuc8-tagged WT or mutant AT1R proteins and β-arrestin-eGFP then treated with AngII, TRV023, or Telm at the indicated concentrations for 10 min before BRET measurement. The net change BRET ratio (eGFP:RLuc8) is expressed as the difference between ligand-treated and untreated groups. Statistical comparisons were performed using two-way ANOVA with Sidak correction for multiple comparisons test. N=5 (AngII and TRV023) or 3 (Telm) independent experiments per treatment group. Data are shown as mean±SD. *P< 0.05, **P<0.001, ligand at highest concentration vs. untreated group.
We next tested the cluster of amino acid residues required to support β-arrestin–mediated internalization of the AT1R following ligand activation as measured by proximity-based (BRET) (Fig. 5, A and B) and enzyme complementation–based (DiscoverX) endocytosis assays (Fig. 5, C and D). Concordant with our observations for β-arrestin recruitment, phosphorylation of both PROX and MID amino acid clusters by either AngII or TRV023 was required for AT1R internalization (Fig. 5, B and D), whereas phosphorylation of the DIST sites was largely dispensable (Fig. 5, B and D). The AT1R-null was unable to support any appreciable β-arrestin internalization following stimulation by either ligand (Fig. 5, C and D; fig. S2, A and B). Taken together, these data indicate that, for both the endogenous agonist AngII and the β-arrestin–biased agonist TRV023, phosphorylation of key residues in both the PROX and the MID regions of the AT1R C-terminal tail is necessary to support full β-arrestin recruitment and internalization.
Fig. 5. AT1R C-terminal tail PROX and MID mutants prevent β-arrestin internalization.
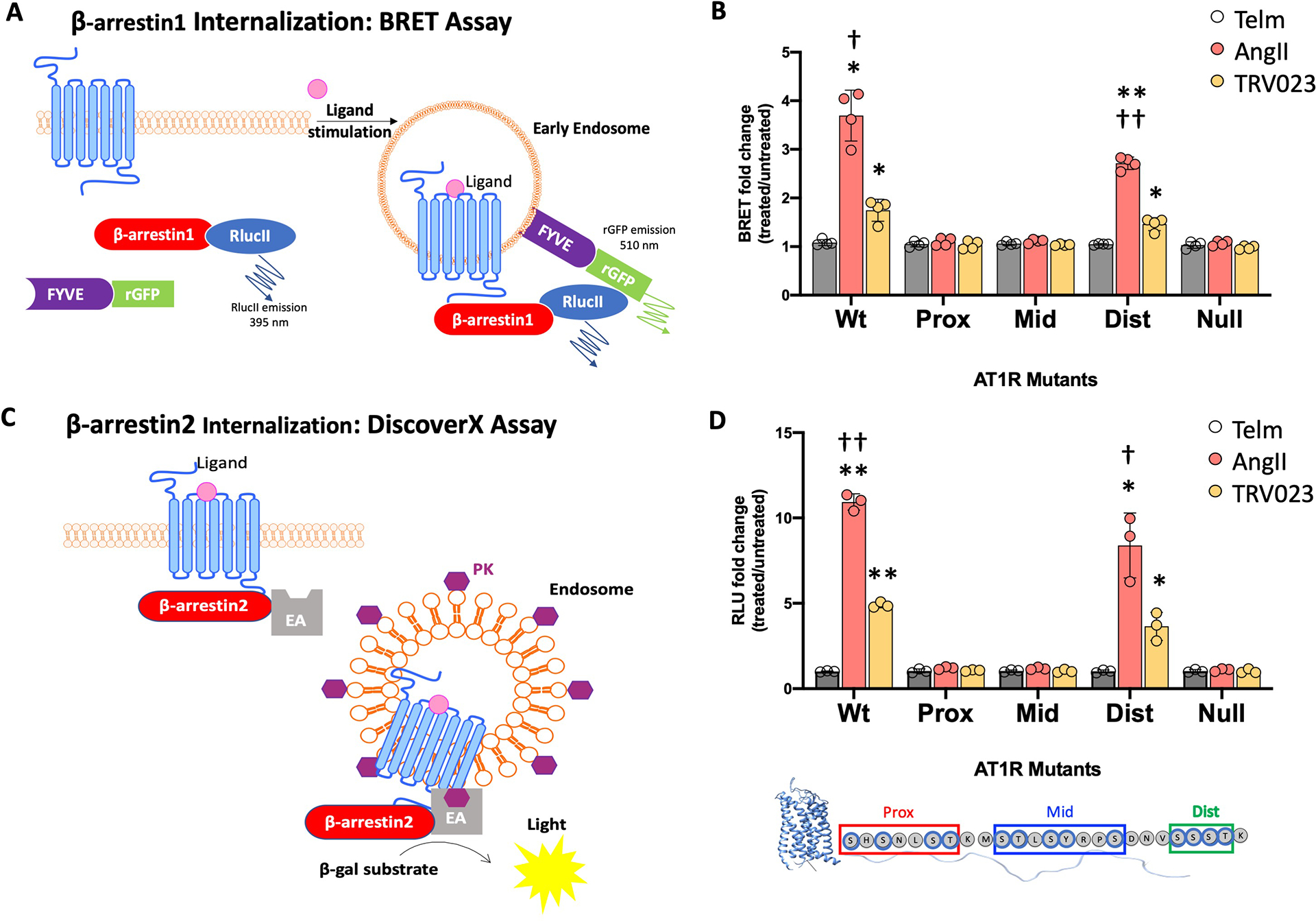
(A) Illustration of the BRET-based assay to monitor β-arrestin internalization. HEK293 cells were cotransfected with AT1R, the BRET donor β-arrestin1-RlucII, and the endosome-localized BRET acceptor FYVE-eGFP. Ligand-induced internalization of AT1R increases the BRET signal. (B) Ligand-induced changes in the BRET ratio in HEK293 cells coexpressing β-arrestin1-RlucII, FYVE-eGFP, and the indicated WT or mutant forms of AT1R. The BRET changes upon treatment with AngII, TRV023, or Telm are expressed as a fold change of the BRET ratio observed in the untreated cells. N = 4 independent experiments per group. Data represent the mean ± SD. (C) Illustration of the DiscoverX assay to monitor β-arrestin internalization. This assay is based on reconstitution of β-galactosidase (β-gal) activity by the interaction of enzyme acceptor (EA) and donor (ED) fragments. AT1R was transfected into U2OS cells that stably express EA-tagged β-arrestin2 and an endosome-localized ProLink ED (PK). Activation of the AT1R induces β-arrestin2 recruitment, followed by internalization of the At1R–β-arrestin-EA complex in PK-tagged endosomes. The resulting functional β-gal enzyme hydrolyzes substrate to generate a chemiluminescent signal. (D) Ligand-induced β-arrestin internalization in U2OS cells stably expressing β-arrestin2-EA and endosomal PK and transfected with WT or mutant AT1R. The changes in luminescence upon treatment with AngII, TRV023, or Telm are expressed as a fold of luminescence observed in untreated cells. N = 3 independent experiments per group. Data represent the mean ± SD. Statistical comparisons were performed using two-way ANOVA with Tukey’s multiple comparisons test. *P< 0.05, **P<0.005, ligand vs. untreated for same receptor mutant. †P< 0.05, ††P< 0.005 AngII vs. TRV023 for WT and DIST mutant.
Distinct ligand-induced β-arrestin conformations depend on the phosphorylation of amino acid clusters in the proximal and middle portions of the AT1R C-terminal tail
To determine the effect that each phosphorylation region plays in directing β-arrestin conformational changes, we used a BRET-based method that utilizes β-arrestin2 fluorescein arsenical hairpin (FlAsH) conformational biosensors [32] (Fig. 6A). In this assay, WT AT1R or mutant AT1R was cotransfected into HEK293 cells with each of six different FlAsH sensors reporting on different β-arrestin conformations [32]. At 48h following transfection, cells were stimulated with AngII, TRV023, or Telm. FlAsH4, which reports on β-arrestin activation[9], showed a significant difference in the BRET signal upon stimulation compared to the NS (vehicle) and Telm groups (Fig. 6, B and C), whereas the other FlAsH biosensors displayed no significant or only a modest change (fig. S3, A and B).
Fig. 6. β-arrestin2 conformations depend on phosphorylation of clusters of residues within the proximal and middle region of the C-tail.
