

Abstract
Metaplastic breast carcinomas (mBrCAs) are a highly aggressive subtype of triple negative breast cancer (TNBC) with histological evidence of epithelial to mesenchymal transition (EMT) and aberrant differentiation. Inactivation of the tumor suppressor gene CCN6 (also known as WISP3) is a feature of mBrCAs, and mice with conditional inactivation of Ccn6 in mammary epithelium (Ccn6-KO) develop spindle mBrCAs with EMT. Elucidation of the precise mechanistic details of how CCN6 acts as a tumor suppressor in mBrCA could help identify improved treatment strategies. Here, we showed that CCN6 interacts with the Wnt receptor FZD8 and co-receptor LRP6 on mBrCA cells to antagonize Wnt-induced activation of β-catenin/TCF-mediated transcription. The histone methyltransferase EZH2 was identified as a β-catenin/TCF transcriptional target in Ccn6-KO mBrCA cells. Inhibiting Wnt/β-catenin/TCF signaling in Ccn6-KO mBrCa cells led to reduced EZH2 expression, decreased histone H3 lysine 27 trimethylation, and deregulation of specific target genes. Pharmacological inhibition of EZH2 reduced growth and metastasis of Ccn6-KO mBrCA mammary tumors in vivo. Low CCN6 is significantly associated with activated β-catenin and high EZH2 in human spindle mBrCAs compared to other subtypes. Collectively, these findings establish CCN6 as a key negative regulator of a β-catenin/TCF-EZH2 axis and highlight inhibition of β-catenin or EZH2 as a potential therapeutic approach for patients with spindle mBrCAs.
Keywords: metaplastic breast carcinoma, metastasis, epithelial-to-mesenchymal transition, triple-negative breast cancer, WNT, CCN6, WISP3, EZH2, H3K27me3, tazemetostat, methyltransferase
Introduction
Metaplastic breast carcinoma (mBrCA) is a subtype of triple-negative breast cancer (TNBC) with histological evidence of epithelial to mesenchymal transition (EMT) and aberrant non-glandular differentiation towards spindle, squamous, or sarcomatoid (e.g., chondroid, osseous, rhabdoid). Genomic analysis showed mBrCAs belong to the mesenchymal TNBC subtype (1). mBrCAs account for 1.5% of all breast cancers but up to 14% of breast cancers in Black women, and the spindle histology is the most frequent (2, 3). mBrCAs remain one of the deadliest breast cancer types (4–7) with a reported 5-year overall survival of 54% compared to 73.3% for non-metaplastic TNBC (8).
Metaplasia, the potentially reversible change from one adult cell identity to another, reflects changes in epithelial-mesenchymal cell states and likely arises from epigenetic reprogramming. Thus, it is not surprising mBrCAs exhibit deregulation of pathways that control cell-type identity (9). Accumulating evidence indicates hyperactive Wnt signaling occurs in association with human mBrCA and the tumors have increased EZH2-mediated transcriptional repression of developmental genes (1, 10, 11). In breast cancer, the Wnt/β-catenin upregulation results chiefly from signaling pathway activation and, less frequently, from gene mutation affecting key Wnt pathway factors (10, 12). Mechanisms linking Wnt and EZH2 activity in mBrCA have not been clarified.
Cellular communication network factor 6, CCN6, also known as WISP3 (Wnt-1 induced secreted protein 3), is a member of the CCN family of matricellular secreted proteins (13). The family is composed of 6 proteins in mammals: CCN1/cysteine-rich protein 61 (Cyr61), CCN2/connective tissue growth factor (CTGF), CCN3/nephroblastoma overexpressed (Nov), CCN4/Wisp1, CCN5/Wisp2, and CCN6/Wisp3 (14). CCN proteins play critical roles in the regulation of normal cell homeostasis in multiple organs in a tissue- and cell-type specific manner, and CCN protein expression is often deregulated in cancer (15, 16). CCN6 is expressed in normal breast epithelial cells but its expression is reduced or lost in 68% of human mBrCA tumors (17). Our previous work has demonstrated CCN6 is secreted by breast epithelial cells into the extracellular matrix to maintain normal breast acinar organization at least in part via regulation of growth factor signaling pathways (e.g., IGF and BMP4 signaling)(18–20). Downregulation of CCN6 in normal mammary epithelium induces EMT, motility, and invasion (19, 21). Conditional, mammary epithelial cell-specific Ccn6 knockout mice (MMTV-Cre;Ccn6fl/fl) develop mammary tumors with bona fide EMT similar to human spindle mBrCA at the pathological and proteogenomic levels, providing direct evidence for a tumor suppressor function of CCN6 (11, 17). While Ccn6-KO mBrCA tumors have upregulated Wnt signaling and reduced expression of the canonical Wnt inhibitor DKK1 (17), definitive insights are lacking into how CCN6 acts as a tumor suppressor in mBrCA.
In the present study we hypothesized CCN6 may suppress spindle mBrCA tumorigenicity by directly antagonizing β-catenin-dependent Wnt signaling. Here, we show the CCN6 protein binds to the Wnt receptor Fzd8 and co-receptor LRP6 in mesenchymal-like TNBC cells. We found inhibition of the Wnt/β-catenin/TCF pathway underlies tumor inhibitory effects of CCN6. We also identify β-catenin/TCF activation of the gene for the histone methyltransferase EZH2 is critical for downstream effects on phenotypes. EZH2 inhibition using a pharmacological approach or shRNA knockdown reduces the growth and metastasis of Ccn6-KO mBrCA tumors in vivo and improves mouse survival. We identify a subset of human spindle mBrCA that harbor a CCN6Low/nuclear β-cat/EZH2High phenotype. Our data delineate a novel CCN6-Wnt/β-catenin/TCF/EZH2 axis underlying mBrCA tumors and highlight potential targets of intervention for patients with mBrCA.
Materials and Methods
Cell culture and transductions.
Breast cancer cell lines MDA-MB-231 and non-tumorigenic breast epithelial cells, HME, were purchased from the American Type Culture Collection and grown under recommended conditions. Ccn6-KO tumor cells were isolated from mammary tumors of MMTV-Cre;Ccn6fl/fl knockout mice (17) using a tumor dissociation kit for mouse (#130–096-730, Miltenyi Biotec, Inc.). Cells were cultured and replicated in DMEM/F12 Gibco medium. Cell lines were authenticated using STR profiling and were tested for mycoplasma infection using Sigma LookOut Mycoplasma PCR Detection Kit (Cat# MP0035). For the preparation of the conditioned medium, cells were cultured in complete DMEM medium, followed by collection and filtration of medium according to the standard procedures. Ponceau stain was used as loading control.
Ectopically expressed flag-tagged full-length CCN6 or a deletion mutant of CCN6 lacking the TSP1 domain (ΔTSP1-Flag) and stable knockdown of CCN6 using two independent shRNAs in lentivirus were achieved as previously described (22). Stable knockdown of EZH2 was performed by lentiviral transduction of stable short-hairpin interfering RNA (MISSION shRNA, Sigma Aldrich) targeting the 3′UTR of Ezh2 (TRCN0000286227), as previously reported (23). Adenovirus expressing MYC-EZH2, MYC-ΔSET and MYC-ΔNLS were developed as described (24). Retrovirus expressing constructs pPGS vector control, Flag-S33Y β-catenin activating mutation, and Flag-tagged dominant negative form of TCF4 lacking the N-terminal 31 amino acids (Flag-dnTCF4) were used for stable transfections (25). All viruses were packaged at the University of Michigan Vector Core. Viral transductions and transfections were carried out as reported (23, 25).
Western blotting, coimmunoprecipitation, and antibodies.
Immunoblot analyses were carried out as previously reported using 50 μg of whole cell extract (23). Briefly, cells were lysed in RIPA lysis buffer (Pierce #89900) with protease and phosphatase inhibitors (Thermo Scientific #1861281). Samples were resolved by SDS–PAGE, transferred onto PVDF membranes, and membranes were blocked and incubated with primary antibodies in 5% BSA (Sigma Aldrich, #A3059) in TBS-T (Bio-Rad, #161–0372 with 0.05% Tween 20) or 5% milk (Bio-Rad #170–6404) in TBS-T at 4°C overnight. Protein signals were detected using enhanced chemiluminescence (Pierce, #32106) as per the manufacturer’s instructions. Immunoprecipitations of flag-tagged proteins were performed using anti-Flag M2 magnetic beads(Sigma-Aldrich #M8823), following the manufacturer’s instructions.
Primary antibodies used include Cell Signaling Technology antibodies EZH2 (#5246, RRID: AB_10694683,1:1000), Histone H3 (#9715, RRID:AB_331563, 1:10 000), myc-tag (#2276, RRID:AB_331783, 1:5000), trimethyl-histone H3 (Lys27) (#9733, RRID:AB_2616029, 1:1000), Snail1 (#3879, RRID:AB_2255011, 1:500), TCF4/TCF7L2 (#2569, RRID:AB_2199816,1:500), Slug (#9585, RRID:AB_2239535,1:500), Cyclin D1 (#55506, RRID:AB_2827374, 1:500), non-phospho (Active) β-catenin (#8814, RRID:AB_11127203, 1:1000), β-catenin (#8480, RRID:AB_11127855, 1:1000), LRP6 (#2560, RRID:AB_2139329 1:500), c-MYC (#13987, RRID:AB_2631168,1:1000). Sigma-Aldrich: FLAG M2 (#A8592, RRID:AB_439702 ,1:1000), and Abcam antibodies: CCN6 (anti-WISP3, #ab224720, 1:50), vimentin (#ab92547, RRID:AB_10562134, 1:1000), cytokeratin-18 (#AB32118, RRID:AB_736394, 1:1000), FZD8 (#ab155093, 1:500). β-actin HRP (Santa Cruz, #sc47778, RRID:AB_626632, 1:5000), was used as for loading control. Secondary antibodies used were Amersham ECL anti-rabbit IgG HRP-linked (GE Healthcare Life Sciences, #NA934, 1:10 000) or Amersham ECL anti-mouse IgG HRP-linked (GE Healthcare Life Sciences, #NA931, 1:10 000).
