

Abstract
Sickle cell disease (SCD) is a common hereditary blood disorder that profoundly impacts individuals’ health, causing chronic pain, anemia, organ damage, increased susceptibility to infections, and social and psychological effects. Over the years, advances in treatment have improved the long-term outcomes of SCD patients. However, problems such as limited access to hematopoietic stem cell transplantation (HSCT) and potential complications associated with the available therapies underscore the importance of continued research and development. The recent FDA approval of Casgevy (Exagamglogene autotemcel), a genetic therapy based on CRISPR/Cas9 technology, demonstrates a comprehensive effort to address the complexity of SCD using new technologies. This review explores the potential of CRISPR/Cas9 for treating SCD and evaluates its efficacy, safety, and long-term outcomes compared to traditional treatment approaches. Long-term research is needed to comprehensively assess the safety, effectiveness, and inclusion of CRISPR/Cas9, ensuring its overall efficacy.
Keywords: casgevy, CRISPR/Cas9, gene editing, hematopoietic stem cell therapy, sickle cell disease
Introduction
Highlights
Cell-based gene therapy with CRISPR/Cas9 for SCD: a transformative approach.
Traditional methods’ limitations: scarcity of donors, graft-versus-host disease risk, and lifelong management.
CRISPR/Cas9 precision: targeting the defective HBB gene.
Overcoming donor availability challenges in traditional HSCT with CRISPR/Cas9.
CRISPR/Cas9 challenges: precise targeting, minimizing off-target effects, and addressing treatment-related hospitalization and chemotherapy.
Ongoing research for confirming the long-term effectiveness and safety of CRISPR/Cas9.
Delivery challenges: carefully considering introducing CRISPR/Cas9 into the nucleus.
Optimization efforts: refining delivery methods for enhanced precision and feasibility.
Importance of recognizing patient perspectives, preferences, and concerns.
Community engagement: integrating insights for successful CRISPR/Cas9 therapy adoption in SCD.
Sickle cell disease (SCD) is a globally prevalent hereditary blood disorder affecting hemoglobin, with a particularly high incidence in sub-Saharan Africa, India, the Mediterranean, and the Middle East1–3. It encompasses various clinically significant hemoglobinopathies, such as sickle cell anemia (SCA), sickle cell syndromes, and other combinations resulting from co-inheritance with the hemoglobin C (Hb C) gene, β-thalassemia genes (β0 or β+), or other β-globin structural variants. The most prevalent SCD type is homozygous hemoglobin SS (Hb SS), characterized by the inheritance of two abnormal β-globin genes4. Homozygous Hb SS, in which both parents contribute to the sickle mutation, commonly leads to sickle cell anemia (SCA) and represents the most severe form of the disease5.
SCD arises from a point mutation within the β-globin gene on the short arm of chromosome 11, leading to the substitution of glutamic acid with valine at the sixth position6. This genetic alteration results in an abnormal form of hemoglobin, known as hemoglobin S (HbS), causing red blood cells to become rigid and sickle-shaped under certain conditions, such as deoxygenation and acidosis. This pathological process contributes to intravascular inflammation and the obstruction of small blood vessels7,8.
Noteworthy clinical features of SCD include acute painful crises and chronic hemolytic anemia9,10. As the pathological processes associated with SCD unfold systemically, potentially life-threatening complications arise11. These complications include acute splenic sequestration, avascular necrosis, retinopathy, aplastic crises, acute chest syndrome (ACS), infections, multisystem organ failure, and stroke12,13. Moreover, complications of SCD during infancy substantially hinder aspects of health-related quality of life, including physical, mental, and psychosocial well-being2.
SCD affects millions of people globally, with an estimated incidence of 300 000 annually worldwide11,14. In the United States, the population of individuals with SCD is ~100 000 and is expected to rise1,3. The carrier frequencies of the mutation are generally below 25%, as higher levels are limited by the profound disadvantages of allelic homozygosity10. Sub-Saharan Africa is particularly impacted by SCD, where SCD accounts for up to 6% of all childhood deaths, and estimates indicate that 50–90% of children born with SCD do not survive beyond 5 years of age10,15. In contrast, higher-resourced nations have experienced a notable decline in the mortality rates associated with SCD over the last five decades. This improvement is attributed in part to the progression of newborn screening and comprehensive care initiatives16.
Studies have shown that the risk of fatal infections in SCD patients is reduced by vaccination and the use of prophylactic antibiotics17,18. Moreover, long-term outcomes have significantly improved with treatments that include blood transfusions and hydroxyurea therapy19. Despite these advancements, there remain complications and toxicities of blood transfusions and hydroxyurea therapy, emphasizing the need for more personalized approaches20,21. The ongoing development of new therapies is crucial to address these limitations and enhance patient care. Unraveling the pathophysiological targets of SCD has provided insights into potential therapeutic avenues and guided clinical trials focusing on anti-platelet, anti-adhesion, and anti-coagulation factor agents to prevent acute vaso-occlusive crisis (VOC) pain in SCD. These include inducers of fetal hemoglobin (HbF) and anti-sickling agents. A notable example is the development of crizanlizumab, an anti-P-selection molecule specifically designed to treat sickle VOC22. Additionally, there are agents targeting free hemoglobin, heme, and iron, as well as modulators of ischemia/reperfusion, oxidative stress, and inflammation. Anti-inflammatory agents and treatments tailored to counteract specific end-organ damage are also being investigated. This multifaceted approach reflects comprehensive efforts to address the complex challenges posed by SCD23.
Currently, available treatments are restricted to transfusions and hydroxyurea, with hematopoietic stem cell transplantation (HSCT) considered the only potentially curative therapy. HSCT for SCD is accessible only to the 18% of SCD patients in the United States who have an HLA-matched sibling donor. The use of unrelated or haploidentical donors increases the risk of mortality and complications. Constraints include graft-versus-host disease and donor scarcity24. An alternative involves using homologous recombination to edit the patient’s own hematopoietic stem cells, thus addressing safety concerns associated with traditional HSCT25,26.
