

Abstract
Background
The use of mobilizing agents for hematopoietic stem cell (HSC) transplantation is insufficient for an increasing number of patients. We previously reported lipid made endocannabinoid (eCB) ligands act on the human bone marrow (hBM) HSC migration in vitro, lacking long term stability to be therapeutic candidate. In this study, we hypothesized if a novel 2-AG-loaded polycaprolactone (PCL)-based nanoparticle delivery system that actively targets BM via phosphatidylserine (Ps) can be generated and validated.
Methods
PCL nanoparticles were prepared by using the emulsion evaporation method and characterized by Zetasizer and scanning electron microscopy (SEM). The encapsulation efficiency and release profile of 2-AG were determined by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The presence of cannabinoid receptors (CBRs) in HSCs and monocytes was detected by flow cytometry. Cell morphology and viability were assessed using transmission electron microscopy (TEM), SEM, and the WST-1 viability assay. The migration efficacy of the 2-AG and 2-AG-loaded nanoparticle delivery system on HSCs and HPSCs (TF-1a and TF-1) and monocytes (THP-1) was evaluated using a transwell migration assay.
Results
The 140–225 nm PCL nanoparticles exhibited an increasing polydispersity index (PDI) after the addition of Ps and 2-AG, with a surface charge ranging from − 25 to -50 mV. The nanoparticles released up to 36% of 2-AG within the first 8 h. The 2-AG-Ps-PCL did not affect cellular viability compared to control on days 5 and 10. The HSCs and monocytes expressed CB1R and CB2R and revealed increased migration to media containing 1 µM 2-AG-Ps-PCL compared to control. The migration rate of the HSCs toward monocytes incubated with 1 µM 2-AG-Ps-PCL was higher than that of the monocytes of control. The 2-AG-Ps-PCL formulation provided a real time mobilization efficacy at 1 µM dose and 8 h time window via a specific CBR agonism.
Conclusion
The newly generated and validated 2-AG-loaded PCL nanoparticle delivery system can serve as a stable, long lasting, targeted mobilization agent for HSCs and as a candidate therapeutic to be included in HSC transplantation (HSCT) protocols following scale-up in vivo preclinical and subsequent clinical trials.
Graphical Abstract

Keywords: Cannabinoid, Endocannabinoid, Hematopoietic stem cell, Transplantation, Bone marrow, Mobilization, Nanoparticle delivery system
Background
Allogeneic hematopoietic stem cell transplantation (HSCT) is the only treatment procedure that offers curative effects or long-term survival for a variety of pediatric and adult patients [1–3]. G-CSF and biosimilars are the sole cytokines used for mobilization according to the European Society for Blood and Marrow Transplantation (EBMT) due to the unavailability of GM-CSF withdrawal [3]. However, the combined use of G-CSF and the CXCR4 antagonist plerixafor can cause mobilization failures in up to 5% of multiple myeloma patients [4–6] and in up to 35% of lymphoma [4, 7, 8] patients and plerixafor is not the first choice in most centers due to its increased cost [9]. Therefore, recent trials (190 studies in the ClinicalTrials.gov database as of May 2024) have focused on improving the application protocols of existing mobilizers [7, 8], encapsulating them to increase efficiency [10–13], and discovering new mobilizers [14, 15].
Rodent and human bone marrow (BM) [16–20] comprises endocannabinoid (eCB) ligands and receptors (CBRs). The CBs AEA and 2-AG act as chemoattractants via CB1Rs when applied to CB2R-overexpressing murine myeloid leukemia cells [18] and human BM mononuclear cells (BM-MNCs) [19]. The addition of the nonselective CB1R and CB2R agonist cannabidiol to a single dose of G-CSF increases granulocyte-macrophage colony-forming unit (GM-CFU) mobilization compared to G-CSF alone in mice [20]. Similarly, eCBs function in mobilization by stress-induced sympathetic hyperactivity in human BM [16–18]. Our group previously reported that human BM LPS-stimulated MSCs secrete AEA and 2-AG and that 2-AG levels are significantly greater in the peripheral blood (PB) of G-CSF-treated donors than in BM plasma. We also reported that AEA and 2-AG specifically induced the migration of human BM hematopoietic stem cells (hBM-HSCs) on a coculture platform simulating peripheral mobilization [19]. The results suggest that 2-AG is a strong candidate for enhancing or facilitating G-CSF-mediated HSC migration under stress conditions in a clinical setting [19]. Despite the promising potential of eCBs for their psychoactive effects [20], their low aqueous solubility, instability and oily resin nature [21, 22] hinder the development of effective eCB formulations. Therefore, eCBs must be encapsulated in a controlled release system that targets the selected tissue. Several untargeted characterization studies revealed increased stability of AEA, CB13, THC, CBD [23–27], and CSR141716, rimonabant, AM251, and URB597 [28, 29] following their incorporation into PCL [23, 27], poly(lactic-co-glycolic acid) (PLGA) [24–26], and liposome [28, 29] nanoparticles for intranasal and oral administration, aiming to alleviate neuropathic pain and induce tumor apoptosis. Some non-CBR mediators passively targeted the BM (nanoparticles less than 400 nm in size) [30] and actively targeted the BM using the mononuclear phagocyte system (MPS) with surface modifications involving phosphatidylserine (Ps) [31–39].
We previously reported CB components involvement in the BM, by revealing endogenous CB ligand 2-AG triggers hBM-HSC migration via CBRs. However, lipid made endocannabinoid ligands lacking long term stability and bioavailability [21, 22] are not suitable to be used as therapeutics as mobilizing agents. We hypothesized that a novel biocompatible PCL nanoparticle delivery system, which provides controlled release of 2-AG and active targeting to the BM, enabling the induction of the mobilization of hBM-HSCs in vitro can be generated and validated. Therefore, we developed a PCL-based nanoparticle delivery system that actively targets the BM via the MPS and slowly releases the encapsulated 2-AG. The results revealed for the first time the long-term mobilizing efficiency of the novel BM targeting PCL-based nanoparticle delivery system on hBM-HSCs and monocytes through a specific CBR agonism at a real time defined 1 µM dose and 8 h time window. The novel formulation has been approved as a patent pending by the Turkish Patent and Trademark Office (Application no: TR2021/013362) and the Patent Cooperation Treaty (PCT) (Application no: PCT/TR2022/050893). The candidate formulation can take place in HSCT protocols in hemato-oncology practice following scale-up preclinical experiments of GLP-manufactured products and GMP-based phase trials.
Methods
We designed a prospective, randomized, controlled in vitro study. The independent variables were time (1, 3, 5, 7 and 10 days), group (control and 2-AG-, 2-AG-PCL- or 2-AG-Ps-PCL-treated or nontreated CD34+/CD38- hBM-HSCs (TF-1), CD34+/CD38 + hBM-HSCs (TF-1a) and monocytes (THP-1)], and the dependent variables were CBR, particle size; PDI; zeta potential measurement of nanoparticles; EE of 2-AG; release profile of nanoparticles; and cell viability and migration. Biological replicates were subjected to power analysis (G-Power v3.1). During the preparation of this work the authors did not use Artificial Intelligence (AI). The experimental setup is illustrated schematically in Fig. 1a-d.
Fig. 1.

Schematic design of the study. A We detected CB1R and CB2R expression in TF-1, TF-1a and THP-1 cells. B We prepared the nanoparticles using the emulsion evaporation method and characterized them by analyzing their PDI, zeta potential, surface charge, size, morphology, EE, release profile and invasion profile using DLS, SEM, LC‒MS/MS and in vitro invasion assays, respectively. C We analyzed the effect of nanoparticles on TF-1, TF-1a and THP-1 cell viability using SEM, TEM and MTT assays. D We detected the migratory effect of nanoparticles on TF-1, TF-1a and THP-1 cells using a transwell migration assay
Preparation and characterization of 2-AG-Ps-PCL nanoparticles
2-AG-loaded PCL nanoparticles were prepared by using the emulsion evaporation method [40]. PCL (0.1% (w/v)) with two different molecular weights (MW: 10 and 45 kDa), 0,005% (w/v) 2-AG and 0,005% (w/v) Ps were dissolved in DCM at 550 rpm. This organic phase was added to an aqueous phase containing 0.5% (w/v) surfactant (Pluronic™ F-68) at a 1:2 (v/v) ratio and homogenized. The Y/S emulsion was evaporated at 100 rpm and 40 °C for 30 min and filtered (0.45 μm pore-size PET filter).