Reporter assays.
The Wnt reporter assay was performed using CCN6 protein (rhCCN6, 0.2 μg/μL and 0.5 μg/μL) (PeproTech, # 120–20), human Wnt-3A (0.04 μg/μL, R&D System, #5036-WN), and DKK-1 (0.2 μg/μL, Enzo,#ENZ-60002-C005) and the Leading light Wnt reporter assay kit (Enzo, #ENZ-61001–001) following manufacturer’s instructions.
For the EZH2 promoter luminescence reporter assay MDA-MB-231-dnTCF4, Ccn6-KO-dnTCF4, HME-shCCN6-dnTCF4, and corresponding control cells were transduced with lentivirus bearing human EZH2 promoter (for MDA-MB-231 and HME cells) (GeneCopoeia, # HPRM38653-LvPG04-H) or mouse EZH2 promoter (for Ccn6-KO cells) (GeneCopoeia, # MPRM33952-LvPG04-H), and controls (GeneCopoeia # NEG-LvPG04-H) constructs. We additionally deleted three E-boxes in the human EZH2 promoter construct (CAAGTG, CACGTG, and CAGCTG) and in the mouse promoter construct (CAGATG, CAATG, CAAGTG) that are sites of TCF4 binding (5’-CANNTG-3’) (UniProt P15884 ITF2). Luminescence was measured following the manufacturer’s instructions of Secrete-Pair™ Dual Luminescence Assay Kit, for parallel bioluminescence assays of Gaussia luciferase (GLuc) and secreted Alkaline Phosphatase (SEAP) (GeneCopoeia, # LF031).
Invasion, cell attachment, and competition assays.
In vitro invasion assays were performed using a 24-well Matrigel invasion chamber (BD Biosciences, #354480) per manufacturer’s instructions. All invasion experiments were carried out in technical triplicates and repeated at least three times with biological replicates. Cells that had invaded through the Matrigel membrane were quantified by Immunofluorescence GFP pictures or fixed with methanol, stained with crystal violet, photographed at high resolution, and counted manually using ImageJ. The representative whole inserts were imaged under the same conditions and are shown after increasing brightness by 20% across all images in Microsoft PowerPoint. Cell attachment assays were performed by trypsinizing 70% confluent cell dishes and seeding 1 × 105 cells in a 12 well plate. After 30 min, non-adherent cells were removed by washing wells with PBS three times. Adherent cells were then imaged, and entire wells were counted using ImageJ.
For the competition assays MDA-MB-231 cells transduced with Flag-CCN6; Flag-ΔTSP1 and controls and Ccn6-KO cells transduced with Flag-CCN6, and controls were seeded at 1×106 cells per 100 mm plate and treated for 6hr in serum free with rhWNT3A (0.3 ng/μl, R&D System, #5036-WN) or rhWNT10B (0.5 ng/μl, R&D System, #7196-WN). Active β-catenin, EZH2, and H3K27me3 expression was detected by WB and quantified using ImageJ as described below.
Immunofluorescence analysis.
Immunofluorescence was performed by seeding cells into 2-well chambered slides (Thermo Fisher Lab-Tek #154461). After 24 hr, cells were fixed with 4% PFA diluted in PBS for 15 min at room temperature, rinsed three times with PBS, and blocked for 1 h using blocking buffer, 5% normal goat serum containing 0.3% Triton X-100 in PBS. Subsequently, slides were incubated with primary antibody diluted in antibody buffer (5% bovine serum albumin containing 0.3% Triton X-100 in PBS) at 4°C overnight. On the next day, slides were washed three times for 5 min with PBS and incubated with fluorescent secondary antibodies Alexafluor goat-anti (Rabbit 488 Cat # A-11008) and rhodamine phalloidin (Thermo Fisher, # R415). Slides were washed three times with PBS and cover slipped using ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher, Cat# P36962). Slides were imaged using Leica SP5 Inverted 2-Photon FLIM Confocal, and image analysis was performed using ImageJ.
Tissue samples and immunohistochemistry.
The studies of human breast tissue samples were carried out with approval from the Institutional Review Board of the University of Michigan (protocol# HUM00050330). We utilized paraffin-embedded tissues from 27 human mBrCA previously characterized for their expression of CCN6 (17) and β-catenin (10) were immunostained for EZH2. Briefly, 5 μm-thick paraffin-embedded sections were de-paraffinized in xylene and rehydrated through graded alcohols to water. Heat Induced Epitope Retrieval (HIER) was performed in the Decloaking Chamber (Biocare Medical) with Target Retrieval, pH 6.0 (DakoCytomation). Slides were incubated in 3% hydrogen peroxide for 5 min to quench endogenous peroxidases. Anti-EZH2 (Cell Signaling, D2C9, catalog no. 5246, RRID:AB_10694683 1:150) was incubated with the tissue sections for overnight at 4°C. Antibodies were detected with Envision+HRP Labeled Polymer (DakoCytomation) for 30 min at room temperature. HRP staining was visualized with the DAB+Kit (DakoCytomation). Negative control slides were run. Slides were counterstained in hematoxylin, blued in running tap water, dehydrated through graded alcohols, cleared in xylene, and then mounted with Permount. Expression of EZH2 was analyzed blindly by two observers, at least twice. Nuclear EZH2 was considered high or low based on staining intensity and percentage of tumor cells staining based on a previously validated scoring system (26).
Animal experiments.
All studies were approved by the University of Michigan UCUCA protocol# PRO00010731. Eight-week-old FVB/NJ (Jackson Laboratories, stock#001800, n=30) were used to examine tumorigenicity. Briefly, Ccn6-KO cells (5×105 resuspended in 50 μl Matrigel) were orthotopically injected into the right inguinal mammary fat pad of female mice. Twenty-four hr after implantation, mice were treated with 120 ml per 30 g of body weight of EPZ-6438 (EZH2i) or vehicle, i.p. daily. Primary tumor growth was monitored daily by caliper measurement in accordance with UCUCA tumor size guidelines (2 cm3), at which point animals were euthanized and underwent necropsy. In an independent experiment, Ccn6-KO cells (1×105 resuspended in 50 ml Matrigel) were orthotopically injected into the right inguinal mammary fat pads of female mice (n=20). When tumors were palpable (0.2–0.3 cm) mice were treated as above.
For the metastasis assays, firefly-luciferase expressing Ccn6-KO cells (1×105) were injected intracardially (n=20). Twenty-four hr after injection mice were treated as described above. Metastasis were monitored using bioluminescence imaging as previously described (23). Briefly, mice were anesthetized and injected i.p. with 75 mg/kg D-Luciferin (Xenogen) resuspended in PBS. Bioluminescence images were acquired using the IVIS imaging system (Xenogen) within ~2–5 min after injection. Analysis was performed using the Living Image software platform (Xenogen) by measuring photon flux measured in photons/s/cm2/sr by using a region of interest (ROI) drawn around the bioluminescence signal to be measured and subtracting background measurements.
Firefly-luciferase expressing Ccn6-KO cells transduced with EZH2 shRNAs or scrambled control (shControl) were orthotopically injected into the right inguinal mammary fat pad of female mice (2×105 cells in 50 ml Matrigel, n=10 mice/group) or intracardially (1×105 cells, n=10 mice/group). Tumor growth and metastasis were monitored as described above.
Primary tumors, lungs, bones, liver, and brains were collected, fixed in 10% neutral buffered formalin, and embedded in paraffin for H&E staining and immunohistochemistry. Histopathology of tumors and metastasis was evaluated under light microscopy. The number of mitoses was analyzed by light-microscopy counting mitotic cells per 10 high power fields in at least 3 independent tumors per condition. Immunohistochemistry of mouse tissues was performed using antibodies against CK-18 (Abcam, #ab181597, RRID:AB_2922417, 1:500), vimentin (Abcam, #ab92547, RRID:AB_10562134, 1:500), ZEB1 (Cell Signaling, #70512, RRID:AB_2935802, 1:200), phospho histone H3 (Cell Signaling, #9701, RRID:AB_331535, 1:100), and cleaved caspase 3 (Cell Signaling, #9661, RRID:AB_2341188,1:400). The percentage of cancer cells expressing each protein and the intensity of immunostaining was quantified under light microscopy using a validated method (27) in 10 high power fields of at least 3 independent tumors and shown in bar graphs representing the mean ± SEM.
Paraffin embedded tissues of orthotopic xenografts of MMTV-Cre;Ccn6fl/fl previously treated with rhCCN6 or vehicle (28) were employed for H&E and immunostaining using the following Cell Signaling antibodies: anti-non phospho (active) β-catenin (#8814, 1:400), anti-c-Myc (#13987, 1:400), anti-EZH2 (#5246, 1:100), anti-H3K27me3 (#9733, 1:250) and CyclinD1 (#55506, 1:250). Percentage of tumor cells and staining intensity were quantified using the IHC score as described above.
Proteomic analysis.
