

Abstract
The mechanisms underlying phenotypic heterogeneity in autism spectrum disorder (ASD) are not well understood. Using a large neuroimaging dataset, we identified three latent dimensions of functional brain network connectivity that predicted individual differences in ASD behaviors and were stable in cross-validation. Clustering along these three dimensions revealed four reproducible ASD subgroups with distinct functional connectivity alterations in ASD-related networks and clinical symptom profiles that were reproducible in an independent sample. By integrating neuroimaging data with normative gene expression data from two independent transcriptomic atlases, we found that within each subgroup, ASD-related functional connectivity was explained by regional differences in the expression of distinct ASD-related gene sets. These gene sets were differentially associated with distinct molecular signaling pathways involving immune and synapse function, G-protein-coupled receptor signaling, protein synthesis and other processes. Collectively, our findings delineate atypical connectivity patterns underlying different forms of ASD that implicate distinct molecular signaling mechanisms.
Individuals with ASD present with a range of difficulties in social interaction and communication, repetitive and ritualistic behaviors, differing levels of intellectual disability and various medical comorbidities. ASD is not a unitary entity. Distinct pathophysiological processes may underlie different forms of ASD and benefit from different types of therapeutic interventions1–4. Phenotypic heterogeneity is thus a major obstacle to defining pathophysiological mechanisms and discovering new therapeutic approaches.
Functional magnetic resonance imaging (fMRI) studies have found that impaired social cognition and language processing in ASD are associated with atypical activity in the thalamus, visual areas and salience network5–7, and that repetitive and ritualistic behaviors are associated with atypical inhibitory control and frontostriatal circuit function8,9. Large-scale, multi-site resting-state fMRI (rsfMRI) datasets have identified–at the group level–robust and reproducible differences in functional connectivity in corticostriatal and frontoparietal networks in ASD10,11. More recently, neuroimaging studies have investigated the neurobiological basis of phenotypic heterogeneity in ASD, showing that anatomically defined subgroups can improve the prediction of ASD symptom severity; that functional connectivity differentiates individuals with ASD from neurotypical controls; and that multiple functional connectivity patterns are found in different subsets of individuals with ASD12–14. Still, whether and how atypical connectivity contributes to individual differences in symptoms and behaviors in ASD is not well understood.
Family exome sequencing studies have estimated over 1,000 genetic variants confer risk for ASD with varying penetrance15–18, and large-scale genome-wide association studies have identified over 500 common variants19,20 associated with a wide range of biological properties. In most cases, risk for ASD is thought to be influenced by the cumulative impact of many common variants, which complicates efforts to model their role in brain function, development and behavior. ASD has also been associated with transcriptional differences in specific brain regions21,22, and recent studies suggest that regional differences in gene expression may regulate network function in the healthy human brain23 as well as structural abnormalities in ASD and schizophrenia24–27. These observations led us to hypothesize that distinct genetic pathways may be important in subsets of individuals, and may confer risk for specific symptoms by modulating functional connectivity in ASD-related brain networks.
To test this hypothesis, we used regularized canonical correlation analysis (RCCA) and resampling methods optimized to reduce overfitting and improve generalizability, and identified three latent brain–behavior dimensions explaining phenotypic heterogeneity in ASD in two large-scale rsfMRI datasets (Autism Brain Imaging Data Exchange (ABIDE) I and II)10,11. These three dimensions described patterns of functional connectivity that predicted individual differences in (1) verbal ability, (2) social affect and (3) repetitive behavior and restricted interests, and were estimated in training samples and validated using held-out data. Hierarchical clustering along these three dimensions identified four distinct subgroups of individuals with ASD that were reproducible in held-out data and associated with differing patterns of functional connectivity and behavior. Finally, by integrating rsfMRI data with normative gene expression data from two transcriptomic brain atlases28,29, we found that regional differences in the expression of ASD-related genes predicted which networks exhibited atypical connectivity in ASD and implicated distinct biological processes and molecular signaling mechanisms in each ASD subgroup.
Results
We began by testing whether functional connectivity in ASD-related brain networks explains individual differences in ASD symptoms in a large, extensively validated, and well-studied neuroimaging sample (ABIDE I and ABIDE II) comprising 432 individuals with ASD with verbal IQ and ADOS-2 calibrated severity scores (CSS) and 1,106 neurotypical controls from 36 research sites. Because head motion and other artifacts can confound analyses of multi-site neuroimaging datasets30,31, we implemented a stringent protocol for controlling for motion artifacts and data quality following or exceeding well-established guidelines32,33 (Supplementary Fig. 1 and Methods). Using an extensively validated functional parcellation atlas34, we estimated whole-brain resting-state functional connectivity (RSFC) maps for each participant. After excluding data that did not meet our data quality inclusion criteria (~21.5% of participants; Methods), all subsequent analyses focused on a sample of 299 participants with ASD and 907 neurotypical controls.
Three brain–behavior dimensions explain individual differences in autism spectrum disorder
We used RCCA to identify latent brain–behavior dimensions explaining individual differences in three ASD-related domains: social affect symptoms, restricted and repetitive behaviors (RRBs) and verbal IQ (Fig. 1a–c). To reduce overfitting35,36, we first used resampled feature selection (1,000 training sets subsampled on 95% of the data) to identify a subset of functional connectivity features that were reliably correlated with one or more ASD behaviors. Next, we used cross-validated RCCA in 1,000 training set/test set replicates with test data (5%) held out from both feature selection and RCCA (Supplementary Fig. 2 and Methods). RCCA identified three latent brain–behavior dimensions (Fig. 1d–f). The first dimension predicted individual differences in verbal IQ and was modestly anticorrelated with social affect and RRB symptoms, as measured by ADOS-2 CSS (Fig. 1d). The second dimension predicted individual differences in the social affect CSS (Fig. 1e). The third dimension was strongly correlated with individual differences in RRB symptoms and moderately correlated with verbal IQ (Fig. 1f). Canonical correlations in the cross-validation test set were statistically significant for all three brain–behavior dimensions (Supplementary Fig. 3a–c; 1: r = 0.269, P < 0.0001, d = 1.119; 2: r = 0.180, P = 0.0005, d = 0.771; and 3: r = 0.115, P = 0.0185, d = 0.484), based on a corrected (and conservative) variance estimator that accounts for correlations between replicates37. Although all three canonical correlations were significant in held-out data, the higher canonical correlations in the training set relative to the test set unsurprisingly indicate some overfitting (a common finding during cross-validation38) and suggest there could still be room for further improvement on held-out test data with, for example, more complex regularization procedures.
Fig. 1 |. Three brain–behavior dimensions explain individual differences in autism spectrum disorder.

a, Schematic summarizing stabilized feature selection and RCCA in N = 299 ASD participants and N = 907 neurotypical controls. b, Glass brain (left) depicting functional parcellation50 spanning the cerebrum, brainstem and cerebellum (colored by functional network). BOLD signal time series extracted from three representative ROIs (middle) and correlation between each ROI and every other ROI (functional connectivity matrix, right) for each participant. c, Glass brain depicting the neuroanatomical distribution of functional connectivity features that were correlated with one or more ASD behaviors, identified in one representative training set (colored by functional network, sized by correlation). d–f, RCCA revealed three dimensions predicting individual differences in verbal IQ (d), social affect (e), and RRB (f) symptoms. Scatterplots depict the association between connectivity scores and behavior scores for each RCCA dimension across participants. Mean scores calculated on held-out data are light, while the average scores calculated on the training set data are dark. Heat maps (left) depict the mean correlation over training sets between each dimension’s behavior scores and each clinical symptom (Fisher z-transformed Spearman correlation coefficients). The canonical correlations (r) for all three dimensions were statistically significant in held-out data (inset also shows Cohen’s d) compared to an empirical null distribution (shuffled held-out test set data; 1,000 shuffles in each of the 1,000 training/test sets). (variate 1: r = 0.269, P <0.0001, d = 1.119; variate 2: r = 0.180, P = 0.0005, d = 0.771; and variate 3: r = 0.115, P = 0.0185, d = 0.484; r indicates mean test set canonical correlation, P indicates P value, and d indicates Cohen’s d). a.u., arbitrary units; BOLD, blood-oxygen-level-dependent; CBL, cerebellum; Cd, caudate; COTC, cerebellar-occipital task control; DMN, default mode network; FPTC, frontoparietal task control; M1, primary motor cortex; MOG, medial orbital gyrus; MTG, middle temporal gyrus; PFC, prefrontal cortex; PPC, posterior parietal cortex; r, canonical correlation; RCCA, regularized canonical correlation; S1, primary somatosensory cortex; SA, social affect.
To better understand how atypical functional connectivity in specific regions and networks underlies individual differences in ASD symptoms, we first examined the RSFC correlates (connectivity score loadings) of each latent brain–behavior dimension. We found that each brain–behavior dimension described a distinct pattern of functional connectivity (Fig. 2a–c and Extended Data Fig. 1). Specifically, we found that the verbal IQ-related dimension was associated with connectivity between brain areas known to be important for language processing and reading ability, including corticothalamic, visual network and striatal connectivity (Fig. 2a), which is consistent with previous work indicating that reading ability is negatively associated with thalamic synchronization39 and functional connectivity involving these areas6,40,41. The social affect-related dimension was associated with connectivity between brain areas known to be important for socio-emotional processing, including connectivity between the salience network, visual network and striatal areas (Fig. 2b). These results are consistent with previous studies showing that the salience network is hyperactive at rest in ASD42; that salience network connectivity is associated with sensory over-responsivity to irrelevant stimuli and social interaction deficits7; and that corticostriatal hyperconnectivity is a commonly replicated feature of ASD43,44 and may contribute to abnormal gating of socially relevant stimuli45. The RRB-related dimension was associated with connectivity between brain areas known to be important for cognitive control, response inhibition and action selection, including corticostriatal connectivity with primary motor areas and the frontoparietal task control network (Fig. 2c). These results are consistent with previous work that shows association of severe RRB symptoms with corticostriatal, frontoparietal and motor cortex connectivity8,46,47 and RRBs with executive function48. Moreover, these three brain–behavior dimensions were robust and stable when calculated in different subsets of participants with ASD (Supplementary Figs. 3 and 4). Further, we replicated key brain–behavior association findings in a subset of the data comprising a narrower age range (ages 8–18) and in a second brain parcellation49 (Supplementary Figs. 5 and 6). Together, these results delineate distinct sets of functional connectivity features that explain individual differences in verbal IQ, social affect and RRB symptoms and align with convergent findings from previous studies, enhancing confidence in the results.
Fig. 2 |. Functional connectivity correlates of autism spectrum disorder symptoms.

a, Mean correlation between RSFC features and behavior scores for verbal IQ-related dimension (on cross-validation (CV) training folds). Heat map (left) labeled by brain region (x axis) and RSFC network (y axis), showing a subset of RSFC features with strong loadings for dimension (maximum RSFC-to-dimension correlation; FDR < 0.05; 247 ROIs mapped to 37 brain regions, collapsed over hemispheres). Chord plot (middle) depicts connectivity score correlations for the most important RSFC features (>1 connection with FDR < 0.001). Glass brain (right) depicts neuroanatomical distribution of RSFC features in chord plot. b, Mean correlation between RSFC features and behavior scores for social affect-related dimension (on CV training folds). c, Mean correlation between RSFC features and behavior scores for RRB-related dimension (on CV training folds). d, Heat map of atypical connectivity in N = 299 individuals with ASD compared to N = 907 neurotypical controls (two-sided Welch’s t-test; FDR < 0.001), showing maximum atypical connectivity statistic between ROIs within each of the 37 brain region groups. See Extended Data Fig. 1 for whole-brain (247 × 247 ROIs) results. e, Venn diagram indicating the number of RSFC features (of 247 × 247) correlated with dimensions in a–c but not atypical (yellow; nRSFC = 1,137; FDR < 0.05) and number of RSFC features not correlated with any dimension but atypical (green; nRSFC = 4,257; FDR < 0.05). Only 13.4% of symptom-predictive RSFC features (overlap; nRSFC = 176 of 1,313; FDR < 0.05) were also atypical. f, Venn diagram indicating number of RSFC features (of 247 × 247) significantly (FDR < 0.05) correlated with each dimension. Each RSFC dimension score was associated with a mostly unique set of RSFC features. ACC, anterior cingulate cortex; antPFC, anterior prefrontal cortex; DLPFC, dorsolateral prefrontal cortex; IOG, inferior orbital gyrus; IPL, inferior parietal lobe; ITG, inferior temporal gyrus; MCC, medial cingulate cortex; NAcc, nucleus accumbens; OFC, orbital frontal cortex; paraHC, parahippocampus; PCC, posterior cingulate cortex; SM, somatomotor; SMA, supplementary motor area; STG, superior temporal gyrus; Temp Pole, temporal pole; VLPFC, ventrolateral prefrontal cortex; VMPFC, ventromedial prefrontal cortex.
To better understand the extent to which RSFC features in distinct versus overlapping brain networks were associated with individual differences in verbal IQ, social affect and RRB symptoms, we tested for overlap between the most important RSFC features in each latent brain–behavior dimension. We found that 1,313 RSFC features correlated with symptom scores in at least one dimension (false discovery rate (FDR)-corrected P < 0.05), and most were specific to one dimension, such that only ten features (representing just 0.03% of all RSFC features brain wide) associated with more than one dimension (Fig. 2f).
Finally, we examined whether individual differences in ASD symptoms were explained predominantly by RSFC features that were abnormal relative to neurotypical controls, or by variation in RSFC within the normal range50–52. ASD was associated with widespread connectivity abnormalities spanning a variety of cortical and subcortical regions (FDR-corrected P < 0.05; Fig. 2d and Extended Data Fig. 1d) and involving 4,433 RSFC features or ~14.6% of all RSFC features throughout the brain. Unexpectedly, only a small minority (13.4%) of the most important symptom-predictive RSFC features were also atypical compared to controls (Fig. 2e). Results were highly similar when this analysis was restricted to age-matched controls (N = 868 neurotypical controls aged 5–35 years; Supplementary Fig. 7). These results suggest that while some abnormal RSFC features are predictors of symptoms, most are not; rather, individual differences in ASD symptoms are explained predominantly by variation in RSFC that falls within the normal range and is associated with ASD symptoms only when it co-occurs with a distinct set of abnormal RSFC features involving the default mode network (especially the middle temporal gyrus), thalamus, primary sensorimotor areas, brainstem (especially the dorsal raphe) and other regions.
Brain–behavior dimensions define four autism spectrum disorder subgroups
While numerous studies have identified consistent and reproducible patterns of atypical connectivity in ASD53,54, it is unclear whether distinct patterns of circuit dysfunction are operative in some subgroups of individuals with ASD but not in others. Having identified three brain–behavior dimensions explaining individual differences in ASD, we tested whether ASD individuals tended to cluster into relatively homogeneous subgroups in this three-dimensional space. Hierarchical clustering along these three dimensions identified an optimal four-cluster solution (Fig. 3a), based on three in-sample and three out-of-sample assessments of the goodness of fit (Supplementary Figs. 8 and 9, Extended Data Fig. 2 and Methods). In a secondary exploratory analysis, we did not observe sex differences between clusters (Supplementary Fig. 8h); however, a limitation of this analysis is that only 15.4% of the ASD participants in the ABIDE ASD dataset were female (Nfemale = 46 females of N = 299 ASD participant). In subsequent analyses, we used the participant’s modal cluster assignment as an ensemble estimate across 1,000 subsamples (Methods).
Fig. 3 |. Hierarchical clustering on brain–behavior dimension scores reveals four autism spectrum disorder subgroups.

