

Abstract
The gut microbiota plays a critical role in maintaining a healthy human body and its dysregulation is associated with various diseases. In this study, we investigated the influence of gut microbiome diversity on the development of chronic lymphocytic leukemia (CLL). Analysis of stool samples from 59 CLL patients revealed individual and heterogeneous microbiome compositions, but allowed for grouping of patients according to their microbiome diversity. Interestingly, CLL patients with lower microbiome diversity and an enrichment of bacteria linked to poor health suffered from a more advanced or aggressive form of CLL. In the Eμ-TCL1 mouse model of CLL, we observed a faster course of disease when mice were housed in high hygiene conditions. Shotgun DNA sequencing of fecal samples showed that this was associated with a lower microbiome diversity which was dominated by Mucispirillum and Parabacteroides genera in comparison to mice kept under lower hygiene conditions. In conclusion, we applied taxonomic microbiome analyses to demonstrate a link between gut microbiome diversity and the clinical course of CLL in humans, as well as the development of CLL in mice. Our novel data serve as a basis for further investigations to decipher the pathological and mechanistic role of intestinal microbiota in CLL development.
Introduction
The gut microbiome, an ecosystem formed by commensal, symbiotic, and pathogenic microorganisms colonizing the gastrointestinal tract, is recognized as an important, life-long partner of the host.1,2 The significance of the gut microbiome in both health and disease is a rapidly growing field of research. Recently, the role and direction of the crosstalk between the gut microbiome and immune cells, and its impact on treatment and disease development, has come into focus.
Homeostasis in the host microbiome is constantly influenced by factors such as diet, medication and stress levels,3 and the reciprocal interactions between the gut microbiome and the immune system are being constantly challenged. Dysbiosis, an imbalance of the gut microbiota often associated with loss of beneficial microbes and blooms of pathogens, may lead to a disruption of the physical integrity of the intestinal barrier and/or function of the immune system. Dysbiosis can disrupt the development and distribution of immune cells, which may affect the immune response to pathogens and the ability to mount an appropriate immune defense.4 Gut dysbiosis has also been associated with susceptibility to cancer.5,6 Within the area of hematologic malignancies, the gut microbiome has been suggested to play important roles in cancer microenvironment alterations and disease progression7 as well as in treatment outcomes by, for example, affecting the efficacy of chimeric antigen receptor T-cell treatment and targeted therapies.8,9 Very little is known about the diversity of the gut microbiome and its interaction with the immune system in patients with chronic lymphocytic leukemia (CLL). In a previous study, we demonstrated that the microbiome in CLL patients is less diverse than that in healthy individuals, with half of the CLL patients demonstrating severe dysbiosis caused by a dominance of Bacteroides.10 We also showed that patients with CLL had a lower abundance of bacterial species belonging to Lachnospiraceae and Ruminococcaceae families, including some of the main producers of short chain fatty acids. Given the antigen-driven nature and inherent immune dysfunction of CLL,11-13 and based on our previous pilot study, we hypothesized that the gut microbiome could contribute to the development of CLL. This might happen by it affecting the immune system through various mechanisms,14 including the production of cytokines triggered by certain bacterial species.15,16 The gut microbiome might also be itself impacted by the immune dysfunction observed in CLL patients, and/or reflect the increased prescription of antimicrobials for this group of patients.17 Here, we investigated a potential association between the composition of the fecal microbiome and CLL development in humans and mice. For this, we used a cohort of patients with CLL, as well as the immunocompetent Eμ-TCL1 transplantation mouse model kept under conditions of low or high hygiene.
Methods
Patients, data, sample collection, sequencing, and profiling
Fecal samples were collected from 60 patients diagnosed with CLL and one patient with T-cell prolymphocytic leukemia (T-PLL) enrolled in the CLL biobank and the PERSIMUNE biobank during regular outpatient visits at Rigshospitalet (Copenhagen, Denmark). The participants in our study were new patients, excluding the 12 from our previous study,10 with each patient contributing one sample. One stool sample had to be excluded from further analysis because of low quality. The patients’ data and antibiotic records were retrieved from the Danish National CLL registry and from manual review of medical health records.18 The project was approved by the national ethics committee (approval number 1804410) and written informed consent was obtained from all patients prior to sampling. As previously described,19 fecal samples were collected using OMNIgene.GUT (DNA Genotek) stabilization tubes according to the manufacturer’s instructions. Briefly, samples were immediately fixed and subsequently frozen within 72 h. All samples were stored at -80°C until shipment for sequencing. Samples underwent shotgun metagenomic sequencing on an Illumina Hi-Seq platform. Read preprocessing and taxonomic profiling were done using an in-house pipeline (Online Supplementary Methods).
Animal models
Six- to 8-week-old female C57BL/6 J mice born and maintained in two different animal facilities of the German Cancer Research Center (DKFZ) were transferred to an experimental animal facility and subjected to adoptive transfer (AT) with leukemia cells from the TCL1 mouse model (TCL1 AT), kindly provided by Dr. Carlo Croce (The Ohio State University, Columbus, OH, USA), as previously described.20-22 Leukemic development was monitored through weekly blood specimens starting at week 2 after TCL1 AT. All animal experiments were carried out according to governmental and institutional guidelines and authorized by the local authorities (permit numbers: DKFZ337, G-16/15).
Murine sample collection and sequencing
Fecal samples were collected at week 0, 1 day before TCL1 AT, and at week 3 after transplantation. Fecal samples were snap-frozen immediately after collection. A QIAmp DNA Stool Mini Kit (Qiagen) was used to extract DNA from fecal samples according to the manufacturer’s instructions. Shotgun sequencing on the Illumina Hi-Seq platform was conducted at the European Molecular Biology Laboratory (EMBL, Heidelberg, Germany). Read preprocessing and taxonomic profiling were done using an in-house pipeline (Online Supplementary Methods). Peripheral blood was drawn from the submandibular vein for weekly flow cytometric analysis (Online Supplementary Methods).