Fresh samples of Ccn6-KO mammary tumors and normal tissues excised from mice treated with rhCCN6 or vehicle (n=2 per condition) were collected following the University of Michigan UCUCA approved protocol# PRO00010731. One ~0.3 cm tumor section was embedded in paraffin, stained by hematoxylin and eosin, and evaluated pathologically for tissue diagnosis. Tandem mass tag (TMT0 labeling was performed for mass spectroscopy (MS) using three consecutive TMT-10-plex isobaric labeling kits (ThermoFisher, Cat #90111) according to the manufacturer’s protocol, and subjected to LC-MS/MS analysis. One proteomic TMT 10-plex experiment identified 85,683 peptides to a depth of 4609 unique proteins across all samples. Data analysis and protein quantification were performed using MSFragger and Philosopher toolkit (v20181119, github.com/Nesvilab/philosopher) and the UniProt mouse protein database (UP000000589, last modified: 5 November 2019; 55,408 proteins) as in our recent publication (11). We performed hierarchical clustering, enrichment and GSEA analyses as in (11).
We used MetaCore (genego.com) to analyze network interactions between deregulated genes of tumor samples. Analysis between the comparison groups were based on an adjusted p-value of <0.05. It was expressed in -log(p-value) and ranked by statistical significance. Changes in expression levels were presented as fold changes. Genes indicated as network objects were used to build network.
Chromatin Immunoprecipitation (ChIP)-sequencing library preparation and sequencing.
ChIP-seq was performed using anti-H3K27me3 (Cell signaling #9733S) on 10 million Ccn6-KO-dnTCF4 and Ccn6-KO-pPGS controls at the BRCF Epigenomics Core. Cells were fixed with 1% formaldehyde, quenched with glycine, and frozen as a pellet. A total of 1 million cells per condition was used. We used the Diagenode ChIPmentation kit with dual indices and followed the manufacturer’s protocol. Briefly, the cells were lysed and resuspended in 130 μl of chromatin shearing. Shearing was done in a Covaris S2 instrument. A 15 μl aliquot of the sheared chromatin was taken for assessment of the shearing size. The remaining sheared chromatin split for immunoprecipitation in the presence of H3K27me3 antibody and IgG as input. Four ug of each antibody was used per immunoprecipitation. Immunoprecipitation was done overnight at 4°C with rotation. The immunoprecipitate were next washed before on-bead tagmentation at 37°C for 10 minutes with agitation. The reaction was stopped by putting the samples on ice and additional washes, before processing for stripping, end filling and reverse crosslinking. qPCR was used to determine the optimal cycle number for enrichment PCR, according to the manufacturer’s protocol. The final amplified libraries were cleaned using AMPure XP beads at a 1.8:1 bead:DNA ratio, quantitated with Qubit HS dsDNA, and fragment size assessed on a TapeStation HS D1000 kit. The libraries were qPCR quantitated for pooling using the KAPA Illumina qPCR quantitation kit, and sequenced 151bp paired-end on a NovaSeq S4 flow cell at the UM Advanced Genomics Core.
ChIP-seq Analysis.
We used FastQC (FastQC) (v0.11.8) to assess the overall quality of each sequenced sample. We use TrimGalore (v0.4.5) and cutadapt (v1.15) with the following parameters: –nextera -e 0.1 –stringency 6 –length 20 –nextseq 20. We align trimmed reads to mm10 with Bowtie2 (v2.3.4.1) using default parameters with the exception of the following flags: -X 2000. Duplicate reads are marked with Picard (v2.20.2). Alignments are filtered with samtools (v1.2) using the flags: -F4 -F8 -f3 -q10 -F1024. Alignments completely overlapping blacklisted regions (ENCODE Blacklist Regions) are removed with bedtools (v2.28.0). Sample-wise peaks are called with macs (v2.1.2) with flags: –format BAMPE –gsize mm.
Peaks over all samples are merged with bedops (v2.4.36) for the purpose of principal component analysis and unsupervised clustering to assess the similarity of samples. Read cross-correlations within samples are plotted using phantompeakqualtools. MultiQC(29) (v1.7) generates a report combining FastQC, trimming, alignment, and duplicate calling over all the samples. The software DeepTools is used to calculate coverage and IP efficiency plots (v3.3.0).
RNA-seq Analysis.
RNA-seq was performed on Ccn6-KO-dnTCF4 or Ccn6-KO-pPGS cells infected with adenovirus containing EZH2-WT, EZH2-ΔSET or adenovirus control in quadruplicate samples. RNASeq samples were subjected to 151bp paired-end sequencing. Reads were aligned to GRCm38 using STAR v 2.7.8a. Differential expression analysis was conducted with DESeq2 and functional enrichment analysis was done with iPathway Guide (Advaita). H3K27me3 differential promotor occupancy was evaluated using the median q-value and q-value-weighted mean fold change for peaks within 100 kbp of the transcription start site. Library prep and next-generation sequencing was carried out in the Advanced Genomics Core at the University of Michigan.
Quantification and statistical analyses
Data are expressed as mean ± SEM. All experiments were repeated at least in triplicate. The number of replicates is indicated in the dot plots in the figures and corresponds to biological replicates. Differences between 2 groups were examined by 2-tailed unpaired Student’s t test. One-way ANOVA or 2-way ANOVA with Tukey’s multiple comparison test were used to compare more than 2 groups, as indicated. P value of less than 0.05 was considered significant.
Data Availability.
All mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium through the PRIDE partner repository with identifier PXD014414. The RNA-seq and ChIP-seq data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE250575 and GSE250574, respectively. Other data generated in the study are available upon request to the corresponding author.
Results
A CCN6Low/nuclear β-catenin/EZH2High phenotype characterizes a subset of human spindle mBrCA tumors
Prior studies from our lab demonstrated that CCN6 protein levels are reduced in 68% and nuclear β-catenin is expressed in 56% of human mBrCA tumors, respectively (10, 17). To identify other major oncogenic pathways linked to CCN6 and Wnt/β-catenin in mBrCA tumors, we performed a functional analysis of our recently reported quantitative proteomics landscape that includes 15 human mBrCA samples (11). We found spindle mBrCA tumors have a predicted CCN6-β-catenin-EZH2 protein network with high significance (PPI enrichment p-value:< 1.0e-16) relative to normal breast (Fig. 1A and B and Supplementary Fig. S1). Compared to normal breast tissue, mBrCAs have differential expression of proteins that mediate EZH2 methyltransferase transcriptional repressor function and EZH2 related oncogenic processes (e.g., EMT, cell adhesion, stemness) (Supplementary Table 1). We next assessed the expression of EZH2 by immunohistochemistry in our cohort of 27 mBrCA previously stained for CCN6 and β-catenin (9, 10, 17). We found 54% of mBrCA with a predominant spindle component have a CCN6low/nuclear β-catenin/EZH2high expression pattern whereas 14% of mBrCA with predominant squamous or sarcomatoid components (chondroid and osseous) displayed such an expression pattern (Fig. 1C).
Figure 1. Human spindle metaplastic carcinomas display an interacting network of CCN6, β-catenin, and EZH2.

A. Protein-protein interaction (PPI) network for differentially expressed proteins in metaplastic breast carcinomas (mBrCA) compared to normal breast. MetaCore network analysis shows β-catenin-EZH2-CCN6 (WISP3) interaction containing 16 up-regulated (red target) proteins and 24 down-regulated (blue target) proteins. Fold change of +/− 1 and a p value of < 1.0e-16 were considered for the analysis.
B. Enrichment analysis using gene ontology (GO) annotations showing the top GO terms based on biological process. Significant proteins were considered using p-value < 0.05, fold change (log2-FC) >1. Barplot shows significant terms of hub genes.
C. Representative images of human mBrCAs of spindle, squamous and sarcomatoid subtypes stained with hematoxylin and eosin (H&E) and immunostained for CCN6, β-catenin, and EZH2 (×600 magnification). Results are tabulated. Low/negative CCN6 is significantly associated with associated with nuclear β-catenin and high nuclear EZH2 in the spindle subtype of mBrCA compared to other subtypes (sarcomatoid and squamous) (Chi-square p < 0.029). Bars, 100 μm.
CCN6 antagonizes Wnt/β-catenin signaling and reduces EZH2 expression
To investigate mechanisms underlying the role of CCN6 in regulating Wnt/β-catenin pathway activity in mBrCA, we employed the human MDA-MB-231 mesenchymal-like TNBC breast cancer cell line as well as Ccn6-KO cells derived from MMTV-Cre;Ccn6fl/fl mBrCAs (17, 30). Our approaches included use of recombinant CCN6 protein (rhCCN6) along with ectopic expression of a full-length CCN6 protein or a deletion-mutant form of CCN6 lacking the TSP1 domain (ΔTSP1-Flag). The TSP1 domain has previously been shown to regulate the invasion suppressive function of CCN6 (19, 22). In addition, we used shRNA approaches to reduce CCN6 expression in HME non-tumorigenic breast cells. Consistent with our previous studies (18, 22), we detected ectopically expressed CCN6 and ΔTSP1-CCN6 in the conditioned medium of the transduced MDA-MB-231 cells, which endogenously express only low levels of CCN6 (21) (Fig. 2A).
Figure 2. CCN6 antagonizes canonical Wnt/β-catenin signaling and reduces EZH2 expression.

A. Immunoblot of the conditioned media (CM) of MDA-MB-231 cells stably transduced with lentivirus containing CCN6-Flag or ΔTSP1-Flag. Ponceau stain serves as loading control.
