

Abstract
The Millennium Eruption of Mt. Baekdu, one of the largest volcanic eruptions in the Common Era, initiated in late 946. It remains uncertain whether its two main compositional phases, rhyolite and trachyte, were expelled in a single eruption or in two. Investigations based on proximal and medial ash have not resolved this question, prompting us to turn to high-resolution ice-core evidence. Here, we report a suite of glaciochemical and tephra analyses of a Greenlandic ice core, identifying the transition from rhyolitic to trachytic tephra with corresponding spikes in insoluble particle fallout. By modeling annual snow accumulation, we estimate an interval of one to two months between these spikes, which approximates the hiatus between two eruptive phases. Additionally, negligible sulfur mass-independent fractionation, near-synchroneity between particle and sulfate deposition, and peak sulfur fallout in winter all indicate an ephemeral aerosol veil. These factors limited the climate forcing potential of the Millennium Eruption.
Subject terms: Palaeoclimate, Natural hazards
The Millennium Eruption of Mt. Baekdu was a multiphase eruption with two main phases separated approximately by one or two months, and was less climatically significant than previously thought, according to high-resolution glaciochemical and tephra analyses of an ice core in Greenland.
Introduction
Large volcanic eruptions can cause surface cooling for several years due to the radiative effects of sulfate aerosols1. The Millennium Eruption (ME) of Mt. Baekdu (Paektu, Changbaishan), which is located on the border of North Korea and China (41.99°N, 128.08°E) (Supplementary Fig. 1), is dated to late 946 CE2–5 and is one of the largest eruptions of the Common Era (CE) with a Volcanic Explosivity Index (VEI) of 6 or 7. ME tephra are widely distributed across Russia and Japan, and have been identified in ice cores from Greenland3,6,7. Two main compositional phases are recognized: an earlier light gray rhyolite (Phase 1), and a subsequent dark gray/black trachyte (Phase 2)8–11. The continuity of the ME spanning these two phases is unclear, but the presence of an intervening lahar deposit11 and grain-size analysis of the Baegdusan-Tomakomai (B-Tm) ash12 have suggested a hiatus of several months. A chronicle from Kofuku-ji temple in Nara (Japan) records white ash-fall on 3 November 946, while the Nihon Kiryaku and Tei-shin Koki chronicles from Kyoto (Japan) note thunderous sounds from the sky on 7 February 9474. The first observation very likely represents the ME, while the second conceivably reflects the detonations of a subsequent phase. Resolving whether or not there was a hiatus, and estimating its duration, if there was one, has important implications for understanding of volcanic hazards at large caldera complexes, notably concerning the question of when to conclude that an eruption is ‘over’.
Measurements of volatile abundances in melt inclusions and matrix glass have yielded estimates of syn-eruptive sulfur emissions from the ME ranging from 2 to 11 Tg7,13,14. However, this ‘petrologic method’ can underestimate total sulfur emissions of volcanic eruptions15,16, especially when sulfur-bearing vapor phases accumulate in the magmatic system prior to eruption (known as pre-eruptive sulfur)14,15. One estimate suggests that up to 42 Tg S may have been available in a pre-eruptive fluid phase corresponding to a total sulfur emission of as much as 45 Tg (including 3 Tg of syn-eruptive sulfur), surpassing estimated emissions for all Common Era eruptions bar Samalas (circa 1257 CE)14. However, more recent work suggests a total ME sulfur release of 2–7 Tg, with most of the pre-eruptive sulfur lost prior to eruption through crystal fractionation processes and bubble migration17. These wide-ranging estimates of sulfur loading propagate uncertainty into evaluations of any climate forcing arising from the ME.
Another factor influencing climate forcing from volcanic eruptions is plume height. In this regard also, estimates vary considerably for the ME. Inversions of grain-size data from proximal fall deposits18 have suggested heights of 25–35 km8,19, while a lower range of 15–20 km was derived from analysis of the distal B-Tm deposit12. More recent model-based estimations constrained by tephra thicknesses indicate plume heights of 30–40 km, i.e. well into the stratosphere20. However, it is well-established that the sulfur injection height can be much lower than the tephra (isopleth) derived plume height21. Resolving the altitude at which the ME sulfate aerosol dispersed is therefore also critical to understanding the climatic impacts of the ME.
While extra-tropical northern hemisphere (NH) winter-time explosive eruptions can have a substantial climatic impact22–24, evidence for transient NH summer cooling following the ME is lacking in tree-ring temperature reconstructions2 and has not been identified in historical records4. This appears consistent with the modest 1.7 ± 0.6 (1σ) Tg of stratospheric sulfur injection estimated from sulfur concentrations associated with ME deposition in Greenland ice25.
Since ice cores offer a highly time-resolved record of deposition from the ME aerosols, we conducted continuous and discrete measurements of a section of the NGRIP1 core of the North Greenland Ice-Core Project. Our aims were to (i) evaluate the timeline of the ME from ultra-high-resolution ion and microparticle concentrations; (ii) shed light on the atmospheric transport pathways of ME sulfur emission from isotopic measurements (32S, 33S, and 34S) and the timing between insoluble particles and sulfate26–28; and (iii) investigate the provenance and other characteristics of populations of tephra particles using glass shard geochemistry.
Results and discussion
Tephra geochemistry, glaciochemical, and insoluble microparticle records
Discrete sections of ice from the NGRIP1 core, T3 (deepest/oldest) through T10 (shallowest/youngest), were sampled for tephra extraction (see ‘Sample collection’ in Methods and Fig. 1). Shard sizes ranged from 4 to 38 μm (Supplementary Figs. 2 and 3), and their morphologies are diverse (Supplementary Data 1, discussed in Supplementary Note 1). The shards in samples T4 and T5 included both trachyte and rhyolite compositions that mostly match those of the ME (Fig. 1 and Supplementary Data 1). Specifically, sample T4 yielded eight rhyolite shards and one trachyte shard, while the overlying T5 sample yielded two rhyolite and four trachyte shards. One rhyolite in T4 plots near the rhyolite/trachyte border, and one rhyolite shard in T5 does not correspond to the ME (Fig. 1). This transition from rhyolite to trachyte shards in the ice mirrors the temporal progression evident in the proximal stratigraphic record of the ME, suggesting minimal reworking of snow at the NGRIP site, and consistent with the high snow accumulation rates and relatively low wind speeds experienced at the topographic divide where the ice core was drilled (in contrast to more coastal sites in Greenland)29.
Fig. 1. Stratigraphy and geochemistry of shards found in NGRIP1 and NEEM-2011-S16 cores.