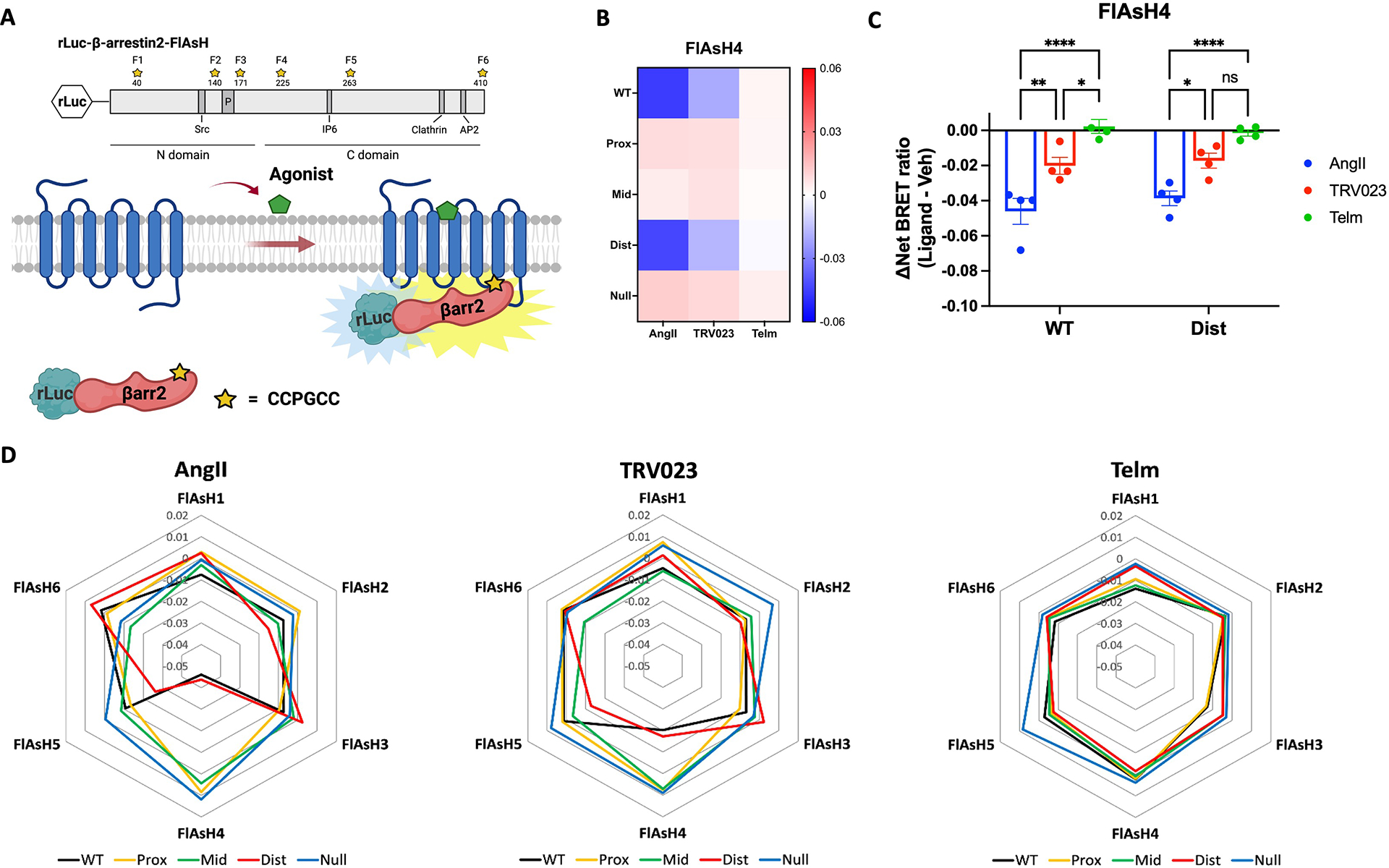
(A) Schematic of rLuc-β-arrestin2-FlAsH BRET reporters (FlAsH 1–FlAsH6, F1–F6) to detect ligand-induced conformational changes of β-arrestin2. The tetracysteine motif CCPGCC was inserted after amino acid residues 40, 140, 171, 225, 263, or 410 of β-arrestin2. This motif binds to an exogenously supplied arsenic-containing fluorescein derivative (FlAsH-EDT2) that acts as the BRET acceptor for rLuc. The change in BRET signal between the rLuc fused to the N-terminus of β-arrestin2 and the CCPGCC-targeted fluorescein arsenical acceptor located at one of the six locations within β-arrestin2 reflects β-arrestin2 conformational change. The Src, inositol hexaphosphate (IP6), clathrin, and AP2 interaction sites in β-arrestin2 are noted as well as the phosphorylation site (P). (B and C) FlAsH4 BRET signals following ligand stimulation of the indicated WT and mutant AT1R proteins. HEK293 cells were transfected with the rLuc-β-arrestin2-FlAsH4 reporter and WT or mutant AT1R. Cells were labeled with FlAsH-EDT2 or HBSS (mock labeling) and then treated with AngII (1μM), TRV023 (10 μM), Telm(10 μM), or vehicle 10 min before BRET measurement. The average BRET ratios from mock-labeled cells (background) were subtracted from those of FlAsH4-labeled cells to obtain the net BRET ratios. The ΔNet BRET ratio was calculated by subtracting the Net BRET ratio of vehicle-treated cells from the Net BRET ratio of ligand-stimulated cells. The heatmap shows the ΔNet BRET ratio for all WT and mutant AT1R proteins (B). The graph shows the ΔNet BRET ratio for WT AT1R an AT1R-DIST only (C). N = 4 independent experiments per group. (D) Radar charts showing β-arrestin2 conformational signatures from the six FlAsH sensors with WT and mutant AT1R proteins upon stimulation with AngII, TRV023, or Telm. N=4 independent experiments per group. Data represents the mean ± SEM. Statistical comparisons were performed using two-way ANOVA with Bonferroni’s multiple comparisons test. For (B), P<0.0001, overall main effect for interaction between ligand and receptor. For (C), *P< 0.05, **P<0.005, and ****P<0.0001, AngII vs. TRV023 vs. Telm group.
These differences were also demonstrated in radar plots of the relative changes in β-arrestin2 conformation as measured by BRET signals induced by different ligands binding to WT AT1R or AT1R mutants (Fig. 6D). For WT AT1R, the AngII stimulation induced a conformation in which FlAsH4 showed a significant decrease in BRET compared to the shape profile induced by Telm (Fig. 6D). TRV023 stimulation induced a shape similar to that induced by AngII but showed a smaller decrease in the FlAsH4 signal (Fig. 6D). This indicates that the β-arrestin2 conformation promoted by TRV023-bound AT1R is similar, but not identical, to the conformation induced by AngII. The FlAsH profiles of AT1R-PROX, AT1R-MID, and AT1R-NULL mutants in cells stimulated with AngII or TRV023 were similar to those in cells treated with the antagonist Telm (Fig. 6D). This is consistent with the previous experiments that indicated that β-arrestin2 recruitment to these mutated receptors was reduced and indicates that the phosphorylation of these regions is of importance in directing β-arrestin conformation and recruitment. Conversely, both AngII and TRV023 induced a β-arrestin2 FlAsH profile for the AT1R-DIST mutant that was more comparable to that of the WT AT1R (Fig. 6D), consistent with the results of this mutant in both β-arrestin recruitment and internalization assays. These data support a model in which proximal phosphorylation sites in the AT1R C-tail are principally responsible for inducing activated conformations of β-arrestin2.
Molecular dynamic simulations show how different phosphorylation patterns affect the structure of the AT1R C-terminal tail–β-arrestin 2 complex
Taking advantage of the above insights regarding the importance of the proximal and middle portions of the AT1R C-terminal tail for arrestin function and other previous reports about key phosphorylation sites [33], we generated a model showing the potential binding mode of the AT1R C-terminal tail:β-arrestin 2 complex (Fig. 7A). Using this complex, we aimed to gain structural insight into why phosphorylation of the proximal and middle C-terminal tail is crucial for β-arrestin 2 recruitment. We simulated three variants of the C-terminal tail, including a variant with a fully phosphorylated C-terminal tail (variant 1), a variant with phosphorylation in the proximal and distal segment (variant 2), and a variant with phosphorylation in the middle and distal segment (variant 3). By plotting polar contacts formed between phosphorylated residues in the fully phosphorylated C-terminal tail and β-arrestin2 (Fig. 7B), we can appreciate that the majority of phosphorylated residues engage in interactions with β-arrestin2. Residues present in the middle segment appear to form the largest number of stable interactions, followed by residues in the proximal segment, and finally those in the distal segment.
Fig. 7. The effect of distinct phosphorylation patterns on the interaction of the AT1R C-terminal tail with β-arrestin 2.
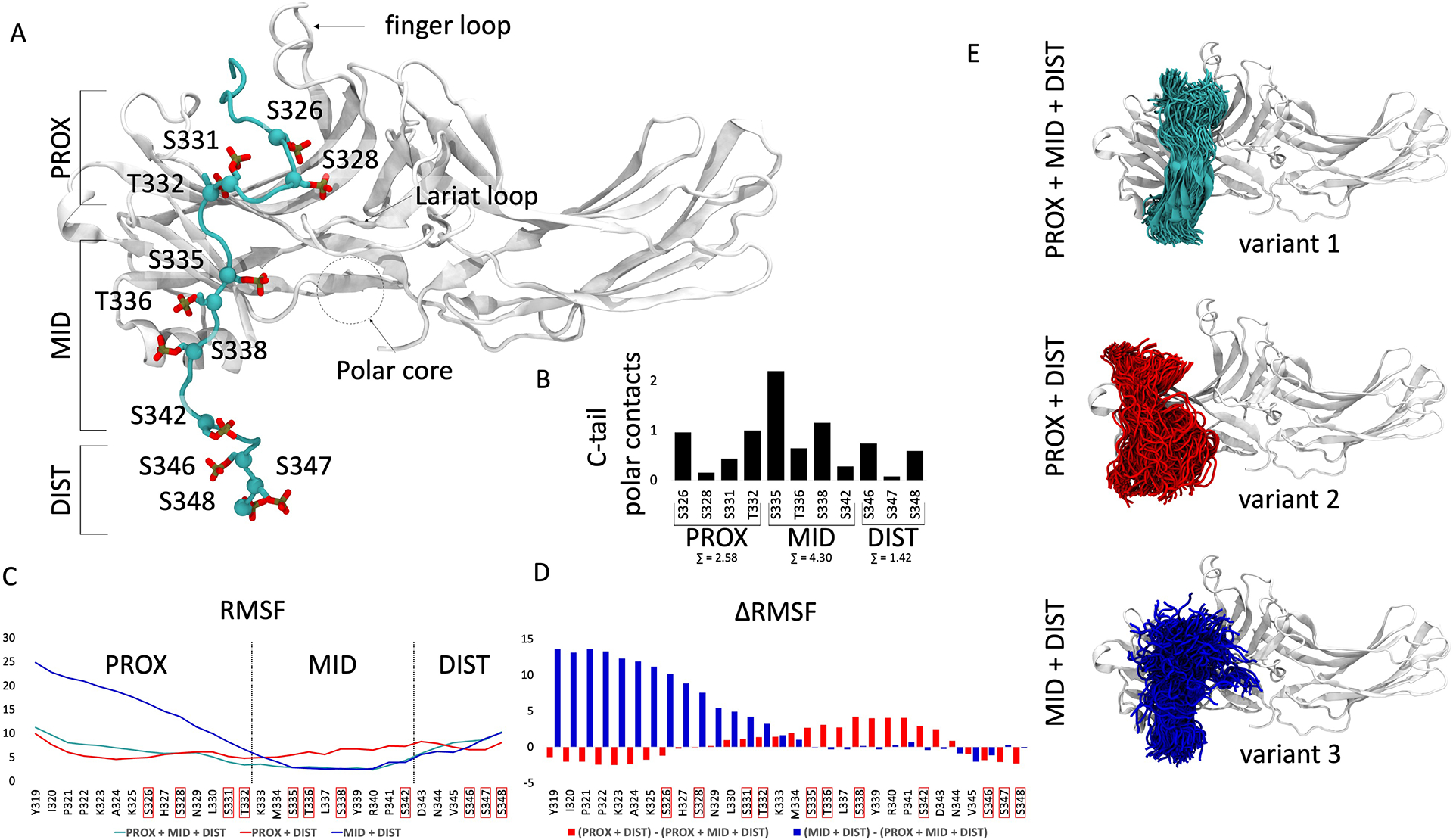
(A) Representative frame of the most populated cluster of the fully phosphorylated AT1R C-terminal tail (cyan) conformation observed in molecular dynamics simulations. Phosphorylatable Ser and Thr residues are indicated in red. Proximal (PROX), middle (MID) and distal (DIST) C-terminal tail segments are labeled. Functionally important structural features of β-arrestin 2 are noted with labels. (B) The stability of polar interactions formed by each Ser and Thr residue in the AT1R C-terminal tail with a β-arrestin 2 residue was measured (0 to 1), then the stability of each interaction formed by a C-terminal tail residue was summed and depicted on the bar plot. The sum of polar interactions formed by each region of the tail is depicted beneath the label. (C) Root mean square fluctuation (RMSF) values calculated for Cα atoms of each residue of every phosphorylation variant of the AT1R C-tail. PROX + MID + DIST, Ser and Thr residues of each segment phosphorylated; PROX +DIST, Ser and Thr residues of the PROX and DIST segments phosphorylated; MID + DIST, Ser and Thr residues of the MID and DIST segments phosphorylated. Higher values indicate less structural stability. (D) Changes in RMSF values calculated for Cα atoms of the AT1R C-terminal tail when losing phosphorylation in the PROX (blue) or MID (red) segments. (E) Snapshots of C-terminal tail conformations observed for each phosphorylation pattern. Variant 1, all PROX, MID, and DIST residues phosphorylated; variant 2, PROX and DIST residues phosphorylated; variant 3, MID and DIST residues phosphorylated. The position of the C-terminal tail was plotted once every 10 ns, resulting in 300 snapshots extracted from 3 μs of accumulated simulation time.