A Malvern Zetasizer Nano ZS (Worcestershire, UK) instrument with dynamic light scattering (DLS) was used to determine the particle size, PDI, zeta potential and surface charge [41, 42]. Briefly, nanoparticle dispersions were diluted to 1:1 (v: v) in ultrapure water and transferred to a folded capillary zeta cell (DTS 1070). The mean diameter and PDI were measured at angles of 173° and 25 °C, respectively, and the surface charge of the nanoparticles at RT was measured at an angle of 12.8° in ultrapure water (n = 3).
SEM was used to quantitively assess the ultrastructural properties of the nanoparticles via DLS [42]. Briefly, lyophilized and gold-palladium (Au/Pd) alloy-coated (10 nm) nanoparticle dispersions were photographed, and the particle sizes were measured at 40,000x. The ultramicrographs were captured with a high-voltage scanning electron microscope (FEI, Quanta 200 F) attached to a digitalized image analysis system (Xt microscope control).
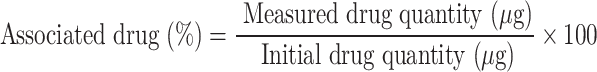
A validated liquid chromatography-tandem mass spectrometry (LC‒MS/MS) method was used to determine the EE of 2-AG and release profile [19] by using a C18 column (Hypersill-ODS4, 50 × 3.0 mm, 2.1 μm) with a mobile phase of acetonitrile and water (both containing 0.1% formic acid) to accomplish chromatographic separation at a 0.3 ml/min flow rate, and gradient elution was applied. Synthetic cannabinoid ACPA was used as an internal standard. A daily calibration curve of 2-AG was prepared at 6 different concentrations (1–1000 ng/ml) and constructed with the peak area ratio of 2-AG to IS.
The EE of 2-AG was expressed as the associated drug percentage (%) and was calculated as follows:
 |
To quantify the amount of nanoparticle-bound 2-AG, the loaded nanoparticles were centrifuged at 3500 rpm. The supernatant was lyophilized and dissolved in dichloromethane and removed under a nitrogen atmosphere. The amount of 2-AG was measured by LC‒MS/MS [19, 43].
The dialysis membrane technique was used to determine the 2-AG release profile under sink conditions at 37 °C. Briefly, nanoparticle dispersions were placed in a dialysis membrane (Cellulose Membrane MWCO: 14 kDa) with 0.1% Tween 80. Samples were taken at 30 min, 1, 4, 8, 12, and 24 h, and the amount of free 2-AG in the release medium was determined by LC‒ESI‒MS/MS [19, 43].
Cell culture
For this study we used hBM-HSC cell line (CD34+/CD38- (TF-1), #CRL2003, ATCC), hBM-HPSCs cell line (CD34+/CD38+ (TF-1a), #CRL2451, ATCC) and human blood monocyte cell line (THP-1, TIB202, ATCC). TF-1 cells were expanded in RPMI-1640 media (ATCC) supplemented with 10% fetal calf serum (FCS, Gibco) and 2 ng/ml recombinant human GM-CSF (#300-03, PeproTech) [44]. TF-1a cells were cultured in RPMI-1640 media supplemented with 10% FCS [45]. Monocytes (THP-1, #TIB202, ATCC) were cultured in RPMI-1640 media (ATCC) supplemented with 10% FCS (GIBCO) and 0.05 mM 2-mercaptoethanol (#60-24-2, Calbiochem) [46]. To be used in the BBB experiment, cerebral endothelial cells (hCMEC/D3 cell line, #SCC066, Merck/Millipore) were seeded in collagen (Collagen Type I, #08-115, Sigma)-coated culture dishes with EndoGRO media (#SCME001, Millipore) supplemented with 1 ng/mL FGF-2 (#GF003, Sigma) [47]. All cells were incubated at 37 °C in 5% CO2, and the media was changed every 3–4 days.
Flow cytometry
The permeabilized (Permeabilizing Solution 2, #340973, BD Biosciences, USA) cells were indirectly immunolabeled using rat anti-human CB1R (#ab3558, Abcam), CB2R (#ab3561, Abcam) and a secondary antibody (goat anti-rabbit IgG FITC, #ab7086, Abcam) [48, 49]. The IF-labeled cells were measured by a Novocyte Adventeon FC (ACEA) and analyzed using Novoexpress software with 10,000 events [50].
Transmission and scanning electron microscopy (TEM and SEM)
For TEM analysis, cells were fixed in 2.5% glutaraldehyde, postfixed in 1% osmium tetroxide and dehydrated in a graded series of ethanol as previously described [51]. Ultrastructural integrity was analyzed on uranyl acetate- and lead citrate-stained thin sections by transmission electron microscopy (TEM) (JEOL, JEM1400, Japan) with an attached digital camera (Orius, Germany) [52, 53].
For SEM analysis, the samples were fixed with 2.5% glutaraldehyde, dehydrated in a graded series of ethanol and sputter-coated (Gatan, PECS-682, USA) with 10 nm thick Au/Pd after drying with a critical point dryer (Tousimis, Autosamdri 815B, USA). The ultramicrographs were captured with a high-voltage scanning electron microscope (FEI, Quanta 200 F) attached to a digitalized image analysis system (Xt microscope control) [42].
Viability assay
WST-1 (#ab155902, Abcam) was used to analyze the effect of the nanoparticle carrier system on THP-1, TF-1, and TF-1a cell viability on days 1, 3, 5, 7, and 10 [54, 55]. As a control, (i) cells in regular growth media and (ii) cells containing 2-AG obtained according to release data were used. The absorbance was measured at an optical density of 450 nm and a reference wavelength of 620 nm by a spectrophotometer (BMG Labtech). The half-maximal inhibitory concentration (IC50) values for 2-AG-Ps-PCL were calculated on day 10 by GraphPad Prism Version 10.1.2 (La Jolla, CA, USA).
In vitro BBB invasion method
hCMEC/D3 cells were expanded on the filter of collagen-coated transwell culture dishes (#3401, Corning) for 5 days. We added Ps-PCL or 2-AG-Ps-PCL nanoparticles to the cells at concentrations of 0.1, 1, 10, and 100 µM. On days 1, 3, 5, and 10, we analyzed the number of nanoparticles that passed to the lower part of the transwell plate using SEM (QUANTA 400 F field emission microscope) (n = 4) [56].
Transwell migration assay
According to the first and second experimental setups (Fig. 1d), we placed TF1, TF1a or THP-1 cells (5 × 104 cells/well) on transwell filters, and 2-AG, 2-AG-PCL or 2-AG-Ps-PCL nanoparticles with or without AM251 and AM630 were placed in the bottom chambers of the transwell plates. For the third experimental setup (Fig. 1d), THP-1 cells were incubated with 2-AG-Ps-PCL nanoparticles for 24 h, and then the bottom chambers of the transwell plates were placed with or without AM251 and AM630. TF1 and TF1a cells were placed in the upper part of the transwell. The cells were allowed to migrate for 4 h at 37 °C. Cell counting was performed using a Bürker slide. We used 4 concentrations of 2-AG (0.1, 1, 10, and 100 µM) w/wo AM281 and AM630 (10 µM, #1115, and #1120, respectively; Tocris Bioscience) [19]. The half-maximal effective concentration (EC50) values for 2-AG-Ps-PCL were calculated for THP-1, TF-1, and TF-1a cells using GraphPad Prism Version 10.1.2.
Statistical analysis
The normality of the data distribution was evaluated by the Shapiro–Wilk test. Multiple comparisons were performed by analysis of variance (ANOVA) and the Kruskal‒Wallis test. The parametric data are presented as the mean ± S.E.M.; the nonparametric data are presented as the median, minimum and maximum values. Analyses were performed at 95% confidence intervals.