Ongoing developments in therapeutic options include gene therapy and editing11. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR/Cas9) and CRISPR-associated protein 9 is a revolutionary gene-editing technology27. Casgevy, a cell-based gene therapy, has recently gained approval for treating SCD in individuals aged 12 years and above who experience recurring VOC. The treatment involves the modification of patients’ hematopoietic (blood) stem cells through genome editing using CRISPR/Cas9 technology28.
This significant achievement marks the first Food and Drug Administration (FDA) approved use of CRISPR/Cas9 technology, reflecting a paradigm shift in genetic interventions for SCD. Its approval as a therapeutic represents a transformation in managing hereditary blood disorders but requires a close examination of its implications. The approval of Casgevy underscores the transformative potential of genetic interventions, particularly CRISPR/Cas9, in addressing hereditary disorders such as SCD, marking a significant milestone in the field of genetic therapies29,30.
This narrative review centers on a fundamental inquiry: What defines the current landscape of cell gene therapy using CRISPR/Cas9 for sickle cell disease, and how does it compare to traditional treatment approaches concerning efficacy, safety, and long-term outcomes? Such an investigation is crucial for informed decision-making and guiding the trajectory of SCD management.
Method
A systematic search was performed in PubMed, Google Scholar, Embase, and Web of Science using a combination of boolean operators. The following search strategy was employed: (“Sickle Cell Anemia” OR “Sickle Cell Anemia/therapy” OR “Hemoglobin, Sickle” OR “Hemoglobin, Sickle/therapy”) AND (“Therapeutics” OR “Gene Therapy” OR “CRISPR-Cas Systems” OR “Gene Editing”). Articles retrieved from the initial search were screened based on their relevance to the objectives of the narrative review. Titles and abstracts were reviewed to identify articles discussing the pathophysiology of SCD, traditional treatment approaches (e.g. hydroxyurea, blood transfusions), and recent advancements in CRISPR/Cas9 gene therapy for SCD. Full-text articles were then assessed for eligibility, with inclusion criteria encompassing studies providing insights into the mechanisms, efficacy, and safety profiles of both traditional and gene therapy interventions for SCD. Exclusion criteria included studies unrelated to the review’s scope and not in English language.
As a narrative review, quality assessment was primarily focused on the credibility and relevance of the included literature to the review’s objectives. While no formal quality assessment tools were employed, efforts were made to critically appraise the methodological rigor, clarity of reporting, and potential biases of individual studies. High-quality studies with robust methodologies were given greater consideration. Through the review process, any gaps in the existing literature were identified, and recommendations for future research directions, improvement areas, and challenges to be addressed in the field of gene therapy for sickle cell disease using CRISPR/Cas9 were provided.
Limitations of this narrative review included the potential for selection bias inherent in the search strategy and the reliance on published literature. The review may not have captured all relevant studies, and the interpretation of findings may be influenced by the subjective nature of narrative synthesis. Ethical considerations were taken into account throughout the review process, including proper citation and attribution of sources to acknowledge the contributions of previous research.
Traditional treatment options
The treatment of SCD can be divided into preventive therapy, symptomatic therapy, and curative therapy31,32. Preventive therapy aims to minimize the chances of life-threatening infections and prevent the sickling of cells. Infections are primarily prevented by administering prophylactic penicillin to affected children, which increases their lifespan and improves their quality of life33. Hydroxycarbamide, also known as hydroxyurea, was first approved in 1998 by the FDA and was the only effective drug available in the market for SCD for nearly 20 years14. The drug is a ribonucleotide reductase inhibitor with multiple effects, including increasing the expression of human fetal hemoglobin (HbF) expression and decreasing RBC adhesion and leukocyte count34,35. The major benefit is the induction of HbF synthesis, which inhibits sickle cell hemoglobin (HbS) polymerization and improves the anemic profile by reducing red blood cell rigidity and hemolysis. RBC adhesion is also reduced by the ability of hydroxyurea to act as a vasodilator via nitric oxide (NO) production. It also prevents vaso-occlusion by decreasing white blood cell (WBC) count3. Although abundantly used, the drug has many side effects, such as hair loss, neutropenia, oligospermia, and bone marrow suppression, and regular blood count monitoring is required to rule out severe neutropenia36. In 2017, L-glutamine became the second SCD drug to reach the market37. The drug reduces the intensity and number of episodic pain episodes and ACS, which is one of the leading causes of hospitalization and death in patients with SCD38. The mechanism mainly involves the antioxidant properties of L-glutamine. L-glutamine administration in sickle cell disease enhances the NAD redox system, mitigating oxidative stress and reducing membrane damage in sickle red blood cells by increasing the antioxidative potential of NAD despite elevated production39. The drug is a potential substitute for patients who are either non-responsive to hydroxyurea therapy or experience severe side effects37. Except for nausea and gastrointestinal (GI) distress, this drug does not have significant safety concerns40. Other drugs introduced more recently include Voxelotor and Crizanlizumab. Voxelotor induces a conformational change in HbS, enhancing its oxygen affinity and reducing the potential for polymerization, ultimately mimicking normal hemoglobin and improving SCD symptoms41. Similarly, Crizanlizumab, a P-selectin inhibitor, has been shown to increase microvascular flow and significantly decrease levels of soluble vascular cell adhesion molecule 1, and was observed to reduce the pain crisis by 45.3% more effectively than with placebo, with no significant side effects42. In addition to pharmacologic agents, ~10% of SCD patients in high-income countries receive chronic red blood cell (RBC) transfusions. Transfusion recipients with SCD may be transfused over ten units per therapy to replace circulating sickle RBCs with donor RBCs43. Alloimmunization, iron overload, and the transmission of infectious agents are major concerns with frequent transfusions44. However, exchange transfusion is the mainstay of SCA treatment. Symptomatic treatments for Sickle cell disease include pain management with analgesics, hydration to maintain proper blood flow, blood transfusions for severe anemia, antibiotics for infection prevention, vaccinations, targeted therapies for pulmonary hypertension, and comprehensive care programs for overall management.
