

Abstract
BACKGROUND
Intracranial arteriosclerosis and cerebral amyloid beta (Aβ) are both involved in the etiology of Alzheimer's disease (AD) dementia, but the direct link between these two pathologies remains elusive.
METHODS
In 633 participants (mean age 69 years, 51% women) from the population‐based Rotterdam Study, we quantified cerebral Aβ accumulation on amyloid positron emission tomography (PET). We assessed calcification of the intracranial internal carotid (ICAC) and vertebrobasilar arteries (VBAC) as proxies of arteriosclerosis on non‐enhanced computed tomography (CT). Using logistic and linear regression, we studied the relationship of presence, burden, and type of calcification with the presence and burden of Aβ.
RESULTS
We found no associations of ICAC [odds ratio (OR): 0.85, 95% confidence interval (CI): 0.43, 1.72] or VBAC [OR: 0.59, CI: 0.26, 1.24] with cerebral Aβ. The results did not vary across ICAC subtypes.
DISCUSSION
Intracranial arteriosclerosis was not associated with cerebral Aβ, underscoring their independence in the etiology of AD dementia.
Highlights
Comprehensive assessment of intracranial arteriosclerosis (e.g., including subtypes).
Intracranial arteriosclerosis in different arteries and cerebral Aβ are not related.
Arteriosclerosis and Aβ likely influence Alzheimer's disease dementia independently.
Keywords: amyloid, arteriosclerosis, dementia, PET‐CT, vascular disease
1. BACKGROUND
Alzheimer's disease (AD) is the most common form of dementia and has a complex multifactorial etiology. 1 , 2 One of the most established risk factors in the etiological framework of AD is the parenchymal deposition of amyloid β (Aβ) plaques. 3 , 4 In the very first pathology study by Alois Alzheimer, 5 Aβ depositions were already described as a common phenomenon in patients with dementia. He also described that these depositions frequently coexisted with vascular disease of the intracranial arteries.
Many decades later, increasing evidence suggests that intracranial arteriosclerosis, that is, vascular disease in the cerebral circulation, substantially increases the risk of cognitive impairment and dementia. 6 − 10 Interestingly, whether or not intracranial arteriosclerosis and Aβ depositions are directly related to one another remains largely unknown. A first study indicated that these two pathologies may occur independently. 11 However, important aspects regarding intracranial arteriosclerosis remain understudied.
More specifically, previous studies did not investigate anterior and posterior intracranial arteriosclerosis separately or did not consider morphological subtypes of arteriosclerosis. 11 , 12 Previous work by van den Beukel and colleagues 6 shows that arteriosclerosis in the posterior circulation substantially contributes to a higher risk of developing dementia. Moreover, severe internal elastic lamina (IEL) calcification particularly increases dementia risk as opposed to a high, atherosclerotic, intimal calcification burden. Thus, these two morphological subtypes may differentially influence the etiology underlying dementia.
In this project, we set out to comprehensively study the association of intracranial arteriosclerosis in the anterior and posterior circulation with cerebral Aβ presence and burden in non‐demented elderly from the general population. By investigating this question in the general population, we aim to explore the interplay between cerebrovascular disease and Aβ, unraveling early mechanisms underlying neurodegeneration in AD dementia. We hypothesized that the presence and burden of intracranial arteriosclerosis will be associated with an increased risk of having Aβ and an increased Aβ burden. Furthermore, we hypothesized that the IEL calcification subtype will be most strongly associated with Aβ presence and an increased Aβ burden.