The motion of residues within the C-terminal tail can be approximated by plotting the root-mean-square fluctuation of RMSD (RMSF) for each residue (Fig. 7C). We found that the fully phosphorylated C-terminal tail (Fig. 7C, cyan line) showed relatively low RMSF values through all segments, which indicates high stability of those regions. Lack of phosphorylation in the middle segment led to destabilization of this region (Fig. 7B, red line), which is evident in increased RMSF values compared to the variants with phosphorylated middle segment residues (Fig. 7C, cyan and blue lines). Furthermore, lack of phosphorylation in the proximal segment resulted in the highest RMSF values and destabilization (Fig. 7C, blue line vs cyan and red lines). The differences in stability can also be appreciated when plotting ΔRMSF values for residues within the C-terminal tail (Fig 7D).
The differences between the AT1R C-terminal tail:β-arrestin 2 complex variant were also reflected when depicting conformations explored by each variant during simulations (Fig. 7E). The conformational space sampled by the variant 1 C-terminal tail was much more restricted, showing the greatest stability, compared to variants 2 and 3.
These data show that the fully phosphorylated C-terminal tail variant promotes tight interactions and forms the most stable complex with β-arrestin 2. This stability was lost for simulated variants 2 and 3 with a lack of phosphorylation in the PROX and MID segments. This was further confirmed when measuring the energy of interactions between interface residues of the C-terminal tail and β-arrestin2 in each variant. For a representative frame from variant 1 simulations, this energy equals −318.586 kcal/mol, whereas this value is less favorable for variants 2 and 3, with −166.284 kcal/mol and −141.643 kcal/mol, respectively.
The BRET-based experiments (Fig. 4B; Fig. 5, B and D) showed that C-terminal tail mutants that lack phosphorylation in the PROX and MID segments were severely impaired in their ability to recruit and internalize β-arrestin 2. Based on MD simulations and BRET-based experiments, we conclude that tight interactions are critical for β-arrestin 2 recruitment. Thus, our results provide a possible explanation for the observed experimental data, which show that loss of phosphorylation in either the PROX or MID segments reduces the ability of AT1R to recruit β-arrestin 2.
Phosphorylation events in the proximal and middle regions of the AT1R C-terminal tail have different effects on downstream ERK signaling
To determine the effect of phosphorylation of amino acid residues in the C-terminal tail on downstream signaling, we quantified ERK phosphorylation in response to treatment of WT AT1R or mutant AT1Rs with AngII, TRV023, or Telm. We transiently transfected HEK293 cells with WT AT1R or mutant constructs and stimulated with each ligand for 10 min. We observed a significant increase in ERK phosphorylation upon stimulation with AngII and a modest increase with TRV023 (Fig. 8A). All phosphorylation-deficient mutants showed a response to AngII and TRV023 that was largely similar to that of the WT AT1R with a somewhat higher activation of ERK phosphorylation for the AT1R-MID mutant.
Fig. 8. Loss of phosphorylation in the proximal and middle regions of the AT1R C-terminal tail alter G protein–mediated ERK phosphorylation.
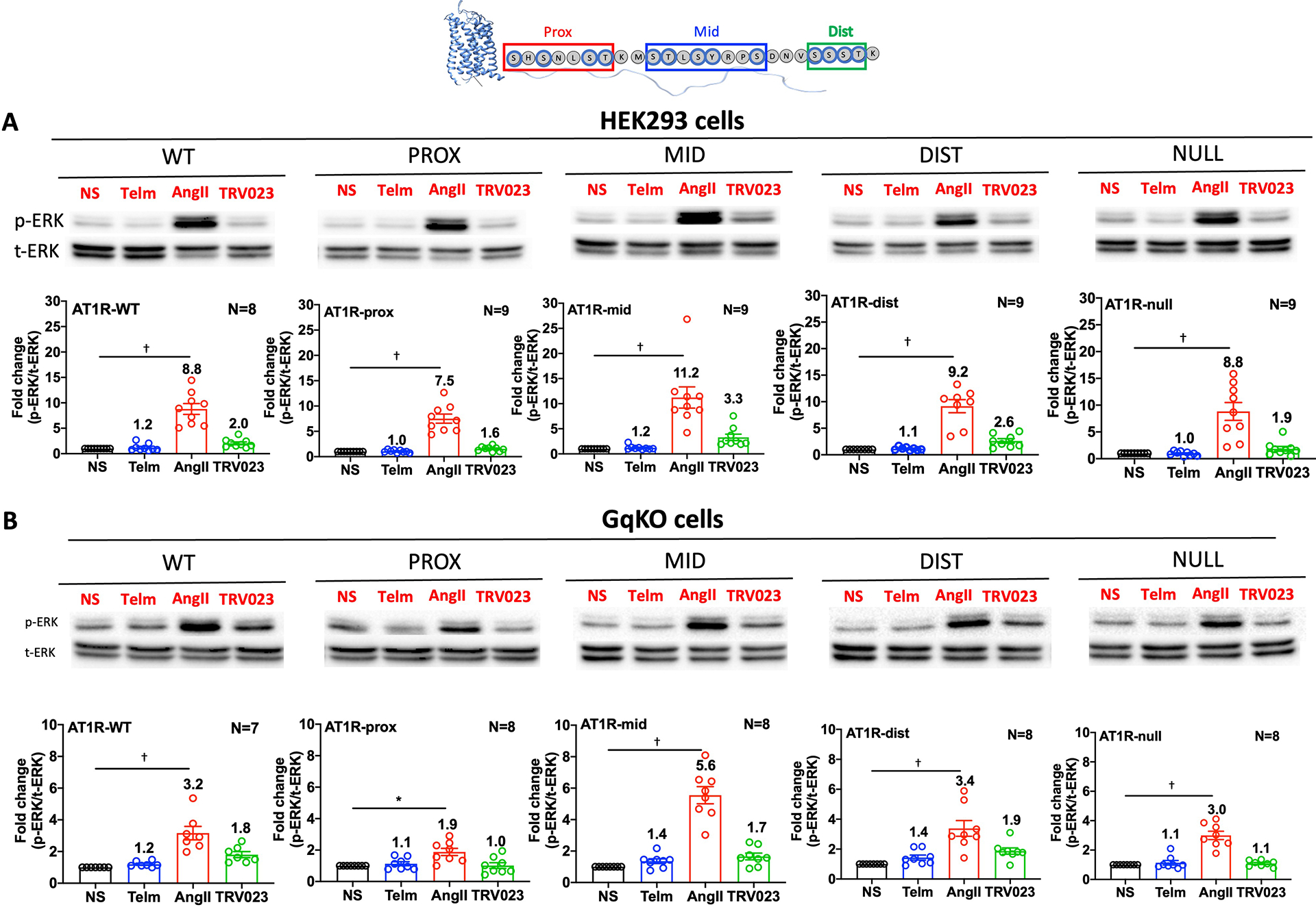
(A and B) Representative Western blots and quantification of phosphorylated ERK (p-ERK) relative to total ERK (t-ERK) in HEK293 cells (A) or HEK293 GqKO cells (B) transiently transfected with AT1R-WT, AT1R-PROX, AT1R-MID, AT1R-DIST or AT1R-NULL and starved for 3h before stimulation with AngII, TRV023 or treatments with the inhibitor Telm. N=8–9 (HEK293 cells) or N=7–8 (GqKO cells) independent experiments per group. * P< 0.05, †P< 0.005, ligand vs. untreated. Data for additional mutants can be found in the Supplementary Materials (fig. S4 and fig. S5).
Given the known promiscuity of the AT1R in using other G proteins, such as Gi, as transducers [34], we next evaluated the amount of ERK phosphorylation in HEK293 cells lacking Gq (GqKO). Activation of ERK by AngII and TRV023 was substantially lower in GqKO cells compared to WT cells, with the PROX mutant showing the most impairment relative to WT cells (Fig. 8B). Treatment with Telm did not induce ERK activation. Data for all AT1R mutants are shown in the Supplementary Materials (fig. S4, fig. S5). To test whether the ERK activation observed in GqKO cells depended on Gi, we pretreated GqKO cells with pertussis toxin (PTX), which prevents Gi from interacting with GPCRs, prior to stimulation with AngII. Inhibiting Gi signaling with PTX largely blocked AngII-induced ERK activation in cells expressing the WT and mutant AT1Rs, indicating a possible rewiring to a Gi-dependent pathway in the absence of Gq, as previously demonstrated in CRISPR KO cells [35] (fig. S6).