QUB-1819c is a shard potentially corresponding to a Japanese volcano (see Supplementary Note 1) (QUB stands for Queen’s University Belfast). a Schematic diagram of the NGRIP1 ice core. b Total alkali versus silica (TAS) diagram60. c Additional major oxide bi-plots for glass shards. Only samples for which analytical totals exceeded 90 % and those that might represent a tephra population are shown. Shards in sample T7 were measured by scanning electron microscope energy dispersive spectroscopy (SEM-EDS); all other shards were measured by wavelength dispersive spectroscopy (WDS). Gray shading characterizes the previously reported range of ME glass geochemistry for both proximal and distal deposits6,9,61,62. Tephra populations in T3 and T7 may correspond to fallout from Mt. Rainier eruptions (Supplementary Note 1). d Location of Mt. Baekdu and NGRIP1 core.
A range of elements, high-resolution liquid conductivity, and size-resolved insoluble particle counts were measured on separate longitudinal samples of the NGRIP1 core using the continuous ice-core analytical system at the Desert Research Institute (DRI) (see ‘Continuous-flow aerosol analyses’ in Methods)23. Sulfur concentrations from discrete and continuous measurements were consistent (Supplementary Fig. 4). Liquid conductivity, which reflects acidity and is dominated by changes in sulfate concentration in Greenland ice, provides an additional high-resolution proxy for volcanic sulfate deposition (Supplementary Data 2). Insoluble particle concentrations increased abruptly at ~218.52 m depth and fell abruptly at 218.49 m, spanning ice samples T4 and T5 (Fig. 2 and Supplementary Data 3), thereby confirming the microparticle peak’s association with the ME. This microparticle peak coincides with seasonal peaks in concentrations of Na (a sea salt proxy) and Ca (a terrestrial dust proxy; Supplementary Fig. 5 and Supplementary Data 4), consistent with the late 946 CE timing of the ME suggested by studies of tree-rings and the NEEM-2011-S1 core from the North Greenland Eemian Ice Drilling (NEEM) site in Greenland2,30.
Fig. 2. Glaciochemistry and microparticle records from the NGRIP1 ice core.
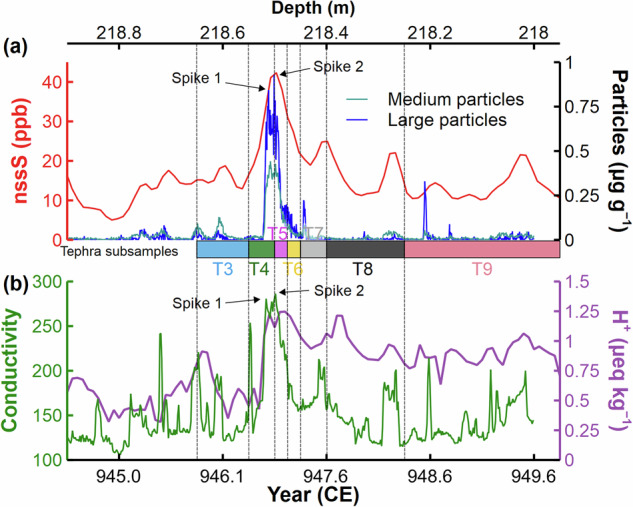
a Non-sea-salt sulfur (nssS) and insoluble particle (large: 4.5–9.5 μm; medium: 2.5–4.5 μm) concentrations from Continuous Flow Analysis (CFA) at the Desert Research Institute (DRI). T3 to T9 represent consecutive ice samples for tephra shard analysis, with corresponding depths shown at the bottom of the panel. b High-resolution liquid conductivity (dimensionless) analyzed at the DRI, and previously published 1-cm resolution H+ measurements based on the electrical conductivity method (ECM)63.
Time interval estimation between the two ME phases
On close inspection, the microparticle peak exhibits two spikes in the size range of 4.5–9.5 μm, which are mirrored in the simultaneously measured continuous liquid conductivity record as well as the electrical conductivity method (ECM)-based H+ measurements (Fig. 2). The depth interval between the spikes is approximately 18 mm in the liquid conductivity and 11 mm for the large insoluble particles. The separation for the lower resolution ECM-based H+ spike is 20–30 mm. Separations greater than the effective depth resolutions of approximately 1.8 mm for liquid conductivity and approximately 3 mm for insoluble particle concentrations mean that the double spikes are sufficiently separated in depth to be considered separate signals. Considering that Spike 1 (the earlier/deeper large insoluble particle peak) is fully covered by sample T4, while Spike 2 is partially shared by samples T4 and T5 (Fig. 2 and Supplementary Fig. 6), we infer that Spike 1 corresponds to rhyolitic deposition, and Spike 2 corresponds to trachytic deposition based on the dominant chemical composition of tephra shards found in samples T4 and T5. Accordingly, we conclude that Spikes 1 and 2 represent the two compositional phases of the ME.
To estimate the time interval between the two spikes in microparticles, we developed a simple annual net accumulation model (Fig. 3) drawing on modern climatology of snowfall recorded from 1991 to 199531 (see ‘Annual net accumulation model’ in Methods). Given that the annual accumulation rate at the NGRIP site during the 1990s is unexceptional in the context of the last 2000 years32, we assume that the seasonal accumulation trend (i.e., a monthly fraction of the annual accumulation rate) of 946–947 CE was comparable to that of the 1990s. We acknowledge that the seasonality of snow accumulation at the NGRIP site is highly variable from year to year, but the 1991–1995 data set is, to our knowledge, the only observed record of sub-seasonal accumulation from northern Greenland. Our accumulation model incorporates both spatial and temporal variability to ensure a conservative interpretation (see ‘Annual net accumulation model’ in Methods). Based on air trajectory calculations (Supplementary Fig. 7) and assuming the first eruption occurred circa 1 November, particle deposition at the NGRIP site is registered around 14 ± 7 days later (see “Forward air trajectory analysis” in Methods). The assumption of first eruption at 1 November aligns with the suggested eruption date for the rhyolite near 2 November, as suggested by the Kofuku-ji chronicle, which records ash fallout in Japan on 3 November2,4. The maximum in the first microparticle spike then dates to 46 ± 21 days after 1 November and the second to 77 ± 23 days after 1 November (Fig. 3). The time interval between spikes is 31 ± 25 days (all uncertainties are 2σ) (see ‘Processes of time interval estimation’ in Methods). We estimate probabilities of the time interval being within a week as 2.5%; within four weeks, 42.7%; and within eight weeks, 97.9% (Supplementary Fig. 8). Even allowing for the vagaries of aerosol transport times from source to Greenland, this timescale likely precludes the possibility that the thunderous sounds heard in Japan on 7 February 947 CE (i.e., 14 weeks after 1 November 946 CE) were related to a second phase of the ME (Fig. 3).
Fig. 3. Annual net accumulation model for north central Greenland between 1991 and 199531.
