

Abstract
PURPOSE:
Early intervention for High-Risk Smoldering Multiple Myeloma (HR-SMM) achieves deep and prolonged responses. It is unclear if beneficial outcomes are due to treatment of less complex, susceptible disease or inaccuracy in clinical definition of cases entered.
EXPERIMENTAL DESIGN:
Here, we interrogated whole genome and whole exome sequencing for 54 patients across two HR-SMM interventional studies (NCT01572480, NCT02279394).
RESULTS:
We reveal that the genomic landscape of treated HR-SMM is generally simple as compared to Newly Diagnosed (ND)MM counterparts with less inactivation of tumor suppressor genes, RAS pathway mutations, MYC disruption, and APOBEC contribution. The absence of these events parallels that of indolent precursor conditions, possibly explaining overall excellent outcomes. However, some patients harboring genomic complexity fail to sustain response and experience resistant, progressive disease. Overall, clinical risk scores do not effectively discriminate between genomically indolent and aggressive disease.
CONCLUSIONS:
Genomic profiling can contextualize the advantage of early intervention in SMM and guide personalization of therapy.
Keywords: whole genome sequencing, mutational signatures, smoldering multiple myeloma, genomics
INTRODUCTION
Multiple Myeloma (MM) is a plasma cell neoplasm that progresses through precursor states before evolution to symptomatic cancer1–6. On the spectrum of precursor conditions is smoldering MM (SMM); a clinically-defined state of asymptomatic expansion of clonal plasma cells for which approximately 60% of affected individuals will experience progression to overt disease within 10 years of diagnosis1,3. Various risk models have been developed to stratify the risk of progression and are thus far based on surrogate clinical markers and indirect measures of disease burden6–9. It has been hypothesized that by intervening before evolution to clinically significant disease (i.e., MM), therapeutic outcomes might be improved by preventing organ damage and by treatment of a less expanded, less complex disease state. Based on these principles, multiple investigations have been developed for treatment of individuals with SMM at high risk (HR-SMM) of progression to MM 6,10–13. In the era of modern drugs, early intervention for HR-SMM has been seen to result in deeper and more prolonged treatment responses compared to newly diagnosed MM (NDMM). However, it is unknown if this benefit is due to treatment of a less complex, susceptible entity in a fit individual or inaccuracy and incongruity with and between differing risk models resulting in trial inclusion and subsequent treatment of more biologically indolent disease14.
The introduction of next generation sequencing (NGS) technologies has promoted a gradual shift from the use of clinical surrogate markers of disease burden to genomic determinants of disease biology5,15–17. Whole genome sequencing (WGS) and whole exome sequencing (WES) have expanded upon and delineated several myeloma-defining genomic events18–22. These events include presence of complex structural variants (SV), APOBEC mutational signatures, mutations involving the MAPK and NFkB pathways, loss of tumor suppressor genes, and events involving MYC17. However, the genomic landscape of clinical HR-SMM that has been subjected to early therapeutic intervention has not been comprehensively explored.
To contextualize the outcomes of contemporary SMM treatment trials, we performed the first WGS-based analysis of patients with HR-SMM treated on an interventional study, using samples and data from a cohort of HR-SMM patients treated with carfilzomib, lenalidomide, and dexamethasone and lenalidomide maintenance (KRd/R) as part of a phase II clinical trial with long-term clinical follow-up10. Furthermore, in our analysis we included information from an additional HR-SMM phase II interventional trial (E-PRISM) and we evaluated genomic lesions and clinical outcomes across the two studies, and compared against those of NDMM. Overall, likely driven by a high level of discordance among different SMM prognostic models used for trial inclusion, a common genomic simplicity was observed across both cohorts, potentially providing a biologic rationale for the more favorable responses observed on these HR-SMM interventional trials compared to NDMM. Conversely, patients harboring complex genomic profiles and distinct known MM genomic drivers generally failed to achieve sustained negativity for minimal residual disease (MRD) and experienced progression of disease, suggesting that early triplet-therapy intervention does not completely overcome the adverse impact of these features on disease evolution and clinical outcomes.
METHODS
Samples and data were obtained and managed in accordance with the Declaration of Helsinki. All patients reported here provided written informed consent for each of the respective studies on which they were enrolled and investigations on human samples were approved on the respective institutional review boards.
All patients had protocolized testing for MRD yearly. We considered any patient achieving MRD-negativity on trial as having MRD-negativity as a best response. We considered sustained MRD-negativity as at least two consecutive MRD-negative measurements at least 12 months apart. For the purposes of our analysis, for a patient to be considered to have sustained MRD-negativity, their MRD measurement at the most recent follow-up visit must have remained negative. Conversely, patients with a conversion from MRD-negativity to MRD-positivity and patients that never achieved MRD-negativity were considered together as patients with failure to sustain MRD-negativity. Finally, we considered both biochemical progression and progression to overt MM requiring treatment as progression of disease.
Fisher’s Exact Test was used for categorical comparisons and the Wilcoxon Ranked Sum test was used for continuous differences between groups. False discovery rate corrections were not employed given our inclusion of only previously established drivers (i.e., driver discovery was not performed). P-values <0.05 were considered statistically significant. Survival data were analyzed and visualized with Kaplan-Meier methods.
Sample Selection from KRD/R (non-randomized; non-blinded; NCT01572480)
27 of the original 54 patients had baseline samples available and amenable for WGS and so were included in this analysis. There was no significant difference (Fisher p>0.05) between the WGS cohort and the trial cohort, at large, in: sex (48% vs 44% female); age (33% vs 28% age 65 or greater); MRD-negativity (70.3% vs 70.4%); progression events [4 biochemical (14.8%) and 1 progression to MM (3.7%) vs 6 biochemical (11.1%) and 2 progressions to (3.7%) MM]; and in high-risk cytogenetics by FISH [del17p, t(4;14), t(14;16), t(14:20), and 1q gain; 40.7% vs 37%]. Inclusion and Exclusion Criteria for the protocol have been previously published10.