a, Heat map and dendrogram depict hierarchical clustering on all 299 ASD individuals (rows) along three dimensions (columns) using cosine similarity (dendrogram) between connectivity scores of ASD participant pairs (heat map; dashed line indicates 80% of maximum cosine distance). b–e, Box plots of the distribution of clinical symptom z-scores (superimposed bar graphs depict the median) for social affect (SA) (b), RRB (c), verbal IQ (VIQ) (d) and total severity (e; N = 69, N = 87, N = 67 and N = 76 ASD participants for subgroups 1–4, respectively; color indicates subgroup). All four measures–social affect, RRB, verbal IQ and total severity–differed by subgroup (Kruskal–Wallis test between subgroups showed significant between-subgroup differences for each symptom: social affect, X2 (3, N = 299) = 115.86, P = 6.02 × 10−25; RRB: X2 (3, N = 299) = 124.52, P = 8.18 × 10−27; VIQ: X2 (3, N = 299) = 138.28, P = 8.88 × 10−30; total severity: X2 (3, N = 299) = 115.22, P = 8.25 × 10−25). Note that higher social affect, RRB and total severity scores and lower verbal IQ indicate greater impairment. f–h, Kernel density estimation plots of participant connectivity scores in two dimensions (lowest iso-proportion level = 0.25). Box plots indicate distributions of subgroup connectivity scores along a single dimension (N = 69, N = 87, N = 67 and N = 76 ASD participants for subgroups 1–4, respectively). For b–h, box bounds indicate the 25th and 75th percentiles; the center line denotes the median; whiskers correspond to ±2.7σ and 99.3% of the data; and outliers are shown as circles. Analyses for b–h use the aggregate clustering assignment described in the main text and Methods.
Next, we tested for differences in ASD symptoms and atypical functional connectivity between the four subgroups and found subgroup differences in both domains. The four subgroups differed strongly with respect to their clinical symptom profiles (Fig. 3b–e). Each subgroup was also associated with distinct patterns of atypical RSFC in ASD-related networks, especially limbic areas (subgroups 2 and 3), the default mode network (subgroup 4) and sensorimotor areas (subgroup 1), among others (Extended Data Fig. 3). These results were stable when we evaluated the distribution of clinical symptoms in subgroups across 1,000 subsamples (of 95% ASD participants; Extended Data Fig. 4a–d) and the mean and standard deviation of the atypical functional connectivity across 1,000 subsamples (of 95% ASD participants; Extended Data Fig. 4e–l and Methods).
A closer pairwise comparison of the relationship between atypical connectivity, dimension-related connectivity and clinical symptom profiles in specific subgroups revealed associations that suggest distinct network-level mechanisms underlying individual differences in each subgroup (Fig. 3b–h and Fig. 4). Subgroup 1 had above-average verbal IQ (Fig. 3d), high connectivity scores in the verbal IQ-related dimension 1 (Fig. 3f,g) and abnormally low RSFC in IQ-related language processing areas compared to neurotypical controls (Fig. 4a). In contrast, subgroup 2 had below-average verbal IQ (Fig. 3d), low connectivity scores in the verbal IQ-related dimension 1 (Fig. 3f,g) and abnormally elevated RSFC in the same language processing areas, as well as multiple other connections that predict individual differences in verbal IQ (Fig. 4b)–a finding not observed in the other subgroups. These results show how abnormally elevated connectivity in verbal IQ-related networks is specific to a subgroup of ASD individuals, and suggest that in other ASD individuals, atypical connectivity in the opposite direction (abnormally reduced) might compensate for other abnormalities, preserving verbal IQ even in the presence of symptoms in other domains.
Fig. 4 |. Autism spectrum disorder subgroups have distinct atypical connectivity patterns in dimension-related RSFC features.

a-l, Chord plots of atypical connectivity (two-sided Welch’s t-test between RSFC of participants with ASD in each subgroup (N = 69, N = 87, N = 67 and N = 76 ASD participants) and N = 907 neurotypical controls; FDR < 0.05) for the most important dimension-related RSFC features identified in Fig. 2a–c. Blue boxes highlight findings discussed in the main text. a–d, Subgroup atypical connectivity in verbal IQ-related RSFC (dimension 1). e–h, Subgroup atypical connectivity in social affect-related RSFC (dimension 2); i–l, Subgroup atypical connectivity in RRB-related RSFC (dimension 3). b, Subgroup 2 had atypical connectivity associated with dimension 1, predicting lower verbal IQ. g, Subgroup 3 had atypical connectivity associated with dimension 2, predicting high social affect symptoms and low RRB symptoms. h, Subgroup 4 had atypical connectivity associated with dimension 2 but in the opposite direction, predicting low social affect symptoms and high RRB symptoms. i, Subgroup 1 had atypical connectivity associated with dimension 3, predicting high RRB symptoms and high verbal IQ. k, Subgroup 3 had atypical connectivity associated with dimension 3 but in the opposite direction, predicting low RRB symptoms. Color bars on the right indicate the direction of atypical connectivity and connection strength (warm indicates increased, while cool indicates decreased relative to neurotypical controls) and functional network (node color). Analyses use the aggregate clustering assignment described in the main text and Methods.
Similarly, subgroup 3 had high social affect symptoms (Fig. 3b), low RRB symptoms (Fig. 3c) and, compared to neurotypical controls, abnormally elevated connectivity associated with social affect-related dimension 2 (Fig. 4g), including anterior cingulate and ventrolateral prefrontal areas of the salience network, among others. In contrast, subgroup 4 had low social affect symptoms (Fig. 3b), high RRB symptoms (Fig. 3c) and atypical connectivity in the same areas but in the opposite direction, with abnormally low connectivity associated with dimension 2 (Fig. 4h). Comparing subgroup 3 (low RRB symptoms, hyperconnectivity between cognitive control areas, the striatum and primary motor cortex; Fig. 4k) and subgroup 1 (high RRB symptoms, abnormally low connectivity in the same areas; Fig. 4i) also revealed potentially compensatory connectivity differences associated with reduced RRB symptoms. Together, these results define four ASD subgroups along three brain–behavior dimensions that differed with respect to both ASD symptom profiles and functional network organization. They are also consistent with the hypothesis that varying and contrasting connectivity patterns may contribute to clinical heterogeneity in ASD, and that similar ASD symptoms may be associated with distinct network-level substrates.
For comparison, we clustered directly on the standardized clinical symptom scores (z-score of scale value; that is, hierarchical clustering using cosine distance with average linkage and splitting the resulting dendrogram at four clusters as in Fig. 3, but on clinical symptoms only). We found that N = 226 of the N = 299 ABIDE participants (75.6%) were assigned to the same clusters as those found using brain functional connectivity (connectivity scores; Supplementary Fig. 10). We interpret this result as indicating that the clustering results share some similarity; however, clustering on the brain–behavior dimensions incorporates additional information that importantly influences the final clustering assignments.
For further validation, we confirmed that connectivity scores by subgroup were not sensitive to small changes in the RCCA parameters and were stable (Supplementary Figs. 3, 4 and 11). Subgroup results were also consistent in a secondary analysis restricted to age-matched controls (N = 868 neurotypical controls aged 5–35 years; Supplementary Fig. 12). Next, we replicated key findings of ASD subgroups, first, in a narrower age-range sample (aged 8–18 years) and, second, in a second brain parcellation49 (Extended Data Figs. 5 and 6 and Supplementary Figs. 13–16). Furthermore, we evaluated the impact of age on these ASD subgroups, and did not detect evidence of developmental heterogeneity within the brain–behavior associations of the ASD subgroups (Supplementary Figs. 17–20 and Supplementary Tables 1 and 2).
Finally, we tested whether the ASD subgroups defined in ABIDE were replicable in an independent, out-of-sample dataset from the National Institute of Mental Health (NIMH) Data Archive (NDA; N = 85 ASD participants; Extended Data Fig. 7, Supplementary Fig. 21 and Methods). To summarize our approach, among a total of N = 113 participants, NNDA = 85 participants (aged 8–39 years; 58 males, 27 females) had usable fMRI data according to the quality-control criteria used in this work. We repeated the analyses exactly as they were implemented in the ABIDE dataset, including feature selection, RCCA and clustering. Despite the relatively small sample size, we identified four ASD subgroups with behavioral profiles and atypical connectivity patterns that were strikingly similar to those observed in the ABIDE ASD subgroups (Extended Data Fig. 7 and Supplementary Fig. 21; NDA subgroup sizes were NNDA_1 = 20, NNDA_2 = 21; NNDA_3 = 27; NNDA_4 = 17).
Transcriptomic correlates of subgroup-specific connectivity
We next hypothesized that common ASD risk variants could modulate ASD pathophysiology by influencing resting-state connectivity in ASD-related brain networks and that distinct genetic pathways may be important in subsets of individuals. To test this hypothesis, we first investigated whether regional differences in normative gene expression patterns explain the spatial pattern of atypical connectivity in the four ASD subgroups identified in Figs. 3 and 4. We mapped normative regional gene expression profiles for 10,438 microarray probes in the Allen Human Brain Atlas (AHBA), including gene expression data for 3,702 samples from 6 healthy adults (N = 5 men, N = 1 woman, aged 24–57 years)28, to the functional parcellation used above (Fig. 1b), preprocessing the AHBA microarray expression dataset following best practices55 (Methods). Next, we used partial least squares (PLS) regression to test for weighted combinations of gene expression probes that covary with the spatial distribution of atypical RSFC (collapsed to regional seeds) in each ASD subgroup (Fig. 5a). PLS regression confirmed that regional differences in gene expression predicted the neuroanatomical distribution of atypical connectivity in all four subgroups, with statistical significance established in both a simple permutation test and a stricter, spatial permutation (‘spin’) test27,56 (Supplementary Table 3 and Methods). To determine the degree to which distinct gene sets were implicated across the four subgroups, we calculated the ranking similarity between the top 1,000 ranked gene weights of the subgroups using rank biased overlap (RBO)57, a ranking similarity measure for non-conjoint lists. The RBO similarity scores indicated that different sets of gene candidates were prioritized for each subgroup (RBO = 0.36–0.59; 1 is perfect similarity; Fig. 5b).
Fig. 5 |. Transcriptomic correlates of atypical connectivity patterns in autism spectrum disorder subgroups.
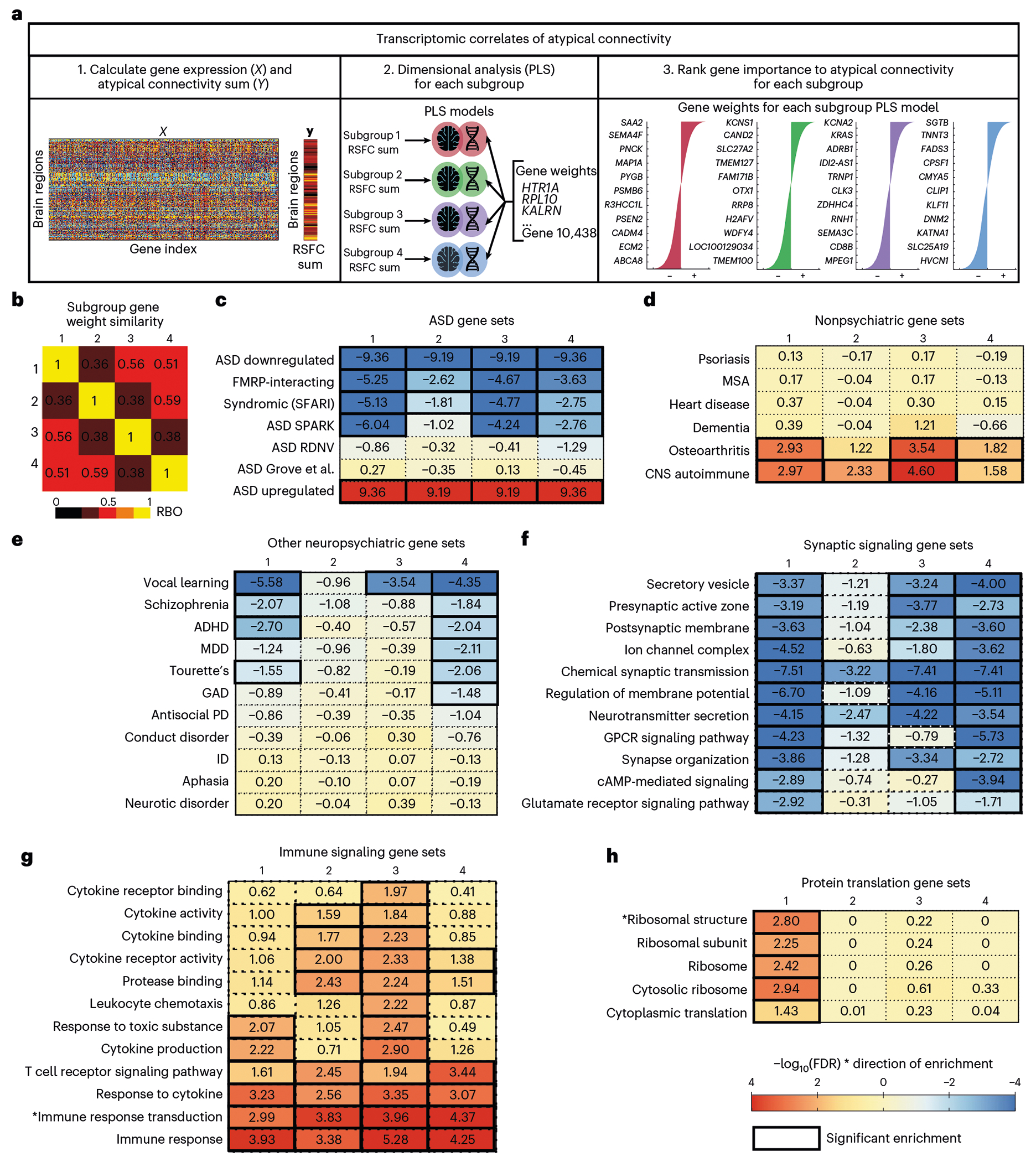
a, Schematic of transcriptomics analysis to test whether gene expression explains atypical connectivity in each subgroup. First, we calculated gene expression at each brain region (ROI) and atypical connectivity (RSFC) summed over ROIs for each subgroup. Second, we performed PLS regression for each subgroup and estimated the significance of each PLS model using a spatial permutation (‘spin’) test27,56. The PLS models for all four subgroups were significant (subgroup 1: P = 0.014; subgroup 2: P < 0.001; subgroup 3: P < 0.001; subgroup 4: P < 0.001; all statistics in Supplementary Table 3). Third, we ranked genes by PLS gene weights in each model. b, Heat map of similarity between subgroup gene rank lists (average of RBO for top 1,000 positively ranked genes and RBO for the top 1,000 negatively ranked genes between subgroups). Each subgroup was associated with a distinct set of genes (RBO = 0.36–0.59, 1 is perfect similarity). c–e, Heat maps of gene set enrichment for each subgroup’s ranked gene weights for ASD-related gene sets (c), nonpsychiatric disease-related gene sets (d), psychiatric disorder-related gene sets (e), synaptic signaling gene sets (f), immune signaling gene sets (g) and protein translation gene sets (h). Full GSEA results are in Supplementary Table 4. All subgroups were enriched for ASD-related gene sets, but not for unrelated diseases. Color indicates strength of negative log-transformed FDR for normalized enrichment score multiplied by the sign of the gene weight (+1 or −1). ADHD, attention-deficit/hyperactivity disorder; CNS, central nervous system; GAD, generalized anxiety disorder; GPCR, G-protein-coupled receptor; ID, intellectual disability; MDD, Major depressive disorder; MSA, multiple system atrophy; PD, personality disorder; RDNV, rare de novo variants.
Next, we tested the prediction that ASD risk variants would be among the most important predictors of atypical RSFC in these gene sets, using weighted fast gene set enrichment analysis (fGSEA)58 to evaluate whether the subgroup gene weights were enriched for gene sets implicated in ASD. All four subgroup PLS models were enriched for multiple ASD-related gene sets (Fig. 5c). Of note, negatively weighted genes in the PLS models (anticorrelated with atypical RSFC) were enriched for genes transcriptionally downregulated in ASD, while positively weighted genes were enriched for genes upregulated in ASD (Fig. 5c), lending further support to a biologically meaningful association between gene expression and atypical functional connectivity.
To establish the specificity of these findings, we also tested for enrichment of published gene sets associated with other disease phenotypes. Importantly, there was no enrichment for genes associated with multiple systems atrophy, dementia, heart disease or psoriasis, which were not expected to have genetic risk overlap with ASD (Fig. 5d). However, negatively weighted genes in subgroups 1 and 4–subgroups with relatively severe RRB symptoms–were enriched for genes associated with Tourette’s disorder and attention-deficit hyperactivity disorder (ADHD; Fig. 5e). Positively weighted genes in all four subgroups were enriched for immune-related diseases known to be comorbid with ASD59,60 (Fig. 5d). We also found that the gene sets for subgroups 1, 3 and 4, which had average to above-average verbal IQ, were enriched for vocal learning-related genes61 (Fig. 5e). Together, these results indicate that regional differences in gene expression predict the spatial distribution of atypical connectivity in the four ASD subgroups identified in Figs. 3 and 4, and that these genes are enriched for ASD risk variants but not for genes associated with other unrelated disorders.