Traditionally, HSCT has been considered the only curative therapy. The long-term survival of patients with β-thalassemia who have undergone HSCT is greater than 90%, indicating the utility and safety of this therapy for SCD. Clinical trials conducted in children with SCD in Europe and the US showed greater than 90% long-term survival. Twenty-two children under 16 years of age with symptomatic SCD received marrow allografts from human leukocyte antigen (HLA)-identical siblings between September 1991 and April 1995. Indications for transplantation included a history of stroke (n=12), recurrent ACS (n=5), and recurrent painful crises (n=5). Twenty patients survived, with a median follow-up of 23.9 months (range, 10.1–51.0), and 16 patients had stable engraftment of donor hematopoietic cells45.
There are many barriers to HSCT, such as donor match, patient age, and the presence of severe sequelae. A major limitation in the use of HSCT for SCD treatment is that a matched sibling donor is available to less than 15% of patients who are otherwise suitable candidates for transplantation. Therefore, gene therapy is an alternative treatment option for SCD. Despite advancements, the number of HSCT cases of SCD is increasing in many countries, including Brazil44. To date, very few transplants have been performed in adults with SCD because morbidity and mortality concerns of HSCT are significantly higher in adults than in children46. The risk of graft-versus-host-disease, infections, infertility, and other long-term complications further limits its widespread use47. ABO mismatch between the patient and donor, which occurs in ~30% of HSCT cases, can cause hemolysis and red cell aplasia44.
Overall, the treatment of SCD encompasses preventive, symptomatic, and curative approaches. While HSCT has traditionally been considered curative, its limited availability and associated risks drive the exploration of alternative options, such as gene therapy, given the challenges and constraints related to HSCT.
CRISPR/Cas9 technology for SCD gene therapy
Genome editing is a new treatment option that offers promising outcomes in managing SCD.
Genome editing involves DNA modification by inserting, deleting, or replacing DNA fragments at the target sites to acquire specific genetic traits, inactivate target genes, and correct pathogenic gene mutations. To enhance gene editing, low cell density cultures, hematopoietic stem cell (HSCs) self-renewal agonists, and cytokines are commonly used. Most studies performing gene editing in hematopoietic stem and progenitor cells (HSPCs) use the CRISPR/Cas9 system, particularly Spy Cas9. Researchers have made certain optimizations to improve safety and efficiency, such as using ribonucleic proteins (RNP). The mechanism behind the improved safety and efficacy of RNPs in CRISPR lies in their ability to reduce off-target effects, eliminate the need for intracellular processes, lower the risk of insertional mutagenesis and immune responses, and enhance genome editing efficacy across different cell types. Researchers are exploring using the CRISPR gRNA/Cas9 RNP complex and DNA donor templates to correct mutations that cause SCD. They evaluated different viral vectors (rAAV6) and single-stranded oligodeoxynucleotides (ssODNs) for delivering the donor template. However, some challenges must be addressed, such as the low homology-directed repair (HDR)/ non-homologous end joining (NHEJ) ratio in long-term reconstituting HSCs and the potential risk of inducing β-thalassemia. Efforts are currently underway to develop in vivo technologies to cure SCD. However, this endeavor comes with many challenges. These include achieving high in vivo delivery and editing efficiency, mitigating potential off-target effects, and determining the optimal percentage of edited HSCs required for cure. CRISPR/Cas9 gene editing raises concerns regarding off-target effects. Strategies, such as RNP delivery and high-fidelity Cas9, aim to minimize these effects. It is important to use robust methods to identify and quantify off-target sites and monitor patients in the long term.
Noteworthy innovations have been made to the CRISPR/Cas9 system. These include the Dead-Cas9 system, in which specific mutations result in a nuclease-dead-Cas9 (dCas9) protein48. This protein does not cleave DNA but has been used for genome imaging and epigenetic modifications. The inactivity of the dCas9 system in terms of DNA cleavage enhances specificity. Unlike the typical Cas9 system, which introduces double-stranded breaks, the dCas9 system functions as a precise DNA-binding tool without altering the DNA sequence. This is advantageous for applications that require targeted DNA binding without inducing genetic changes such as epigenetic modifications. Moreover, the dCas9 system performs exceptionally well at delivering cargo in the form of functionally active domains. These functionally active domains can activate or suppress gene expression, investigate chromatin structure, and direct three-dimensional remodeling of chromatin structure49. A prime editing system can also modify DNA templates without cleaving DNA molecules. This involves a catalytically impaired Cas9 protein binding to the reverse transcriptase enzyme and guide RNA. It modifies genes through substitution, deletion, and insertion50,51.
Traditionally, HSCT is a definitive treatment for β-hemoglobinopathies such as SCD and β-thalassemia. However, the limitations of this procedure include a shortage of matching donors and graft-versus-host diseases. Since 1985, efforts have been made to genetically modify patients’ stem cells by a process called homologous recombination (HR) or HDR29. Another process that works closely with HR to ensure precise genomic modification is called NHEJ. Both HR and NHEJ mechanisms occur naturally in mammalian cells, and manipulation of these mechanisms to precisely modify the defective hemoglobin subunit beta (HBB) gene forms the foundation of genome editing technologies for SCD. CRISPR/Cas9 can be used to precisely target the HBB gene in patient-derived human induced pluripotent stem cells (iPSCs), cleave the DNA at a precise location, and use the above-mentioned repair mechanisms to modify the faulty HBB gene. The CRISPR technique is the most used genome editing technology owing to its simplicity of design, affordability, and good repeatability, and is more efficient than other less commonly used genome editing technologies, including transcription activator-like effector nucleases (TALENs) and zinc finger nucleases (ZFNs)50.
Targeted iPSCs demonstrate intact integrity with minimal adverse effects and successfully differentiate into mature erythrocytes carrying the corrected HBB gene52.