E-PRISM Sample Selection
The trial accrued 51 patients. As previously reported, 37 samples at WXS performed. Six samples had tumor purity <15% and were excluded. We were able to retrieve data on 28 of these cases. As per the Methods below, 1 was excluded in our analysis for purity concerns leaving us with the final dataset of 27 exomes from E-PRISM. No known bias explains the original selection of samples for exome sequencing. Samples were prepared and processed as described in the primary publication13.
Sample Preparation
For patients on NCT01572480, available samples from pre-treatment bone marrow were thawed and CD138+ microbeads were used to sort plasma cells from frozen bone marrow mononuclear cells. Normal match was selected for each patient from peripheral blood mononuclear cells.
Sequencing and Analytical Methods
Sequencing was performed at New York Genome Center. Following quantification via PicoGreen and quality control by Agilent Bioanalyzer, ~500 ng of genomic DNA was sheared (LE220-plus Focused-ultrasonicator; Covaris, catalog no., 500569) and sequencing libraries were prepared using a modified KAPA Hyper Prep Kit (Kapa Biosystems, KK8504). Briefly, libraries were subjected to a 0.5 × size select using aMPure XP Beads (Beckman Coulter, catalog no., A63882) after post-ligation cleanup. Libraries that were not amplified by PCR (07652_C) were pooled equivolume. Libraries amplified with five cycles of PCR (07652_D, 07652_F, and 07652_G) were pooled equimolar. Samples were run on a NovaSeq 6000 in a 150 bp/150 bp paired-end run, using the NovaSeq 6000 SBS v1 kit and an S4 Flow Cell (Illumina), as described previously23. Target coverage depth was 80x for tumor and 40x for normal.
Whole-genome analysis pipeline.
Coverage for tumor and normal samples are reported in Supplemental Table S1. Short insert paired-end reads were aligned to the reference genome (GRCh38) using the Burrows–Wheeler Aligner (v0.5.9; ref. 17). All samples were uniformly analyzed by the following bioinformatic tools: somatic mutations were identified by MuTect2, VarScan2, and Strelka24–26; indels were identified by VarScan2, MuTect2, SvABA, and Strelka24–27; copy number analysis and tumor purity (i.e., cancer cell fraction) were evaluated using ASCAT28; IgCaller was used to identify translocations at the immunoglobulin loci29; structural variants were defined by Manta, DELLY, and SvABA27,30,31 and passed through additional quality filters, and were manually curated to define complex events (i.e., templated insertions, chromothripsis, and chromoplexy) as described previously32. Chromothripsis was called from WXS data, additionally, using methods described by Maclachlan et al33. SV hotspots were identified and called using previously described methods20. An SV hotspot was considered to be involved by structural variation in a given sample if either an SV breakpoint fell within the hotspot or withing 1MB of its start or end. We also considered effect and impact. For example: a deleterious SV within an amplification hotspot would not be considered.
The exomes data downloaded from public repositories were aligned to the reference human genome (GRCh37) using Burrows-Wheeler Aligner, BWA (v0.7.17; RRID:SCR_010910). Deduplicated aligned BAM files were analyzed using FACETS (v0.5.6, https://github.com/mskcc/facets) for copy number variants, and otherwise subjected to the same pipeline as for genomes above – with the exception of structural variant calling.
Sample Quality Control and Purity Estimation
As quality control, we did exclude samples with low purity considered to be sequencing failures, which included 3 cases from E-PRISM. We performed this quality control by estimating sample purity by orthogonal methods: 1) sample purity was generated by ASCAT estimation; 2) purity was validated by multiplying by two the median [clonal] variant allele frequency from diploid regions. The purity reported in Supplemental Tables 1 and 2 is a composite (i.e., average of the first 2 criteria). Samples with low purity, no copy number changes, low mutational burden at WGS level (< 500) and no myeloma-specific lesions (as below in Copy Number Aberrations and Driver Mutations) were excluded. Of the original 28 WXS samples available from EPRISM, 1 was excluded on this basis.
Mutational Signatures.
Mutational signatures were analyzed across all whole genomes. To estimate the activity of mutational signatures, we first employed a three step process of de novo extraction, assignment, and fitting34. For the first step, we ran SigProfiler for SBS signatures35. All extracted signatures were then compared with the latest Catalogue of Somatic Mutations in Cancer (COSMIC) reference (https://cancer.sanger.ac.uk/cosmic/signatures/SBS; RRID:SCR_002260) to identify the known mutational processes active in the cohort. For SBS, we applied mmsig (https://github.com/UM-Myeloma-Genomics/mmsig)36, a fitting algorithm, to confirm the presence and estimate the contribution of each mutational signature in each sample guided by the catalog of signatures extracted for each individual sample by SigProfiler’s de novo refit.
For unbiased comparisons of mutational signatures, HR-SMM WGS were compared to WGS from patients with NDMM treated with KRd +/− Dara37. SBS2 and SBS13 contribution were summed across WGS samples and hyper-APOBEC samples were defined as those with expression above the 10th decile. The same analysis for HR-SMM WXS and CoMMpass WXS data was performed.
Copy Number Aberrations and Driver Mutations
Given the size of the SMM cohort, GISTIC peak discovery was not performed. Instead, reference GISTIC peaks previously identified as significant in MM were used as reference20. The analysis was executed using Gene Pattern web interface (http://genepattern.broadinstitute.org; RRID:SCR_000151).
We used the dN/dScv package to annotate genes in our cohort38. Given the size of the SMM cohort, driver discovery was not performed. Instead, a reference driver gene list was used to designate driver status to identified variants39. Similarly, SV hotspots, recurrent CNV and GISTIC peaks were referenced from prior work20,39.
We then developed a locus-based classification scheme, combining all of focal and large CNA, GISTIC peaks, and SNV to detect dysregulation at driver loci39. These methods allow for identification of common biological processes affected by various genomic aberrations (https://github.com/UM-Myeloma-Genomics/GCP_MM). CNA were used as a proxy for SV given the combination of both WGS and WXS in this study. Sensitivity analysis was performed with removal of 2 HR-SMM cases that would be reclassified as MM per IMWG 2014 diagnostic criteria40. Overall and progression-free survival annotations were pulled from CoMMpass clinical annotations39.