We further tested the effect of the AT1R mutants on ERK signaling by using a previously reported BRET-based extracellular signal–regulated kinase activity reporter (EKAR) biosensor to monitor nuclear ERK activity following 5 min AngII stimulation with or without PTX pretreatment in HEK293T cells[36]. Although AT1R WT and all AT1R mutants were able to promote an increase in ERK activation, the PROX mutant showed a significantly impaired ERK response compared to WT receptor (fig. S7A). Furthermore, inhibition of Gi with PTX blocked about 50% of nuclear ERK stimulated by AngII and TRV023 at WT and mutant receptors, suggesting a substantial role for Gi in mediating nuclear ERK activity (fig. S7B).
Distinct effect of full and biased AT1R ligands on GRK recruitment to AT1R C-terminal tail phosphorylation mutants
To evaluate the effect of phosphorylation of residues in the C-tail of AT1R on GRK recruitment, we used BRET to assess the recruitment of yellow fluorescent protein (YFP)-tagged GRK2, GRK3, GRK5, or GRK6 to WT or AT1R-RLuc8 mutants in response to 5 min stimulation with AngII or TRV023 in HEK293 cells lacking of all 4 of these GRKs (GRKΔ4 cells). Whereas GRK2 demonstrated robust recruitment to AT1R WT and the PROX mutant in response to AngII or TRV023, recruitment to the MID and DIST mutants was significantly impaired following AngII stimulation and more robustly impaired following TRV023 stimulation (fig. S8, A and B). GRK3 recruitment to the MID mutant was impaired in response to TRV023, but not AngII, treatment, but the NULL mutant lacking all phosphorylatable residues abrogated most GRK2 and GRK3 recruitment by either ligand. Because GRK5 and GRK6 are primarily localized to the plasma membrane [37, 38], the assay was only able to capture the dissociation of GRK5 or GRK6 from the receptor as it internalizes into endosomes, resulting in a negative net BRET signal, and therefore cannot be used to measure the effect of AT1R phosphorylation mutants on GRK5 or GRK6 recruitment. Together, these data confirm the known critical role for GRKs in the phosphorylation of Ser and Thr residues in the C-terminal tail of AT1R and demonstrate that ensembles of specific residues on the PROX and MID portion of the C-tail are needed to promote GRK2 and GRK3 recruitment.
The proximal and middle regions of the AT1R C-terminal tail have differing effects on Gq protein activation and Ca2+ signaling
To determine whether phosphorylation of the C-terminal tail of AT1R influences G protein activation, we measured loss of the BRET signal between the Gαq-RLuc8 donor and Gγ-GFP2 acceptor following dissociation of the Gαβγ heterotrimeric complex after AngII stimulation (fig. S9A). Over a broad range of AngII concentrations, maximal G protein activation was significantly reduced with the AT1R-PROX mutant and significantly enhanced with mutation of the 4 MID amino acids of the C-terminal tail (fig. S9, B and C). No difference in EC50 was observed for any of the AT1R mutants (fig. S9D). Consistent with the reduction in G protein activation, the AT1R-PROX mutant also displayed a reduced maximal Ca2+ response to AngII, with no difference in EC50 (fig. S10, A to D). These data indicate that phosphorylation in the proximal and middle area of the C-terminal tail is not only important for β-arrestin functionality, but also for maximal G protein activation in response to a full agonist.
Lastly, to assess whether TRV023 stimulates the phosphorylation of distinct ensembles of phosphorylation sites on the C-tail of the AT1R, or rather similar residues but with less efficacy than does AngII, we performed a Principle Component Analysis of the MS phosphoisomer relative abundance data. While PC2 was a smaller component of the data than PC1, TRV023 showed a distinct contribution from PC2 compared to Telm or AngII. This is consistent with TRV023 acting as a biased agonist compared to AngII in terms of receptor phosphorylation (fig S11).
Discussion
Here, we found that the endogenous ligand AngII and the β-arrestin–biased agonist TRV023 induced two distinct patterns of phosphorylation clusters or ensembles of Ser and Thr residues along the entire C-terminal tail of AT1R. Mutation of either the proximal four Ser and Thr or the middle 4 Ser and Thr residues largely abolished β-arrestin recruitment and internalization, whereas loss of phosphorylation of the four distal C-terminal tail Ser and Thr residues had no effect of β-arrestin function. AT1R stimulation by AngII and TRV023 induced distinct β-arrestin conformations that depended on phosphorylation of this cluster of 8 proximal and middle Ser and Thr amino acid residues. Molecular dynamics simulations showed that loss of phosphorylation in either the proximal or middle segments of the C-tail promoted destabilization of the ATR1-β-arrestin interaction, consistent with our experimental data showing reduced ability of AT1R to recruit β-arrestin 2. Finally, AngII- and TRV023-stimulated ERK signaling was modestly impaired when the proximal four Ser and Thr residues of the AT1R tail were mutated in WT cells but was more substantially diminished in cells lacking Gq.
The concept of phosphorylation clusters, or a “barcode” of phosphorylated serine and threonine residues, as a driver of β-arrestin–GPCR interaction stability was first proposed by Kim et al. [5] and supported experimentally for the β2AR, rhodopsin, CXCR4, and the dopamine D1 receptor[7, 16, 39–41]. Subsequent work has been based on molecular simulation data[18], but detailed experimental evidence for the complete ensemble of phosphorylation barcodes by endogenous and biased agonists and their effects on signaling has been lacking. Here, we demonstrated that different ensembles of phosphopeptides—not single phosphosites—determine the “barcode” for AT1R. TRV023 induced a pattern of phosphorylation in the C-terminal tail of AT1R similar to that induced by AngII, but with lower intensity, consistent with earlier bias studies[42, 43] that showed the TRV compounds have a nearly complete loss of G protein efficacy and partial loss of β-arrestin efficacy compared to AngII.
In our model, the relative abundance of phosphorylation site clusters along the entire C-terminal tail promoted differential β-arrestin engagement and subsequent signaling. Our proteomics data, and functional studies of AT1R mutants, identified the proximal and middle phosphorylation sites found in previous studies to be critical for β-arrestin and receptor internalization [15, 16, 20, 44–47]. These experiments indicated that sites in the proximal and middle regions of the C-terminal tail of AT1R were required for both AngII- and TRV023-driven β-arrestin recruitment and internalization, whereas those in the distal region were largely dispensable. The sites in the proximal region is unlikely to be driven by only Thr332 because earlier studies found single Thr332 mutants had little effect on receptor internalization [45, 47] and implicate the importance of at least one or more of the other proximal sites (Ser326, Ser328, Ser331). Although the recruitment of high amounts of β-arrestin to AT1R depended on C-terminal tail phosphorylation, lower amounts of β-arrestin recruitment and ERK activation are also possible through interaction of β-arrestin solely with the transmembrane core of AT1R (AT1R C-tail truncation mutant) [48].
The interaction of a GPCR with a specific ligand stabilizes distinct conformational states of the receptor to promote the recruitment and interaction of specific transducers to initiate a wide range of cellular responses [49]. Indeed, at endogenous amounts of the receptor, TRV023 does not activate Gq signaling[50], likely due to the structurally distinct AT1R active conformations induced by AngII and the biased ligand TRV023[51]. Our data showing differences in β-arrestin recruitment and downstream Gq signaling depending on the sites of C-tail phosphorylation likely relate to both transducer bias (Gq and β-arrestin for AngII vs β-arrestin only for TRV023) and the degree of ligand efficacy (partial vs full recruitment of β-arrestin). AngII has a somewhat stronger allosteric interaction with β-arrestin than does TRV023 under conditions where the receptor is “pre-phosphorylated.”[43].
The MS data together with the β-arrestin conformational analysis using FlAsH biosensors, demonstrate the dependence of the proximal and middle 8 phosphorylatable residues to induce at least two distinct active β-arrestin conformations depending on whether stimulation was by the endogenous agonist, AngII, or the β-arrestin-biased agonist, TRV023. Consistent with the β-arrestin functional data, mutation of the distal Ser and Thr sites showed little influence on inducing an active β-arrestin conformation. The interaction between β-arrestin and a GPCR can exist as a high-affinity “core” state or a low-affinity “hanging” state as dictated by the subclass of the GPCR and affecting the stability and duration of the GPCR–β-arrestin complex[12]. The distinction between these two β-arrestin binding states is largely determined by the amount of phosphorylation in the C-terminal tail and/or intracellular loops of the GPCR following activation or potentially even in the inactive state. Additionally, by inducing different amounts of receptor phosphorylation, individual ligands can drive specific β-arrestin conformations to the same GPCR[52]. This is apparent in our conformational studies of β-arrestin (Fig. 6, C and D; fig. S3B) wherein stimulation of WT AT1R with AngII or TRV023 induced distinct β-arrestin conformations.
Structural insights[30, 53] into activation of signaling pathways[29, 34, 54] have substantially improved our understanding of the mechanisms involved in ligand-receptor-G protein interactions. Conformational heterogeneity has been observed in certain domains, such as the transmembrane helices (TM) 3, 5, and 7 and intracellular loop 2 (ICL2), in response to different ligands, with a wide range of conformational changes of the receptor[49, 55], the transducer[14, 54, 56], and the entire receptor-transducer complex [3].
Previous studies identified AT1R-stimulated ERK activation to occur through both G protein and β-arrestin–mediated pathways[21, 57, 58]. Although a truncated tail AT1R mutant shows enhanced basal and ligand-stimulated Gq activation, suggesting some inhibitory effect of the C-tail (or part of it)[21], other data suggest that C-tail phosphorylation could affect β-arrestin sequestration and therefore, accessibility of the core to G proteins[59]. Our data are consistent with these observations, showing that unphosphorylated proximal residues are inhibitory to Gq activation whereas basal phosphorylation of middle residues may release that inhibition (fig. S9C), likely through movement of the C-tail sterically affecting the accessibility of the receptor core to G proteins.
Our data are not able to identify unique site-specific patterns in the C-terminal tail or determine the minimum quantity or specific location of phosphates required for β-arrestin binding and therefore the redundancy and individual contributions of each site were not determined. However, our data do show that AngII-induced AT1R phosphorylation occurs most robustly in a region that is in close proximity to the receptor and includes Ser326, Ser328, Ser331, and Thr332 and confirmed that these sites were functionally important in receptor signaling. These findings are consistent with a molecular mechanism in which AngII stimulation recruits GRK2/3 and GRK5/6, inducing a “full engagement” of β-arrestin with activated AT1R[24] because the proximal phosphorylation region occupies a binding site that alters the finger loop of arrestin[16]. In contrast, TRV023 stimulation mainly recruits GRK5/6[24], hence does not have as robust phosphorylation in the proximal region and therefore likely induces a lower affinity “hanging” arrestin conformation, which drives the different signaling outcomes between ligands.