B. Ectopic CCN6 interacts with endogenous Lrp6 and Fzd8. Proteins from whole cell extracts of cells in A were pulled down using Flag magnetic beads followed by WB.
C. Immunofluorescence of MDA-MB-231 cells transduced with Flag-CCN6 or Flag-Vector with antibodies against active hypo-phosphorylated β-catenin and DAPI. Bars show the quantification of nuclear active β-catenin. Bars: mean ± SEM of triplicate experiments. Scale bar, 50 μm.
D. Immunofluorescence of Ccn6-KO cells treated rhCCN6 (0.5ng/μl), rhWNT10B (0.3ng/μl), or combined and stained with antibodies against active β-catenin and DAPI. Bars show quantification of nuclear active β-catenin. Scale bar, 50 μm.
E. Immunoblot for the indicated proteins of Ccn6-KO cells transduced with CCN6-Flag or ΔTSP1-Flag treated with rhWNT3A (0.3ng/μl) (left) and immunoblot of Ccn6-KO cells treated with rhCCN6 (0.5ng/μl) or rhWNT10B (0.3ng/μl) (right).
F. Leading light Wnt reporter assay in cells treated as indicated and shown as response to untreated. rhCCN6 antagonizes canonical Wnt transcriptional activity to similar levels than the Wnt inhibitor Dkk1.
G. Immunoblot for the indicated proteins of HME cells transduced with lentivirus containing sh-Control or two independent shRNAs against CCN6.
H. Bars show quantification of immunofluorescence staining for nuclear active β-catenin in HME cells treated as indicated.
I. Immunoblot of HME-shCCN6 cells treated with rhCCN6 (0.5ng/μl) or WNT3A (0.3ng/μl) for the indicated proteins.
For all panels *P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001
Prior studies have shown zWisp3, the zebrafish ortholog of CCN6, interacts with Fzd8 and Lrp6 to inhibit Wnt signaling (31). However, this potential mechanism has not been substantiated in cancer. Our combined immunoprecipitation and immunoblotting studies showed full length CCN6, but not ΔTSP1-CCN6, interacts with endogenous Fzd8 and Lrp6 in MDA-MB-231 cells (Fig. 2B).
We next tested if CCN6 reduces cellular responses to canonical Wnt ligands in breast cancer cells. Immunofluorescence and immunoblot studies showed ectopic full length CCN6 or rhCCN6 reduced levels of active β-catenin in the nucleus of MDA-MB-231 and Ccn6-KO cells (Fig. 2C and D; Supplementary Figs. S2A–S2D and S3). Ectopic CCN6 or rhCCN6 protein attenuated canonical Wnt (Wnt3A and Wnt10B)-mediated upregulation of active β-catenin levels and increased expression of EZH2 and its transcriptional repression mark, H3K27me3 (Fig. 2E; Supplementary Figs. S2B–S2D and S3). Functionally, rhCCN6 treatment significantly reduced Wnt ligand-induced β-catenin transcriptional activity in a manner akin to that seen with the known Wnt inhibitor DKK1, based on data obtained with the Leading Light Wnt reporter cell line (Fig. 2F; Supplementary Fig. S4A). Also, rhCCN6 treatment reduced the invasion-promoting function of canonical Wnt pathway ligands (Supplementary Fig. S4B).
We also implicated CCN6 as a key regulator of Wnt signaling and EZH2 levels in mammary epithelial cells by showing a CCN6 shRNA knockdown approach in HME cells led to increased expression of active β-catenin, as well as changes in the levels of EZH2, H3K27me3, c-Myc and Cyclin D1 (Fig. 2G; Supplementary Fig. S4C and S4D). In the HME-shCCN6 cells, treatment with rhCCN6 protein reduced the increased active β-catenin and EZH2/H3K27me3 levels. In HME cells, treatment with rhCCN6 also antagonized the effect of Wnt3A and Wnt10B on β-catenin nuclear localization and on EZH2/H3K27me3 upregulation (Fig. 2H and 2I; Supplementary Fig. S4E and S4F).
Collectively, these data demonstrate CCN6 antagonizes the effect of canonical Wnt ligands on β-catenin activation, EZH2 expression, and invasion function in breast cancer cells, at least in part by interacting with Wnt receptors. Further, our data show the TSP1 domain of CCN6 is important for these effects.
CCN6 regulates a β-catenin/EZH2 pathway in mBrCA tumors in vivo
We have recently shown rhCCN6 treatment is sufficient to reduce the growth of MMTV-Cre;Ccn6fl/fl (Ccn6-KO) mammary mBrCA tumors implanted in the mammary fat pad of syngeneic FVB mice (28). Here, we employed this model of mBrCA to test if rhCCN6 antagonizes β-catenin activity and EZH2 expression in the tumor cells in vivo. Consistent with the in vitro data, rhCCN6 treatment significantly reduced nuclear β-catenin and led to cytoplasmic/membrane β-catenin expression in the resected mBrCA tumors compared to vehicle treatment (Fig. 3A). The rhCCN6 treatment significantly reduced the expression of the canonical Wnt targets c-Myc and Cyclin D1, as well as the levels of nuclear EZH2 and its trimethylation substrate H3K27me3, compared to vehicle treatment (Fig. 3B). To gain a better understanding of CCN6-dependent effects in an unbiased manner, we performed a tandem mass tag (TMT)-based proteomic analysis of fresh samples of Ccn6-KO tumors treated with rhCCN6 or vehicle. Of the 192 significant differentially expressed proteins, 37 (19.3%) form a significant β-catenin-EZH2 protein-protein (PPI) interaction network (Fig. 3C). The STRING analysis also showed a combined confidence of interaction between β-catenin and EZH2 pathway proteins of 0.934 and a combined score of functional interaction of 0.986. Consistent with the major cancer-related signatures that we identified in human mBrCA (Supplementary Table 1), the CCN6-regulated β-catenin-EZH2 network proteins have roles in cell differentiation, EMT, regulation of cell cycle, mitosis, immune modulation, and metabolic processes (Fig. 3D).
Figure 3. CCN6 regulates a β-catenin/EZH2 pathway in mBrCA tumors in vivo.

A. Paraffin embedded orthotopic tumors of MMTV-Cre;Ccn6fl/fl previously treated with rhCCN6 or vehicle were employed for H&E and for active β-catenin immunostaining. Bars show percentage of cells with nuclear active β-catenin per 10 high power fields ± SEM. Scale bar, 100 μm.
B. Immunohistochemistry of tumors in A, stained with the indicated antibodies. Bars: mean IHC score calculated using the percentage of positive cells and intensity of staining per 10 high power fields ± SEM. Scale bar, 100 μm.
C. MetaCore network analysis of quantitative TMT-proteomics of tumors in A reveals a protein-protein interaction (PPI) network of 18 up-regulated (red) proteins and 14 down-regulated (blue) proteins highlighting β-catenin-EZH2 involvement. P value <1.0e-16.
D. Gene Ontology (GO) functional annotation of statistically significant differential proteins in the CCN6-β-catenin-EZH2 PPI network.
*P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001
Supporting the ability of EZH2 to mediate at least in part the CCN6-dependent effects, inhibition of EZH2 via an shRNA approach rescued the CCN6-dependent decrease in E-cadherin and the increased invasion seen in cells where CCN6 levels were antagonized by an shRNA approach (Supplementary Fig. S4G and S4H). Together, these data demonstrate that CCN6 expression regulates the protein expression landscape in mBrCA and CCN6 specifically downregulates a novel β-catenin-EZH2 protein network, with resultant effects on H3K27me3 in vivo and in vitro.
Ccn6-KO-induced β-catenin/TCF activation mediates EZH2 transcriptional upregulation and H3K27me3 repressive marks
In the canonical pathway, Wnt transduces downstream signals by stabilizing β-catenin and promoting β-catenin binding to the TCF family of transcription factors, resulting in activation of β-catenin-dependent gene expression (32). To assess the relevance of the canonical Wnt pathway on the EMT, invasion, and adhesion properties of Ccn6-KO mBrCA phenotype, Ccn6-KO cancer cells derived from MMTV-Cre;Ccn6fl/fl mammary tumors were transduced with a dominant-negative TCF4 construct (25) (Fig. 4A). As expected, dnTCF4 expression in Ccn6-KO cells significantly reduced known Wnt/β-catenin/TCF target genes Myc, Cyclin-D1, CD44, LRP5, BMP4, AXIN2 compared to pPGS vector (Supplementary Fig. S5A). Under these conditions, Ccn6-KO cells display reduced invasion, increased adhesion, and protein expression changes towards an epithelial phenotype compared to controls (Fig. 4B and C). Similar results were observed in MDA-MB-231 cells, which have low or negligible levels of endogenous CCN6 protein expression (Supplementary Fig. S5B and S5C).
Figure 4. The canonical WNT pathway mediates EZH2 transcriptional activity and invasive phenotype of Ccn6-KO mBrCA.