One study utilized CD34+ HSPCs from healthy donors and individuals with SCD. These cells were then edited using CRISPR/Cas9 and gRNA-68, resulting in sustained on-target editing without off-target mutations. Following in vitro differentiation or xenotransplantation into immunodeficient mice, edited cells produced high levels of HbF. The study also involved three participants who received autologous OTQ923 after undergoing myeloablative conditioning. The participants were monitored for 6–18 months. By the end of the follow-up period, all participants had engraftment and stable induction of fetal hemoglobin (at a percentage of total hemoglobin ranging from 19.0 to 26.8%). HbF is distributed broadly in red cells (F cells as a percentage of red cells ranging from 69.7 to 87.8%)53.
Over the last decade, extensive tests have been conducted using CRISPR/Cas9 technology in vitro, in vivo, and human trials. These tests have shown promising outcomes in various human illnesses. Khlidj (2023) comprehensively summarizes some of the most noteworthy achievements and proof-of-concept evidence from human trials to in vivo and in vitro studies. CRISPR/Cas9 technology has been successful in the treatment of β-thalassemia and SCD. A human phase I trial targeting BCL11A resulted in increased concentrations of HbF54.
These groundbreaking studies demonstrate the versatility and potential of CRISPR/Cas9 for addressing a range of genetic and acquired disorders.
Demirci and colleagues delves into various strategies to increase HbF expression as a potential treatment for SCD and β-thalassemia. The focus is on CRISPR/Cas9 genome editing and the significance of disrupting regulatory sequences and chromosomal configuration to achieve robust HbF synthesis. Different methodologies, such as large deletional mutations and base editing, have been explored to induce HbF expression without generating double-strand breaks (DSBs). Some important findings of this study include the discovery of the BCL11A binding site located at position -115 in the HBG promoter, which plays a pivotal role in controlling HbF expression. The study also found that CRISPR/Cas9 editing of this site is effective in reactivating HbF in progenitor and erythroid cells, thereby reducing sickling in the edited RBC. Furthermore, the study explored other targets, such as ZBTB7A and the LRF binding site, for their involvement in HbF regulation. This study highlights the need for precise and cell-specific editing to avoid adverse hematological effects55.
Lattanzi and colleagues aimed to correct the HBB gene in hematopoietic stem and progenitor cells from healthy donors and patients with SCD. The gene correction strategy involves the use of CRISPR/Cas9, specifically a high-fidelity Cas9 variant, to target HBB6.
Gene-editing strategies for SCD have been comprehensively reviewed by Park & Bao (2021). Many preliminary studies concentrate on gene editing of HSPCs, followed by transplantation in immunodeficient mouse models56. It is of utmost importance to evaluate the long-term engraftment potential of these gene-edited HSCs to ensure that autologous HSCT remains durable.
In March 2021, the FDA approved the first trial using CRISPR to correct the genetic mutations causing SCD. Research at the Innovative Genomics Institute demonstrated that CRISPR/cas9 gene editing can modify major HbF repressors, promoting HbF production. The first phase (phase 1) clinical trial showed promising results, indicating a significant restoration of functional hemoglobin, reduced sickle cell-associated pain, and slowed disease progression. By targeting genes like BCL11A, this genetic technology has the potential to transform disease management for SCD patients and may also apply to those with β-thalassemia57. CRISPR/Cas9, though a relatively new technique, has been extensively studied for the treatment of SCD, as shown by the studies referenced here. It is also important to recognize that despite the extensive research, each study has its own credibility, strengths, and limitations (Table 1).
Table 1.
The strength and credibility of the previously discussed studies concerning CRISPR/cas9 technique for treatment of SCD
| Study ID | Study method | Strength and credibility |
|---|---|---|
| Sharma et al., 202353 | Multicenter, phase 1–2 clinical study | The strengths of the study are Innovative technology, Comprehensive screening, rigorous pre-clinical tests, comprehensive monitoring, and ethical oversight. The limitations include limited participants, selection bias, short follow-up, and manufacturing and logistic challenges. |
| Khlidj, 202354 | literature review | The strengths of the study include a comprehensive overview, broad data sources, an empirical focus to provide concrete evidence, and a longitudinal perspective. The limitations include scope being limited to existing data, no new data has been incorporated, selection bias, publication bias, and depth of analysis on methodologies. |
| Demirci et al., 202155 | narrative review | The strengths of the study include a comprehensive overview of the study which includes the current and relevant information, clear presentation, practical focus, and interdisciplinary insights. The limitations include limited participant data, selection bias, and short follow-up. |
| Park & Bao, 202156 | Pre-clinical experimental study | The strength of the study includes the translation potential as it uses nonhuman primates, an Innovative approach of CRISPR technology, and evaluates multiple aspects. The limitation of the study is its pre-clinical nature, off-target effects, and durability of gene editing. |
| Huang, 202357 | Clinical trial study | The strengths are the innovative treatment approach of gene editing, targeted gene editing, promising preliminary results, shift towards personalized medicine. The limitations are that a phase 1 trial is limited to a safety feasibility study that requires validation in the long run, potential off-target effects, complexity, and accessibility of the procedure. |
| Lattanzi et al., 20216 | Pre-clinical experimental study | Strengths of the study include high correction efficiency as it provides up to 60% allelic correction and 20% gene correction, potential for clinical translation. Limitations of the studies are its pre-clinical stage complex manufacturing process, Variability in human responses, and potential off-target effects. |
SCD, sickle cell disease.
These studies conclude that SCD poses significant health challenges and the need for genetic disease treatment advancements. Genome editing, using CRISPR technology, is a promising approach to address the underlying causes of SCD. The versatility of CRISPR/Cas9 in precisely modifying genetic sequences, especially those related to HbF repression, is a significant breakthrough in disease management.
The FDA recently approved the cell-based gene therapy for SCD, Casgevy, for use in patients aged 12 and older. Casgevy is the first therapy to use CRISPR/Cas9 technology for the treatment of SCD. Casgevy has been approved by the FDA for the prevention of severe VOCs. In an ongoing clinical trial involving 44 patients with SCD, Casgevy demonstrated its efficacy by achieving freedom from severe VOC episodes for at least 12 consecutive months in 93.5% of evaluable patients. Common side effects included low platelet and white blood cell levels, mouth sores, nausea, musculoskeletal pain, abdominal pain, vomiting, febrile neutropenia, headache, and itching28. The trial for SCD conducted by the Innovative Genomics Institute shows positive results in restoring functional hemoglobin, reducing pain associated with SCD, and slowing disease progression. The approvals were granted to Vertex Pharmaceuticals Inc. for Casgevy. Long-term studies will assess the safety and effectiveness of these therapies.