To transcend constraints of clinically applied disease definitions, clinical annotations were removed, and cases were clustered together (701 NDMM and 54 HR-SMM) to determine the relationship of genomic lesions to underlying disease state. We used the R Package, pheatmap, to perform hierarchical clustering to report on patterns of co-occurrence (https://github.com/raivokolde/pheatmap).
Data Availability
The dataset used for this paper is derived from public and newly sequenced sources: 27 SMM exomes were imported from dbGaP: phs001323.v3.p1. 701 MM exomes (from patients with RNA-seq and low coverage WGS data) were imported from the CoMMpass trial; IA 13 (dbGap: phs001323.v3.p1, https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001323.v3.p1). Data from the prior KRd +/− Dara NDMM patients along with 27 newly sequenced HR-SMM samples are available on EGA: EGAS00001007404.
Code Availability
Analyses conducted using previously published code are detailed and available via github links her and in the Methods. Facets for Copy Number Calling: https://github.com/mskcc/facets. Mmsig for mutational signatures fitting: https://github.com/UM-Myeloma-Genomics/mmsig. GISTIC peak algorithm: http://genepattern.broadinstitute.org.
RESULTS
To gain biologic insight into treatment outcomes, we performed WGS of available baseline samples for 27 of 54 treated patients with HR-SMM (Methods). Patients received 8 cycles (32 weeks) of carfilzomib 20/36 mg/m2 on days 1, 2, 8, 9, and 15; lenalidomide 25mg days 1-21; and dexamethasone 20mg twice weekly for cycles 1-4 and 10mg for cycles 5-8; followed by 2 years of maintenance lenalidomide (KRd/R; NCT01572480)10. Clinical records were updated from prior publication and reviewed to correlate trial outcomes with genomic features. We pooled genomic features with whole exome sequencing data (WXS) from a second cohort of 27 patients with HR-SMM treated with Elotuzumab-R+/−d (Elo-Rd; E-PRISM; NCT02279394; Methods, Supplemental Methods)13 and compared to 701 patients with NDMM from CoMMpass (NCT01454297) with available WGS and WXS. Additional comparators included WGS from 60 patients with NDMM treated with daratumumab(dara)-KRd (NCT03290950) and KRd (NCT02937571)37,41. Comparisons are summarized in Supplemental Figure S1.
Clinical outcomes for patients with HR-SMM treated with KRd/R
Clinical features for the KRd/R cohort are listed in Supplemental Table S1. After a median follow-up of 52.8 months, median PFS was unreached for patients treated with KRd/R (Figure 1a–b). After 8 cycles of KRd, 19 (70.3%) achieved MRD-negativity (Flow-MRD; LOD 10−5, Figure 1a). At time of data cutoff, June 13, 2023, 14 patients (51.9%) achieved sustained MRD-negativity at last follow up, defined as at least two consecutive MRD-negative assessments measured 1 year apart and MRD-negative at last follow-up. Six patients (22.2%) lost an initial MRD-negative response and 5 patients experienced progression of disease [18.5%; 4 biochemical and 1 progression to MM (NIH018A; Supplemental Table S1, Methods)].
Figure 1: Clinical summary of the KRd/R cohort.
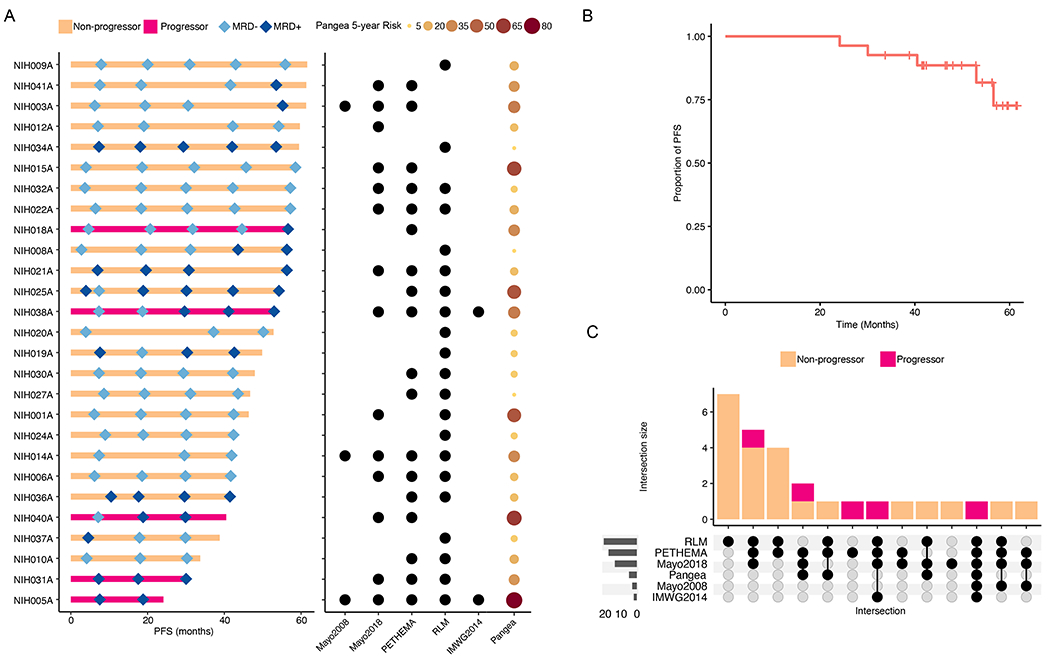
A. Swimmer plot of progression-free survival (PFS) annotated with serial MRD measurements (left panel) and pre-treatment high-risk status used as enrolment criteria by relevant clinical risk scores (right panel). Mayo2018 = 20/2/20 criteria. B. PFS curve for the KRD/R cohort generated with Kaplan-Meier methods. C. Upset plot demonstrating incongruity between clinical risk scores as they pertain to outcomes after treatment with KRD/R.