While the gene sets explaining atypical connectivity were enriched for ASD-related risk variants in all four subgroups, the results in Fig. 5b indicate that distinct combinations of these genes were important in different subgroups. To further understand whether distinct biological pathways were implicated in each subgroup, we used fGSEA and Gene Ontology analysis to test for enrichment of genes associated with specific cellular components, molecular functions and biological processes62 as well as cell types (Fig. 5f–h, Supplementary Table 4 and Supplementary Figs. 22 and 23). Three patterns stood out. First, genes explaining atypical connectivity were enriched for synaptic signaling gene sets, but to differing degrees. Genes related to subgroup 2 connectivity patterns were only enriched for 2 of the 11 synaptic signaling gene sets, while subgroups 1, 3 and 4 were enriched for 11, 8 and 11 of the 11 synaptic signaling gene sets, respectively. Second, genes explaining atypical connectivity were also enriched for immune signaling gene sets, but again, to differing degrees. Genes related to subgroup 3 connectivity patterns were enriched for all 12 immune signaling gene sets, while subgroups 1, 2 and 4 were only enriched for 6, 8 and 6 of the immune signaling gene sets accordingly. This suggests that the well-established role of immune signaling in ASD63,64 may be more important in specific subgroups, at least with respect to their impact on brain network connectivity. Third, genes explaining atypical connectivity were enriched for protein translation gene sets only in subgroup 1, including ribosomal gene sets, which have been implicated in ASD65–67. Importantly, we also found that gene set enrichment results in the cross-validation analyses were highly similar to results from the full dataset analysis (Supplementary Fig. 24), lending further confidence in the findings.
For additional validation, we repeated the PLS and GSEA in a separate gene expression dataset, the BrainSpan Atlas of the Developing Human Brain29 (N = 13 individuals (7 males), aged 8–40 years; Methods). Overall, we observed highly similar results across the two gene expression datasets (Extended Data Fig. 8), providing evidence that our gene set enrichment results for the ASD clusters generalize across a developmental age range that recapitulates the age range of our neuroimaging samples. Finally, to better understand the relationship between atypical connectivity, gene expression and ASD symptoms, we conducted secondary analyses using data from the remainder of the ASD sample, that is, participants with usable fMRI data who were excluded from our primary analyses due to incomplete behavioral assessments. We tested for and found associations between verbal IQ, social affect and RRB symptoms that resembled those observed in the four subgroups defined above (Extended Data Fig. 9). Together, these analyses provide converging evidence for associations between atypical connectivity, ASD symptom domains and specific gene sets.
Subgroup-specific protein–protein interactions linked to autism spectrum disorder behaviors
To conclude, we investigated the relationship between the gene sets identified by the PLS regression models in Fig. 5 and the subgroup-specific symptom profiles identified in Figs. 3 and 4, as a means of further validating the results. We hypothesized that if the highly ranked genes in each subgroup-specific gene set played an important role in modulating pathophysiological connectivity and ASD-related behavior, then an analysis of protein–protein interactions (PPIs) derived from each gene set would reveal molecular signaling pathways that are particularly relevant in each subgroup; are enriched for ASD risk genes; and have been associated with subgroup-specific, ASD-related behaviors in previous studies.
To test these predictions, we first identified highly ranked genes that were at least modestly associated with atypical connectivity in each subgroup (P < 0.01) and differentiated genes shared by all four subgroups versus genes associated specifically with one or two subgroups (Fig. 6 and Methods). We next performed a graph-based network analysis using the STRING PPI database and identified the zero-order PPI (fully-connected graph between seed connectivity-related genes) for each subgroup (Fig. 6b–e, Supplementary Table 5 and Methods) and for the overlap between subgroups (Extended Data Fig. 10). The connectivity-related PPI of each subgroup identified numerous hub genes and functional modules (Fig. 6b–e). Notably, the PPI results for subgroup 1 consisted of only a single module related to protein synthesis and multiple ribosomal genes (Fig. 6b). The results for subgroups 2–4 contained multiple significant functional modules (Fig. 6c–e), associated with G-protein-coupled receptor signaling (subgroups 2–4), potassium channels (subgroup 2), synapse function and signal transduction (subgroup 3) and gastrin–CREB signaling (subgroups 2 and 4). Of note, the results also include multiple hub genes that are known to be transcriptionally altered in ASD, as well as numerous GWAS-confirmed ASD risk genes, lending further confidence in the results.
Fig. 6 |. Protein-protein interaction networks reveal distinct connectivity-related genes with textual associations to autism spectrum disorder-related behaviors.
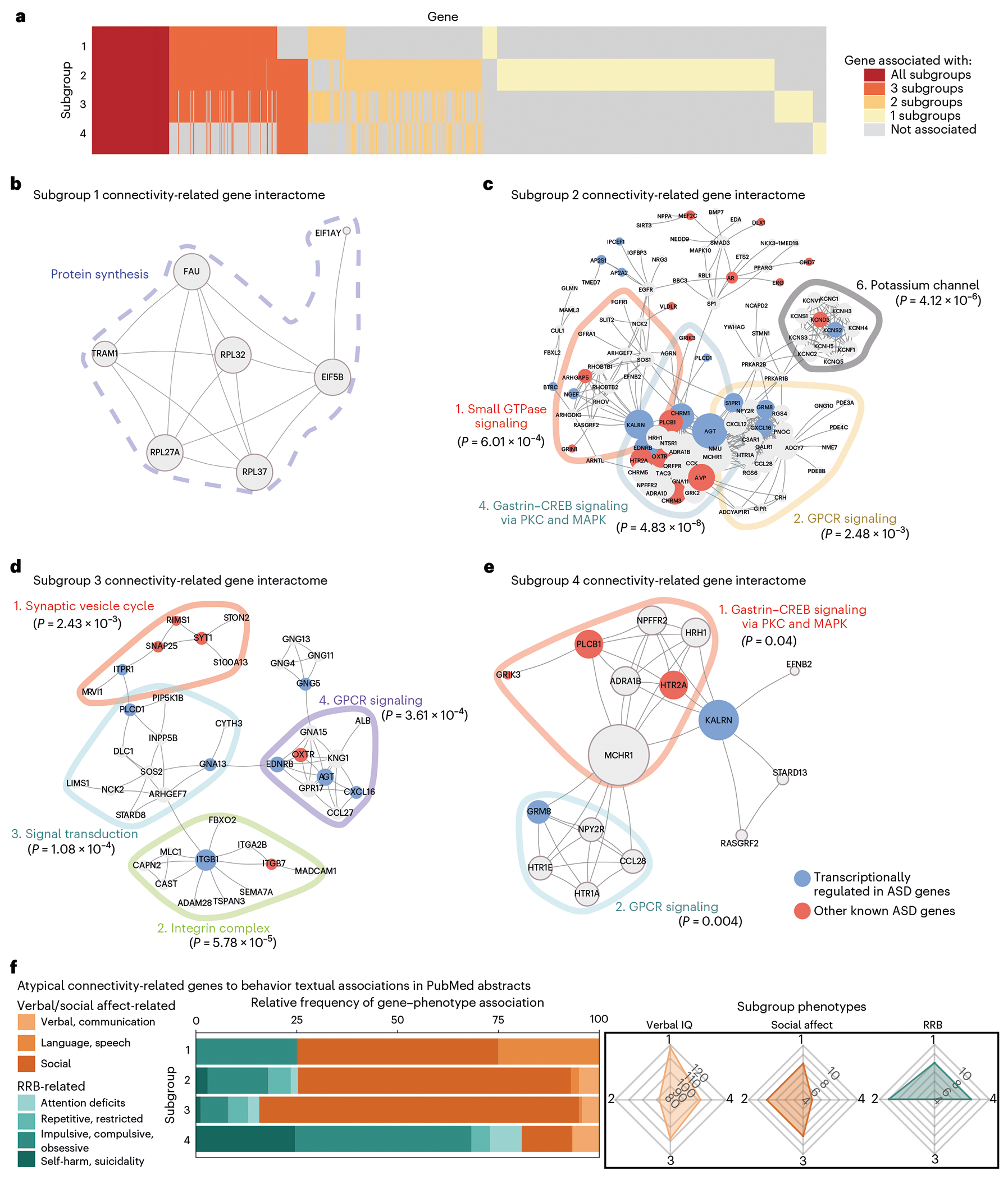
a, Heat map of overlap between genes (y axis) significantly associated with each subgroup’s atypical connectivity (P < 0.01; Methods). Some genes were significantly associated with all four subgroups (red), while others were associated with just one, two or three subgroups (yellow, pale orange or orange). b–e, Zero-order PPI networks for genes associated with each subgroup and no more than one other subgroup (STRING database; Methods). b, Subgroup 1’s interactome was associated with protein synthesis-related genes (dashed line around genes). c–e, Subgroups 2–4 had multiple significant functional modules as determined by the Walktrap algorithm (Supplementary Table 5), delineated by colored lines around each module, and implicating G-protein-coupled receptor signaling (subgroups 2–4), transforming growth factor-beta (TGF-β) signaling (subgroup 2), synapse function and signal transduction (subgroup 3) and gastrin–CREB signaling (subgroup 4), among others. The significance of each PPI module is the two-sample Wilcoxon rank-sum test (unpaired, two-sided) of within-module degrees versus cross-module degrees (no adjustments for multiple comparisons of modules). For each gene in the module, the within-module degree is the number of connected genes within the module and the cross-module degree is the number of connected genes outside the module. f, Nested bar graph of relative frequency (for each subgroup, number of keyword-associated abstracts divided by total number of abstracts) of PubMed abstract associations between subgroup-specific hub genes (up to ten top-connected genes in subgroup PPI) and behavioral keywords. Radar charts (right) show relative distribution of clinical symptom severity between subgroups (Fig. 3b–e). Subgroup 4 (severe RRB, mild social affect) connectivity-associated hub genes were most strongly associated with RRB-related keywords (RRB-related terms for S4 = 80.85%). Subgroups 1–3 showed the opposite relationship (social affect-related terms are S1 = 75.00%, S2 = 74.71% and S3 = 84.35%). Keywords are defined in Supplementary Table 6. In g, “immune response transduction’ abbreviates the ‘immune response-activating signal transduction’ gene set and in h, ‘*ribosomal structure’ abbreviates the ‘structural constituent of ribosome’ gene set.
Finally, to implement a more conclusive, unbiased validation of the association between the subgroup-specific PPI networks and the ASD-related symptoms and behaviors associated with each subgroup, we performed a text mining analysis68 of biomedical abstracts from the PubMed/MEDLINE database. We tested for associations between the most connected genes in each PPI network (‘hub genes’) and behavioral keywords related to social affect and RRB symptom domains (see schematic in Supplementary Fig. 25, Supplementary Table 6 and Methods). We found that the frequency of RRB-related keywords relative to social affect-related keywords was much higher for genes associated with subgroup 4 (80.85%), which had severe RRB symptoms and minimal social affect impairment (Fig. 6f). In contrast, the opposite relationship was found for genes associated with subgroup 3 (84.35%), which had minimal RRB symptoms and severe social affect impairment, providing an important unbiased validation of this approach to linking functional connectivity, genes and behavior. In summary, these analyses reveal multiple testable hypotheses implicating ASD-related genetic pathways in modulating the functional organization of specific brain networks and behaviors–hypotheses that are plausible in the context of the existing literature and implicate specific pathways in specific subsets of individuals. They also provide further validation that the ASD subgroups identified in Figs. 3–5 represent distinct forms of ASD associated with distinct biological processes.
Discussion
We identified and cross-validated a low-dimensional description of ASD that can disambiguate individual differences in patterns of functional connectivity and clinical behaviors and identify clinically meaningful subgroups. These brain–behavior dimensions and the associated ASD subgroups were stable across different subsets of participants, reproducible in held-out test data, and replicated in out-of-sample ASD participants from the NDA (Extended Data Fig. 7 and Supplementary Fig. 21), demonstrating the robustness of the latent model of ASD and generalizability of the subgroups. These putative ASD subtypes were associated with distinct gene expression patterns and biological processes, many of which have previously been implicated in ASD at the group level.
Our limited understanding of the neural substrates underlying ASD heterogeneity has impeded the development of therapeutic interventions. Our approach to subtyping individuals with ASD suggests testable hypotheses about how different biochemical, genetic and cellular processes may shape distinct clinical phenotypes and functional connectivity in ASD. Interestingly, two subgroups (1 and 2) separated primarily along one connectivity-related dimension (that is, the verbal intelligence quotient (VIQ)-related dimension 1). While both subgroups were highly impaired for core ASD symptoms, they differed in verbal intellectual ability, atypical connectivity and gene expression associations. The high-VIQ subgroup 1 was associated with decreased atypical connectivity between cerebellar-to-visual network regions of interest (ROIs) and somatomotor-to-prefrontal network ROIs, and our analyses highlight a correlation between atypical connectivity and gene sets involved in protein translation in this subgroup. In contrast, subgroup 2 (with low VIQ) showed atypically strengthened visual network and corticothalamic connectivity and was not correlated with protein translation gene sets. These results suggest the testable hypothesis that in at least some individuals, decreased connectivity in these networks and abnormal expression of protein translation genes might be neurobiological substrates of ASD symptoms in the setting of high VIQ but not low VIQ. These results are consistent with findings linking cerebellar connectivity to verbal IQ41; prefrontal networks to semantic processing69; corticothalamic connectivity to visual-auditory predictive coding70 impairments in ASD71,72; atypically increased corticothalamic connectivity to impaired verbal cognition in premature-born infants73 (a risk factor for ASD74); and ribosomal genes to intellectual disability in ASD66.
The other subgroups (3 and 4) had average verbal intellectual ability but differed in the ratio of impairment in the two core ASD symptoms–social affect and RRB symptoms–consistent with reports of imbalances in symptom severity in these two domains in some individuals with ASD75–77. In subgroup 3 (with social affect > RRB symptoms), we observed atypically strengthened connectivity between the visual and salience networks, and our analyses implicated immune-related gene sets—consistent with previous reports implicating these regions in reward processing in ASD78. In contrast, in subgroup 4 (with RRB > social affect symptoms), we observed atypically weakened connectivity between these networks, and our analyses implicated serotonergic hub genes–consistent with known associations between serotonin and RRBs in ASD79,80. These results suggest that the hypothesis that in at least some individuals, atypical visual-to-salience network connectivity, immune-related gene sets and serotonergic genes might be neurobiological substrates of ASD symptoms subserving social affect and RRB symptoms (with atypical connectivity in overlapping networks but with opposing changes in connectivity defining different subgroups).
A text mining analysis of hub genes associated with subgroup-specific patterns of atypical connectivity provided additional support for these associations. This is useful because our genomic and proteomic analyses identified genes associated with a subgroup using only the subgroup’s atypical connectivity, and thus did not directly measure associations between subgroup-associated genes and behavior. Our text mining analysis therefore serves as a bridge for this gene-to-behavior inference. For example, subgroup 3’s connectivity-predictive genes were frequently associated with social affect-related keywords in published biomedical abstracts (84.35%, relative to RRB-related keywords). In contrast, subgroup 4’s connectivity-predictive genes were frequently associated with RRB-related keywords (80.85%, relative to social affect-related keywords).
The ASD subgroups identified here provide insight into the biological mechanisms that may regulate changes in brain function that lead to ASD behaviors, and identify multiple testable hypotheses that could be explored in future studies. For example, in subgroup 4 (high RRB and low social affect), atypical connectivity was linked to decreased expression of HTR1A, a gene encoding a serotonin receptor associated with severe repetitive behaviors and restricted interests. HTR1A expression is known to be downregulated in ASD81 and is associated with stress and anxiety82, and dysfunctional serotonin signaling has been implicated in altered reward processing83,84 and sensorimotor impairments during development85 that contribute to RRBs. Of note, atypical functional connectivity compared to typical controls is associated with higher RRB scores86 and drugs that target serotonin signaling may be beneficial for reducing RRBs in some individuals with ASD79,80. In a Shank3 mouse model of ASD, tandospirone reduced repetitive self-grooming and learning deficit80.