Results
PCL nanoparticles were characterized
The particle size of the PCL nanoparticles was determined to be between 140 and 225 nm (Fig. 2a). The addition of Ps increased the particle size (p < 0.05). Compared with the 2-AG-unloaded nanoparticles, the 2-AG-loaded nanoparticles did not change the diameter of the nanoparticles (p > 0.05). The addition of Ps and 2-AG to the PCL nanoparticles increased the PDI (p < 0.05). PCL MW did not affect the critical quality parameters of the blank, Ps-coated or 2-AG-loaded nanoparticles (p > 0.05) (Fig. 2a). The surface charges of the nanoparticles ranged from − 25 mV to -50 mV (Fig. 2a). SEM revealed that the diameter of the nanoparticles was approximately 150–500 nm, as globular particles containing an electron lucent halo at their periphery (Fig. 2b-g).
Fig. 2.

The 2-AG-Ps-PCL system provides sustained release. A Particle size, PDI and zeta potential of the nanoparticles (n = 3). B,C,D,E,F,G SEM micrographs of nanoparticle formulations (B) blank PCL (MW: 10 kDa), (C) 2-AG-PCL (MW: 10 kDa), (D) 2-AG-Ps-PCL (MW: 10 kDa), (E) blank PCL (MW: 45 kDa), (F) 2-AG-PCL (MW: 45 kDa), and (G) 2-AG-Ps-PCL (MW: 45 kDa). NPs were observed in the range of approximately 200–500 nm as globular particles with an electron lucent halo around the periphery (white arrows). Scale bar: 40 μm, ×2.500. H, i (h) Associated 2-AG (%) and (I) cumulative 2-AG release of nanoparticle formulations (n = 3, ±SD)
The amount of nanoparticle-bound 2-AG was similar for each formulation (10 and 45 kDa MW PCL w/wo Ps) (p > 0.05). The difference between the EE of nanoparticles prepared with two different MWs was not statistically significant, as was the case for other critical quality attributes (p > 0.05). The addition of Ps to the formulations decreased the EE of 2-AG (p < 0.05, Fig. 2h). The highest release of 2-AG was observed for the PCL nanoparticles (MW 45 kDa), with 36% released in the first 8 h. The release of 2-AG from the Ps-coated nanoparticles appeared to be slower than that from the nanoparticles without Ps (Fig. 2i). In the BBB invasion analysis, nanoparticles were not observed in the samples by SEM on days 1, 3, 5 and 10 or at different concentrations of nanoparticles. Although the MW of PCL did not affect the critical quality parameters of the nanoparticles, 10 kDa MW PCL nanoparticles were used in subsequent experiments because the stability (PDI values, Fig. 2a) of the nanoparticles was greater.
THP-1, TF-1 and TF-1a cell viability was maintained in culture with 2-AG-Ps-PCL nanoparticles
The THP-1 cells incubated with the 2-AG-Ps-PCL nanoparticles had a typical monocyte appearance with a euchromatic round nucleus and cytoplasm rich in lysosomes (Fig. 3a). Apoptotic bodies were not observed in any of the experimental groups (Fig. 3a). SEM revealed that the cells presented a typical monocyte character and had a healthy appearance. We did not observe any apoptotic bodies in the evaluation (Fig. 3b).
Fig. 3.

Ultrastructural morphology of THP-1 cells and viability of THP-1, TF-1 and TF-1a cells maintained in culture with 2-AG-Ps-PCL nanoparticles. A TEM micrographs of THP-1 monocytes incubated with 2-AG-Ps-PCL nanoparticles on days 1, 3 and 5. Vesicles in the control cells are indicated by white arrows, and electron-dense vesicles in the experimental groups are shown in micrographs with black arrows at 12,000×. B SEM micrographs of THP-1 monocytes incubated with 2-AG-Ps-PCL nanoparticles on days 1, 3 and 5. C The effects of the 2-AG-Ps-PCL nanoparticles and control groups on THP-1, TF-1 and TF-1a hematopoietic cell viability are shown in a bar graph, *p < 0.05 by one-way analysis of variance (ANOVA) (n = 3, ±SD). D Dose-inhibition response curve and logIC50, IC50 and R2 values for 2-AG and 2-AG-Ps-PCL in the THP-1, TF-1 and TF-1a cell lines (n = 3, ±S.E.M.)
On days 1, 3 and 7, the viability of THP-1 cells in the group treated with 2-AG-Ps-PCL (PCL MW 10 kDa) did not differ from that in either the control group or the other groups at any concentration (p > 0.05, Fig. 3c). On day 5, compared with that in the control ‘Cell’ group, the viability of monocytes in the medium containing 2-AG and the 2-AG-containing nanoparticle carrier system (2-AG-Ps-PCL) at a concentration of 100 µM was adversely affected (p < 0.05). On day 10, medium containing 10 µM active 2-AG decreased THP-1 cell viability (Fig. 3c).
Compared with the control and other treatments, 2-AG-Ps-PCL did not affect the viability of TF-1 cells on days 1, 3, or 5 at any concentration. On day 7, compared with the control, 10 µM 2-AG decreased TF-1 cell viability (p < 0.05, Fig. 3c). This decrease was also observed in media containing 2-AG on day 10. Compared with that in the control ‘Cell’ group, the viability of TF-1 cells in medium containing 0.1, 10 or 100 µM 2-AG was adversely affected (Fig. 3c). When the effect of the nanoparticle carrier system on TF-1a cell viability was examined at all-time points, the group containing 2-AG-Ps-PCL did not significantly change cell viability compared to that of the controls, both for the control and the other groups, at each concentration (Fig. 3c).
The IC50 values in THP-1, TF-1 and TF-1a cells on the 10th day were 4.56, 12.10 and 40.51 µM for 2-AG and 20.7, 155.3 and 2.4 µM for 2-AG-Ps-PCL, respectively (Fig. 3d).
Human BM-HSCs express high levels of CB1Rs and low levels of CB2Rs
TF1 cells expressed 80.32% CB1R and 13.93% CB2R (Fig. 4a). TF1a cells expressed 41.62% CB1R and 18.75% CB2R (Fig. 4b).
Fig. 4.

The nanoparticle carrier system has a dose-dependent effect on CBR-mediated HSC migration. A CB1R and CB2R labeling of TF1 hematopoietic cells using only secondary antibodies was detected at rates of 0.01% and 1.68%, respectively. A total of 80.32% of the CB1R-labeled cells and 13.93% of the CB2R-labeled cells were labeled. B CB1R and CB2R labeling of TF1a hematopoietic cells without using only secondary antibodies was detected at rates of 0.05% and 1.99%, respectively. A total of 41.62% of the CB1R-labeled cells and 18.75% of the CB2R-labeled cells were labeled. C,D,E The migratory effects of (C) 2-AG, (D) 2-AG-PCL and (D) 2-AG-Ps-PCL on human monocyte THP-1, TF-1 and TF-1a hematopoietic cells. F,G,H Migration of (F) THP-1, (G) TF-1 and (h) TF-1a hematopoietic cells to different 2-AG concentrations and nanoparticles in different formulations containing different 2-AG concentrations. I Dose-migration response curves and EC50 values for 2-AG, 2-AG-PCL and 2-AG-Ps-PCL in THP-1, TF-1 and TF-1a cells (n = 3, ±S.E.M.). J,K The effects of (J) TF-1 and (K) TF-1a hematopoietic cells on the migration of THP-1 cells incubated with 2-AG-Ps-PCL are shown by a bar graph; *p < 0.05 according to one-way analysis of variance (ANOVA) (n = 3, ±SD)
The nanoparticle carrier system is effective in CBR-mediated HSC migration in a dose-dependent manner
THP-1 cells migrated to media containing 2-AG, 2-AG-PCL and 2-AG-Ps-PCL at a concentration of 1 µM at a higher rate; however, they migrated at a lower rate to the other concentration groups (0.1, 10, 100 µM) compared to the control ‘media’ group (Fig. 4c). This migration effect was significantly inhibited by CBR antagonists (AM281 and AM630) (p < 0.05). The applied doses provided a similar migration capacity for the 2-AG, 2-AG-PCL and 2-AG-Ps-PCL groups (ns, Fig. 4f).