Metabolic potential profiles
As previously described,23 gut metabolic modules (GMM) were used to profile the functional potential of the bacterial community present in the stool samples. In short, GMM profiling was performed by length normalizing the IGC count profiles and summing the values for each KEGG gene ontology term,24 which were taken from IGC_catalog-v1.0.0.emapper. annotations-v2.tsv. The values were then normalized to 16sRNA and turned into GMM profiles using omixer-RPM.25
Bioinformatics and statistical analysis
Descriptive analyses were performed for both the human and mouse CLL cohorts with relative bacterial abundance as input data. Unless stated otherwise, Wilcoxon rank-sum testing was used to identify significant differences between subgroups and the Benjamini-Hochberg method was used for multiple-testing correction; a Benjamini-Hochberg adjusted P value of <0.05 was considered statistically significant. Permutational multivariate analysis of variance (PERMANOVA) was used to test variations in the microbial composition among groups of patients’ samples (such as gender or antibiotic usage) or different groups of murine samples (such as cage effect). α diversity measures (richness, Shannon index) were calculated at species level using the vegan R package.26
The inter-individual dissimilarities in human gut microbiota composition (β diversity) were assessed by calculating a dissimilarity matrix. Hierarchical clustering was applied on the distance matrix, dissimilarities were explored using principal coordinate analysis (gl.pcoa),27 and the first three components of this analysis were visualized using a three-dimensional plot. Differential abundance of bacterial species in the fecal microbiome between clusters of CLL patients was assessed using R implementation of SIAMCAT.28 Further details on bioinformatics analyses are provided in the Online Supplementary Methods.
A generalized linear model was used to visualize the relationship between two independent binary variables, TCL1 AT and hygiene of the animal facilities, and one dependent variable, relative abundance of bacteria.
Results
Microbial composition and diversity are heterogeneous in patients with chronic lymphocytic leukemia
Fifty-nine patients diagnosed with CLL and one patient diagnosed with T-PLL (Pt ID=16) delivered stool samples between June 2017 and July 2020. Forty-four stool samples were collected prior to any treatment; 16 stool samples were collected from patients who received treatment before microbiome sampling. The characteristics of the patients are provided in Table 1.
We evaluated the heterogeneity of microbiome composition in our patients by β-diversity estimates, clustering, and statistical testing (PERMANOVA). We observed that the microbial composition was heterogeneous within the cohort of CLL patients. Unsupervised hierarchical clustering revealed three distinct clusters based on the microbial composition of all patients’ samples (Figure 1A). The separation is visualized in a three-dimensional plot using the results of principal coordinate analysis (Figure 1B). The dissimilarity between microbial communities estimated by β diversity was evaluated and significant differences were observed between the three clusters (R=0.09, P=0.001). The differences remained significant after adjusting for gender, age at sampling, and body mass index (R=0.086, P=0.001). No significant differences in the microbiome structure were observed with regard to age at microbiome sampling alone (R=0.017, P=0.444), nor after adjustment for gender and body mass index (R=0.017, P=0.409). To evaluate differences in the microbial communities between the clusters, diversity was assessed. The Shannon diversity for cluster 1 (C1) was lower than for clusters 2 and 3 (C2 and C3) (C1 vs. C2: median, 1.80 vs. 3.50, P=2.4x10-5; C1 vs. C3: median, 1.80 vs. 2.80, P=8.1x10-5) (Figure 1C).
Besides the observed differences in diversity between clusters, a high variability of the microbiome composition was observed within and between clusters. At the genus level, Bacteroides was the most abundant genus across the 60 samples. Additionally, there was a trend of Bacteroides acquiring bacterial dominance (>30% relative abundance) in seven out of 12 samples in C1, and Prevotella dominating the composition of nine samples in C2 and C3 while being completely depleted in all samples in C1. A detailed visualization of the microbiome composition of all 60 patients at the genus level can be explored through an interactive web application at: https://terezafait.shinyapps. io/microbiome_composition/. Instructions and examples of how to navigate in the application can be found in the Online Supplementary Methods. An overview of the bacterial classification into six major taxonomic levels is provided in Online Supplementary Table S1.
Table 1.
Patients’ characteristics.

Figure 1.

Assessment of microbiome (dis)similarity and diversity in patients with chronic lymphocytic leukemia. The (dis)similarity was measured by a distance matrix constructed using Robust Aitchison distances. (A) Hierarchical clustering over bacterial taxa. Hierarchical clustering (hclust function in R) with the Ward minimum variance method was run on the distance matrix calculated based on robust Aitchison distances. The clustering approach used was purely data-driven and the number of resulting clusters was not specified in advance. Cutting a hierarchical clustering tree at the point of the largest distance (“cut”), resulted in three clusters: cluster 1 (C1), purple; cluster 2 (C2), blue; and cluster 3 (C3), orange. Patient N. 16 was diagnosed with T-cell prolymphocytic leukemia (T-PLL) and is represented in black. (B) Principal coordinate analysis representation of the (dis) similarity of the cohort of patients with chronic lymphocytic leukemia. Each dot in the principal coordinate analysis plot represents one sample. Samples ordinated closer to one another are more similar than those ordinated further away. The patient diagnosed with T-PLL showed average microbiome values and has been marked by a black circle. (C) Shannon α diversity in CLL samples grouped according to clusters from (A). The α diversity measures include richness - representing observed number of genera, and Shannon index - representing evenness of species in a community. In the box plots, box edges represent the 25th and 75th percentiles, the center line shows the median and whiskers extend from the box edges to the most extreme data point. The P values (adjusted for multiple testing with the Benjamini-Hochberg procedure) obtained upon Wilcoxon rank-sum tests are indicated (****). Values <0.05 were considered statistically significant.