2. METHODS
2.1. Setting and study sample
The current study is embedded in the population‐based Rotterdam Study which was implemented in 1990 to assess the prevalence and determinants of common age‐related diseases in Rotterdam in the Netherlands. 13
Between 2018 and 2021, 639 participants underwent an amyloid positron emission tomography–computed tomography (PET‐CT) scan. The selection procedure has been described elsewhere in detail. 14 In brief, only participants who were 60 years or older and had a previous brain MRI scan between 2011 and 2016 were included. Exclusion criteria were contraindications for PET‐CT, insufficient quality of the previous MRI, large cortical infarcts, or a clinical diagnosis of dementia. To ensure that the burden of cerebrovascular pathology in the PET‐CT sub‐study was representative of that in the entire Rotterdam Study, we invited participants by randomly selecting them from quartiles of the white matter hyperintensity volume distribution, as quantified on previous MRI. 14 In the current study, we excluded four participants due to missing apolipoprotein E (APOE) ε4 genotyping and two participants due to imaging artifacts on CT. Thus, our final sample was comprised of 633 participants.
RESEARCH IN CONTEXT
Systematic review: We performed a systematic literature search on PubMed to explore existing knowledge on intracranial arteriosclerosis, Alzheimer's disease (AD), and amyloid β (Aβ). One previous study investigated the relationship between intracranial arteriosclerosis and cerebral Aβ and did not find an association. Furthermore, no other study incorporated morphological subtypes of intracranial arteriosclerosis into their analyses.
Interpretation: We did not find an association between intracranial arteriosclerosis and cerebral Aβ, indicating that these two factors likely influence the development of dementia and AD independently. Instead of causing cerebral Aβ accumulation, intracranial arteriosclerosis may contribute to neurodegeneration through hemodynamic and ischemic changes. Previous research indicates that intracranial arteriosclerosis can precede changes in cerebrovascular reactivity and pulsatility which may contribute to cell death.
Future directions: Our research further supports a multifactorial approach to AD dementia etiology. Therefore, future studies should investigate the etiology underlying AD holistically.
2.2. Amyloid PET CT imaging
PET imaging was performed using 300 MBq (±20%) of 18F‐florbetaben (Neuraceq, Life Molecular Imaging GmbH). Approximately 90 to 110 min after injection, participants were scanned for 20 min in list mode on a Siemens Biograph mCT PET‐CT (Siemens Healthineers, Erlangen, Germany). Prior to the PET scan, all participants received a low‐dose CT, which is intended for anatomical referencing and attenuation correction. The scan was obtained at 120 kVp and 40 mAs with a slice thickness of 2 mm.
2.2.1. Arteriosclerosis assessment
Using the anatomical low‐dose CT scan from the amyloid PET‐CT, we quantified intracranial internal carotid artery calcification (ICAC), and vertebrobasilar artery calcification (VBAC), as markers of intracranial arteriosclerosis in the anterior and posterior circulation. ICAC was quantified bilaterally starting at the horizontal segment of the petrous internal carotid artery (segment C2) and ending at the sella turcica (segment C7). VBAC was quantified by assessing calcification in the vertebral arteries as well as in the basilar artery itself. Vertebral artery calcification was assessed between the dura and the level where the arteries merge to form the basilar artery. Basilar artery calcification was measured from the merge of the vertebral arteries to the tip of the basilar artery. Quantification was performed using a validated, previously described semiautomatic scoring method. 15 In brief, regions of interest were manually drawn around the calcifications, after which calcification volumes were calculated by multiplying the number of pixels above a prespecified threshold of 130 Hounsfield Units, with the pixel size and increment. All volumes are expressed in mm3.
The ICAC subtypes were qualitatively evaluated by assessing the circularity, thickness, and morphology of the calcifications. 16 Based on this evaluation, ICAC subtypes were categorized into four groups (no calcification, predominantly intimal calcification, predominantly IEL calcification, and mixed calcification). 17 In contrast to ICAC, VBAC subtypes and according to standardized evaluation tools are not validated yet. Therefore, only ICAC subtypes have been included in our final analysis. Examples of the ICAC and VBAC assessments are provided in Figure 1.
FIGURE 1.