Gene therapy via CRISPR/Cas9 versus traditional approaches for SCD
HSCT has been the only curative therapy for a long time to treat SCD, but there have been many limitations and improvements over the years. Analysis of the retrospective study reported overall survival (OS) of 92.9% and event-free survival (EFS) of 91.4% in 1000 children who underwent HSCT. However, EFS, an indicator of the outcome of treatment, is significantly driven by the age at the time of HSCT and the type of donor58. Age is also a contributory factor that decides the outcome and post-transplant results. The age of the donor is also known to predict the fate of transplants. A study conducted by Brazauskas et al. 59 indicated that patients younger than 12 years receiving HSCT have the best outcomes in terms of EFS at 92% for 3 years among the 1425 who received HSCT for SCD. Thus, CRISPR promises a successful treatment option for people aged 12 years and above with SCD, which has been a hindering factor for traditional SCD treatment methods.
HSCT requires finding a suitably matched donor, and more than 80 percent of patients with SCD do not find a donor60. Pre-treatment conditioning with a myeloablative conditioning regimen along with two courses of immunosuppression is required to prevent rejection in the case of haploidentical (a type of allogeneic transplant that uses healthy cells from a half-matched donor) and unrelated donor HSCT for SCD58,61. Complications like graft-versus-host-disease (GVHD) and infections due to immunotherapy also pose risks. Prophylaxis for GVHD to avoid transplant failure and its complications is practiced using multiple regimens62. CRISPR/Cas9 gene therapy for SCD may overcome the limitations of strict requirements for matched donors for HSCT. With CRISPR/Cas9 gene therapy, patients may receive autografts alleviating dependency on a donor. This allows a larger population of SCD patients to benefit from curative treatment, as finding a matched donor for HSCT is rare. Pre-treatment conditioning has also been modified by the emergence of CRISPR, sparing patients from the hazards of a combination of post-chemotherapy immune deficiency and radiation. Furthermore, the autogenic nature of CRISPR eliminates the possibility of GVHD. CRISPR involves simple infusion of the treated cells, avoiding surgical intervention. Current recommendations also require a prolonged follow-up to look for complications arising from HSCT62. While these studies offer valuable insights into the traditional treatment options for SCD and their risks and benefits, assessing their credibility, strengths, and limitations is crucial before drawing any conclusions from them (Table 2).
Table 2.
The strength and credibility of the previously discussed studies concerning traditional treatment options for SCD
| Study ID | Study method | Strength and credibility |
|---|---|---|
| Gluckman et al., 201758 | Observational and retrospective | The strength of the study is the large sample size with a long follow-up period, International collaboration which includes 106 centers in 23 countries, and comprehensive analysis. The limitations of the studies are retrospective study subject to selection bias, limited to HLA identical siblings transplant, heterogeneity of treatment protocols |
| Brazauskas et al., 202059 | international, retrospective, registry-based survey | The study’s strengths include specific inclusion criteria and a large sample size with a long follow-up period. The limitations of the study are selection bias, reliance on registry-based data, and the potential of missing data. |
| Baronciani et al., 201660 | retrospective non-interventional study | The strength of the data includes a large sample size with long follow-up and multicenter data and standardized data collection. The limitations of the study include variability in clinical practices, limited information on secondary transplants, |
| Pawlowska et al., 201861 | retrospective observational study | The strength of the study includes innovative approach such as novel pretransplant immunosuppressive strategy (PTIS) combined with T cell–replete grafts and PTCY-based GVHD prophylaxis and detailed case analysis. The limitations of the study include a small sample size, a short follow-up period, and a single-center study. |
| Krishnamurti, 202162 | Observational study | The strength of the study includes comprehensive data collection, relevance to clinical practice, and data from a diverse population. The limitations of the study include lack of a control group and Potential Socioeconomic and Regional Disparities. |
GVHD, graft-versus-host-disease; HLA, human leukocyte antigen; PTCY, post transplantation cyclophosphamide; SCD, sickle cell disease.
The costs of traditional methods of treatment and management for SCD have been steadily growing, making it hard for under-resourced populations to afford treatments and concurrently burdening the healthcare system. The lifetime expenses of a patient at 50 years of age with SCD are estimated to be $8 million. There has been a documented rise in patient fees from $200 000 for ages 0-5 years to over $7 million for the age group 17–50 years63. HSCT carries a significant financial cost in the first year. The reported cost of HSCT for adults with malignant or nonmalignant conditions in the first year ranges from $96 000 to $204 000. This cost varies based on the conditioning regimen, allograft type, and donor source. In contrast, the median estimated transplant cost in children during the transplant year is ~$413 000, suggesting that factors unique to pediatric populations confer increased costs compared with adults64. CRISPR-based gene therapy to treat SCD is predicted to cost ~$2 million to treat one patient. Murky regulations regarding coverage of expenses by Medicaid and other insurance limit the availability of gene therapy via CRISPR/Cas9 to a larger portion of the SCD population, which is already underserved.
Significant discrepancies emerge when comparing the treatment durations of CRISPR/Cas9 and traditional SCD therapy. Traditional SCD therapies may include continuing or lifetime efforts to manage symptoms and avoid complications. Medication such as hydroxyurea, for example, may be required continually to maintain therapeutic benefits19. In contrast, CRISPR/Cas9 treatment for SCD offers the potential to provide a more decisive and perhaps curative strategy. While further research and clinical studies are required to completely confirm its long-term effectiveness and safety, CRISPR/Cas9 therapy has the potential to be used as a single treatment that targets the underlying etiology of SCD56. If effective, this might considerably reduce or eliminate the requirement for continuous symptom management and supportive care that is common with existing therapy methods and may remove the need for HSCT. It is important to note that while CRISPR/Cas9 holds great promise for treating genetic disorders like SCD, there are still challenges related to its clinical implementation, including ensuring precise targeting and minimizing off-target effects65. In addition to the off-target effects, CRISPR requires patients to be hospitalized and receive chemotherapy, potentially exposing them to risks like deadly infections. Introducing the CRISPR/Cas9 into the cytosol and then the nucleus can be challenging and requires variable delivery methods, which have their limitations. Additionally, ethical considerations and regulatory approval processes will play crucial roles in determining the widespread adoption of CRISPR/Cas9 therapies for SCD.