TRV023 promotes less internalization and recruitment of β-arrestin than does AngII, and it also induces less conformational change of β-arrestin although the patterns across AT1R mutants are similar with lower efficacy. Still, there are notable differences, such as the signal noted at FlAsH4, which results in an overall difference in conformation (Fig. 6B). The weaker FlAsH signal of TRV023 reflects less conformational change of arrestin, and because the other FlAsH reporters have similar signals between AngII and TRV023, this reflects a true difference in arrestin conformation and is not due to differential arrestin recruitment.
A number of limitations are important to acknowledge. The MS data represent a snapshot in time of the phosphorylation ensembles at the C-tail of the receptor following ligand stimulation, and therefore it is possible we did not identify some phosphoisomer clusters that would have occurred with more prolonged stimulation. Because we did not identify any peptide with more than three phosphorylated sites, it is possible that phosphorylation was evenly distributed along the tail region or the phospho-enrichment procedure limited the detection of peptides with four or more phosphorylation sites. We cannot exclude the possibility that the functional impact observed for the mutated AT1Rs (PROX, MID, DIST) were the result of effects beyond the loss of phosphorylation at the altered sites. The mutations themselves could affect the conformational distribution of AT1R and transducer coupling and phosphorylation of nearby Ser and Thr residues that remain intact. We used a cell system with overexpressed receptors for the MS and signaling experiments but relied on endogenous GRKs to phosphorylate the C-terminal tail of WT and mutant AT1Rs. It is possible that the difference in stoichiometry between physiological amount of AT1Rs and GRKs would yield different phosphorylation ensembles. Lastly, differences in cell lines and/or culture conditions can influence the phosphorylation and signaling patterns we observed in this study.
In conclusion, we show that, of the 12 Ser and Thr residues within the C-terminal tail of AT1R, the 4 proximal and 4 middle residues are required for full β-arrestin functionality in response to the endogenous ligand AngII or a β-arrestin-biased ligand. This supports a model in which different GPCR C-terminal tail phosphorylation ensembles, consisting of different populations of phosphoisomers of specific regions (“barcodes”), function to regulate GPCR signaling by β-arrestins and G proteins.
Materials and Methods
Expi293F AT1R cell lines
Expi293F™ cells were grown in suspension culture at 37°C in a humidified 8% CO2 incubator in Expi293™ Expression Medium with shaking at 110 rpm. Expi293F™ cell line was maintained in the presence of 10 μg/mL blasticidin, which was removed prior to transfection. Previously described N-terminal-FLAG-tagged human WT AT1R (FLAG-AT1R) cells [30] were maintained in the presence of 10 μg/mL blasticidin and 10 μg/mL zeocin.
Ligand-induced AT1R phosphorylation
FLAG-AT1R cells were grown in suspension culture to a cell density of ~2.5 × 106. AT1R overexpression was induced using doxycycline (4 μg/mL doxycycline and 5 mM sodium butyrate) 30–36 hrs prior to experimentation, and the cells were separated into 100 mL portions. Telmisartan (Telm) (10 μM final concentration) was added to its respective flask 24 hrs prior to the experiment. Prior to the experiment an aliquot of cells was reserved to confirm high AT1R expression levels using radioligand binding[60]. The remaining cell aliquots were then stimulated with 1 μM angiotensin II peptide (AngII), 10 μM TRV023, or left untreated. The efficacy of the stimulation has been confirmed by Western blotting on proteins extracted by a small aliquot of the sample. After 10 mins, cell pellets were collected at 2.2k rpm for 5 mins, flash frozen using liquid nitrogen and stored at −80°C.
AT1R purification
All solutions and buffers used for the pull-down experiments were freshly made. Frozen cells were resuspended in 20 mM HEPES pH 7.4, 500 mM NaCl, 10 mM MgCl2, 0.5% Maltose-Neopentyl Glycol (MNG), 0.05% Cholesteryl Hemisuccinate (CHS) 2.5 U/mL benzonase (Sigma), 1 μM losartan for receptor stabilization, benzamidine, leupeptin, and PhosSTOP (Roche). The samples were then dounced at least 12 times on ice and incubated for 2 hrs at 4°C. The lysate was centrifuged at 12,000 g for 20 mins and filtered to remove insoluble material. CaCl2 was added to a final concentration of 2 mM, and an aliquot of each sample was saved for Western blotting. The receptor was bound to M1 Anti-FLAG beads in batch format for 2 hrs at 4°C. Beads were then washed with 5x column volume of Wash Buffer (20 mM HEPES pH 7.4, 500 mM NaCl, 0.01% MNG, 0.01% CHS, 2 mM CaCl2 with benzamidine, leupeptin, and PhosSTOP (Roche), and eluted (Wash Buffer minus CaCl2, plus 5 mM EDTA and 0.3 mg/mL Flag peptide). Eluted protein was then precipitated by addition of 100% TCA to bring the overall sample to 20% TCA and incubated on ice for 1 hr. Purified protein was pelleted at 13.2k rpm for 15 mins at 4°C, and the supernatant was discarded. The pellet was then washed with ice cold acetone three times and air dried. Samples were then stored at −20°C until needed.
AT1R digestion and MS sample preparation
AT1R pellets were reconstituted in 8M urea, 100mM EPPS pH 8.5. Samples were reduced with 5mM TCEP for 15 mins and alkylated with 10mM iodoacetamide for 15 mins in the dark at RT. The reaction was quenched by adding 10mM of DTT and samples were diluted 10-fold with 100mM EPPS pH 8.5. Trypsin stock (Promega, 0.5mg/mL) was added 1:200 (v/v) and incubated for 6 hours at 37°C. Digests were acidified with TFA and peptides were desalted by C18 solid phase extraction before being dried in a vacuum centrifuge. Digested peptides were redissolved in 200mM EPPS, 30% acetonitrile and labeled with TMT reagents (Thermo Fisher Scientific) for 90 mins at room temperature. Reactions were quenched with hydroxylamine, and 2% of each sample was analyzed for digestion and labeling efficiency. Samples were then desalted by C18 solid phase extraction before and after using Thermo Fisher Scientific’s High-Select Fe-NTA kit to enrich for phosphopeptides before mass spectroscopy analysis.
MS and Data Analysis
Data were collected by an SPS MS3 TMT method[61] using Orbitrap Fusion Lumos mass spectrometers coupled to a Proxeon EASY-nLC 1000 liquid chromatography system. A capillary column with an inner diameter of 100μm and was packed with Accucore C18 resin 2.6μm, 150Å. The 10 most intense MS2 product ion peaks were isolated to produce the reporter ions used for relative quantification. Peptides were searched on Comet release 2021.01 using a single protein database along with common contaminants with a 3Da precursor mass tolerance and no enzyme specificity. Fixed modifications included TMT reporters on both N-terminal and lysine, and carbamidomethylation and variable modifications included methionine oxidation and S/T/Y phosphorylation. Finally, only fully trypsin-digested peptides with 5 ppm mass accuracy were exported for further analysis.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE[62] partner repository with the dataset identifier PXD032672.
Post-database searching analysis was completed using custom python scripts. In brief, identical peptides reported during the same MS run were combined, peptides were required to have a minimum TMT channel sum of 100, phosphoisomers were identified, and then fold difference compared to the unstimulated sample was calculated both directly and on a log2 scale for graphing. A “single-site” analysis was performed on the same data set to highlight which site was most often found phosphorylated upon a specific stimulation. In this case, each phosphoisomer has been virtually divided into its components, and the value of the phosphorylation obtained from the previous analysis was duplicated and assigned to each site. For instance, a peptide found to be phosphorylated in the MS with a score of 5 fold/NS and containing two phosphorylation sites (i.e., Ser328 and a Thr331) was counted twice: an arbitrary score of 5 (based on the fold change over NS) was assigned to the Ser and to the Thr. The same sites (i.e., Ser 328) but belonging to different phosphoisomers were merged to generate a summed score value.
AT1R Mutants Cloning
AT1R Mutants have been obtained by Q5® Site-Directed Mutagenesis Kit (NEB) following standard protocol. For mutagenic primers design the software NEBaseChanger™ was used. Three different regions of the AT1R C-terminal tail have been mutated to generate phosphorylatable deficient mutants (arbitrarily defined as Proximal (PROX), Middle (MID), Distal (DIST)); in each one all the Ser, Thr and Tyr present in the desired region were changed in Ala. A Null mutant was generated that lacked all 12 phosphorylation sites in the c-tail. The following mutagenic primers were used with the WT FLAG-AT1R as template to obtain the region PROX, MID and DIST:
PROX For- CCTTGCAGCAAAAATGAGCACGCTTTCC
PROX Rev- TTTGCGTGGGCTTTGGCTTTTGGGGGAAT
MID For- TACCGCCCCGCAGATAATGTAAGCTCATCCAC
MID Rev- GGCAAGCGCGGCCATTTTTGTTGAAAGGTTTGAG
DIST For- GCCGCCAAGAAGCCTGCACCATGT
DIST Rev- TGCGGCTACATTATCTGAGGGGCG
For the “null-mutant” a two-step mutagenesis was required. First, the Proximal region was mutated in the “MID FLAG-AT1R” backbone. In a second step, the distal part of the C-terminal tail was mutated using the “PROX/MID FLAG-AT1R” as backbone. For both reactions, a standard protocol has been followed. The primers used were the following:
PROX to MID For- AATGGCCGCGCTTGCCTACCGCCCCGCAGAT
PROX to MID Rev- TTTGCTGCAAGGTTTGCGTGGGCTTTGGCTTTTG
DIST to MID For- GCCGCCAAGAAGCCTGCACCATGTTTT
DIST to MID Rev- TGCGGCTACATTATCTGCGGGGCG
Cell transfection
HEK293T, HEK293 GqKO[63] and HEK293 cells lacking all 4 GRKs (GRKΔ4)[24] were cultured in minimum essential medium (MEM) (Corning) supplemented with 10% fetal bovine serum (Sigma) and 1% penicillin and streptomycin (Sigma). Transfections were carried out using lipofectamine 2000 (ThermoFisher Scientific) for plasmid DNA according to the manufacturer’s protocol. The ratio between plasmid and transfection reagent is 1:2.5. Cells were transfected 48 hours before experiments. Transfection amount of DNA for each plasmid was adjusted to match receptor expression and confirmed by radioligand binding in all the different cell lines (table S1).