Accrual on NCT01572480 began in 2012 and, as such, 2 patients would today be reclassified as MM per 2014 IMWG criteria on the basis of bone marrow plasma cells ≥60%40. Both of these patients (NIH005A and NIH038A) experienced biochemical disease progression on study. Otherwise, 3 (11.1%) were HR by Mayo2008 criteria, 14 (51.9%) retrospectively by Mayo 20/2/20, 18 (66.7%) by PETHEMA, and 21 (77.8%) by Rajkumar/Landgren/Mateos criteria6–9. 18 (66.7%) met criteria by 2 or more scores (Figure 1c) and the median 5-year risk of progression per the Pangea model was 18.6% (range 4.8-82.1)42. Overall, though all patients who were treated on trial were considered high-risk, there is clear discordance between competing clinical risk models14. Though the models are designed for prediction of spontaneous progression to MM, we see here that no one risk model has high-risk status associated with progression after triplet intervention (p>0.05). E-PRISM sample and clinical details are listed in Supplemental Table S2.
The genomic landscape of treated HR-SMM
To contextualize the overall favorable outcomes of treated HR-SMM, we sought to qualify genomic features across studies by pooling WGS data with available WXS from the E-PRISM interventional study13. Genomic data from treated HR-SMM were then compared to 701 patients with NDMM from the CoMMpass study. For the purposes of this comparison, we removed the two cases that would today be considered as MM. As an initial check for bias towards less aggressive disease, there were no differences in the frequency of high-risk FISH-based cytogenetic lesions [(t(4;14), t(14;16), del(17p)] between treated HR-SMM and NDMM, aside from higher frequency of gain of 1q in NDMM (p = 0.045).
Within the KRd/R HR-SMM WGS cohort, the median mutational burden was 4134 (range, 2275-8346), which was significantly lower compared to WGS from NDMM treated with KRd +/− dara (median, 5179; range, 1157-14471, p = 0.033). To investigate differences in mutational processes involved in HR-SMM and NDMM we profiled the mutational signatures landscape of the KRd/R cohort (Figure 2a, Supplemental Table S3), NDMM treated with KRd +/− dara (Supplemental Figure S2), and WXS from the E-PRISM cohort (Figure 2b). Overall HR-SMM contained the same mutational signatures detected in NDMM and in prior studies of myeloma precursor disease (Methods) 17,21. However, HR-SMM patients enrolled in both the KRd/R and the E-PRISM studies had a significantly higher proportion of patients without evidence of APOBEC (SBS2+SBS13) mutagenesis compared to NDMM [WGS 44% vs 86.7%, p < 0.001; WXS 14.8% vs 45.2% (CoMMpass), p < 0.001; Figure 2C–D]. Similarly, cases of hyper-APOBEC mutagenesis (SBS2+SBS13>11%; Methods), which has been associated with poor prognosis in MM,43 were under-represented in these treated HR-SMM (1.9% vs 11.3%; p = 0.033). Overall, these findings are consistent with prior observations that APOBEC activity is lower in SMM and increases across the continuum of progression to MM17,43. Importantly, however, APOBEC activity has been identified in over 82–85% of myeloma precursors destined for progression and its relative absence here is suggestive that despite clinical high-risk status, many of these cases enrolled for therapeutic intervention may have represented more indolent conditions17. Sequencing further confirmed no difference in frequency between MAF- and MAFB- translocated cases between NDMM and HR-SMM cohorts (Supplemental Table S3).
Figure 2: Mutational Signatures Landscape of treated HR-SMM.

A. Mutational signatures contribution for each case treated with KRD/R. Asterisks Colored sample-names denote MAF(B)-translocated cases. There were no MAF translocations in the E-PRISM cohort. SBS: Single Base Substitution. B. Mutational signatures contribution for each case treated on E-PRISM. C. Comparison of APOBEC (SBS2 + SBS13) contribution between NDMM treated with (dara)-KRd vs. HR-SMM treated with KRd/R. D. Comparison of APOBEC contribution between NDMM from CoMMpass (i.e., WXS calls) vs. HR-SMM treated on E-PRISM.
We next interrogated the landscape of driver events across the cohorts using a catalog of 91 known genes involved by mutations and focal copy number aberrations derived from de novo driver discovery across 1933 NDMM39. We then combined mutations in driver genes with copy number aberrations (CNA; Supplemental Figure S3a–b), including known GISTIC hotspots20,39, to define inactivation of tumor suppressor genes (Supplemental Figure S4, Methods). Consistent with their later role in MM evolution16,44, a large number of tumor suppressor genes were less frequently inactivated, including CDKN2C, CYLD, TENT5C, FUBP1, NCOR1, NF1, NFKBIA, RB1, RPL5, and TRAF3 (p < 0.05; Supplemental Table S4). Restricting scope only to biallelic inactivations of tumor suppressor genes, TRAF3 was more frequently inactivated in MM (69/701 vs 0/52; p = 0.05) and importantly, the only HR-SMM case was one (NIH038A) that would be reclassified as MM with current IMWG criteria. This finding reinforces the role of the NF-ΚB pathway as a late driver in the pathogenesis of MM45. Consistent with their later role in the genomic evolution from SMM to MM, mutations in RAS pathway genes (NRAS, KRAS, BRAF; p = 0.011) and amplifications at 8q24 (i.e., MYC; p < 0.001) were more frequent in MM17,46 . Finally, a sensitivity analysis was performed by adding back both cases that would today be reclassified as MM per IMWG 2014 criteria40, with little change in observations (Supplemental Table S4).
For the available full coverage WGS samples (KRd/R HR-SMM and KRd +/− dara NDMM) we compared the landscape of structural variants (SV). There were no significant differences in the frequency of involvement of SV hotspots between HR-SMM and NDMM (Supplemental Table S5). Though there was not a significant difference in the frequency of chromothripsis events in the KRD/R genomes, alone, using CN signatures to estimate the presence of chromothripsis from the E-PRISM WXS, there was a lower frequency of chromothripsis events in pooled HR-SMM (p = 0.028), consistent with the SV’s association with complex genomes and poor outcomes (Methods, Supplemental Figure S5 A–B)20,33. We additionally performed a sensitivity analysis with removal of the 7 patients who met inclusion criteria only by RLM features and saw little change in results (Supplemental Table S4).