The use of CRISPR/Cas9 to treat SCD presents various ethical concerns. One of the key concerns is the possibility of off-target consequences, which occur when unwanted modifications are produced to the genome. This might have unintended repercussions for the patient and future generations. Furthermore, there are ethical considerations associated with germline editing, as any alterations made to an individual’s DNA might be passed on to their progeny66. The ethical use of CRISPR/Cas9 in SCD therapy necessitates careful examination of these possible hazards and advantages. In contrast, the common curative treatment for SCD, HSCT, does not entail genetic manipulation67. The cost of CRISPR is high, but in the long run, it is expected to decrease the disease burden by treating SCD and save lifelong expenses of hospitalization and management. There has also been no clarity regarding the coverage of treatment expenses by Medicaid. The cost of treatment may prevent its use in areas where the disease burden is highest, as in Africa, which is healthcare resource-limited68.
The advantages and limitations of traditional treatment options and gene therapy via SCD are shown in Figure 1.
Figure 1.
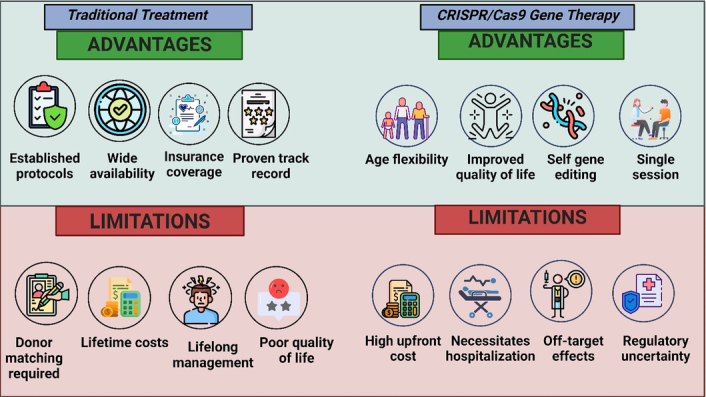
Advantages and limitations of traditional treatment options and gene therapy with CRISPR/Cas9 for sickle cell disease. (Created with biorender.com).
Conclusion
Cell-based gene therapy using CRISPR/Cas9 for the treatment of SCD promises a potentially transformative management approach in contrast to traditional treatments. Traditional methods such as pharmacological treatments, blood transfusions, and bone marrow transplants have limitations, including the scarcity of matching donors, the risk of graft-versus-host disease, and the need for lifelong management. CRISPR/Cas9 technology offers a precise and versatile method to target the underlying genetic cause of SCD by modifying the defective HBB gene. Importantly, CRISPR/Cas9 addresses the challenge of donor availability, which has been a significant hurdle in traditional HSCT. While CRISPR/Cas9 holds great potential, challenges remain, including ensuring precise targeting, minimizing off-target effects, and addressing the need for hospitalization and chemotherapy during treatment. Ongoing research and clinical studies must further confirm long-term effectiveness and safety. Additionally, the introduction of CRISPR/Cas9 into the nucleus poses delivery challenges that require careful consideration. Efforts should also be directed towards refining delivery methods to enhance the precision of CRISPR/Cas9 and minimize associated challenges, such as hospitalization and chemotherapy. This optimization will contribute to the overall feasibility and acceptance of CRISPR/Cas9 therapy as a viable treatment option for SCD. Moreover, recognizing and understanding patient perspectives, preferences, and concerns regarding gene therapy is paramount. Engaging with the affected communities and integrating their insights into the development and implementation processes will contribute to the successful adoption of CRISPR/Cas9 therapy for SCD.
Ethical approval
Not applicable.
Consent
Informed consent was not required for this review.
Source of funding
None.
Author contribution
Concept and outline: A.A., M.H.K., A.J. Initial draft: H.T., F.K., M.H.K., A.D., A.Z., W.R., A.J., D.K., S.K., A.A. Figures: M.H.K., A.A. Critical revision of draft: A.A. Revision of final draft: H.T., F.K., M.H.K., A.D., A.Z., W.R., A.J., D.K., S.K., A.A.
Conflicts of interest disclosure
The authors declare no conflicts of interest.
Research registration unique identifying number (UIN)
Not applicable.
Guarantor
Not applicable.
Data availability statement
Not applicable.
Provenance and peer review
Not applicable.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Published online 14 August 2024
Contributor Information
Hamza Tariq, Email: hamzatariq0222@gmail.com.
Fatima Khurshid, Email: fatimakhurshid61@yahoo.com.
Muhammad Hamza Khan, Email: ihamzakhan145@gmail.com.
Aamna Dilshad, Email: aamnadilshad8@gmail.com.
Ahmad Zain, Email: ahmadzain99@gmail.com.
Warda Rasool, Email: wardarasoolisb@gmail.com.
Alishba Jawaid, Email: alishjawaid00@gmail.com.
Digbijay Kunwar, Email: digbijaykunwar@gmail.com.
Sneha Khanduja, Email: snehakhanduja30@gmail.com.
Anum Akbar, Email: anum.akbar@unmc.edu.