Whole-cell saturation binding assays
Non-transfected and transfected HEK293T Cells with AT1R mutants were detached from 6 well plates using 0.25% Trypsin for 5 mins. Cells were collected, counted using a Countess III (Invitrogen) and centrifuged for 7 mins at 300g. The pellet was resuspended in Assay Buffer (50mM Tris-HCl, 150mM NaCl, 12.5mM Mg2Cl) and 2×105 cells were added to each well in a 96 well plate in a final volume of 200μl per well. Cells were incubated with 2nM to 16nM of tritiated Olmesartan (3H-Olm) for 2 hours to reach equilibrium. Telmisartan (10μM) was used to define nonspecific binding. After incubation, cells were harvested (Brandell) into filter paper and soaked in 5ml of scintillation fluid overnight. Radioactivity was measured using a TRI-CARB liquid scintillation counter (Perkin Elmer). Specific Binding was obtained by subtracting total binding from nonspecific binding.
G Protein activation: TRUPATH assay
G protein activation was measured by Bioluminescence Resonance Energy Transfer (BRET). Cells were plated in 6-well plate at a density of 500,000 cells per well. Cells were transfected 24hrs later, using a 1:1:1 DNA ratio of Gαq-RLuc8:Gβ:Gγ-GFP2 (100ng per construct for 6-well plates); the mutant receptors were co-transfected and the amount of plasmid adjusted for the mutant receptor expression. The next day, cells were harvested from the plate using trypsin and plated in white, clear-bottom 96-well assay plates at a density of 100,000 cells per well. 24hrs after plating in 96-well assay plates, the growth media was aspirated out and a white backing applied to the plate bottom. Then were added 60 μl of assay buffer (1× Hank’s balanced salt solution (HBSS) + 20 mM HEPES, pH 7.4), followed by a 20 μl addition of freshly prepared 25 μM coelenterazine 400a (Nanolight Technologies). After 5 min 20 μl of each drug were added for a final volume of 100 μl and incubated for 10 mins. Plates were read with the plate reader with 395 nm for RLuc8-coelenterazine 400a and 510 nm for GFP2, at integration times of 1 s per well. Plates were read serially six times, and measurements from the sixth read were used in all analyses. The ratio of the GFP2 emission to RLuc8 emission were the BRET2 ratio.
DiscoverX Assay
β-arrestin2 endocytosis was measured by the active endocytosis assay (DiscoverX) as described[30] with slight modifications. 4 μg AT1R or AT1R mutants of comparable gene expression were transiently transfected into U2OS cells stably expressing β-arrestin2 with an Enzyme Acceptor tag and endosome-localized ProLink tag protein in 10 cm dish. On the following day, transfected cells were plated at a density of 35,000 cells per well. Two days post-transfection, cells were treated with ligands for three hours at 37 °C. Following the addition of reagents from the PathHunter Detection kit (DiscoverX) and an additional 1-hour incubation at room temperature, chemiluminescence arising from the complementation of β-galactosidase fragments (Enzyme Acceptor and ProLink) within endosomes due to β-arrestin endocytosis was detected on a Neo2 (BioTek) microplate reader.
β-arrestin1 Endocytosis BRET Assay
The level of β-arrestin1 endocytosis was measured by the endocytosis BRET assay as described with minor modifications[64]. HEK293 cells in 10 cm dish were transfected with 2 μg AT1R or AT1R mutants to produce comparable gene expression along with 250 ng of β-arr1-RlucII and 1000 ng of rGFP-FYVE. The day following transfection, cells were trypsinized and re-seeded onto poly-L-ornithine coated, white, 96-well plates at a density of ~25,000 cells per well. The next day, cells were pre-incubated with assay buffer (HBSS, 20 mM HEPES and pH 7.4) for 20 min, and then stimulated with specific ligands (1μM AngII, 10μM TRV023, 10μM Telm) in assay buffer for 25 min at 37 °C. The cell-permeable substrate, coelenterazine 400a (final concentrations of 5 μM) was added 2 min before BRET measurements. The BRET measurements were performed in 96-wells plate using a Neo2 (BioTek) microplate reader with a filter set (center wavelength/band width) of 410/80 nm (donor) and 515/30 nm (acceptor).
β-arrestin recruitment assay BRET measurement
Plasmids containing Rluc8 constructs were obtained from Addgene (#87121). AT1R-mutants-Rluc8 constructs were generated via HiFi DNA assembly (New England Biolabs). PcDNA-Rluc8 backbone were linearized by PCR (F: ggtggaggaggtagtggcggtggtggaagcgccaccatggcttccaaggtg, R: gcttgggtctccctatagtg). Mutants insert sequence was amplified by PCR (F: taatacgactcactatagggagacccaagcgccaccatgaagacgatcatc, R: gcttccaccaccgccactacctcctccaccctcaacctcaaaacatggtg). There was a flexible (GGGGS)2 linker between AT1R mutants construct and Rluc8 construct. Backbone and insert constructs were co-incubated with HiFi master mix and transformed into Top10 E. coli (ThermoFisher Scientific). HEK293 cells in 10 cm dish were transfected with 200 ng AT1R-RLuc8 or AT1R mutants carrying RLuc8 tag along with 1000 ng of β-arrestin2-eGFP. The day following transfection, cells were trypsinized and re-seeded onto poly-L-ornithine coated, white, 96-well plates at a density of ~25,000 cells per well. The next day, cells were pre-incubated with assay buffer (HBSS, 20 mM HEPES and pH 7.4) for 20 min, and then stimulated with specific ligands (1μM AngII, 10μM TRV023, 10μM Telm) in assay buffer for 10 min at 37 °C. The cell-permeable substrate coelenterazine 400a (final concentrations of 5 μM) was added 2 min before BRET measurements. The BRET measurements were performed using a Neo2 (BioTek) microplate reader with a filter set (center wavelength/band width) of 410/80 nm (donor) and 515/30 nm (acceptor).
Intramolecular FlAsH BRET using the rLuc–β-arrestin2–FlAsH constructs
HEK293 cells in 6-well plates were co-transfected with 1.5μg of AT1R, either WT or any of the mutants, and 0.1 μg of rLuc–β-arrestin2–FlAsH construct using Fugene HD as previously described[32]. At 48h after transfection, cells were collected in 600μl of HBSS. TC-FlAsH II In-Cell Tetracysteine detection reagent was added at 2.5 μM final concentration and the cells were incubated at RT for 30 min, then they were washed using 1× BAL buffer from the TC-FlAsH kit, resuspended in BRET buffer and placed in white-wall clear-bottom 96-well plates at a density of 100,000 cells per well. Background and total TC-FlAsH fluorescence were read on an Optiplate microplate reader (Perkin-Elmer) with 485 nm excitation and 525–585 nm emission filters. Cells were stimulated with different ligands (1 μM AngII, 10 μM TRV023, 10 μM Telm) for 10 min, then coelenterazine was added at a final concentration of 5 μM. Six consecutive readings of luciferase (440–480 nm) and TC-FlAsH (525–585 nm) emissions were taken, and the BRET ratio (emission eYFP/emission Rluc) calculated using Berthold Technologies Tristar 3 LB 941. The Δ net change in intramolecular BRET ratio for each of the six rLuc–β-arrestin2–FlAsH constructs was calculated by background subtracting the BRET ratio measured for cells in the same experiment stimulated with vehicle only.
Nuclear ERK BRET Biosensor Assay
HEK293 cells seeded in 6-well plates were transiently transfected using polyethylenimine (PEI) in a ratio of PEI transfection reagent to plasmid DNA of 1:3. WT or mutant AT1Rs were co-transfected with 50 ng of EKAR biosensor tagged with a nuclear localization signal. The EKAR BRET biosensor for nuclear ERK1/2 activity was generated using the EKAR FRET ERK1/2 biosensors previously published[65] by removing the N-terminal mCerulean through restriction digest and inserting a NanoLuciferase. Transfection amount of DNA for each receptor plasmid was adjusted to match receptor expression. 24 hours after transfection cells were washed with PBS, harvested from the plate using trypsin and plated into 96-well assay plates with a density of 50,000 cells/well in clear starvation MEM supplemented with 0.5% FBS and 1% penicillin/streptomycin. For PTX treatment, cells were plated in starvation MEM with 200 ng/mL PTX. The following day, the media was removed, and cells were incubated at room temperature with 80 μL coelenterazine h (2.5 μM final concentration) in assay buffer (HBSS supplemented with 20 mM HEPES) for 5 minutes before stimulated with 20 μL AngII (1 μM final concentration) or HBSS vehicle. Plates were read with a BioTek Synergy Neo2 plate reader at 37°C using a 480 nm filter (donor) and 530 nm filter (acceptor). BRET ratios were calculated by dividing the 530 nm acceptor signal by the 480 nm donor signal. Net BRET ratios were calculated by subtracting the vehicle BRET ratio from the ligand-stimulated BRET ratio. The percent of Gi-mediated nuclear ERK activity over total nuclear ERK was calculated by subtracting the PTX-treated net BRET from the non-PTX-treated net BRET (total ERK), dividing the difference by the non-PTX-treated net BRET and multiplying by 100.
GRK Recruitment BRET Assay
HEK293 cells lacking all 4 GRKs (GRKΔ4 cells)[24] were seeded in 6-well plates and transiently transfected with WT or mutant AT1R-Rluc8 and 1 μg of GRK2, GRK3, GRK5, or GRK6 tagged with C-terminal YFP using PEI transfection protocol described above. Transfection amount of DNA for each receptor plasmid was adjusted to match receptor expression. 24 hours later, transfected cells were plated into 96-well assay plates with a density of 50,000 cells/well in clear starvation MEM supplemented with 2% FBS, 1% penicillin/streptomycin, 10mM HEPES, 1x GlutaMax, and 1x Antibiotic-Antimycotic (Gibco). The next day, the media was aspirated, and cells were incubated at room temperature with 80 μL coelenterazine h (2.5 μM final concentration) in HBSS assay buffer for 5 minutes followed by stimulation with 20 μL of specific ligands (1 μM AngII, 10 μM TRV023) or HBSS vehicle. Plates were read with a BioTek Synergy Neo2 plate reader at 37°C using a 480 nm filter (donor) and 530 nm filter (acceptor). BRET ratios were calculated by dividing the 530 nm acceptor signal by the 480 nm donor signal. Net BRET ratios were calculated by subtracting the vehicle BRET ratio from the ligand-stimulated BRET ratio.