Overall, we see that across cohorts of HR-SMM, genomic features associated with NDMM (i.e., Myeloma-defining genomic events) are significantly underrepresented. As clinical HR-SMM should theoretically be at risk for imminent progression to MM (i.e., 2-5 years), most genomic drivers should already have been acquired on its course to overt disease17,19,46. This suggests that clinical risk scores, based on surrogate markers of disease burden, have limitations in accurately recapitulating the underlying disease biology that contributes to disease aggression and imminent progression and that their use in trial settings may lead to inadvertent capture and treatment of indolent disease states.
Genomic features associated with outcomes for HR-SMM treated with triplet therapy
We next sought to investigate the genomic lesions associated with clinical outcomes following trial intervention with potent triplet therapy. Sustained MRD-negativity and disease progression (either biochemical or clinical) during intervention with KRd/R were the key endpoints. Currently, there is not a definitive endpoint established for interventional studies targeting HR-SMM. Biochemical/clinical progression (indicator of disease aggression and treatment resistance) was an obvious choice of endpoints. Given the evolving landscape of HR-SMM intervention towards curative strategies47,48, we also reasoned that sustained MRD-negativity could be a proxy for prolonged and deep suppression of disease and included it as appropriate for analysis. Deletions of chromosome 13q14.2 (RB1) and inactivation of MAX and HIST1H2BK (p < 0.05) were each associated with disease progression, as was any presence of APOBEC mutagenesis (p = 0.016; Supplemental Table S6). Examining SV, chromothripsis, known to be associated with poor outcomes in MM, was likewise seen here to be a risk factor for disease progression (p = 0.002) as were SV involving TENT5C (p=0.028; Methods, Supplemental Table S5)20. In fact, progressors were more likely to have a higher number of involved SV hotspots, signifying the role of co-occurrence of multiple disease drivers (i.e., genomic complexity) as a main contributor to therapy resistance and continued clinical disease evolution (Supplemental Figures 5A–C).
We next examined genomic features within the context of clearance of MRD following treatment with KRd/R. No genomic features emerged as significantly associated with achievement of MRD-negativity following 8 cycles of KRd (primary endpoint). On the KRd/R study, patients had protocolized yearly MRD testing and we observed that gain of 1q, inactivation of MAX, and t(4:14) were each associated with failure to sustain MRD-negativity at last follow-up (Supplemental Table S6). We likewise performed sensitivity analyses with removal of the cases that would today be considered as MM per SLiM-CRAB criteria with little change in results (Supplemental Table S6).
As HR-SMM is a clinical definition and given the wide heterogeneity and relatively simplicity of the genomic landscape of most of the HR-SMM enrolled in KRd/R and E-PRISM, we sought to determine what genomic lesions shared with NDMM were more closely representative of biologically progressive, clonally mature disease. In an orthogonal approach, we removed clinical annotations and performed hierarchical clustering of genomic features from all 701 NDMM from CoMMpass and all 54 HR-SMM (Supplemental Figure S6, Methods). In parallel to the prior clinical comparisons, APOBEC activity, gain1q, and loss of tumor suppressor genes all clustered together and often co-occurred as features of genomic complexity that predict poor outcomes in terms of PFS and sustained MRD-negativity. Furthermore, the majority of cases enrolled and treated in the KRd/R HR-SMM clustered within the most indolent and genomically uncomplicated group of NDMM.
Finally, a summary of all significant genomic features was compiled and considered (Figure 3A). Altogether a catalogue of lesions underlying de-regulation of MYC and the NF-ΚB pathway, genomic instability in the form of APOBEC activity and chromothripsis, and t(4;14) emerged as risk factors for resistance to early intervention with triplet therapies and eventual disease evolution (Supplemental Table S7)46. Co-occurrence of many of these lesions, in parallel (i.e., genomic complexity), was a common theme amongst those with progressive disease. We finally assessed the effect on KRd/R outcome, considering any one or more of the [high-risk] features associated with treatment outcomes (Supplemental Table S6) and observe that co-occurrence of any 2 features in the KRD/R dataset was associated with progression despite intervention with potent triplet therapy (p = 0.034; Figure 3b, Supplemental Figure S7). We also examined outcomes within the EPRISM study and similarly found that genomic complexity was associated with clinical progression to MM, but not biochemical progression (Supplemental Figure S8A and 8B). The latter may be due to the different interventional strategies between the studies.
Figure 3: Genomic lesions associated with clinical outcomes in HR-SMM.

A. Heatmap of genomic features found at significantly different frequency in comparison between NDMM and HR-SMM and within the KRd/R cohort associated with disease progression or failure to sustain MRD-negativity. B. PFS curves for the KRd/R-treated cohort stratified by presence of features associated with PFS or failure to sustain MRD-negativity.
DISCUSSION
With a growing armamentarium of active, tolerable therapies, there is increasing interest in the early treatment of patients with myeloma precursor disease. However, there is incongruity in currently available clinical risk stratification models for progression from SMM to MM14. These discrepancies can result in the inclusion of patients who are not at immediate risk of progression and/or those with overall biological low-risk, indolent precursors. Inclusion of relatively less aggressive disease states may lead to deeper observed responses during early intervention, potentially impacting the interpretation of SMM clinical trials. For instance, a sustained MRD-negativity rate of 70% with KRD in absence of autologous stem cell transplantation is higher than what is typically observed in NDMM patients treated with a similar approach49,50. It is unclear whether this heightened response is solely due to the benefits of early intervention where the disease is potentially more susceptible to protocol therapy or if it is influenced by the fact that the patient population being treated is inherently less aggressive/complex than NDMM counterparts, or both. On the other hand, improvement in outcomes in a randomized setting could be due to inclusion of patients with genomic features already consistent with MM (myeloma-defining genomic lesions), where anti-MM therapy would be beneficial compared to surveillance12.