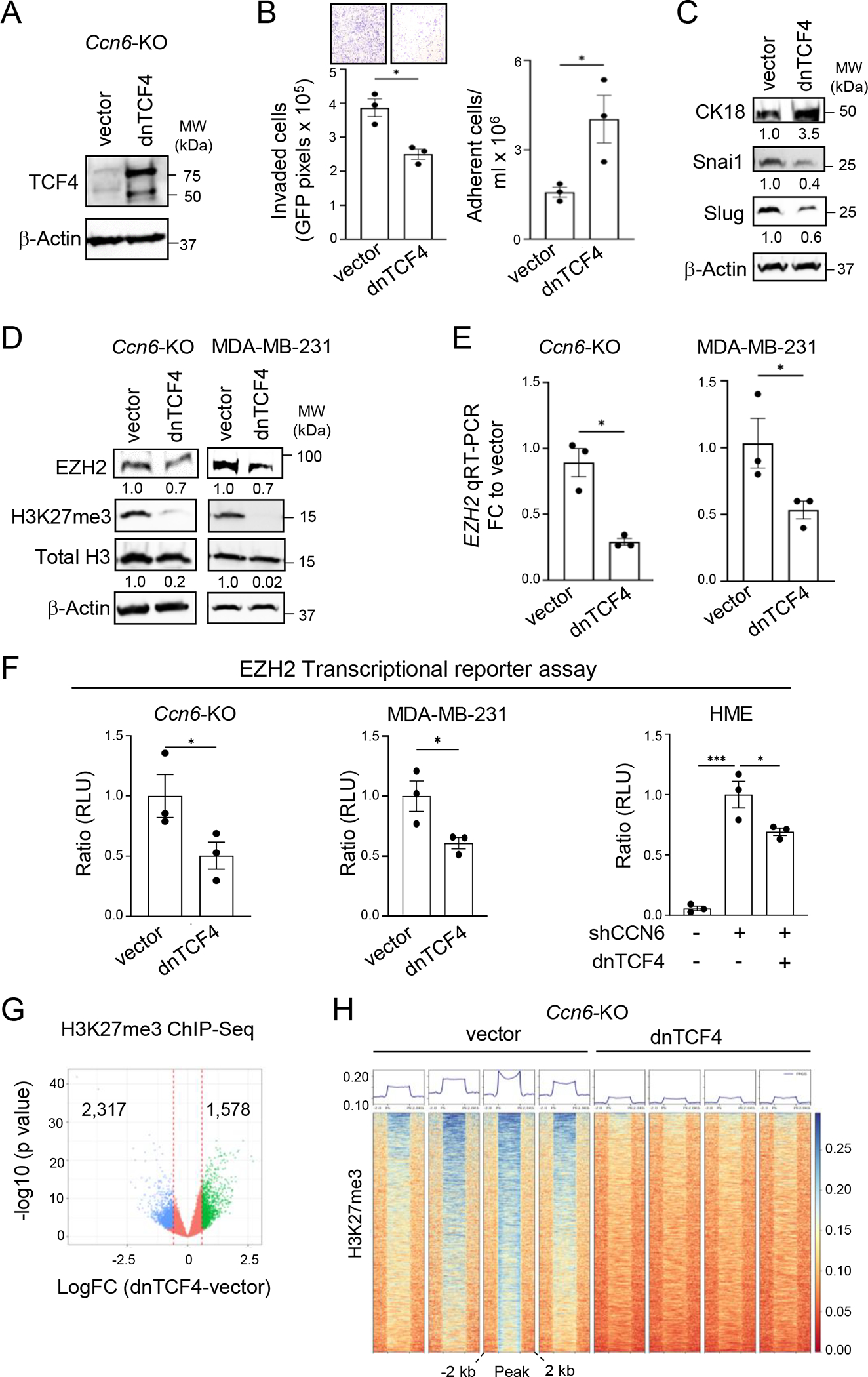
A. Western blot (WB) of MMTV-Cre;Ccn6fl/fl tumor-derived cells (Ccn6-KO) transduced with a dominant-negative TCF4 (dnTCF4) construct or pPGS vector control.
B. Cells in A were used for cell invasion and adhesion assays.
C. WB of Ccn6-KO transduced with dnTCF4 or pPGS control for the indicated proteins. The numbers underneath the bands represent the fold change of dnTCF4 to vector.
D. WB of Ccn6-KO and MDA-MB-231 cells transduced with dnTCF4 or pPGS control show that dnTCF4 leads to reduced EZH2 and H3K27me3 expression. The numbers underneath the bands represent the fold change of dnTCF4 to vector.
E. qRT-PCR of cells in A shows that dnTCF4 significantly reduces EZH2 mRNA expression levels in Ccn6-KO and in MDA-MB-231 cells. Bars: mean ± SEM of triplicate experiments.
F. EZH2 promoter activity using an EZH2 transcriptional reporter assay in Ccn6-KO, MDA-MB-231, and HME-shCCN6 cells transduced with dnTCF4 or pPGS control. Bars: mean promoter activity measured as RLU ± SEM of triplicate experiments.
G. H3K27me3 ChIP-sequencing analyses of Ccn6-KO cells transduced with dnTCF4 or pPGS vector. Volcano plot of the log fold change (dnTCF4 vs. pPGS) showing all tested loci. A locus is considered differentially bound if FDR < 0.05 and |logFC| > 0.585. Points are colored according to the direction of differential binding. The vertical lines correspond to the logFC cut-off.
H. Profile plot and binding heatmap for H3K27me3 ChIP-seq signal shows the binding within each sample to target loci split among regions differentially bound in dnTCF4 and pPGS.
For all panels *P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001
To independently assess whether β-catenin/TCF function is required for the invasion suppression function of CCN6, we transfected Ccn6-KO and MDA-MB-231 cells with a stabilized, phosphorylation-resistant mutant form of β-catenin containing an S33Y substitution (25), and treated the cells with rhCCN6 or vehicle. The S33Y-transfected cells displayed increased levels of β-catenin and active β-catenin, and the cells were more invasive compared to controls (Supplementary Fig. S5D–S5G). In contrast to control cell lines, the S33Y-transduced cells showed sustained invasion in the presence of rhCCN6, indicating that the invasion suppression function of CCN6 requires inhibition of β-catenin levels and functional activity (Supplementary Fig. S5E and S5G). We also found that S33Y-β-catenin was sufficient to rescue the protein profile, and the diminished invasion and increased cell adhesion in HME CCN6-shRNA cells (Supplementary Fig. S6A–S6C). Together, these results show that induction of canonical Wnt pathway transcriptional activity is critical to the CCN6 KO EMT and invasive phenotype.
We next sought to elucidate whether the upregulated expression of EZH2 due to CCN6 KO requires Wnt pathway activation. Ectopic expression of dnTCF4 reduced EZH2 and H3K27me3 proteins and EZH2 mRNA levels in Ccn6-KO cells, MDA-MB-231, and in HME CCN6-shRNA cells compared to pPGS vector (Fig. 4D and E; Supplementary Fig. S6A). Next, we used a lentivirus based EZH2 transcriptional reporter assay to evaluate the effect of inhibition of canonical Wnt pathway on EZH2 promoter activity. Blockade of TCF signaling significantly reduced EZH2 promoter activity in Ccn6-KO, MDA-MB-231, and HME CCN6-shRNA cells (Fig. 4F), and deletions of the TCF4 binding sites at the EZH2 promoter reduced promoter activity (Supplementary Fig. S6D). Providing independent validation for a β-catenin-TCF-EZH2 axis in CCN6 depleted cells, S33Y-β-catenin was unable to rescue the reduced EZH2 promoter activity due to dnTCF4 in HME CCN6-shRNA cells (Supplementary Fig. S6E).
As the enzymatic component of the Polycomb repressive complex 2 (PRC2), EZH2 catalyzes H3K27me3 to maintain transcriptional memory and cell-type identity (33). To investigate the function of CCN6 KO-induced β-catenin/TCF in EZH2 transcriptional repressor function, we performed H3K27me3 chromatin immunoprecipitation sequencing (ChIP-seq) of Ccn6-KO mBrCA cells transduced with dnTCF4 or pPGS vector. ChIP-seq analyses revealed that Ccn6-KO-dnTCF4 cells display significantly reduced genome-wide H3K27me3 transcriptional repressive mark deposition compared to Ccn6-KO-pPGS (n=1578 vs. 2317, p <0.05) (Fig. 4G and H; Supplementary Fig. S7A). Taken together, these data show the induction of Wnt canonical signaling is critical to the CCN6-deficient EMT and invasion program and identify EZH2 methyltransferase as a downstream effector of this pathway.
The Ccn6-KO-induced Wnt/β-catenin invasion program requires EZH2 catalytic activity
To discover canonical Wnt targets that depend on EZH2 methyltransferase activity in mBrCA cells we performed RNA sequencing (RNA-seq) studies. We infected Ccn6-KO-dnTCF4 and control cells with adenovirus containing vector, wild-type EZH2 or ΔSET-EZH2, an EZH2 mutant with deletion of the SET methyltransferase domain previously characterized in our lab (24). RNA-seq analyses identified 452 genes differentially expressed (1% FDR, 1.5 linear fold change) between Ccn6-KO-dnTCF4 and pPGS vector that are rescued by wild-type EZH2 (Fig. 5A). Of these, 64 genes are rescued by wild-type EZH2 but not by ΔSET-EZH2, indicating that their expression depends on EZH2 methyltransferase activity (Fig. 5A green bars). Gene set enrichment analysis using STRING databases highlight a cluster of 17 genes that form the strongest predicted interaction network with EZH2 (Fig. 5B yellow circles; Supplementary Fig. S7B). The most significant GO pathways represented by the 17 genes are mitosis and focal adhesion processes (Fig. 5C). Taken together, these data show that in Ccn6-KO mBrCA cells, β-catenin/TCF regulates EZH2 methyltransferase activity on a subset of gene targets.
Figure 5. EZH2 transcriptional repressor activity is required for Ccn6-KO-induced β-catenin/TCF4-mediated invasion.