TF1 and TF-1a cells migrated to media containing 2-AG, 2-AG-PCL and 2-AG-Ps-PCL at a concentration of 1 µM at a high rate; however, they migrated at a low rate to the other concentrations (0.1, 10, 100 µM) (Fig. 4d, e). In addition, TF-1a cells migrated to media containing 10 µM 2-AG or 2-AG-Ps-PCL at a high rate (Fig. 4e). This migration effect was significantly inhibited by the CBR antagonists AM281 and AM630 (p < 0.05). There were no significant differences in migration at any of the tested doses (ns, Fig. 4g, h). The half-maximum effective concentration (EC50) values of 2-AG-Ps-PCL in TF-1 and TF-1a cells were 6.1 and 0.1 µM, respectively (n = 3, mean ± S.E.M.) (Fig. 4ı).
TF-1 cells migrated to monocytes after incubation with 1 or 10 µM 2-AG-Ps-PCL at a high rate (p < 0.05) or with 0, 1 or 100 µM 2-AG-Ps-PCL at a low rate compared to those in the control ‘Media’ group (Fig. 4j). CBR antagonists (AM281 and AM630) significantly inhibited the migration of TF-1 cells at a concentration of 1 µM (p < 0.05) (Fig. 4j). Compared with control ‘Media’, TF-1a cells migrated to THP-1 cells incubated with 1 µM 2-AG-Ps-PCL at a higher rate and with 0, 1, 10 and 100 µM 2-AG-Ps-PCL at a lower rate (Fig. 4k). CBR antagonists (AM281 and AM630) significantly inhibited the migration of TF-1a cells at a concentration of 1 µM (p < 0.05, Fig. 4k).
Discussion
In this study, we developed an active BM-targeted, 2-AG-loaded PCL-based nanoparticle delivery system and demonstrated its effectiveness in vitro to eliminate side effects and ensure its stability as a candidate for clinical use. FDA-approved PCL nanoparticles have been the focus of several drug delivery studies due to their biocompatibility and tailored release properties, especially for lipophilic molecules [57–59]. We preferred the Y/S single emulsion method, considering that 2-AG, a lipophilic active substance, would be used and that the nanoparticles would be coated with Ps, which is insoluble in solvents such as ethanol and acetone [60]. We determined the particle size of the PCL nanoparticles to be between 140 and 225 nm, suggesting a favorable passive targeting ability and longer circulation time [23, 30, 33]. The addition of Ps increases the particle size, and thus, an effective surface coating of nanoparticles with Ps has been determined [61]. However, 2-AG did not change the diameter of the nanoparticles, suggesting that 2-AG is mostly encapsulated within the nanoparticle matrix, which was also confirmed by the nano-sized globular ultrastructure. Previously, several particles with various active groups and materials that were passively or actively targeted to the BM reached a range between 75 and 400 nm [11, 30, 32–37], and a particle size below 250 nm was considered appropriate for passive targeting [30, 33]. Since the sizes of the nanoparticles we synthesized were between 140 and 225 nm, we can theoretically target the BM. Similar to our system, AEA loaded with 83.52 ± 21.38 nm PCL nanoparticles did not increase the particle size by the light scattering method, thus maintaining the particle size that provides passive targeting to the BM [23]. Our group previously loaded the CB1R ligand ACPA into PCL nanoparticles using a nanoprecipitation method, revealing a mean particle size of 162.2 ± 2.3 nm for APCA-PCL nanoparticles [51]. The fact that particle sizes are different despite using the same polymer may be due to the synthesis of nanoparticles using different methods [62]. However, the smaller size of the particles we produce provides an advantage for active targeting [30, 33]. We developed a nanoparticle delivery system for 2-AG with promising characteristics and an appropriate particle size for passive targeting to the BM and effective surface coating using Ps, which is consistent with previous literature on nanoparticle-based drug delivery systems using CBs [23, 51] and targeting the BM [11, 30, 32, 34–37].
In this study, we targeted nanoparticles with a low PDI (below 0.5). Previously, changes in the MW of PCL derivatives were shown to affect the average particle size of nanoparticles [63]. In particular, nanoparticles prepared with a PCL MW of 10 kDa had smaller particle sizes than nanoparticles prepared with a PCL MW of 45 kDa. The addition of Ps affected the PDI, and 2-AG made the nanoparticle dispersion more heterogeneous than that of nanoparticles without Ps but still within the acceptable range (below 0.7) for drug delivery [64]. In this study, the surface charges of the prepared nanoparticles ranged from − 25 mV to -50 mV. Nanoparticles with a zeta potential between − 25 mV and + 25 mV present stable colloidal systems [65]. Thus, the nanoparticles obtained in the present study have a net negative charge in terms of surface charge, and their aggregation risk is low. Previous studies revealed that PCL nanoparticles maintain their particle size for 30 days [66, 67]. In line with these studies, in our study, non-Ps-coated PCL (with MWs of 10 and 45 kDa) nanoparticles remained stable for 30 days, and the particle sizes did not exceed 250 nm. In a previous study by our group, the PDI and zeta potential of APCA-loaded PCL nanoparticles (PCL MW 80 kDa) were 0.251 ± 0.008 and − 29.4 ± 0.6 mV, respectively [51]. In parallel with this study, the PDI and zeta potential were above 0.4 and − 25 mV, respectively. This difference was attributed to the Ps in the structure. Consequently, by showing that the nanoparticles we developed remained stable for 7 days, we proved that we could determine the time interval at which the 2-AG content in the BM was released by targeting the BM via the MPS system of the nanoparticular system containing 2-AG, which can be applied to PB [31].
Here, we report that all formulations released no more than 36% of the 2-AG they encapsulated during the first 8-hour period. In parallel with our study, the EEs were 96.05 ± 1.77% and 39.9 ± 14.7% for AEA [23] and ACPA [51], respectively, in PCL nanoparticles according to HPLC. Similar to our data (2-AG-Ps-PCL (MW 45 kDa)), 50% of the AEA was released at the end of day 1, the stability of the AEA in suspension increased [23], and the sustained cumulative release of ACPA from the nanoparticles was 63.9% for a period of 7 days [51]. In our study, it was determined that there was 25% release on day 1 and 30% release on the 6th day. The low EE and cumulative release values were attributed to the lipophilic nature of AEA and ACPAs, such as 2-AG, and therefore, the low stability and absence of a lipid core in the PCL nanoparticles [26, 29]. Also, in our study the release of 2-AG from the Ps-coated nanoparticles appeared to be slower than that from the nanoparticles without Ps. This resulted in lower release profile compared to previous studies. However, the EE of cannabidiol- or THC-loaded PCL nanoparticles was 99.09 ± 5.14% and 84.55 ± 13.6%, respectively [27]. Although the amount of nanoparticle-bound 2-AG was similar for each formulation, we observed that the EE of 2-AG decreased with the addition of Ps to the formulations. The release appears to be slower in Ps-coated nanoparticles than in nanoparticles without Ps. Similar results were obtained in a study with Ps-coated PLGA nanoparticles, where Ps-coated nanoparticles encapsulated a lower amount of a lipophilic substance, α-tocopherol, than non-Ps-coated nanoparticles [68]. This can be attributed to the lipophilic nature and negative surface charge of Ps. Ps may hinder encapsulation/adsorption and slow the release of 2-AG, which is also a lipophilic molecule with a strong affinity for PCL [69–71].
Using an in vitro BBB invasion model, we determined that the nanoparticles did not cross the BBB. Theoretically, 2-AG-containing nanoparticles cannot pass through the BBB due to their size of 50–250 nm [72]. Considering the studies carried out to increase the half-life of G-CSF, previous groups preferred to use PEGylated dextran-PLGA microspheres [11], liposomes [10, 13] and polymeric micelles [12]. PEGylated nanoparticles remain in circulation longer than non-PEGylated nanoparticles; however, they do not actively direct them to the BM [10–13]. Since our method of targeting the BM involves active targeting of monocytes via Ps, this drug delivery system is expected to promote monocyte accumulation in the BM along with sites of destruction [31, 68, 73]. In this study, we reported for the first time that a novel nanoparticle release system did not cross the BBB under in vitro conditions and provided preliminary data for in vivo studies.