In this study, to investigate these critical questions, we used WGS and WXS to characterize the genomic landscape of HR-SMM in two parallel phase II interventional trials. We found that compared to NDMM, patients included in these studies had a lower frequency of established myeloma-defining genomic lesions; particularly those associated with poorer clinical outcomes and genomic complexity including a lower frequency and contribution of APOBEC-induced mutagenesis, and fewer disruptions of the MYC locus. Notably, these features are generally considered to be later events in the clonal evolution of disease progression to MM, but they are also two highly prevalent and powerful prognostic marker for predicting SMM progression16–18,46. Though it could be argued that it would be expected for HR-SMM to be less genomically complex than MM, recent WGS studies have revealed that these late lesions are still generally acquired many years prior to clinically active disease and generally before the 2- to 5-year time-to-progression window expected for patients classified clinically as high-risk6–9,18,19,46. Though the relative absence of these complex genomic features in the clinically defined HR-SMM cohorts does not imply that these patients would not have had early spontaneous progression into MM in the absence of study intervention, it does suggest that, at least at time of enrolment, they had genomically more indolent disease, or they were at lower risk of imminent progression in contrast to what is implied by their high-risk clinical risk scores, or a combination of both. In fact, patients on the KRd/R study who obtained deep and sustained responses (i.e. sustained MRD-negativity appear genomically similar to patients with non-progressive monoclonal gammopathy and SMM under observation (i.e., stable myeloma precursor conditions) or to a small group of MM with very favorable outcomes (Supplemental Figure S6)17,51.
Aside from the overall genomic simplicity observed in treated clinically defined HR-SMM patients, there is a set of genomic features that portends sub-optimal outcome despite study intervention. Combinations of APOBEC-induced mutagenesis, deregulation of MYC and the NF-ΚB pathway, and chromothripsis are associated with disease persistence (i.e., failure to achieve or sustain MRD-negativity), and progression from SMM to MM despite effective triplet therapy, as paralleled in treatment of NDMM. Whether these SMM patients with high genomic complexity would have had worse outcome without early intervention or whether early intervention changed the course of their disease are unanswered questions. Possibly, these lesions are representative of disease biology not susceptible to currently available interventional therapies provided on these clinical trials. For example, it has recently been seen in another study that for patients with clinically defined high-risk SMM, even intensive early treatment with KRd followed by autologous stem cell transplantation and maintenance therapy, only half are able to achieve MRD-negativity48. It seems reasonable to conjecture that myeloma precursor disease with genomically complex, high-risk disease biology (i.e., myeloma-to-be), may be better served by alternate diagnostic and novel therapeutic strategies.
We do acknowledge that the risk scores used as inclusion criteria in these studies are dated, but note that there continues to be vast heterogeneity in the inclusion criteria of past and present interventional studies. Prior seminal studies and currently accruing trials do not enroll patients on uniform clinical risk criteria11,12,47, such that the comprehensive genomic and clinical annotation here provides valuable insights into biologic determinants of therapeutic outcomes in a disease area of continuous clinical revision. Further, we acknowledge the limitation of a lack of WGS at time of progression as a comparator, the general lower purity of SMM vs NDMM samples, and the availability of only single-site sampling which may under-represent spatial heterogeneity of high-risk subclones52. Future translational studies are needed to fully address these questions. However, given the elective nature of treatment of myeloma precursor conditions, our results support the use of genomic profiling to contextualize advantages of early intervention and to better identify patients who are both likely to progress in the absence of early intervention and likely to require alternative therapeutic approaches to achieve optimal outcomes.
Supplementary Material
Statement of Translational Relevance:
Combining next generation sequencing across two interventional studies for high-risk smoldering multiple myeloma, we demonstrate that use of clinical risk scores results in inclusion of genomically indolent disease states that may influence trial outcomes. For the few with poor outcomes, genomic high-risk features predominate calling for alternate early interventional strategies.
ACKNOWLEDGEMENTS
This work was supported by the Paula and Rodger Riney Multiple Myeloma Research Program Fund, the Tow Foundation, the intramural NCI program, and the Biospecimen Shared Resource (SCR022889) under the Sylvester Comprehensive Cancer Center NCI Core Grant (P30 CA 240139). B.D. is supported by the Sylvester K12 Calabresi Clinical Oncology Research Career Development Program and the American Cancer Society. FM is supported by International Myeloma Society, NIH, and American Society of Hematology.
CONFLICT OF INTEREST
B.D. has received honoraria from advisory boards from Sanofi and Janssen and Independent Data Review Committee for Janssen.
D.K. acknowledges research funding from: NCI/NIH, FDA, MMRF, DoD-PROMETHEUS (Murtha Cancer Center Research Program), Amgen, Celgene, Janssen, Karyopharm; has received honoraria for advisory boards and presentations for: Alphasights, Aptitude Health, Arcellx, BMS, Bridger Consulting Group, Curio Science, Karyopharm, MJH Life Sciences, MMRF, Plexus Communications, Sanofi; and served on Independent Data Monitoring Committees (IDMC) for: Aperture Medical Technology, Arcellx.
O.L. acknowledges research funding from: NCI/NIH, FDA, LLS, Rising Tide Foundation, Memorial Sloan Kettering Cancer Center, MMRF, IMF, Paula and Rodger Riney Foundation, Myeloma Solutions Fund, Perelman Family Foundation, Amgen, Celgene, Janssen, Takeda, Glenmark, Seattle Genetics, Karyopharm; has received honoraria for scientific talks/participated in advisory boards for: Adaptive, Amgen, Binding Site, BMS, Celgene, Cellectis, Glenmark, Janssen, Juno, Pfizer; and served on Independent Data Monitoring Committees (IDMC) for international randomized trials by: Takeda, Merck, Janssen, Theradex.
S.U. acknowledges research funding from Amgen, Array Biopharma, BMS, Celgene, GSK, Janssen, Merck, Pharmacyclics, Sanofi, Seattle Genetics, SkylineDX, Takeda; honoraria/consulting from Abbvie, Amgen, BMS, Celgene, EdoPharma, Genentech, Gilead, GSK, Janssen, Oncopeptides, Sanofi, Seattle Genetics, SecuraBio, SkylineDX, Takeda, TeneoBio; grant support from NCI, LLS, MMRF.
None of the other Authors have conflict of interest to disclose.