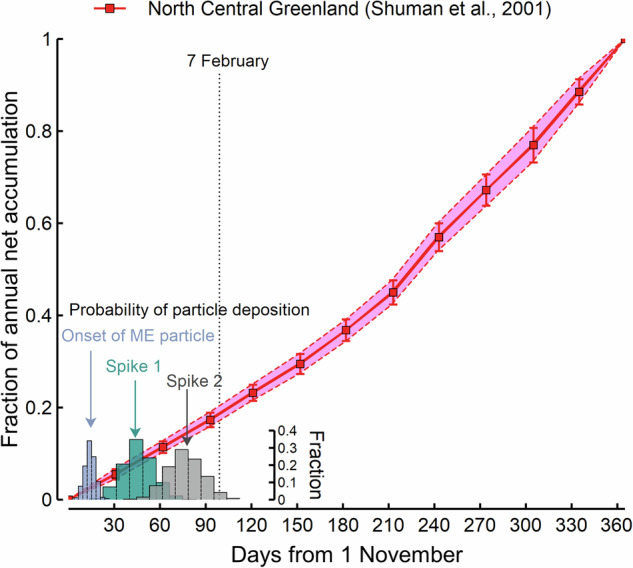
Error bars indicate 1 σ uncertainty. Histograms are for the timing of microparticle deposition (onset and Spikes 1 and 2) and represent probability density distributions based on 1000 Monte Carlo simulations in each case. See ‘Processes of time interval estimation’ in Methods for details.
Transport timescales for the two phases may well have differed, reflecting different injection heights and meteorology, but atmospheric residence times for tephra shards exceeding one to two months are unlikely33. Accordingly, our analysis does support the hiatus hypothesis rather than a single eruptive episode. More accurate constraints on the monthly accumulation history in northern Greenland could improve the estimation of the time between microparticle peaks.
Atmospheric transport of the ME sulfate aerosol and limited climate impact of the eruption
When volcanic sulfur dioxide (SO2) penetrates the stratospheric ozone layer and is exposed to shortwave ultraviolet (UV) radiation, the resulting sulfate exhibits a sulfur mass-independent fractionation (S-MIF, i.e., non-zero Δ33S)26. SO2 that does not reach the ozone layer undergoes mass-dependent fractionation (resulting in near-zero Δ33S)26. For example, ice-core sulfate derived from both the 43 BCE (Before Common Era) eruption of Okmok (Alaska) and that of Tambora (Indonesia) in 1815 CE has a stratospheric S-MIF signal (∆33S of 0.17–0.20 ‰ and −0.84 to 1.68 ‰, respectively)23,34. On the other hand, ice-core sulfate sourced from the upper tropospheric/lower stratospheric (UT/LS) aerosol from Cerro Hudson (Chile) in 1991 CE, and Laki (Iceland) in 1783 CE lacks a S-MIF signal (∆33S of around −0.09 ‰ and −0.13–0.02 ‰, respectively)26,35. The NGRIP1 ice samples spanning the ME similarly lack a S-MIF signal, and there is no hint of the positive-to-negative Δ33S evolution typical of large stratospheric sulfur injections27,34,36,37 (Fig. 4) (Supplementary Table 1). Therefore, sulfate formation during the ME mostly occurred below the stratospheric ozone layer.
Fig. 4. Measured δ34S and Δ33S, and estimated volcanic (background corrected) δ34S and Δ33S with continuous nssS concentrations.
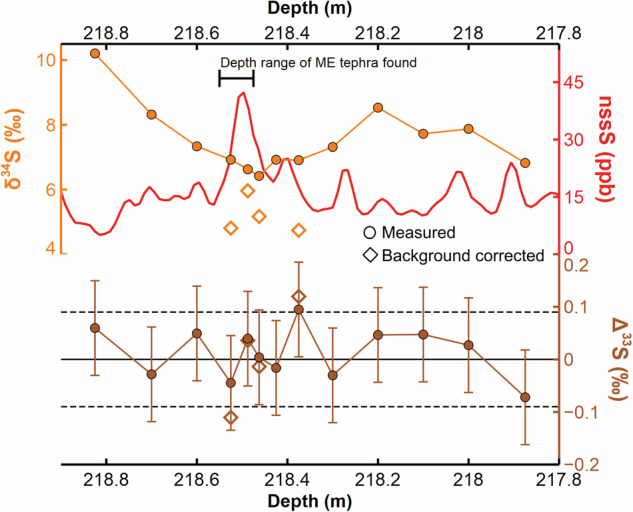
Background correction is applied when volcanic sulfate accounts for over 60 % (fvolc > 0.60). Solid line at Δ33S = 0 ‰. Dashed lines indicate Δ33S ± 0.09 bracketing range of S-MIF signals. Error bars represent 2σ uncertainty.
Fine volcanic ash (1–15 μm) has an atmospheric lifetime of a few days to weeks33,38,39. Submicron sulfate aerosols in the mid-to-high stratosphere have a longer atmospheric lifetime of one to three years. This difference in residence time explains the delayed deposition of sulfate against microparticles in the polar regions following past explosive eruptions that generated stratospheric aerosol veils, as recognized in high-resolution ice-core records (Supplementary Fig. 9)23,27,34,40. However, volcanic sulfate in the lower stratosphere (13–16 km altitude) from extra-tropical eruptions experiences shorter lifetimes of a few months22. The insoluble microparticle and sulfur deposition in Greenland associated with the 1783 CE Laki eruption reveals only a few months delay28 consistent with model simulations22. For the 2011 CE Puyehue-Cordón Caulle (PCC) eruption (VEI 5) whose plume only reached the tropopause, sulfur and microparticle fallout in Antarctica were simultaneous (Supplementary Fig. 9)41. The near-synchronous fallout of insoluble particles and sulfur associated with the ME evident in the NGRIP1 core (Fig. 2a) is consistent with UT/LS sulfate transport and the lack of an S-MIF signal. The discrepancy between the maximum plume height of 30–40 km20 and the sulfur injection height of UT/LS might be attributed to the estimated high mass eruption rate20 of approximately 4 × 108 kg s−1. This inference is based on a previous study that compiled data on mass eruption rates, maximum plume heights and sulfur injection heights for past eruptions21. According to the study, there can be a difference of >15 km between the maximum plume height and the sulfur injection height for eruptions with a mass eruption rate exceeding 108 kg s−1.
The ME sulfate deposition at the NGRIP site corresponds to 11 kg km−2 (see ‘Processes of sulfate deposition estimation’ in Methods), similar to that estimated from the NEEM record6 (9 kg km−2), identical to a previous estimate from the NGRIP1 core based on discrete measurements42. This is much lower than the 1815 CE Tambora eruption (40 kg km−2)5, reflecting lower sulfur yield and lower sulfur injection height of the ME20 compared with the Tambora eruption34. Furthermore, our accumulation model suggests sulfate deposition to the ice spanned 232 ± 42 days (2σ), and based on a 1 November start date (see “Processes of time interval estimation” in Methods), this implies the ME sulfate aerosols persisted mainly in the boreal winter and spring. Consequently, there was minimal sulfate aerosol remaining in the atmosphere during the summer of 947 CE, a further factor limiting any climate forcing.