Chronic lymphocytic leukemia is associated with low microbiome diversity
Given that dysbiosis, often interpreted as loss of diversity, has been documented to play a role in the development and progression of hematologic diseases,29 we explored the individual course of the disease for all patients and focused on comparing patients from C1 and C2, representing those with lowest and highest gut microbiome diversity. In contrast to C2 patients, patients in C1 exhibited a more advanced and progressive CLL. This was evidenced by an extended duration from CLL diagnosis to microbiome sampling (C1: median 5.3 years, interquartile range [IQR]: 0.3-9.2 years, C2: median 0.3 years, IQR: 0.3-9.2 years; P=0.47), a higher proportion of patients who needed treatment for CLL before and/or after microbiome sampling (C1: 92%; C2: 50%; P value based on Kaplan-Meier analysis from diagnosis to first-line treatment: 0.21), and a higher occurrence of patients who underwent hematopoietic stem cell transplantation or developed Richter transformation (1 had Richter transformation, 1 received a transplant, 1 had both transformation and a transplant) as illustrated in Figures 2 and 3. Patients in C3 (intermediate microbiome diversity) demonstrated a greater similarity to those in C2 than to those in C1 with regard to time from CLL diagnosis to microbiome sampling (median: 0.4 years, IQR: 0.2-5.9 years) and proportion of patients in need of CLL treatment (47%). We further monitored the most recent antimicrobial prescriptions for all patients, finding that 58% of C1 patients, 25% of C2 patients, and 29% of C3 patients received antimicrobial treatment within 6 months prior to microbiome sampling (Figure 3, Online Supplementary Table S2). With regard to IGHV mutational status, all CLL patients, irrespective of diversity cluster, exhibited a comparable percentage of mutated CLL (C1: 33%, C2: 37.5%, C3: 32%).
Figure 2.

Swimmer plot illustrating the clinical course of disease of patients with chronic lymphocytic leukemia. Patients from clusters 1 and 2 in Figure 1 are included in the swimmer plot. All included patients were alive at the end of the follow-up period (September 15, 2022). The time points of diagnosis of chronic lymphocytic leukemia, treatment for the leukemia, antimicrobial treatment, and microbiome sampling are shown in the swimmer plot. X-axis: time before and after microbiome sampling: non-continuous parts of the time scale are represented by dashed lines. Y-axis: each subject’s number, colored according to the clusters they belonged to (purple: cluster 1, low diversity; blue: cluster 2, high diversity). L: line of treatment; m: months; y: years; HSCT: hematopoietic stem cell transplantation; CLL: chronic lymphocytic leukemia; R-CHOP + HD-MTX: rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone plus high-dose methotrexate.
Differential abundance of bacterial taxa illustrates heterogeneity among patients with chronic lymphocytic leukemia
Having observed a correlation between the clinical course of CLL and gut microbiome composition, we further aimed to identify groups of bacterial taxa differing significantly between the clusters of patients. In total, the abundance of 30 bacterial genera was significantly different between C1 and C2 (log2 fold change >1) as determined by SIAMCAT (Online Supplementary Table S3). Of these, Hungatella, Anaerotruncus, Dialister, Erysipelatoclostridium/Clostridiales, Lachnoclostridium and Flavonifractor were more abundant among C1 (low diversity) patients, while Parabacteroides, Barnesiella, Odoribacter and Bilophila, among others, were noticeably enriched among C2 (high diversity) patients (Figure 4). Interestingly, patient N 2 from the C2 cluster was clinically similar to patients in C1 (diagnosis 3 years prior to microbiome sampling and antimicrobial treatment prior to sampling), and also showed a similar microbiome composition to that of C1 patients. Along the same line, patient N 4 from C1, clinically similar to patients in C2 with regard to time from diagnosis to microbiome sampling (<1 month), showed an enrichment of bacteria with higher abundance in C2 patients. Bacterial genera differentially abundant between C1 and C3 partly overlapped with those identified as differentially abundant between C1 and C2 (Online Supplementary Figure S4), whereas no taxa were identified to be significantly different between C2 and C3 (Online Supplementary Table S3).
At the species level, 110 bacterial species were identified to be differentially abundant between C1 and C2 patients’ samples (Online Supplementary Table S3). All detected bacterial species were enriched in C2 and depleted in C1 (Online Supplementary Figure S5), which is likely due to strong differences in abundance of individual species and not groups of species as demonstrated at the genus level above. Intriguingly, bacteria such as Prevotella copri, Dorea longicatena, and Bifidobacterium adolescentis which belong to a healthy microbiome signature30 were enriched among C2 (high diversity) patients.
Figure 3.

Heatmap representation of clinical outcomes over time for all patients. The color-coded cells in the heatmap depict different temporal intervals: time from diagnosis to sampling, time from diagnosis to initiation of first-line treatment (1L), time from 1L to sampling, time from sampling to 1L for patients without prior 1L before microbiome sampling, and time from sampling to progression for patients who received 1L before microbiome sampling. For instance, the time from diagnosis to 1L: in cluster 1, 92% of patients required 1L within a median period of 3.7 years; in cluster 2, 50% of patients required 1L within a median period of 0.8 years; in cluster 3, 45% of patients required 1L within a median timeframe of 3.7 years. Annotation of each patient’s sample: cluster affiliation based on results from Figure 1, microbiome sample obtained before receiving 1L (yes/no), IGHV mutation status (mutated/unmutated), antibiotic treatment within the 6 months preceding microbiome sampling (yes/no), hematopoietic stem cell transplantation, and/or Richter transformation. White color represents missing values. IGHV: immunoglobulin heavy-chain variable region gene; 1L: first-line treatment; AB: antibiotics; HSCT: hematopoietic stem cell transplantation;2L/3L/4L: second-/third-/fourth-line treatment; M-CLL: chronic lymphocytic leukemia with mutated IGHV; U-CLL: chronic lymphocytic leukemia with unmutated IGHV.
Clustering of patients does not reflect shared metabolic functions
It has been shown that different bacterial species can have similar metabolic function.31 Hence, an assessment of functional bacterial groups might be more informative than the bacterial composition itself. Thus, we used the bacterial genes identified in a stool sample that were annotated to metabolic functions by omixer-RPM25 as estimates of a potential function of the bacterial community, i.e. GMM. The most abundant GMM in samples from CLL patients was lactose degradation (Table 2). Despite the differences in diversity and composition of the microbiome, no clear pattern in GMM between C1 (low diversity) and C2 (high diversity) could be detected. The potentially clinically relevant GMM related to production of overall beneficial short chain fatty acids,32 and a variety of indole derivatives promoting fortification of the gut epithelial barrier33 were detected in many samples, but were not different between clusters of patients (Table 2). A detailed visualization of GMM grouped according to Vieira-Silva et al.34 (Online Supplementary Table S4) in all 60 patients’ samples can be explored through an interactive web application accessible via this link: https://terezafait.shinyapps.io/gmm_modules/.
Figure 4.
