

Glucose is an essential source of energy in human diets. After ingestion, blood glucose levels rapidly increase but soon return to baseline due to the regulatory effects of insulin [1]. The glucose absorbed by cells is primarily converted into glycogen in the liver for storage. This glycogen is gradually released as needed to maintain blood glucose levels between meals. Excess glucose can also be transformed into fatty acids and stored as triglycerides [2, 3]. Unlike glycogen, which can be mobilized to sustain blood glucose levels, stored fat is primarily oxidized in mitochondria to provide energy and cannot contribute to maintaining blood glucose levels. When the body requires energy, it primarily utilizes glucose, including that stored as glycogen. Once glucose is converted into fat, it is not easily eliminated and tends to accumulate, potentially leading to health issues such as fatty liver and obesity [4].
Hepatocytes can store glucose energy through either glycogenesis or lipogenesis. Despite the critical roles of these two metabolic pathways, the mechanisms underlying the selection between glycogenesis and lipogenesis for glucose carbon storage in hepatocytes have not been fully elucidated. It is evident that the liver has an intrinsic ability to control the choice between glycogen and fat synthesis, yet our understanding of how this regulation occurs remains incomplete. Recently, Chen et al. provided new insights into this regulatory decision-making process [5]. The authors discovered that liver cells preferentially convert glucose into glycogen by utilizing uridine diphosphate glucose (UDPG), an intermediate metabolite of glycogen synthesis, to block fat synthesis (Fig. 1).
Fig. 1.
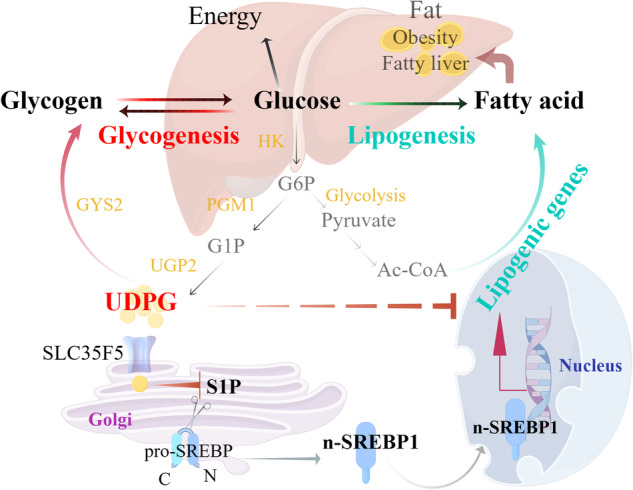
Hepatic glycogenesis antagonizes lipogenesis by obstructing S1P-mediated SREBP activation through UDPG. Hepatocytes can opt to store glucose energy through either glycogenesis or lipogenesis. During glycogenesis, glucose is first converted into glucose-6-phosphate (G6P) by the enzyme hexokinase (HK). It is then transformed into glucose-1-phosphate (G1P) by phosphoglucomutase 1 (PGM1). Subsequently, G1P is converted into UDP-glucose (UDPG) by UDP-glucose pyrophosphorylase 2 (UGP2), which acts as the activated glucose precursor for glycogen synthesis. Glycogen synthase 2 (GYS2) utilizes UDPG to elongate existing glycogen chains. During lipogenesis, G6P is metabolized through glycolysis to yield pyruvate, which is then converted into citrate in the mitochondria via the pyruvate dehydrogenase (PDH) complex and the tricarboxylic acid (TCA) cycle. This citrate is transported into the cytoplasm and cleaved by citrate lyase (ACLY) to produce acetyl-CoA (Ac-CoA), which is crucial for fatty acid synthesis. Additionally, UDPG, a key intermediate in glycogen synthesis, is transported to the Golgi apparatus via SLC35F5, where it interacts with sphingosine-1-phosphate (S1P). This interaction alters the spatial structure of S1P, promoting its ubiquitination and degradation. This inhibits the cleavage of sterol regulatory element-binding protein 1 (SREBP1), leading to reduced production of the active form of SREBP1 (n-SREBP1) and ultimately suppressing the expression of genes involved in lipogenesis. The figure was created with FigDraw
To synthesize glycogen, glucose is first converted into glucose-6-phosphate (G6P) by the enzyme hexokinase (HK) and then into glucose-1-phosphate (G1P) by phosphoglucomutase 1 (PGM1). G1P is subsequently transformed into UDPG by UDP-glucose pyrophosphorylase 2 (UGP2). UDPG acts as an activated glucose donor in glycogen synthesis. Glycogen synthase 2 (GYS2) incorporates UDPG into an existing glycogen chain, thereby extending it [4] (Fig. 1). Conversely, excess glucose can also be directed toward fatty acid synthesis. During lipogenesis, G6P is metabolized through glycolysis to yield pyruvate, which is then converted into citrate in the mitochondria via the pyruvate dehydrogenase (PDH) complex and the tricarboxylic acid (TCA) cycle. This citrate is transported into the cytoplasm and cleaved by citrate lyase (ACLY) to produce acetyl-CoA (Ac-CoA), which is crucial for fatty acid synthesis [6] (Fig. 1).
Using 13C-glucose isotope tracing analysis in primary liver cells, researchers observed a preference for glycogen production over fat synthesis when liver cells encountered conditions of excess glucose. By manipulating the key enzyme PGM1 involved in glycogen synthesis, they demonstrated that the glycogenesis pathway inhibits lipogenesis. Further investigations revealed that UDPG, an intermediate metabolite in glycogen synthesis, directly suppresses the expression of genes involved in fatty acid synthesis, thereby inhibiting fat production from glucose.