REFERENCES
- 1.Kyle RA, Larson DR, Therneau TM, Dispenzieri A, Kumar S, Cerhan JR, et al. Long-term follow-up of monoclonal gammopathy of undetermined significance. New England journal of medicine. 2018;378(3):241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kyle RA, Remstein ED, Therneau TM, Dispenzieri A, Kurtin PJ, Hodnefield JM, et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. New England Journal of Medicine. 2007;356(25):2582–2590. [DOI] [PubMed] [Google Scholar]
- 3.Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, Offord JR, et al. Prevalence of monoclonal gammopathy of undetermined significance. New England Journal of Medicine. 2006;354(13):1362–1369. [DOI] [PubMed] [Google Scholar]
- 4.Landgren O, Weiss B. Patterns of monoclonal gammopathy of undetermined significance and multiple myeloma in various ethnic/racial groups: support for genetic factors in pathogenesis. Leukemia. 2009;23(10):1691–1697. [DOI] [PubMed] [Google Scholar]
- 5.Maura F, Bolli N, Rustad EH, Hultcrantz M, Munshi N, Landgren O. Moving from cancer burden to cancer genomics for smoldering myeloma: a review. JAMA oncology. 2020;6(3):425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rajkumar SV, Landgren O, Mateos M-V. Smoldering multiple myeloma. Blood, The Journal of the American Society of Hematology. 2015;125(20):3069–3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lakshman A, Rajkumar SV, Buadi FK, Binder M, Gertz MA, Lacy MQ, et al. Risk stratification of smoldering multiple myeloma incorporating revised IMWG diagnostic criteria. Blood cancer journal. 2018;8(6):59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dispenzieri A, Kyle RA, Katzmann JA, Therneau TM, Larson D, Benson J, et al. Immunoglobulin free light chain ratio is an independent risk factor for progression of smoldering (asymptomatic) multiple myeloma. Blood, The Journal of the American Society of Hematology. 2008;111(2):785–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pérez-Persona E, Vidriales M-B, Mateo G, García-Sanz R, Mateos M-V, de Coca AG, et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood, The Journal of the American Society of Hematology. 2007;110(7):2586–2592. [DOI] [PubMed] [Google Scholar]
- 10.Kazandjian D, Hill E, Dew A, Morrison C, Roswarski J, Korde N, et al. Carfilzomib, lenalidomide, and dexamethasone followed by lenalidomide maintenance for prevention of symptomatic multiple myeloma in patients with high-risk smoldering myeloma: a phase 2 nonrandomized controlled trial. JAMA oncology. 2021;7(11):1678–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lonial S, Jacobus S, Fonseca R, Weiss M, Kumar S, Orlowski RZ, et al. Randomized trial of lenalidomide versus observation in smoldering multiple myeloma. Journal of Clinical Oncology. 2020;38(11):1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mateos M-V, Hernández M-T, Giraldo P, de la Rubia J, de Arriba F, Corral LL, et al. Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. New England Journal of Medicine. 2013;369(5):438–447. [DOI] [PubMed] [Google Scholar]
- 13.Sklavenitis-Pistofidis R, Aranha MP, Redd RA, Baginska J, Haradhvala NJ, Hallisey M, et al. Immune biomarkers of response to immunotherapy in patients with high-risk smoldering myeloma. Cancer Cell. 2022;40(11):1358–1373. e1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hill E, Dew A, Morrison C, Yuan C, Stetler-Stevenson M, Landgren O, et al. Assessment of discordance among smoldering multiple myeloma risk models. JAMA oncology. 2021;7(1):132–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nature reviews Clinical oncology. 2017;14(2):100–113. [DOI] [PubMed] [Google Scholar]
- 16.Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood, The Journal of the American Society of Hematology. 2018;132(6):587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oben B, Froyen G, Maclachlan KH, Leongamornlert D, Abascal F, Zheng-Lin B, et al. Whole-genome sequencing reveals progressive versus stable myeloma precursor conditions as two distinct entities. Nature communications. 2021;12(1):1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolli N, Maura F, Minvielle S, Gloznik D, Szalat R, Fullam A, et al. Genomic patterns of progression in smoldering multiple myeloma. Nature communications. 2018;9(1):3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maura F, Bolli N, Angelopoulos N, Dawson KJ, Leongamornlert D, Martincorena I, et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nature communications. 2019;10(1):3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rustad EH, Yellapantula VD, Glodzik D, Maclachlan KH, Diamond B, Boyle EM, et al. Revealing the impact of structural variants in multiple myeloma. Blood cancer discovery. 2020;1(3):258–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rustad EH, Yellapantula V, Leongamornlert D, Bolli N, Ledergor G, Nadeu F, et al. Timing the initiation of multiple myeloma. Nature communications. 2020;11(1):1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diamond B, Yellapantula V, Rustad EH, Maclachlan KH, Mayerhoefer M, Kaiser M, et al. Positive selection as the unifying force for clonal evolution in multiple myeloma. Leukemia. 2021;35(5):1511–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones D, Raine KM, Davies H, Tarpey PS, Butler AP, Teague JW, et al. cgpCaVEManWrapper: simple execution of CaVEMan in order to detect somatic single nucleotide variants in NGS data. Current protocols in bioinformatics. 2016;56(1):15.10.11–15.10.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology. 2013;31(3):213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome research. 2012;22(3):568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor–normal sample pairs. Bioinformatics. 2012;28(14):1811–1817. [DOI] [PubMed] [Google Scholar]
- 27.Wala JA, Bandopadhayay P, Greenwald NF, O’Rourke R, Sharpe T, Stewart C, et al. SvABA: genome-wide detection of structural variants and indels by local assembly. Genome research. 2018;28(4):581–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Loo P, Nordgard SH, Lingjærde OC, Russnes HG, Rye IH, Sun W, et al. Allele-specific copy number analysis of tumors. Proceedings of the National Academy of Sciences. 2010;107(39):16910–16915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nadeu F, Mas-de-Les-Valls R, Navarro A, Royo R, Martín S, Villamor N, et al. IgCaller for reconstructing immunoglobulin gene rearrangements and oncogenic translocations from whole-genome sequencing in lymphoid neoplasms. Nature communications. 2020;11(1):3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Källberg M, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics. 2016;32(8):1220–1222. [DOI] [PubMed] [Google Scholar]