In the present study, at a concentration of 1 µM, the phagocytosed nanoparticles did not interfere with THP-1 cell viability, which was determined to be a safe dose. Additionally, the nanoparticles in the monocyte cytoplasm were stained darkly and clearly separated from the vesicles in the nanoparticle-untreated control cells, and no apoptotic bodies were observed following 2-AG-Ps-PCL application. In a study targeting a mechanism similar to the HSC migration pathway we targeted through monocytes, PCL composite nanoparticles were incubated with cells for 2 days, and as a result of the MTT analysis, it was determined that cell viability was not affected [74]. In our study, we showed that our release system did not affect cell viability on days 1 and 3 with MTT analysis. In another study with a similar hypothesis, the treatment of atherosclerosis was aimed at providing mannitol and rapa active substances to macrophages (RAW264.7) via PHEA-g-RhB-g-SUCC-PCL nanoparticles. After the incubation of nanoparticles at 3 different concentrations for 1 day, MTS analysis was performed, and it was determined that cell viability was not affected [75]. Similarly, in our study, we determined that the nanoparticle release system containing 4 different doses did not affect cell viability on the first day. When the literature information was compared with the data obtained within the scope of this study, it was concluded that the results obtained were compatible and thus we have proven that we have developed a nanoparticle delivery system that can be used safely in terms of cell viability for our main goal, HSC migration.
In this study, TF-1 and TF-1a human HSCs expressed 80.32% and 41.62% of CB1R and 13.93% and 18.75% of CB2R, respectively. There are limited publications reporting CB1R and/or CB2R distribution in BM-HSCs [19, 76]. Our group previously showed that human primary BM-HSCs express 59.2 ± 27.7% of CB1Rs and 43.2 ± 23.0% of CB2Rs according to FC, which was confirmed by RT‒PCR [19]. In the previous study, BM-HSCs were selected as CD34 + and CD38+, which correspond to TF-1 cells in our study and their CBR expression rates are compatible. Rodent BM-MNCs express 27.4% of CB1Rs via FC [76]. Since BM-MNCs contain a wide variety of cell types, including stromal cells, a lower percentage of CBR expression compared to BM-HSCs is an expected result [19]. Our data are in line with previous studies investigating CB1R and CB2R expression in primary hBM-HSCs [19] and rodent BM-MNCs [76] and support that CB1R is expressed at higher levels than CB2R in human primary BM-HSCs [19]. With the results we obtained, we determined the presence of CBR in two different subtypes of HSCs, which is the first step in achieving the migratory effect we aimed to achieve in our study.
In this study, NPs containing 2-AG and 2-AG at a concentration of 1 µM (EC50 values ranging between 0.31 and 1.35 µM) had a migratory effect on human HSCs in a CBR-mediated and dose-dependent manner, as in our previous study [19]. Additionally, we showed that the Ps content added to the nanoparticles also supported the migration effect. Thus, we not only tested our own results but also analyzed the migration of HSCs as well as monocytes, which has not been previously reported in the literature. We previously observed an increase in the number of HSCs migrating toward 1 µM 2-AG compared to the migration facilitated by the SDF-1 chemoattractant [19]. The results showed that 2-AG dose-dependently and specifically increased HSC migration mainly through CB2Rs [19]. When 1 or 0.25 µM 2-AG was applied to CB2R-overexpressing rodent-derived myeloid leukemia cells and mouse BM-MNCs in vitro in a transwell assay, the cells migrated after 4 h [77, 78]. The dose of 2-AG providing migration was the same in this study. In another in vitro study using the transwell assay, 2-AG at a dose of 0.25 µM allowed the migration of C3H/HeJ mouse BM MNCs in 4 h [78]. In our study, we used 2-AG at a concentration of 0.1 µM as the lowest dose, and although we achieved a migratory effect at this concentration, we observed the greatest effect at a concentration of 1 µM. Overall, the effective dose range for 2-AG appears to be in the range of 0.25 to 10 µM [19, 77, 78]. The Ki values of 2-AG for CB1 and CB2 receptors, of which 2-AG is a full agonist, are 3.42 ± 3.3 and 1.20 ± 0.33 µM, respectively [79]. In our study, we found that the EC50 values for CB1R and CB2R ranged between 0.31 and 1.35 µM. Therefore, the EC50 values included the Ki values of 2-AG for CB1R and CB2R. Taken together, 2-AG and 2-AG at a concentration of 1 µM had a migratory effect on human HSCs in a CBR-mediated and dose-dependent manner.
These preclinical study results are somewhat limited because they were derived from an in vitro study due to ethical considerations. One of the main aims of this study was to minimize the psychoactive effects of CBs on the CNS. Before clinical studies can be conducted, in vivo animal experiments should be performed to examine the possible distribution of nanoparticles in the body by labeling them with markers and determining whether they pass the BBB, which we identified in vitro in this study. Although we measured the low EE of 2-AG-Ps-PCL [68], we determined that with a sufficient release profile of 2-AG, the migration effect that we obtained in in vitro studies can be achieved in in vivo studies [23, 26, 29, 51]. In this study, we chose to use HSC and HPSC cell lines (TF-1a and TF-1, respectively) instead of human bone marrow-derived primary cells for several reasons. Ethical concerns and the limited availability of human samples posed significant challenges. The methods used required a large number of cells, which would be difficult to obtain from primary human samples. As this study is a preliminary in vitro investigation, we aimed to evaluate the effectiveness of our developed delivery system using standard and homogeneous cells, thereby avoiding individual differences such as gender, age and drug use [80]. These limitations, however, do not constrain future in vivo and clinical studies because statistical accuracy was validated at the beginning of the study. Additionally, in light of these data, further in vitro and in vivo studies can investigate the possible roles of CBs in the prevention and treatment of GVHD [81–83] and in the treatment and prevention of metastasis of various cancers [51, 54] including leukemia [84].
Conclusion
Within the scope of this study, a PCL-based nanoparticle delivery system containing 2-AG and BM was developed to mobilize BM-HSCs, and its migratory effect was tested in vitro. Thus, following in vivo validation, this clinically usable form of the CBR agonist could be a strong migratory agent candidate for use alone or in combination with current migratory agents, which are currently inadequate for HSC migration protocols in HSCT and/or unaffordable.
Acknowledgements
The authors declare that artificial intelligence is not used in this study.
Abbreviations
- Au/Pd
Gold-Palladium
- BBB
Blood Brain Barrier
- BM
Bone Marrow
- BM-MNC
Bone Marrow Mononuclear Cell
- CBR
Cannabinoid Receptor
- DLS
Dynamic Light Scattering
- EBMT
European Society for Blood and Marrow Transplantation
- EC50
Half-Maximal Effective Concentration
- eCB
Endocannabinoid
- EE
Encapsulation Efficacy
- FC
Flow Cytometry
- FCS
Fetal Calf Serum
- G-CSF
Granulocyte Colony-Stimulating Factor
- GM-CFU
Granulocyte-Macrophage Colony-Forming Unit
- GM-CSF
Granulocyte-Macrophage Colony-Stimulating Factor
- HSC
Hematopoietic Stem Cell
- HSCT
Hematopoietic Stem Cell Transplantation
- IC50
Half-maximal Inhibitory Concentration
- LC-MS/MS
Liquid Chromatography-Tandem Mass Spectrometry
- MPS
Mononuclear Phagocyte System
- PB
Peripheral Blood
- Ps
Phosphatidylserine
- PCL
Polycaprolactone
- PDI
Polydispersity Index
- PLGA
Poly(lactic-Co-Glycolic Acid)
- SEM
Scanning Electron Microscopy
- TEM
Transmission Electron Microscopy
- WST-1
Water Soluble Tetrazolium-1
- MW
Molecular Weights
Author contributions
S.K., E.B. and P.K. performed study concept and design; S.K., C.V., S.Ö., E.N. and P.K., performed development of methodology and writing, review and revision of the paper; S.K., C.V. and S.Ö. provided acquisition, analysis and interpretation of data, and statistical analysis. All authors read and approved the final paper.