Our study has several limitations. First, it is limited by the datasets available. The ABIDE I and II cohorts were collected at 36 research sites that utilized different MRI scanners and scanning protocols. Clinical phenotyping data, including both verbal IQ and ADOS-2 scale scores, were limited to a subset of the ASD participants, and participant-level genotypes were not accessible in the publicly available ABIDE datasets. To address these potential confounds, we implemented a stringent protocol to remove head motion and scanner-related artifacts based on best practices and control for site effects by interquartile normalization of each individual’s functional connectivity matrix. At least one recent report suggests that accounting for individual differences in functional topology might further enhance the performance of our models87.
Second, we found that unexpectedly, the relatively small sample size available in NDA was sufficient to implement feature selection, RCCA and clustering and yield similar clustering results. However, a sample of this size is not sufficient to identify atypical connectivity patterns associated with each cluster (with just 17 to 27 participants per cluster), especially in a whole-brain analysis. Instead, in our analysis of atypical connectivity compared to neurotypical control participants, we were able to identify similarities to the results derived from the ABIDE sample by: (1) leveraging a very large neurotypical control sample for contrast (N = 907 participants); (2) identifying qualitative convergences by focusing on RSFC features that were shown to be significantly altered in the ABIDE sample; and (3) confirming that atypical connectivity patterns associated with the NDA subgroups were significantly more similar to the ABIDE subgroups than expected by chance. We also note that the feature selection, RCCA and clustering results in the NDA sample represent a fully independent replication of the corresponding analyses in the ABIDE sample, but the comparison of RSFC in the NDA subgroups versus neurotypical controls is not fully independent because they both relied on the same neurotypical control sample.
Third, although we did not find evidence of developmental differences within or across cluster, it should be noted that this dataset is not optimal for evaluating developmental differences for multiple reasons, including that verbal IQ is inversely correlated with age in this ABIDE dataset, and thus it would be challenging to parse results if we did observe differences. Also, our observation that there was no measurable developmental heterogeneity in the particular functional connectivity features that explained individual differences in ASD symptoms does not rule out the possibility of developmental changes in other functional connectivity features that were not important in our analysis. On the contrary, a large body of pioneering studies have characterized developmental effects on functional connectivity in both ASD and typically developing populations88–91.
Fourth, the AHBA microarray dataset we used contains brain-wide gene expression measurements for 3,702 brain region samples from the postmortem brains of only six healthy adults. Despite this limitation, the statistical methods used in this study linking functional connectivity differences to gene expression have previously been shown to be statistically robust27,55,56. The GSEAs in this study found that the genes whose expression predicted patterns of atypical connectivity were enriched for ASD gene sets, but not for unrelated diseases and that molecular enrichments differed between the four groups. While spatial autocorrelation can be a concern in spatial transcriptome enrichment analyses92,93, we bootstrapped PLS gene weights over brain regions before ranking genes and implemented the weighted version of GSEA. The clear subgroup differences in gene set enrichment patterns indicate spatial autocorrelation was not a major factor in enrichment findings (because spatial autocorrelation would be similar across subgroups). Furthermore, we replicated our findings using the developmental transcriptome from the BrainSpan Atlas of the Developing Human Brain, which contains gene expression measurements from 26 brain regions of 42 individuals varying in age between 8 postconceptional weeks and 40 years (although it has a missing data rate of ~52% among these individuals/brain region samples)29. Lending further confidence in our results, enrichment findings replicated in cross-validation analyses, and an analysis of PubMed abstracts supported phenotypic differences between subgroups, together enhancing confidence that these associations were not artifactual92. Finally, there was a large age range in the dataset used in this study (ages 5–65 years, although >72% of participants spanned a narrower range of 8–16 years and >81% spanned the narrower range of 8–18 years). Repeating key analyses in the smaller sample limited to ages 8–18 years showed results consistent with the main analyses (Supplementary Figs. 5, 13 and 14 and Extended Data Fig. 5). To minimize the impact of age-related heterogeneity, we converted ADOS-2 scores to the CSS, which control for age effects and differences in verbal ability. We chose not to regress out the effect of age on RSFC, because there may be multiplicity between age effects and functional connectivity relevant to ASD, and thus regressing out age could remove biological information relevant to ASD. Importantly, the subgroups we identified did not differ substantially by age (median ages differed by 2 years or less; Supplementary Fig. 8g), indicating that age was not a driving factor of the observed subgroup differences.
In summary, we identified four subgroups within the autism spectrum that may represent distinct functional connectome phenotypes in which genotype manifests as intermediate phenotypes of atypical brain function that give rise to the clinical heterogeneity of the behavioral manifestations of autism. Our dimensional and subgroup results provide testable hypotheses that could be assessed in animal models and future clinical studies. They suggest distinct alterations in brain function that could be targeted using circuit-based neuromodulation, and they predict distinct biological pathways that could help inform studies of pharmacotherapeutic targets specific to each ASD phenotype. Future efforts to test these hypotheses will benefit from prospective samples comprising larger cohorts of ASD individuals and neurotypical controls with deeper phenotyping and associated genomic data.
Methods
Research protocols for the ABIDE and NDA datasets were approved by the Institutional Review Boards (or equivalent for international sites) at all sites as indicated in the original studies and consortium documentation. Participants provided informed consent, received a small cash reward in some studies and all regulatory guidelines were followed as described in the original reports. Data collection and analysis followed the procedures outlined in the reports from the original studies and were not randomized or blinded for collection of resting-state neuroimaging data. No data points were excluded from the analyses following the initial study exclusion criteria as outlined in ‘Participants’. Of note, all results reported in our paper were from retrospective analyses of existing datasets, not prospectively designed experiments involving randomization and blinding. As such, no power analyses or other statistical methods were used to predetermine sample sizes.
Participants
Autism Brain Imaging Data Exchange.
The ABIDE I and ABIDE II datasets contain 1,031 ASD participants and 1,139 neurotypical controls from 41 different scan sites. Following the study exclusion criteria, there were 299 ASD participants from 15 sites and 907 neurotypical controls from 35 sites for a total of 1,206 participants from 36 different sites. The 299 ASD participants had an age range of 5.13–34.76 years at time of the scan, a verbal IQ of 42–153, an ADOS-2 total severity CSS of 2–10, an ADOS-2 social affect CSS of 1–10 and an ADOS-2 RRB CSS of 1, 4–10. The neurotypical controls ranged from ages 5.89 to 64 years and had a verbal IQ of 67–156. In total, 782 ASD participants and 907 typical control participants had functional connectivity matrices that passed the exclusion criteria; they had at least 180 s of time remaining following rsfMRI scan preprocessing with motion censoring and had voxels with a temporal signal-to-noise ratio (TSNR) < 75 in all 247 power ROIs used in the study. A total of 299 of the 782 ASD participants also had all the clinical measures used in this study: verbal IQ, ADOS-2 total severity CSS, ADOS-2 social affect CSS and ADOS-2 RRB CSS. The ASD sample used in the analyses included N = 299 (aged 5–35 years); Nmale = 253 male (aged 5–27 years); Nfemale = 46 female (aged 5–35 years) and controls sample N = 907 (aged 5–64 years); Nmale = 688 male (aged 5–64 years); Nfemale = 219 female (aged 5–47 years).
Imaging parameters and preprocessing.
Because the participants were scanned at 36 different sites, imaging parameters varied between sites with repetition time (TR) values of 2, 2.5, 2.7 and 3 and scan durations of 190–580 s (for details, see refs. 10–11).We implemented preprocessing steps to standardize data and increase the SNR including: (1) aligning all participants’ rsfMRI scans to standard space, (2) applying slice-timing correction, (3) removing scanner and physiological noise and (4) correcting for motion-induced artifacts. We preprocessed rsfMRI images using a custom pipeline with commands in the open-source processing environments, FMRIB Software Library (FSL, version 6.0) and Analysis of Functional NeuroImages (AFNI, version 17.1.11)94,95. We extracted the voxels corresponding to brain tissue by creating a brain mask using FSL BET96 from the structural T1 scan, linearly registered rsfMRI data to structural scans using FSL FLIRT97,98, and applied the brain masks to the registered rsfMRI using FSL fslmaths with all non-brain voxels set to 0. Next, we registered the rsfMRI data to the standard anatomical Montreal Neurological Institute (MNI) of McGill University Health Centre atlas space99,100. First, we aligned the anatomical T1 scan to the anatomical MNI scan using linear (FSL FLIRT) and nonlinear (FSL FNIRT) registration101. We then applied the resulting transformation matrices with FSL applywarp to the rsfMRI data.
Next, we implemented standard denoising measures: (1) despiking (AFNI 3dDespike); (2) motion parameter estimation and correction (AFNI 3dvolreg); (3) slice-timing correction (AFNI 3dTshift); (4) spatial smoothi ng (4-mm FWHM Gaussian kernel); (5) temporal bandpass filtering (0.01–0.1 Hz) AFNI 3dBandpass); (6) nuisance signal regression for 12 motion parameters (AFNI 3dDeconvolve, 3dTproject) and (7) local and global hardware artifact removal using AFNI ANATICOR102.
Motion correction.
We implemented stringent preprocessing to reduce motion artifacts that includes volume realignment and motion estimate regression (AFNI 3dvolreg). We removed high motion volumes (>0.3 mm framewise displacement due to head movement) and the volumes immediately preceding and following these volumes32. Following motion censoring, we selected participants with at least 3 min of quality data, leaving remaining participants with scans ranging from 182.5 to 590 s in duration.
NIMH Data Archive out-of-sample dataset.
From the NDA database (https://www.nimh.nih.gov/), we identified NNDA = 113 ASD participants with rsfMRI data and the same three clinical behaviors as used in our study (ADOS-2 social affect and RRB, which we converted to CSS and verbal IQ). From these 113 available participants, we identified NNDA = 85 participants with usable rsfMRI data (after excluding scans with poor scan quality for example, <120 frames/180 s as with ABIDE). NNDA = 85 participants were aged 8–39 years; Nmale = 58 male (aged 8–39 years) and Nfemale = 27 female (aged 8–18 years). We repeated all the main analyses using the NDA dataset (NNDA = 85 ASD participants), including feature selection followed by RCCA and hierarchical clustering on RCCA-defined connectivity scores (brain–behavior dimensions/ canonical variates). We found that the ASD subgroups identified in the NDA dataset replicated the key findings for clinical symptoms, atypical connectivity and gene set enrichment in the ABIDE dataset (NABIDE = 299), providing evidence that the ASD subgroups we identified in the ABIDE dataset generalize to a new group of individuals with ASD (Extended Data Fig. 7 and Supplementary Fig. 21). Despite the smaller sample size, we found the behavioral distributions and atypical connectivity patterns in the NDA ASD clusters were highly similar to the ABIDE. Atypical connectivity was more similar than expected by chance when we compare to atypical connectivity measured using the same cluster sizes as NDA, but with random subsets of NDA participants not assigned to a given cluster (Supplementary Fig. 21i–l).
Feature extraction and functional connectivity measurement.
To extract RSFC measurements from the preprocessed rsfMRI scans, we implemented dimensionality reduction by parcellating the brain into 277 functionally defined spherical ROIs from the Power atlas, extracting the residual BOLD time series from these regions, and then correlating the BOLD signal between each region and every other region using AFNI 3dNetCorr, excluding voxels with low TSNR < 75 (ref. 103). ROIs that were missing for ten or more of the remaining ASD participants due to poor coverage or high TSNR signal were excluded from the study, resulting in 247 ROIs included in all subsequent analyses. Each participant’s 247 × 247 RSFC matrix was standardized by subtracting the median connectivity value and dividing by the interquartile range, the difference between the 75th and the 25th percentiles of the matrix of connectivity values. Before the calculation, NaN values were set to 0.
Feature selection and dimensionality reduction
Statistical clustering methods often work best when they are performed on a low-dimensional feature space involving a relatively small number of features that are relevant to the desired clustering outcome. As described below, we implemented additional steps to (1) select a subset of connectivity features that are important in ASD and (2) define a low-dimensional representation of those connectivity features, while taking precautions to avoid overfitting due to the variable selection step.
Robust feature selection.
To determine the strength and direction of the monotonic relationship between the clinical symptoms and functional connectivity measures, we used the Spearman correlation between the three normalized (z-score) clinical measures and the 247 × 247 functional connectivity (interquartile range-normalized) features. In 100 replicates, we subsampled 95% (N = 284) of participants and measured the Spearman correlation between the 30,3081 unique connectivity features and the three clinical symptoms. RSFC features were then ranked by the number of replicates in which they had significant correlations, yielding a robust rank list of important RSFC features in ASD.
Regularized canonical correlation analysis.
We ranked these RSFC features by how frequently they were selected as statistically significant in the robust feature selection step, and used this rank list as input into RCCA to estimate sets of linear combinations of connectivity features that best relate to linear combinations of ASD-related clinical symptoms. We used three behavioral measures from the ABIDE datasets: VIQ and the ADOS-2 subscale measures, social affect and RRB. To remove the effects of age and verbal ability, we standardized the ADOS-2 scores by converting them to CSS using the established lookup tables104,105. We note that only four ASD participants of the 299 participants had an age of ≥ 20.
To avoid potential overfitting, we utilized L2 RCCA (with an L2 or ‘ridge’ or ‘Tikhonov’ penalty) to handle multicollinearity in the input variable sets. We opted to utilize the ridge penalty and not the L1 or lasso penalty because, while L1 penalties yield sparse solutions that may aid interpretation, they are unstable when input variables are correlated106,107 and our goal was to increase stability. We note that L2 regularization has a ‘shrinkage’ effect, biasing CCA coefficients towards zero; however, we are not concerned with this particular source of bias as these are input into our clustering algorithm rather than interpreting their estimated magnitudes (and regularization improves categorical predictions like classification and clustering108,109).
We performed RCCA using the ranked RSFC features list and the three behavioral measures. We split the dataset (N = 299) into 30 RCCA-training (N = 284)/RCCA-validation sets (N = 15) and for each RCCA-training set performed robust feature selection with 20 replicates followed by an RCCA parameter grid search. For each parameter combination, we applied the RCCA-training set canonical equations to the held-out RCCA-validation set and calculated the held-out canonical correlation. We calculated the mean held-out canonical correlation values across the 30 replicates and chose the parameters that maximized the mean held-out canonical correlation. An L2-penalty (λ) was set for both variable sets (RSFC features as X and clinical measures as Y). After an initial course grid search, we settled on a hyperparameter grid of λX = [1,2,3,4,5] and λY = [0.001,0.01]. To determine the optimal number of features (Nfeatures) to include in the RCCA, we repeated this grid search on the 100 to 400 top-ranked features increasing by groups of 10. After identifying the optimal hyperparameter triplet (λX, λY and Nfeatures), we performed feature selection with 100 replicates and RCCA with these parameters in the full dataset (N = 299). The RSFC features and RCCA parameters for the 1,000 training sets were recalculated and optimized separately in each training set, so as not to bias the cross-validation results. The median RCCA parameters over the 1,000 training set replicates were λX = 5, λY = 0.001 and Nfeatures = 340, and the RCCA parameters from the full dataset analysis (N = 299) were λX = 1, λY = 0.001 and Nfeatures = 280. This yielded three canonical variate pairs (connectivity scores and behavior scores) with values for all 299 ASD participants and a canonical correlation for each variate pair. We calculated the Pearson correlation between the three behavior scores and the clinical symptoms as well as the Pearson correlation between the connectivity scores and functional connectivity followed by FDR correction.
RCCA cross-validation analyses.
To evaluate reproducibility, we tested whether the brain–behavior dimensions calculated using the training sets generalized to test set data that was completely held out from feature selection and RCCA fitting in 1,000 training/test set replicates. We split the 299 ASD participants into 1,000 random training set (N = 284) and test set (N = 15) replicates (see schematic in Supplementary Fig. 2). For bootstrapped feature selection and RCCA parameter optimization, each training set was further split into 30 RCCA-training (N = 269) and RCCA-validation (N = 15) sets with 20 replicates for robust feature selection in each RCCA-training set. As described above, each RCCA-validation set was held out from feature selection and RCCA fitting along the parameter grid. Robust feature selection was repeated in each full training set and the optimal hyperparameter triplet was used to fit RCCA. Next, in each replicate, the training set RCCA coefficients were applied to the completely held-out test set data yielding test set canonical correlations used to calculate the significance of each brain–behavior dimension. We identified three brain–behavior dimensions measured in each training set that were significant in held-out test data. Both the training set and test set connectivity scores (per subgroup; see clustering methods below) and both training set and test set correlations between connectivity scores and RSFC were also stable and consistent with analysis in the full dataset results (Supplementary Figs. 3, 4 and 11).