Western Blotting
To measure ERK activation, on day 0 cells were transfected with WT and mutant AT1R receptor constructs using lipofectamine 2000 in 6-well plates. On day 1, the media replaced with fresh one (+FBS). On day 2 (48h from transfection) the cells were starved (by using media with no FBS no P/S) for 3 hours. Then the ligands were added: AngII (1μM final concentration), TRV023 (10μM final concentration), Telm (10μM final concentration). After 10 minutes, the media was removed, and the cells were washed with PBS. Then the cells were collected by adding 90ul of RIPA directly to the dry well. Cells were lysed in RIPA lysis buffer (Sigma) containing 1mM PMSF, 10μg/ml aprotinin, and 5μg/ml leupeptin and phosphatase inhibitors with gentle agitation for 1 h at 4°C and centrifuged for 15 min at 13 200 rpm. Total protein concentrations were assayed with Bio-Rad protein assay reagent. After standard SDS-PAGE, proteins were transferred to PVDF membranes 2h at 4°C, blocked with 5% skim-milk in 1X TBST at RT for 1h. Membranes were incubated with Phospho ERK1/2 (1:1000 Cell Signaling, Danvers, MA) or total ERK1/2 as the loading control (1:2000, EMD Millipore, Billerica, MA) antibody overnight with gentle agitation at 4°C, and then Horseradish peroxidase-conjugated secondary antibody for 1 hour at RT. Protein bands were visualized by ChemiDoc XRS+ (Bio-Rad) and quantified by densitometry using image lab software. For the Flag-AT1R pull down evaluation blotting, total protein was not normalized as above and equal sample volumes were loaded into each well. Blotting followed the protocol described above but used Monoclonal Anti-Flag M2 Peroxidase HRP antibody from mouse (Sigma-Aldrich).
Purification Gel Staining
Samples saved during receptor pull down were loaded in equal volume onto a precast Invitrogen Novex Tris-Glycine 4–20% gel using standard SDS-PAGE. The gel was then carefully removed from the case and fixed for 1 hr at room temperate using 7% acetic acid/40% methanol in water. The gel was then stained for 4 hrs using Brilliant Blue G (Invitrogen) with shaking then washed twice for 30 mins with deionized water followed by overnight shaking in deionized water. Gels were imaged using a Syngene Imager.
Molecular modeling and simulations
The initial AT1R-β-arrestin 2 complex was modeled based on the β-arrestin 2-CXCR7 phosphopeptide complex [PDB 6K3F][66]. The AT1R phosphorylated C-terminal tail (residues 319 to 348) was docked to the N-domain of β-arrestin2 using the HADDOCK webserver[67, 68]. The docking site in β-arrestin2 was defined using positively charged residues that are canonically associated with C-terminal tail recognition (ARG: 8 26 63 77 100 104 148 166, LYS: 11 12 108 158 161) in the N-domain. The docking was carried out with default settings, i.e. 1000 initial complexes were generated through rigid docking and then the top 200 scoring ones were submitted to a semi-flexible refinement. The resulting complexes were clustered based on Fraction of Common Contacts (FCC). After the clustering, we selected the top-scoring complex from a cluster with binding characteristics of known β-arrestin/C-terminal tail complexes (PDB codes 4JQI, 5W0P, 6K3F). This includes C-terminal tail interactions with positively charged residues present in the finger loop region, lariat loop as well as stacking against the β-strand I of β-arrestin2. To study how different phosphorylation patterns impact the structure of the complex we focused on 3 segments of the C-terminal tail explored in this study: the PROX segment (residues 326 to 333), the MID segment (residues 334 to 342) and the DIST segment (residues 343 to 350). Based on this division, three phosphorylation-variants of the C-terminal tail were generated: variant 1 (where all phosphorylatable Ser and Thr residues are phosphorylated), variant 2 (where all phosphorylatable Ser and Thr residues from the MID and DIST segments are phosphorylated) and variant 3 (where all phosphorylatable Ser and Thr residues from the PROX and DIST segments are phosphorylated). Due to very low readouts in the TMT mass spectrometry, the phosphorylation of Tyr339 was omitted within this study. Similarly, we did not simulate a variant of the C-terminal tail where phosphorylation of the DIST segment is omitted, as mutational experiments suggest little impact of those residues on β-arrestin 2 recruitment.
To prepare the complexes for molecular dynamics, they were solvated (TIP3P water) and neutralized using NaCl ions (0.15 concentration). The simulations were carried out using ACEMD3[69]. Simulation parameters were obtained from the Charmm36M forcefield[70]. The system underwent a 100 ns equilibration in conditions of constant pressure (NPT ensemble, pressure maintained with Berendsen barostat, 1.01325 bar), using a timestep of 2fs. During this stage restraints were applied to the backbone. Subsequently we carried out 3 × 1μs in conditions of constant volume (NVT ensemble) using a timestep of 4fs for each system. The temperature (310K) was maintained using the Langevin thermostat, hydrogen bonds were restrained using the RATTLE algorithm. Non-bonded interactions were cut-off at a distance of 9Ȧ, with a smooth switching function applied at 7.5Ȧ. Simulation data have been deposited to the GPCRmd repository (https://www.gpcrmd.org/dynadb/publications/1521/) [43] [71]. Simulations were analyzed using the VMD package[72]. Interaction energy between C-terminal tail variants and β-arrestin2 was calculated using the Protein Contact module in the MOE package using default settings. The representative frames for each simulation were obtained by clustering frames based on the RMSD of the C-terminal tail (RMSD cutoff 5.5). A representative frame from the main cluster was subsequently used for analysis. RMSF values (standard deviation of RMSD values explored by a specific selection of atoms during a simulation) were calculated for Cα atoms of the AT1R C-terminal tail. To calculate the values, simulations were aligned in reference to the β-arrestin2 structure backbone.
FLIPR® (Fluorometric Imaging Plate Reader) Ca2+ Assay
HEK293T cells were seeded in a 6-well plate at a density of 500,000 cells per well in growth media (1X MEM + 10% FBS + 1% P/S). Cells were transfected 24 hours later with either WT AT1R or adjusted amounts of the AT1R-PROX or AT1R-MID mutants that generated equal expression of each receptor (table S1). On the following day, cells were trypsinized and re-plated in a black, clear-bottom 96-well plate at a density of 50,000 cells per well in 100 μL growth media and incubated overnight. Ca2+ mobilization was evaluated using the FLIPR Calcium 6 kit (Molecular Devices) according to the manufacturer’s protocol. Briefly, one aliquot of the calcium-sensitive fluorescent dye (FLIPR Component A) was equilibrated to room temperature, fully dissolved in 10 mL of assay buffer (1X Hank’s Balanced Salt Solution + 20 mM HEPES) for a 2X solution, added to each well of the cells in growth media, and incubated for 2 hours at 37°C. Cells were then stimulated with serial doses of AngII at 37°C while the fluorescent signal was simultaneously recorded for 2 minutes at an interval of 1.5 seconds (excitation 485 nm; emission 525 nm) using the FLexStation 3 microplate reader (Molecular Devices). The raw fluorescent signal was baseline-subtracted and the area under the curve was plotted as a function of [AngII] and fit to a log(agonist) versus response model (three parameters) using GraphPad Prism 9. Statistics were performed using one-way ANOVA with Tukey’s multiple comparisons test of n=5 independent experiments done in at least duplicate wells per condition.
Supplementary Material
Acknowledgements:
We are deeply saddened by the sudden loss of Xinyu Xiong, PhD, who made important experimental contributions to this study. We thank Weili Zou for her expert technical assistance, Dr. Andrew C. Kruse for providing the AT1R-APEX2 stable cell line and Dr. Alem Kahsai for technical consultation and helpful discussions.
Funding:
This work was supported by National Institutes of Health grants HL056687 and HL075443 to H.A.R, GM132129 to J.A.P., GM67945 to S.P.G., GM122798 to S.R.; an Institutional NIH training grant T32HL007101 to C.T.P.; and a Whitehead Scholar award to L.M.W. T.M.S received support from a Sara Borrell grant CD22/00007 funded by the Institute of Health Carlos III (ISCIII) and grant 2021 SGR 00046 funded by Agència de Gestió d’Ajuts Universitaris i de Recerca Generalitat de Catalunya (AGAUR). J.S. received support from the Instituto de Salud Carlos III FEDER (PI18/00094) and the ERA-NET NEURON & Ministry of Economy, Industry, and Competitiveness (AC18/00030).