- 31.Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics. 2012;28(18):i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rustad EH, Yellapantula VD, Glodzik D, Maclachlan KH, Diamond B, Boyle EM, et al. Revealing the impact of structural variants in multiple myeloma. Blood cancer discovery. 2020;1(3):258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maclachlan KH, Rustad EH, Derkach A, Zheng-Lin B, Yellapantula V, Diamond B, et al. Copy number signatures predict chromothripsis and clinical outcomes in newly diagnosed multiple myeloma. Nature communications. 2021;12(1):5172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maura F, Degasperi A, Nadeu F, Leongamornlert D, Davies H, Moore L, et al. A practical guide for mutational signature analysis in hematological malignancies. Nature communications. 2019;10(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Ng AWT, Wu Y, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rustad EH, Nadeu F, Angelopoulos N, Ziccheddu B, Bolli N, Puente XS, et al. mmsig: a fitting approach to accurately identify somatic mutational signatures in hematological malignancies. Communications biology. 2021;4(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Landgren O, Hultcrantz M, Diamond B, Lesokhin AM, Mailankody S, Hassoun H, et al. Safety and effectiveness of weekly carfilzomib, lenalidomide, dexamethasone, and daratumumab combination therapy for patients with newly diagnosed multiple myeloma: the MANHATTAN nonrandomized clinical trial. JAMA oncology. 2021;7(6):862–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, et al. Universal patterns of selection in cancer and somatic tissues. Cell. 2017;171(5):1029–1041. e1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maura F, Rajanna AR, Derkach A, Ziccheddu B, Weinhold N, Maclachlan KH, et al. Individualized Treatment-Adjusted Risk Stratification in Newly Diagnosed Multiple Myeloma. Blood. 2022;140(Supplement 1):1561–1563. [Google Scholar]
- 40.Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos M-V, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. The lancet oncology. 2014;15(12):e538–e548. [DOI] [PubMed] [Google Scholar]
- 41.Maura F, Boyle E, Coffey D, Maclachlan KH, Diamond B, Blaney P, et al. Genomic Determinants of Resistance in Newly Diagnosed Multiple Myeloma Treated with Targeted-Immunotherapy. Blood. 2022;140(Supplement 1):1137–1139. [Google Scholar]
- 42.Cowan A, Ferrari F, Freeman SS, Redd R, El-Khoury H, Perry J, et al. Personalised progression prediction in patients with monoclonal gammopathy of undetermined significance or smouldering multiple myeloma (PANGEA): a retrospective, multicohort study. The Lancet Haematology. 2023;10(3):e203–e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maura F, Petljak M, Lionetti M, Cifola I, Liang W, Pinatel E, et al. Biological and prognostic impact of APOBEC-induced mutations in the spectrum of plasma cell dyscrasias and multiple myeloma cell lines. Leukemia. 2018;32(4):1043–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maura F, Rustad EH, Yellapantula V, Łuksza M, Hoyos D, Maclachlan KH, et al. Role of AID in the temporal pattern of acquisition of driver mutations in multiple myeloma. Leukemia. 2020;34(5):1476–1480. [DOI] [PubMed] [Google Scholar]
- 45.Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng W-J, et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer cell. 2007;12(2):131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bustoros M, Sklavenitis-Pistofidis R, Park J, Redd R, Zhitomirsky B, Dunford AJ, et al. Genomic profiling of smoldering multiple myeloma identifies patients at a high risk of disease progression. Journal of Clinical Oncology. 2020;38(21):2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kumar SK, Alsina M, Laplant B, Badros AZ, Abdallah A-O, Abonour R, et al. Fixed duration therapy with daratumumab, carfilzomib, lenalidomide and dexamethasone for high risk smoldering multiple Myeloma-Results of the ascent trial. Blood. 2022;140(Supplement 1):1830–1832. [Google Scholar]
- 48.Mateos M-V, Martínez-López J, Rodríguez-Otero P, San-Miguel J, Gonzalez-Calle V, Gonzalez MS, et al. Curative strategy (GEM-CESAR) for high-risk smoldering myeloma (SMM): post-hoc analysis of sustained undetectable measurable residual disease (MRD). Blood. 2022;140(Supplement 1):292–294. [Google Scholar]
- 49.Gay F, Musto P, Rota-Scalabrini D, Bertamini L, Belotti A, Galli M, et al. Carfilzomib with cyclophosphamide and dexamethasone or lenalidomide and dexamethasone plus autologous transplantation or carfilzomib plus lenalidomide and dexamethasone, followed by maintenance with carfilzomib plus lenalidomide or lenalidomide alone for patients with newly diagnosed multiple myeloma (FORTE): a randomised, open-label, phase 2 trial. The Lancet Oncology. 2021;22(12):1705–1720. [DOI] [PubMed] [Google Scholar]
- 50.Kumar SK, Jacobus SJ, Cohen AD, Weiss M, Callander N, Singh AK, et al. Carfilzomib or bortezomib in combination with lenalidomide and dexamethasone for patients with newly diagnosed multiple myeloma without intention for immediate autologous stem-cell transplantation (ENDURANCE): a multicentre, open-label, phase 3, randomised, controlled trial. The Lancet Oncology. 2020;21(10):1317–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Samur MK, Samur AA, Fulciniti M, Szalat R, Han T, Shammas M, et al. Genome-wide somatic alterations in multiple myeloma reveal a superior outcome group. Journal of Clinical Oncology. 2020;38(27):3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rasche L, Chavan S, Stephens O, Patel P, Tytarenko R, Ashby C, et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nature communications. 2017;8(1):268. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The dataset used for this paper is derived from public and newly sequenced sources: 27 SMM exomes were imported from dbGaP: phs001323.v3.p1. 701 MM exomes (from patients with RNA-seq and low coverage WGS data) were imported from the CoMMpass trial; IA 13 (dbGap: phs001323.v3.p1, https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001323.v3.p1). Data from the prior KRd +/− Dara NDMM patients along with 27 newly sequenced HR-SMM samples are available on EGA: EGAS00001007404.