To assess the robustness of the brain–behavior dimensions in the cross-validation analysis, we calculated the correlation between the training set brain–behavior dimension scores and the behavior or connectivity data, and compared these with those calculated in the full dataset (Supplementary Figs. 3 and 4). These revealed the correlations of the brain–behavior dimensions to behavior and connectivity data were highly stable across training sets and to analysis performed in the full dataset using all 299 ASD participants. We also measured the stability of the brain–behavior dimensions between training sets by calculating the RV-coefficient (1 is perfect correlation) and cosine angle (0° is perfect correlation) between training sets. The connectivity scores and behavioral scores had high median RV-coefficients (0.9 and 1)110 and low median cosine angles (21.3° and 1.2°) between the training sets and the corresponding participants when RCCA was calculated in the full dataset. Together, these two validation approaches demonstrated the brain–behavior dimension scores were significant in held-out data and were stable across different subsets of participants.
Evaluating significance of brain–behavior dimensions in test set.
Significance of canonical variates (brain–behavior dimensions) was calculated by comparing the canonical correlations on held-out test data to those obtained on row-permuted data. For each of the 1,000 training/test set replicates, we further split the training set into a training and validation set (see schematic of RCCA cross-validation scheme in Supplementary Fig. 2), ran feature selection in this training set and chose hyperparameters using model performance on the validation set. Then, given the feature rank list and optimal model hyperparameters from the training/validation sets, we followed a bootstrapped cross-validation approach suggested by de Torrenté & Hastie111 to generate robust ensemble estimates and an empirical null distribution to compare them against. This procedure bootstrapped the training set (N = 284 training participants with 1,000 bootstraps with replacement) and calculated the RCCA coefficients in each bootstrap training set for the optimal hyperparameters. These 1,000 sets of coefficients were then used to project the held-out test set 1,000 times, and the mean of these test set canonical correlations was taken as an ensemble estimate of test set canonical correlations. This ensemble estimate procedure was repeated for each of the 1,000 training set/test set replications, yielding a distribution of 1,000 robust ensemble estimates of test set canonical correlations111.
To generate paired null test statistics for these ensemble estimates, we next calculated the ensemble test set canonical correlations using 1,000 row-permuted (‘shuffled’) training sets (using the same feature rank list and optimal hyperparameter triplet). For each of the 1,000 training/test set replicates, we randomly permuted the behavior dataset row indices and then subsetted the dataset into permutation training sets (N = 284) and test sets (N = 15). We then calculated the same 1,000 bootstrap estimates and their robust ensemble estimate for test set canonical correlation on the shuffled data111. In Supplementary Fig. 3a–c, we present both (a) the histogram of observed ensemble canonical correlations over 1,000 splits and (b) a paired histogram of 1,000 shuffled ensemble results (‘empirical null distribution’ in gray). These paired observed and shuffled distributions use a paired Welch’s t-test to establish significance of the ensemble test set canonical variates (again using a corrected variance estimator designed to account for overlapping data across replicates37, and testing the one-sided alternative hypothesis that the mean difference between paired observations between the observed and permuted distributions was greater than 0). We also calculated the Cohen’s d value as an estimate of the effect size. All three brain–behavior dimensions were significant in the held-out test canonical correlations (Fig. 1g–i; variate 1: r = 0.269, P < 0.0001, d = 1.119; variate 2: r = 0.180, P = 0.0005, d = 0.771; and variate 3: r = 0.115, P = 0.0185, d = 0.484; r indicates mean test set canonical correlation, P indicates P value and d indicates Cohen’s d).
Hierarchical clustering and subgroup assignment
Next, we used these ASD-related RSFC components (termed connectivity scores or canonical variates) to cluster the ASD participants into functionally distinct subgroups. First, we calculated the cosine similarity of the connectivity scores between the 299 ASD participants and used this cosine similarity matrix to hierarchically cluster participants using the average linkage. Next, we used six statistical heuristics to evaluate the goodness-of-fit performance for 2–10 clusters using cluster criterion values (Calinski Harabasz, mean Silhouette value, and Davies Bouldin; Supplementary Fig. 8a–c) and cluster stability measures (the Rand index and adjusted Rand index with leave-one-out; Supplementary Fig. 8d–f). The cluster heuristic metrics indicated a four-cluster solution was optimal and this yielded cluster assignments for the 299 individuals with ASD when clustering was performed in all participants. To enhance robustness of participant clustering, we also performed hierarchical clustering on the training set participants from each of the 1,000 training sets from the cross-validation analysis described above choosing the four-cluster solution in each training set. We took the mode of participant assignments to clusters across 1,000 subsamples as an aggregate measure of the central tendency and used these robust cluster assignments for all subsequent analyses of ASD subgroups.
For cross-validation, we compared the results for the mode ASD subgroups from Figs. 3–6 to those when the ASD subgroups were calculated separately in the 1,000 training set cluster assignments (Extended Data Fig. 4 and Supplementary Fig. 24). Results for behavior scores across subgroups, atypical connectivity (Welch’s t-test between functional connectivity of ASD subgroup participants and neurotypical controls) and gene set enrichment (described below) were highly similar, increasing confidence in the mode ASD subgroup results shown in the main text.
In our primary analysis of group-level and subgroup-level atypical connectivity, we compared group-level or subgroup-level atypical connectivity to all controls with usable neuroimaging data regardless of age (that is, N = 907 typical control participants with high-quality fMRI data; see ‘Participants’ for details). However, to rule out age confounds, we also performed a secondary analysis restricted to control participants who were age matched to the ASD sample age range (N = 868 TC participants, aged 5–35 years). We replicated all key findings of group-level and subgroup-level results with the age-matched controls (Supplementary Fig. 12), validating our primary analysis. Next, we replicated key findings of the ASD subgroups (by repeating the RCCA and clustering analyses) first in a narrower age-range sample (aged 8–18 years) and second in a second brain parcellation (Craddock 200; ref. 49). We found the subgroup results for clinical symptoms/behaviors, atypical connectivity and gene expression were consistent with those for the main analyses presented in Figs. 3 and 4 (Extended Data Figs. 5 and 6 and Supplementary Figs. 13–16). Finally, we evaluated the impact of age on these ASD subgroups, and did not detect evidence of developmental heterogeneity within the brain–behavior associations of the ASD subgroups (Supplementary Figs. 17–20 and Supplementary Tables 1 and 2).
Allen Human Brain Atlas dataset
Participants and data collection.
We used the AHBA28,112, which contains brain-wide microarray samples from 3,702 brain regions. The samples were collected from six neurotypical adult brains, sampling from both hemispheres in two brains and one hemisphere in the remaining four brains, and include T1 MRI data with MNI sample coordinates. RNA-sequencing (RNA-seq) data are also available for two of the brains involving 112 brain regions.
Preprocessing.
Preprocessing of microarray data consisted of two steps: probe to gene assignment (step 1) and anatomical sample location to Power ROI assignment (step 2). For step 1, we (a) reannotated probes to include updated Entrez ID assignments, (b) filtered out microarray probe expression that did not exceed background levels (due to nonspecific hybridization), (c) measured correspondence between microarray probe expression data and RNA-seq gene expression, (d) selected one probe per gene (the probe with expression most similar to RNA-seq expression with a threshold of at least 0.2 correlation; removing unmapped/below threshold genes) and (e) standardizing gene symbols to the Hugo Gene Nomenclature Committee (HGNC) nomenclature. This resulted in 10,438 gene expression values for each of the microarray samples.
For step 2, for each AHBA participant, we assigned the microarray samples to the 247 functionally defined Power ROIs as follows: (a) by assigning microarray samples and Power ROIs to major anatomical parcels (left and right cortex, subcortex, cerebellum and brainstem); (b) for each anatomical parcel, measuring the Euclidean distance from the MRI coordinates of each microarray sample in the parcel to the centroid of each Power atlas ROI in that parcel; (c) based on this distance measure, assigning each microarray sample to the closest Power ROI within 15 mm (average distance of mapped sample to ROI was 4.84 ± 1.48 mm) and (d) averaging expression for each ROI over all assigned microarray samples for each gene. With this distance threshold, this resulted in 2,857 AHBA samples from the six brains assigned to 230 of the 247 ROIs used in subgroup discovery. Following assignment, gene expression was standardized by the z-score of the log2 of each value. The result of microarray sample preprocessing and sample-to-ROI assignment was a 230-ROI by 10,438-gene matrix.
BrainSpan Atlas of the developing human brain developmental transcriptome (BrainSpan) dataset.
We used the BrainSpan dataset29 that contains RNA-seq expression data collected in 26 brain regions from postmortem brains of 42 individuals varying in age between 8 postconceptional weeks and 40 years of age. There is a missing data rate of ~52% among these individuals/brain region samples, such that only a subset of the brain regions was sampled among different individuals. We first excluded BrainSpan participants <5 years of age because this does not overlap with the age range of ABIDE and most of those samples are from <1-year-old brains with markedly different brain anatomy. This resulted in gene expression measurements from 13 BrainSpan donors aged 8–40 years (N = 6 females aged 11–40 years; N = 7 males aged 8–37 years), and these gene expression measurements spanned 16 distinct BrainSpan regions across the cortex, subcortex and cerebellum. We next mapped these BrainSpan brain structures onto the Power atlas by manually comparing the BrainSpan brain region labels to ROIs; that is, we upsampled to the Power atlas by assigning gene expression from BrainSpan samples to the Power ROIs (for example, Power amygdala ROIs were assigned BrainSpan amygdala gene expression). This mapping of BrainSpan onto the Power atlas resulted in 15/16 BrainSpan brain regions mapped onto 106/230 Power atlas from our PLS analysis (that is, 1/16 BrainSpan brain region was not sampled by the Power atlas and 124/230 Power atlas ROIs were not sampled by BrainSpan). We only included BrainSpan expression for the 9,648 genes that overlapped with the 10,438 genes from the AHBA PLS analyses. Thus, the input gene expression matrix (X) in the Brainspan PLS analysis was 106 ROIs × 9,648 genes and the input net atypical RSFC vector (y) was a vector of length 106 ROIs. The X in BrainSpan was calculated as the average RNA-seq gene expression (z-score(log2(RPKM))) across the 13 donors aged 8–40 years (N = 6 female aged 11–40 years, N = 7 male aged 8–37 years). We next performed bootstrapped PLS and gene expression analysis following the procedure outlined for the AHBA atlas below. The results are depicted in Extended Data Fig. 8.
Identifying autism spectrum disorder subgroup-associated genes
Partial least squares regression models.
To investigate whether brain-wide gene expression from the AHBA atlas predicts ASD-related changes in functional connectivity, we utilized PLS using the SIMPLS algorithm (MATLAB plsregress) with the 230 brain regions as samples, the predictors (X) as the 10,438 gene expression values across these samples, and one response variables (y): the net atypical connectivity (sum of positive atypical connectivity to each ROI minus the absolute value of the sum of negative atypical connectivity to each ROI). Atypical connectivity was calculated as the t-test between the RSFC of ASD participants in each subgroup and neurotypical controls (Fig. 5a). This resulted in two input matrices: (1) X: 10,438 genes × 230 brain regions and (2) y: atypical connectivity vector 1 × 230 brain regions (one per subgroup). A separate PLS model was calculated for each subgroup.
Each PLS model output two sets (one for X and one for y) of score vectors (230 samples × 1 component) and loading weights (10,438 gene weights × 1 component and 1 atypical connectivity sum weights × 1 component) as well as the variance explained in X by y and in y by X. PLS score vectors are the weighted linear sum of the variables over all brain regions and loading weights are calculated to maximize the covariance between the two variable sets (here, gene expression and atypical connectivity). We bootstrapped gene weights to reduce gene expression measures from a subset of brain samples dominating the model, and to increase generalization of the set of genes that significantly explained atypical connectivity when different combinations of brain regions are sampled. Each bootstrap sample contained, on average, 145 of the 230 ROIs for AHBA (and 67 of the 106 ROIs for the BrainSpan replication analysis). For the gene loading weights, the magnitude indicates the relative importance of each gene’s expression to explaining atypical connectivity. We calculated the correlation between the PLS gene scores and net atypical connectivity. Thus, positively weighted genes were positively correlated with net atypical connectivity and negatively weighted genes negatively correlated with net atypical connectivity.
Gene weight stability, significance testing and overlap testing.
To improve the stability of the PLS model predictions, we bootstrapped each model 100 times (without replacement) across different sets of ROIs and recalculated the gene expression loading weight vector output for each model by dividing by the standard deviation of the bootstrap distribution (this is similar to a z-test when the null hypothesis is that the gene has a weight equal to 0). We ranked genes based on the bootstrapped PLS loading weight. We tested the significance of the first component of the PLS model by permuting the samples 10,000 times, recalculating the PLS model, and comparing the actual variance explained by the PLS model to the permutation values. We implemented both a simple permutation test and a stricter, spatial permutation (‘spin’) test27,56 with a threshold of significance at P < 0.05. We compared gene weight similarity between the PLS models for the four subgroups using RBO of the top 1,000 positively weighted and top 1,000 negatively weighted genes, an indefinite rank similarity measure that handles non-conjoint ranking lists57.
Functional enrichment of candidate gene sets
Gene set lists.
We extracted the following gene set lists from the cited articles and databases: Grove et. al, ASD common variants19, ASD cell types113, ASD transcriptionally upregulated (asdM16 from ref. 114) or downregulated (asdM12 from ref. 114) extracted from ref. 115, ASD rare de novo116, ASD SPARK117, FMRP-interacting118, intellectual disability (‘ID All’)119, vocal learning61, psoriasis120, Simons Foundation Autism Research Initiative (syndromic SFARI)121, Comparative Toxicogenomics Database122 (schizophrenia), heart disease123, DISEASES database124 (ADHD, antisocial personality disorder, aphasia, conduct disorder, generalized anxiety disorder, major depressive disorder, neurotic disorder and Tourette’s syndrome), RGD disease ontology125 (CNS autoimmune, dementia, osteoarthritis and multiple system atrophy) and the PANTHER 16.0 GO slim Gene Ontology database62,126,127.
Gene set enrichment analysis.
Using the bootstrapped gene weight rank lists for each subgroup, we used the weighted fGSEA R package58 to evaluate whether they were enriched for genes related to ASD, but not to unrelated diseases, and whether subgroup models differed in enrichment for Gene Ontology gene sets (molecular function, cellular components and biological processes). The fGSEA algorithm compares the enrichment score of gene weights to a null distribution by permuting gene weight assignments. Genes are sorted by weight and then a running sum is calculated as the enrichment score and normalized by the gene set size to obtain the normalized enrichment score. The algorithm finds the vertex point where the second derivative or rate of change of the first derivative (slope) of the enrichment score equals 0. This vertex is the position of maximum overlap between the ranked gene set and gene set of interest. Next the algorithm calculates the significance of enrichment by comparing the vertex enrichment score to the permuted null distribution. The P values were calculated by comparing normalized enrichment scores to the empirical null distribution, FDR-corrected using Benjamini–Hochberg correction and thresholded for FDR < 0.05.
Analysis of relationships between genetic variants, functional connectivity and behavior.
We designed an analysis in which our goal was to assess the relationship between atypical connectivity, clinical symptoms/behaviors in ASD, and gene expression and to make use of more of the ASD sample that had usable RSFC data but incomplete behavioral assessments. N = 782 of the ASD sample had usable RSFC but incomplete behavior; however, NVIQ = 590 of these had VIQ (but not ADOS-2 social affect/RRB) measurements, while NAD0S-2 = 353 had ADOS-2 measures of social affect and RRB (but not VIQ). To assess relationships between gene expression with atypical connectivity and behavior in larger usable samples, we split the NVIQ = 590 sample with VIQ into VIQ bins (participants with VIQ > 120, 85–120 or <85) and used PLS (in the same way as in the main analysis) to assess the relationship of these VIQ-binned participants’ atypical connectivity with gene expression. As subgroups 3 and 4 differed in the ratio of social affect to RRB, we also aimed to assess the relationship of social affect/RRB to atypical connectivity and gene expression. We split the NADOS-2 = 353 participants with ADOS-2 assessment into bins of social affect > RRB and RRB > social affect and used PLS (as in the main analysis) to assess the relationship of these ADOS-2-binned participants’ atypical connectivity with gene expression. Our results revealed interesting relationships between atypical connectivity related to behavioral class and gene expression, consistent with our results in the subgroup-level PLS and GSEA that we outline further in the Results (Extended Data Fig. 9 and Supplementary Discussion).