Despite its great magnitude, ice-core sulfate deposition and isotopes are consistent with a limited stratospheric sulfur loading from the ME. Reasons for this may include the loss of pre-eruptive sulfur17 and/or the low altitude of the sulfate aerosol veil and its accelerated scavenging. Our high-resolution glaciochemical records and cryptotephra results also add weight to the hypothesis of a hiatus between the rhyolitic and trachytic phases of the ME. The conductivity peak and width of Spike 2 appear to be greater than those of Spike 1 (Fig. 2b). This may suggest that more SO2 was released during Phase 2 of the ME, which may help in understanding the sulfur budgets of the two magma compositions in respect of eruptive volumes and degassing histories17,20. Taken together with understanding of the radiative and dynamical impacts of boreal winter, extra-tropical NH eruptions23,43–45, our findings reinforce the evidence from tree-ring summer temperature reconstructions2 for negligible cooling after the ME, and highlight the importance of evaluating the role of volcanism in climate variability and human history on a case-by-case basis.
Conclusions
We conducted high-resolution analyses of cryptotephras, glaciochemical records (e.g. water-soluble ion concentrations, conductivity, multiple sulfur isotopes), and insoluble particles within the NGRIP1 core from Greenland. We identified a transition from rhyolitic to trachytic tephra, associated with the ME, corresponding with discrete spikes in insoluble particle concentrations. Based on a simple annual snow accumulation model for northern Greenland, the insoluble particle spikes suggests a hiatus between the rhyolitic and trachytic phases of the ME of order one month. However, our accumulation model is based on a relatively short data set and more accurate reconstructions of monthly accumulation in Greenland could lead to improvements and reductions in uncertainty. Our findings also revealed negligible sulfur mass-independent fractionation, near-synchronous deposition of insoluble particles and sulfate, and peak sulfur deposition occurring in winter. Based on these observations, the muted climate forcing arising from the ME likely reflects a short atmospheric lifetime of the volcanic sulfate aerosol veil, along with boreal winter timing of aerosol formation and modest sulfur yield. This refined picture of the timeline of the ME and sulfur injection heights can inform risk assessment at restless calderas worldwide.
Methods
Sample collection
Our investigation included both discrete and continuous analyses of the NGRIP1 core. Two longitudinal samples with approximately 10 cm2 cross-sections were cut from the archived core. For the discrete analyses, one of the longitudinal samples was subsampled at a resolution of 2.5 to 5 cm over the depth range corresponding to the volcanic fallout and 10 to 15 cm during background periods in the year 2022. These samples were decontaminated by shaving their surfaces by approximately 1 mm with a clean ceramic knife. The samples were then sent to Seoul National University and stored at −20 °C before analysis. For the continuous analyses, the second set of longitudinal samples was cut several years earlier in 2013 and sent frozen to the DRI (Nevada, USA) and stored at −20 °C prior to analysis. The NGRIP1 core has been stored at −24 °C in a freezer facility in Copenhagen since it was drilled in 1996, and subsequently, at −30 °C since 2019.
Continuous-flow aerosol analyses
High-resolution liquid conductivity, elemental chemistry, and insoluble particle concentrations of NGRIP1 were measured using the well-established continuous flow ice-core melter system at DRI46 with effective depth resolutions of approximately 1.8 mm, 20 mm, and 3 mm, respectively. Briefly, longitudinal samples of the archived NGRIP1 ice core were analyzed for a range of ~30 elements, as well as several chemical species, water isotopes, size-resolved insoluble particles, and other parameters. Ridges engraved into a ceramic heated melter plate split meltwater from different parts of the sample into three continuously flowing sample streams: innermost 10%, middle 20%, and outer 70%. Elemental measurements, including sulfur, sodium, and calcium were made on the innermost 10% using two Element2 Inductively Coupled Mass Spectrometers operating in parallel. Semi-quantitative, size-resolved insoluble particle concentrations and liquid conductivity were measured in high depth resolution on the middle 20% melt stream47 using a laser-based Abakus particle counter48 and Amber Scientific low-volume conductivity cell, respectively. The outer 70% was discarded due to potential contamination.
Sea-salt sodium (ssNa) was calculated from measured sodium and calcium concentrations following standard procedures49. Sea-salt sulfur (ssS) was calculated from ssNa using a sea water mass ratio of 905/10770, and the ssS was subtracted from the total measured sulfur to yield the non-sea-salt sulfur (nssS) concentration49–51.
Tephra shards
The sample preparation and analysis of ice-core tephra shards was conducted at the University of St Andrews. Discrete samples were melted into acid-cleaned bottles, and these were then centrifuged, and the supernatant pipetted off for sulfur concentration and isotope measurements. The remaining water (approximately 2 to 3 mL) was pipetted into the center of a plastic ring, which was placed on a Kapton tape-covered hotplate (80 °C) for evaporation. Once fully dry, the ring mounts were filled with epoxy resin. The surface was then lightly polished (for one to two minutes) using 1 μm diamond paste and 0.3 μm alumina, prior to carbon coating.
Tephra shards were identified and chemically analyzed using a JEOL (Japan Electron Optics Laboratory) iSP100 scanning electron microscope (SEM) and Electron Probe Microanalyzer (EPMA) at the University of St Andrews. Chemical composition of tephra shards was measured using an energy dispersive spectrometers (EDS) and five wavelength dispersive spectrometers (WDS) (Supplementary Data 1). For EPMA we used a 15 kV of accelerating voltage. A 5 nA of beam current and 5 μm of beam diameter was used for large shards (>10 μm), and a 1 nA of beam current and 3 μm of beam diameter for small shards (<10 μm). Counting times at each peak position are as follows: 30 s for Si, Fe, Mg, Ca, K, P, Mn, and Ti; 20 s for Al; 10 s for Na for the 5 μm setup and 8 s for Na in 3 μm setup. Background counts were half of the peak counts. Semi-quantitative measurements using SEM-EDS were aquired using a 15 kV of accelerating voltage, 1 nA of beam current, and 20 s live time. Three international secondary glass standards were measured for each analytical session: Mauna Loa tholeiitic basalt glass (ML3B), St. Helens andesitic ash glass (StHs6/80-G), and an obsidian from Lipari Island52,53. The average measurements of our standards were generally within 2σ uncertainty of the reference values (Supplementary Data 5).