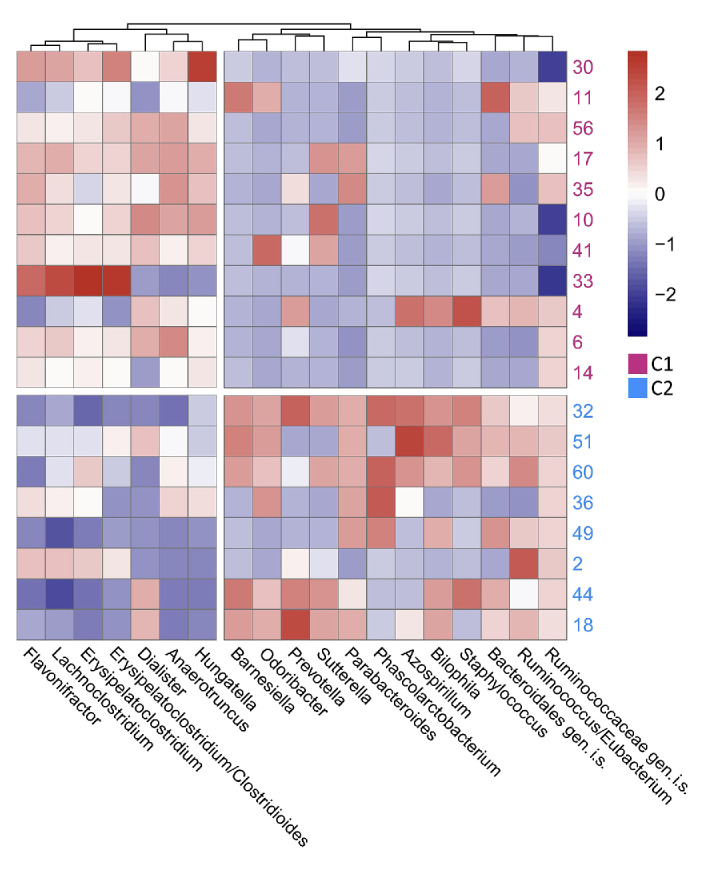
Heatmap of differential abundance of bacteria in samples from patients in clusters 1 and 2. Relative abundances of all genera with log fold-change >1 in samples from patients in cluster 1 and 12 genera with highest log fold-change in samples from patients in cluster 2 are visualized. Color scale: centered log ratio transformed relative abundance of bacterial genera, scaled by columns in pheatmap function in R. Subject 5 (from cluster 1) is omitted from the visualization due to extremely low relative abundances across all genera. C1: patient cluster 1; C2: patient cluster 2 according to Figure 1; gen.i.s.: genus incertae sedis; for instance, Ruminococcaceae gen.i.s.: reads could not be certainly classified as Ruminococcus (genus), but were classified as Ruminococcaceae (family).
Hygiene level influences the progression of chronic lymphocytic leukemia in mice
Adoptive transfer of Eμ-TCL1 leukemia (TCL1 AT) in C57BL/6 mice housed in two animal facilities at the German Cancer Research Center with different hygiene status revealed differences in the development of CLL. In order to elucidate whether the gut microbiome causally contributed to this observation, we performed TCL1 AT with C57BL/6 mice that were born and kept in either a closed breeding facility with altered Schaedler flora (high hygiene, HH)35 or an experimental barrier with individually ventilated cages (low hygiene, LH). One day before TCL1 AT, mice were brought into a common experimental facility with LH conditions and kept there for the rest of the experiment (Figure 5A). Mice originating from the HH facility developed CLL more rapidly compared to LH mice, reaching higher percentages and absolute numbers of CLL cells in the blood over time (week 2: LH=398.5 cells/μL vs. HH=1,390 cells/μL, P=0.0037; and week 4: LH=17,489 cells/μL vs. HH=31,918 cells/μL, P=0.0274) (Figure 5B, C). To assess potential differences in the immune system at a similar stage of leukemia development, HH mice were euthanized at 4 weeks after TCL1 AT, and mice from the LH group were euthanized 5 days later (Figure 5A). At these time points, similar tumor burden in the spleen was achieved in both groups (Online Supplementary Figure S1A). Immunophenotyping of splenic immune cell populations, specifically of the T-cell compartment, revealed no differences between the two groups (Online Supplementary Figures S1 and S2).
Hygiene level and microbiome diversity in mice are inversely proportional
We hypothesized that mice kept in the HH environment would develop a less diverse microbiome than mice kept in the LH facility.36 To confirm this in the setup described above, we analyzed the gut microbiome by shotgun DNA sequencing of fecal samples. One day before TCL1 AT (TP1), corresponding to the untouched microbiome status of the mice maintained in the two different facilities, lower diversity was seen in the HH mice as determined by richness and diversity index (Figure 6A). Three weeks after TCL1 AT (TP2) and co-housing of mice in the same LH facility, which was necessary to allow for experimental interventions to the mice, the microbiome diversity of the two groups became more similar, with a massive increase in diversity in the HH group, and only a minor change towards higher diversity in the LH group (Figure 6A).
Table 2.
Abundance of the seven most abundant gut metabolic modules and seven gut metabolic modules selected a priori.

Focusing on TP1 as the baseline condition for CLL onset in the mice, we explored differences of the gut microbiome between the two groups (Figure 6B). Major differences were observed between the composition of HH and LH microbiomes, with Mucispirillum and Muribaculaceae dominating (i.e., constituting more than 30% of sequencing reads) the microbiomes of HH and LH, respectively. We further explored relative abundances of specific bacterial species in relation to two predictor variables, HH condition and TCL1 AT, by running a generalized linear model. Each bacterial species is represented as a dot weighted according to the predictor variable values (Figure 6C). β values, coefficients obtained from analysis of the generalized linear model, represent the potential influence of the HH condition and TCL1 AT on the relative abundance of each bacterial species. For instance, we observed Clostridium to have a β value of 1.9 on the x-axis (representing the HH condition as correlated with high abundance) and a β value close to zero on the y-axis (representing no clear correlation with TCL1 AT). In summary, the results of our study clearly link, for the first time, clinical course of CLL in patients and development of CLL in mouse models with the diversity of the gut microbiome, where higher microbiome diversity is associated with slower disease progression. Our in-depth characterization of bacterial species in groups of patients with a difference in outcome provides relevant data to study the role and pathological function of these microorganisms, with implications for stratification and therapy of CLL patients.