References
- 1.Sedrak A, Kondamudi NP. Sickle Cell Disease. StatPearls StatPearls Publishing Copyright © 2023. StatPearls Publishing LLC; 2023. [Google Scholar]
- 2.Brousse V, Rees DC. Sickle cell disease: more than a century of progress. where do we stand now? Indian J Med Res 2021;154:4–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kavanagh PL, Fasipe TA, Wun T. Sickle cell disease: a review. JAMA 2022;328:57–68. [DOI] [PubMed] [Google Scholar]
- 4.Prevention CDC. Sickle Cell Disease (SCD). Centers for Disease Control and Prevention. 2023. https://www.cdc.gov/sickle-cell/about/
- 5.Mangla A, Ehsan M, Agarwal N, et al. Sickle Cell Anemia. StatPearls StatPearls Publishing Copyright © 2023. StatPearls Publishing LLC; 2023. [Google Scholar]
- 6.Lattanzi A, Camarena J, Lahiri P, et al. Development of β-globin gene correction in human hematopoietic stem cells as a potential durable treatment for sickle cell disease. Sci Transl Med 2021;13:eabf2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manwani D, Frenette PS. Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood 2013;122:3892–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Henry ER, Cellmer T, Dunkelberger EB, et al. Allosteric control of hemoglobin S fiber formation by oxygen and its relation to the pathophysiology of sickle cell disease. Proc Natl Acad Sci U S A 2020;117:15018–15027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.National Academies of Sciences E, Medicine, Health, et al . In: Martinez RM, Osei-Anto HA, McCormick M, eds. Addressing Sickle Cell Disease: A Strategic Plan and Blueprint for Action. National Academies Press (US) Copyright 2020 by the National Academy of Sciences. All rights reserved; 2020. [PubMed] [Google Scholar]
- 10.Chakravorty S, Williams TN. Sickle cell disease: a neglected chronic disease of increasing global health importance. Arch Dis Child 2015;100:48–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ware RE, de Montalembert M, Tshilolo L, et al. Sickle cell disease. Lancet 2017;390:311–323. [DOI] [PubMed] [Google Scholar]
- 12.Tanabe P, Spratling R, Smith D, et al. CE: Understanding the complications of sickle cell disease. Am J Nurs 2019;119:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abboud MR. Standard management of sickle cell disease complications. Hematol/Oncol Stem Cell Ther 2020;13:85–90. [DOI] [PubMed] [Google Scholar]
- 14.Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers 2018;4:18010. [DOI] [PubMed] [Google Scholar]
- 15.Oron AP, Chao DL, Ezeanolue EE, et al. Caring for Africa’s sickle cell children: will we rise to the challenge? BMC Med 2020;18:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dua M, Bello-Manga H, Carroll YM, et al. Strategies to increase access to basic sickle cell disease care in low- and middle-income countries. Expert Rev Hematol 2022;15:333–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Délicat-Loembet LM, Baraïka MA, Bougoudogo F, et al. Bacterial infection in the sickle cell population: development and enabling factors. Microorganisms 2023;11:859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rankine-Mullings AE, Owusu-Ofori S. Prophylactic antibiotics for preventing pneumococcal infection in children with sickle cell disease. Cochrane Database Syst Rev 2021;3:Cd003427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McGann PT, Ware RE. Hydroxyurea therapy for sickle cell anemia. Expert Opin Drug Saf 2015;14:1749–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vichinsky EP. Current issues with blood transfusions in sickle cell disease. Semin Hematol 2001;38(1 suppl 1):14–22. [DOI] [PubMed] [Google Scholar]
- 21.Reeves SL, Peng HK, Wing JJ, et al. Changes in hydroxyurea use among youths enrolled in medicaid with sickle cell anemia after 2014 revision of clinical guidelines. JAMA Netw Open 2023;6:e234584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salinas Cisneros G, Thein SL. Recent advances in the treatment of sickle cell disease. Rev Front Physiol 2020;11:435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Telen MJ, Malik P, Vercellotti GM. Therapeutic strategies for sickle cell disease: towards a multi-agent approach. Nat Rev Drug Discov 2019;18:139–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shenoy S, Eapen M, Panepinto JA, et al. A trial of unrelated donor marrow transplantation for children with severe sickle cell disease. Blood 2016;128:2561–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koniali L, Lederer CW, Kleanthous M. Therapy development by genome editing of hematopoietic stem cells. Cells 2021;10:1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Badat M, Davies J. Gene therapy in a patient with sickle cell disease. N Engl J Med 2017;376:2093–2094. [DOI] [PubMed] [Google Scholar]
- 27.Kato-Inui T, Takahashi G, Hsu S, et al. Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 with improved proof-reading enhances homology-directed repair. Nucleic Acids Res 2018;46:4677–4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.U.S. Food and Drug Administration. (2023, December 8). FDA Approves First Gene Therapies to Treat Patients with Sickle Cell Disease. 2024. https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapies-treat-patients-sickle-cell-disease
- 29.Dever DP, Bak RO, Reinisch A, et al. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature 2016;539:384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med 2015;21:121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ballas SK. Sickle cell disease: classification of clinical complications and approaches to preventive and therapeutic management. Clin Hemorheol Microcirc 2018;68:105–128. [DOI] [PubMed] [Google Scholar]
- 32.Joshua J, Feild EPV. Overview of the management and prognosis of sickle cell disease. 2024. https://www.uptodate.com/contents/overview-of-the-management-and-prognosis-of-sickle-cell-disease
- 33.Gaston MH, Verter JI, Woods G, et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial N Engl J Med 1986;314:1593–1599. [DOI] [PubMed] [Google Scholar]
- 34.Platt OS, Orkin SH, Dover G, et al. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J Clin Invest 1984;74:652–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferster A, Vermylen C, Cornu G, et al. Hydroxyurea for treatment of severe sickle cell anemia: a pediatric clinical trial. Blood 1996;88:1960–1964. [PubMed] [Google Scholar]