Annual net accumulation model
We developed an annual net snow accumulation model based on the seasonal variation of net snow accumulation in north-central Greenland from 1991 to 199531. First, we digitized Figure 6a from Shuman et al.31 to obtain monthly net accumulation values (mm in water equivalent (w.e.)) for four sites. These values were averaged by month for each site, and then across all sites to produce the overall monthly average net accumulation (Supplementary Fig. 10). Error bars shown in Supplementary Fig. 10 represent standard deviations for each month. However, we argue that this is not an appropriate average and error estimate since it is based on a relatively short data set that is not normally distributed. In addition, the accumulation for January has only one data point available, which makes the standard deviation meaningless (Supplementary Fig. 10). To overcome this problem, we generated 1000 random values from the monthly net accumulation range, assuming a uniform distribution. The minimum and maximum value for net accumulation of January, where only one data point was available, was assumed to be the same as the average of the minimum and maximum net accumulation of November, December, February, and March. The resulting annual net accumulation distribution (sum of the monthly net accumulation) was near-normal, estimated at 191 ± 22 mm w.e. (2σ). The averages and standard deviations of these 1000 monthly net accumulation simulations are provided in Supplementary Table 2. In order to establish the annual net accumulation model (Fig. 3), the monthly net accumulations were divided by the annual net accumulation in each random generation to determine the monthly fractions (averages and standard deviations are reported in Supplementary Table 2).
Processes of time interval estimation
In order to estimate the time interval between the two spikes from the observed depth difference, we used the accumulation model to derive the time elapsed since the assumed eruption date of 1 November 946. According to the 240-hour forward air trajectory, the ash from the ME could have reached Greenland within a few days to weeks after the eruption started (Supplementary Fig. 7). In this study, we assumed that the ash would have taken 14 ± 7 days to reach the NGRIP site in Greenland after 1 November (see the next “Forward air trajectory analysis” section). We assumed a normal distribution for the estimated accumulation and ash travel timing. Using the developed annual net accumulation model, we assigned a ‘fraction of annual net accumulation’ value (i.e., y axis value) to the ash travel timing, which is 14 ± 7 days from 1 November (i.e., x axis value). This process was repeated 1000 times. As a result, the duration of 14 ± 7 days corresponds to a ‘fraction of annual net accumulation’ of 0.0258 ± 0.0245. This value is converted to 0.005 ± 0.005 m by multiplying it with the NGRIP average annual layer ice thickness between 940 and 949 CE (0.183 ± 0.023 m)54. Afterward, we determined the depth that corresponds to 1 November 946 within the NGRIP1 core. The large insoluble particles associated with the ME start at a depth of 218.5225 m in the NGRIP1 core (Supplementary Data 3). To calculate the depth for 1 November 946, we added 0.005 ± 0.005 to 218.5225, resulting in a depth of 218.527 ± 0.005 m. The depth differences between 1 November 946 and the double spikes of the large particle concentrations are calculated as 0.016 ± 0.005 m for Spike 1 and 0.026 ± 0.005 m for Spike 2 (Supplementary Data 3). These values were then divided by the NGRIP average annual layer ice thickness of 940–949 CE (0.183 ± 0.023 m) to derive the ‘fraction of annual net accumulation’; 0.085 ± 0.031 for Spike 1 and 0.143 ± 0.035 for Spike 2. Finally, we assigned the days after 1 November (i.e. x-axis value) for the double spikes using the annual net accumulation model (Fig. 3) and estimated the time interval between the double spikes. The first microparticle spike was deposited 46 ± 21 days after 1 November, and the second 77 ± 23 days after. Accordingly, the time interval between spikes was 31 ± 25 days. The uncertainties are quoted to 2σ and were achieved by 1000 Monte Carlo simulations under normal distribution.
For estimating the scavenging of the ME sulfate aerosols in the atmosphere, we determined the sulfur in the NGRIP1 core associated with the ME ends at a depth of 218.431 m (Supplementary Data 4). The ‘fraction of annual net accumulation’ (y axis value) for the end of sulfur associated to the ME was 0.525 ± 0.130 (2σ), by following the same procedure described above. Finally, the days after 1 November of 232 ± 42 days (2σ) were assigned using the annual net accumulation model (x axis value). The uncertainties were achieved by 1000 Monte Carlo simulations under normal distribution.
Forward air trajectory analysis
Considering that volcanic fine ash (1–15 μm) has an atmospheric lifetime of a few days to weeks33,38,39, a 240-hour (10-day) forward air trajectory was calculated using the Hybrid Single-Particle Lagrangian Integrated Trajectory (HYSPLIT) model of the National Oceanic and Atmospheric Administration (NOAA) in the READY (Real-time Environmental Applications and Display System) website to find how fast the volcanic ash of the ME had transferred to Greenland through the atmosphere (Supplementary Fig. 7)55,56. Although the forward air trajectory model may not exactly account for the ash dispersion and deposition processes, it provides a tentative estimate of the time taken for the ash to travel to Greenland. For the calculation, we utilized the meteorological data from 00 Coordinated Universal Time (UTC) on 1 November, ranging from 2005 to 2023, obtained from the Global Data Assimilation System (GDAS) at a resolution of 1°. The forward air trajectories shown in this study were initiated from Mt. Beakdu, with starting heights of 8 and 14 km above mean sea level (AMSL), considering that the ME was likely a UT/LS eruption.
According to the calculation, some air parcels started at 8 km AMSL (upper troposphere) may not reach Greenland during 10 days of transport, while most of that started at 14 km AMSL (lower stratosphere) may reach Greenland within 10 days (Supplementary Fig. 7). Hence, we roughly assumed that ME tephra might have deposited on the Greenland surface within 14 ± 7 (2σ) days after the eruption.
Sulfur isotopes
The sample preparation and analysis for sulfur isotopes were conducted at the University of St Andrews. First, a 1.5 mL aliquot of each melted ice sample was analyzed by ion chromatography for sulfate concentration. Based on concentrations, the volume required for 20 nmol of sulfate was dried down in Teflon vials on a hotplate. The residue was re-dissolved in 100 μL of 0.01% v/v distilled HCl. The solutions were then transferred to auto-sampler vials for processing by an automated sulfate purification system (Elemental Scientific prepFAST MC), and 9 μL of Milli-Q water was added to each sample for every hour it would be waiting before loading on the column to prevent complete evaporation during the sample processing. Sulfate from the samples was purified by using the prepFAST MC with AG1X8 anion-exchange resin (Dry mesh: 100–200) on a 50 μL column. The resin was cleaned with 350 μL of 10% HNO3, 350 μL of 10% HCl, and 350 μL of 0.5% HCl. Then, samples were loaded, the column was rinsed with 200 μL of Milli-Q water three times, and sulfate was eluted by 3 100 μL of 0.5 M HNO3. The eluted samples were dried down in Teflon vials once again on a hotplate. The residue was diluted with 0.5 M HNO3 containing NaOH to match the sulfate and sodium concentration of the in-house Na2SO4 bracketing standard. Sulfate in the in-house secondary standards Switzer Falls and seawater samples were also purified for checking accuracy and reproducibility. A blank sample was also processed (sulfur blank was 0.04 nmol and had a δ34S = 4.53 ‰). The reproducibility using the prepFAST method for our in-house Switzer Falls standard was δ34S = 4.23 ± 0.11 ‰ and Δ33S = 0.04 ± 0.09 ‰ (n = 21, 2σ), and seawater was δ34S = 21.13 ± 0.15 ‰ and Δ33S = 0.04 ± 0.09 ‰ (n = 12, 2σ), which agrees well with previous studies: δ34S = 4.17 ± 0.11 ‰ and Δ33S = 0.01 ± 0.10 ‰ for Switzer Falls34,57 and δ34S = 21.24 ± 0.88 ‰ and Δ33S = 0.050 ± 0.014 ‰ for seawater58.