Discussion
There is increasing evidence of an important role of gut microbiota in human physiology, arguing for a critical role of the microbiome to maintain a healthy state.37 A recent update of Hanahan’s hallmarks of cancer has included the microbiota as an important player in carcinogenesis.38 This is based on results from several studies showing that the microbiome contributes to the development of several cancer entities, such as colorectal, gastric, and biliary cancer, and studying the underlying mechanisms will help in the development of novel therapies.39-41 In this study, lower diversity of the gut microbiome was linked to more aggressive and/or more progressive disease development in patients with CLL and TCL1 AT mice. The study of human stool samples showed that, upon unsupervised clustering of patients with CLL based on gut bacterial distribution, a group of patients with lower microbiome diversity had a more severe clinical course. The severe disease course was characterized by longer time from diagnosis to microbiome sampling signaling more advanced disease, higher frequency of CLL treatment and disease progression implying more aggressive disease, as well as increased antimicrobial usage either implying pre-existing immune system impairment or being a cause of the identified microbiota disruption. By applying TCL1 AT in immunocompetent C57BL/6 mice with basal differences in microbiome diversity, co-housed during the development of CLL, we provide evidence for a causal link between lesser gut microbiome diversity at onset of disease and faster development of CLL. Our novel data identify the microbiome as a driver of disease progression and, therefore, as a potential target to impact the course of CLL development.
Figure 5.

Leukemia development after adoptive transfer of TCL1 leukemia in mice from facilities with low or high levels of hygiene. (A) Schematic of the experimental design of adoptive transfer of TCL1 leukemia of mice from a low hygiene facility (N=9) or high hygiene facility (N=8). Mice without engraftment of TCL1 cells (N=1 in the low hygiene group, N=2 in the high hygiene group) were removed from the study. Figure created with BioRender.com. (B) Percentage of chronic lymphocytic leukemia cells in peripheral blood out of CD45+ viable cells 2, 3 and 4 weeks after adoptive transfer of TCL1 leukemia. (C) Number of leukemic cells/ µL of peripheral blood at the same timepoints. Statistics: one independent study including two groups of ten mice. Mann-Whitney non-parametric test for each timepoint. NS: not statistically significant (P>0.05), *P<0.05, **P<0.01. LH: low hygiene; HH: high hygiene; d: day; TP1: timepoint 1; w: week; TP2: timepoint 2; CLL: chronic lymphocytic leukemia; PB: peripheral blood.
In line with our findings suggesting more advanced or more aggressive CLL correlating with a less diverse, dysbiotic microbiome, a study of B-cell lymphomas demonstrated an association between gut microbiome composition and disease severity, where patients with indolent lymphomas presented greater microbiome diversity and enrichment of certain bacterial genera when compared to patients with diffuse large B-cell lymphoma.42 Similarly, the majority of patients with low microbiome diversity were treated for CLL either prior to or 1 month after microbiome sampling, illustrating the link between a dysbiotic microbiome and more advanced CLL. Several studies, mainly focusing on chemotherapy regimens, found changes in the gut microbiota after treatment, some of which persist for years, which could also be part of the mechanism for dysbiosis in patients having received CLL treatment prior to collection of the microbiome sample.43-45 However, further studies and randomized clinical trials are needed to elucidate the influence of combination and targeted therapies on CLL microbiomes. As signs of dysbiosis among CLL patients we documented the loss of diversity (Shannon diversity <2.0) as well as blooms of bacteria associated with poor health. Several clinically important bacterial taxa enriched in low diversity patients (C1) included Flavonifractor, Anaerotruncus and Dialister genera, whose members were among the top 40 microbial species associated with disease by Gacesa et al.30 Flavonifractor plautii was recently shown to be associated with young-onset colorectal cancer,46 and together with Anaerotruncus colihominis was strongly associated with disease and smoking;30 Dialister invisus was a common bacterium in individuals with poor dietary habits.30,47 Patients with higher diversity (C2) showed significant abundance of bacterial species such as Prevotella copri, Dorea longicatena and Bifidobacterium adolescentis, which are known to produce short chain fatty acids through fermentation of dietary fibers,32,48 also overlapping with the pattern of the healthy-like microbiome described by Gacesa et al.30 Thus, we speculated that the bacterial composition in patients with greater microbiome diversity might lead to beneficial outcomes during the course of their CLL. While results regarding the function of gut bacteria are sometimes contradictory and isolated effects of specific bacteria are difficult to prove due to complex interactions,49 we hypothesized that exploring taxonomic distributions reflected in the gut metabolic potential profiles, representing metabolic function of individual gut microbiomes,25 might be more informative. A study using gut metabolic potential profiles in patients undergoing hematopoietic stem cell transplantation revealed that the conditioning regimen is associated with the degree of changes in metabolic potential of their gut microbiomes.23 We focused on production of short chain fatty acids such as propionate, and butyrate, the crucial gut microbiome metabolites with known ability to have immunomodulatory effects, and on starch degradation metabolism, which shows largely consistent health-promoting effects. However, the abundances of metabolic pathways directly involved in the production of short chain fatty acids and other compounds were not substantially different between subgroups of patients. It may be that the gut metabolic potential profiles will still reveal differences between patients with CLL as compared to patients with other diseases and healthy volunteers. We are currently undertaking such studies to extend our previous exploration of a CLL gut microbiome signature.10 Studying the impact of the gut microbiome on cancer development in mouse models remains a challenging but essential task. Most studies of tumor mouse models are performed in facilities with various and often unknown levels of hygiene and microbiome status. In our study, we used mice that were born and maintained in either a HH or LH facility which assured that the two groups were distinct in terms of their gut microbiome. The clear difference in microbiome diversity that we observed in these mice impacted CLL development. This approach does, however, come with the limitation of not using littermates in our study and the risk of a slightly different genetic background in the two groups. To overcome this limitation, future experiments should include animals from germ-free facilities that fully block the exposure of mice to any microorganisms.50 Exposure of these mice to defined gut bacteria of interest will help to clarify the impact of the bacteria on tumor development. A crucial and unequivocal takeaway from the findings in this study is that when conducting tumor development studies, it is imperative to use animals that are co-housed and possess identical microbial compositions.