Funding
This work was supported by grants from the Scientific and Technological Research Council of Turkey (TUBITAK; #119S182).
Data availability
All data are included in the text and supplementary materials. Data details are available from the corresponding author on request.
Declarations
Ethical approval
The study entitled “Development and in vitro analysis of a bone marrow-targeted endocannabinoid-loaded nanoparticular carrier system” was approved on 12/09/2019 by The Scientific and Technological Research Institution of Turkey (reference number 119S182). All patients gave written informed consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Henig I, Zuckerman T. Hematopoietic stem cell transplantation-50 years of evolution and future perspectives. Rambam Maimonides Med J. 2014;5(4):e0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Juric MK, Ghimire S, Ogonek J, Weissinger EM, Holler E, van Rood JJ, et al. Milestones of hematopoietic stem cell transplantation - from First Human studies to current developments. Front Immunol. 2016;7:470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carreras E, Dufour C, Mohty M, Kroger N, editors. The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies. 7th ed. Cham (CH)2019. [PubMed]
- 4.Wuchter P, Ran D, Bruckner T, Schmitt T, Witzens-Harig M, Neben K, et al. Poor mobilization of hematopoietic stem cells-definitions, incidence, risk factors, and impact on outcome of autologous transplantation. Biol Blood Marrow Transpl. 2010;16(4):490–9. [DOI] [PubMed] [Google Scholar]
- 5.Giralt S, Costa L, Schriber J, Dipersio J, Maziarz R, McCarty J, et al. Optimizing autologous stem cell mobilization strategies to improve patient outcomes: consensus guidelines and recommendations. Biol Blood Marrow Transpl. 2014;20(3):295–308. [DOI] [PubMed] [Google Scholar]
- 6.Pusic I, Jiang SY, Landua S, Uy GL, Rettig MP, Cashen AF, et al. Impact of mobilization and remobilization strategies on achieving sufficient stem cell yields for autologous transplantation. Biol Blood Marrow Transpl. 2008;14(9):1045–56. [DOI] [PubMed] [Google Scholar]
- 7.Hosing C, Saliba RM, Ahlawat S, Korbling M, Kebriaei P, Alousi A, et al. Poor hematopoietic stem cell mobilizers: a single institution study of incidence and risk factors in patients with recurrent or relapsed lymphoma. Am J Hematol. 2009;84(6):335–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Basak GW, Urbanowska E, Boguradzki P, Torosian T, Halaburda K, Wiktor-Jedrzejczak W. Booster of plerixafor can be successfully used in addition to chemotherapy-based regimen to rescue stem cell mobilization failure. Ann Transpl. 2010;15(4):61–7. [PubMed] [Google Scholar]
- 9.Shaughnessy P, Chao N, Shapiro J, Walters K, McCarty J, Abhyankar S, et al. Pharmacoeconomics of hematopoietic stem cell mobilization: an overview of current evidence and gaps in the literature. Biol Blood Marrow Transpl. 2013;19(9):1301–9. [DOI] [PubMed] [Google Scholar]
- 10.Kiafar F, Siahi Shadbad MR, Valizadeh H. Filgrastim (G-CSF) loaded liposomes: mathematical modeling and optimization of encapsulation efficiency and particle size. Bioimpacts. 2016;6(4):195–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu G, Hong X, Jiang M, Yuan W. Sustained-release G-CSF microspheres using a novel solid-in-oil-in-oil-in-water emulsion method. Int J Nanomed. 2012;7:4559–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harada M, Ohuchi M, Hayashi T, Kato Y. Prolonged circulation and in vivo efficacy of recombinant human granulocyte colony-stimulating factor encapsulated in polymeric micelles. J Control Release. 2011;156(1):101–8. [DOI] [PubMed] [Google Scholar]
- 13.Yatuv R, Carmel-Goren L, Dayan I, Robinson M, Baru M. Binding of proteins to PEGylated liposomes and improvement of G-CSF efficacy in mobilization of hematopoietic stem cells. J Control Release. 2009;135(1):44–50. [DOI] [PubMed] [Google Scholar]
- 14.Domingues MJ, Nilsson SK, Cao B. New agents in HSC mobilization. Int J Hematol. 2017;105(2):141–52. [DOI] [PubMed] [Google Scholar]
- 15.Hosoba S, Waller EK. New molecule for mobilizing marrow stem cells. Blood. 2014;123(3):310–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Katayama Y, Battista M, Kao WM, Hidalgo A, Peired AJ, Thomas SA, Frenette PS. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell. 2006;124(2):407–21. [DOI] [PubMed] [Google Scholar]
- 17.Spiegel A, Shivtiel S, Kalinkovich A, Ludin A, Netzer N, Goichberg P, et al. Catecholaminergic neurotransmitters regulate migration and repopulation of immature human CD34 + cells through wnt signaling. Nat Immunol. 2007;8(10):1123–31. [DOI] [PubMed] [Google Scholar]
- 18.Beiermeister KA, Keck BM, Sifri ZC, ElHassan IO, Hannoush EJ, Alzate WD, et al. Hematopoietic progenitor cell mobilization is mediated through beta-2 and beta-3 receptors after injury. J Trauma. 2010;69(2):338–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kose S, Aerts-Kaya F, Kopru CZ, Nemutlu E, Kuskonmaz B, Karaosmanoglu B, et al. Human bone marrow mesenchymal stem cells secrete endocannabinoids that stimulate in vitro hematopoietic stem cell migration effectively comparable to beta-adrenergic stimulation. Exp Hematol. 2018;57:30–41. e1. [DOI] [PubMed] [Google Scholar]
- 20.Gurney SM, Scott KS, Kacinko SL, Presley BC, Logan BK. Pharmacology, Toxicology, and adverse effects of Synthetic Cannabinoid drugs. Forensic Sci Rev. 2014;26(1):53–78. [PubMed] [Google Scholar]
- 21.Aizpurua-Olaizola O, Elezgarai I, Rico-Barrio I, Zarandona I, Etxebarria N, Usobiaga A. Targeting the endocannabinoid system: future therapeutic strategies. Drug Discov Today. 2017;22(1):105–10. [DOI] [PubMed] [Google Scholar]
- 22.Pertwee RG, Ross RA. Cannabinoid receptors and their ligands. Prostaglandins Leukot Essent Fat Acids. 2002;66(2–3):101–21. [DOI] [PubMed] [Google Scholar]
- 23.Aberturas MR, Hernan P, de la Ossa D, Gil ME, Ligresti A, De Petrocellis L, Torres AI, et al. Anandamide-loaded nanoparticles: preparation and characterization. J Microencapsul. 2011;28(3):200–10. [DOI] [PubMed] [Google Scholar]
- 24.Martin-Banderas L, Alvarez-Fuentes J, Duran-Lobato M, Prados J, Melguizo C, Fernandez-Arevalo M, Holgado MA. Cannabinoid derivate-loaded PLGA nanocarriers for oral administration: formulation, characterization, and cytotoxicity studies. Int J Nanomed. 2012;7:5793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alvarez-Fuentes J, Martin-Banderas L, Munoz-Rubio I, Holgado MA, Fernandez-Arevalo M. Development and validation of an RP-HPLC method for CB13 evaluation in several PLGA nanoparticle systems. ScientificWorldJournal. 2012;2012:737526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durán-Lobato M, Martín-Banderas L, Gonçalves LMD, Fernández-Arévalo M, Almeida AJ. Comparative study of chitosan- and PEG-coated lipid and PLGA nanoparticles as oral delivery systems for cannabinoids. J Nanopart Res. 2015;17(2).