Multiple sulfur isotopes (32S, 33S, and 34S) were measured using a Neptune Plus Multi Collector Inductively Coupled Plasma Mass Spectrometer (MC-ICP-MS)34,57,59. All sulfur isotopic ratios are blank-corrected and reported relative to Vienna Canyon Diablo Troilite (V-CDT) in standard delta notation (Eq. (1)). The Δ33S values were then calculated by following Eq. (2).
| 1 |
| 2 |
Sulfate concentration and sulfur isotope ratios of the background sample, prior to the volcanic eruption, were used to determine the sulfur isotope values of the volcanic sulfate following Equation (3)27,37.
| 3 |
The fbackground is the mass fraction of sulfate that is attributed to the background (fbackground = [SO42−]background/[SO42−]measured), and fvolc is the mass fraction of sulfate that is attributed to the volcanic eruption (fvolc = 1−fbackground). The δxSvolc was then used to calculate Δ33Svolc. Uncertainties for the results were estimated through Monte Carlo simulation34.
Processes of sulfate deposition estimation
The NGRIP average accumulation rate between 940 and 949 CE (0.197 m ice yr−1)32 was multiplied with high-resolution nss-sulfate concentration of NGRIP1 ice core to estimate the sulfate flux (mg m−2 yr−1) (Supplementary Data 4). Then, the sulfate flux was multiplied by the time interval of an adjacent depth (in DRI_NGRIP chronology) to estimate the sulfate deposition. Because the latter half of the main sulfur concentration peak area of the ME slightly overlaps with a shoulder sulfur peak in around 218.4 m (Fig. 2a), background corrected-sulfate depositions corresponding only to the first half of the main sulfur peak area were summed up, and then doubled to estimate the entire sulfate deposition of the ME (Supplementary Data 4). Note that this is not the ideal choice because the volcanic aerosol formations from the gas precursors have an e-folding residence time of several months depending on latitude33 and thus would actually have a tailing of sulfur concentrations following the peak. The nss-sulfate flux value prior to the ME sulfur was defined as the background sulfate flux. The background sulfate flux was multiplied with the time interval corresponding to the first half of the ME sulfur peak area and then doubled for the background correction.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
We thank Thomas Blunier, Helen Innes, N. Chellman, and O. Maselli for laboratory assistance and technical support. The authors gratefully acknowledge the NOAA Air Resources Laboratory (ARL) for the provision of the HYSPLIT transport and dispersion model and/or READY website (https://www.ready.noaa.gov) used in this publication. This work was supported by the National Research Foundation of Korea (NRF-2018R1A5A1024958, 2020M1A5A1110607); the Basic Research Project (GP2021-006) of the Korea Institute of Geoscience and Mineral Resources (KIGAM) funded by the Ministry of Science, ICT of Korea; the EPSRC Light Element Analysis Facility Grant (EP/T019298/1); the Strategic Equipment Resource Grant (EP/R023751/1); U.S. National Science Foundation grants (1023672 and 1925417); Villum Investigator Project IceFlow grant (16572); a Philip Leverhulme prize in Earth Sciences (PLP-2021-167); UKRI Future Leaders Fellowship (MR/S033505/1); the European Research Council under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 820047).
Author contributions
G.L. and J.A. conceptualized the idea and wrote the manuscript with contributions from all co-authors. G.L., A.B., W.H., P.S., and C.S. performed sulfur isotope and tephra analyses, and contributed to the discussion of the data. J.R.M. was responsible for the high-resolution liquid conductivity, ion concentrations, and insoluble particle concentrations of the NGRIP1 ice core. M.S. provided advice on the interpretation of the cryptotephra results and sulfur isotopes. C.O. provided advice on the time interval estimation and the overall interpretation of the study by editing and revising the original writings. S.O.R. provided advice on the dating and accumulation model. J.P.S. provided access to the NGRIP1 ice core and contributed to the overall interpretation. S.R.L. provided financial support for the project and contributed to the overall interpretation.
Peer review
Peer review information
Communications Earth & Environment thanks Ivan Sunyè Puchol and Laura Wainman for their contributions to the peer review of this work. Primary Handling Editors: Domenico Doronzo and Carolina Ortiz Guerrero. A peer review file is available.
Data availability
The data used for manuscript figures are available on Figshare, with the Supplementary Data under identifier 10.6084/m9.figshare.26549344.v1 and the Supplementary Tables under identifier 10.6084/m9.figshare.26550304.v1. Supplementary Tables are also available in the Supplementary Information.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s43247-024-01713-z.