Protein–protein interaction network.
We performed a graph-based gene network analysis in R with NetworkAnalyst128–130 using the set of genes that were strongly weighted in the PLS analysis for that subgroup and no more than one other subgroup (Fig. 6a). Strongly weighted genes were genes whose bootstrapped weight magnitude was greater than the distribution of null bootstrapped weight magnitudes from a permutation analysis in which the rows of atypical connectivity were shuffled (1,000 shuffles, using P < 0.01 without FDR correction as a heuristic threshold). Subgroup gene sets were next mapped to the STRING PPI database131, and a search algorithm identified proteins that directly interacted with candidate gene seeds (confidence score cutoff > 900 for STRING functional associations and physical associations from experimental data). The seeds and interaction partners were used to build a zero-order PPI subnetwork. We calculated the degree (number of connections) for each gene in the PPI networks and identified the overlap between genes in the PPI networks and genes known to be transcriptionally upregulated or downregulated in ASD114,115 or, if not transcriptionally regulated, then known to be an ASD risk gene in the SFARI database121,132 (Fig. 6b–e). The degree was used to plot the relative size of the gene node, and the gene overlap with ASD risk gene sets was used to color the nodes. Subgroup 2 was thresholded for nodes with a degree of 10 or greater due to PPI network size.
To identify molecular modules associated with ASD-related connectivity, we first calculated the zero-order PPI network using all subgroup-associated candidate genes that were associated with atypical functional connectivity patterns as the seed nodes. We next used the Walktrap algorithm to highlight the independent components within the graph that likely represent distinct biological functional modules important to the ASD phenotype. We labeled the modules by colored lines surrounding each module along with a textual description of the biological property and the module significance calculated by NetworkAnalyst (also see Supplementary Table 5 for PPI module significance). The significance of each PPI module is the two-sample Wilcoxon rank-sum test (unpaired, two-sided) of within-module degrees versus cross-module degrees (no adjustments for multiple comparisons of modules). For each gene in the module, the within-module degree is the number of connected genes within the module and the cross-module degree is the number of connected genes outside the module.
Text mining analysis.
To provide an additional, unbiased validation of the association between the subgroup-specific PPI networks and the ASD-related symptoms/behaviors associated with each subgroup, we performed a text mining analysis68,133–135 of biomedical abstracts from PubMed for associations between the most connected genes in each PPI (‘hub genes’) and behavioral keywords related to social affect and RRB symptom domains (see schematic in Supplementary Fig. 25). We asked whether the top genes in the PPI network (most interconnected) for each subgroup had known associations with behaviors that were related to the differing clinical behavioral patterns found in each ASD subgroup. In each subgroup’s PPI network, we identified genes with a degree of at least 1, and selected the top ten genes for the text mining analysis (or all genes if less than ten had degree > 1, as occurred in subgroup 1, which had only seven genes exceeding this threshold). For each subgroup-specific gene set, the resulting subnetwork represents an atypical functional connectivity PPI network.
Taking these top hub genes for each subgroup, we identified hub gene-associated biomedical literature. We used the MeSH IDs corresponding to the hub genes to query the PubTator Central database of annotated biomedical entities136,137. We downloaded and tokenized the abstracts, standardized the token words; removed stop words using the ‘tm’ and ‘quanteda’ libraries in R138,139; and created a phenotypic keyword dictionary for social affect-related terms (verbal, communication, language, speech and social interaction) and RRB-related terms (attention deficits, repetitive or restricted behaviors/interests, compulsive, impulsive and obsessive behaviors, and self-harm/suicidality; Supplementary Table 6).
For each subgroup, we calculated the relative frequency of each keyword being included in MEDLINE/PubMed abstracts associated with the subgroup-specific hub gene set (or MeSH synonyms of these genes). Relative frequency was calculated separately for each subgroup as the number of abstracts containing the keyword divided by the total number of abstracts matched to any keyword in the dictionary. This resulted in a statistical measure of how associated each phenotypic keyword was to the hub genes in a subgroup relative to the other keywords, and allowed us to test the hypothesis that the relative frequency of social affect-related terms as compared to repetitive behavior and restricted interest-related terms would align with the phenotypic composition of each subgroup. The text mining results supported the hypothesis that key subgroup-associated genes may lead to the distinct ASD-related behavioral phenotypes in the subgroups via interactions with the intermediate atypical functional brain connectivity patterns that define each ASD subgroup.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Connectivity score loadings on RSFC and atypical RSFC in 247 × 247 heatmaps.
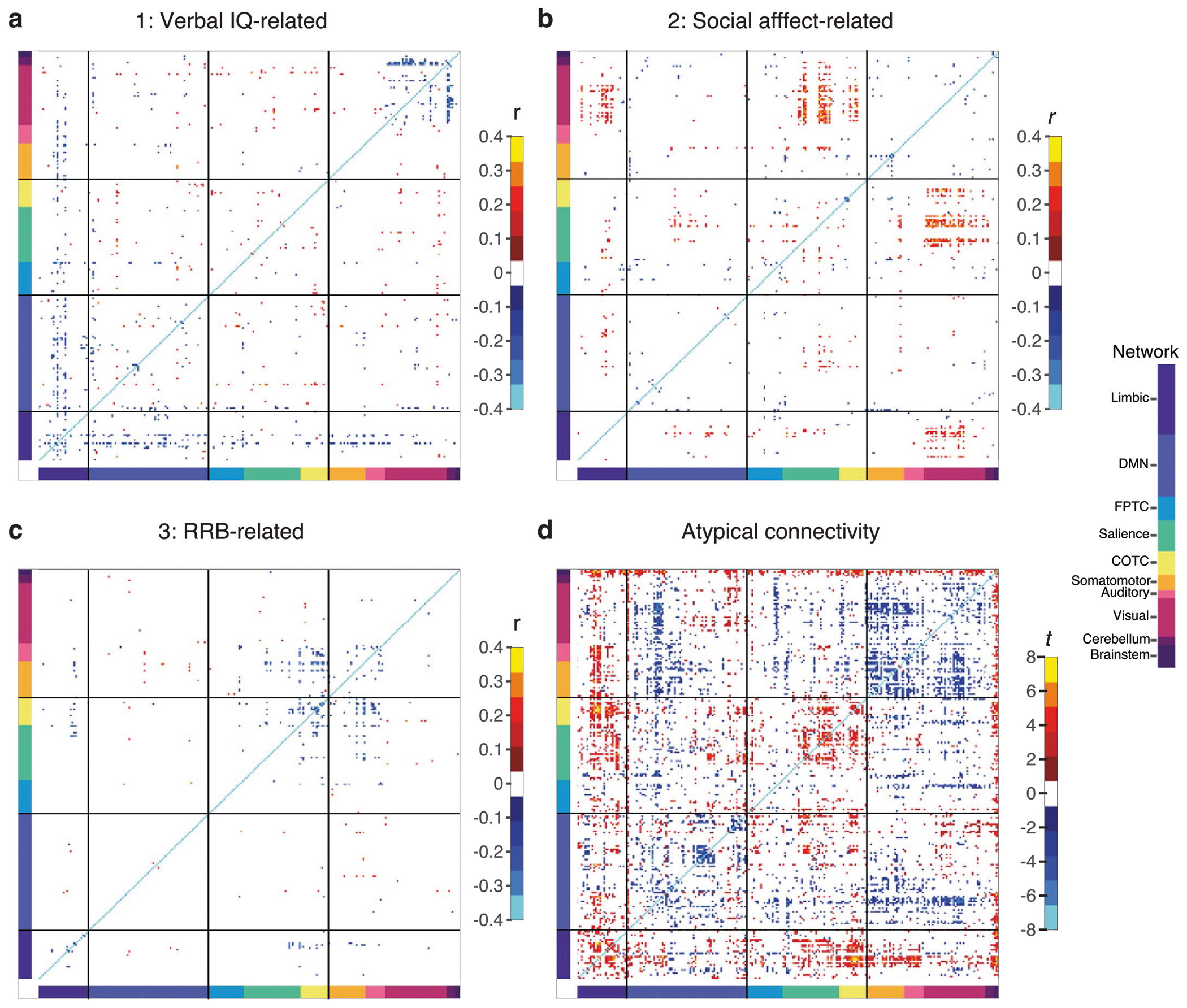
Heatmaps of 247 × 247 regions of interest (ROIs) corresponding to panels in Fig. 2 sorted and labeled by functional network. (a) Correlation between verbal IQ-related dimension (dimension 1) and RSFC (FDR < 0.05; see Fig. 2a). (b) Correlation between social affect-related dimension (dimension 2) and RSFC (FDR < 0.05; see Fig. 2b). (c) Correlation between RRB-related dimension (dimension 3) and RSFC (FDR < 0.05; see Fig. 2c). (d) Atypical connectivity in ASD subjects versus controls (Welch’s t-test; FDR < 0.05; see Fig. 2d). Abbreviations described previously in Figs. 1–2.
Extended Data Fig. 2 |. Autism spectrum disorder subgroups replicate when using different clustering methods.

(a, b) K-means clustering with cosine distance, (c, d) spectral clustering with cosine distance, and (e, f) hierarchical clustering with Euclidean distance and Ward linkage across 1,000 training set replicates (N = 284). In (g, h) we show the original analysis using hierarchical clustering with cosine distance and average linkage (see Methods for more details). Boxplots show distribution of clinical symptom z-scores (superimposed bar graphs depict the median) for social affect, repetitive, restrictive behaviors and interests (RRB), verbal IQ, and total severity (color indicates subgroup). Plots include 284 subjects x 1,000 training sets to indicate distribution of clinical behaviors across all 1,000 training set cluster assignments. Box bounds: [25th,75th percentile]; center: median; whiskers: 99.3% data in + /−2.7 σ; outliers: circles). Heatmaps show patterns of mean atypical connectivity across replicates in each subgroup across brain regions (rows) and functional networks (columns), and were thresholded for significant atypical connectivity (two-sided Welch’s t-test, mean FDR < 0.05), evaluated relative to N = 907 neurotypical controls. See additional comparisons in Supplementary Fig. 9.
Extended Data Fig. 3 |. Functional connectivity differences reveal subgroup-specific atypical connectivity.

Subgroups were defined as the modal subgroup assignment over the 1,000 training set replicates, which is used in the main text for Figs. 3–6. (a-d) Heatmaps show patterns of atypical connectivity in each subgroup across brain regions (rows) and functional networks (columns). Thresholded for significant atypical connectivity (two-sided Welch’s t-test, FDR < 0.05), evaluated in N = 69 ASD subjects in subgroup 1, N = 87 ASD subjects in subgroup 2, N = 67 ASD subjects in subgroup 3, N = 76 ASD subjects in subgroup 4, relative to N = 907 neurotypical controls.
Extended Data Fig. 4 |. Cross-validation of the clinical symptom and atypical connectivity differences between subgroups.

To cross-validate the clinical symptom and atypical connectivity differences between subgroups in Figs. 3–4 and Extended Data Fig. 3, we first subsampled 95% of the data in 1,000 replicates. Second, we calculated canonical variates (connectivity score and clinical score for each brain–behavior dimension) in each replicate. Third, in each replicate, we hierarchically clustered on connectivity scores using cosine similarity distance and average linkage and identified four subgroups. Fourth, we used the Hungarian method to match clusters between replicates (numerical assignment of subgroups can change without changing subject composition in cluster). Fifth, we calculated the distribution of clinical symptom z-scores for each subgroup across replicates. Sixth, in each replicate, we calculated atypical connectivity per subgroup versus N = 907 neurotypical controls (two-sided Welch’s t-test). Seventh, we calculated the mean and standard deviation (σ) of atypical connectivity (t) on RSFC over 1,000 subsampled replicates. (a-d) Note similarity to Fig. 3b–e: Subgroups differ with respect to clinical symptoms, similar to subgroup differences identified when subgroups were calculated as modal cluster assignment across 1,000 training sets (mode analysis) shown in Fig. 3b–e. Plots include 284 subjects x 1,000 training sets to indicate distribution of clinical behaviors across all 1,000 training set cluster assignments. Box bounds: [25th,75th percentile]; center: median; whiskers: 99.3% data in + /−2.7 σ; outliers: circles). (e-h) Heatmaps show patterns of mean atypical connectivity across replicates in each subgroup across brain regions (rows) and functional networks (columns), and were thresholded for significant atypical connectivity (two-sided Welch’s t-test, mean FDR <0.05). (i-l) Heatmaps show patterns of the standard deviation of atypical connectivity across replicates in each subgroup across brain regions (rows) and functional networks (columns).
Extended Data Fig. 5 |. RCCA and clustering analysis using narrower age range (ages 8–18) yields ASD subgroups with clinical symptoms and atypical connectivity consistent with main analysis.
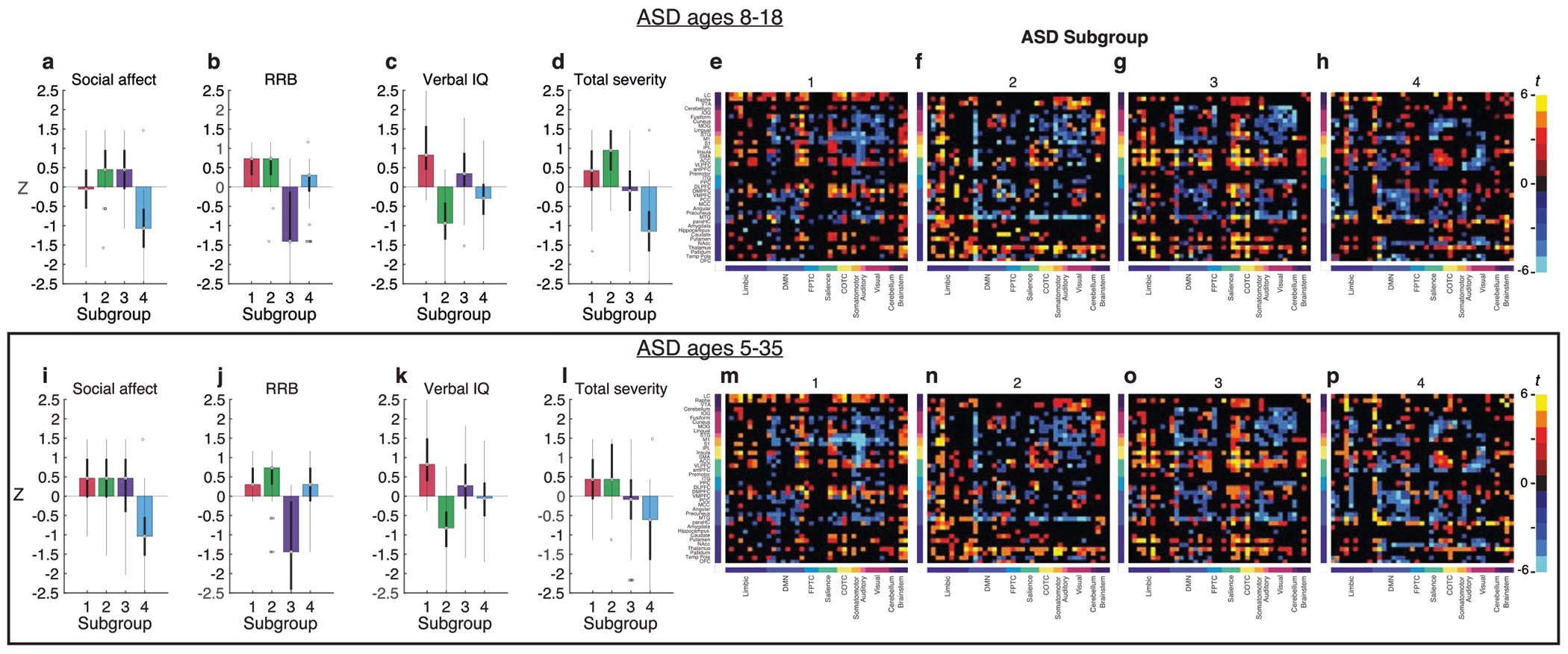
We repeated all the main analyses (shown in box, (i-p) using a smaller age range, including only ASD and neurotypical individuals of ages 8–18 (shown in a-d and i-l). This reduced our ASD sample from N = 299 ages 5–35 to N = 243 ages 8–18 and reduced our neurotypical sample from N = 907 to N = 573. In this secondary analysis, we found similar clinical symptom profiles associated with each subgroup (a-d vs. i-l). Boxplots of the distribution of clinical symptom z-scores (superimposed bar graphs depict the median) for (a,e) social affect, (b,f) repetitive, restrictive behaviors and interests (RRB), (c,g) verbal IQ, and (d,h) total severity (color indicates subgroup). Note that higher social affect, RRB, and total severity scores and lower verbal IQ indicate greater impairment. Box bounds: [25th,75th percentile]; center: median; whiskers: 99.3% data in + /−2.7 σ; outliers: circles). Next, we found similar atypical connectivity associated with each subtype (e-h vs. m-p). (e-h) Atypical connections that were significant (P < 0.05) in the narrower age range, thresholded for significant atypical connectivity (two-sided Welch’s t-test, FDR < 0.05). (m-p) Atypical connections that were significant (P < 0.05) in the full age range, thresholded for connections that were significant in the main analysis (two-sided Welch’s t-test, FDR < 0.05). Heatmaps show patterns of atypical connectivity in each subgroup across brain regions (rows) and functional networks (columns). Thresholded for significant atypical connectivity (two-sided Welch’s t-test, FDR < 0.05), evaluated relative to N = 907 neurotypical controls. For additional results, see Supplementary Figs. 13, 14 and 17–20.