- 27.Hernan P, de la Ossa D, Lorente M, Gil-Alegre ME, Torres S, Garcia-Taboada E, Aberturas Mdel R, et al. Local delivery of cannabinoid-loaded microparticles inhibits tumor growth in a murine xenograft model of glioblastoma multiforme. PLoS ONE. 2013;8(1):e54795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Esposito E, Ravani L, Drechsler M, Mariani P, Contado C, Ruokolainen J, et al. Cannabinoid antagonist in nanostructured lipid carriers (NLCs): design, characterization and in vivo study. Mater Sci Eng C Mater Biol Appl. 2015;48:328–36. [DOI] [PubMed] [Google Scholar]
- 29.Esposito E, Drechsler M, Cortesi R, Nastruzzi C. Encapsulation of cannabinoid drugs in nanostructured lipid carriers. Eur J Pharm Biopharm. 2016;102:87–91. [DOI] [PubMed] [Google Scholar]
- 30.Schettini DA, Ribeiro RR, Demicheli C, Rocha OG, Melo MN, Michalick MS, Frezard F. Improved targeting of antimony to the bone marrow of dogs using liposomes of reduced size. Int J Pharm. 2006;315(1–2):140–7. [DOI] [PubMed] [Google Scholar]
- 31.Sou K, Goins B, Takeoka S, Tsuchida E, Phillips WT. Selective uptake of surface-modified phospholipid vesicles by bone marrow macrophages in vivo. Biomaterials. 2007;28(16):2655–66. [DOI] [PubMed] [Google Scholar]
- 32.Sou K, Goins B, Leland MM, Tsuchida E, Phillips WT. Bone marrow-targeted liposomal carriers: a feasibility study in nonhuman primates. Nanomed (Lond). 2010;5(1):41–9. [DOI] [PubMed] [Google Scholar]
- 33.Porter CJ, Moghimi SM, Illum L, Davis SS. The polyoxyethylene/polyoxypropylene block co-polymer poloxamer-407 selectively redirects intravenously injected microspheres to sinusoidal endothelial cells of rabbit bone marrow. FEBS Lett. 1992;305(1):62–6. [DOI] [PubMed] [Google Scholar]
- 34.Swami A, Reagan MR, Basto P, Mishima Y, Kamaly N, Glavey S, et al. Engineered nanomedicine for myeloma and bone microenvironment targeting. Proc Natl Acad Sci U S A. 2014;111(28):10287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harris TJ, Green JJ, Fung PW, Langer R, Anderson DG, Bhatia SN. Tissue-specific gene delivery via nanoparticle coating. Biomaterials. 2010;31(5):998–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mann AP, Tanaka T, Somasunderam A, Liu X, Gorenstein DG, Ferrari M. E-selectin-targeted porous silicon particle for nanoparticle delivery to the bone marrow. Adv Mater. 2011;23(36):H278–82. [DOI] [PubMed] [Google Scholar]
- 37.Chi B, Park SJ, Park MH, Lee SY, Jeong B. Oligopeptide delivery carrier for osteoclast precursors. Bioconjug Chem. 2010;21(8):1473–8. [DOI] [PubMed] [Google Scholar]
- 38.Szabo R, Peiser L, Pluddemann A, Bosze S, Heinsbroek S, Gordon S, Hudecz F. Uptake of branched polypeptides with poly[L-lys] backbone by bone-marrow culture-derived murine macrophages: the role of the class a scavenger receptor. Bioconjug Chem. 2005;16(6):1442–50. [DOI] [PubMed] [Google Scholar]
- 39.Allen TM, Austin GA, Chonn A, Lin L, Lee KC. Uptake of liposomes by cultured mouse bone marrow macrophages: influence of liposome composition and size. Biochim Biophys Acta. 1991;1061(1):56–64. [DOI] [PubMed] [Google Scholar]
- 40.Varan C, Bilensoy E. Cationic PEGylated polycaprolactone nanoparticles carrying post-operation docetaxel for glioma treatment. Beilstein J Nanotechnol. 2017;8:1446–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Manaia EB, Abucafy MP, Chiari-Andreo BG, Silva BL, Oshiro Junior JA, Chiavacci LA. Physicochemical characterization of drug nanocarriers. Int J Nanomed. 2017;12:4991–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rasouliyan F, Eskandani M, Jaymand M, Akbari Nakhjavani S, Farahzadi R, Vandghanooni S, Eskandani M. Preparation, physicochemical characterization, and anti-proliferative properties of lawsone-loaded solid lipid nanoparticles. Chem Phys Lipids. 2021;239:105123. [DOI] [PubMed] [Google Scholar]
- 43.Onen S, Atik AC, Gizer M, Kose S, Yaman O, Kulah H, Korkusuz P. A pumpless monolayer microfluidic device based on mesenchymal stem cell-conditioned medium promotes neonatal mouse in vitro spermatogenesis. Stem Cell Res Ther. 2023;14(1):127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tanabe M, Hosokawa K, Nguyen MAT, Nakagawa N, Maruyama K, Tsuji N, et al. The GPI-anchored protein CD109 protects hematopoietic progenitor cells from undergoing erythroid differentiation induced by TGF-beta. Leukemia. 2022;36(3):847–55. [DOI] [PubMed] [Google Scholar]
- 45.Georgantas RW 3rd, Hildreth R, Morisot S, Alder J, Liu CG, Heimfeld S, et al. CD34 + hematopoietic stem-progenitor cell microRNA expression and function: a circuit diagram of differentiation control. Proc Natl Acad Sci U S A. 2007;104(8):2750–5. [DOI] [PMC free article] [PubMed]
- 46.Song Y, Yen S, Southam K, Gaskin S, Hoy RF, Zosky GR. The aryl hydrocarbon receptor pathway is a marker of lung cell activation but does not play a central pathologic role in engineered stone-associated silicosis. J Appl Toxicol. 2024. [DOI] [PubMed]
- 47.Zhang Y, Li T, Miao J, Zhang Z, Yang M, Wang Z, et al. Gamma-Glutamyl transferase 5 overexpression in cerebrovascular endothelial cells improves brain pathology, cognition, and behavior in APP/PS1 mice. Neural Regen Res. 2025;20(2):533–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fathi E, Azarbad S, Farahzadi R, Javanmardi S, Vietor I. Effect of rat bone marrow derived-mesenchymal stem cells on Granulocyte differentiation of mononuclear cells as Preclinical Agent in Cellbased Therapy. Curr Gene Ther. 2022;22(2):152–61. [DOI] [PubMed] [Google Scholar]
- 49.Farahzadi R, Fathi E, Mesbah-Namin SA, Vietor I. Granulocyte differentiation of rat bone marrow resident C-kit(+) hematopoietic stem cells induced by mesenchymal stem cells could be considered as new option in cell-based therapy. Regen Ther. 2023;23:94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Onen S, Kose S, Yersal N, Korkusuz P. Mesenchymal stem cells promote spermatogonial stem/progenitor cell pool and spermatogenesis in neonatal mice in vitro. Sci Rep. 2022;12(1):11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boyacioglu O, Bilgic E, Varan C, Bilensoy E, Nemutlu E, Sevim D, et al. ACPA decreases non-small cell lung cancer line growth through Akt/PI3K and JNK pathways in vitro. Cell Death Dis. 2021;12(1):56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bal E, Hanalioglu S, Kopru CZ, Kose S, Basak AT, Pehlivan SB, et al. Effect of mesenchymal stem cells therapy in experimental kaolin induced syringomyelia model. J Neurosurg Sci. 2022;66(1):40–8. [DOI] [PubMed] [Google Scholar]
- 53.Yersal N, Kose S, Horzum U, Ozkavukcu S, Orwig KE, Korkusuz P. Leptin promotes proliferation of neonatal mouse stem/progenitor spermatogonia. J Assist Reprod Genet. 2020;37(11):2825–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Onay O, Kose S, Suslu N, Korkusuz P, Nemutlu E, Aydin C, Hosal S. Human laryngeal squamous cell carcinoma cell line release of endogenous anandamide and 2-arachidonoylglycerol, and their antiproliferative effect via exogenous supplementation: an in vitro study. Cell Tissue Bank. 2022;23(1):93–100. [DOI] [PubMed] [Google Scholar]
- 55.Rafat A, Dizaji Asl K, Mazloumi Z, Movassaghpour AA, Talebi M, Shanehbandi D, et al. Telomerase inhibition on acute myeloid leukemia stem cell induced apoptosis with both intrinsic and extrinsic pathways. Life Sci. 2022;295:120402. [DOI] [PubMed] [Google Scholar]