A. Heatmap of RNA-seq analyses of Ccn6-KO cells transduced with pPGS vector or dnTCF4 and rescued with adenovirus containing MYC-tagged wild-type EZH2 (MYC-EZH2), ΔSET-EZH2 (MYC-ΔSET), or adenovirus vector. The heatmap shows differential gene expression of Ccn6-KO-dnTCF4-vector vs. Ccn6-KO-dnTCF4-EZH2 (n=452 differentially expressed genes, DEGs, from 13,710 genes with measured expression). Green bars indicate genes that are rescued by MYC-EZH2 but not by MYC-ΔSET (n=64).
B. Volcano plot of the 452 significant DEGs between Ccn6-KO-dnTCF4-vector and Ccn6-KO-dnTCF4-EZH2. The x-axis shows the measured expression of DEGs and y-axis shows the significance of the change as negative log (base 10) of the p-value. The dotted lines represent the thresholds used to select DEGs. Upregulated genes are red, downregulated genes are blue. We found that 64 genes are rescued by wild-type EZH2 but not by ΔSET-EZH2. Highlighted in yellow are 17 most significant DEGs that form the strongest network with EZH2.
C. GO functional annotation of the 17 genes highlighted in yellow in B.
D. Immunoblot of Ccn6-KO cells transduced with pPGS vector or dnTCF4 and rescued with MYC-tagged wild type EZH2, ΔSET-EZH2, ΔNLS-EZH2, or vector.
E. Cells in E were subjected to invasion (left) and adhesion (right) assays. Bars show mean of triplicate experiments ± SEM. *P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001.
To identify gene expression changes related to EZH2 methyltransferase repressor activity, we examined RNA expression levels of the genes with decreased H3K27me3 peaks in the promoter region in Ccn6-KO cells with dnTCF4 compared to pPGS. We observed concordant decreased H3K27me3 peaks and increased expression of genes that regulate transcriptional processes (e.g., Zfp518b, Zfp704), cell adhesion and migration (e.g., Cyp1b1, Mfap3l, Shtn1, Megf10), Wnt inhibitory functions (Shisa2), and metabolic pathway genes (e.g., Pde4b, Pde4c, Cyp1b1) (Supplementary Fig. S7C). Genomic snapshots of gene loci confirmed decreased H3K27me3 in promoter regions of these genes, most of which constitute novel EZH2 targets (Supplementary Fig. S8). Underscoring the relevance of EZH2 enzymatic activity in the regulation of these gene targets, treatment of Ccn6-KO cells with an EZH2i (EPZ-6438/tazemetostat, a clinically used potent and selective EZH2 methyltransferase inhibitor (34, 35) significantly upregulated their expression levels (Supplementary Fig. S9A).
To delineate the function of EZH2 in mediating an invasive phenotype in the CCN6-deficient state, Ccn6-KO-pPGS vector and Ccn6-KO-dnTCF4 cells transduced with wild-type EZH2, ΔSET-EZH2, or vector control were subjected to immunoblot for EMT proteins, invasion, and cell adhesion assays. As an additional control for the nuclear function of EZH2, we also used ΔNLS-EZH2, an EZH2 mutant with deletion of the nuclear localization signal that we have shown to be localized to the cytoplasm (23, 24). Expression of wild-type EZH2 but not that of the ΔSET or ΔNLS mutants was sufficient to reverse the increased expression of luminal cytokeratin (CK18) and to increase Snai1, Cyclin D1 and c-Myc expression in Ccn6-KO-dnTCF4 cells compared to controls (Fig. 5D). Consistent with a critical role of the EZH2 methyltransferase activity in mediating the invasiveness of the CCN6-deficient/β-catenin/TCF mBrCA phenotype, only wild-type EZH2 completely reversed the invasive, adhesive, and proliferative properties of Ccn6-KO-dnTCF4 cells (Fig. 5E; Supplementary Fig. S9B). Collectively, these analyses identify a previously unknown effect of CCN6 deficiency in mBrCA tumors, which involves activation of the β-catenin/TCF/EZH2 axis to regulate transcription and tumor cell invasion.
EZH2 methyltransferase catalytic activity inhibition reduces the tumorigenicity of mBrCA CCN6 deficient tumors in vivo
To assess if EZH2 methyltransferase activity inhibition could be of potential benefit in treatment of spindle mBrCAs, we employed our well characterized murine spindle mBrCA model (17). MMTV-Cre;Ccn6fl/fl tumor cells were transplanted in the mammary fat pads of FVB mice to develop syngeneic tumors, and the mice were treated with EZH2i (EPZ-6438/Tazemetostat) or vehicle. Treatment with the EZH2i at the time of injection or after tumors became palpable, significantly reduced tumor volume compared to vehicle (Fig. 6A; Supplementary Fig. S10A) and improved tumor-free survival (Fig. 6B). Immunoblot analysis of the resected tumors showed that EZH2i effectively reduced H3K27me3 levels (Supplementary Fig. S10B). In histopathological studies, vehicle-treated tumors were larger and highly invasive into the skeletal muscle (Fig. 6C, arrowhead). EZH2i treatment induced areas of tumor necrosis and scar, significantly reduced the number of mitoses (Fig. 6C; Supplementary Fig. S10C) and increased the percentage of tumor cell apoptosis (Supplementary Fig. S10D). EZH2i treatment also increased expression of the luminal differentiation protein CK18 and reduced expression of the mesenchymal cell marker vimentin and the EMT transcription factor ZEB1 compared to controls (Fig. 6D).
Figure 6. EZH2 methyltransferase activity inhibition reduces tumor growth and distant metastasis in Ccn6-KO mBrCA tumors.

A. Primary tumor growth curves of FVB mice orthotopically implanted with syngeneic Ccn6-KO cells expressing GFP-Luciferase treated with vehicle or with EZH2i (EPZ-6438/tazemetostat, daily i.p 120 μl / 30 gm of body weight, n=15 mice/condition) starting 24 h after orthotopic implantation. Primary tumor growth measured with caliper, shown as mean ± SEM.
B. Kaplan-Meier plot of tumor-free survival of mice in A, as determined by the detection of palpable tumors.
C. Representative histopathological images of the resected primary tumors at necropsy of mice treated with EZH2i or vehicle stained with H&E. At 20x magnification, arrowhead shows skeletal muscle invasion in a vehicle treated tumor, and an area of treatment effect in an EZH2i treated tumor (asterisk) (scale bar, 500 μm). At 400x, vehicle treated tumors display a spindle cell morphology which contrasts with the polygonal epithelial cell shape of EZH2i-treated tumors (scale bar, 100 μm). Bars quantify the number of mitoses per 10 high power fields, mean ± SEM.
D. Representative images of orthotopic tumors immunostained for cytokeratin 18 (CK18), vimentin, and ZEB1. Bars: mean IHC score calculated using the percentage of positive cells and intensity of staining per 10 high power fields ± SEM. Scale bar, 100 μm.
E. Representative bioluminescence images of distally disseminated Ccn6-KO cells expressing GFP-Luciferase injected intracardially in FVB mice, treated with EZH2i or vehicle (n=10 mice/condition) starting 24 h after injection. Images were obtained at the indicated timepoints after treatment initiation. Bars quantify metastatic burden assessed by measuring photon flux at the indicated timepoints using Live Image Pro. Data are presented as means ± SEM. *P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001.
F. Working model of CCN6 tumor suppressor function in mBrCA.
Patients with mBrCA tumors have higher rates of metastatic relapse than non-metaplastic TNBC (2, 4). To specifically investigate if EZH2 methyltransferase activity inhibition might be useful in the metastatic setting in the absence of primary tumor, we injected Luciferase-labeled Ccn6-KO mBrCA cells intracardially in FVB mice followed by treatment with EZH2i or vehicle. EZH2i significantly reduced the metastatic burden compared to vehicle (Fig. 6E). Providing further support, EZH2 shRNA knockdown in Ccn6-KO mBrCA cells significantly reduced tumor growth and distant metastasis compared to shRNA control (Supplementary Fig. S10E–S10F). Together, these data show that inhibition of EZH2 catalytic activity or downregulation diminishes primary tumor growth and metastasis in an immune competent mouse model that recapitulates human spindle mBrCA. Our working model is shown in Fig. 6F.
Discussion
Relative to breast cancer overall and even other forms of TNBC, mBrCA has unique pathological as well as clinical features, including chemoresistance and frequent early recurrences. There is a critical need to improve our understanding of the molecular pathogenesis of mBrCA to design more effective therapies and improve patient outcomes.
Our recent proteogenomic landscape study of human mBrCA elucidated deregulated protein pathways in spindle mBrCAs including EMT, ribosomal, transcriptional, and translational processes (11). We found CCN6 functions as tumor suppressor protein in mBrCA and that MMTV-Cre;Ccn6fl/fl tumors share histopathology and proteogenomic features seen in human mBrCA tumors (11, 17). In this study, we sought to clarify the detailed mechanisms underlying the aggressive behavior of mBrCA. We showed CCN6 deficiency initiates a β-catenin/TCF-EZH2 signaling axis that regulates spindle mBrCA invasion and metastasis. We also demonstrated inhibition of EZH2 methyltransferase activity reduces mBrCA progression in vivo and may constitute a new therapeutic approach in these tumors.