Extended Data Fig. 6 |. RCCA and clustering analysis using the Craddock 200 atlas yields ASD subgroups with clinical symptoms and atypical connectivity consistent when analyzed using the Power atlas.

We reparcellated the brains using the Craddock 200 atlas69, recalculated functional connectivity for each subject, and repeated the full analysis following the original pipeline (feature selection, RCCA, clustering, and PLS). Key findings from the primary analysis using the Power parcellation replicate in this secondary analysis using the Craddock atlas. Here we plot the clinical symptom scores (boxplots as in Extended Data Fig. 5) for each subgroup when (a-d) we used the Craddock 200 parcellation for functional connectivity versus (i-l) the Power parcellation for functional connectivity (main text analysis). Next, we measured atypical connectivity using the Craddock parcellation and mapped it onto the Power atlas for visual comparison between the two parcellations. We plot the atypical connectivity for each subgroup for (e-h) the analysis in the Craddock 200 parcellation thresholded the significant connections from the Power parcellation, and (m-p) the analysis in the Power atlas. Heatmaps show patterns of atypical connectivity in each subgroup across brain regions (rows) and functional networks (columns). Thresholded for significant atypical connectivity (two-sided Welch’s t-test, FDR < 0.05), each evaluated separately relative to N = 907 neurotypical controls. For additional results, see Supplementary Figs. 15, 16.
Extended Data Fig. 7 |. Out-of-sample replication of ASD subgroup clinical symptoms and atypical connectivity in NDA dataset (NNDA = 85 ASD subjects).

We repeated the main analyses to define ASD subgroups using the NDA dataset (RCCA and clustering). This analysis replicated key results from ABIDE, such that the four NDA subgroups (NNDA_1 = 20, NNDA_2 = 21; NNDA_3 = 27; NNDA_4 = 17) exhibited clinical symptom / behavior profiles and atypical connectivity patterns that were highly similar to those observed in the ABIDE subgroups (NABIDE_4 = 69, NABIDE_2 = 87; NABIDE_3 = 67; NABIDE_4 = 76). In this summary figure, we plot the clinical symptom scores (NDA: a-d, ABIDE: i-l; boxplots as in Extended Data Fig. 5) and atypical connectivity patterns for each subgroup (NDA: e-h, ABIDE: m-p). As expected, statistical power to detect significant atypical connectivity was reduced due to the smaller sample size of NDA. Here, the heatmaps show atypical functional connectivity in NDA and ABIDE subgroups, with the NDA subgroups thresholded by significance from ABIDE for comparison (that is, we set elements in the NDA heatmaps with FDR < 0.05 from a connectivity (two-sided Welch’s t-test in ABIDE heatmaps to 0). However, we confirmed that compared to an empirical null (100 shuffles, see Methods for details), atypical connectivity patterns in the NDA ASD subgroups were more correlated with ABIDE ASD subgroups than expected by chance (P1 = 0.0099, P2 = 0.0297, P3 = 0.0099, P4 = 0.0198). Note that the P values correspond to the probability of obtaining the observed sum of ranks statistic (sum of observed ranks across a range of FDR thresholds, FDR in {1, 0.05, 0.01, 0.005, 0.001, 0.0005, 0.0001, 0.00005}) under the empirical null. For additional results, see Supplementary Fig. 21.
Extended Data Fig. 8 |. Replication of transcriptomic correlates of subgroup atypical connectivity using BrainSpan gene expression.

We mapped data from the BrainSpan gene expression atlas to the Power atlas, and repeated the PLS and gene set enrichment analyses described in the main text. We found similar results to the original analysis in which we had used the AHBA gene expression dataset, including highly similar transcriptomic correlates of subgroup atypical connectivity. For the PLS analysis, we first calculated gene expression at each brain region (ROI) and atypical connectivity (RSFC) summed over ROIs for each subgroup. Second, we performed PLS regression for each subgroup. Third, we ranked genes by PLS gene weights in each model. The results were highly similar to those observed in the original analysis using the AHBA gene expression atlas. Heatmaps of gene set enrichment for each subgroup’s ranked gene weights for (a vs. b) ASD-related gene sets, (c vs. d) nonpsychiatric disease-related gene sets, (e vs. f) psychiatric disorder-related gene sets, (g vs. h) synaptic signaling gene sets, (i vs. j) immune signaling gene sets, and (k vs. l) protein translation gene sets. All subgroups were enriched for ASD-related gene sets, but not for unrelated diseases. Color indicates strength of negative log transformed FDR for normalized enrichment score multiplied by sign of gene weight (+1 or −1). The P values were calculated and FDR-corrected as in Fig. 5.
Extended Data Fig. 9 |. Transcriptomic correlates of atypical connectivity patterns associated with ASD-related behaviors.

To further assess relationships between gene expression with atypical connectivity and behavior in larger useable samples (that is, now including subjects with usable fMRI data who were excluded from primary analyses due to incomplete behavioral assessments) we started with the N = 782 subjects with usable scan data, and split the NVIQ = 590 subjects with VIQ into VIQ bins (ASD subjects with [NVIQ>120 = 127] VIQ > = 120, [N85≤VIQ≤120 = 383] VIQ 85–120, or [NVIQ<85 = 80] VIQ < = 85). We also split the NADOS-2 = 353 subjects with ADOS-2 assessment into bins by calculating social affect divided by RRB. The social affect > RRB bin (social affect / RRB > 1) had NSA>RRB = 113 ASD subjects and the RRB > social affect bin (social affect / RRB >1) had NSA<RRB = 171 ASD subjects; the NSA=RRB = 69 ASD subjects with SA/RRB = 1 were not included in either ADOS-2 bin. The overlap of subjects between the NVIQ = 590 subjects with VIQ and NADOS-2 = 353 subjects with ADOS-2 was the NVIQ;ADOS-2 = 299 ASD subjects in the main analysis. We used the same PLS and gene set enrichment procedure as in Fig. 5 (see b,d,f,h,j,l in box) to assess the relationship of these binned subjects’ atypical connectivity with gene expression. Heatmaps of gene set enrichment for each subgroup’s ranked gene weights for (a-b) ASD-related gene sets, (c-d) nonpsychiatric disease-related gene sets, (e-f) psychiatric disorder-related gene sets, (g-h) synaptic signaling gene sets, (i-j) immune signaling gene sets, and (k-l) protein translation gene sets. Color indicates strength of negative log transformed FDR for normalized enrichment score multiplied by sign of gene weight (+1 or −1). The results were consistent with our predictions: gene set enrichments for the low-VIQ bin resembled those for subgroup 2 (featured low Verbal IQ) and enrichments for the high-VIQ bin resembled those for subgroup 1 (featured above-average VIQ). See further description of results in Supplementary Discussion. The P values were calculated and FDR-corrected as in Fig. 5.
Extended Data Fig. 10 |. Zero-order protein-protein interaction (PPI) networks for genes associated with multiple subgroups.

Zero-order protein-protein interaction (PPI) networks for (a) genes associated with all four subgroups and (b) genes associated with at least 3 subgroups (STRING database; see Methods). Blue genes are known to be transcriptionally regulated in ASD while red genes are genes not known to be transcriptionally regulated but that have been associated with ASD in the SFARI database. The significance of each PPI module is the two-sample Wilcoxon rank sum test (unpaired, two-sided) of within-module degrees versus cross-module degrees (no adjustments for multiple comparisons of modules). For each gene in the module, the within-module degree is the number of connected genes within the module and the cross-module degree is the number of connected genes outside of the module.
Supplementary Material
Acknowledgements
This work was supported by grants from the NIMH (MH118388, MH114976, MH123154, MH118451, MH109685 and MH109685-04S1), the National Institute on Drug Abuse (DA047851), the Hope for Depression Research Foundation, the Pritzker Neuropsychiatric Disorders Research Consortium, the Klingenstein–Simons Foundation Fund, the One Mind Institute, the Rita Allen Foundation, the Dana Foundation, the Foundation for OCD Research, the Hartwell Foundation and the Brain and Behavior Research Foundation (NARSAD).
Footnotes
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41593-023-01259-x.
Code availability
Code packages used are indicated in the Methods. Custom code for the RCCA is included in the Supplementary Information.
Competing interests
C.L. is listed as an inventor for Cornell University patent applications on neuroimaging biomarkers for depression that are pending or in preparation. C.L. has served as a scientific advisor or consultant to Compass Pathways, Delix Therapeutics, Magnus Medical and Brainify. AI. The authors declare no other competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41593-023-01259-x.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41593-023-01259-x.
Data availability
The data that support the findings of this study are publicly available. The neuroimaging datasets are available from ABIDE I and ABIDE II (https://fcon_1000.projects.nitrc.org/indi/abide/) and the the NDAR database (https://nda.nih.gov/). Users must register with the NITRC and 1000 Functional Connectomes Project to gain access to ABIDE I and ABIDE II. Users must be affiliated with a National Institutes of Health (NIH)-recognized research institution that maintains active Federalwide Assurance, be registered on NIH’s eRA Commons and complete and submit a Data Use Certification that is reviewed by the Data Access Committee to gain access to NDAR. The gene expression datasets are available from the AHBA (https://human.brain-map.org/static/download) and BrainSpan (https://www.brainspan.org/static/download.html).
References
- 1.Lombardo MV, Lai M-C & Baron-Cohen S Big data approaches to decomposing heterogeneity across the autism spectrum. Mol. Psychiatry 24, 1435–1450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Insel T. et al. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am. J. Psychiatry 167, 748–751 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Jeste SS & Geschwind DH Disentangling the heterogeneity of autism spectrum disorder through genetic findings. Nat. Rev. Neurol 10, 74–81 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lord C, Elsabbagh M, Baird G & Veenstra-Vanderweele J Autism spectrum disorder. Lancet 392, 508–520 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kana RK, Keller TA, Cherkassky VL, Minshew NJ & Just MA Sentence comprehension in autism: thinking in pictures with decreased functional connectivity. Brain 129, 2484–2493 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koyama MS et al. Resting-state functional connectivity indexes reading competence in children and adults. J. Neurosci 31, 8617–8624 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Green SA, Hernandez L, Bookheimer SY & Dapretto M Salience network connectivity in autism is related to brain and behavioral markers of sensory overresponsivity. J. Am. Acad. Child Adolesc. Psychiatry 55, 618–626 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kana RK, Keller TA, Minshew NJ & Just MA Inhibitory control in high-functioning autism: decreased activation and underconnectivity in inhibition networks. Biol. Psychiatry 62, 198–206 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shafritz KM, Dichter GS, Baranek GT & Belger A The neural circuitry mediating shifts in behavioral response and cognitive set in autism. Biol. Psychiatry 63, 974–980 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martino DA et al. The autism brain imaging data exchange: towards a large-scale evaluation of the intrinsic brain architecture in autism. Mol. Psychiatry 19, 659–667 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martino A et al. Enhancing studies of the connectome in autism using the autism brain imaging data exchange II. Sci. Data 4, 170010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hong S-J, Valk SL, Di Martino A, Milham MP & Bernhardt BC Multidimensional neuroanatomical subtyping of autism spectrum disorder. Cereb. Cortex 28, 3578–3588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yahata N. et al. A small number of abnormal brain connections predicts adult autism spectrum disorder. Nat. Commun 7, 11254 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Easson AK, Fatima Z & R MA Functional connectivity-based subtypes of individuals with and without autism spectrum disorder. Netw. Neurosci 3, 344–362 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Roak BJ et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485, 246–250 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Rubeis S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu JM et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat. Genet 54, 1320–1331 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hashem S. et al. Genetics of structural and functional brain changes in autism spectrum disorder. Transl. Psychiatry 10, 229 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grove J. et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet 51, 431–444 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matoba N. et al. Common genetic risk variants identified in the SPARK cohort support DDHD2 as a candidate risk gene for autism. Transl. Psychiatry 10, 265 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anitha A. et al. Brain region-specific altered expression and association of mitochondria-related genes in autism. Mol. Autism 3, 12 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhubi A. et al. Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Transl. Psychiatry 4, e349 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richiardi J. et al. BRAIN NETWORKS. Correlated gene expression supports synchronous activity in brain networks. Science 348, 1241–1244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romme IAC, de Reus MA, Ophoff RA, Kahn RS & van den Heuvel MP Connectome disconnectivity and cortical gene expression in patients with schizophrenia. Biol. Psychiatry 81, 495–502 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Rafael R-G, Warrier V, Bullmore ET, Simon B-C & Bethlehem RAI Synaptic and transcriptionally downregulated genes are associated with cortical thickness differences in autism. Mol. Psychiatry 24, 1053–1064 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morgan SE et al. Cortical patterning of abnormal morphometric similarity in psychosis is associated with brain expression of schizophrenia-related genes. Proc. Natl. Acad. Sci. USA 116, 9604–9609 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seidlitz J. et al. Transcriptomic and cellular decoding of regional brain vulnerability to neurogenetic disorders. Nat. Commun 11, 3358 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hawrylycz MJ et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature 489, 391–399 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.BrainSpan Atlas of the Developing Human Brain [Internet]. Funded by ARRA Awards 1RC2MH089921-01, 1RC2MH090047-01 and 1RC2MH089929-01. Available from https://brainspan.org/ (2011).