- 56.Hatherell K, Couraud PO, Romero IA, Weksler B, Pilkington GJ. Development of a three-dimensional, all-human in vitro model of the blood-brain barrier using mono-, co-, and tri-cultivation transwell models. J Neurosci Methods. 2011;199(2):223–9. [DOI] [PubMed] [Google Scholar]
- 57.Unal S, Can Ozturk S, Bilgic E, Yanik H, Korkusuz P, Aktas Y, et al. Therapeutic efficacy and gastrointestinal biodistribution of polycationic nanoparticles for oral camptothecin delivery in early and late-stage colorectal tumor-bearing animal model. Eur J Pharm Biopharm. 2021;169:168–77. [DOI] [PubMed] [Google Scholar]
- 58.Unal S, Aktas Y, Benito JM, Bilensoy E. Cyclodextrin nanoparticle bound oral camptothecin for colorectal cancer: Formulation development and optimization. Int J Pharm. 2020;584:119468. [DOI] [PubMed] [Google Scholar]
- 59.Varan C, Wickstrom H, Sandler N, Aktas Y, Bilensoy E. Inkjet printing of antiviral PCL nanoparticles and anticancer cyclodextrin inclusion complexes on bioadhesive film for cervical administration. Int J Pharm. 2017;531(2):701–13. [DOI] [PubMed] [Google Scholar]
- 60.Giri TK, Choudhary C, Ajazuddin, Alexander A, Badwaik H, Tripathi DK. Prospects of pharmaceuticals and biopharmaceuticals loaded microparticles prepared by double emulsion technique for controlled delivery. Saudi Pharm J. 2013;21(2):125–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sahin A, Esendagli G, Yerlikaya F, Caban-Toktas S, Yoyen-Ermis D, Horzum U, et al. A small variation in average particle size of PLGA nanoparticles prepared by nanoprecipitation leads to considerable change in nanoparticles’ characteristics and efficacy of intracellular delivery. Artif Cells Nanomed Biotechnol. 2017;45(8):1657–64. [DOI] [PubMed] [Google Scholar]
- 62.Alshamsan A. Nanoprecipitation is more efficient than emulsion solvent evaporation method to encapsulate cucurbitacin I in PLGA nanoparticles. Saudi Pharm J. 2014;22(3):219–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Esim O, Bakirhan NK, Yildirim N, Sarper M, Savaser A, Ozkan SA, Ozkan Y. Development, optimization and in vitro evaluation of oxaliplatin loaded nanoparticles in non-small cell lung cancer. Daru. 2020;28(2):673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bhattacharjee S. DLS and Zeta potential - what they are and what they are not? J Control Release. 2016;235:337–51. [DOI] [PubMed] [Google Scholar]
- 65.Sapsford KE, Tyner KM, Dair BJ, Deschamps JR, Medintz IL. Analyzing nanomaterial bioconjugates: a review of current and emerging purification and characterization techniques. Anal Chem. 2011;83(12):4453–88. [DOI] [PubMed] [Google Scholar]
- 66.Alex AT, Joseph A, Shavi G, Rao JV, Udupa N. Development and evaluation of carboplatin-loaded PCL nanoparticles for intranasal delivery. Drug Deliv. 2016;23(7):2144–53. [DOI] [PubMed] [Google Scholar]
- 67.Kumar N, Chaurasia S, Patel RR, Khan G, Kumar V, Mishra B. Atorvastatin calcium encapsulated eudragit nanoparticles with enhanced oral bioavailability, safety and efficacy profile. Pharm Dev Technol. 2017;22(2):156–67. [DOI] [PubMed] [Google Scholar]
- 68.Hosain MZ, Yuzuriha K, Khadijah, Takeo M, Kishimura A, Murakami Y, et al. Synergic modulation of the inflammatory state of macrophages utilizing anti-oxidant and phosphatidylserine-containing polymer-lipid hybrid nanoparticles. Medchemcomm. 2017;8(7):1514–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roy MT, Gallardo M, Estelrich J. Influence of size on Electrokinetic Behavior of Phosphatidylserine and phosphatidylethanolamine lipid vesicles. J Colloid Interface Sci. 1998;206(2):512–7. [DOI] [PubMed] [Google Scholar]
- 70.Kay JG, Grinstein S. Sensing phosphatidylserine in cellular membranes. Sens (Basel). 2011;11(2):1744–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hourani W, Alexander SPH. Cannabinoid ligands, receptors and enzymes: pharmacological tools and therapeutic potential. Brain Neurosci Adv. 2018;2:2398212818783908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gaumet M, Vargas A, Gurny R, Delie F. Nanoparticles for drug delivery: the need for precision in reporting particle size parameters. Eur J Pharm Biopharm. 2008;69(1):1–9. [DOI] [PubMed] [Google Scholar]
- 73.Sou K, Goins B, Oyajobi BO, Travi BL, Phillips WT. Bone marrow-targeted liposomal carriers. Expert Opin Drug Deliv. 2011;8(3):317–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liang S, Zheng J, Wu W, Li Q, Saw PE, Chen J, et al. A robust nanoparticle platform for RNA interference in macrophages to suppress Tumor Cell Migration. Front Pharmacol. 2018;9:1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Craparo EF, Cabibbo M, Conigliaro A, Barreca MM, Musumeci T, Giammona G, Cavallaro G. Rapamycin-loaded polymeric nanoparticles as an Advanced Formulation for Macrophage Targeting in Atherosclerosis. Pharmaceutics. 2021;13(4). [DOI] [PMC free article] [PubMed]
- 76.Jiang S, Zagozdzon R, Jorda MA, Parmar K, Fu Y, Williams JS, et al. Endocannabinoids are expressed in bone marrow stromal niches and play a role in interactions of hematopoietic stem and progenitor cells with the bone marrow microenvironment. J Biol Chem. 2010;285(46):35471–8. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 77.Jorda MA, Verbakel SE, Valk PJ, Vankan-Berkhoudt YV, Maccarrone M, Finazzi-Agro A, et al. Hematopoietic cells expressing the peripheral cannabinoid receptor migrate in response to the endocannabinoid 2-arachidonoylglycerol. Blood. 2002;99(8):2786–93. [DOI] [PubMed] [Google Scholar]
- 78.Patinkin D, Milman G, Breuer A, Fride E, Mechoulam R. Endocannabinoids as positive or negative factors in hematopoietic cell migration and differentiation. Eur J Pharmacol. 2008;595(1–3):1–6. [DOI] [PubMed] [Google Scholar]
- 79.McPartland JM, Glass M, Pertwee RG. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: interspecies differences. Br J Pharmacol. 2007;152(5):583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mirabelli P, Coppola L, Salvatore M. Cancer Cell lines are useful Model systems for Medical Research. Cancers (Basel). 2019;11(8). [DOI] [PMC free article] [PubMed]
- 81.Aerts-Kaya F, Ulum B, Mammadova A, Kose S, Aydin G, Korkusuz P, Uckan-Cetinkaya D. Neurological regulation of the bone marrow niche. Adv Exp Med Biol. 2020;1212:127–53. [DOI] [PubMed] [Google Scholar]
- 82.Kose S, Yersal N, Onen S, Korkusuz P. Comparison of hematopoietic and spermatogonial stem cell niches from the Regenerative Medicine aspect. Adv Exp Med Biol. 2018;1107:15–40. [DOI] [PubMed] [Google Scholar]
- 83.Aerts-Kaya F, Kilic E, Kose S, Aydin G, Cagnan I, Kuskonmaz B, Uckan-Cetinkaya D. G-CSF treatment of healthy pediatric donors affects their hematopoietic microenvironment through changes in bone marrow plasma cytokines and stromal cells. Cytokine. 2021;139:155407. [DOI] [PubMed] [Google Scholar]
- 84.Fathi E, Farahzadi R, Sheervalilou R, Sanaat Z, Vietor I. A general view of CD33(+) leukemic stem cells and CAR-T cells as interesting targets in acute myeloblatsic leukemia therapy. Blood Res. 2020;55(1):10–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are included in the text and supplementary materials. Data details are available from the corresponding author on request.