Figure 6.

Microbiome composition and diversity in the mouse model of adoptive transfer of TCL1 leukemia. (A) Fecal α diversity in murine samples from the study described in Figure 5 collected at two different timepoints (before and after adoptive transfer of TCL1 leukemia [TCL1 AT]; TP1 and TP2, respectively). Mice for which the quality of microbiome sequencing was low upon quality control were excluded from the analysis (N=3 from the low hygiene group, N=5 from the high hygiene group). Boxplots were constructed as described in Figure 1C. (B) The relative abundance of bacterial genera in murine samples taken at TP1. Bacterial genera with abundance <1% in a sample were omitted from the plot. Sequences that could not be assigned to a genus were grouped as Unclassified. (C) Generalized linear model for every bacterial genus representing its abundance based on two predictor variables: hygiene and TCL1 AT. Center log ratio-transformed relative abundance data at genus level were used as input. The position of a point is given by coefficients (α values), where α represents the weight assigned to the predictor variables. In other words, each of the points illustrates to what degree the relative abundance of the bacterial genus is influenced by the two predictor variables. A positive (negative) value on the x-axis indicates that mice initially housed in a high hygiene barrier will have higher (lower) relative abundance of a bacterial genera compared to mice initially housed in low hygiene barrier. A positive (negative) value on the y-axis indicates that mice transplanted with chronic lymphocytic leukemia (CLL) cells will have higher (lower) relative abundance of a bacterial genus than mice before transplantation of CLL cells. As a concrete example: Helicobacter is positioned at coordinates x: -0.95 and y: -0.25, which can be interpreted as Helicobacter’s relative abundance is more influenced by the hygiene of the barrier than by the CLL cell transplantation. Also, based on this model, the relative abundance of Helicobacter will be lower in mice kept in a high hygiene barrier and slightly lower in mice transplanted with CLL cells. NS: not statistically significant (P>0.05), **P<0.01.
Among the bacteria that were detected in the LH but not the HH group, Muribaculaceae has been described as an immune-protective bacterial family in a CT26 melanoma mouse model.51 Helicobacter, also highly present in the LH mice, is widely known for its correlation with the occurrence of gastric cancer. Its metabolites are known to drive macrophages into an anti-inflammatory state.52,53 In our study, the presence of these bacteria in the LH mice could, however, have had beneficial effects, perhaps by shaping myeloid cells into a phenotype that was less supportive of CLL growth. Lastly, Bacteroidales, also upregulated in this group and considered to be beneficial for gut health, but also correlated with worse disease outcome in lung cancer patients,54,55 could be priming the gut-associated immune system in the LH mice and contributing to its immune-protective action against leukemia development.
In HH mice, we detected an enrichment of Mucispirillum, which has been described as cancer-promoting due to its induction of lipopolysaccharide production, which enhances inflammation.56 Parabacteroides, also highly enriched among HH mice, is a bacterial genus generally considered as anti-inflammatory.57 Such species present in the HH mice could contribute to enhancing immune suppression and thereby promote CLL development. Importantly, linking the presence of specific bacteria in the gut of these mice with specific functions according to the literature is not straightforward, partly because of the multiple effects that bacteria can have in different settings.
Our study is limited by only assessing the microbiome on the DNA level, whereas a more precise way would be the inclusion of metatranscriptomics and/or metabolomics focused on the microbiome. Other limitations of our study are the lack of consecutive samples per patient, which would allow us to describe the microbiome changes during disease progression more precisely, and small numbers in the extreme clusters (C1 and C2), which most likely prevent us from achieving statistical significance. A study set-up in which stool samples are collected before and after treatment initiation has been applied as translational studies adjoined to several clinical trials (NCT04008706, NCT04639362, NCT04608318); thus, elucidation of microbiome dynamics throughout treatment will be the focus of upcoming studies. The observed association between microbiome low diversity and advanced CLL may be influenced by sampling bias with potential overrepresentation of patients starting treatment in planned clinical trials. Additionally, antibiotic exposures accompanying CLL treatment could confound our findings, influencing microbial composition and diversity. Given the purpose of this study, while a descriptive overview of the patient and mouse data is itself insightful, a translational and functional comparison of our findings in humans and mice would be ideal. However, exploration of the microbial overlap between human and mouse showed that 85% of bacterial genera found in the murine microbiome are not present in humans.58 These impressive differences might be caused by the obvious dissimilarity between the murine and human systems, as well as by external factors. Therefore, translating conclusions from murine to human data remains challenging. In conclusion, taxonomic analyses of gut microbiota provide evidence for a link between microbiome diversity and CLL aggressiveness and development in patients with CLL and mouse models, respectively. In the study of patients, we grapple with a classic chicken-and-egg dilemma as it remains unclear whether the microbiome dysbiosis is a result of the CLL, its treatment, and antibiotic use, or whether it represents an underlying condition driving the disease’s development. However, in the mouse study, we provide evidence through the TCL 1 AT that the microbiome alterations are not just a consequence but indeed play a significant role in the progression of the disease. Furthermore, we provide a complete overview of the taxonomic and functional composition identified in patients’ samples. Lastly, we attempted to apply metabolic potential analysis to provide a superior understanding of the biological processes underlying gut dysbiosis in this cohort of patients. However, profound taxonomic changes were not reflected in changes in the gut metabolic potential. It is thus appealing to investigate further whether intestinal microbial composition and function could serve as potential predictors of CLL development.
Supplementary Material
Acknowledgments
The authors would like to express sincere thanks to the patients who provided samples for the study and made this kind of research possible. This research would not have been possible without our collaborator, PERSIMUNE Center of Excellence, which provided the infrastructure as well as financial support and exceptional expertise. A special thank you to Mette Jørgensen for providing access and training for an in-house taxonomic profiling pipeline. The staff at the Hematology Department and at the PERSIMUNE biobank at Rigshospitalet were essential for this study as they organized and collected the samples for our research. Computerome team provided a computational platform and technical support. The authors would like to thank Dr. Emma Philips for editing the manuscript for English grammar and punctuation.
Funding Statement
Funding: The study was funded by the Danish Cancer Society (grant R269-A15924) and the Alfred Benzon Foundation. The infrastructure for sampling and analyses was within the Danish National Research Foundation-funded PERSIMUNE project (grant 126).