Our studies demonstrate that in mesenchymal-like TNBC cells CCN6 interacts with Wnt receptor FZD8 and co-receptor LRP6 to antagonize the effects of canonical Wnt ligands on EMT and invasion. A rhCCN6 protein or ectopically expressed CCN6 secreted from transduced cells into the conditioned medium effectively antagonize Wnt ligand-mediated activation of β-catenin/TCF signaling, leading to a change from a mesenchymal-like to an epithelial phenotype as well as inhibition of the invasion program seen in the TNBC cells. These data are consistent with previous studies from our lab and others demonstrating CCN6, a matricellular protein, has major roles in the maintenance of the epithelial phenotype through regulation of growth factor signaling (18–20, 31). We have previously shown CCN6 antagonizes the pro-tumorigenic effects of IGF1 and BMP4 on the breast epithelium (18–20). A clue to the possible role of CCN6 in regulating β-catenin/TCF signaling was that zWisp3, the zebrafish CCN6 ortholog, had been reported to interact with LRP6 and Fzd8 to inhibit canonical Wnt signaling in the developing zebrafish (31). Also, morpholino-mediated inhibition of zWisp3 protein expression in developing zebrafish affected pharyngeal cartilage size and shape, and human CCN6 inhibited Wnt signaling in HEK293T cells (31). However, the apparent role of CCN6 on Wnt/β-catenin/TCF signaling had not been explored in cancer. Here, we provide evidence that CCN6 also has Wnt pathway inhibitory activity in breast cancer cells by binding to the cell surface coreceptors LRP6 and Fzd8. Furthermore, we show inhibition of β-catenin/TCF4 activity is required for CCN6 tumor suppressor function. While further studies are necessary, this biological activity of CCN6 may be useful to develop novel therapeutics against spindle mBrCA tumors.
CCN proteins have an evolutionary conserved protein structure consisting of several domains which share homology with other proteins including IGF-binding proteins (IGFBPs), von Willebrand factor (VWC), thrombospondin 1 (TSP1), and proteins that contain carboxyterminal cystine knots (CTs), such as Noggin, SOST, WISE, Cerberus, and members of the TGF-β superfamily (36–38). In our previous studies we found the TSP1 domain is essential for CCN6 invasion and stem cell-like regulatory functions (22). Here, we demonstrate that deletion of the TSP1 domain of CCN6 affected the interaction with Wnt coreceptors and Wnt inhibitory function, suggesting that development of a TSP1 peptide may be a potential strategy to inhibit Wnt pathway in mBrCA cells.
A characteristic pathological feature of mBrCA is aberrant tumor differentiation towards spindle, squamous, and mesenchymal elements (11, 39). This led us to hypothesize that EZH2, a major regulator of cell type identify through epigenetic transcriptional repression, may play a central role in their pathogenesis of these tumors. Our data demonstrate that EZH2 is a downstream effector of the CCN6-Wnt pathway in spindle mBrCA tumors. We identified that EZH2 induces H3K27me3 transcriptional repressor function of a set of 64 specific gene targets of the CCN6-Wnt pathway that mediate cell communication, adhesion, and division functions including CYP1B1, MEGF10, MAP3K1, NR3C2, and PDE4C. These data are supported by recent multi-omics studies, that have identified increased EZH2 methyltransferase activity on specific gene promoters in mesenchymal TNBC tumors (designated spindle mBrCA in pathology practice) (1).
Our studies suggest CCN6 functions to inhibit β-catenin-dependent Wnt signaling in a similar manner to that of the secreted protein Dickkopf-1 (DKK1), for which there are oncology candidates in preclinical and clinical development (40). Furthermore, PRC2 has been shown to repress DKK1 expression in multiple malignancies (41–43). Our RNA sequencing analyses of MMTV-Cre;Ccn6fl/fl tumors revealed significant downregulation of DKK1 compared to normal breast epithelium (17). These data suggest the hypothesis that in CCN6-deficient mBrCA, the upregulated Wnt/β-catenin/EZH2 axis may maintain low levels of DKK1 leading to enhanced Wnt activity, which warrants further detailed investigation. These observations also support the potential therapeutic utility of CCN6 in this setting.
Emerging data from our lab and other investigators show EZH2 has canonical as well as non-canonical functions that do not depend on EZH2 histone methyltransferase activity (23, 44–46). We have recently showed p38-mediated EZH2 phosphorylation at threonine 367 (T367) leads to EZH2 cytoplasmic accumulation and methylation of p38 as well as direct interaction with vinculin and other regulators of cell-cell adhesion (23, 45). Here, we show rhCCN6 reduces nuclear EZH2 and H3K27me3 expression in Ccn6-KO mBrCA tumors in vivo, supporting the notion that CCN6 regulates the nuclear function of EZH2. In CCN6-deficient spindle mBrCA, we found upregulated β-catenin/TCF controls EZH2 H3K27me3-dependent function primarily on genes that regulate mitosis, focal adhesion, and invasion processes. Together, these data illustrate the Ccn6-KO/β-catenin/TCF4 axis regulates EZH2 histone methyltransferase activity in spindle mBrCA.
An important result of our study is that inhibition of EZH2 expression and specifically EZH2 methyltransferase activity using a selective pharmacological inhibitor currently in the clinic, effectively reduced growth and dissemination of spindle mBrCA cells in an immune-competent mouse model. The anti-tumorigenic activity of EZH2 inhibitors has been previously shown in refractory B-cell non-Hodgkin’s lymphoma and epithelioid sarcomas, for which EPZ-6438/Tazemetostat has been approved for clinical use (35, 47). The data available in breast cancer so far has not been conclusive. Treatment with EPZ-6438 reduced the growth of 4T1 syngeneic xenografts and enhanced the efficacy of taxane chemotherapy (1). Using MDA-MB-231 cells we have shown that EPZ-6438 significantly reduces tumor growth and metastasis and noted a synergistic effect with p38 inhibitors(45). GSK-126, another EZH2 methyltransferase inhibitor significantly reduced spontaneous metastasis but no primary tumor growth of a mesenchymal and a basal-like 1 (BL-1) TNBC PDX models (48). The differences in outcomes among these models may be multifactorial including differences in the immunological state of the mice, the use of cells of different breast cancer subtypes and of different inhibitors. Recent data show that EZH2 regulates the breast cancer immune microenvironment (1) and also that immune activation is essential for the antitumor activity of EZH2 in urothelial carcinoma (49). These data may explain at least in part the significant reduction of primary tumor growth and distant metastasis observed in our immune competent syngeneic mouse model. Several studies including ours have demonstrated that EZH2 expression is higher in TNBC tumors, mainly in mBrCA (mesenchymal TNBC) and in BL-1 subtypes (26, 48, 50). In clinical samples we demonstrated that over half of spindle mBrCA display a CCN6Low/nuclear β-catenin/EZH2High phenotype, which is detectable using immunohistochemistry in paraffin embedded samples. Together, our findings support that EZH2 inhibition may be effective in reducing tumorigenicity of spindle mBrCA tumors and illustrate the relevance of mechanistically guided treatment strategies. In addition, detection of CCN6Low/nuclear β-catenin/EZH2High in mBrCA patients’ samples may identify tumors for which strategies aimed at CCN6 restoration or β-catenin and/or EZH2 signaling inhibition may have therapeutic benefit.
Supplementary Material
Significance Statement.
CCN6 deficiency drives metaplastic breast carcinoma growth and metastasis by increasing Wnt/β-catenin activation to upregulate EZH2, identifying EZH2 inhibition as a mechanistically guided treatment strategy for this deadly form of breast cancer.
Acknowledgments.
We are grateful to members of the Kleer laboratory for helpful discussion and suggestions during this project. We thank Prof. Bernard Perbal for helpful discussions. This work was supported by National institutes of Health (NIH) grants R01CA125577 and R01CA107469 (CGK), Department of Defense Breast Cancer Research Program grant W81XWH-19-1-0093 (CGK) and University of Michigan Rogel Cancer Center support grant P30CA046592. CGK is a Rogel Scholar. We acknowledge support from the Bioinformatics Core of the University of Michigan Medical School’s Biomedical Research Core Facilities (RRID:SCR_019168).
Footnotes
Conflict of Interest Statement: The authors declare no conflicts of interest.