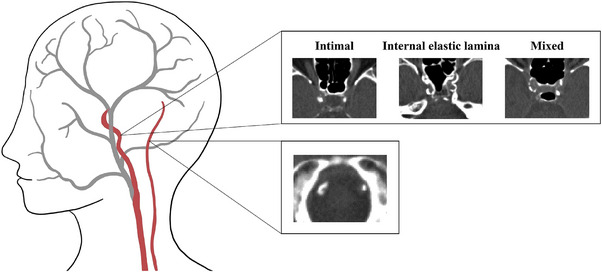
Examples of intracranial internal carotid artery calcification (top) and vertebrobasilar artery calcification (bottom) as seen on non‐contrast computed tomography (CT).
2.2.2. Classification of Aβ burden
Aβ burden was assessed using an algorithm that combines qualitative visual reads with quantitative assessments expressed as the standardized uptake value ratio (SUVR). 18 The average cortical SUVR was calculated by dividing the tracer uptake in four brain areas (lateral temporal, frontal, posterior cingulate, and parietal cortices), by that of the cerebellar reference region. Participants with an SUVR ≥ 1.24 were classified as Aβ‐positive and those with an SUVR < 1.10 as Aβ‐negative. Participants with an SUVR between 1.10 and 1.24 were only considered Aβ‐positive when their scan was read positive by at least two trained and independent raters. 14
2.3. Assessment of covariables
Information on age, sex, as well as cardiovascular and genetic risk factors was also obtained. These factors included body mass index (BMI), hypertension, diabetes, dyslipidemia, smoking status (never, current, former), alcohol intake (grams/day), and APOE ε4 carriership (number of ε4 alleles). BMI was measured as weight (in kg)/height2 (in m). Hypertension was classified as present if one or more of the following characteristics were met: systolic blood pressure ≥140 mmHg, diastolic blood pressure ≥90 mmHg, use of antihypertensive medication. Participants were rated as diabetic if fasting serum glucose levels were ≥7 mmol/L and/or if they were using antidiabetic medication. Dyslipidemia was defined as using lipid‐lowering medication and/or having a total cholesterol concentration of at least 6.2 mmol/L. Hypertension, diabetes, and dyslipidemia were scored if participants had at least one available blood or medication measure.
2.4. Data analysis and statistics
Both calcification volumes and Aβ burden were strongly skewed and, therefore, log‐transformed to approximate a normal distribution. To account for calcification volumes of 0, we added + 1 mm3 to all individual volumetric scores before transforming the data. 19 In the following step, we standardized all log‐transformed estimates to ensure an interpretation per standard deviation and to allow for meaningful comparisons of regression coefficients. One participant had missing smoking data. Therefore, we used the last observation carried forward method and imputed this value using the most recent available observation, which was collected in 2007.
First, we calculated ICAC and VBAC prevalence. The prevalence of ICAC subtypes was additionally investigated across 5‐year interval age strata.
Second, we investigated the association of the presence and volume of ICAC and VBAC with the presence of Aβ using logistic regression models. In the first model, we adjusted for age, sex, and number of APOE ε4 alleles. In the second model, we additionally adjusted for BMI, hypertension, diabetes, dyslipidemia, smoking status, and alcohol intake. Interactions of ICAC and VBAC with APOE ε4 status were taken into account and removed from the model if they were not statistically significant.
Third, we studied the association of the presence and volume of ICAC and VBAC with continuous Aβ burden (SUVR) using two linear regression models with the same adjustments as above.
Fourth, we evaluated the association of dummy‐coded ICAC subtypes on Aβ presence and burden using logistic and linear regression, respectively. In logistic regression analyses, we first compared the three different calcification subtypes (intima, IEL, mixed) to no calcification. Then, the analysis was repeated only focusing on intimal and IEL calcification, hence, excluding participants with mixed calcification patterns and no calcification. The adjustments used in both models were identical to those mentioned above. We used logistic and multiple linear regression to determine the effect of ICAC subtypes on Aβ presence and burden, respectively.