- 36.Okocha EC, Gyamfi J, Ryan N, et al. Barriers to therapeutic use of hydroxyurea for sickle cell disease in nigeria: a cross-sectional survey. Original Res Front Genet 2022;12:765958. doi: 10.3389/fgene.2021.765958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niihara Y, Miller ST, Kanter J, et al. A phase 3 trial of l-glutamine in sickle cell disease. N Engl J Med 2018;379:226–235. [DOI] [PubMed] [Google Scholar]
- 38.Vichinsky EP, Neumayr LD, Earles AN, et al. Causes and outcomes of the acute chest syndrome in sickle cell disease. N Engl J Med 2000;342:1855–1865. [DOI] [PubMed] [Google Scholar]
- 39.Zerez CR, Lachant NA, Lent KM, et al. Decreased pyrimidine nucleoside monophosphate kinase activity in sickle erythrocytes. Blood 1992;80:512–516. [PubMed] [Google Scholar]
- 40.Rastogi B, Rani S, Mayuzumi M, et al. Safety profile of L-glutamine in patients with sickle cell disease: data from post-marketing surveillance. Blood 2021;138(suppl 1):4184. [Google Scholar]
- 41.Glaros AK, Razvi R, Shah N, et al. Voxelotor: alteration of sickle cell disease pathophysiology by a first-in-class polymerization inhibitor. Therap Adv Hematol 2021;12:20406207211001136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med 2016;376:429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Culp-Hill R, Srinivasan AJ, Gehrke S, et al. Effects of red blood cell (RBC) transfusion on sickle cell disease recipient plasma and RBC metabolism. Transfusion 2018;58:2797–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Santis GC, Costa TCM, Santos FLS, et al. Blood transfusion support for sickle cell patients during haematopoietic stem cell transplantation: a single-institution experience. Br J Haematol 2020;190:e295–e297. [DOI] [PubMed] [Google Scholar]
- 45.Walters MC, Patience M, Leisenring W, et al. Bone marrow transplantation for sickle cell disease. N Engl J Med 1996;335:369–376. [DOI] [PubMed] [Google Scholar]
- 46.Abdel-Hadi L, Ventura Carmenate Y, Castillo-Aleman YM, et al. Treatment of sickle cell disease—options and perspective. Am J Blood Res 2023;13:61–70. [PMC free article] [PubMed] [Google Scholar]
- 47.Kassim AA, Sharma D. Hematopoietic stem cell transplantation for sickle cell disease: the changing landscape. Hematol/Oncol Stem Cell Ther 2017;10:259–266. [DOI] [PubMed] [Google Scholar]
- 48.Saifaldeen M, Al-Ansari DE, Ramotar D, et al. CRISPR FokI Dead Cas9 system: principles and applications in genome engineering. Cells 2020;9:2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brezgin S, Kostyusheva A, Kostyushev D, et al. Dead Cas systems: types, principles, and applications. Int J Mol Sci 2019;20:6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu Y, Li Z. CRISPR-Cas systems: overview, innovations and applications in human disease research and gene therapy. Comput Struct Biotechnol J 2020;18:2401–2415. doi: 10.1016/j.csbj.2020.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Anzalone AV, Randolph PB, Davis JR, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019;576:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang X, Wang Y, Yan W, et al. Production of gene-corrected adult beta globin protein in human erythrocytes differentiated from patient iPSCs after genome editing of the sickle point mutation. Stem Cells 2015;33:1470–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma A, Boelens JJ, Cancio M, et al. CRISPR-Cas9 editing of the HBG1 and HBG2 promoters to treat sickle cell disease. N Engl J Med 2023;389:820–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Khlidj Y. What did CRISPR-Cas9 accomplish in its first 10 years. Biochem Med (Zagreb) 2023;33:030601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Demirci S, Leonard A, Essawi K, et al. CRISPR-Cas9 to induce fetal hemoglobin for the treatment of sickle cell disease. Mol Ther Methods Clin Dev 2021;23:276–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Park SH, Bao G. CRISPR/Cas9 gene editing for curing sickle cell disease. Transfus Apher Sci 2021;60:103060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huang H. First approved CRISPR trial on sickle cell disease treatment. RCSIsmj, 15:10–11. http://www.rcsismj.com/wp-content/uploads/RCSIsmj_2022_Final-Proof.pdf [Google Scholar]
- 58.Gluckman E, Cappelli B, Bernaudin F, et al. Sickle cell disease: an international survey of results of HLA-identical sibling hematopoietic stem cell transplantation. Blood 2017;129:1548–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brazauskas R, Scigliuolo GM, Wang HL, et al. Risk score to predict event-free survival after hematopoietic cell transplant for sickle cell disease. Blood 2020;136:623–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baronciani D, Angelucci E, Potschger U, et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry, 2000-2010. Bone Marrow Transplant 2016;51:536–541. [DOI] [PubMed] [Google Scholar]
- 61.Pawlowska AB, Cheng JC, Karras NA, et al. HLA haploidentical stem cell transplant with pretransplant immunosuppression for patients with sickle cell disease. Biol Blood Marrow Transplant 2018;24:185–189. [DOI] [PubMed] [Google Scholar]
- 62.Krishnamurti L. Hematopoietic cell transplantation for sickle cell disease: updates and future directions. Hematology 2021;2021:181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leonard A, Tisdale JF. Stem cell transplantation in sickle cell disease: therapeutic potential and challenges faced. Expert Rev Hematol 2018;11:547–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kassim AA, Leonard A. Debating the future of sickle cell disease curative therapy: haploidentical hematopoietic stem cell transplantation vs. gene therapy. J Clin Med 2022;11:4775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu W, Li L, Jiang J, et al. Applications and challenges of CRISPR-Cas gene-editing to disease treatment in clinics. Precis Clin Med 2021;4:179–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ayanoğlu FB, Elçin AE, Elçin YM. Bioethical issues in genome editing by CRISPR-Cas9 technology. Turk J Biol 2020;44:110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gamage U, Warnakulasuriya K, Hansika S, et al. CRISPR gene therapy: a promising one-time therapeutic approach for transfusion-dependent β-thalassemia—CRISPR-Cas9 gene editing for β-thalassemia. Thalassemia Rep 2023;13:51–69. [Google Scholar]
- 68.https://www.sciencenews.org/article/first-crispr-therapy-sickle-cell-fda Garcia de Jesús, E. The first CRISPR therapy approved in the U.S. will treat sickle cell disease. Science News, 2024.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.