Sterol regulatory element-binding proteins (SREBPs) play pivotal roles in hepatic lipid synthesis. SREBP1c, the predominant form of SREBP1 in the liver, is crucial for lipogenesis. Under normal conditions, it resides as a full-length precursor tethered to the endoplasmic reticulum (ER) membrane, where it interacts with SREBP cleavage-activating protein (SCAP). Under conditions of low cholesterol, SCAP transports SREBP to the Golgi apparatus via COPII vesicles. Within the Golgi, site-1 protease (S1P) and site-2 protease (S2P) cleave SREBP. This releases the N-terminal fragment (n-SREBP), which activates genes essential for fatty acid synthesis [7, 8]. In this study, they identified SLC35F5, a solute carrier superfamily member, as the transporter responsible for shuttling UDPG into the Golgi apparatus. Within the Golgi apparatus, UDPG interacts with S1P. This interaction modifies the spatial conformation of S1P, promoting its ubiquitination and subsequent degradation. These changes inhibit the cleavage of SREBP1, thereby decreasing the production of its active form n-SREBP1 and ultimately suppressing the expression of genes involved in lipogenesis (Fig. 1).
Importantly, in high-fat diet-induced nonalcoholic fatty liver disease (NAFLD) animal models, UDPG reduced overall body weight and the levels of triglycerides and cholesterol in the liver and serum without affecting crucial indicators of glucose metabolism, such as glucose levels, ketone bodies, or insulin signaling. Additionally, UDPG inhibited hepatocyte ballooning and intracellular lipid droplet formation during liver pathogenesis. Analysis of primary liver tissues from NAFLD patients revealed that the glycogen synthesis pathways were suppressed and UDPG production was reduced in affected human liver cells. Furthermore, through in vitro models utilizing normal human liver cells, fatty acid-induced steatotic cells, and oleic acid-induced steatotic organelles, researchers have demonstrated that exogenous supplementation with UDPG promotes glycogen synthesis while inhibiting fat generation in both normal and steatotic liver cells. These results confirm that supplementation with UDPG can serve as a potential treatment for human NAFLD.
NAFLD is now widely recognized by experts as metabolic dysfunction-associated steatotic liver disease (MASLD) and has become the most common chronic liver condition worldwide in recent decades. Further exploration into the reprogramming of glucose and lipid metabolism in MASLD liver cells, as well as the underlying molecular regulatory mechanisms, is crucial not only for understanding how metabolic disorders contribute to the onset and progression of MASLD but also for developing innovative therapeutic strategies for its treatment. The significance of this research lies in its identification of a new mechanism for glucose processing within liver cells. This not only provides a novel theoretical foundation for understanding the pathogenesis of fatty liver but also opens avenues for developing new treatment strategies. Modulating UDPG levels to influence metabolic balance within liver cells may prove to be an effective therapeutic approach, potentially alleviating or reversing the progression of fatty liver disease. As an endogenous metabolite, UDPG possesses inherent advantages as a drug candidate due to its minimal side effects. The authors also confirmed that while UDPG effectively inhibits lipid synthesis, it has a negligible impact on carbohydrate metabolism. However, the progression of MASLD involves various factors leading to hepatic lipid accumulation, such as disturbances in fatty acid synthesis, absorption, transport, and degradation [9]. The effectiveness of using UDPG to treat MASLD caused by different etiologies requires further experimental validation.
In conclusion, this study revealed a previously unexplored aspect of glucose metabolism, providing valuable insights into the complex relationship between glycogenesis and lipogenesis. The intermediate product of glycogen synthesis, UDPG, can inhibit the synthesis of fatty acids, ensuring that glucose is preferentially metabolized into glycogen and preventing lipid accumulation in the liver. These findings significantly enhance our understanding of the pathogenesis of fatty liver disease and offer new therapeutic possibilities for individuals suffering from MASLD.
Competing interests
The authors declare no competing interests.
References
- 1.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414:799–806. [DOI] [PubMed] [Google Scholar]
- 2.Shao W, Espenshade PJ. Sugar makes fat by talking to SCAP. Cancer Cell. 2015;28:548–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cheng C, Ru P, Geng F, Liu J, Yoo JY, Wu X, et al. Glucose-mediated N-glycosylation of SCAP is essential for SREBP-1 activation and tumor growth. Cancer Cell. 2015;28:569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang H, Ma J, Tang K, Huang B. Beyond energy storage: roles of glycogen metabolism in health and disease. FEBS J. 2020;288:3772–83. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Zhou Y, Liu Z, Lu Y, Jiang Y, Cao K, et al. Hepatic glycogenesis antagonizes lipogenesis by blocking S1P via UDPG. Science. 2024;383:eadi3332. [DOI] [PubMed] [Google Scholar]
- 6.Menendez JA, Lupu R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer. 2007;7:763–77. [DOI] [PubMed] [Google Scholar]
- 7.Luo J, Yang H, Song B-L. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol. 2019;21:225–45. [DOI] [PubMed] [Google Scholar]
- 8.Sun L-P, Seemann J, Goldstein JL, Brown MS. Sterol-regulated transport of SREBPs from endoplasmic reticulum to Golgi: Insig renders sorting signal in Scap inaccessible to COPII proteins. Proc Natl Acad Sci USA. 2007;104:6519–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Powell EE, Wong VW-S, Rinella M. Non-alcoholic fatty liver disease. Lancet. 2021;397:2212–24. [DOI] [PubMed] [Google Scholar]