Fifth, we performed a sensitivity analysis to investigate potential selection bias. In this analysis, we repeated all aforementioned analyses stratified by white matter lesion volume quartiles. All analyses have been carried out in R version 4.2.2. 20
3. RESULTS
3.1. Sample characteristics
A descriptive overview of sample characteristics is presented in Table 1. Approximately half of the sample was female (51.2%) and the mean age was 69.3 years (SD = 5.46 years). In total, 16.4% were Aβ positive. The prevalence of ICAC was 78.4%. Among those with ICAC, intimal calcification was most prevalent (59.1%), followed by IEL (26.4%) and mixed calcification (14.5%). ICAC subtypes stratified across age groups are presented in Figure S1. Overall, intimal calcification was most prevalent in participants aged 65 to 70 and decreased in older individuals. IEL calcification followed a reversed pattern, characterized by lower prevalence in younger participants that increased until the age of 80 and stabilized afterwards. Mixed calcification followed a less stable trajectory with prevalence ranging from just under 10% to approximately 25%. Finally, the absence of calcification decreased linearly until the age of 85. All individuals aged 85 and older presented with at least one of the different calcification subtypes. The overall prevalence of VBAC was 13.3%. A scatterplot showing calcification volumes and Aβ SUVRs is provided in Figure S2.
TABLE 1.
Descriptive and demographic characteristics of the study sample.
| Characteristics | Participants (N = 633) |
|---|---|
| Sex (female) | 324 (51.2) |
| Age (years) | 69.3 (5.46) |
| APOE ε4 | |
| None | 444 (70.2) |
| 1 allele | 168 (26.5) |
| 2 alleles | 21 (3.3) |
| Prevalent stroke | 10 (1.6%) |
| BMI (kg/m2) | 27.4 (4.15) |
| Hypertension (yes) | 352 (55.6) |
| Diabetes (yes) | 59 (9.3) |
| Dyslipidemia (yes) | 336 (53.1) |
| Smoking history | |
| Never | 218 (34.4) |
| Current | 112 (17.7) |
| Former | 302 (47.7) |
| Missing | 1 (0.2) |
| Alcohol intake (gr/day) | 8.38 (9.14) |
| ICAC prevalence | 496 (78.4) |
| ICAC volume (mm3) a | 50.8 (177) |
| ICAC subtype b | |
| Intimal | 293 (59.1) |
| IEL | 131 (26.4) |
| Mixed | 72 (14.5) |
| VBAC prevalence | 84 (13.3) |
| VBAC volume (mm3) c | 0, 304 |
| Amyloid prevalence | 104 (16.4) |
| Amyloid PET SUVR a | 0.986 (0.0951) |
Note: Continuous values are reported as mean (standard deviation) and categorical variables as number (percentage).
Measure is presented as median (interquartile range).
Based on N = 496 participants with prevalent ICAC.
Measure is presented as minimum, maximum.
Abbreviations: APOE, apolipoprotein E; BMI, body mass index; ICAC, internal carotid artery calcification; IEL, internal elastic lamina; PET, positron emission tomography; SUVR, standardized uptake value ratio; VBAC, vertebrobasilar artery calcification.
3.2. Association of intracranial calcification with Aβ deposition
The point estimates and associated confidence intervals of the regression models are presented in Figure 2. Furthermore, the corresponding regression table has been summarized in Table S1. We found no associations of either ICAC or VBAC presence [ICAC: odds ratio (OR) = 0.85, 95% confidence interval (CI) = 0.43, 1.72); VBAC: OR = 0.59, 95% CI = 0.26, 1.24] and volume [ICAC: OR = 0.77, 95% CI = 0.58, 1.02; VBAC: OR = 0.91, 95% CI = 0.69, 1.16] with Aβ presence. We also found no association of ICAC and VBAC presence [ICAC: β per SD increase (β) = −0.07, 95% CI = −0.24, 0.10; VBAC: β = −0.08, 95% CI = −0.28, 0.12] and volume [ICAC: β = −0.06, 95% CI = −0.14, 0.01]; VBAC: β = −0.02, 95% CI = −0.09, 0.05] with Aβ burden (SUVR).
FIGURE 2.