Data-sharing statement
The datasets generated and analyzed during this study are derived from patients treated in Denmark. The datasets contain sensitive patient data governed by GDPR and Danish law. Due to Danish legislation (Act number 502 of May 23, 2018) and approvals granted by the Danish Data Protection Agency, it is not possible to upload raw data to a publicly available database. However, access to these data can be made available from the corresponding author on reasonable request, provided a data transfer agreement is entered into according to current regulations. Taxonomic profiling data of the murine cohort as well as bioinformatics analysis implemented in R are available at: https://github.com/PERSIMUNE/PAC2023Faitova_Human_Mice_CLL
References
- 1.Valdes AM, Walter J, Segal E, Spector TD. Role of the gut microbiota in nutrition and health. BMJ. 2018;361:k2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorkiewicz G, Moschen A. Gut microbiome: a new player in gastrointestinal disease. Virchows Arch. 2018;472(1):159-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodríguez JM, Murphy K, Stanton C, et al. The composition of the gut microbiota throughout life, with an emphasis on early life. Microb Ecol Health Dis. 2015;26:26050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoo JY, Groer M, Dutra SV, Sarkar A, McSkimming DI. Gut microbiota and immune system interactions. Microorganisms. 2020;8(10):1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brüssow H. Problems with the concept of gut microbiota dysbiosis. Microb Biotechnol. 2020;13(2):423-434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonnet M, Buc E, Sauvanet P, et al. Colonization of the human gut by E. coli and colorectal cancer risk. Clin Cancer Res. 2014;20(4):859-867. [DOI] [PubMed] [Google Scholar]
- 7.Wang R, Yang X, Liu J, et al. Gut microbiota regulates acute myeloid leukaemia via alteration of intestinal barrier function mediated by butyrate. Nat Commun. 2022;13(1):2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vicente-Dueñas C, Janssen S, Oldenburg M, et al. An intact gut microbiome protects genetically predisposed mice against leukemia. Blood. 2020;136(18):2003-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park JS, Gazzaniga FS, Wu M, et al. Targeting PD-L2–RGMb overcomes microbiome-related immunotherapy resistance. Nature. 2023;617(7960):377-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faitová T, Svanberg R, da Cunha-Bang C, et al. The gut microbiome in patients with chronic lymphocytic leukemia. Haematologica. 2022;107(9):2238-2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caligaris-Cappio F. Inflammation, the microenvironment and chronic lymphocytic leukemia. Haematologica. 2011;96(3):353-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niemann CU, Wiestner A. B-cell receptor signaling as a driver of lymphoma development and evolution. Semin Cancer Biol. 2013;23(6):410-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Svanberg R, Janum S, Patten PEM, Ramsay AG, Niemann CU. Targeting the tumor microenvironment in chronic lymphocytic leukemia. Haematologica. 2021;106(9):2312-2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aghamajidi A, Maleki Vareki S. The effect of the gut microbiota on systemic and anti-tumor immunity and response to systemic therapy against cancer. Cancers (Basel). 2022;14(15):3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schulthess J, Pandey S, Capitani M, et al. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity. 2019;50(2):432-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Modoux M, Rolhion N, Lefevre JH, et al. Butyrate acts through HDAC inhibition to enhance aryl hydrocarbon receptor activation by gut microbiota-derived ligands. Gut Microbes. 2022;14(1):2105637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andersen MA, Rostgaard K, Niemann CU, Hjalgrim H. Antimicrobial use before chronic lymphocytic leukemia: a retrospective cohort study. Leukemia. 2021;35(3):747-751. [DOI] [PubMed] [Google Scholar]
- 18.da Cunha-Bang C, Geisler CH, Enggaard L, et al. The Danish National Chronic Lymphocytic Leukemia Registry. Clin Epidemiol. 2016;8:561-565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ilett EE, Jørgensen M, Noguera-Julian M, et al. Associations of the gut microbiome and clinical factors with acute GVHD in allogeneic HSCT recipients. Blood Adv. 2020;4(22):5797-5809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanna BS, McClanahan F, Yazdanparast H, et al. Depletion of CLL-associated patrolling monocytes and macrophages controls disease development and repairs immune dysfunction in vivo. Leukemia. 2016;30(3):570-579. [DOI] [PubMed] [Google Scholar]
- 21.Bichi R, Shinton SA, Martin ES, et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci U S A. 2002;99(10):6955-6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McClanahan F, Hanna B, Miller S, et al. PD-L1 checkpoint blockade prevents immune dysfunction and leukemia development in a mouse model of chronic lymphocytic leukemia. Blood. 2015;126(2):203-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jørgensen M, Nørgaard JC, Ilett EE, et al. Metabolic potential of the gut microbiome is significantly impacted by conditioning regimen in allogeneic hematopoietic stem cell transplantation recipients. Int J Mol Sci. 2022; 23(19):11115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Darzi Y, Falony G, Vieira-Silva S, Raes J. Towards biome-specific analysis of meta-omics data. ISME J. 2016;10(5):1025-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oksanen J, Simpson GL, Blanchet FG, et al. vegan: Community Ecology Package. R package version 2.6-2. https://github.com/vegandevs/vegan. Accessed 23.09.2022. [Google Scholar]
- 27.Gruber B, Unmack PJ, Berry OF, Georges A. dartr: an r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol Ecol Resour. 2018;18(3):691-699. [DOI] [PubMed] [Google Scholar]
- 28.Wirbel J, Zych K, Essex M, et al. Microbiome meta-analysis and cross-disease comparison enabled by the SIAMCAT machine learning toolbox. Genome Biol. 2021;22(1):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uribe-Herranz M, Klein-González N, Rodríguez-Lobato LG, Juan M, de Larrea CF. Gut microbiota influence in hematological malignancies: from genesis to cure. Int J Mol Sci. 2021;22(3):1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gacesa R, Kurilshikov A, Vich Vila A, et al. Environmental factors shaping the gut microbiome in a Dutch population. Nature. 2022;604(7907):732-739. [DOI] [PubMed] [Google Scholar]