References
- 1.Robock, A. Volcanic eruptions and climate. Rev. Geophys.38, 191–219 (2000). [Google Scholar]
- 2.Oppenheimer, C. et al. Multi-proxy dating the ‘Millennium Eruption’ of Changbaishan to late 946 CE. Quat. Sci. Rev.158, 164–171 (2017). [Google Scholar]
- 3.Yang, Q. et al. The Millennium eruption of Changbaishan Tianchi volcano is VEI 6, not 7. Bull. Volcanol.83, 74 (2021). [Google Scholar]
- 4.Yun, S.-H., Lee, J. & Oppenheimer, C. A re-assessment of historical records pertaining to the activity of Mt. Baekdu (Paektu, Tianchi) volcano. Geosci. Lett.10, 30 (2023). [Google Scholar]
- 5.Sigl, M. et al. Timing and climate forcing of volcanic eruptions for the past 2,500 years. Nature523, 543–549 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Sun, C. et al. Ash from Changbaishan Millennium eruption recorded in Greenland ice: Implications for determining the eruption’s timing and impact. Geophys. Res. Lett.41, 694–701 (2014). [Google Scholar]
- 7.Coulter, S. E. et al. Holocene tephras highlight complexity of volcanic signals in Greenland ice cores. J. Geophys. Res. Atmos.117, D21303 (2012). [Google Scholar]
- 8.Horn, S. & Schmincke, H.-U. Volatile emission during the eruption of Baitoushan volcano (China/North Korea) ca. 969 AD. Bull. Volcanol.61, 537–555 (2000). [Google Scholar]
- 9.Chen, X.-Y. et al. Clarifying the distal to proximal tephrochronology of the Millennium (B-Tm) eruption, Changbaishan volcano, northeast China. Quat. Geochronol.33, 61–75 (2016). [Google Scholar]
- 10.Pan, B. et al. The VEI-7 Millennium eruption, Changbaishan-Tianchi volcano, China/DPRK: new field, petrological, and chemical constraints on stratigraphy, volcanology, and magma dynamics. J. Volcanol. Geotherm. Res.343, 45–59 (2017). [Google Scholar]
- 11.Pan, B., de Silva, S. L., Xu, J., Liu, S. & Xu, D. Late Pleistocene to present day eruptive history of the Changbaishan-Tianchi volcano, China/DPRK: New field, geochronological and chemical constraints. J. Volcanol. Geotherm. Res.399, 106870 (2020). [Google Scholar]
- 12.Utkin, I. V. Reconstructing the setting for deposition of distal tephra in the Sea of Japan basin: a catastrophic eruption of Baitoushan Volcano. J. Volcanol. Seismol.8, 228–238 (2014). [Google Scholar]
- 13.Guo, Z. et al. The mass estimation of volatile emission during 1199–1200 AD eruption of Baitoushan volcano and its significance. Sci. China Ser. D: Earth Sci.45, 530–539 (2002). [Google Scholar]
- 14.Iacovino, K. et al. Quantifying gas emissions from the “Millennium Eruption” of Paektu volcano, Democratic People’s Republic of Korea/China. Sci. Adv.2, e1600913 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westrich, H. R. & Gerlach, T. M. Magmatic gas source for the stratospheric SO2 cloud from the June 15,1991, eruption of Mount Pinatubo. Geology20, 867–870 (1992). [Google Scholar]
- 16.Wallace, P. J. Volcanic SO2 emissions and the abundance and distribution of exsolved gas in magma bodies. J. Volcanol. Geotherm. Res.108, 85–106 (2001). [Google Scholar]
- 17.Scaillet, B. & Oppenheimer, C. Reassessment of the sulfur and halogen emissions from the Millennium Eruption of Changbaishan (Paektu) volcano. J. Volcanol. Geotherm. Res.442, 107909 (2023). [Google Scholar]
- 18.Carey, S. & Sparks, R. S. J. Quantitative models of the fallout and dispersal of tephra from volcanic eruption columns. Bull. Volcanol.48, 109–125 (1986). [Google Scholar]
- 19.Wei, H. et al. Three active volcanoes in China and their hazards. J. Asian Earth Sci.21, 515–526 (2003). [Google Scholar]
- 20.Costa, A. et al. Eruption plumes extended more than 30 km in altitude in both phases of the Millennium eruption of Paektu (Changbaishan) volcano. Commun. Earth Environ.5, 6 (2024). [Google Scholar]
- 21.Aubry, T. J. et al. New insights into the relationship between mass eruption rate and volcanic column height based on the IVESPA data set. Geophys. Res. Lett.50, e2022GL102633 (2023). [Google Scholar]
- 22.Toohey, M. et al. Disproportionately strong climate forcing from extratropical explosive volcanic eruptions. Nat. Geosci.12, 100–107 (2019). [Google Scholar]
- 23.McConnell, J. R. et al. Extreme climate after massive eruption of Alaska’s Okmok volcano in 43 BC and its effects on the civil wars of the late Roman Republic. Proc. Natl. Acad. Sci. USA.117, 15443–15449 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Burke, A. et al. High sensitivity of summer temperatures to stratospheric sulfur loading from volcanoes in the Northern Hemisphere. Proc. Natl. Acad. Sci. USA.120, e2221810120 (2023). [Google Scholar]
- 25.Toohey, M. & Sigl, M. Volcanic stratospheric sulfur injections and aerosol optical depth from 500 BCE to 1900 CE. Earth Syst. Sci. Data9, 809–831 (2017). [Google Scholar]
- 26.Savarino, J., Romero, A., Cole-Dai, J., Bekki, S. & Thiemens, M. H. UV induced mass-independent sulfur isotope fractionation in stratospheric volcanic sulfate. Geophys. Res. Lett.30, 2131 (2003). [Google Scholar]
- 27.Baroni, M., Thiemens, M. H., Delmas, R. J. & Savarino, J. Mass-independent sulfur isotopic compositions in stratospheric volcanic eruptions. Science315, 84–87 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Plunkett, G., Sigl, M., McConnell, J. R., Pilcher, J. R. & Chellman, N. J. The significance of volcanic ash in Greenland ice cores during the Common Era. Quat. Sci. Rev.301, 107936 (2023). [Google Scholar]
- 29.Steffen, K. & Box, J. Surface climatology of the Greenland ice sheet: Greenland climate network 1995–1999. J. Geophys. Res. Atmos.106, 33951–33964 (2001). [Google Scholar]
- 30.Yin, J., Jull, A. J. T., Burr, G. S. & Zheng, Y. A wiggle-match age for the Millennium eruption of Tianchi volcano at Changbaishan, northeastern China. Quat. Sci. Rev.47, 150–159 (2012). [Google Scholar]
- 31.Shuman, C. A., Bromwich, D. H., Kipfstuhl, J. & Schwager, M. Multiyear accumulation and temperature history near the north Greenland ice core project site, north central Greenland. J. Geophys. Res. Atmos.106, 33853–33866 (2001). [Google Scholar]
- 32.Andersen, K. K. et al. Retrieving a common accumulation record from Greenland ice cores for the past 1800 years. J. Geophys. Res. Atmos.111, D15106 (2006). [Google Scholar]
- 33.Niemeier, U. et al. Initial fate of fine ash and sulfur from large volcanic eruptions. Atmos. Chem. Phys.9, 9043–9057 (2009). [Google Scholar]