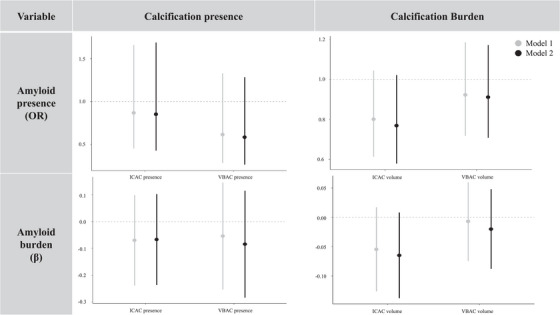
Association of calcification presence and burden with amyloid β (Aβ) presence and burden. The bars represent regression coefficients and 95% confidence intervals. The top row shows logistic regression coefficients that were transformed into odds ratios (OR). OR = odds of Aβ positivity for calcification presence versus absence and calcification volumes (standardized ln(volume in mm3 + 1)). The bottom row shows linear regression coefficients (β). β = change in standardized Aβ burden (ln(Aβ SUVR)) for calcification presence versus absence and calcification volumes (standardized ln(volume in mm3 + 1)). ICAC, internal carotid artery calcification; SUVR, standardized uptake value ratio; VBAC, vertebrobasilar artery calcification.
3.3. Relationship between ICAC subtypes and global Aβ deposition
As can be seen in Figure 3, there were no differences in the association of ICAC with cerebral Aβ across calcification subtypes. Neither the comparison of ICAC subtypes with no calcification nor the comparison of IEL with intimal calcification yielded significant results. Hence, the absence of an effect of ICAC on Aβ presence and burden was consistent across all morphological subtypes.
FIGURE 3.
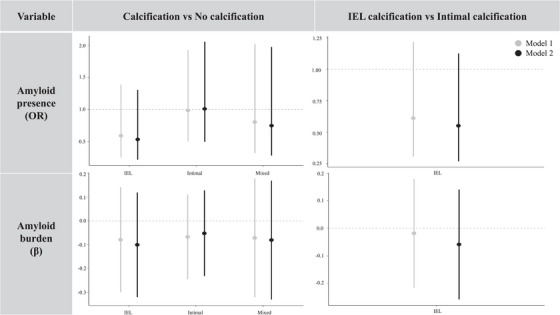
Association of internal carotid artery calcification (ICAC) subtypes with amyloid β (Aβ) presence and burden. The bars represent regression coefficients and 95% confidence intervals. The top row shows logistic regression coefficients that were transformed into odds ratios (OR). OR = odds of Aβ positivity across ICAC subtypes. The bottom row shows linear regression coefficients (β). β = change in standardized Aβ burden (ln(Aβ SUVR)) across ICAC subtypes. IEL, internal elastic lamina; SUVR, standardized uptake value ratio.
3.4. Sensitivity analyses
To rule out selection bias due to the sampling strategy of the PET‐CT study – randomly selecting participants from each quartile of the white matter hyperintensity volume distribution (see 14 ) – we carried out sensitivity analyses by stratifying the sample according to these white matter hyperintensity volume quartile groups. The results are summarized in Tables S2 and S3. In line with the main analysis, neither ICAC nor VBAC presence nor volume were associated with cortical Aβ presence and burden across quartiles of white matter hyperintensity burden.
4. DISCUSSION
In this study including 633 community‐dwelling elderly from the general population, the majority of the participants had prevalent ICAC while VBAC was less prevalent. Overall, we found no evidence for an association of intracranial arteriosclerosis with cerebral Aβ deposition. Neither calcification presence and volume nor intracranial calcification subtypes were associated with cerebral Aβ presence and burden.
In line with earlier findings from the Atherosclerosis Risk in Communities Study, 11 we did not find a relationship between intracranial arterial calcification and cerebral Aβ depositions. Our study further investigated this relationship by assessing the differential impact of calcification subtypes on Aβ presence and burden. Intimal and IEL calcification vary greatly in histology. 16 While intimal calcification is associated with stenosis and a potentially increased risk for thromboembolic events, IEL calcification is related to stiffening of blood vessels and resulting changes in blood flow and pulsatility. 17 We hypothesized that IEL calcification would relate to cerebral Aβ accumulation more strongly since previous research showed that arterial stiffness could contribute to Aβ accumulation through hemodynamic changes. 21 However, Aβ burden did not vary across intracranial arteriosclerosis subtypes in the current study.