- 31.Deleu S, Machiels K, Raes J, Verbeke K, Vermeire S. Short chain fatty acids and its producing organisms: an overlooked therapy for IBD? EBioMedicine. 2021;66:103293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fusco W, Lorenzo MB, Cintoni M, et al. Short-chain fatty-acid-producing bacteria: key components of the human gut microbiota. Nutrients. 2023;15(9):2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roager HM, Licht TR. Microbial tryptophan catabolites in health and disease. Nat Commun. 2018;9(1):3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vieira-Silva S, Falony G, Darzi Y, et al. Species–function relationships shape ecological properties of the human gut microbiome. Nat Microbiol. 2016;1(8):16088. [DOI] [PubMed] [Google Scholar]
- 35.Dewhirst FE, Chien CC, Paster BJ, et al. Phylogeny of the defined murine microbiota: altered Schaedler flora. Appl Environ Microbiol. 1999;65(8):3287-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ege MJ. The hygiene hypothesis in the age of the microbiome. Ann Am Thorac Soc. 2017;14(Supplement_5):S348-S353. [DOI] [PubMed] [Google Scholar]
- 37.Mohajeri MH, Brummer RJM, Rastall RA, et al. The role of the microbiome for human health: from basic science to clinical applications. Eur J Nutr. 2018;57(Suppl 1):1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12(1):31-46. [DOI] [PubMed] [Google Scholar]
- 39.Saus E, Iraola-Guzmán S, Willis JR, Brunet-Vega A, Gabaldón T. Microbiome and colorectal cancer: Roles in carcinogenesis and clinical potential. Mol Aspects Med. 2019;69:93-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Baba Y, Ishimoto T, et al. Gut microbiome in gastrointestinal cancer: a friend or foe? Int J Biol Sci. 2022;18(10):4101-4117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wheatley RC, Kilgour E, Jacobs T, et al. Potential influence of the microbiome environment in patients with biliary tract cancer and implications for therapy. Br J Cancer. 2022;126(5):693-705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diefenbach CS, Peters BA, Li H, et al. Microbial dysbiosis is associated with aggressive histology and adverse clinical outcome in B-cell non-Hodgkin lymphoma. Blood Adv. 2021;5(5):1194-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Montassier E, Gastinne T, Vangay P, et al. Chemotherapy-driven dysbiosis in the intestinal microbiome. Aliment Pharmacol Ther. 2015;42(5):515-528. [DOI] [PubMed] [Google Scholar]
- 44.Galloway-Peña JR, Shi Y, Peterson CB, et al. Gut microbiome signatures are predictive of infectious risk following induction therapy for acute myeloid leukemia. Clin Infect Dis. 2020;71(1):63-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rajagopala SV, Singh H, Yu Y, et al. Persistent gut microbial dysbiosis in children with acute lymphoblastic leukemia (ALL) during chemotherapy. Microb Ecol. 2020;79(4):1034-1043. [DOI] [PubMed] [Google Scholar]
- 46.Yang Y, Du L, Shi D, et al. Dysbiosis of human gut microbiome in young-onset colorectal cancer. Nat Commun. 2021;12(1):6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vals-Delgado C, Alcala-Diaz JF, Molina-Abril H, et al. An altered microbiota pattern precedes type 2 diabetes mellitus development: from the CORDIOPREV study. J Adv Res. 2021;35:99-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19(1):29-41. [DOI] [PubMed] [Google Scholar]
- 49.Shetty SA, Kuipers B, Atashgahi S, Aalvink S, Smidt H, de Vos WM. Inter-species metabolic interactions in an in-vitro minimal human gut microbiome of core bacteria. NPJ Biofilms Microbiomes. 2022;8(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodriguez-Palacios A, Aladyshkina N, Ezeji JC, et al. ‘Cyclical bias’ in microbiome research revealed by a portable germ-free housing system using nested isolation. Sci Rep. 2018;8(1):3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tomasi M, Dalsass M, Beghini F, et al. Commensal Bifidobacterium strains enhance the efficacy of neo-epitope based cancer vaccines. Vaccines (Basel). 2021;9(11):1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Danne C, Ryzhakov G, Martínez-López M, et al. A large polysaccharide produced by Helicobacter hepaticus induces an anti-inflammatory gene signature in macrophages. Cell Host Microbe. 2017;22(6):733-745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang F, Meng W, Wang B, Qiao L. Helicobacter pylori-induced gastric inflammation and gastric cancer. Cancer Lett. 2014;345(2):196-202. [DOI] [PubMed] [Google Scholar]
- 54.Peters BA, Pass HI, Burk RD, et al. The lung microbiome, peripheral gene expression, and recurrence-free survival after resection of stage II non-small cell lung cancer. Genome Med. 2022;14(1):121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Giri S, Mangalam A. Chapter 4. The gut microbiome and metabolome in multiple sclerosis. In: Faintuch J, Faintuch S, editors. Microbiome and Metabolome in Diagnosis, Therapy, and other Strategic Applications. Academic Press; 2019. p. 333-340. [Google Scholar]
- 56.Daniel SG, Ball CL, Besselsen DG, Doetschman T, Hurwitz BL. Functional changes in the gut microbiome contribute to transforming growth factor β-deficient colon cancer. mSystems. 2017;2(5):e00065-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koh GY, Kane A, Lee K, et al. Parabacteroides distasonis attenuates toll-like receptor 4 signaling and Akt activation and blocks colon tumor formation in high-fat diet-fed azoxymethane-treated mice. Int J Cancer. 2018;143(7):1797-1805. [DOI] [PubMed] [Google Scholar]
- 58.Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102(31):11070-11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nguyen TL, Vieira-Silva S, Liston A, Raes J. How informative is the mouse for human gut microbiota research? Dis Model Mech. 2015;8(1):1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during this study are derived from patients treated in Denmark. The datasets contain sensitive patient data governed by GDPR and Danish law. Due to Danish legislation (Act number 502 of May 23, 2018) and approvals granted by the Danish Data Protection Agency, it is not possible to upload raw data to a publicly available database. However, access to these data can be made available from the corresponding author on reasonable request, provided a data transfer agreement is entered into according to current regulations. Taxonomic profiling data of the murine cohort as well as bioinformatics analysis implemented in R are available at: https://github.com/PERSIMUNE/PAC2023Faitova_Human_Mice_CLL