- 30.Satterthwaite TD et al. Impact of in-scanner head motion on multiple measures of functional connectivity: relevance for studies of neurodevelopment in youth. Neuroimage 60, 623–632 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caballero C, Mistry S, Vero J & Torres EB Characterization of noise signatures of involuntary head motion in the autism brain imaging data exchange repository. Front. Integr. Neurosci 12, 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Power JD, Barnes KA, Snyder AZ, Schlaggar BL & Petersen SE Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. Neuroimage 59, 2142–2154 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan C-G, Craddock RC, Zuo X-N, Zang Y-F & Milham MP Standardizing the intrinsic brain: towards robust measurement of inter-individual variation in 1000 functional connectomes. Neuroimage 80, 246–262 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Power JD et al. Functional network organization of the human brain. Neuron 72, 665–678 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grosenick L. et al. Functional and optogenetic approaches to discovering stable subtype-specific circuit mechanisms in depression. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 4, 554–566 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mihalik A, Adams RA & Huys Q Canonical correlation analysis for identifying biotypes of depression. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 5, 478–480 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Nadeau C & Bengio Y Inference for the generalization error. Mach. Learn 52, 239–281 (2003). [Google Scholar]
- 38.Hastie T, Tibshirani R & Friedman J The Elements of Statistical Learning: Data Mining, Inference, and Prediction, 2nd edn. 10.1007/978-0-387-84858-7 (Springer Science & Business Media, 2009). [DOI] [Google Scholar]
- 39.Koyama MS, Molfese PJ, Milham MP, Mencl WE & Pugh KR Thalamus is a common locus of reading, arithmetic, and IQ: analysis of local intrinsic functional properties. Brain Lang. 209, 104835 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Achal S, Hoeft F & Bray S Individual differences in adult reading are associated with left temporo-parietal to dorsal striatal functional connectivity. Cereb. Cortex 26, 4069–4081 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dryburgh E, McKenna S & Rekik I Predicting full-scale and verbal intelligence scores from functional connectomic data in individuals with autism spectrum disorder. Brain Imaging Behav. 14, 1769–1778 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uddin LQ et al. Salience network-based classification and prediction of symptom severity in children with autism. JAMA Psychiatry 70, 869–879 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martino A. et al. Aberrant striatal functional connectivity in children with autism. Biol. Psychiatry 69, 847–856 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cerliani L. et al. Increased functional connectivity between subcortical and cortical resting-state networks in autism spectrum disorder. JAMA Psychiatry 72, 767–777 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sinclair D, Oranje B, Razak KA, Siegel SJ & Schmid S Sensory processing in autism spectrum disorders and Fragile X syndrome—from the clinic to animal models. Neurosci. Biobehav. Rev 76, 235–253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abbott AE et al. Repetitive behaviors in autism are linked to imbalance of corticostriatal connectivity: a functional connectivity MRI study. Soc. Cogn. Affect. Neurosci 13, 32–42 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Supekar K, Ryali S, Mistry P & Menon V Aberrant dynamics of cognitive control and motor circuits predict distinct restricted and repetitive behaviors in children with autism. Nat. Commun 12, 3537 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iversen RK & Lewis C Executive function skills are linked to restricted and repetitive behaviors: three correlational meta analyses. Autism Res. 14, 1163–1185 (2021). [DOI] [PubMed] [Google Scholar]
- 49.Craddock RC, James GA, Holtzheimer PE 3rd, Hu XP & Mayberg HS A whole-brain fMRI atlas generated via spatially constrained spectral clustering. Hum. Brain Mapp 33, 1914–1928 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mennes M. et al. Inter-individual differences in resting-state functional connectivity predict task-induced BOLD activity. Neuroimage 50, 1690–1701 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finn ES et al. Functional connectome fingerprinting: identifying individuals using patterns of brain connectivity. Nat. Neurosci 18, 1664–1671 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seitzman BA et al. Trait-like variants in human functional brain networks. Proc. Natl. Acad. Sci. USA 116, 22851–22861 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zikopoulos B & Barbas H Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front. Hum. Neurosci 7, 609 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maximo JO, Cadena EJ & Kana RK The implications of brain connectivity in the neuropsychology of autism. Neuropsychol. Rev 24, 16–31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Arnatkeviciute A, Fulcher BD & Fornito A A practical guide to linking brain-wide gene expression and neuroimaging data. Neuroimage 189, 353–367 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Vértes PE et al. Gene transcription profiles associated with inter-modular hubs and connection distance in human functional magnetic resonance imaging networks. Philos. Trans. R. Soc. Lond. B Biol. Sci 371, 20150362 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Webber W, Moffat A & Zobel J A similarity measure for indefinite rankings. ACM Trans. Inf. Syst. Secur 28, 1–38 (2010). [Google Scholar]
- 58.Korotkevich G. et al. Fast gene set enrichment analysis. Preprint at bioRxiv 10.1101/060012 (2016). [DOI] [Google Scholar]
- 59.Enstrom AM, Van de Water JA & Ashwood P Autoimmunity in autism. Curr. Opin. Investig. Drugs 10, 463–473 (2009). [PMC free article] [PubMed] [Google Scholar]
- 60.Mannion A & Leader G An investigation of comorbid psychological disorders, sleep problems, gastrointestinal symptoms and epilepsy in children and adolescents with autism spectrum disorder: a two-year follow-up. Res. Autism Spectr. Disord 22, 20–33 (2016). [Google Scholar]
- 61.Pfenning AR et al. Convergent transcriptional specializations in the brains of humans and song-learning birds. Science 346, 1256846 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mi H, Muruganujan A, Ebert D, Huang X & Thomas PD PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 47, D419–D426 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suzuki K. et al. Microglial activation in young adults with autism spectrum disorder. JAMA Psychiatry 70, 49–58 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Zhan Y. et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat. Neurosci 17, 400–406 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Darnell JC et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Porokhovnik L. Individual copy number of ribosomal genes as a factor of mental retardation and autism risk and severity. Cells 8, 1151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lombardo MV Ribosomal protein genes in post-mortem cortical tissue and iPSC-derived neural progenitor cells are commonly upregulated in expression in autism. Mol. Psychiatry 26, 1432–1435 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rebholz-Schuhmann D, Oellrich A & Hoehndorf R Text-mining solutions for biomedical research: enabling integrative biology. Nat. Rev. Genet 13, 829–839 (2012). [DOI] [PubMed] [Google Scholar]
- 69.Nozari N & Thompson-Schill SL Chapter 46 - left ventrolateral prefrontal cortex in processing of words and sentences. in Neurobiology of Language (eds. Hickok G & Small SL) 569–584 10.1016/B978-0-12-407794-2.00046-8 (Academic Press, 2016). [DOI] [Google Scholar]
- 70.Antunes FM & Malmierca MS Corticothalamic pathways in auditory processing: recent advances and insights from other sensory systems. Front. Neural Circuits 15, 721186 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gonzalez-Gadea ML et al. Predictive coding in autism spectrum disorder and attention deficit hyperactivity disorder. J. Neurophysiol 114, 2625–2636 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van Laarhoven T, Stekelenburg JJ, Eussen ML & Vroomen J Atypical visual-auditory predictive coding in autism spectrum disorder: electrophysiological evidence from stimulus omissions. Autism 24, 1849–1859 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Menegaux A. et al. Aberrant cortico-thalamic structural connectivity in premature-born adults. Cortex 141, 347–362 (2021). [DOI] [PubMed] [Google Scholar]
- 74.Crump C, Sundquist J & Sundquist K Preterm or early term birth and risk of autism. Pediatrics 148, e2020032300 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Happé F & Ronald A The “fractionable autism triad”: a review of evidence from behavioural, genetic, cognitive and neural research. Neuropsychol. Rev 18, 287–304 (2008). [DOI] [PubMed] [Google Scholar]
- 76.Georgiades S. et al. Investigating phenotypic heterogeneity in children with autism spectrum disorder: a factor mixture modeling approach. J. Child Psychol. Psychiatry 54, 206–215 (2013). [DOI] [PubMed] [Google Scholar]
- 77.Bertelsen N. et al. Imbalanced social-communicative and restricted repetitive behavior subtypes of autism spectrum disorder exhibit different neural circuitry. Commun. Biol 4, 574 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fuccillo MV Striatal circuits as a common node for autism pathophysiology. Front. Neurosci 10, 27 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chugani DC et al. Efficacy of low-dose buspirone for restricted and repetitive behavior in young children with autism spectrum disorder: a randomized trial. J. Pediatr 170, 45–53 (2016). [DOI] [PubMed] [Google Scholar]
- 80.Dunn JT, Mroczek J, Patel HR & Ragozzino ME Tandospirone, a partial 5-HT1A receptor agonist, administered systemically or into anterior cingulate attenuates repetitive behaviors in Shank3b mice. Int. J. Neuropsychopharmacol. 23, 533–542 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yahya SM, Gebril O, Abdel Raouf ER & Elhadidy ME A preliminary investigation of HTR1A gene expression levels in autism spectrum disorders. Int. J. Pharm. Pharm. Sci 11, 1–3 (2019). [Google Scholar]
- 82.Kieran N, Ou X-M & Iyo AH Chronic social defeat downregulates the 5-HT1A receptor but not Freud-1 or NUDR in the rat prefrontal cortex. Neurosci. Lett 469, 380–384 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dölen G, Darvishzadeh A, Huang KW & Malenka RC Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature 501, 179–184 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kohls G, Yerys BE & Schultz RT Striatal development in autism: repetitive behaviors and the reward circuitry. Biol. Psychiatry 76, 358–359 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Langen M. et al. Changes in the development of striatum are involved in repetitive behavior in autism. Biol. Psychiatry 76, 405–411 (2014). [DOI] [PubMed] [Google Scholar]
- 86.Wilkes BJ & Lewis MH The neural circuitry of restricted repetitive behavior: magnetic resonance imaging in neurodevelopmental disorders and animal models. Neurosci. Biobehav. Rev 92, 152–171 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dickie EW et al. Personalized intrinsic network topography mapping and functional connectivity deficits in autism spectrum disorder. Biol. Psychiatry 84, 278–286 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Geschwind DH & Levitt P Autism spectrum disorders: developmental disconnection syndromes. Curr. Opin. Neurobiol 17, 103–111 (2007). [DOI] [PubMed] [Google Scholar]
- 89.Zuo X-N et al. Growing together and growing apart: regional and sex differences in the lifespan developmental trajectories of functional homotopy. J. Neurosci 30, 15034–15043 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gee DG et al. A developmental shift from positive to negative connectivity in human amygdala-prefrontal circuitry. J. Neurosci 33, 4584–4593 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Menon V. Developmental pathways to functional brain networks: emerging principles. Trends Cogn. Sci 17, 627–640 (2013). [DOI] [PubMed] [Google Scholar]
- 92.Fulcher BD, Arnatkeviciute A & Fornito A Overcoming false-positive gene-category enrichment in the analysis of spatially resolved transcriptomic brain atlas data. Nat. Commun 12, 2669 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arnatkeviciute A. et al. Genetic influences on hub connectivity of the human connectome. Nat. Commun 12, 4237 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Smith SM et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 23, S208–S219 (2004). [DOI] [PubMed] [Google Scholar]
- 95.Cox RW AFNI: Software for analysis and visualization of functional magnetic resonance neuroimages. Comput. Biomed. Res 29, 162–173 (1996). [DOI] [PubMed] [Google Scholar]
- 96.Smith SM Fast robust automated brain extraction. Hum. Brain Mapp 17, 143–155 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jenkinson M, Bannister P, Brady M & Smith S Improved optimization for the robust and accurate linear registration and motion correction of brain images. Neuroimage 17, 825–841 (2002). [DOI] [PubMed] [Google Scholar]
- 98.Jenkinson M & Smith S A global optimisation method for robust affine registration of brain images. Med. Image Anal 5, 143–156 (2001). [DOI] [PubMed] [Google Scholar]
- 99.Collins LD, Holmes CJ, Peters TM & Evans AC Automatic 3D model-based neuroanatomical segmentation. Hum. Brain Mapp 3, 190–208 (1995). [Google Scholar]
- 100.Mazziotta J. et al. A probabilistic atlas and reference system for the human brain: International Consortium for Brain Mapping (ICBM). Philos. Trans. R. Soc. Lond. B Biol. Sci 356, 1293–1322 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Andersson JLR, Jenkinson M, Smith S & Andersson J Non-linear registration, aka spatial normalisation. FMRIB Technial Report TR07JA2. https://www.fmrib.ox.ac.uk/datasets/techrep/tr07ja2/tr07ja2.pdf (2007). [Google Scholar]
- 102.Jo HJ, Saad ZS, Simmons WK, Milbury LA & Cox RW Mapping sources of correlation in resting-state fMRI, with artifact detection and removal. Neuroimage 52, 571–582 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Murphy K, Bodurka J & Bandettini PA How long to scan? The relationship between fMRI temporal signal to noise ratio and necessary scan duration. Neuroimage 34, 565–574 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gotham K, Pickles A & Lord C Standardizing ADOS scores for a measure of severity in autism spectrum disorders. J. Autism Dev. Disord 39, 693–705 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hus V & Lord C The autism diagnostic observation schedule, module 4: revised algorithm and standardized severity scores. J. Autism Dev. Disord 44, 1996–2012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zou H & Hastie T Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B Stat. Methodol 67, 301–320 (2005). [Google Scholar]
- 107.Grosenick L, Klingenberg B, Katovich K, Knutson B & Taylor JE Interpretable whole-brain prediction analysis with GraphNet. Neuroimage 72, 304–321 (2013). [DOI] [PubMed] [Google Scholar]
- 108.Friedman JH Regularized discriminant analysis. J. Am. Stat. Assoc 84, 165–175 (1989). [Google Scholar]
- 109.Tibshirani R, Hastie T, Narasimhan B & Chu G Class prediction by nearest shrunken centroids, with applications to DNA microarrays. Stat. Sci 18, 104–117 (2003). [Google Scholar]
- 110.Robert P & Escoufier Y A unifying tool for linear multivariate statistical methods: the RV-coefficient. J. R. Stat. Soc. Ser. C. Appl. Stat 25, 257–265 (1976). [Google Scholar]
- 111.de Torrenté L & Hastie T Does cross-validation work when p≫n? https://hastie.su.domains/Papers/does_cross-validation_work.pdf (2012).
- 112.Allen Institute for Brain Science. Allen Human Brain Atlas. Available from: http://human.brain-map.org
- 113.Velmeshev D et al. Single-cell genomics identifies cell type-specific molecular changes in autism. Science 364, 685–689 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Voineagu I et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Parikshak NN et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sanders SJ et al. A framework for the investigation of rare genetic disorders in neuropsychiatry. Nat. Med 25, 1477–1487 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.SPARK Consortium. SPARK: a US Cohort of 50,000 families to accelerate autism research. Neuron 97, 488–493 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Steinberg J & Webber C The roles of FMRP-regulated genes in autism spectrum disorder: single- and multiple-hit genetic etiologies. Am. J. Hum. Genet 93, 825–839 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Parikshak NN et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nair RP et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-κB pathways. Nat. Genet 41, 199–204 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Abrahams BS et al. SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 4, 36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Davis AP et al. The comparative toxicogenomics database: update 2019. Nucleic Acids Res. 47, D948–D954 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pua CJ et al. Development of a comprehensive sequencing assay for inherited cardiac condition genes. J. Cardiovasc. Transl. Res 9, 3–11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pletscher-Frankild S, Pallejà A, Tsafou K, Binder JX & Jensen LJ DISEASES: text mining and data integration of disease-gene associations. Methods 74, 83–89 (2015). [DOI] [PubMed] [Google Scholar]
- 125.Shimoyama M et al. The Rat Genome Database 2015: genomic, phenotypic and environmental variations and disease. Nucleic Acids Res. 43, D743–D750 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 47, D330–D338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ashburner M et al. Gene Ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Xia J, Benner MJ & Hancock REW NetworkAnalyst—integrative approaches for protein-protein interaction network analysis and visual exploration. Nucleic Acids Res. 42, W167–W174 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xia J, Gill EE & Hancock REW NetworkAnalyst for statistical, visual and network-based meta-analysis of gene expression data. Nat. Protoc 10, 823–844 (2015). [DOI] [PubMed] [Google Scholar]
- 130.Zhou G et al. NetworkAnalyst 3.0: a visual analytics platform for comprehensive gene expression profiling and meta-analysis. Nucleic Acids Res. 47, W234–W241 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Szklarczyk D et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43, D447–D452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Banerjee-Basu S & Packer A SFARI Gene: an evolving database for the autism research community. Dis. Models Mech 3, 133–135 (2010). [DOI] [PubMed] [Google Scholar]
- 133.Müller H-M, Kenny EE & Sternberg PW Textpresso: an ontology-based information retrieval and extraction system for biological literature. PLoS Biol. 2, e309 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jensen LJ, Saric J & Bork P Literature mining for the biologist: from information retrieval to biological discovery. Nat. Rev. Genet 7, 119–129 (2006). [DOI] [PubMed] [Google Scholar]
- 135.Singhal A, Simmons M & Lu Z Text mining genotype—phenotype relationships from biomedical literature for database curation and precision medicine. PLoS Comput. Biol 12, e1005017 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wei C-H, Kao H-Y & Lu Z PubTator: a web-based text mining tool for assisting biocuration. Nucleic Acids Res. 41, W518–W522 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wei C-H, Allot A, Leaman R & Lu Z PubTator central: automated concept annotation for biomedical full text articles. Nucleic Acids Res. 47, W587–W593 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Feinerer I, Hornik K & Meyer D Text mining infrastructure in R. J. Stat. Softw. Artic 25, 1–54 (2008). [Google Scholar]
- 139.Benoit K et al. quanteda: an R package for the quantitative analysis of textual data. J. Open Source Softw 3, 774 (2018). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are publicly available. The neuroimaging datasets are available from ABIDE I and ABIDE II (https://fcon_1000.projects.nitrc.org/indi/abide/) and the the NDAR database (https://nda.nih.gov/). Users must register with the NITRC and 1000 Functional Connectomes Project to gain access to ABIDE I and ABIDE II. Users must be affiliated with a National Institutes of Health (NIH)-recognized research institution that maintains active Federalwide Assurance, be registered on NIH’s eRA Commons and complete and submit a Data Use Certification that is reviewed by the Data Access Committee to gain access to NDAR. The gene expression datasets are available from the AHBA (https://human.brain-map.org/static/download) and BrainSpan (https://www.brainspan.org/static/download.html).