The absence of an association between these two pathologies indicates that intracranial calcification and Aβ may influence the development of AD dementia independently. Previous research indicates that calcification of the intracranial vasculature is associated with hemodynamic and ischemic changes in cerebrovascular reactivity and pulsatility. 22 , 23 These changes, in turn, may promote neurodegeneration and dementia. Calcification of the arteries could, thus, both independent of and in interplay with Aβ, disrupt brain homeostasis and contribute to cell death. 22 Therefore, future studies should investigate how intracranial arteriosclerosis influences AD dementia via different etiological pathways. As indicated by previous studies, 24 − 26 potential links could be made to genetics, other biomarkers, and environmental factors.
An alternative explanation of the absence of an effect between intracranial arteriosclerosis and cerebral Aβ could be the timing of the biomarker assessment. Aβ plaque development is estimated to take between 15 and 40 years, suggesting that it is preceded by complex brain changes that may already take place in middle adulthood. 27 It is, thus, possible that calcification and Aβ were measured after an initial cascade of events had already been triggered. Future research should, therefore, extend to younger populations as well.
4.1. Limitations
Strengths of this study include the large sample size compared to previous PET‐CT studies 11 , 28 and the population‐based setting in which we concurrently assessed arterial calcification and cerebral Aβ. However, important limitations need to be considered.
First, the external validity of our conclusions is limited due to a lack of replication. Without replication, uncertainty remains regarding the consistency of the observed associations in different contexts. It is worth noting that our results align with a prior study, 11 therefore, indicating a certain degree of robustness.
Second, the absolute number of participants with prevalent amyloid and prevalent VBAC was somewhat lower in our study than in previous epidemiological studies, 29 , 30 potentially affecting our statistical power. Future studies with larger cohorts or meta‐analyses combining data from multiple studies may be necessary to overcome this limitation and provide additional robust statistical inferences.
Third, it is possible that our study was object to selection bias. The prevalence of intracranial arteriosclerosis was lower in this sample compared to previous Rotterdam Study cohorts (81.6% for ICAC and 21.0% for VBAC). 17 , 29 Given that the current sample was predominantly stroke‐free and healthy enough to attend a PET‐CT scan, it is possible that the selected participants were, on average, healthier limiting the generalizability of our findings.
Finally, our study design does not allow to draw conclusions regarding the association between intracranial arteriosclerosis and amyloid deposition in individuals with dementia. While we established that no such association exists in non‐demented elderly, this does not imply the absence of a relationship in dementia patients. Consequently, further research is necessary to explore both earlier and later mechanisms in the development of AD dementia.
4.2. Conclusion
To conclude, we found no significant associations between intracranial arteriosclerosis and cerebral Aβ presence and burden. Given the known association of both pathologies with AD dementia risk, this finding indicates that intracranial arteriosclerosis and Aβ influence AD dementia independently. Therefore, future research should aim at unraveling their links to other neuropathological processes to gain insights into the etiology underlying the disease.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest. Author disclosures are available in the Supporting information.
CONSENT STATEMENT
This study was approved by the Erasmus MC Medical Ethics Committee (MEC‐2018‐085). Approval is based on the Declaration of Helsinki concerning medical research with human subjects. Approval for the Rotterdam study has been obtained by the Erasmus MC Medical Ethics Committee as well as by the Dutch Ministry of Health, Welfare, and Sport (see Ikram et al. 13 for details).