References
- 1.Lehmann BD, Colaprico A, Silva TC, Chen J, An H, Ban Y, et al. Multi-omics analysis identifies therapeutic vulnerabilities in triple-negative breast cancer subtypes. Nat Commun. 2021;12(1):6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abouharb S, and Moulder S. Metaplastic breast cancer: clinical overview and molecular aberrations for potential targeted therapy. Curr Oncol Rep. 2015;17(3):431. [DOI] [PubMed] [Google Scholar]
- 3.Pang J, Toy KA, Griffith KA, Awuah B, Quayson S, Newman LA, et al. Invasive breast carcinomas in Ghana: high frequency of high grade, basal-like histology and high EZH2 expression. Breast Cancer Res Treat. 2012;135(1):59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.El Zein D, Hughes M, Kumar S, Peng X, Oyasiji T, Jabbour H, et al. Metaplastic Carcinoma of the Breast Is More Aggressive Than Triple-negative Breast Cancer: A Study From a Single Institution and Review of Literature. Clin Breast Cancer. 2017;17(5):382–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luini A, Aguilar M, Gatti G, Fasani R, Botteri E, Brito JA, et al. Metaplastic carcinoma of the breast, an unusual disease with worse prognosis: the experience of the European Institute of Oncology and review of the literature. Breast Cancer Res Treat. 2007;101(3):349–53. [DOI] [PubMed] [Google Scholar]
- 6.Jung SY, Kim HY, Nam BH, Min SY, Lee SJ, Park C, et al. Worse prognosis of metaplastic breast cancer patients than other patients with triple-negative breast cancer. Breast Cancer Res Treat. 2010;120(3):627–37. [DOI] [PubMed] [Google Scholar]
- 7.Wong W, Brogi E, Reis-Filho JS, Plitas G, Robson M, Norton L, et al. Poor response to neoadjuvant chemotherapy in metaplastic breast carcinoma. NPJ Breast Cancer. 2021;7(1):96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song Y, Liu X, Zhang G, Song H, Ren Y, He X, et al. Unique clinicopathological features of metaplastic breast carcinoma compared with invasive ductal carcinoma and poor prognostic indicators. World J Surg Oncol. 2013;11:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Toy KA, and Kleer CG. Metaplastic breast carcinomas are enriched in markers of tumor-initiating cells and epithelial to mesenchymal transition. Mod Pathol. 2012;25(2):178–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayes MJ, Thomas D, Emmons A, Giordano TJ, and Kleer CG. Genetic changes of Wnt pathway genes are common events in metaplastic carcinomas of the breast. Clin Cancer Res. 2008;14(13):4038–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Djomehri SI, Gonzalez ME, da Veiga Leprevost F, Tekula SR, Chang HY, White MJ, et al. Quantitative proteomic landscape of metaplastic breast carcinoma pathological subtypes and their relationship to triple-negative tumors. Nat Commun. 2020;11(1):1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng CKY, Piscuoglio S, Geyer FC, Burke KA, Pareja F, Eberle CA, et al. The Landscape of Somatic Genetic Alterations in Metaplastic Breast Carcinomas. Clin Cancer Res. 2017;23(14):3859–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perbal BaT M CCN proteins: A new family of cell growth and differentiation regulators. London: World Scientific Publishers; 2005. [Google Scholar]
- 14.Monsen VT, and Attramadal H. Structural insights into regulation of CCN protein activities and functions. J Cell Commun Signal. 2023;17(2):371–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yeger H, and Perbal B. The CCN axis in cancer development and progression. J Cell Commun Signal. 2021;15(4):491–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen CC, and Lau LF. Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol. 2009;41(4):771–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin EE, Huang W, Anwar T, Arellano-Garcia C, Burman B, Guan JL, et al. MMTV-cre;Ccn6 knockout mice develop tumors recapitulating human metaplastic breast carcinomas. Oncogene. 2017;36(16):2275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleer CG, Zhang Y, Pan Q, and Merajver SD. WISP3 (CCN6) is a secreted tumor-suppressor protein that modulates IGF signaling in inflammatory breast cancer. Neoplasia. 2004;6(2):179–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pal A, Huang W, Li X, Toy KA, Nikolovska-Coleska Z, and Kleer CG. CCN6 modulates BMP signaling via the Smad-independent TAK1/p38 pathway, acting to suppress metastasis of breast cancer. Cancer Res. 2012;72(18):4818–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lorenzatti G, Huang W, Pal A, Cabanillas AM, and Kleer CG. CCN6 (WISP3) decreases ZEB1-mediated EMT and invasion by attenuation of IGF-1 receptor signaling in breast cancer. J Cell Sci. 2011;124(Pt 10):1752–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang W, Zhang Y, Varambally S, Chinnaiyan AM, Banerjee M, Merajver SD, et al. Inhibition of CCN6 (Wnt-1-induced signaling protein 3) down-regulates E-cadherin in the breast epithelium through induction of snail and ZEB1. Am J Pathol. 2008;172(4):893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang W, Martin EE, Burman B, Gonzalez ME, and Kleer CG. The matricellular protein CCN6 (WISP3) decreases Notch1 and suppresses breast cancer initiating cells. Oncotarget. 2016;7(18):25180–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anwar T, Arellano Garcia C, Ropa J, Chen YC, Kim HS, Yoon E, Grigsby S, Basrur V, Nesvizhshkii AI, Muntean A, Gonzalez ME, Kidwell KM, Nikolovska-Coleska Z, Kleer CG. p38-mediated phosphorylation at T367 induces EZH2 cytoplasmic localization to promote breast cancer metastasis. Nat Commun. 2018;9:2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonzalez ME, Moore HM, Li X, Toy KA, Huang W, Sabel MS, et al. EZH2 expands breast stem cells through activation of NOTCH1 signaling. Proc Natl Acad Sci U S A. 2014;111(8):3098–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kolligs FT, Hu G, Dang CV, and Fearon ER. Neoplastic transformation of RK3E by mutant beta-catenin requires deregulation of Tcf/Lef transcription but not activation of c-myc expression. Mol Cell Biol. 1999;19(8):5696–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100(20):11606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fitzgibbons PL, Dillon DA, Alsabeh R, Berman MA, Hayes DF, Hicks DG, et al. Template for reporting results of biomarker testing of specimens from patients with carcinoma of the breast. Arch Pathol Lab Med. 2014;138(5):595–601. [DOI] [PubMed] [Google Scholar]
- 28.McMullen ER, Gonzalez ME, Skala SL, Tran M, Thomas D, Djomehri SI, et al. CCN6 regulates IGF2BP2 and HMGA2 signaling in metaplastic carcinomas of the breast. Breast Cancer Res Treat. 2018;172(3):577–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ewels P, Magnusson M, Lundin S, and Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32(19):3047–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Augimeri G, Gonzalez ME, Paoli A, Eido A, Choi Y, Burman B, et al. A hybrid breast cancer/mesenchymal stem cell population enhances chemoresistance and metastasis. JCI Insight. 2023;8(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakamura Y, Weidinger G, Liang JO, Aquilina-Beck A, Tamai K, Moon RT, et al. The CCN family member Wisp3, mutant in progressive pseudorheumatoid dysplasia, modulates BMP and Wnt signaling. J Clin Invest. 2007;117(10):3075–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reya T, and Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434(7035):843–50. [DOI] [PubMed] [Google Scholar]
- 33.Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298(5595):1039–43. [DOI] [PubMed] [Google Scholar]
- 34.Knutson SK, Warholic NM, Wigle TJ, Klaus CR, Allain CJ, Raimondi A, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci U S A. 2013;110(19):7922–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Italiano A, Soria JC, Toulmonde M, Michot JM, Lucchesi C, Varga A, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 2018;19(5):649–59. [DOI] [PubMed] [Google Scholar]
- 36.Perbal B CCN proteins: multifunctional signalling regulators. Lancet. 2004;363(9402):62–4. [DOI] [PubMed] [Google Scholar]
- 37.Brunkow ME, Gardner JC, Van Ness J, Paeper BW, Kovacevich BR, Proll S, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68(3):577–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Avsian-Kretchmer O, and Hsueh AJ. Comparative genomic analysis of the eight-membered ring cystine knot-containing bone morphogenetic protein antagonists. Mol Endocrinol. 2004;18(1):1–12. [DOI] [PubMed] [Google Scholar]
- 39.Oberman HA. Metaplastic carcinoma of the breast. A clinicopathologic study of 29 patients. Am J Surg Pathol. 1987;11(12):918–29. [DOI] [PubMed] [Google Scholar]
- 40.Kagey MH, and He X. Rationale for targeting the Wnt signalling modulator Dickkopf-1 for oncology. Br J Pharmacol. 2017;174(24):4637–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hussain M, Rao M, Humphries AE, Hong JA, Liu F, Yang M, et al. Tobacco smoke induces polycomb-mediated repression of Dickkopf-1 in lung cancer cells. Cancer Res. 2009;69(8):3570–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, et al. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40(5):499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen H, Hou G, Yang J, Chen W, Guo L, Mao Q, et al. SOX9-activated PXN-AS1 promotes the tumorigenesis of glioblastoma by EZH2-mediated methylation of DKK1. J Cell Mol Med. 2020;24(11):6070–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nie L, Wei Y, Zhang F, Hsu YH, Chan LC, Xia W, et al. CDK2-mediated site-specific phosphorylation of EZH2 drives and maintains triple-negative breast cancer. Nat Commun. 2019;10(1):5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gonzalez ME, Naimo GD, Anwar T, Paoli A, Tekula SR, Kim S, et al. EZH2 T367 phosphorylation activates p38 signaling through lysine methylation to promote breast cancer progression. iScience. 2022;25(8):104827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23(6):839–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gounder M, Schoffski P, Jones RL, Agulnik M, Cote GM, Villalobos VM, et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: an international, open-label, phase 2 basket study. Lancet Oncol. 2020;21(11):1423–32. [DOI] [PubMed] [Google Scholar]
- 48.Yomtoubian S, Lee SB, Verma A, Izzo F, Markowitz G, Choi H, et al. Inhibition of EZH2 Catalytic Activity Selectively Targets a Metastatic Subpopulation in Triple-Negative Breast Cancer. Cell Rep. 2020;30(3):755–70 e6. [DOI] [PubMed] [Google Scholar]
- 49.Piunti A, Meghani K, Yu Y, Robertson AG, Podojil JR, McLaughlin KA, et al. Immune activation is essential for the antitumor activity of EZH2 inhibition in urothelial carcinoma. Sci Adv. 2022;8(40):eabo8043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bachmann IM, Halvorsen OJ, Collett K, Stefansson IM, Straume O, Haukaas SA, et al. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J Clin Oncol. 2006;24(2):268–73. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All mass spectrometry proteomics data were deposited to the ProteomeXchange Consortium through the PRIDE partner repository with identifier PXD014414. The RNA-seq and ChIP-seq data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE250575 and GSE250574, respectively. Other data generated in the study are available upon request to the corresponding author.