Footnotes
Data and materials availability:
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://www.proteomexchange.org) through the PRIDE [62] partner repository with the dataset identifier PXD032672. Simulation data for molecular dynamics have been deposited to the GPCRmd repository (https://www.gpcrmd.org/dynadb/publications/1521/) [43] [71]. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
References and Notes
- 1.Wang JG,C; Rockman HA, G-Protein–Coupled Receptors in Heart Disease. Circ res, 2018: p. 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauser AS, et al. , Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov, 2017. 16(12): p. 829–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomsen ARB, et al. , GPCR-G Protein-beta-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell, 2016. 166(4): p. 907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eiger DS, et al. , Biased agonism at chemokine receptors. Cell Signal, 2021. 78: p. 109862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kim J, et al. , Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc Natl Acad Sci U S A, 2005. 102(5): p. 1442–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wisler JW, et al. , Recent developments in biased agonism. Curr Opin Cell Biol, 2014. 27: p. 18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nobles KN, et al. , Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal, 2011. 4(185): p. ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Latorraca NR, et al. , Molecular mechanism of GPCR-mediated arrestin activation. Nature, 2018. 557(7705): p. 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee MH, et al. , The conformational signature of beta-arrestin2 predicts its trafficking and signalling functions. Nature, 2016. 531(7596): p. 665–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pfeiffer CT, et al. , Mapping Angiotensin II Type 1 Receptor-Biased Signaling Using Proximity Labeling and Proteomics Identifies Diverse Actions of Biased Agonists. J Proteome Res, 2021. 20(6): p. 3256–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abraham DM, et al. , beta-Arrestin mediates the Frank-Starling mechanism of cardiac contractility. Proc Natl Acad Sci U S A, 2016. 113(50): p. 14426–14431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shukla AK, et al. , Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature, 2014. 512(7513): p. 218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim YJ, et al. , Crystal structure of pre-activated arrestin p44. Nature, 2013. 497(7447): p. 142–6. [DOI] [PubMed] [Google Scholar]
- 14.Shukla AK, et al. , Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature, 2013. 497(7447): p. 137–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oakley RH, et al. , Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis*. J Biol Chem, 2001. 276(22): p. 19452–60. [DOI] [PubMed] [Google Scholar]
- 16.Zhou XE, et al. , Identification of Phosphorylation Codes for Arrestin Recruitment by G Protein-Coupled Receptors. Cell, 2017. 170(3): p. 457–469 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flock T, et al. , Selectivity determinants of GPCR-G-protein binding. Nature, 2017. 545(7654): p. 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Latorraca NR, et al. , How GPCR Phosphorylation Patterns Orchestrate Arrestin-Mediated Signaling. Cell, 2020. 183(7): p. 1813–1825 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dwivedi-Agnihotri H, et al. , Distinct phosphorylation sites in a prototypical GPCR differently orchestrate beta-arrestin interaction, trafficking, and signaling. Sci Adv, 2020. 6(37). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qian H, Pipolo L, and Thomas WG, Association of beta-Arrestin 1 with the type 1A angiotensin II receptor involves phosphorylation of the receptor carboxyl terminus and correlates with receptor internalization. Mol Endocrinol, 2001. 15(10): p. 1706–19. [DOI] [PubMed] [Google Scholar]
- 21.Wei H, et al. , Stable interaction between beta-arrestin 2 and angiotensin type 1A receptor is required for beta-arrestin 2-mediated activation of extracellular signal-regulated kinases 1 and 2. J Biol Chem, 2004. 279(46): p. 48255–61. [DOI] [PubMed] [Google Scholar]
- 22.Gagnon L, et al. , Genetic code expansion and photocross-linking identify different beta-arrestin binding modes to the angiotensin II type 1 receptor. J Biol Chem, 2019. 294(46): p. 17409–17420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drube J, et al. , GPCR kinase knockout cells reveal the impact of individual GRKs on arrestin binding and GPCR regulation. Nat Commun, 2022. 13(1): p. 540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kawakami K, et al. , Heterotrimeric Gq proteins act as a switch for GRK5/6 selectivity underlying beta-arrestin transducer bias. Nat Commun, 2022. 13(1): p. 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holloway AC, et al. , Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol Pharmacol, 2002. 61(4): p. 768–77. [DOI] [PubMed] [Google Scholar]
- 26.Thomas WG, et al. , Agonist-induced phosphorylation of the angiotensin II (AT(1A)) receptor requires generation of a conformation that is distinct from the inositol phosphate-signaling state. J Biol Chem, 2000. 275(4): p. 2893–900. [DOI] [PubMed] [Google Scholar]
- 27.Premont RT and Gainetdinov RR, Physiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol, 2007. 69: p. 511–34. [DOI] [PubMed] [Google Scholar]
- 28.Rakesh K, et al. , beta-Arrestin-biased agonism of the angiotensin receptor induced by mechanical stress. Sci Signal, 2010. 3(125): p. ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wingler LM, et al. , Distinctive Activation Mechanism for Angiotensin Receptor Revealed by a Synthetic Nanobody. Cell, 2019. 176(3): p. 479–490 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wingler LM, et al. , Angiotensin Analogs with Divergent Bias Stabilize Distinct Receptor Conformations. Cell, 2019. 176(3): p. 468–478 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Navarrete-Perea J, et al. , Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. J Proteome Res, 2018. 17(6): p. 2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoffmann C, et al. , A FlAsH-based FRET approach to determine G protein-coupled receptor activation in living cells. Nat Methods, 2005. 2(3): p. 171–6. [DOI] [PubMed] [Google Scholar]
- 33.Baidya M, et al. , Key phosphorylation sites in GPCRs orchestrate the contribution of beta-Arrestin 1 in ERK1/2 activation. EMBO Rep, 2020. 21(9): p. e49886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, et al. , Mechanoactivation of the angiotensin II type 1 receptor induces beta-arrestin-biased signaling through Galphai coupling. J Cell Biochem, 2018. 119(4): p. 3586–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luttrell LM, et al. , Manifold roles of beta-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci Signal, 2018. 11(549). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eiger DS, et al. , Location bias contributes to functionally selective responses of biased CXCR3 agonists. Nat Commun, 2022. 13(1): p. 5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pitcher JA, Freedman NJ, and Lefkowitz RJ, G protein-coupled receptor kinases. Annu Rev Biochem, 1998. 67: p. 653–92. [DOI] [PubMed] [Google Scholar]
- 38.Pronin AN, Carman CV, and Benovic JL, Structure-function analysis of G protein-coupled receptor kinase-5. Role of the carboxyl terminus in kinase regulation. J Biol Chem, 1998. 273(47): p. 31510–8. [DOI] [PubMed] [Google Scholar]
- 39.Kaya AI, et al. , Phosphorylation barcode-dependent signal bias of the dopamine D1 receptor. Proc Natl Acad Sci U S A, 2020. 117(25): p. 14139–14149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Busillo JM, et al. , Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J Biol Chem, 2010. 285(10): p. 7805–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Prihandoko R, et al. , Determination of GPCR Phosphorylation Status: Establishing a Phosphorylation Barcode. Curr Protoc Pharmacol, 2015. 69: p. 2 13 1–2 13 26. [DOI] [PubMed] [Google Scholar]
- 42.Rajagopal S, et al. , Quantifying ligand bias at seven-transmembrane receptors. Mol Pharmacol, 2011. 80(3): p. 367–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strachan RT, et al. , Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR). J Biol Chem, 2014. 289(20): p. 14211–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balmforth AJ, et al. , Functional domains of the C-terminus of the rat angiotensin AT1A receptor. Eur J Pharmacol, 1995. 291(2): p. 135–41. [DOI] [PubMed] [Google Scholar]
- 45.Hunyady L, et al. , Identification of a cytoplasmic Ser-Thr-Leu motif that determines agonist-induced internalization of the AT1 angiotensin receptor. J Biol Chem, 1994. 269(50): p. 31378–82. [PubMed] [Google Scholar]
- 46.Smith RD, et al. , Agonist-induced phosphorylation of the angiotensin AT1a receptor is localized to a serine/threonine-rich region of its cytoplasmic tail. Mol Pharmacol, 1998. 54(6): p. 935–41. [DOI] [PubMed] [Google Scholar]
- 47.Thomas WG, et al. , Phosphorylation of the angiotensin II (AT1A) receptor carboxyl terminus: a role in receptor endocytosis. Mol Endocrinol, 1998. 12(10): p. 1513–24. [DOI] [PubMed] [Google Scholar]
- 48.DeWire SM, et al. , Beta-arrestins and cell signaling. Annu Rev Physiol, 2007. 69: p. 483–510. [DOI] [PubMed] [Google Scholar]
- 49.Staus DP, et al. , Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature, 2016. 535(7612): p. 448–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li A, et al. , Loss of biased signaling at a G protein-coupled receptor in overexpressed systems. PLoS One, 2023. 18(3): p. e0283477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wingler LM, et al. , Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science, 2020. 367(6480): p. 888–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sente A, et al. , Molecular mechanism of modulating arrestin conformation by GPCR phosphorylation. Nat Struct Mol Biol, 2018. 25(6): p. 538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang H, et al. , Structural Basis for Ligand Recognition and Functional Selectivity at Angiotensin Receptor. J Biol Chem, 2015. 290(49): p. 29127–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zimmerman B, et al. , Differential beta-arrestin-dependent conformational signaling and cellular responses revealed by angiotensin analogs. Sci Signal, 2012. 5(221): p. ra33. [DOI] [PubMed] [Google Scholar]
- 55.Kahsai AW, et al. , Multiple ligand-specific conformations of the beta2-adrenergic receptor. Nat Chem Biol, 2011. 7(10): p. 692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eichel K, et al. , Catalytic activation of beta-arrestin by GPCRs. Nature, 2018. 557(7705): p. 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ahn S, et al. , Desensitization, internalization, and signaling functions of beta-arrestins demonstrated by RNA interference. Proc Natl Acad Sci U S A, 2003. 100(4): p. 1740–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wei H, et al. , Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc Natl Acad Sci U S A, 2003. 100(19): p. 10782–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Asher WB, et al. , GPCR-mediated beta-arrestin activation deconvoluted with single-molecule precision. Cell, 2022. 185(10): p. 1661–1675 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tang W, et al. , Allosteric modulation of beta-arrestin-biased angiotensin II type 1 receptor signaling by membrane stretch. J Biol Chem, 2014. 289(41): p. 28271–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McAlister GC, et al. , MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal Chem, 2014. 86(14): p. 7150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Perez-Riverol Y, et al. , The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res, 2022. 50(D1): p. D543–D552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Inoue A, et al. , Illuminating G-Protein-Coupling Selectivity of GPCRs. Cell, 2019. 177(7): p. 1933–1947 e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Namkung Y, et al. , Monitoring G protein-coupled receptor and beta-arrestin trafficking in live cells using enhanced bystander BRET. Nat Commun, 2016. 7: p. 12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harvey CD, et al. , A genetically encoded fluorescent sensor of ERK activity. Proc Natl Acad Sci U S A, 2008. 105(49): p. 19264–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Min K, et al. , Crystal Structure of beta-Arrestin 2 in Complex with CXCR7 Phosphopeptide. Structure, 2020. 28(9): p. 1014–1023 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Honorato RV, et al. , Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front Mol Biosci, 2021. 8: p. 729513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Zundert GCP, et al. , The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J Mol Biol, 2016. 428(4): p. 720–725. [DOI] [PubMed] [Google Scholar]
- 69.Harvey MJ, Giupponi G, and Fabritiis GD, ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J Chem Theory Comput, 2009. 5(6): p. 1632–9. [DOI] [PubMed] [Google Scholar]
- 70.Huang J, et al. , CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat Methods, 2017. 14(1): p. 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rodriguez-Espigares I, et al. , GPCRmd uncovers the dynamics of the 3D-GPCRome. Nat Methods, 2020. 17(8): p. 777–787. [DOI] [PubMed] [Google Scholar]
- 72.Humphrey W, Dalke A, and Schulten K, VMD: visual molecular dynamics. J Mol Graph, 1996. 14(1): p. 33–8, 27–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (https://www.proteomexchange.org) through the PRIDE [62] partner repository with the dataset identifier PXD032672. Simulation data for molecular dynamics have been deposited to the GPCRmd repository (https://www.gpcrmd.org/dynadb/publications/1521/) [43] [71]. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.