- 34.Burke, A. et al. Stratospheric eruptions from tropical and extra-tropical volcanoes constrained using high-resolution sulfur isotopes in ice cores. Earth Planet. Sci. Lett.521, 113–119 (2019). [Google Scholar]
- 35.Lanciki, A., Cole-Dai, J., Thiemens, M. H. & Savarino, J. Sulfur isotope evidence of little or no stratospheric impact by the 1783 Laki volcanic eruption. Geophys. Res. Lett.39, L01806 (2012). [Google Scholar]
- 36.Gautier, E., Savarino, J., Erbland, J. & Farquhar, J. SO2 oxidation kinetics leave a consistent isotopic imprint on volcanic ice core sulfate. J. Geophys. Res. Atmos.123, 9801–9812 (2018). [Google Scholar]
- 37.Crick, L. et al. New insights into the ~ 74 ka Toba eruption from sulfur isotopes of polar ice cores. Clim. Past17, 2119–2137 (2021). [Google Scholar]
- 38.Baran, A. J. & Foot, J. S. New application of the operational sounder HIRS in determining a climatology of sulphuric acid aerosol from the Pinatubo eruption. J. Geophys. Res. Atmos.99, 25673–25679 (1994). [Google Scholar]
- 39.Guo, S., Rose, W. I., Bluth, G. J. S. & Watson, I. M. Particles in the great Pinatubo volcanic cloud of June 1991: the role of ice. Geochem. Geophy. Geosyst.5, Q05003 (2004). [Google Scholar]
- 40.Koffman, B. G., Kreutz, K. J., Kurbatov, A. V. & Dunbar, N. W. Impact of known local and tropical volcanic eruptions of the past millennium on the WAIS Divide microparticle record. Geophys. Res. Lett.40, 4712–4716 (2013). [Google Scholar]
- 41.Koffman, B. G. et al. Rapid transport of ash and sulfate from the 2011 Puyehue-Cordón Caulle (Chile) eruption to west Antarctica. J. Geophys. Res. Atmos.122, 8908–8920 (2017). [Google Scholar]
- 42.Plummer, C. T. et al. An independently dated 2000-yr volcanic record from Law Dome, East Antarctica, including a new perspective on the dating of the 1450s CE eruption of Kuwae, Vanuatu. Clim. Past.8, 1929–1940 (2012). [Google Scholar]
- 43.Kravitz, B. & Robock, A. Climate effects of high-latitude volcanic eruptions: role of the time of year. J. Geophys. Res. Atmos116, D01105 (2011). [Google Scholar]
- 44.Paik, S. et al. Impact of volcanic eruptions on extratropical atmospheric circulations: review, revisit and future directions. Environ. Res. Lett.18, 063003 (2023). [Google Scholar]
- 45.Stevenson, S., Fasullo, J. T., Otto-Bliesner, B. L., Tomas, R. A. & Gao, C. Role of eruption season in reconciling model and proxy responses to tropical volcanism. Proc. Natl. Acad. Sci. USA.114, 1822–1826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McConnell, J. R. et al. Synchronous volcanic eruptions & abrupt climate change ~17.7ka plausibly linked by stratospheric ozone depletion. Proc. Natl. Acad. Sci. USA.114, 10035–10040 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jensen, B. J. L. et al. Transatlantic distribution of the Alaskan white river ash. Geology42, 875–878 (2014). [Google Scholar]
- 48.Ruth, U., Wagenbach, D., Steffensen, J. P. & Bigler, M. Continuous record of microparticle concentration and size distribution in the central Greenland NGRIP ice core during the last glacial period. J. Geophys. Res. Atmos.108, 4098 (2003). [Google Scholar]
- 49.Bowen, H. J. M. Environmental Chemistry of the Elements (Academic Press, London, 1979).
- 50.McConnell, J. R., Lamorey, G. W., Lambert, S. W. & Taylor, K. C. Continuous ice-core chemical analyses using inductively coupled plasma mass spectrometry. Environ. Sci. Technol.36, 7–11 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Sigl, M. et al. A new bipolar ice core record of volcanism from WAIS Divide and NEEM and implications for climate forcing of the last 2000 years. J. Geophys. Res. Atmos.118, 1151–1169 (2013). [Google Scholar]
- 52.Jochum, K. P. et al. MPI-DING reference glasses for in situ microanalysis: new reference values for element concentrations and isotope ratios. Geochem. Geophys. Geosyst.7, Q02008 (2006). [Google Scholar]
- 53.Kuehn, S. C., Froese, D. G., Shane, P. A. & Participants, I. I. The INTAV intercomparison of electron-beam microanalysis of glass by tephrochronology laboratories: Results and recommendations. Quatern. Int.246, 19–47 (2011). [Google Scholar]
- 54.Rasmussen, S. O., Svensson, A. M., & Vinther, B. M. Greenland ice-core chronology 2005 (GICC05) annual layer depths for various Greenland ice cores. PANGAEA, 10.1594/PANGAEA.943195 (2022).
- 55.Stein, A. F. et al. NOAA’s atmospheric transport and dispersion modeling system. Bull. Am. Meteorol. Soc.96, 2059–2077 (2015). [Google Scholar]
- 56.Rolph, G., Stein, A. & Stunder, B. Real-time environmental applications and display system: READY. Environ. Modell. Softw.95, 210–228 (2017). [Google Scholar]
- 57.Burke, A. et al. Sulfur isotopes in rivers: insights into global weathering budgets, pyrite oxidation, and the modern sulfur cycle. Earth Planet. Sci. Lett.496, 168–177 (2018). [Google Scholar]
- 58.Tostevin, R. et al. Multiple sulfur isotope constraints on the modern sulfur cycle. Earth Planet. Sci. Lett.396, 14–21 (2014). [Google Scholar]
- 59.Paris, G., Sessions, A. L., Subhas, A. V. & Adkins, J. F. MC-ICP-MS measurement of δ34S and Δ33S in small amounts of dissolved sulfate. Chem. Geol.345, 50–61 (2013). [Google Scholar]
- 60.Le Bas, M. J., Le Maitre, R. W., Streckeisen, A. & Zanettin, B. A chemical classification of volcanic rocks based on the total alkali-silica diagram. J. Petrol.27, 745–750 (1986). [Google Scholar]
- 61.Machida, H., Moriwaki, H. & Zhao, D.-C. The recent major eruption of Changbai volcano and its environmental effects. In: Geographical Reports of Tokyo Metropolitan University25, 1–20, http://hdl.handle.net/10748/3613 (1990).
- 62.McLean, D. et al. Identification of the Changbaishan ‘Millennium’ (B-Tm) eruption deposit in the Lake Suigetsu (SG06) sedimentary archive, Japan: Synchronisation of hemispheric-wide palaeoclimate archives. Quat. Sci. Rev.150, 301–307 (2016). [Google Scholar]
- 63.Rasmussen, S. O. et al. Concentration of hydrogen ions measured with the electrical conductivity method (ECM) on the NGRIP ice core. PANGAEA, 10.1594/PANGAEA.831526 (2013).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
The data used for manuscript figures are available on Figshare, with the Supplementary Data under identifier 10.6084/m9.figshare.26549344.v1 and the Supplementary Tables under identifier 10.6084/m9.figshare.26550304.v1. Supplementary Tables are also available in the Supplementary Information.