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
We are thankful to Dr Roelf Valkema, for his help in initiating this study and for providing clinical expertise and assistance. Moreover, we thank the entire staff of the Nuclear Medicine department for their help in acquiring the amyloid PET data, including but not limited to Dennis Kuijper, Annelies Schipper, Pieter Meppelink, and Jean‐Baptiste Aarssen for their coordinating roles. We would also like to acknowledge the immense contribution of the data management team of the Rotterdam Study, with Jolande Verkroost‐van Heemst in particular, and of the Imaging Trialbureau. Lastly, we would like to thank our study participants for their contribution. D.B., J.N., M.W.V., and A.M.S. were supported by a Cure Alzheimer's Fund. Furthermore, this project has received funding from the European Union's Horizon 2020 Research and Innovation Programme (MSCA‐IF‐GF no.101032288 to J.N.), ZonMW Memorabel grant 733050817 (to M.W.V.), Alzheimer's Association Research Grant (AARG‐22‐972229 to M.W.V. and J.N.), ABOARD, which is a public‐private partnership receiving funding from ZonMW (no. 73305095007) and Health∼Holland, Topsector Life Sciences & Health (PPP‐allowance; no. LSHM20106 to M.W.V.) as well as TAP‐dementia, a ZonMw Funded Project (no. 10510032120003 to M.W.V.) in the context of the Dutch National Dementia Strategy. D.B. was supported by the BrightFocus Foundation (A2017424F) and Alzheimer's Association (AARG‐21‐846504). The Rotterdam Study is supported by the Erasmus Medical Center and Erasmus University, Rotterdam, the Netherlands; the Organization for Scientific Research; the Netherlands Organization for Health Research and Development; the Research Institute for Diseases in the Elderly; the Netherlands Genomics Initiative; the Ministry of Education, Culture, and Science; the Ministry of Health, Welfare, and Sports; the European Commission (DG XII); and the Municipality of Rotterdam. The funding sources that contributed to the Rotterdam Study or to the authors had no role in study design, data collection, data analysis, data interpretation, or writing of the report.
Streiber AM, Neitzel J, Nguyen Ho PT, Vernooij MW, Bos D. Intracranial arteriosclerosis is not associated with cerebral amyloid deposition. Alzheimer's Dement. 2024;16:e70005. 10.1002/dad2.70005
REFERENCES
- 1. Gouilly D, Rafiq M, Nogueira L, et al. Beyond the amyloid cascade: an update of Alzheimer's disease pathophysiology. Rev Neurol (Paris). 2023. [DOI] [PubMed] [Google Scholar]
- 2. Cummings JL. Alzheimer's disease. N Engl J Med. 2004;351:56‐67. [DOI] [PubMed] [Google Scholar]
- 3. Frisoni GB, Molinuevo JL, Altomare D, et al. Precision prevention of Alzheimer's and other dementias: anticipating future needs in the control of risk factors and implementation of disease‐modifying therapies. Alzheimers Dement. 2020;16:1457‐1468. [DOI] [PubMed] [Google Scholar]
- 4. Hampel H, Hardy J, Blennow K, et al. The amyloid‐β pathway in Alzheimer's disease. Mol Psychiatry. 2021;26:5481‐5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alzheimer A. Uber eigenartige Erkrankung der Hirnrinde. All Z Psychiatr. 1907;64:146‐148. [Google Scholar]
- 6. van den Beukel TC, Wolters FJ, Siebert U, et al. Intracranial arteriosclerosis and the risk of dementia: a population‐based cohort study. Alzheimers Dement. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roher AE, Tyas SL, Maarouf CL, et al. Intracranial atherosclerosis as a contributing factor to Alzheimer's disease dementia. Alzheimers Dement. 2011;7:436‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sabayan B, Goudarzi R, Ji Y, et al. Intracranial atherosclerosis disease associated with cognitive impairment and dementia: systematic review and meta‐analysis. J Am Heart Assoc. 2023;12:e032506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boyle PA, Yu L, Leurgans SE, et al. Attributable risk of Alzheimer's dementia attributed to age‐related neuropathologies. Ann Neurol. 2019;85:114‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dearborn JL, Zhang Y, Qiao Y, et al. Intracranial atherosclerosis and dementia: the Atherosclerosis Risk in Communities (ARIC) Study. Neurology. 2017;88:1556‐1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gottesman RF, Mosley TH, Knopman DS, et al. Association of intracranial atherosclerotic disease with brain β‐amyloid deposition: secondary analysis of the ARIC study. JAMA Neurol. 2020;77:350‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rahmani F, Nguyen M, Chen CD, et al. Intracranial internal carotid artery calcification is not predictive of future cognitive decline. Alzheimers Res Ther. 2022;14:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ikram MA, Kieboom BCT, Brouwer WP, et al. The Rotterdam Study. Design update and major findings between 2020 and 2024. Eur J Epidemiol. 2024:1‐24. [DOI] [PubMed] [Google Scholar]
- 14. van Arendonk J, Neitzel J, Steketee RME, et al. Diabetes and hypertension are related to amyloid‐beta burden in the population‐based Rotterdam Study. Brain. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bos D, van der Rijk MJM, Geeraedts TEA, et al. Intracranial carotid artery atherosclerosis: prevalence and risk factors in the general population. Stroke. 2012;43:1878‐1884. [DOI] [PubMed] [Google Scholar]
- 16. Kockelkoren R, Vos A, Van Hecke W, et al. Computed tomographic distinction of intimal and medial calcification in the intracranial internal carotid artery. PLoS One. 2017;12:e0168360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van Den Beukel TC, Van Der Toorn JE, Vernooij MW, et al. Morphological subtypes of intracranial internal carotid artery arteriosclerosis and the risk of stroke. Stroke. 2022;53:1339‐1347. [DOI] [PubMed] [Google Scholar]
- 18. Pontecorvo MJ, Arora AK, Devine M, et al. Quantitation of PET signal as an adjunct to visual interpretation of florbetapir imaging. Eur J Nucl Med Mol Imaging. 2017;44:825‐837. [DOI] [PubMed] [Google Scholar]
- 19. Osborne J. Notes on the use of data transformations. Pract Assess Res Eval. 2002;8. [Google Scholar]
- 20. R Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2022. [Google Scholar]
- 21. Hughes TM, Kuller LH, Barinas‐Mitchell EJM, et al. Arterial stiffness and β‐amyloid progression in nondemented elderly adults. JAMA Neurol. 2014;71:562‐568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klohs J. An integrated view on vascular dysfunction in Alzheimer's disease. Neurodegener Dis. 2020;19:109‐127. [DOI] [PubMed] [Google Scholar]
- 23. Iadecola C. The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol. 2010;120:287‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hoogmartens J, Cacace R, Van Broeckhoven C. Insight into the genetic etiology of Alzheimer's disease: a comprehensive review of the role of rare variants. Alzheimers Dement. 2021;13:e12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang X‐X, Tian Y, Wang Z‐T, Ma Y‐H, Tan L, Yu J‐T. The epidemiology of Alzheimer's disease modifiable risk factors and prevention. J Prev Alzheimers Dis. 2021;8:313‐321. [DOI] [PubMed] [Google Scholar]
- 26. Blennow K, Zetterberg H. Biomarkers for Alzheimer's disease: current status and prospects for the future. J Intern Med. 2018;284:643‐663. [DOI] [PubMed] [Google Scholar]
- 27. Patterson BW, Elbert DL, Mawuenyega KG, et al. Age and amyloid effects on human central nervous system amyloid‐beta kinetics. Ann Neurol. 2015;78:439‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jack CR, Barrio JR, Kepe V. Cerebral amyloid PET imaging in Alzheimer's disease. Acta Neuropathol. 2013;126:643‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. van der Toorn JE, Engelkes SR, Ikram MK, et al. Vertebrobasilar artery calcification: prevalence and risk factors in the general population. Atherosclerosis. 2019;286:46‐52. [DOI] [PubMed] [Google Scholar]
- 30. Jansen WJ, Janssen O, Tijms BM, et al. Prevalence estimates of amyloid abnormality across the Alzheimer disease clinical spectrum. JAMA Neurol. 2022;79:228‐243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information